DOI:
10.1039/D4RA03041A
(Paper)
RSC Adv., 2024,
14, 22195-22208
Peroxymonosulfate activation by cobalt-doped ferromanganese magnetic oxides through singlet oxygen and radical pathways for efficient sulfadiazine degradation†
Received
24th April 2024
, Accepted 9th July 2024
First published on 15th July 2024
Abstract
In this paper, cobalt-doped MnFe2O4 (CMFO-0.4) with oxygen vacancies was successfully synthesised by the sol–gel method and applied as a high-performance catalyst for the activation of peroxomonosulfate (PMS). The catalyst showed an excellent catalytic effect for the degradation of sulfadiazine (SDZ) by activated PMS, and the degradation rate can reach 100% in 10 minutes. The effects of different conditions on the degradation of SDZ were investigated, and it was determined that the optimal concentrations of catalyst and PMS were 0.2 g L−1 and 1 mM, respectively, and had good degradation effects in the pH 5–11 range. Free radical quenching experiments, XPS, and electron paramagnetic resonance (EPR) analyses revealed the presence of hydroxyl radicals (˙OH), sulphate radicals (SO4˙−), singlet oxygen (1O2), and superoxide radicals (˙O2−) in the CMFO-0.4/PMS system, with 1O2 being the main reactive oxygen species (ROS). In addition, CMFO-0.4 has good reusability and adaptability to the presence of other substances.
1. Introduction
The utilization of sulfonamides (SAs) in poultry and aquaculture is prevalent, mostly driven by their affordability, with a particular emphasis on the application of sulfadiazine (SDZ).1 The presence of SDZ has been observed in several aquatic ecosystems, such as river water, groundwater, and wastewater treatment facilities, exhibiting amounts ranging from ppb to ppm.2 The biodegradability of SDZ is low and exhibits high stability.3 Consequently, the accumulation of SDZ residues in the environment surpasses the natural decomposition capacity, resulting in the proliferation of drug-resistant genes and posing a significant risk to bacterial communities. Therefore, there is an immediate requirement for a novel technology to treat wastewater containing SDZ efficiently.
The efficacy of conventional advanced oxidation techniques (AOPs) in the elimination of developing pollutants has been demonstrated, mostly through the action of hydroxyl radicals (˙OH). The ˙OH exhibits a redox potential of 1.8–2.7 V and possesses the capability to disrupt the molecular arrangement of organic compounds.4 Recently, the sulphate radical advanced oxidation technologies (SR-AOPs) have garnered considerable attention for their effectiveness in addressing challenging organic pollution treatment.5 SR-AOPs involve using different methods to catalytically activate persulfate, resulting in the production of sulfate radicals (SO4˙−) with a standard reduction potential (E0) ranging from 2.5 to 3.1 volts. They have high oxidative activity with a long half-life, a wide range of pH adaptability6 and selective oxidation properties.7 Furthermore, SR-AOPs can produce reactive substances including ˙OH, ˙O2− and 1O2. These reactive substances exhibit significant efficacy in the degradation of antibiotics present in water.8 Numerous techniques exist for inducing the activation of PMS to generate SO4˙−, such as ultrasonic activation,9 UV activation,10 thermal,11 homogeneous12 and non-homogeneous phase catalysts.13 Compared with alternative catalytic approaches, non-homogeneous phase catalysis demonstrates significant advantages in the process of SR-AOPs. These advantages include facile separation and reusability, reduced secondary pollutants, and broad flexibility to various reaction conditions.14 Among the applied non-homogeneous catalysts, iron, copper, cobalt, manganese, nickel, and zinc, along with their respective oxides, are widely recognized as efficient activators for PMS.15
In recent times, the compound MnFe2O4 has attracted considerable interest as a potential catalyst for the activation of PMS owing to its exceptional stability and magnetic characteristics.16,17 Nevertheless, there is still a need for further enhancement in the catalytic effectiveness of MnFe2O4.8 Hence, it is imperative to research efficacious approaches aimed at enhancing the catalytic activity of MnFe2O4. Numerous approaches have been investigated to improve the catalytic efficiency of metal catalysts including the incorporation of specific cations by doping, the incorporation of other materials through compounding, and the alteration of the morphology and crystal phases.18 For instance, Wang19 successfully synthesized g-C3N4/MnFe2O4 using a self-combustion sol–gel technique, resulting in the production of a recyclable catalyst for the breakdown of silica chloroform. Remarkably, silica chloroform was completely decomposed in 60 minutes. Recently the doping of transition metal ions has received much attention due to its advantages of simplicity and effectiveness. For example, Sun20 degraded tetracycline hydrochloride with Cu-doped MnFe2O4 magnetic ferrite with excellent removal effect even when the pH was extended to 11, revealing that the catalytic activity was improved because of the synergistic impact observed among the Fe, Mn, and Cu ions. Lu prepared magnetic MnFe2O4/ZIF-67 nanocomposites, which were used to activate PMS to efficiently degrade tetracycline hydrochloride (TCH), and 82.6% of TCH was removed within 15 minutes.21 Based on this, it can be inferred that more active metals can be introduced by doping Co to activate PMS, and at the same time, different metal ion radii in spinel structures may lead to some defects and distortions, thus generating oxygen vacancies to further improve the catalytic activity of MnFe2O4.
In this paper, a magnetic Co-doped MnFe2O4 (CMFO-0.4) catalyst enriched with OVs was prepared by the sol–gel method and used as an activator for PMS. The catalytic activity of CMFO-0.4 for PMS was evaluated by selecting SDZ as the target contaminant. The crystal structure, physical shape and chemical composition of the synthesised catalysts were analysed. To evaluate the catalytic efficiency of CMFO-0.4, this study further delved into the effect of each variable on the degradation process. Repeat experiments were also conducted to evaluate the durability and reusability of the catalyst. Furthermore, the intermediates involved in the degradation process were identified by liquid chromatography-mass spectrometry (LC-MS) and the degradation pathways of SDZ were analysed and determined. The active chemicals involved in the reaction were investigated by quenching experiments and EPR analysis, and a rational catalytic mechanism was proposed. The reproductive toxicity, developmental toxicity and teratogenicity of the degraded intermediates were analysed by T.E.S.T. technique.
2. Materials and methods
2.1. Chemicals and reagents
The compounds Mn(NO3)2·4H2O, Co(NO3)2·6H2O, Fe(NO3)3·9H2O, humic acid (HA), formic acid, furfuryl alcohol (FFA), methanol (MeOH), p-benzoquinone (p-BQ), and acetonitrile (C2H3N) were procured from Shanghai Maclin Biochemical Technology Co., Ltd. The following chemicals were procured from National Medicine Group Chemical Reagent Co., Ltd: hydrochloric acid (HCl), anhydrous ethanol (EtOH), sulfuric acid (H2SO4), sodium hydroxide (NaOH), nitric acid (HNO3), and PMS (2KHSO5·KHSO4·K2SO4, 4.5% active oxygen). The aforementioned reagents possess a level of purity that is either analytically pure or exceeds it.
2.2. Preparation of catalysts
Firstly, weighed amounts of Co(NO3)2·6H2O, Mn(NO3)2·4H2O, and Fe(NO3)3·9H2O were dissolved in 50 mL of purified water. 30 mmol of citric acid was added, and the mixture was sonicated and stirred at 60 °C for 2 hours. The solution was then heated to evaporate the solvent until it reached a gel-like state, followed by further evaporation in an oven at 105 °C. The brownish-yellow solid material was pulverized into powder, subjected to a temperature gradient of 5 °C min−1 in a tube furnace until reaching 400 °C, heated in a nitrogen atmosphere for 1 hour, and subsequently allowed to cool to room temperature. The resulting catalyst was denoted as CMFO-x (x = Co/Fe), and various Co/Fe materials were synthesized by adjusting the quantity of Co(NO3)2·6H2O introduced. MFO was obtained without adding Co(NO3)2·6H2O, CFO was obtained without adding Mn(NO3)2·4H2O, and CMO was obtained without adding Fe(NO3)3·9H2O.
