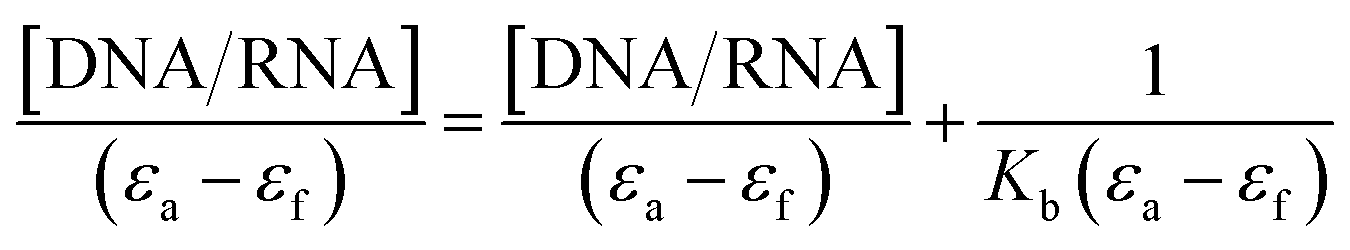DOI:
10.1039/D4RA02738H
(Paper)
RSC Adv., 2024,
14, 19512-19527
Palladium(II), platinum(II), and silver(I) complexes with 3-acetylcoumarin benzoylhydrazone Schiff base: Synthesis, characterization, biomolecular interactions, cytotoxic activity, and computational studies†
Received
12th April 2024
, Accepted 2nd June 2024
First published on 18th June 2024
Abstract
New Pd(II) (C1), Pt(II) (C2), and Ag(I) (C3) complexes derived from 3-acetylcoumarin benzoylhydrazone (HL) Schiff base were synthesized and characterized by FTIR, 1H NMR, UV-visible spectroscopies along with elemental analysis (C, H, N), magnetic, molar conductivity measurements, and DFT calculations. The obtained results suggested that the ligand had different behaviors in the complexes: mono-negative tridentate (C1) and neutral tridentate (C2) as an ONO-donor and neutral bidentate (C3) as an ON-donor. Quantum chemistry calculations were performed to validate the stability of the suggested geometries and indicated that all the complexes possess tetra-coordinated metal ions. The binding affinity of all the compounds toward calf thymus (ctDNA), yeast (tRNA), and bovine serum albumin (BSA) was evaluated by absorption/emission spectral titration studies, which revealed the intercalative binding to ctDNA and tRNA and static binding upon complex formation with BSA. Molecular insights into the binding affinity of the characterized complexes were provided through conducting molecular docking analysis. Moreover, the cytotoxic activity (in vitro) of the compounds was screened against human cancerous cell lines and a non-cancerous lung fibroblast (WI38) one using cis-platin as a reference drug. The IC50 and selective index (SI) values indicated the higher cytotoxic activity of all the metal complexes compared to their parent ligand. Among all the compounds, the complex C2 showed the highest activity. These results confirmed the improvement of the anticancer activity of the ligand by incorporating the metal ions. In addition, flow cytometry results showed that complexes C1 and C2 induced cell cycle arrest at S and G1/S, respectively.
1. Introduction
Coumarins (2H-1-benzopyran-2-one) are a crucial class of oxygen-based heterocyclic compounds of natural origin and can be also found in synthetic medicinally active compounds.1,2 They have been used in flavoring foods and in cosmetic products as a fragrant.3 Coumarin-containing compounds have attracted considerable interest in biological applications including in vitro/in vivo anticancer,4,5 antimicrobial, antioxidant, and anti-inflammatory activities.1 In comparison to free coumarin-based ligands, the anticancer and antimicrobial activities have been enhanced by coordination with metal ions.6–8 Recently, coumarin-derived Schiff bases along with associated metal complexes have been considered to be the most efficient classes of compounds that exhibit a wide range of biological activities.7 It has been found that the C3-position of coumarin is an effective position that can improve the cytotoxic efficacy against several human cell lines.9–11 In particular, 3-acetylcoumarin has demonstrated high reactivity to form remarkably stable Schiff base ligands such as hydrazone (ONO-donor ligands) and the synthesis of their metal complexes.12
On the other hand, hydrazone Schiff bases have also attracted considerable scientific attention owing to their unique structural and biological/pharmacological properties.13–15 The keto–enol tautomerism of the hydrazone structure gives them the possibility to coordinate metal ions through azomethine nitrogen, or ketonic or enolic amide oxygen. Additionally, the coordination site from the substituent attached to the hydrazide amine group offers diverse coordination modes.16–20 In fact, a series of hydrazide/hydrazine coumarin-based derivatives have been reported for their potential antitumor activity.21 Moreover, hydrazone-based complexes were reported to improve the binding affinity to DNA, as well as the antitumor activity.22,23 Therefore, the structural improvement of hydrazone/coumarin may lead to an important category of compounds with utility for designing and developing new potential drugs.24,25 Indeed, the literature is rich with numerous coumarin-derived complexes with therapeutic applications due to their anticancer, antimicrobial, and antioxidant effects and as enzyme inhibitors.7,26 For instance, a series of 3-acetylcoumarin hydrazone Schiff base complexes containing nickel(II), cobalt(II), copper(II),27 ruthenium(II), Rh(III), and Ir(III)28 have been reported for their anticancer and antibacterial activities.
Based on the reported biological activities of coumarin, hydrazone Schiff bases, and their metal chelates, and also on a continuation of our research on acetylcoumarin and hydrazone Schiff bases,29–31 we sought to develop novel metal complexes containing Pd(II), Pt(II), and Ag(I) based on 3-acetylcouamrin benzoylhydrazone Schiff base. Moreover, we evaluated their interaction with biomolecules (ctDNA, tRNA, and BSA), and in addition, their in vitro cytotoxic activity against human breast carcinoma (MCF7), cervical carcinoma (HeLa), and normal lung fibroblast (WI38) cell lines. Moreover, the cell death mechanism was elucidated by flow cytometry. Later, DFT calculations were performed to further support the proposed structures while molecular docking was carried out to provide deep insights into their interaction with BSA.
2. Experimental
2.1. Materials and equipment
Details on the materials and physical measurements are presented in the ESI materials (ESI,† Section 1.1).
2.2. Synthesis
2.2.1. Synthesis of 3-acetylcoumarin benzoyl hydrazone ligand (HL). A methanolic solution of 3-acetylcoumarine (0.94 g, 5 mmol) was added to benzyhydrazide (0.68 g, 5 mmol, 10 mL) in methanol in the presence of a few drops of glacial acetic acid. The mixture was refluxed for 3 h. The white product was filtered off, recrystallized from methanol, then dried in vacuo. Yield: 80%, m.p.: 158–160 °C. Elemental analysis for C18H14N2O3 (306.3): calcd (found): C, 70.58 (70.30); H, 4.61 (4.41); N, 9.15 (9.0). FTIR (KBr, cm−1): ν (NH) 3192, ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)lactone 1721, ν(CO)hydrazone1664, ν(CN) 1611, ν(N–N) 963 cm−1. 1H NMR (400 MHz, DMSO-d6, δ, ppm): δ 10.85 (s, 1H, NH), δ 8.26 (s, 1H, H4), δ 7.88 (d, 2H, H6,9), δ 7.67 (t, 2H, H7,8), δ 7.53 (t, 2H, H16,18), δ 7.47 (d, 2H, H15,19), δ 7.41 (t, 1H, H17), δ 2.35 (s, 3H, H12). 13C NMR (100 MHz, DMSO-d6, δ, ppm): C(2) 159.7, C(3) 127.3, C(4) 142.4, C(5) 119.5, C(6) 116.5, C(7) 125.3, C(8) 133.5, C(9) 128.7, C(10) 151.6, C(11) 153.9, C(13) 128.3, C(14) 134.5, C(15,19) 130, C(16, 18) 129.7, C(17) 128.8, C(CH3) 16.7. UV-vis (DMSO, 2.5 × 10−5 M): λmax (nm) (ε, M−1 cm−1): 232 (100
O)lactone 1721, ν(CO)hydrazone1664, ν(CN) 1611, ν(N–N) 963 cm−1. 1H NMR (400 MHz, DMSO-d6, δ, ppm): δ 10.85 (s, 1H, NH), δ 8.26 (s, 1H, H4), δ 7.88 (d, 2H, H6,9), δ 7.67 (t, 2H, H7,8), δ 7.53 (t, 2H, H16,18), δ 7.47 (d, 2H, H15,19), δ 7.41 (t, 1H, H17), δ 2.35 (s, 3H, H12). 13C NMR (100 MHz, DMSO-d6, δ, ppm): C(2) 159.7, C(3) 127.3, C(4) 142.4, C(5) 119.5, C(6) 116.5, C(7) 125.3, C(8) 133.5, C(9) 128.7, C(10) 151.6, C(11) 153.9, C(13) 128.3, C(14) 134.5, C(15,19) 130, C(16, 18) 129.7, C(17) 128.8, C(CH3) 16.7. UV-vis (DMSO, 2.5 × 10−5 M): λmax (nm) (ε, M−1 cm−1): 232 (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), 336 (90000).
000), 336 (90000).
2.2.2. Synthesis of [Pd(L)Cl] (C1) and [Pt(HL)Cl]Cl (C2). To a methanolic solution (10 mL) of the ligand HL (0.092 g, 0.3 mmol), an aqueous solution (2 mL) of K2PdCl4 (0.098 g, 0.3 mmol) or K2PtCl4 (0.125 g, 0.3 mmol) was added. The above solution was refluxed for 3 h. The precipitate was filtered off, washed with methanol followed by diethyl ether, and then dried.[Pd(L)Cl] (C1): Yellow precipitate, yield: 75%, m.p.: 190–192 °C. Elemental analysis for C18H13ClN2O3Pd: calcd (found): C, 48.35 (48.03); H, 2.93 (2.80); N, 6.26 (6.14). FTIR (cm−1): ν(CO)lactone 1620, ν(C–O)new 1221, ν(CN) 1589, ν(CN)new 1554, ν(N–N) 975, ν(M–O) 575, ν (M–N) 442. 1H NMR (400 MHz, DMSO-d6, δ, ppm): δ 8.53 (s, 1H, H4), δ, 8.02, 7.85 (d, 2H, H6, 9), δ 7.66 (t, 2H, H7,8), δ 7.32 (t, 2H, H16,18), δ 7.42 (d, 2H, H15,19), δ 7.03 (t, 1H, H17), δ 2.66 (s, 3H, H12). UV-vis (DMSO, 2.5 × 10−5 M): λmax (nm) (ε, M−1 cm−1): 276 (127000), 330 (52000). Molar conductivity (10−3 M, DMSO), Λm = 2.0 Ω m2 mol−1.
[Pt(HL)Cl]Cl (C2): Orange precipitate, yield: 64%, m.p.: 200–202 °C. Elemental analysis for C18H14Cl2N2O3Pt, calcd (found): 37.78 (37.86); H, 2.47 (2.39); N, 4.89 (4.77). FTIR (KBr, cm−1): ν (NH) 3278, ν(CO)lactone 1724, ν(CO)hydrazone 1663, ν(CN) 1575, ν(N–N) 1021, ν(M–O) 529, ν (M–N) 435. 1H NMR (400 MHz, DMSO-d6, δ, ppm): δ 10.72 (s, 1H, NH), δ 8.84 (s, 1H, H4), δ 8.13 (d, 2H, H6,9), δ 7.89 (t, 2H, H7,8), δ 7.60 (t, 2H, H16,18), δ 7.77 (d, 2H, H15,19), δ 7.50 (t, 1H, H17), δ 2.63 (s, 3H, H12). UV-vis (DMSO, 2.5 × 10−5 M): λmax (nm) (ε, M−1 cm−1): 276 (33500), 327 (25700). Molar conductivity (10−3 M, DMSO), Λm = 65 Ω m2 mol−1.
2.2.3. [Ag(HL)2]NO3 (C3). To a methanolic solution of the ligand HL (0.092 g, 0.3 mmol), an aqueous solution of AgNO3 (0.05 g, 0.3 mmol) was added. The above solution was stirred under ambient conditions for 1 h. A grayish white precipitate formed was filtered, washed with methanol followed by diethyl ether, and then dried. Grayish white powder, yield: 70%, m.p.: 190–192 °C. Elemental analysis for C36H28AgN5O9: calcd (found): 55.26 (54.95); H, 3.61 (3.24); N, 8.95 (8.65). FTIR (KBr, cm−1): ν (NH) 3277, ν(CO)lactone 1698, ν(CO)hydrazone 1664, ν(CN) 1608, ν(N–N) 971, ν(M–O) 545, ν (M–N) 439. 1H NMR (400 MHz, DMSO-d6, δ, ppm): δ 11.05 (s, 1H, NH), δ 8.29 (s, 1H, H4), δ 7.90 (d, 2H, H6,9), δ 7.68 (t, 2H, H7,8), δ 7.56 (t, 2H, H16,18), δ 7.48 (d, 2H, H15,19), δ 7.42 (t, 1H, H17), δ 2.38 (s, 3H, H12). 13C NMR (100 MHz, DMSO-d6, δ, ppm): C(2) 161.6, C(3) 127.7, C(4) 142.5, C(5) 119.2, C(6) 116.7, C(7) 125.2, C(8) 133.3, C(9) 128.6, C(10) 151.6, C(11) 154.1, C(13), 128.7, C(14) 134.0, C(15,19) 129.7, C(16, 18) 129.5, C(17) 128.6, C(CH3) 16.7. UV-vis (DMSO, 2.5 × 10−5 M): λmax (nm) (ε, M−1 cm−1): 226 (230000), 332 (70000). Molar conductivity (10−3 M, DMSO), Λm = 53 Ω m2 mol−1.
2.3. Solution stability
First, 50 μL (1.0 × 10−3 M) of the test compounds (in DMSO) was added to 1950 μL phosphate buffered saline (PBS, pH 7.2) in a UV-visible cuvette, until the final concentration reached 2.5 × 10−5 M. The electronic spectra were measured at set time intervals (0, 20, 40, 60 min, 24, and 48 h).
2.4. Biological studies
2.4.1. ctDNA, tRNA, and BSA interaction and MTT in vitro cytotoxic activity studies. Details on the experimental procedures are provided in the (ESI,† Section 1.2).
2.5. Theoretical calculations
2.5.1. DFT calculations. Density functional theory (DFT) calculations, as implemented in Gaussian 16 software, were utilized to obtain the most favorable structures for the three studied complexes. The popular DFT functional B3LYP, which has been successfully used to study various metal-containing chemical systems, was used.32–36 All the atoms were treated with the 6-311++G(3df,3pd) basis set except the metal ion that was represented based on the effective core potential (ECP) and LANL2DZ basis set. To further validate the identity of the obtained stable geometries as stationary points, frequency calculations at the optimization level of theory were performed on the optimized complexes, and no imaginary frequency was detected. Gaussian 16 utilities were also used to obtain the frontier molecular orbitals (FMOs) for the studied molecules, which helps to better explain their chemical reactivity. The determined FMOs, including the highest occupied molecular orbitals (HOMOs) and lowest unoccupied molecular orbitals (LUMOs), were visualized using Gaussian view.
2.5.2. Docking methodology. To provide atomistic insights into the binding between the characterized complexes and the albumin protein, molecular docking calculations were performed using Autodock4 tools.37,38 The structure of bovine serum albumin was retrieved from PDB ID: 6QS939 and refined by deleting the crystalized water molecules, assigning hydrogens to the standard amino acids, and adjusting the Kollman charges. Then, the default settings available in Autodock tools were utilized to assign the overall forcefield for the standard amino acids and then the protein was converted into pdbqt format. The optimized geometries for the complexes obtained from our DFT calculations were used as ligands for the docking analysis and were converted into pdbqt format while the charges were computed by the Gasteiger model. To accurately identify the most favorable binding pocket, MGL tools were used for blind docking by constructing a grid box that spread over the entire protein obtained from ref. 37. The genetic Algorithm protocol was selected to perform our molecular docking and the number of runs was set to 20 runs. The energetically favorable binding modes that lead to the highest binding scores were chosen for deeper structural analysis. Chimera software was then used to obtain detailed figures for the bound protein ligands.40
3. Results and discussion
3.1. Synthesis
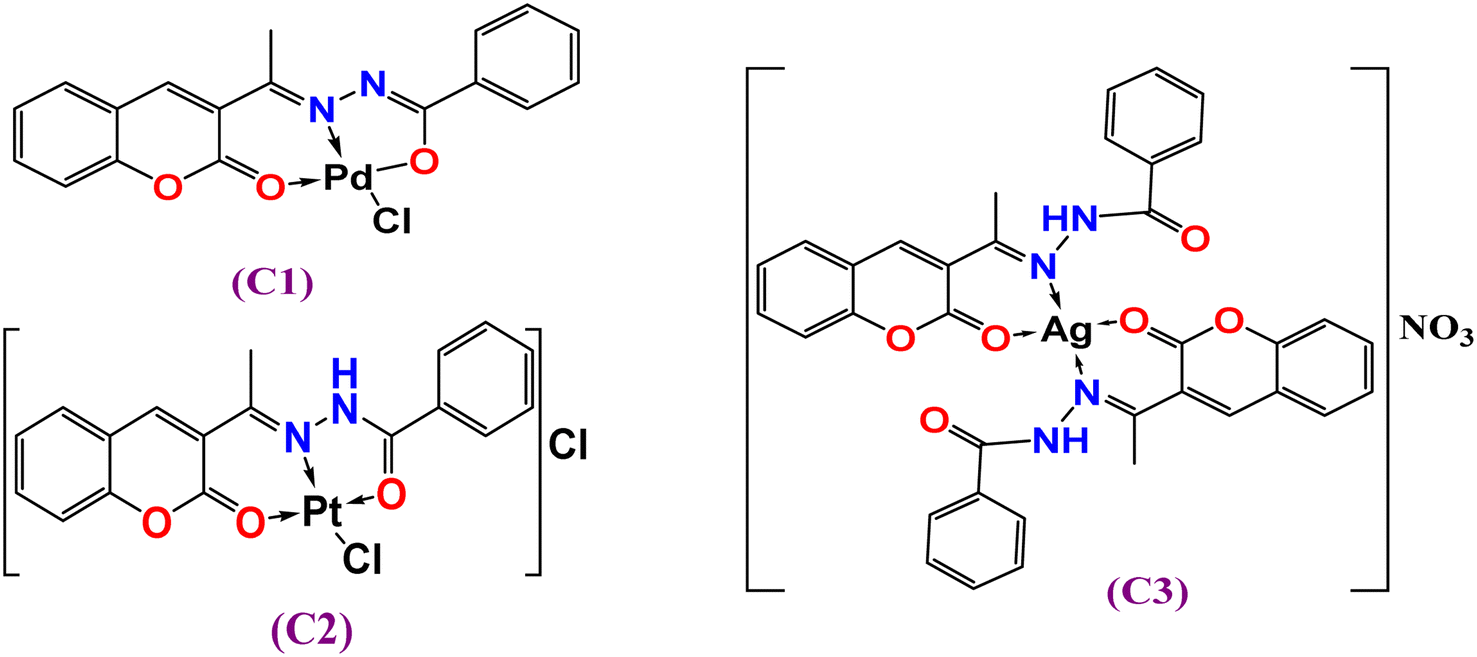
The ligand 3-acetylcomarin benzoylhydrazone Schiff base (HL) and its complexes (C1)–(C3) were prepared and characterized, as shown in the Experimental section (Section 2.2). The analytical data revealed that the molar ratio (M:L) of the complexes was 1:1 metal-to-ligand except for [Ag(HL)2], which seemed to have a 1:2 ratio. All the complexes were completely soluble in DMF, and DMSO but sparingly soluble in common organic solvents (methanol, ethanol, CH2Cl2, CH3CN, etc.). The molar conductance values of 10−3 M in DMSO suggested the neutrality of the [Pd(L)Cl] complex (C1) (ΛM = 2.0 Ω−1 cm2 mol−1), while the 1:1 electrolytic nature41 of the other complexes Pt(II) (C2) ((ΛM = 65 Ω−1 cm2 mol−1) and Ag(I) (C3) were in the range of ΛM = 53 Ω−1 cm2 mol−1). The analytical data suggest that the complexes C1–C3 were four coordinates, as indicated in Fig. 1.
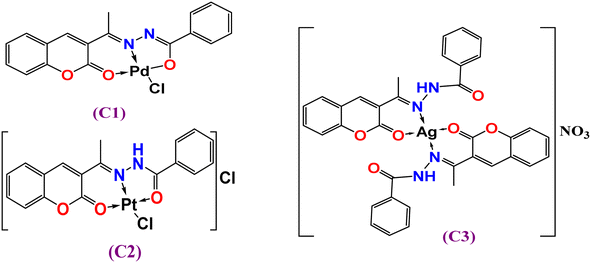 |
| | Fig. 1 Suggested structures of complexes C1–C3. | |
3.2. Characterization
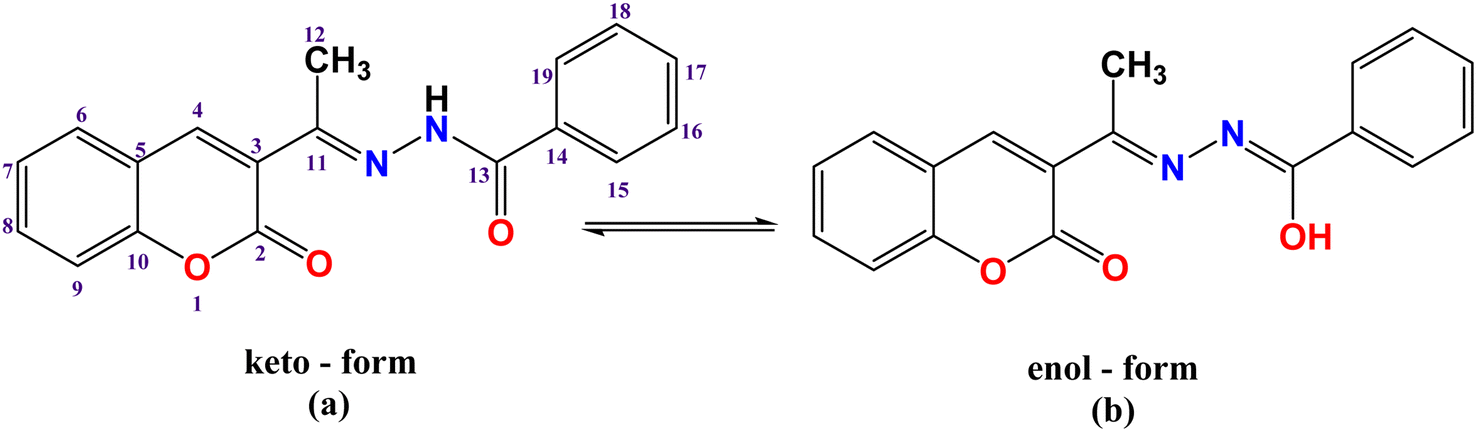
3.2.1. Vibrational spectra. The FTIR spectral bands of 3-acetylcoumarin benzoylhydrazone Schiff base (HL) and its complexes C1–C3 (ESI, Fig S1a–d†) were assigned as detailed in the Experimental section (Section 2.2) and Table S1.† The FTIR spectrum of the HL ligand displayed strong bands at 3192 and 1664 cm−1 due to the stretching vibrations of ν(NH) and carbonyl oxygen ν(CO) of the hydrazone moiety, respectively, suggesting that the ligand displayed an amido keto-form in its solid state (Scheme 1a). In addition, the strong bands that appeared at 1721 cm−1 were attributed to ν(CO) of the lactone ring. The strong band at 1611 cm−1 was due to azomethine ν(CN) stretching vibrations.42 Moreover, bands assigned to amide(II), amide(III), and ν(N–N) were observed at 1536, 1273, and 963 cm−1, respectively.43,44
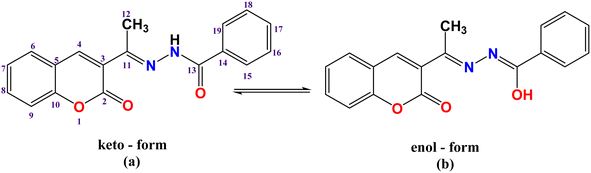 |
| | Scheme 1 (a) Keto-form, and (b) enol form of the HL ligand with the atom labeling scheme. | |