2.3. Characteristics of catalysts
The comprehensive analysis of the prepared materials can be found in Text S1.†
2.4. Experimental procedures
All degradation studies were carried out in a 200 mL reactor at a controlled temperature of 25 ± 1 °C. To evaluate the performance of catalyst activated PMS for SDZ degradation, a certain amount of CMFO-x powder was added to 100 mL of SDZ solution, sonicated for 1 min, and stirred for 10 min to bring the mixture to an equilibrium state of adsorption and desorption and then the reaction was initiated by rapid addition of PMS. At predetermined time intervals, a 0.5 mL sample of the SDZ reaction solution was withdrawn from the degradation system at a time and quickly transferred to a centrifuge tube previously spiked with 0.5 mL of methanol (to prevent the reaction from continuing). The SDZ content was determined by high-performance liquid chromatography after filtration. At the end of the reaction, the CMFO-x powder was recovered by an external magnetic field and subsequently washed several times using ultrapure water. The samples were subjected to a drying process at 60 °C for recovery tests. In this study, the effects of PMS concentration, catalyst dosage, solution pH and initial SDZ concentration on SDZ degradation and PMS activation were investigated using a controlled variable technique.
Furthermore, to examine the degradation mechanism of SDZ, a total of six sets of quenching experiments were conducted to analyze the generation of free radicals in the CMFO-0.4/PMS/SDZ system. These experiments utilized TBA, EtOH, FFA, and p-BQ as the scavengers in separate trials. The quantification of ROS in the degrading system was conducted through the utilization of EPR, as indicated in Text S2.†
2.5. Chemical analysis
Analysis methods for SDZ concentration, identification of intermediate products that may be produced during SDZ degradation, metal leaching, and determination of total organic carbon (TOC) are shown in Text S3.†
3. Results and discussion
3.1. Physical and chemical properties of catalysts
This study involved the production of several catalysts through the introduction of varying amounts of cobalt (Co). Fig. 1a displays the XRD spectra of MFO, CFO, CMO, and samples with varying Co/Fe molar ratios (CMFO-x). The diffraction pattern of MFO displays five different characteristic peaks at specific angles: 30.1°, 35.3°, 43.2°, 57.2°, and 62.6°, corresponding to the (220), (311), (400), (511), and (440) crystallographic planes, respectively.22 The introduction of Co causes the above peaks to change weaker sharpness, lower peak intensity and wider peak width, which may be due to the proximity of the MFO lattice parameters to the CFO's.23 CMFO-0.3 exhibits inferior crystalline morphology compared to CMFO-0.4 and CMFO-0.5, likely attributed to the reduced Co incorporation and weaker binding to Fe.
 |
| | Fig. 1 XRD patterns of the as-prepared catalysts. | |
The determination of the specific surface area, pore volume, and pore size of CMFO-0.4 and MFO was conducted through the utilization of N2 adsorption–desorption isotherms, as depicted in Fig. 2b and Table 1. The isotherms exhibited type IV adsorption branching and H3 hysteresis return lines within the pressure range of 0 to 1.0 (P/P0).8 Additionally, the pore size distributions, measured using the BJH method, revealed the existence of mesopores in both materials. The specific surface area of CMFO-0.4 decreased after Co-doped, and the pore size and volume of CMFO-0.4 were larger than that of MFO. These results show that codoping improves the mesoporous properties of the catalyst. The magnetic properties of the catalysts MFO and CMFO-0.4 were evaluated using a vibrating sample magnetometer (VSM) as shown in Fig. 2b and Table S1.† The coercivities of MFO and CMFO-0.4 were 32.9414 Oe and 1236.99 Oe, respectively. The higher coercivity of the material indicates stronger magnetism,24 which indicates that CMFO-0.4 is more magnetic than MFO. These results suggest that the catalyst CMFO-0.4 exhibits superior magnetic characteristics, which enhance its efficiency in the process of separation for subsequent reuse (Fig. 2b inset).25
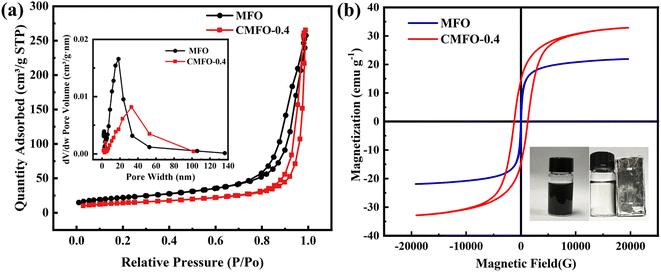 |
| | Fig. 2 (a) N2 physisorption isotherm and pore size distribution (inset), (b) Magnetic hysteresis loops of MFO and CMFO-0.4. The inset in Fig. 2(b) shows the magnetic separation process of the CMFO-0.4. | |
Table 1 Specific surface area, pore size and pore volume of MFO and CMFO-0.4
| |
Surface area (m2 g−1) |
Pore size (nm) |
Pore volume (cm3 g−1) |
| MFO |
78.8956 |
20.2092 |
0.398519 |
| CMFO-0.4 |
50.8630 |
30.8090 |
0.410463 |
The SEM images of CMFO-0.4 (Fig. 3a–c) showed that the catalyst particles appeared as spherical nanoparticles interspersed with octahedral spinels, with spherical particle sizes of 30–50 nm. MFO was mainly in the form of nanoparticle clusters (Fig. S1a†), the morphology of the catalysts changed with the introduction of Co. CMFO-0.3 (Fig. S1b†) and CMFO-0.5 (Fig. S1c†) showed smaller nanoparticle sizes but larger degree of agglomeration compared to MFO (Fig. S1a†), and it was inferred that the introduction of Co led to the enhancement of the catalysts' magnetic properties. The morphological SEM elemental mapping of CMFO-0.4 (Fig. 3d–g) confirms that the catalyst mainly consists of four elements, namely, Mn, Fe, Co, and O and each element is uniformly distributed throughout the composite CMFO-0.4. The content of each element can be obtained from Fig. 3i.
 |
| | Fig. 3 (a–c) SEM images of CMFO-0.4, (d–h) corresponding elemental mapping of Co, Mn, Fe and O, (i) element content map based on EDS data. | |
The surface composition of CMFO-0.4 and CMFO-0.4 after use was analyzed using XPS, and the corresponding findings are presented in Fig. 4. The C1s peak at 284.8 eV was employed as the established reference point, and all binding energy estimates were then calibrated accordingly. The XPS spectrum of CMFO-0.4 (Fig. 4a) exhibits distinct peaks corresponding to the Co 2p, Fe 2p, Mn 2p, and O1s energy levels, confirming the coexistence of the elements Co, Mn, Fe and O, in agreement with the EDX results (Fig. 3i). The energy values of the Mn 2p3/2 and Mn 2p1/2 peaks are 641 eV and 653.07 eV, respectively (Fig. 4b). A difference of 12.07 eV in spin–orbit binding energy was discovered between the two peaks. The Mn 2p3/2 orbital exhibits three distinct peaks in the Mn2p spectrum, with binding energies measured at 640.59 eV, 642.36 eV, and 645.45 eV. These peaks correspond to the presence of Mn(II), Mn(III), and Mn(IV), respectively, as reported in ref. 26. The composition of unused CMFO-0.4 consisted of 45.94% Mn(II), 36.46% Mn(III), and 17.61% Mn(IV).
 |
| | Fig. 4 (a) X-ray photoelectron spectroscopy (XPS) survey, (b) Mn 2p XPS spectra, (c) Co 2p XPS spectra, (d) Fe 2p XPS spectra, (e) O 1s XPS spectra of CMFO-0.4 and (f) EPR spectra of CMFO-0.4 before and after the reaction. | |
Fig. 4c shows the two major peaks of Co 2p3/2 at binding energies 780.60 eV and 783.50 eV (Co(III) and Co(II)) and the two major peaks of Co 2p1/2 at binding energies 796.12 and 798.50 eV (Co(III) and Co(II)). Additionally, two distinct satellite peaks corresponding to the Co 2p3/2 energy level are detected with binding energies of 786.80 eV and 802.97 eV. According to prior research,27 the spectral peaks observed at 780.60 eV and 796.12 eV can be attributed to octahedral and tetrahedral Co(II) species. Similarly, the peaks observed at 783.50 eV and 798.50 eV correspond to octahedral and tetrahedral Co(II) species, respectively. The distribution of Co(II) and Co(II) in the unused CMFO-0.4 was found to be 56.2% and 43.8%, respectively.