Generally, the azomethine stretching band ν(CN) was shifted to a lower frequency, suggesting the participation of azomethine-N in coordination with the metal center; however, a diversity of coordination modes was shown by the complexes. For example, in complex [Pd(L)Cl] (C1) (Fig. 1), the HL ligand behaved in a mono-negative tridentate (ONO) manner. This is illustrated by the lower frequency shifts of both the azomethine (CN) and lactone (CO) groups. Also, there was a disappearance of the ν(NH) and ν(CO)hydrazinic through coordination (deprotonation after enolization (Scheme 1b), as supported by the appearance of a new ν(CN) group45 near 1589 cm−1, besides the shift of the ν(N–N) band to a higher frequency by 7–58 cm−1. This, in turn, indicated that the bonding occurred through enolate oxygen, accordingly, and new ν(C–O) bands were obtained at 1221 and 1213, respectively. This assumes that the ligand behaves as a mono-negative tridentate using azomethine-N, carbonyl-O, and lactone-O.45,46 In the FTIR spectrum of [Pt(HL)Cl]Cl (C2), the presence of ν(NH) and ν(CO)hydrazinic bands, and the lower frequency shifts of azomethine (CN) and lactone (CO) indicate that the coordination took place in a neutral tridentate (ONO) manner. In the FTIR spectrum of [Ag(HL)2]NO3 (C3), the ν(NH) band was shifted to a higher frequency (3277 cm−1), while the ν(HCN) and lactone-O bands were shifted to lower frequencies; hence the ligand coordinated in a neutral bidentate manner (ON-donor). In addition, the strong absorption near 1365 cm−1 was attributed to the stretching vibration of ν(NO3).47 For all the complexes, new medium bands were observed at 435–449 and 529–575 cm−1, assigned to ν(M–N) and ν(M–O), respectively.47
3.2.2. NMR spectra. 1H NMR spectra of all the compounds (ESI, Fig. S2a–d†) were recorded in DMSO-d6 and assigned in the Experimental section (Section 2.2) and Table S2.† The atom labeling scheme of the ligand is presented in Scheme 1. The 1H NMR spectrum of the free ligand (HL) (Fig. S2a†) showed singlet signals at δ 10.85, 8.26, and 2.35 ppm, assignable to NH, H(4), and methyl protons H(12), respectively, while the doublets observed at δ 7.88, 7.47 ppm were attributed to four protons of H6,9 and H15,19. In addition, triplets appeared at δ 7.67, 7.53, and 7.41 ppm and were assigned to H7,8, H16,18, and H17, respectively. In the spectrum of [Pd(L)Cl] (C1) complex (Fig. S2b†), the absence of the –NH signal proved that HL binds to the metal centers in its enolic form48,49 (Scheme 1). Conversely, the presence of the –NH signal in the 1H NMR spectra of both [Pt(HL)Cl]Cl (C2) (Fig. S2c†) and [Ag(HL)2]NO3 (C3) (Fig. S2d†) indicates that HL coordinated through its keto-form, confirming the data proposed by the FTIR spectra. Also, all the other protons adjacent to the coordination sites were shifted upfield to some extent.50,51 Generally, for silver(I) complexes, the spectra are not so different when compared to the free ligand, particularly, when the ligand coordinates as a neutral species, or the d10 electronic configuration of Ag(I) ion.52The 13C NMR of HL (Fig. S3a and Table S3†) showed signals at 153.9 and 159.7 and 16.7 ppm assigned to C(11) and C(2) and the methyl carbon CH3 group, respectively similar to an earlier reported ligand.45 These signals were slightly shifted downfield in the spectrum of the Ag(I) complex (Fig. S3b†). This downfield shift by ∼2 ppm indicated the contribution of lactone carbonyl oxygen in the coordination. Moreover, the upfield shift of azomethine-C indicates that the ligand coordinated as an ON-donor through lactone carbonyl oxygen, and azomethine nitrogen. Noteworthily, the 1H and 13C of the silver complex exhibited very little shift in comparison to the parent ligand. This was anticipated due to the coordination of the ligand in its neutral mode and the Ag(I) ion's d10 electronic configuration.
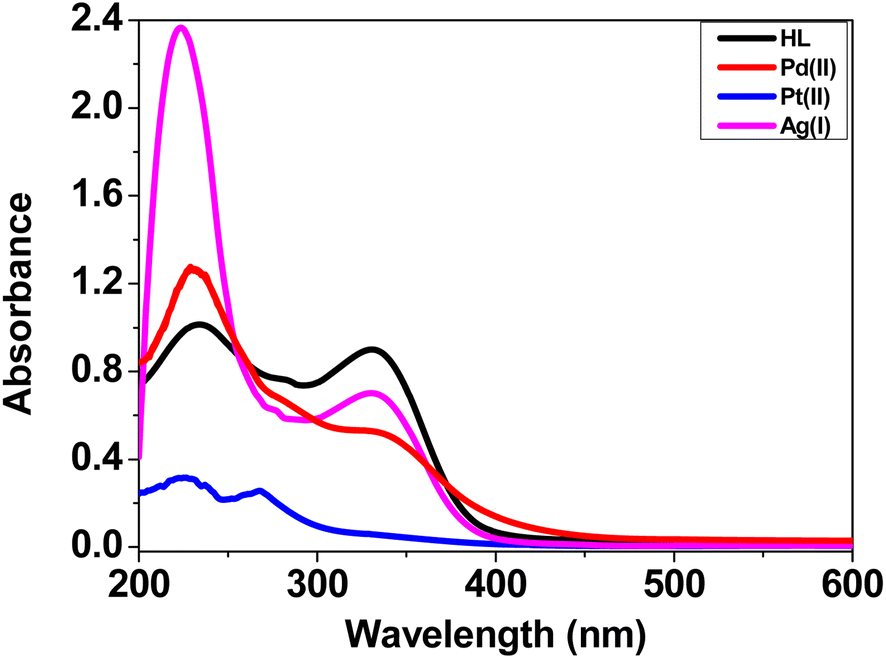
3.2.3. Electronic spectra. The UV-visible spectra of the ligand (HL) and its complexes C1–C3 (10−5 M) (Fig. 2) were recorded in DMSO solvent (Experimental section, Section 2.3, Table S4†). The UV-visible spectrum of the ligand showed bands at 232 and 336 nm attributed to the (π–π*) transition of phenyl rings, and (n–π*) transition of –CN– and –CO– moieties,53,54 respectively. The electronic spectra of the complexes exhibited a hypsochromic (blue-shift) of these transition bands, along with changes in the absorbance intensity, which were due to the coordination to the center of the metal ions.55,56
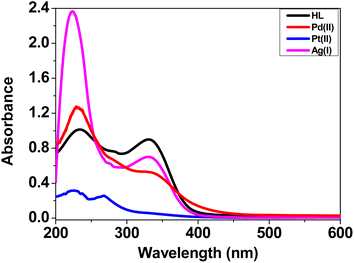 |
| | Fig. 2 UV-visible spectra of HL and complexes C1–C3 (10−5 M) in DMSO. | |
3.2.4. Solution stability. The solution stability study of the metal complex is a very important step to decide the reactivity of a metal complex in biological applications. The solution stability of the compounds was studied in an aqueous solution (PBS, pH 7.2) using UV-visible spectroscopy analysis at set time intervals (0, 30 min, 1 h, 1.5 h, 2 h, 3 h, 1 day, and 2 days) as shown in Fig. S4.† It was observed that the ligand showed insignificant changes up to the first 3 h then suffered hydrolysis in the aqueous media through the next 24 h. No distinctive changes were observed for the complexes (C1–C3) during the first 6 h. These results indicated the stability of these complexes under physiological conditions in buffer–DMSO solution.57 After 24 h, a slight decrease in absorption intensity of the complexes (C1 and C3) was observed, which may be attributed to the solvent exchange (DMSO/H2O) with chloride and nitrate ions, respectively; however complex (C2) showed insignificant changes during the 48 h test.58,59
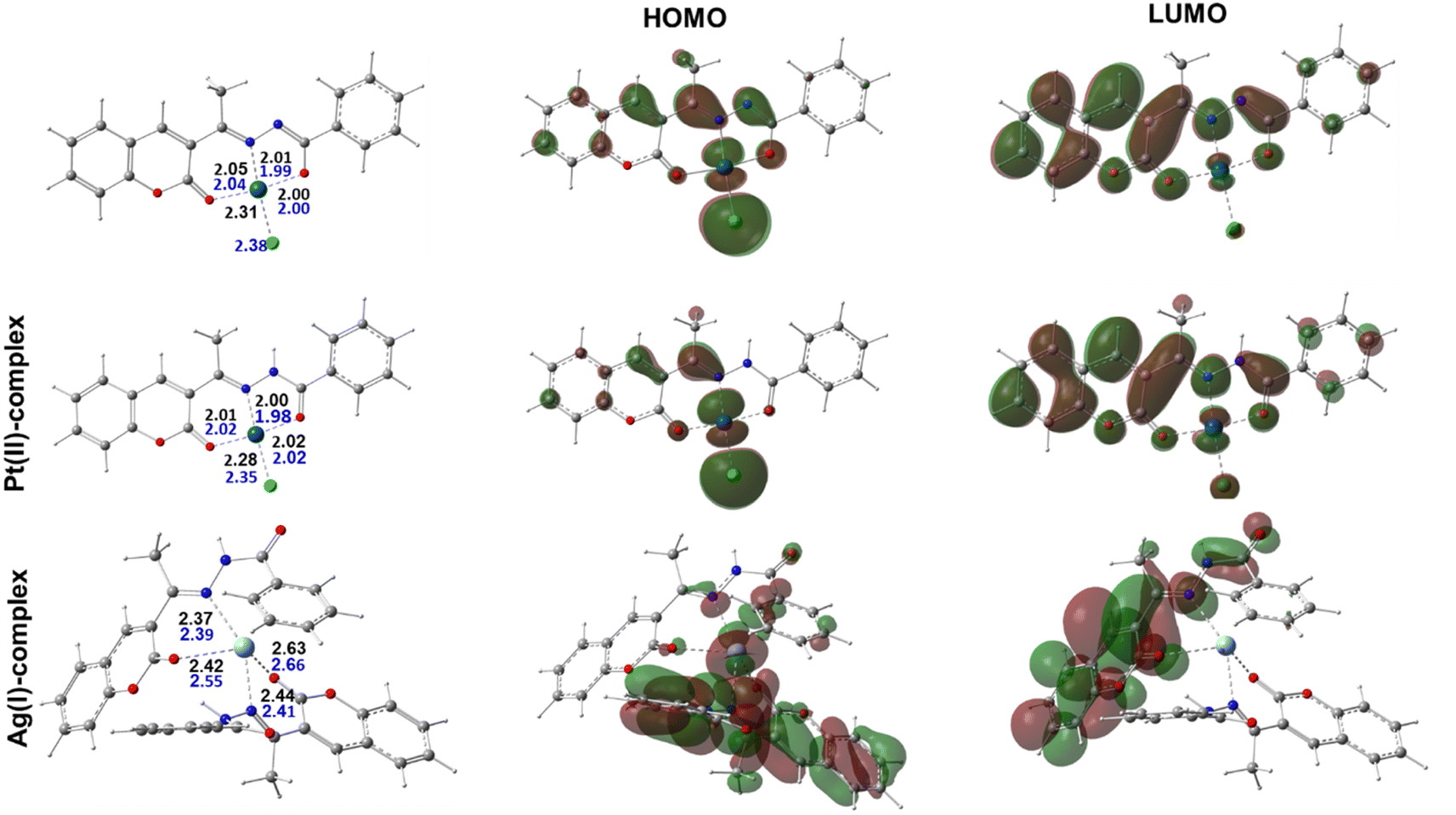
3.2.5. DFT calculations. The geometries for the synthesized complexes were characterized in both vacuum and a water medium. Notably, both Pt(II) and Pd(II) metal ions adopted an identical square planner geometry with a tri-coordinate HL ligand while the chloride ion ligand fulfilled the tetra-coordinated sphere, Fig. 3. The coordinating distances for both geometries are quite similar with an average distance of 2.0 Å where N or O centers are the coordinating atoms. Meanwhile, the M⋯Cl chelating distance was found to be the longest, with an average distance of 2.30 Å. Meanwhile, the obtained Ag(I) coordination geometry resulting from the 2:1 ratio was subtly different with a double ligation from each HL molecule. Its coordination distances were slightly longer than the corresponding ones in Pt(II) and Pd(II) complexes with an average distance of 2.47 Å. Interestingly, the calculated geometries for the three complexes were in accordance with the proposal from the experimental results.