Fig. 4d displays distinct peaks at energy levels of 724.6 eV, 717.38 eV, and about 711.1 eV, which can be attributed to Fe 2p1/2, the satellite peaks, and Fe 2p3/2, respectively.28 The spectra of Fe 2p3/2 exhibit three distinct peaks, these peaks are observed at 710.20 eV, 711.60 eV, and about 713.60 eV, and can be attributed to octahedral Fe(II) (Otc. Fe2+), octahedral Fe(III) (Otc. Fe3+), and tetrahedral Fe(III) (Tet. Fe3+) coordination environments, respectively.18 The proportions of Otc. Fe2+, Otc. Fe3+ and Tet. Fe3+ in unused CMFO-0.4 were 28.56%, 29.81% and 41.63%, respectively.
The O1s peaks observed in Fig. 4e exhibit binding energies of 529.70 eV, 531.30 eV, and 532.50 eV, which can be attributed to lattice oxygen (Olatt), oxygen vacancy (OVs) and other oxygen species Oother (hydroxyl oxygen or loose-surface adsorbed oxygen species).29 The ratio of the three is 17.36%, 62.39%, 20.24%. It can be seen that the doping of Co in MnFe2O4 will release its lattice oxygen, thus promoting the formation of oxygen vacancy in CMFO-0.4. OVs will act as electron reservoirs for PMS activation, thus facilitating the transfer of electrons from PMS to the surface of the catalyst, which will make the electrons more active during the reaction. Therefore, the higher content of OVs in the catalyst is more favourable to the oxidation reaction.30 To verify the conclusions about OVs in XPS, the results were subjected to electron paramagnetic resonance (EPR) testing. As shown in Fig. 4f, the CMFO-0.4 catalyst showed a distinct characteristic peak at the g = 2.003 position, which is since this is due to the unpaired electrons being trapped on the surface oxygen vacancies resulting in the characteristic peak of the OVs.31 The EPR results confirmed the presence of oxygen vacancies in CMFO-0.4, which is in agreement with the XPS results.
Fig. S3† displays the spectrum of the catalyst as obtained using FTIR. The spectral peaks seen at wavenumbers 3420 cm−1 can be attributed to the presence of the functional groups O–H.28 The peak observed at a wavenumber of 1560 cm−1 can be ascribed to the tensile vibration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond that arises during the process of carbonization of the material.32 The C–O stretching vibration is detected at approximately 1320 cm−1. The peaks observed at around 565 cm−1 and 495 cm−1 can be ascribed to the stretching of metallic oxygen, providing evidence for the presence of Fe–O/Co–O and Mn–O compounds.16,28
C bond that arises during the process of carbonization of the material.32 The C–O stretching vibration is detected at approximately 1320 cm−1. The peaks observed at around 565 cm−1 and 495 cm−1 can be ascribed to the stretching of metallic oxygen, providing evidence for the presence of Fe–O/Co–O and Mn–O compounds.16,28
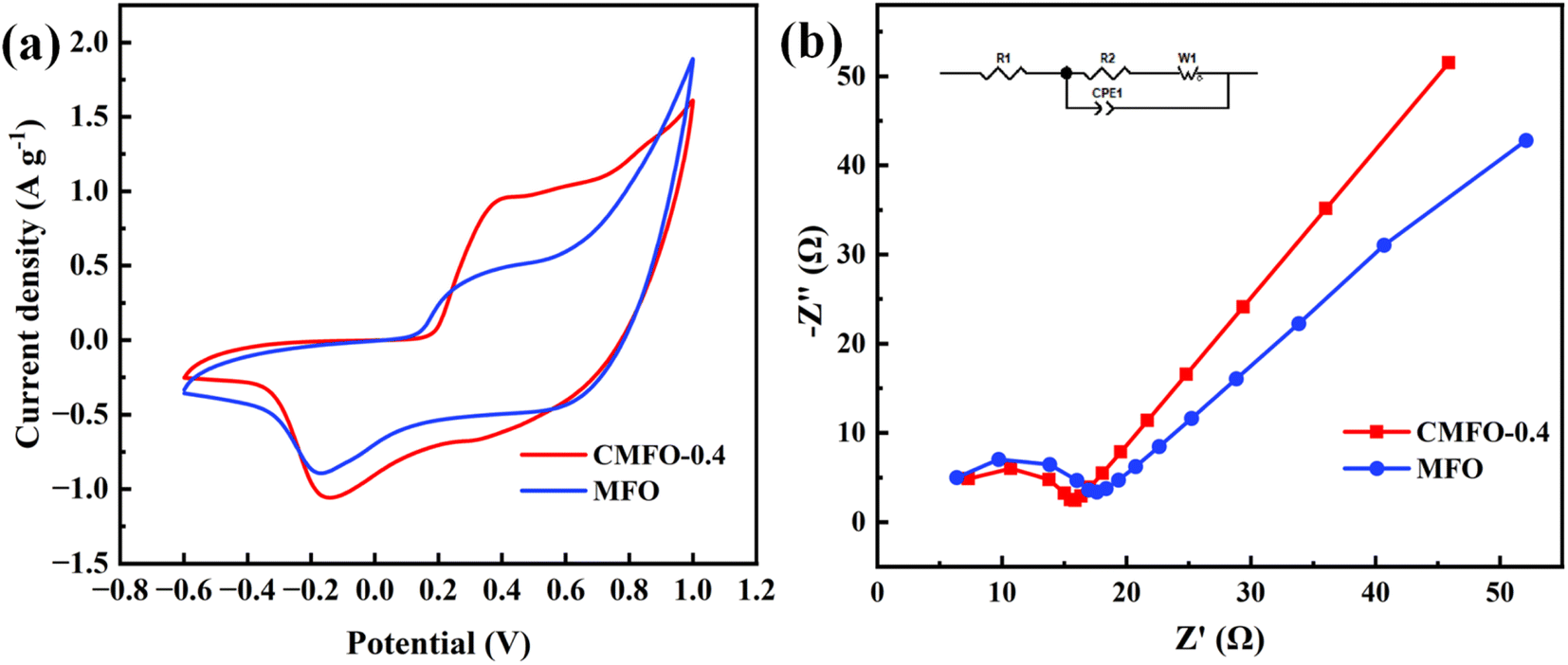
An electrochemical assessment was carried out to examine the transfer charge capability and redox potential of the catalyst. According to the data presented in Fig. 5a, it can be observed that CMFO-0.4 exhibits a higher redox capacity compared to MFO.33 In Fig. 5b, it is evident that CMFO-0.4 presents a smaller semicircle than MFO. In the domain of high frequencies of the EIS Nyquist plot, CMFO-0.4 exhibits smaller resistance and higher conductivity in the non-homogeneous phase reaction, which is favourable for charge transfer for PMS activation.34
 |
| | Fig. 5 Comparison between CMFO-0.4 and MFO: (a) CV curve at a scanning rate of 100 mV s−1; (b) EIS Nyquist maps. | |
3.2. Catalytic properties of the degradation of SDZ
In different systems, the removal effect of the prepared catalyst as a PMS activator to catalyze the degradation of SDZ was evaluated. Fig. 6a shows the degradation effect of SDZ in different systems. The effects of the autooxidation of PMS and the self-adsorption of catalyst on the degradation of SDZ were preliminarily investigated. It can be seen that the removal efficiency of SDZ is less than 7% and 5%, respectively, indicating that the effects of the two on the degradation of SDZ are small and can be ignored. In MFO/PMS reaction system, the degradation rate of SDZ within 16 min is only 23.37%, in CMO/PMS and CFO/PMS reaction system, the degradation rate of SDZ within 16 min is 79.07% and 98.36%, respectively. When CMFO-0.4 is used as a catalyst, the degradation rate of SDZ can reach 100% within 10 min, and the reaction kinetic constants are increased 23.39 times, 4.03 times and 1.65 times, respectively, which conforms to the quasi-first-order reaction kinetics. Therefore, the ability of CMFO-0.4 to activate PMS to degrade SDZ is much higher than that of MFO, CMO and CFO.
 |
| | Fig. 6 (a) The degradation rate of SDZ in different systems, (b) reaction rate constants of different reaction systems. Reaction conditions: [catalyst] = 0.2 g L−1, [PMS] = 1.0 mM, [SDZ] = 20 mg L−1, pH = 5.6. | |
In addition, the influence of the Co doping ratio (Co/Fe = 0.3, 0.4, 0.5) on the degradation effect of SDZ was also studied. It can be seen from Fig. 6a that the increase of Co doping has a significant positive effect on the removal rate of SDZ, but CMFO-0.4 and CMFO-0.5 both have a good degradation effect on SDZ. The reaction can reach 100% within 10 minutes. To save cost, we chose CMFO-0.4 to continue the follow-up experiment. In addition, to clarify the excellent catalytic performance of CMFO-0.4, the effect of CMFO-0.4 on activating PMS was compared with that of other reported catalysts, and the results were shown in Table S2,† which demonstrates that the reaction rate constants of CMFO-0.4 are significantly greater than those seen in other studies. The results indicate that CMFO-0.4 is an efficient catalyst for PMS activation.
3.3. Influence of several key parameters
In order to investigate the catalyst's catalytic performance, the effects of PMS and catalyst dosing on the SDZ removal rate of the CMFO-0.4/PMS system were investigated. The experimental results in Fig. 7a show that the degradation rate of SDZ was 88.89% and 97.3% when the PMS was 0.5 and 0.7 mM, respectively, which was due to insufficient PMS. Increasing the amount of PMS to 1 and 1.25 mM, the degradation rate of SDZ could reach 100% in both cases due to the sufficiency of active substances. The catalytic activation of PMS raises the production of active species, which in turn accelerates the breakdown rate of SDZ as the concentration of PMS rises.35 Furthermore, Fig. 7b demonstrates a positive association between the degradation rate of SDZ and the dosage of the catalyst. The correlation between this connection can be ascribed to a sufficient quantity of active sites on the catalyst, which facilitates the activation of PMS to produce free radicals. The involvement of free radicals is essential for the successful degradation of SDZ. For the ensuing tests, a catalyst dosage of 0.1 g L−1 and a PMS dosage of 1 mM was selected to enable the examination of the impacts of additional parameters.
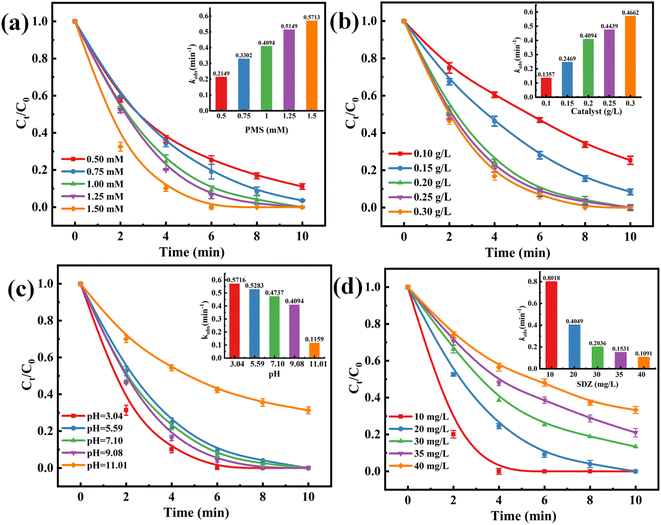 |
| | Fig. 7 Effects of (a) PMS concentration, (b) catalyst dosage, (c) initial solution pH and (d) initial SDZ concentration on SDZ removal in CMFO-0.4/PMS system. The inset in Fig. 7 shows the pseudo-first-order kinetic constants under different conditions. Reaction conditions: [CMFO] = 0.2 g L−1 (for a, c, d), [PMS] = 1.0 mM (for b, c, d), [SDZ] = 20 mg L−1 (for a, b, c), pH = 5.6 (for a, b, d). | |
The influence of pH on the catalytic degradation of non-homogeneous catalysts has been widely acknowledged.6 The pH value of the uncorrected SDZ solution was determined to be 5.60 ± 0.1. Fig. S5† illustrates the pH-dependent zeta potential of CMFO-0.4, with a zero potential point of 4.31. Based on the aforementioned data, it can be inferred that the surface of CMFO-0.4 exhibits a positive charge when the pH is below 4.31, while a negative charge is observed when the pH exceeds 4.31. The investigation focused on the catalytic degradation of SDZ by CMFO-0.4 in the presence of PMS within the pH range of 3.0 to 11.0. In Fig. 7c, there was little difference in the degradation rate of SDZ by CMFO-0.4 in the range of pH 3–9, and the degradation rate reached 100% in 10 min, but at pH 3.04, SDZ was completely removed in 6 min. The possible reason is that the H+ in the solution under an acidic medium leads to a positive charge on the surface of the CMFO-0.4 catalyst, which enhances the adsorption of PMS on the catalyst;36 on the other hand, the phenomenon of metal ion leaching is seen to be more pronounced when exposed to highly acidic environments (Fig. S6†). However, the removal of SDZ decreased significantly to 63.29% at pH 11.01, and the reaction rate constant rapidly decreased to 0.1159 min−1 (illustration in Fig. 7c). This is due to the fact that under alkaline (9.0–11.0) conditions, most of the PMS exists in the form of anionic SO52−, the negatively charged surface of CMFO-0.4 and the SO52− of the PMS experience a notable electrostatic repulsion due to their opposite charges.37 In addition, an excessive concentration of OH− can undergo a reaction with PMS ions (HSO5−) resulting in the formation of SO42− and water molecules (eqn (1)).38 Fig. S6† illustrates the variation in solution pH throughout the removal process of SDZ under various initial pH values. Throughout the reaction, the pH of the solution exhibited a decline, the observed phenomenon may be ascribed to the breakdown of PMS in the aquatic environment.
| | |
HSO5− + OH− + SO5− → 2SO42− + O2 + H2O
| (1) |
To examine the impact of substrate concentration on the rate of SDZ elimination, the concentrations of the substrate [SDZ] were manipulated to values of 10, 20, 30, 35 and 40 mg L−1. It was ensured that the fundamental reaction conditions of the system were kept constant throughout the experiment. Fig. 7d demonstrates a noticeable drop in the degradation rate when the starting concentration of SDZ increased. When the concentration of SDZ increases, the degradation rate of SDZ becomes slower due to the higher concentration of the substrate and the competition between organic pollutants, which is negatively correlated with the increase of its starting concentration. This is because under the same reaction conditions, the active sites on the surface of the catalyst and the active oxide species produced in the solution are limited, so the higher the concentration of organic matter solution, the slower the degradation reaction rate.
3.4. Influence of coexistence of inorganic anions and HA
Actual water bodies usually contain a wide range of inorganic ions and natural organics, and these co-existing inorganic anions and natural organics are of interest because of their competing relationships with the target pollutants or otherwise affecting the catalytic reaction process. In light of the influence of the aforementioned constituents of water quality on the catalytic process, we have opted to examine the impacts of commonly occurring inorganic anions, namely chloride (Cl−), sulfate (SO42−), and nitrate (NO3−), as well as organic HA, on the elimination of SDZ in the CMFO-0.4/PMS system.