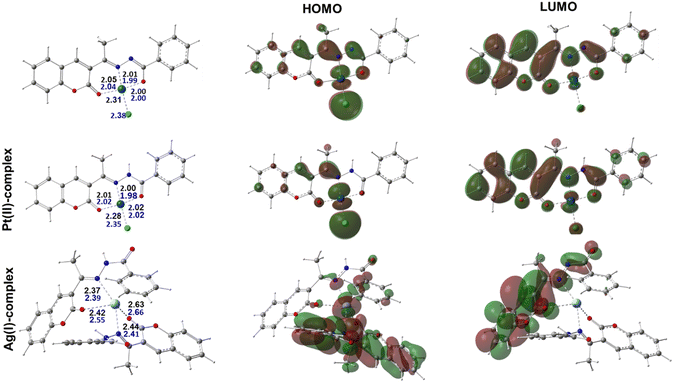 |
| | Fig. 3 Optimized molecular geometries for the three complexes representing the synthesized complexes together with selected bond distances in (Å) for optimization in gas (blue) and aqueous media (black), and their respective HOMOs and LUMOs. | |
Moreover, the most stable geometries for the studied complexes were fully optimized in aqueous medium using a similar functional and basis set combination to further validate their stability in solution. A comparison between the obtained geometries from these later calculations demonstrated there were insignificant changes in both their overall structure and the ligation distances, which agreed well with the experimental observations, Fig. 3. To better explain the chemical reactivity of the three complexes, the energy gap parameter, ΔE, was determined for each complex from the difference between the energy of the highest occupied molecular orbital (EHOMO) and the energy of the lowest unoccupied molecular orbital (ELUMO). In fact, the ΔE value is a stronger procurer for the chemical reactivity of the compounds and the smaller the gap the higher the reactivity of the complex. According to our calculations, the determined energy gap values were found to be 0.0902, 0.0974, and 0.135 au for the Pt(II)-, Pd(II)-, and Ag(I)-containing complexes. Moreover, the softness quantum chemical parameter (σ = 1/ΔE) was calculated and the obtained order for their values was Pt(II) > Pd(II) > Ag(I), with the Pt(II)-containing complex demonstrating the highest value. Therefore, the obtained order of the reactivity from our calculation from both the ΔE and σ values agreed well with the experimental observations.
The HOMO and LUMO were also displayed to better represent the delocalization and localization behavior of the electrons over the characterized complexes, Fig. 3. It was also noted that the HOMO of the Pt(II)- and Pd(II)-containing complexes behaved similarly and mainly localized over both the metal centers and the ligated chloride ions. It is also important to point out that a minor contribution from the ligated atom in the Pd(II) complex in the HOMO was also noticed. For the Ag(I) complex, however, in addition to the localization over the metal center, a significant participation of the aromatic motifs for one of the chelated ligands was observed. Meanwhile, the delocalization of the LUMO also showed some similarity among the three complexes in being distributed over conjugated π-systems of the aromatic rings in the ligand. Overall, this later observation could suggest that the geometry around the metal center together with the conjugated bonds over the aromatic motifs in the ligand are key players in the activity of the studied complexes.
4. Biological applications
4.1. ctDNA and tRNA interaction studies
Transition metal complexes have a very important function in metallodrug design. They can interact with DNA/RNA via different binding modes: intercalation (insertion), groove binding (major or minor), or electrostatic interaction.60 The intercalative binding is considered the most important one,61 because the molecules are inserted among the base pairs of the DNA double helix, forming π–π stacking interactions that induce elongation and unwinding of the DNA double helix due to the separation of the base pairs to accommodate the drug, and consequently, the DNA binding is responsible for various biological activities; for example, antitumor, antimicrobial, antiviral, anti-inflammatory, and antimalarial properties.62,63 In this section, we evaluated the ctDNA/tRNA binding affinity to the test compounds using both absorption and emission spectroscopies.
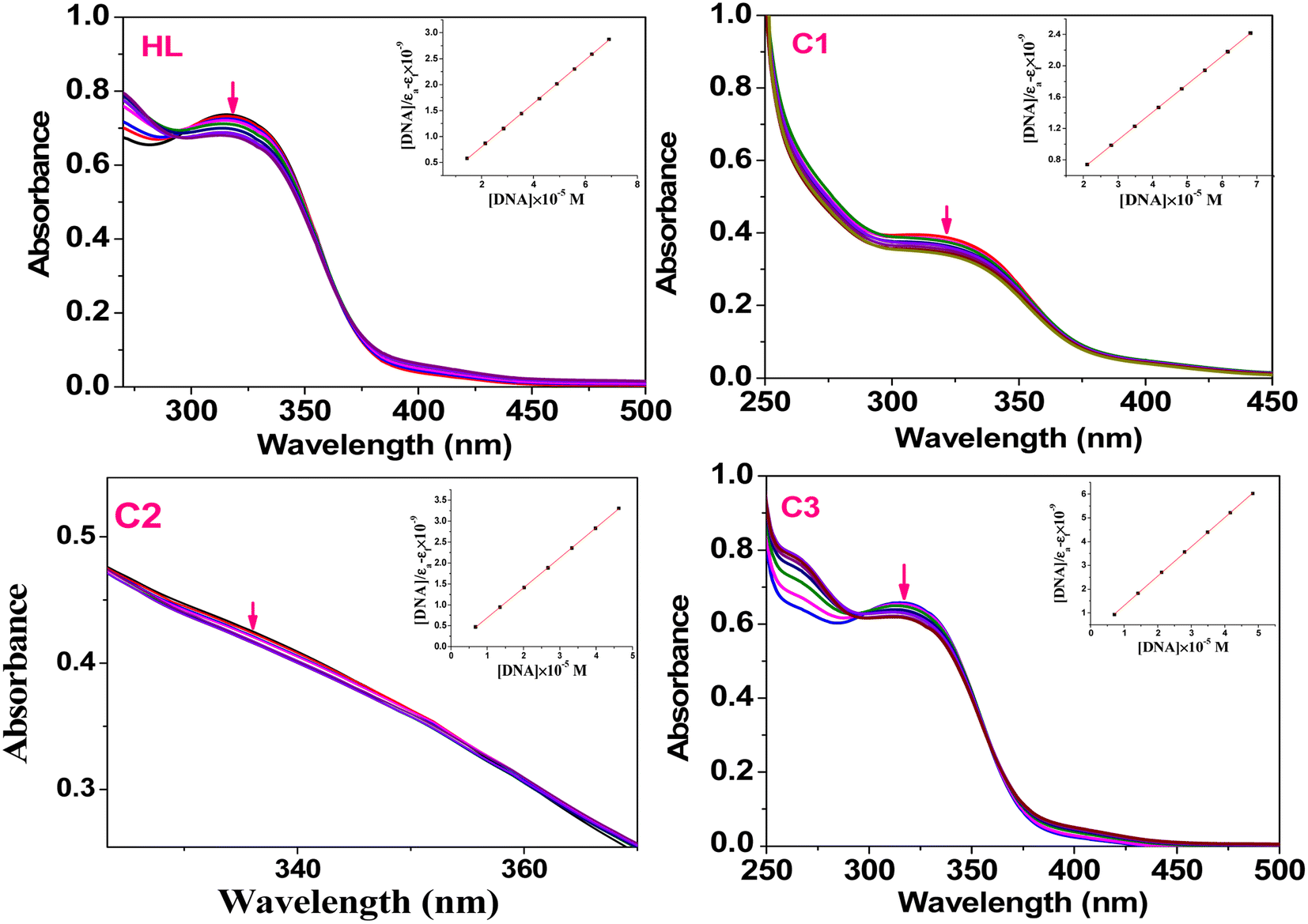
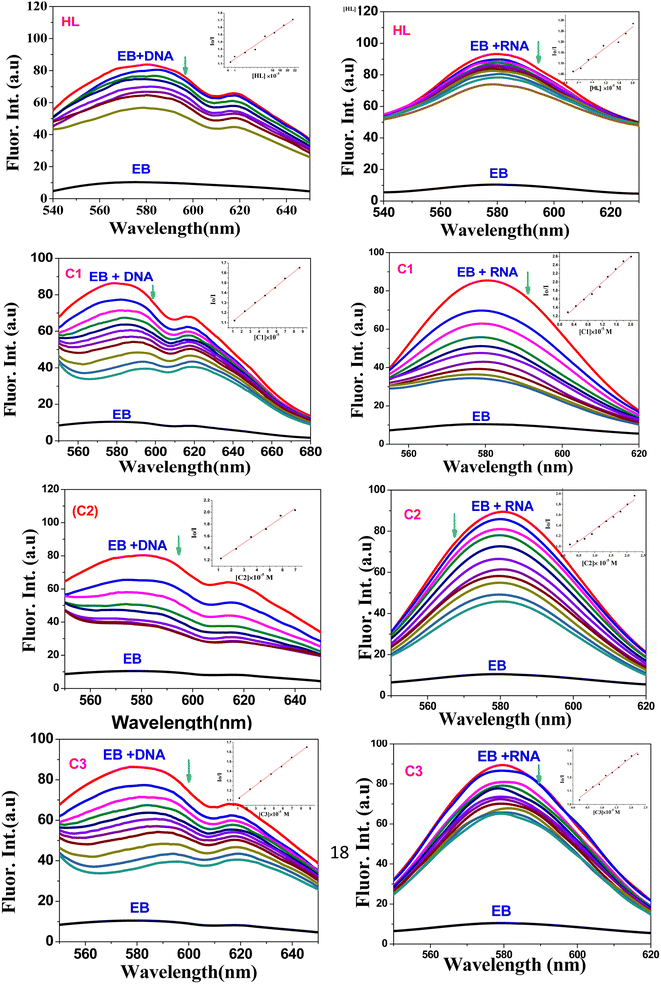
4.1.1. Absorption studies. The absorption spectral changes of the HL and its complexes (C1–C3) upon the addition of variable concentrations of ctDNA or tRNA to the test compound were monitored using UV-visible spectroscopy in Tris–HCl buffer (pH 7.2). The electronic spectra were first recorded for the free compound. The addition of different concentrations of the biomolecules may result in a slight red/blue-shift in the wavelength along with a hypochromic (a decrease in absorption intensity) or hyperchromic (increase in absorption intensity) shift.64 In our compounds (Fig. 4), the free ligand (HL) showed a hypochromic shift at λmax = 314 nm (intraligand π–π* transition), while the complexes (C1–C3) showed hypochromic shifts at 320, 329, and 319 nm, respectively (Fig. 4); this change suggests that the ligand and its complexes behaved with the same binding mode (intercalation). Also, the presence of the isosbestic point (change from hypochromism to hyperchromism) at 293 nm (HL), 319 nm (C2), and 295 nm (C3) indicated that there were only two absorbing species (free nucleic acid and nucleic acid–complex) with the same absorbance in equilibrium in the solution media.65
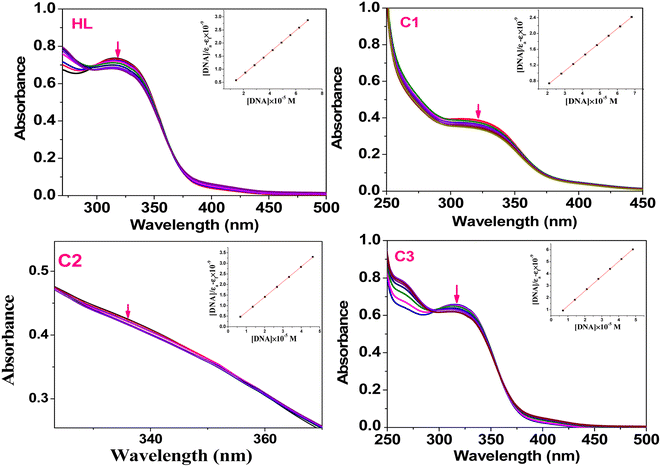 |
| | Fig. 4 Absorption spectra of the ligand (HL) and C1–C3 complexes (50 μM) in the presence of different concentrations of ct DNA (5−50 μM) in Tris–HCl buffer (pH 7.2); (inset): linear plot of ([DNA]/εa–εf) vs. [DNA]. | |
The presence of the aromatic chromophore existing in the ligand aided the interaction of the compounds with the CT DNA bases via non-covalent π–π* stacking interactions, wherein the π*-orbital of the ligand in the complexes could couple with the π-orbital of the DNA base pairs. The binding constants (Kb) was determined using the Wolfe–Shimer eqn (1):66
| |
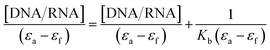 | (1) |
where [DNA] and [RNA] are the molar concentrations of ctDNA and tRNA, respectively,
εa =
Aobs/[compound] is the apparent absorption coefficient, and
εf and
εb correspond to the extinction coefficients of the free and bound compound. The
Kb (slope–intercept ratio) values were obtained from the plot of [DNA]/(
εa–
εf) against [DNA]. The
Kb values of the ligand and its metal complexes were found to be in the order of 10
6 M
−1. The binding constants of the metal complexes followed the order (
C1) > (
C2) > (
C3) > (
HL), revealing a relatively high binding constant of the Pd(
II) complex (
C1) compared to the free ligand and other complexes, as shown in
Table 1. This conclusion indicates that complexation enhanced the binding ability of the metal complexes with the nucleic acid (ctDNA and tRNA). The UV-visible absorption spectra of complexes with tRNA are presented in the ESI materials (Fig. S5
†).