As shown in Fig. 8a, the addition of Cl− caused the CMFO-0.4/PMS system to show a promoting effect on the degradation of SDZ, corresponding to an increasing k value. This phenomenon can be attributed to the fact that the added Cl− generates reactive chlorine such as HOCl and Cl2 through a double electron transfer reaction(eqn (2) and (3)).39 Fig. 8b shows the introduction of SO42− resulted in a modest decrease in the removal of SDZ. However, the extent of the inhibitory activity was not considerably affected by the concentration of SO42−. The potential cause for the hindered removal of SDZ could be ascribed to the competing influence of SO42− ions on the adsorption sites of PMS. The enhanced affinity of SO42− towards the catalyst leads to a more favourable occupation of the active sites located on the catalyst's surface.40 According to the data presented in Fig. 8c, the presence of NO3− in the CMFO-0.4/PMS system exerted an inhibitory impact on SDZ degradation, which was attributable to the depletion of SO4˙− and ˙OH (eqn (4) and (5)). At lower doses, NO3− exhibited little inhibitory effect, while the inhibitory impact on SDZ degradation became more pronounced as the concentration of NO3− increased.27
| | |
HSO5− + 2Cl− + H+ → SO42− + Cl2 + H2O
| (2) |
| | |
HSO5− + Cl− → SO42− + HOCl
| (3) |
| | |
SO4˙− + NO3− → NO3˙− + SO42−
| (4) |
| | |
˙OH + NO3− → NO3˙− + OH−
| (5) |
 |
| | Fig. 8 Effects of (a) chloride (Cl−), (b) sulfate (SO42−), (c) nitrate (NO3−) and (d) HA on SDZ removal in the CMFO/PMS system. The inset Fig. 8 shows the pseudo-first-order kinetic constants under different conditions. Reaction conditions: [catalyst] = 0.2 g L−1, [PMS] = 1.0 mM, [SDZ] = 20 mg L−1, pH = 5.6. | |
HA was employed as a surrogate to investigate the impact of organic coexistence conditions on the degrading efficacy of SDZ. Fig. 8d demonstrates that the administration of HA effectively suppresses the degrading impact of SDZ, which can be ascribed to the effect of HA on both the targeted pollutant and the catalyst. HA as a competitor of organic matter itself participates in chemical oxidation reactions, consuming reactive oxides;41 Moreover, HA is characterized by a substantial presence of phenolic hydroxyl and carboxyl groups. These groups have the ability to adsorb onto the catalyst's surface, leading to the inhibition of SDZ and PMS adsorption in water by the catalyst. Consequently, the removal efficiency of SDZ is notably diminished.
3.5. The stability of CMFO-0.4 and SDZ mineralization rate
To assess the reusability of CMFO-0.4, a set of five cyclic experiments were performed under consistent experimental conditions. After the conclusion of each experiment, the CMFO-0.4 material underwent magnetic separation followed by multiple washes using anhydrous ethanol and deionized water. Following this, the substance underwent a drying process within a vacuum drying oven at 60 °C.
According to the data presented in Fig. 9a, the elimination of SDZ was seen to be 100%, 87.86%, 75.67%, 69.81% and 66.37% during five successive cycles in the study. The observed decline in SDZ removal during the cycling studies might potentially be attributed to several factors, including the leaching of metal ions from the CMFO-0.4 surface, catalyst loss, and reduction of catalyst active sites caused by the adsorption of intermediates.42,43 The solution underwent total organic carbon (TOC) examination using a TOC analyzer both before and following the reaction. As shown in Fig. 9b, it is evident that the initial cycle resulted in a TOC removal rate of 26.85% and the TOC removal rate also decreased with the increase of the number of cycles. The concentration of metal leaching in the solution was determined after each cycle using ICP-OES. The leaching rates of Co, Mn and Fe of CMFO-0.4 were higher in the first cycle. Subsequently, as the number of cycles rose, the concentrations of these metal ions reduced (Fig. S7†). Hence, the presence of elements such as cobalt, manganese, and iron in leaching processes continues to be a significant constraint on their application in the field of water treatment.44
 |
| | Fig. 9 Degradation effect of SDZ (a) and (b) degradation rate of TOC during five consecutive cycles. Reaction conditions: [catalyst] = 0.2 g L−1, [PMS] = 1.0 mM, [SDZ] = 20 mg L−1, pH = 5.6. | |
3.6. Involved oxidizing species in CMFO-0.4/PMS system
As indicated by a prior investigation,8 the utilization of heterogeneous catalysts in the activation of PMS has the potential to yield a wide range of active species, including SO4˙−, ˙OH, ˙O2− and 1O2. To elucidate the process by which ROS are generated in the CMFO-0.4/PMS system and their impact on the degradation of SDZ, a competitive radical burst test was conducted. TBA (50, 100 mmol L−1), EtOH (50, 100 mmol L−1), p-BQ (5 mmol L−1) and FFA (5 mmol L−1) were added to the SDZ aqueous solution as quenchers to capture SO4˙−, ˙OH, ˙O2− and 1O2. EtOH served as the scavenger for SO4˙− and ˙OH, while TBA acted as a scavenger specifically for ˙OH.45 The scavengers utilized for single linear oxygen and superoxide radicals were FFA and p-BQ,46,47 respectively. As depicted in Fig. 10a and Table S4,† the degradation efficiency and reaction rate constants of SDZ were hindered by both EtOH and TBA. However, the inhibitory effect of EtOH on SDZ degradation was more significant, and this inhibition was further intensified with higher dosages of EtOH. This occurrence was ascribed to the rivalry between EtOH and SDZ for SO4˙− and ˙OH. While the inhibitory effect of EtOH on SDZ degradation was consistently more pronounced than that of TBA, irrespective of the concentration. Hence, it can be concluded that the active components in the CMFO-0.4/PMS system were determined to be SO4˙− and ˙OH, with SO4˙− being identified as the primary active species. The reduction of SDZ removal was seen when 5 mM of p-BQ or 5 mM of FFA were introduced into the reaction system, respectively. The degradation efficiency experienced a decrease of 21.88% in the system containing p-BQ and 85.69% in the system containing FFA, in comparison to the system without any added quencher. This suggests that the formation of ˙O2− and 1O2 took place during the breakdown process of SDZ by activated PMS, with 1O2 playing a significant role in the reaction process. Therefore, according to the experimental findings, it may be deduced that the degradation of SDZ by CMFO-0.4-activated PMS is a result of the combined influence of several reactive ROS (SO4˙−, ˙OH, ˙O2− and 1O2), with 1O2 playing a significant role.
 |
| | Fig. 10 (a) The impact of several radical scavengers on the degradation of SDZ in the CMFO-0.4/PMS system. Reaction conditions: [catalyst] = 0.2 g L−1, [PMS] = 1.0 mM, [SDZ] = 20 mg L−1, pH = 5.6. The EPR spectra of (b) DMPO-˙OH and DMPO-SO4˙−, (c) DMPO-˙O2− and (d) TMPO-1O2 in CMFO/PMS system. | |
To confirm the validity of the quenching experiment outcomes, the EPR technique was employed to analyze the CMFO-0.4/PMS system. Fig. 10b exhibits distinct peaks that can be attributable to the DMPO-SO4˙− adduct (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1:1:1:1:1:1:1) and DMPO-˙OH adduct (1:2:2:1),48 this demonstrates the existence of SO4˙− and ˙OH in the CMFO-0.4/PMS system. The magnitude of peak strength exhibited a negative correlation with time, this observation suggests that the generated free radicals were utilized in the process of SDZ breakdown. The signal of the DMPO-˙O2− adduct is observable in Fig. 10c,49 suggesting the generation of ˙O2− in the CMFO-0.4/PMS system. Fig. 10d shows three characteristic TEMP-1O2 peaks (1:1:1), illustrating the process of 1O2 generation by the CMFO/PMS system.50 The intensity of the 1O2 adduct peaks was enhanced at 6 min compared with 3 min, and the production of 1O2 can be ascribed to the self-decomposition of HSO5−. Thus, the degradation of SDZ through the CMFO-0.4/PMS system is a synergistic effect of multiple reactive species of 1O2, ˙OH, O2˙− and SO4˙−.
:1:1:1:1:1:1:1:1) and DMPO-˙OH adduct (1:2:2:1),48 this demonstrates the existence of SO4˙− and ˙OH in the CMFO-0.4/PMS system. The magnitude of peak strength exhibited a negative correlation with time, this observation suggests that the generated free radicals were utilized in the process of SDZ breakdown. The signal of the DMPO-˙O2− adduct is observable in Fig. 10c,49 suggesting the generation of ˙O2− in the CMFO-0.4/PMS system. Fig. 10d shows three characteristic TEMP-1O2 peaks (1:1:1), illustrating the process of 1O2 generation by the CMFO/PMS system.50 The intensity of the 1O2 adduct peaks was enhanced at 6 min compared with 3 min, and the production of 1O2 can be ascribed to the self-decomposition of HSO5−. Thus, the degradation of SDZ through the CMFO-0.4/PMS system is a synergistic effect of multiple reactive species of 1O2, ˙OH, O2˙− and SO4˙−.