Table 1 Binding constant data (Kb, Ksv) along with % hypochromism (% H) obtained from the interaction study of the compounds with ctDNA and tRNA
| Compd |
λmax |
Absorption spectroscopy |
Emission spectroscopy (at λem = 580 nm) |
| (Kb), × 106 |
% H* |
(Ksv),×104 |
% H |
| ctDNA |
tRNA |
ctDNA |
tRNA |
ctDNA |
tRNA |
ctDNA |
tRNA |
| HL |
314 |
1.10 |
1.36 |
8.35 |
7.20 |
0.34 |
1.19 |
32.4 |
26.1 |
| (C1) |
320 |
4.71 |
10.3 |
12.7 |
8.67 |
4.91 |
7.52 |
55.4 |
61.5 |
| (C2) |
329 |
2.58 |
8.96 |
10.4 |
7.60 |
1.42 |
4.51 |
54.2 |
48.8 |
| (C3) |
319 |
1.22 |
1.51 |
6.81 |
6.66 |
0.52 |
1.6 |
50.1 |
26.9 |
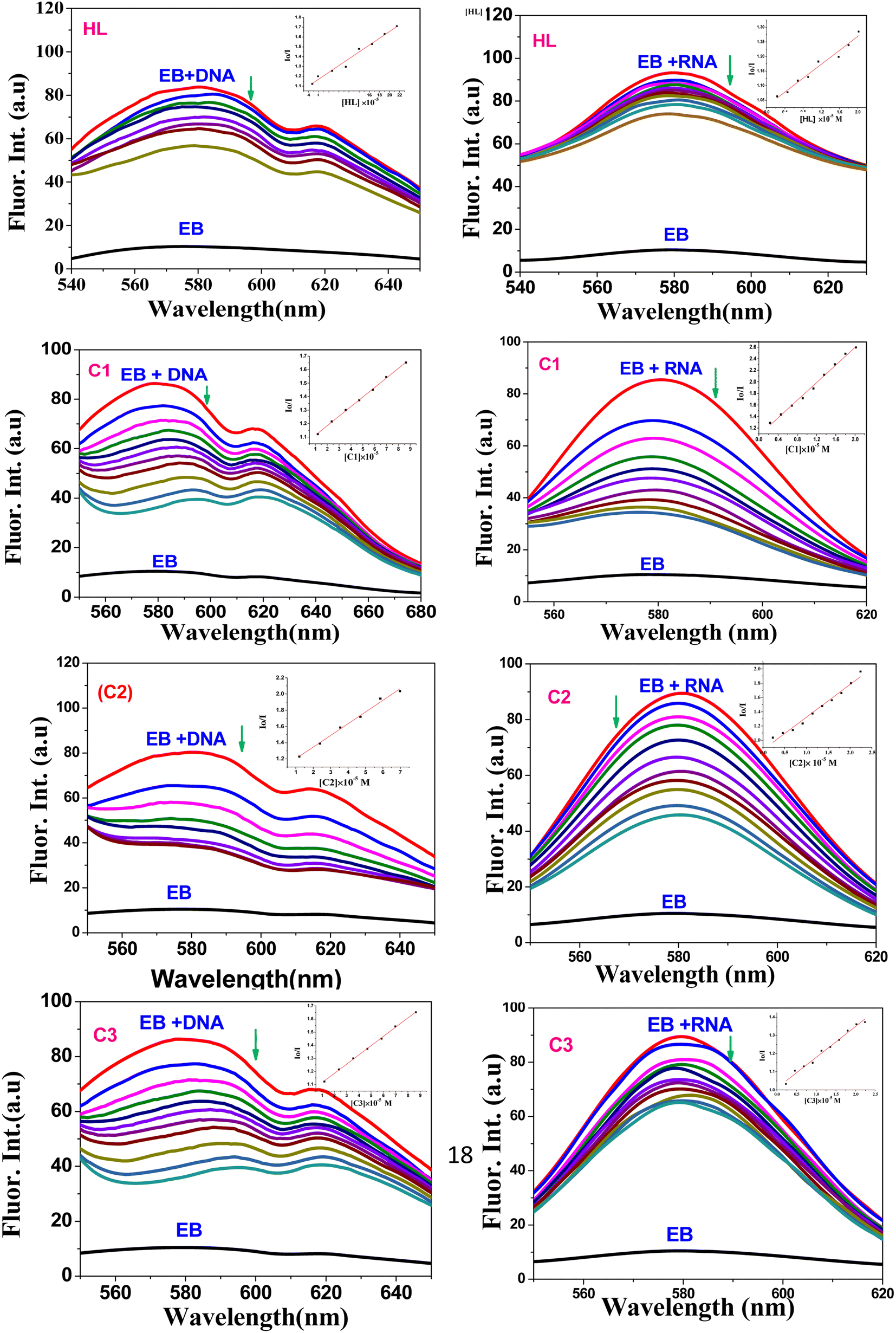
4.1.2. Emission spectroscopy. As the compounds lack fluorescent properties, we used the ethidium bromide assay (EB) to further confirm the intercalation of the small molecules to ctDNA and tRNA, whereby EB increases the fluorescence intensity (at λem = 580 nm) when bound to ctDNA or tRNA after intercalating between their base pairs. At this wavelength, the fluorescence quenching spectra (Fig. 5) of the complexes (C1–C3) and their free ligand (HL) showed that the compounds were able to displace EB from DNA-EB/RNA-EB adducts (the reduction in the emission intensity indicates the binding of the compound with ctDNA or tRNA due to the formation of a non-fluorescent DNA/RNA-complex) and confirmed that ligands/complexes had intercalated onto nucleic acid base pairs.
 |
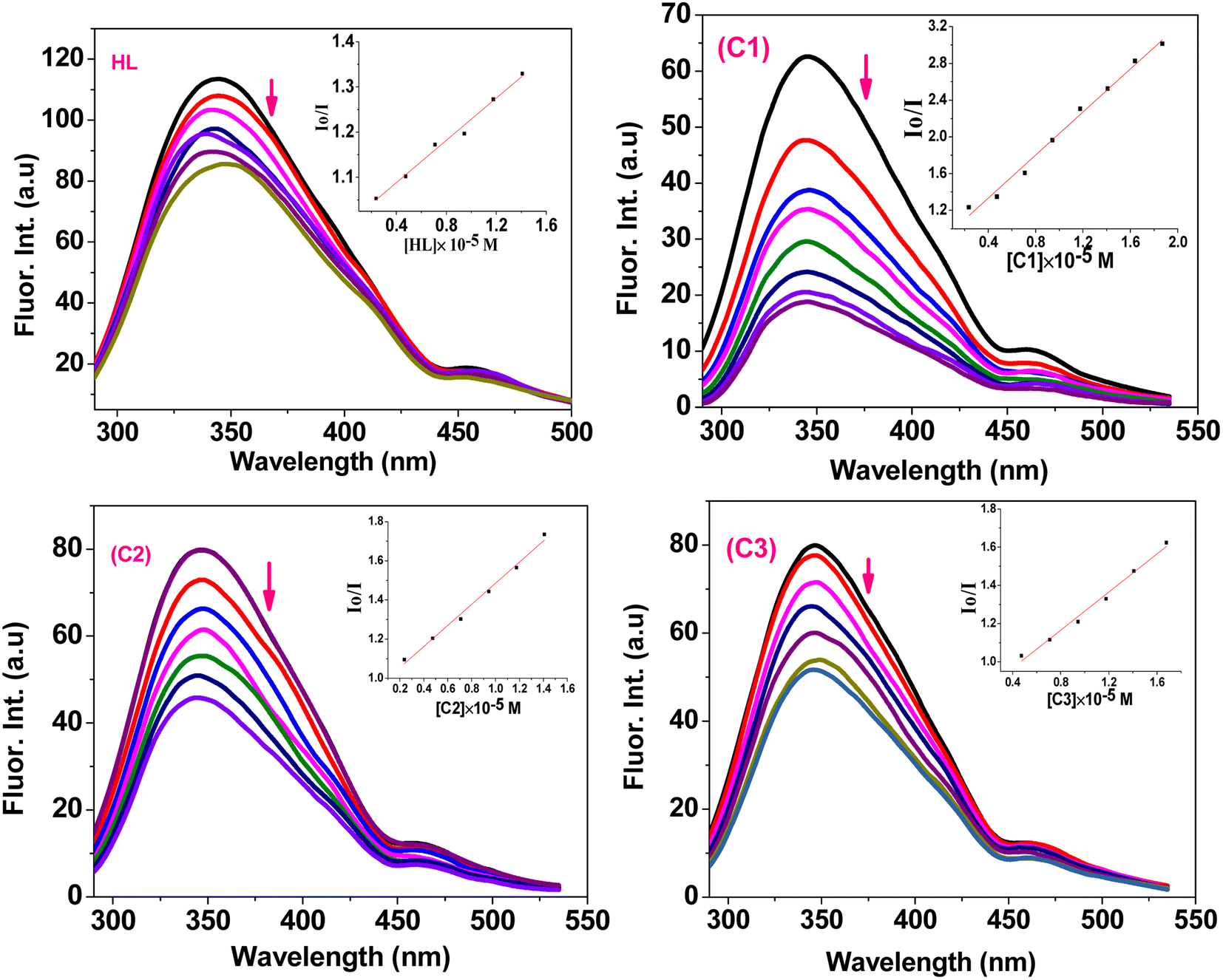
| | Fig. 5 Fluorescence quenching spectra of EB-ctDNA/EB-tRNA adduct in the presence of HL and C1–C3 compounds; [EB] = 5 μM; Left: [DNA] = 50 μM, [Q] = 10–200 μM, Rright: [RNA] = 50 μM, [Q] = 10–200 μM; (inset): plot of Io/I vs. [Q]. | |
The quenching constant (Ksv) values (Table 1) were determined by the Stern–Volmer eqn (2):67
where
Io and
I are the emission intensities of the test compound in the absence and presence of ctDNA/ tRNA , respectively,
KSV is the Stern–Volmer constant, and [
Q] is the concentration of the compound.
4.2. Bovine serum albumin (BSA) interaction studies
The protein interaction with metal complexes has become an interesting research area, where anticancer drugs are mostly administrated via intravenous injection into the bloodstream, where they are bound to plasma protein.68 The drug–protein binding strength displays a significant role in its accessibility to diffuse from the blood to the target.69 In the following section, we evaluated the binding affinity of BSA protein (human serum albumin analog) by employing absorption and emission measurements.
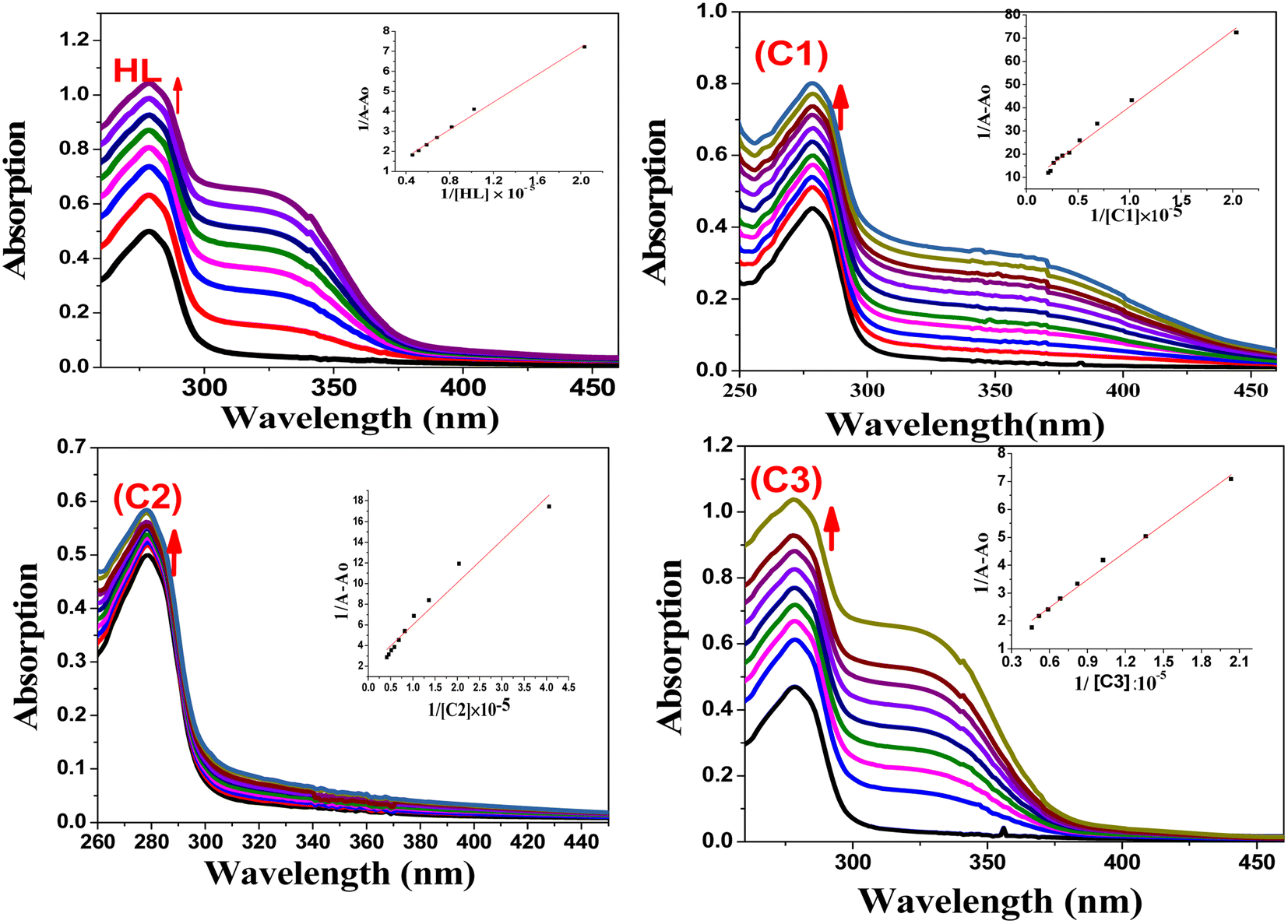
4.2.1. Absorption studies. Absorption spectroscopy is one of the most helpful techniques to examine the structural changes and quenching type of BSA by drug molecules. The absorption titrations of HL and its complexes (C1–C3) with BSA in PBS at physiological pH 7.2 are shown in Fig. 6. Generally, the interaction should occur in a static or dynamic mode. In the static mechanism, a non-fluorescent BSA-complex was formed in the ground state before excitation of any electrons and resulted in changes in the absorption profile. However, in the dynamic mechanism (collisional), the fluorophore interacts with the quencher during the temporary existence of the excited state. Additionally, the dynamic mechanism affects only the excited state, with no alternation on the absorption spectrum.70 In the case of our complexes, with the increasing amounts of the HL ligand or its complexes, the absorption intensity increased (with 11.9–40.74% hyperchromism) together with a blue-shift of 2–4 nm in the range of 260–450 nm. This is an indication of the formation of the BSA–complex in the ground state via a static interaction. These results also showed that the structural changes may be attributed to the non-covalent interactions; for instance, electrostatic and hydrogen bonding interactions between the HL ligand and the complexes with the BSA.71,72 The binding constants were evaluated by eqn (3)73,74 and are tabulated in Table 2.| | |
1/(Ao − A) = 1/Ao + 1/(K × Ao × CQ)
| (3) |
where Ao is the absorbance of unbound BSA, A is the absorbance of bound BSA (after the addition of different concentrations of the test compounds), respectively, K is the binding constant, and CQ is the compound concentration. The K value can be obtained from the linear plot of 1/(Ao − A) against 1/[comp.] from the intercept-to-slope ratio.
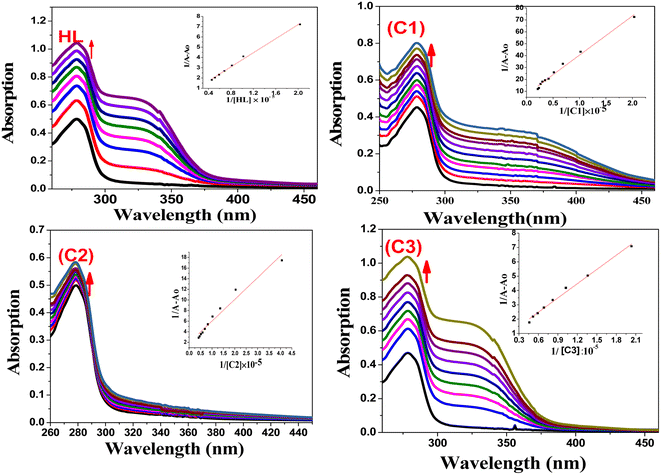 |
| | Fig. 6 Electronic absorption spectra of BSA (50 μM) in the presence of different concentrations of HL and C1–C3 complexes (0–50 μM) pH 7.2 (Tris–HCl buffer); (inset): linear plot of 1/[(A − Ao)] vs. 1/[compound]. | |
Table 2 Binding (Kb) and quenching constant (Kq) data and the number of binding sites (n) of BSA interaction with the titled compoundsa
| Absorption spectroscopy λmax = 278 nm |
Emission spectroscopy λmax = 345 nm |
| Compound |
(Kb), × 104 |
% hyper |
% hypo |
(Ksv), × 104 |
(Kq), × 1012 |
(Kb) |
n |
| The observed BSA binding constant (K) values for all the compounds are presented in (2). It was found that the C1 complex exhibited the highest binding strength with BSA in the order of C1 > C2 > C3 > HL. |
| HL |
1.0 |
39.03 |
27.18 |
2.33 |
2.33 |
8.9 × 103 |
0.93 |
| (C1) |
4.6 |
36.05 |
69.44 |
5.68 |
5.68 |
4.0 × 105 |
1.61 |
| (C2) |
2.5 |
11.90 |
42.63 |
5.39 |
5.39 |
3.9 × 104 |
1.03 |
| (C3) |
1.52 |
40.74 |
35.69 |
4.98 |
4.98 |
2.6 × 104 |
0.92 |
4.2.2. Emission studies. Emission spectroscopy is one of the most efficient tools to study the quenching mechanism, and binding interaction mode the compounds with BSA.75,76 The BSA solution is excited at 278 nm due to the presence of tyrosine and tryptophan residues in proteins, emitting a strong peak at 345 nm. Fig. 7 shows that the emission intensity (I) of BSA decreases upon addition of different amounts upon complexes (C1–C3) or the ligand HL with a slight shift in the peak position (∼2 nm). The results indicate that the BSA intrinsic fluorescence decreases due to the formation of a non-fluorescent complex with BSA. The Stern–Volmer eqn (2)77,78 mentioned above (Io/I = 1 + Ksv [Q]) was used to evaluate the fluorescence quenching constant. The Stern–Volmer quenching constant (Ksv) was obtained from the slope of the plot of Io/I vs. [compound]. The quenching rate constant, Kq was determined using eqn (4)where Kq is the quenching rate constant, and τ0 is the average lifetime of protein in absence of the quencher and it is taken as 10−8 s for BSA.79
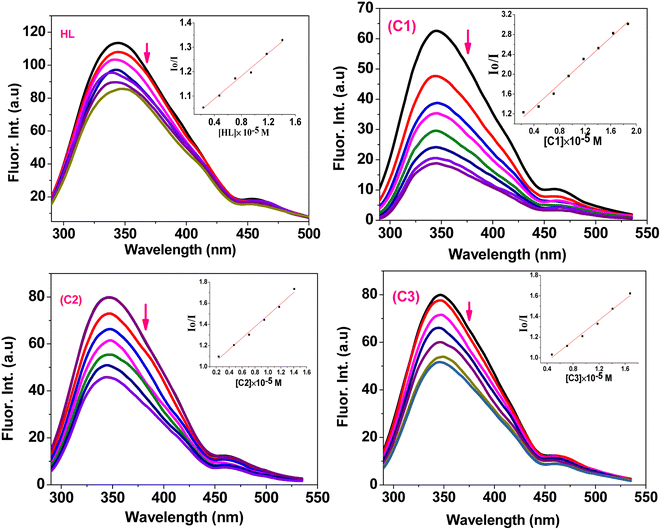 |
| | Fig. 7 Emission spectra of BSA (50 μM) upon addition of different concentrations of HL and complexes (C1–C3) (0–50 μM) in DMSO/PBS solution. (Inset): plots of Io/I vs. [compound]. | |
The maximum collision quenching constant (Kq) for biomacromolecules with various quenchers is 1010 M−1 s−1.80 In our study, the resulting Kq values were 0.5–6.1 × 1012 M−1 s−1, which were higher than 1010 M−1 s−1, confirming the static quenching (rather than dynamic) because of the formation of a ground state complex between BSA and the quenchers,81 which confirmed the data obtained from the absorption spectra.