3.7. Activation mechanisms of PMS
Drawing upon the preceding characterization and empirical findings, we put forth a potential activation pathway for PMS, as seen in Fig. 11. The XPS results (Fig. 4b and c) indicate that, in comparison to fresh CMFO-0.4, the Fe2p and Co2p binding energy values exhibit a minor negative shift.20 This shift suggests a change in the valence states of Fe and Co. For Mn 2p (Fig. 4b), the proportion of Mn2+/Mn3+/Mn4+ in CMFO-0.4 changed from 45.3%, 36.46% and 17.61% to 47.78%, 32.72% and 19.50%, with little change before and after use. These proportions remained relatively consistent before and after usage, indicating a potential process of transition transformation between Mn2+, Mn3+, and Mn4+ the ratio of Mn2+/Mn3+/Mn4+ does not change much. As shown in Fig. 4c the proportion of Co2+ and Co3+ in CMFO-0.4 changed from 43.8% and 56.2% to 28.59% and 71.41%, respectively, which was due to the transformation between Co2+ and Co3+ in the catalyst. The proportions of Otc. Fe2+, Otc. Fe3+ and Tet. Fe3+ in CMFO-0.4 changed from 41.63%, 29.81% and 28.56% to 31.79%, 44.79% and 23.45% (Fig. 4d). This suggests that both Fe3+/Fe3+ and Co3+/Co2+ redox pairs are involved in the activation of PMS by CMFO-0.4, whereas manganese is involved in the reaction to a lesser extent, mainly acting as an adsorption site as well as accelerating the cycling of ions. From Fig. 4e it is evident that the peak intensity of O1s of the reacted material is weakened and the ratio of the three peaks of O1s changes from 17.36%, 62.39% and 20.24% to 18.61%, 53.44% and 24.14%. The increase of hydroxyl oxygen on its surface was attributed to the adsorption of organic intermediates and mineralisation products when the catalyst was added to the solution to participate in the chemical reaction, and the formation of metal-hydroxyl groups M-OH (M = Co, Mn or Fe).51 The decrease of OVs suggests that the oxygen vacancies of the catalysts after participation in the reaction may be occupied by the reaction intermediates leading to their decrease. To further explore the changes of OVs after the reaction, the post-reaction CMFO-0.4 was tested using EPR, which showed a weakening of the characteristic peaks of OVs after the reaction, which is consistent with the XPS results.
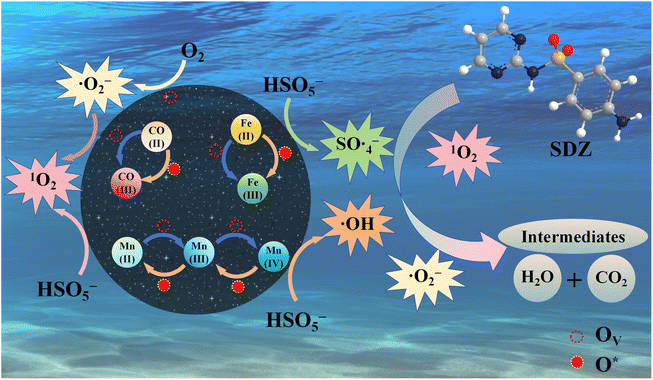 |
| | Fig. 11 The mechanism of CMFO-0.4 activating PMS to degrade SDZ was proposed. | |
According to the results of EPR, free radical quenching experiment and XPS characterization, it is concluded that the reaction mechanism of CMFO-0.4 activated PMS to degrade SDZ is as follows: during the catalytic reaction, HSO5− ions undergo adsorption on the catalyst surface and MnII/MnIII/MnIV, CoII/CoIII and FeII/FeIII redox cycles are of significant importance in the production of SO4˙− or ˙OH (eqn (6) and (7)).26,29 At the same time, OVs (Ov) are converted to reactive oxygen species (O*) by charge transfer. Subsequently, the reaction between O* and PMS to generate 1O2 occurs as described by eqn (9).29,52 The OV on the surface of CMFO-0.4 transfers electrons to adsorbed oxygen to produce ˙O2− through eqn (10),53 and a small portion of the generated ˙O2− is converted to 1O2 through eqn (11) and (12).27 The oxygen missing from the solution can be replaced by formula eqn (13) and (14). In addition, HSO5− is hydrolysed to produce ˙OH via eqn (15). Finally, the generated ROS is strongly oxidising and reacts with SDZ molecules via eqn (16).
| | |
CoII/MnII/FeII + HSO5− + OV → CoIII/MnIII/FeIII + SO4˙− + OH− + O*
| (6) |
| | |
CoII/MnII/MnIII/FeII + HSO5− + H2O + OV → CoIII/MnIII/MnIV/FeIII + SO42− + 2H+ +·OH + O*
| (7) |
| | |
CoIII/MnIII/FeIII + HSO5− + H+ + O* → CoIIMnII/FeII + SO5˙− + H2 + OV
| (8) |
| | |
O* + HSO5− → 1O2 + HSO4−
| (9) |
| | |
˙O2− + ˙OH → 1O2 + OH−
| (11) |
| | |
2˙O2− + 2H2O → 1O2 + H2O2 + 2OH−
| (12) |
| | |
2SO5˙− → 2SO4˙− + O2
| (13) |
| | |
˙O2− + HSO5− → O2 + SO4˙− + OH−
| (14) |
| | |
1O2/˙OH/SO4˙−/˙O2− + SDZ → several steps → CO2 + H2O + SO42−
| (16) |
3.8. Possible degradation pathways of SDZ
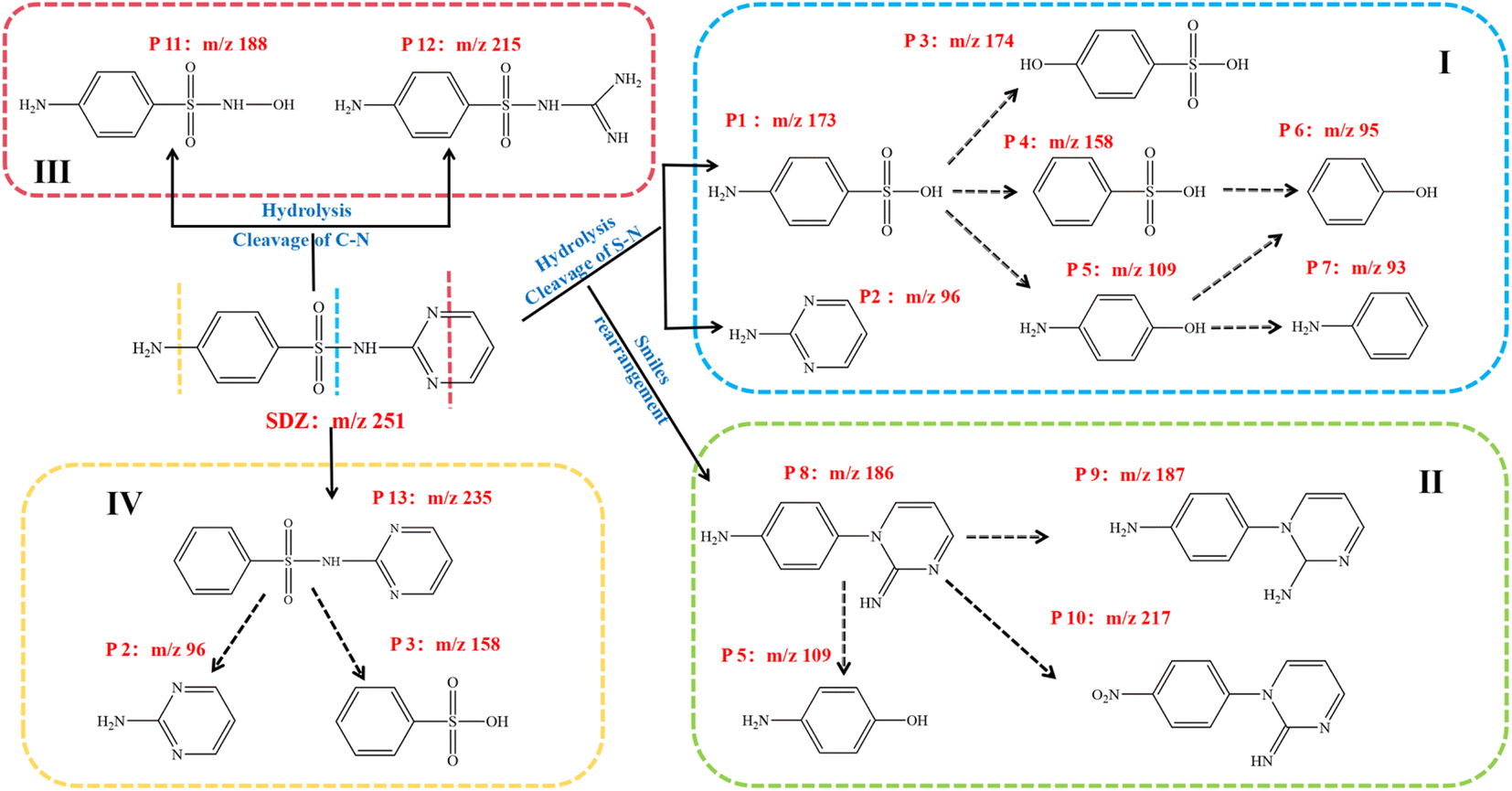
The key intermediates were identified using HPLC-MS and their mass fragment ions (m/z), molecular formulas and structures are described in Table S5.† Four hypothetical degradation routes of SDZ in the CMFO-0.4/PMS system were hypothesized based on the identification of transformation products using HPLC-MS (Fig. 12). The m/z value of the original SDZ is 251. The S–N bond represents the vulnerable point within the structure of SDZ. In Pathway I, the species SO4˙−, ˙OH, O2˙− and 1O2 are capable of directly initiating an attack on the S–N bond of SDZ to form 4-aminobenzenesulfonic acid (P1) and pyrimidine-amine (P2) intermediates. P1 can be fragmented into smaller components, namely P4, P5 (pathway I) or the –NH2 of P1 was further oxidized to –OH (P3). P4 and P5 were further decomposed into P6 and P7. The second potential pathway involves the Smiles-type rearrangement of six-membered heterocyclic sulfonamides under photocatalytic or persulfate-based advanced oxidation processes (AOP). This rearrangement leads to the generation of P8 through the extrusion of OSO and subsequent intermolecular rearrangement. P8 can then undergo further oxidation to yield the smaller fragment P5. Typically, P8 is prone to oxygenation at the –NH2 and NH groups by ROS. The presence of P9 and P10 indicates the potential for additional oxidation of P8. When strong oxidising substances attack the two C–N bonds on the SDZ pyridine ring, the ring can be opened to produce P11 and P12 intermediates (pathway III).54 The fourth possible pathway (pathway IV) involves the direct oxidative cleavage of the –NH2 group to produce P13 under the action of free radicals or 1O2 active substances, and then the active substances attack the S–N bond to produce the same intermediates P2 and P3 as pathway I.