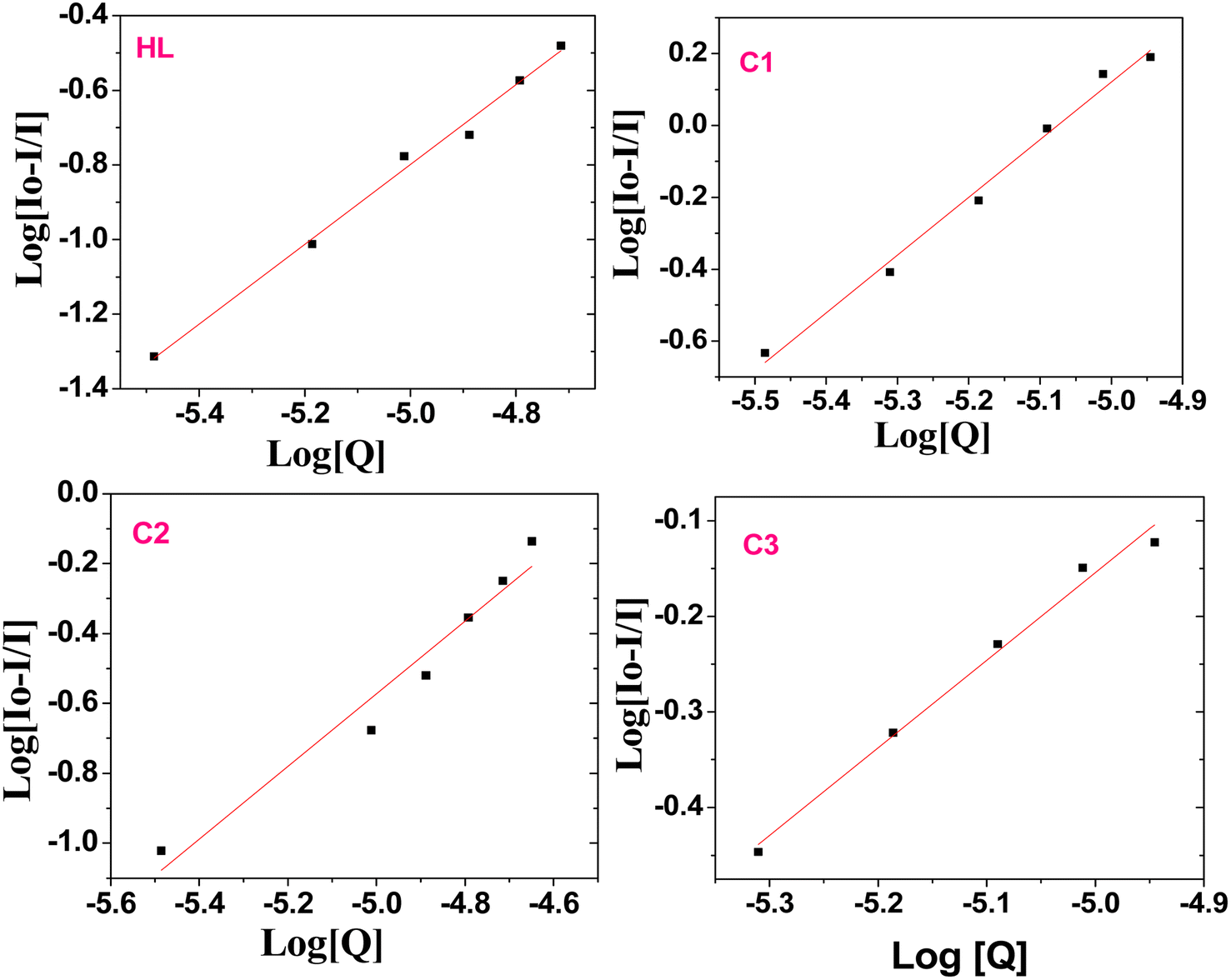
The static binding constant (Kb) and the number of binding sites can be further calculated from the modified Stern–Volmer eqn (5):69,82,83
| | |
log[(Io − I)/I] = logKb + nlog[Q]
| (5) |
Kb and
n can be determined from a plot of log[(
Io − I)/
I]
vs. log[
Q] (
Fig. 8), specifically from the slope and intercept values, respectively. The
n values for the serum albumin-complex were around the 1.0 value (
Table 2), indicating the presence of single binding site in BSA for the compound. Here, we can conclude that the binding ability of the compounds to the biomolecules would be very useful in drug design, where it can enhance the drug stability and toxicity during chemotherapeutic processes.
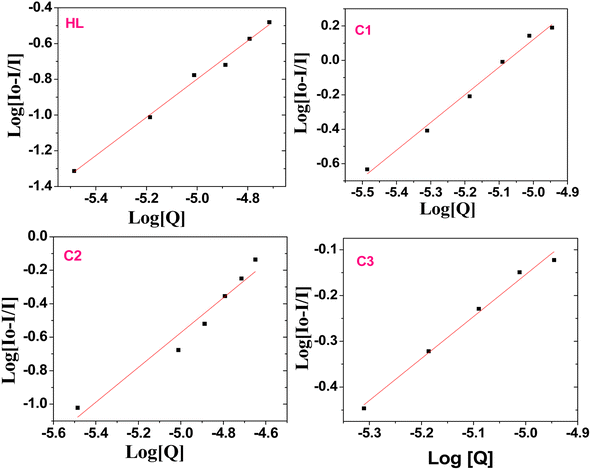 |
| | Fig. 8 Plot of log((Io − I)/I) versus log[Q] after adding various amounts of the ligand (HL) and complexes C1–C3 to BSA. | |
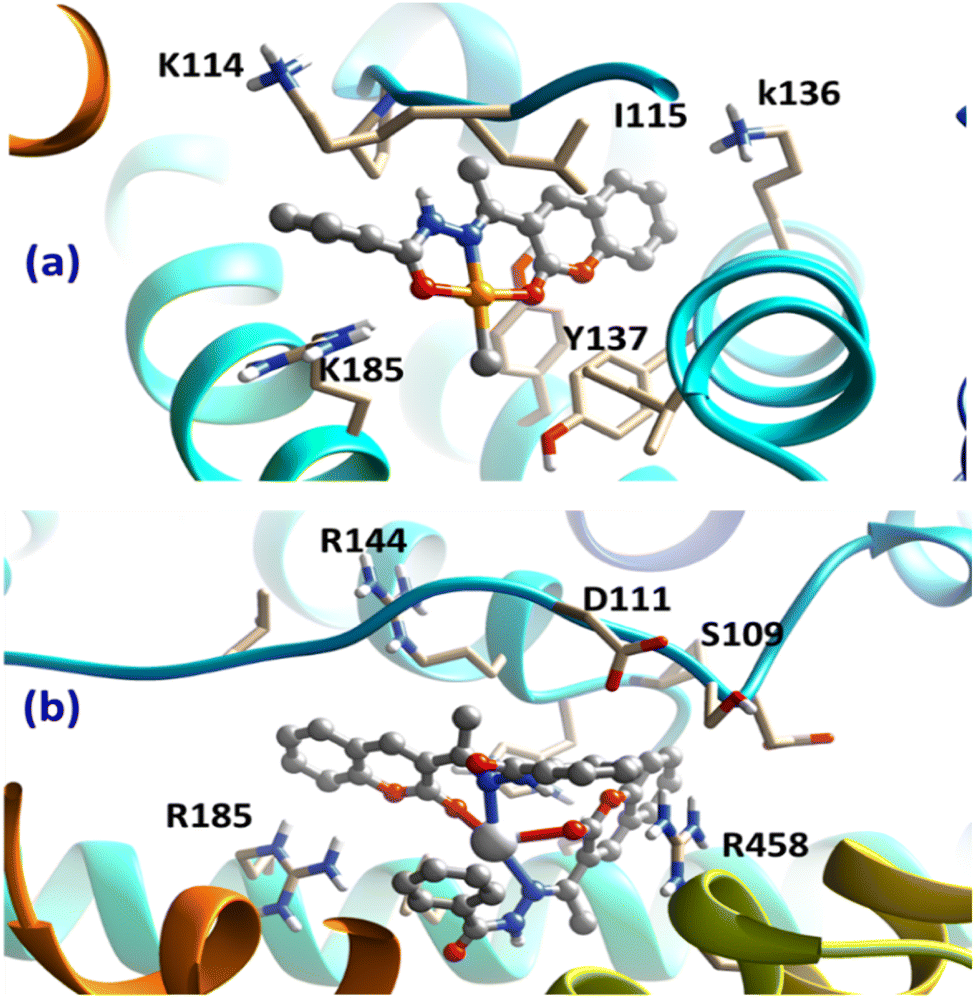
4.2.3. Molecular docking results. Our docking calculations for both the Pt(II)- and Ag(I)-containing complexes on the targeted BSA protein revealed the affinity of our complexes to the biomolecules. In particular, both complexes showed high binding ability with binding scores of −8.62 and −7.97 kJ mol−1 for the Pt(II) and Ag(I) complexes, respectively, Fig. 9.
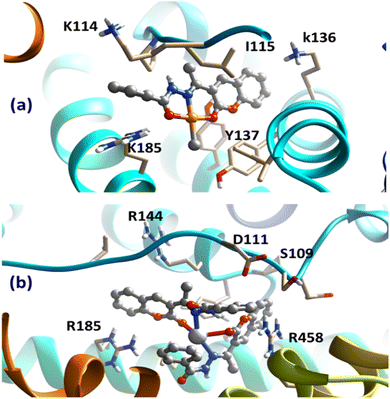 |
| | Fig. 9 Obtained bound complexes for (a) Pt(II) and (b) Ag(I) complexes with the BSA target. The key amino acids participating in the interactions are highlighted. | |
Despite binding to distinct locations over the BSA target, both ligands were involved in different types of interaction with the surrounding amino acids of the protein. Most importantly, the cation–π interaction between cationic amino acids, arginine and lysine in particular, with the aromatic domains of the chelating HL were the most dominant interaction. Interestingly, the subtle difference in the obtained binding scores of the two complexes suggested the binding preference of Pt(II)-containing compound over the Ag(I) one, in accordance with the experimental binding results. These obtained findings from the docking analysis further support the validity of the synthesized complexes to act as potential drugs.
4.3. In vitro anticancer activity
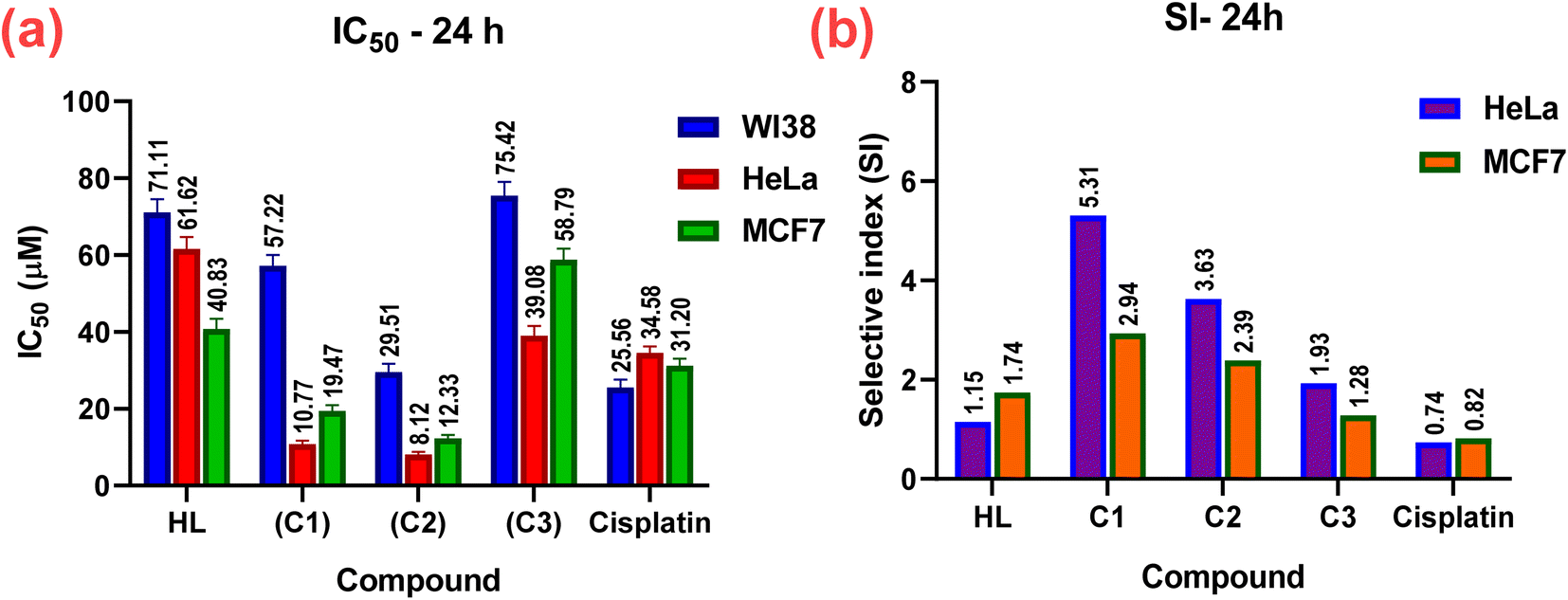
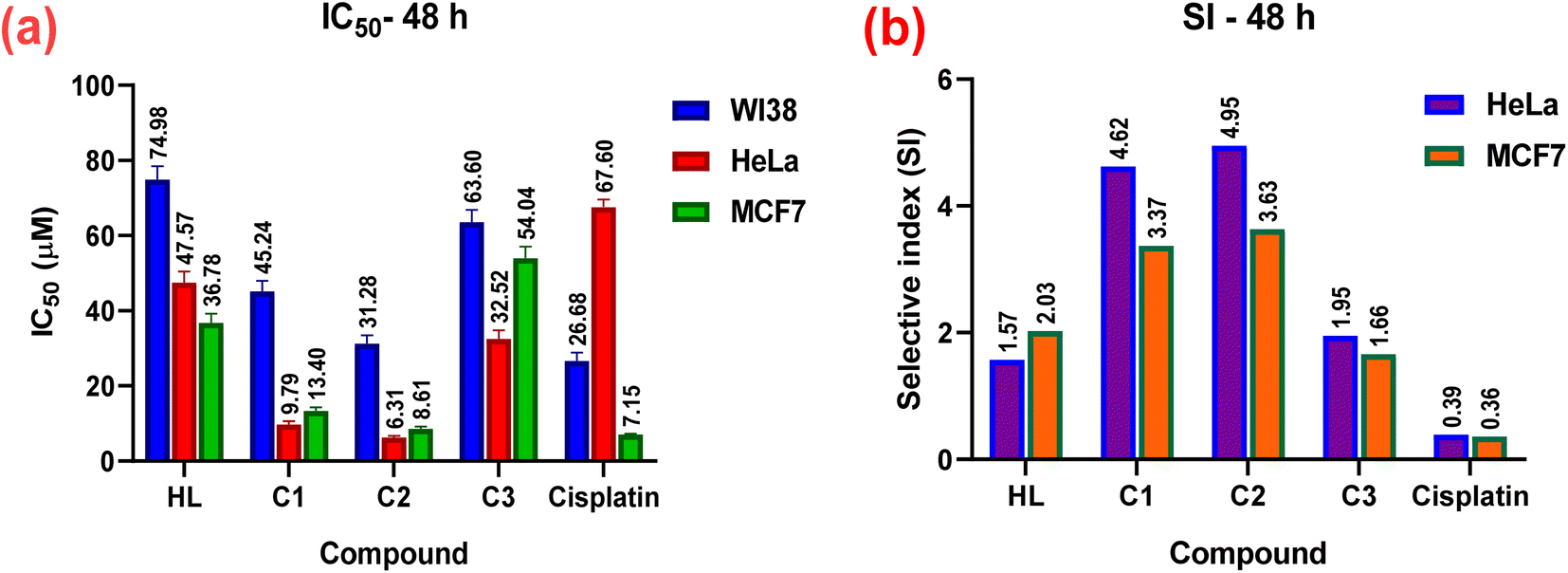
The in vitro cytotoxic effects of HL and its complexes (C1–C3) were evaluated by the colorimetric MTT assay against two human cancer cell lines, namely hormone-dependent breast (MCF7) and cervical (HeLa), in addition to a non-cancerous one, namely the human lung fibroblast (WI38), using cis-platin as a standard drug with exposure times of 24 and 48 h. The results of this study (Table 3, Fig. 10 and 11) showed that all the complexes possessed higher antitumor activity than their parent ligand based on the IC50 and selective index (SI) values. The data revealed that the HeLa cell lines were more sensitive to the complexes than the MCF7 cell lines, as observed from the IC50 and SI values (Table 3). Moreover, all the complexes were found to be more toxic than cis-platin in the HeLa and MCF7 cell lines.