 |
| | Fig. 12 Potential degradation pathway of SDZ in CMFO/PMS system. | |
3.9. Toxicity evaluation
Since toxicity evaluation is a key technique for evaluating ecological concerns, a software tool called Toxicity Estimation Software tool (T. E. S. T.) was employed to make predictions regarding the toxicity of SDZ parent and its degradation intermediates3 (Fig. 13). The biotoxicity of SDZ and its breakdown intermediates was evaluated by the determination of the LC50 (lethal concentration for 50% of the population) in the blackhead minnow (Fig. 13a). For the intermediate products of SDZ, except for the toxicity of P8, P9 and P10 to become higher “Extremely toxic”, the rest of the toxicity is “Toxic” or lower “Harmful”. The severity of the environmental impact is directly proportional to the magnitude of the bioaccumulation factor. The bioaccumulation factor (BAF) of SDZ was determined to be 1.64. The levels of degradation products P2, P11, and P12 saw a decline, whilst the bioaccumulation factors of the other intermediates demonstrated an increase (Fig. 13b). The developmental toxicity of all intermediates was reduced except for P8, P10, P12 and P1 (Fig. 13c). Fig. 13d shows that all were “mutagenic negative” except P8, P9 and P10 which were “mutagenic positive”. From the above results, it can be seen that CMFO-0.4 activated PMS degradation of SDZ could produce smaller compounds, but some intermediates were slightly more toxic.
 |
| | Fig. 13 (a) Acute toxicity LC50 (96 h), (b) bioconcentration factor, (c) developmental toxicity and (d) mutagenicity. | |
4. Conclusion
In this paper, the CMFO-0.4 catalyst with oxygen vacancies was efficiently synthesised by sol–gel method and its performance in activating PMS was investigated. It was found by characterisation that CMFO-0.4 has more magnetic, redox and charge transfer capabilities compared to MFO. Besides, the introduction of Co made the catalyst rich in OVs, which play a key role in enhancing the PMS catalyst and accelerating the redox cycle of metal ions. The catalyst showed excellent catalytic effect in activating PMS for SDZ degradation. The CMFO-0.4/PMS system exhibited excellent SDZ degradation in a wide range of pH values, including acidic, neutral and basic. The EPR analyses and quenching experiments provided evidence that the degradation of SDZ in the CMFO-0.4/PMS system was a synergistic effect of multiple actives of 1O2, ˙OH, O2˙− and SO4˙−, with 1O2 playing a key role. In addition, the intermediates were examined by LC-MS, a reasonable synthetic route was proposed and the toxicity of the intermediates was analysed. We found that the activation of CMFO-0.4 for the degradation of SDZ by PMS could produce compounds with less toxicity and smaller molecules. In addition, the CMFO-0.4 catalyst showed enhanced tolerance to inorganic anions (Cl−, SO42− and NO3−) prevalent in the SDZ solution, and the presence of Cl− even facilitated the SDZ degradation process. The results of the cycling studies showed that the removal of SDZ decreased with the increasing number of cycles due to precipitation of metal ions and loss of catalyst. Therefore, our future work will focus on investigating ways to improve the stability of this catalyst.
Data availability
The data supporting this article have been included as part of the ESI.†
Author contributions
Fengchun Li: conceptualization, data curation, formal analysis, visualization, writing-original draft. Yawei Gu: investigation, supervision, software, funding acquisition. Luwei Zhai: methodology, data curation. Xuan Zhang: data curation, validation. Ting Wang: project administration. Xia Chen: methodology, validation. Chongqing Xu: funding acquisition. Guihuan Yan: methodology. Funding acquisition. Wenqiang Jiang: funding acquisition, supervision, validation, writing review and editing.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This research was supported by the Natural Science Foundation of Shandong Province (ZR2021QE042, ZR2020MB141 and ZR2023MC167), the Science, Education and Industry Integration Pilot Projects of Qilu University of Technology (Shandong Academy of Sciences) (2023PX049), the Talent and Scientific Research Project of Qilu University of Technology (Shandong Academy of Sciences) (2023RCKY113) and the Major Innovation Projects of Science, Education, and Industry Integration of Qilu University of Technology (Shandong Academy of Science) (2022JBZ02-05).
References
- T. Wang, J. Lu, J. Lei, Y. Zhou, H. Zhao, X. Chen, M. Faysal Hossain and Y. Zhou, Sep. Purif. Technol., 2023, 307, 122755 CrossRef CAS.
- B. Wang, B.-J. Ni, Z. Yuan and J. Guo, Water Res., 2020, 173, 115592 CrossRef CAS PubMed.
- M. Zhu, X. Chen, Y. Tang, S. Hou, Y. Yu and X. Fan, J. Hazard. Mater., 2022, 437, 129359 CrossRef CAS.
- J. L. Wang and L. J. Xu, Crit. Rev. Environ. Sci. Technol., 2012, 42, 251–325 CrossRef CAS.
- J. Wang and S. Wang, Chem. Eng. J., 2018, 334, 1502–1517 CrossRef CAS.
- J. Ren, L. Jiang, Y. Li and G. Zhang, Sep. Purif. Technol., 2021, 275, 119100 CrossRef CAS.
- T. Guo, L. Jiang, K. Wang, Y. Li, H. Huang, X. Wu and G. Zhang, Appl. Catal., B, 2021, 286, 119883 CrossRef CAS.
- R. Zhou, S. Liu, F. He, H. Ren and Z. Han, Chemosphere, 2021, 285, 131433 CrossRef CAS PubMed.
- L. Y. Xu, X. Zhou, G. L. Wang, L. Zhou and X. K. Sun, Sep. Purif. Technol., 2021, 276, 119417 CrossRef CAS.
- Q. Wang, P. Rao, G. Li, L. Dong, X. Zhang, Y. Shao, N. Gao, W. Chu, B. Xu, N. An and J. Deng, Ecotoxicol. Environ. Saf., 2020, 187, 109779 CrossRef CAS PubMed.
- Y. Ji, C. Dong, D. Kong, J. Lu and Q. Zhou, Chem. Eng. J., 2015, 263, 45–54 CrossRef CAS.
- Q. Zhu, K. Guo, S. Ma, S. Wang, X. Tang, R. Duan, Y. Huang, J. Wang, G. Cheng, S. Xu and X. Zhuang, Chem. Eng. J., 2023, 455 Search PubMed.
- X. Duan, H. Sun, Z. Ao, L. Zhou, G. Wang and S. Wang, Carbon, 2016, 107, 371–378 CrossRef CAS.
- L. Zhu, Z. Shi, L. Deng and Y. Duan, Colloids Surf., A, 2021, 609 Search PubMed.
- Y. Ren, L. Lin, J. Ma, J. Yang, J. Feng and Z. Fan, Appl. Catal., B, 2015, 165, 572–578 CrossRef CAS.
- J. Deng, M. Xu, C. Qiu, Y. Chen, X. Ma, N. Gao and X. Li, Appl. Surf. Sci., 2018, 459, 138–147 CrossRef CAS.
- J. Deng, S. Feng, X. Ma, C. Tan, H. Wang, S. Zhou, T. Zhang and J. Li, Sep. Purif. Technol., 2016, 167, 181–189 CrossRef CAS.
- R. Yu, J. Zhao, Z. Zhao and F. Cui, J. Hazard. Mater., 2020, 390, 121998 CrossRef CAS.
- J. Wang, M. Yue, Y. Han, X. Xu, Q. Yue and S. Xu, Chem. Eng. J., 2020, 391 CAS.
- Y. Sun, J. Zhou, D. Liu, X. Li and H. Liang, Chemosphere, 2022, 297 CAS.
- S. Lu, S. You, J. Hu, X. Li and L. Li, RSC Adv., 2024, 14, 7528–7539 RSC.
- J. Judith Vijaya, G. Sekaran and M. Bououdina, Ceram. Int., 2015, 41, 15–26 CrossRef CAS.
- G. Chen, X. Zhang, Y. Gao, G. Zhu, Q. Cheng and X. Cheng, Sep. Purif. Technol., 2019, 213, 456–464 CrossRef CAS.
- S. Gautam, P. Shandilya, B. Priya, V. P. Singh, P. Raizada, R. Rai, M. A. Valente and P. Singh, Sep. Purif. Technol., 2017, 172, 498–511 CrossRef CAS.
- A. V. Narendra Kumar and W. S. Shin, Chem. Eng. J., 2023, 465, 142922 CrossRef CAS.
- L. Zhang, X. Zhao, C. Niu, N. Tang, H. Guo, X. Wen, C. Liang and G. Zeng, Chem. Eng. J., 2019, 362, 851–864 CrossRef CAS.
- J. Guo, Y. Chen, W. Chen, Z. Chen, F. Gao, J. Wang and Y. Fu, J. Alloys Compd., 2023, 960, 170868 CrossRef CAS.
- Z. Zhi, D. Wu, F. Meng, Y. Yin, B. Song, Y. Zhao and M. Song, Sci. Total Environ., 2022, 828, 154275 CrossRef CAS PubMed.
- L. Yue, L. Hao, J. Zhang, X. Piao and C. Chen, J. Water Proc. engineering, 2023, 53, 103807 CrossRef.
- M. Li, Z. He, H. Zhong, L. Hu and W. Sun, Environ. Sci.: Nano, 2022, 9, 1037–1051 RSC.
- J. Zhao, Z. Zhao, N. Li, J. Nan, R. Yu and J. Du, Chem. Eng. J., 2018, 353, 805–813 CrossRef CAS.
- Z. Li, F. Wang, Y. Zhang, Y. Lai, Q. Fang and Y. Duan, Chem. Eng. J., 2021, 423 Search PubMed.
- X. Duan, C. Su, J. Miao, Y. Zhong, Z. Shao, S. Wang and H. Sun, Appl. Catal., B, 2018, 220, 626–634 CrossRef CAS.
- C. Chen, L. Liu, Y. Li, W. Li, L. Zhou, Y. Lan and Y. Li, Chem. Eng. J., 2020, 384, 123257 CrossRef CAS.
- J. Liang and L. Fu, Appl. Surf. Sci., 2021, 563, 150335 CrossRef CAS.
- M. M. Mian and G. Liu, Chem. Eng. J., 2020, 392, 123681 CrossRef CAS.
- Y. Yu, H. Quan, Z. Zhang, Q. Zhang, H. Wang, D. Chen and J. Zou, Sep. Purif. Technol., 2023, 322, 124336 CrossRef CAS.
- J. Chen, K. Chu, S. Sun, H. Chen, B. Song, J. Wang, Z. Liu and L. Zhu, J. Environ. Chem. Eng., 2023, 11, 109230 CrossRef CAS.
- L. Wu, Y. Lin, Y. Zhang, P. Wang, M. Ding, M. Nie, C. Yan and S. Chen, RSC Adv., 2021, 11, 33626–33636 RSC.
- X.-C. Zhou, S.-Q. Chen, M.-J. Zhou, M. Li, S. Lan and T. Feng, Rare Met., 2023, 42, 1160–1174 CrossRef CAS.
- L. Zhai, F. Li, Y. He, X. Chen, X. Zhang, Y. Gu, C. Xu, G. Yan, X. Feng and W. Jiang, Chem. Eng. J., 2023, 478, 147378 CrossRef CAS.
- D. Ding, C. Liu, Y. Ji, Q. Yang, L. Chen, C. Jiang and T. Cai, Chem. Eng. J., 2017, 308, 330–339 CrossRef CAS.
- X. Liu, Y. Yao, J. Lu, J. Zhou and Q. Chen, Chemosphere, 2023, 311, 136972 CrossRef CAS PubMed.
- Y. Xu, J. Ai and H. Zhang, J. Hazard. Mater., 2016, 309, 87–96 CrossRef CAS.
- T. Zeng, X. Zhang, S. Wang, H. Niu and Y. Cai, Environ. Sci. Technol., 2015, 49, 2350–2357 CrossRef CAS.
- X. Luo, Y. You, M. Zhong, L. Zhao, Y. Liu, R. Qiu and Z. Huang, J. Hazard. Mater., 2022, 426, 127803 CrossRef CAS PubMed.
- X. Liu, F. Qu, S. Gao, Z. Bai and J. Tian, Chem. Eng. J., 2023, 475, 146291 CrossRef CAS.
- F. Chen, F. Yang, H. Liu, S. Che, G. Zhang, C. Xu and Y. Li, Chem. Eng. J., 2022, 430, 132904 CrossRef CAS.
- H. Xu, D. Wang, J. Ma, T. Zhang, X. Lu and Z. Chen, Appl. Catal., B, 2018, 238, 557–567 CrossRef CAS.
- P. Yang, S. Li, L. Xiaofu, A. Xiaojing, D. Liu and W. Huang, Sep. Purif. Technol., 2022, 285, 120288 CrossRef CAS.
- Y. Ren, L. Lin, J. Ma, J. Yang, J. Feng and Z. Fan, Appl. Catal., B, 2015, 165, 572–578 CrossRef CAS.
- J. Miao, X. Duan, J. Li, J. Dai, B. Liu, S. Wang, W. Zhou and Z. Shao, Chem. Eng. J., 2019, 355, 721–730 CrossRef CAS.
- J. Lee, U. von Gunten and J. H. Kim, Environ. Sci. Technol., 2020, 54, 3064–3081 CrossRef CAS.
- D. Ma, Y. Yang, B. Liu, G. Xie, C. Chen, N. Ren and D. Xing, Chem. Eng. J., 2021, 408, 127992 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Ting Wangc,
Xia Chena,
Chongqing Xub,
Guihuan Yanb and
Wenqiang Jiang
a,
Ting Wangc,
Xia Chena,
Chongqing Xub,
Guihuan Yanb and
Wenqiang Jiang