Table 3 In vitro cytotoxic activity (IC50) and selective indices (SI) values of the ligand and its complexes (C1–C3) against WI38, HeLa, and MCF7 cell lines for 24 and 48 h
| Compound no. |
In vitro cytotoxicity IC50 (μM) and selective indices values |
| WI38 |
HeLa |
SI |
MCF7 |
SI |
| 24 h |
| HL |
71.11 ± 3.4 |
61.62 ± 3.1 |
1.15 |
40.83 ± 2.6 |
1.74 |
| (C1) |
57.22 ± 2.8 |
10.77 ± 0.9 |
5.31 |
19.47 ± 1.5 |
2.94 |
| (C2) |
29.51 ± 2.2 |
8.12 ± 0.7 |
3.63 |
12.33 ± 0.9 |
2.39 |
| (C3) |
75.42 ± 3.6 |
39.08 ± 2.5 |
1.93 |
58.79 ± 2.9 |
1.28 |
| Std. |
25.56 ± 2.1 |
34.58 ± 1.63 |
0.74 |
31.2 ± 1.8 (ref. 84) |
0.82 |
|
| 48 h |
| HL |
74.98 ± 3.5 |
47.57 ± 2.9 |
1.57 |
36.78 ± 2.4 |
2.03 |
| (C1) |
45.24 ± 2.7 |
9.79 ± 0.8 |
4.62 |
13.40 ± 0.9 |
3.37 |
| (C2) |
31.28 ± 2.2 |
6.31 ± 0.4 |
4.95 |
8.61 ± 0.6 |
3.63 |
| (C3) |
63.60 ± 3.3 |
32.52 ± 2.3 |
1.95 |
54.04 ± 3.0 |
1.66 |
| Cisplatin |
26.68 ± 2.2 |
67.6 ± 2.0 (ref. 85) |
0.39 |
72.38 ± 0.1 (ref. 86) |
0.36 |
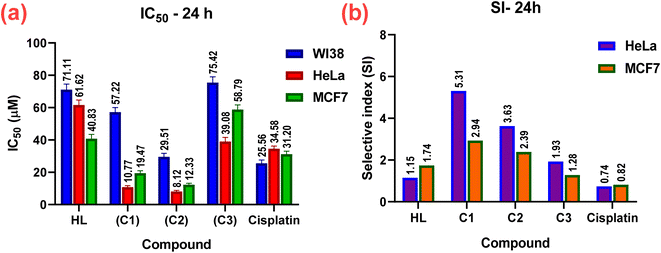 |
| | Fig. 10 In vitro cytotoxicity; (a) IC50 and (b) SI values of the complexes (C1–C3), their parent ligand (HL), and cis-platin (standard) against normal WI38, and cancerous HeLa, and MCF7 human cell lines at 24 h. | |
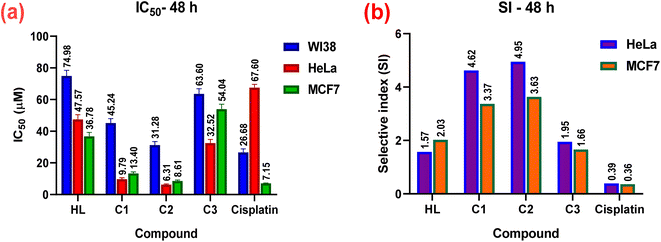 |
| | Fig. 11 In vitro cytotoxicity; (a) IC50 and (b) SI values of the complexes (C1–C3), their parent ligand (HL), and cis-platin against normal WI38, and cancerous HeLa, and MCF7 human cell lines at 48 h. | |
The cytotoxicity and the mechanism of metal complexes mainly depend on their geometry, oxidation state of the metal ion, and redox potential.87–89 According to the IC50 values, the Pt(II) complex (C2) showed the highest cytotoxic activity (i.e., a lower IC50, specifically, IC50 = 8.12 ± 0.7 for HeLa, and 12.33 ± 0.9 for MCF7), which may be due to the cationic nature of the Pt(II)86 rather than the neutral Pd(II) complex, where IC50 = 10.77 ± 0.9 for HeLa and 19.47 ± 1.5 for MCF7. It has been reported that Pd(II) complexes are less stable than Pt(II) ones, because they have a higher aquation rate.90–92 However, the SI values of the Pd(II) complex(C1) (SI = 5.31 for HeLa and 2.94 for MC7) were higher than those of the Pt(II) complex (C2) (SI = 3.63 for HeLa and 2.39 for MCF7), indicating that the Pd(II) complex was more selective and safe for both cell lines. The silver(I) complex had less effect, which may be attributed to the steric hindrance90,93,94 from the introduction of two ligands around the Ag(I) ion (or reduced electrophilicity), which retards the intercalation of the silver(I) complex with DNA (as shown from the binding study, mentioned before), and hence decreased its cytotoxic activity (higher IC50) for both cell lines, as shown in Table 3. It also demonstrated higher selectivity toward both cell lines. Among all the complexes, complex C1 showed the highest selectivity for the tumorous HeLa and MCF7 cells, so, it will be selected in the future for further investigations for better understanding the mechanism of action.
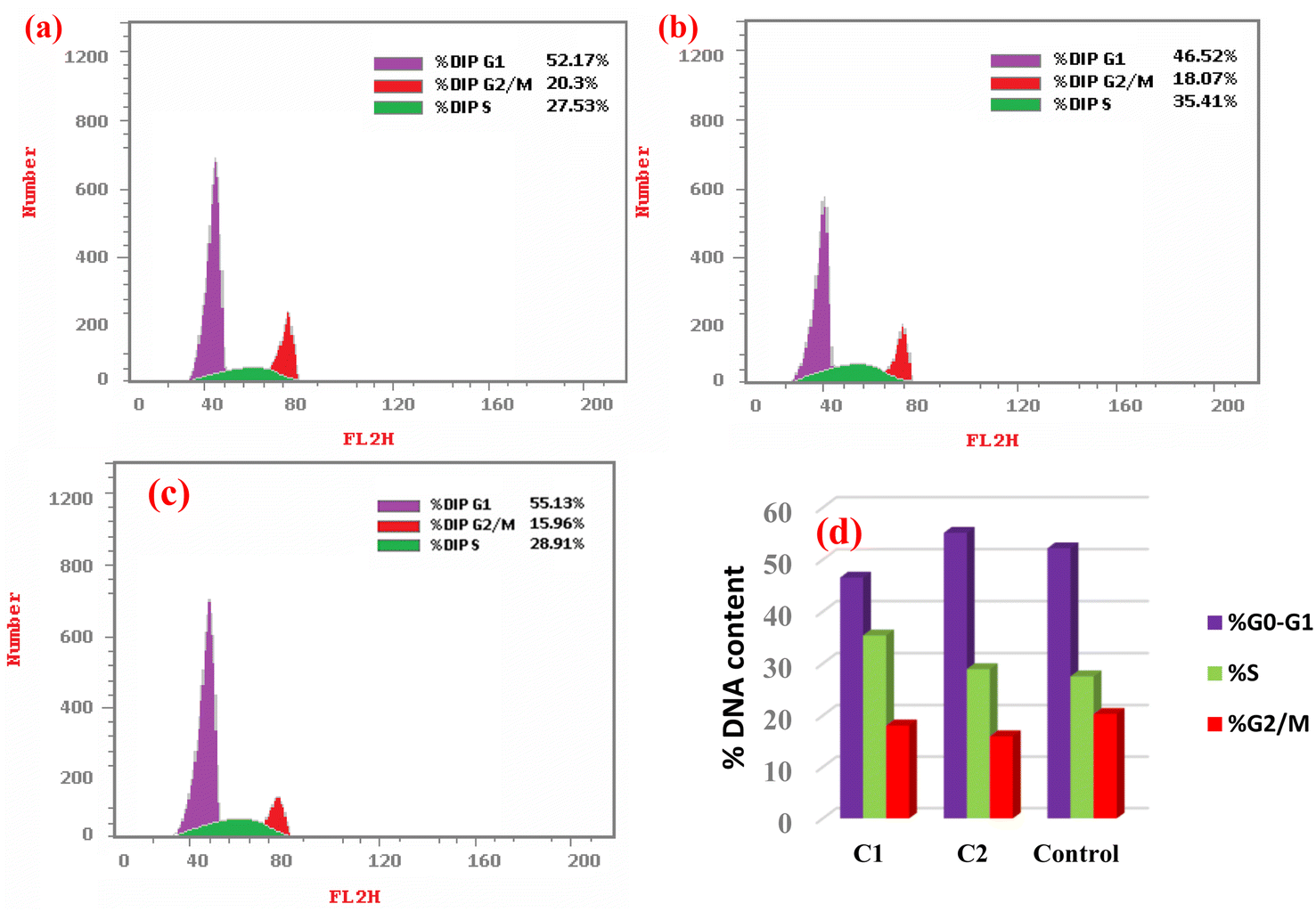
4.3.1. Cell cycle arrest by C1 and C2. Based on the binding affinity and cytotoxicity results, the complexes C1 and C2 were subsequently selected for further investigation on HeLa cells. The cell cycle distribution of HeLa cells was investigated by flow cytometry after being treated with the IC50 concentrations of either complex C1 or C2 (10.77 and 8.12 μM, respectively) for 24 h, as shown in Fig. 12. Cell cycle progression analysis showed that treatment of HeLa cells with complex C1 resulted in a significant decrease in the number of cells in the G0/G1 phase (46.52% compared to the control cells' 52.17%, p = 0.003). The G2/M phase did not show any significant change (18.07% compared to the control cells' 20.3%, p = 0.155). These findings suggest that the treatment with complex C1 prohibited the entrance of HeLa cells into a new cycle. Furthermore, there was a notable increase in the S-phase population percentage (35.41% compared to the control cells' 27.53%, p = 0.027) in HeLa cells treated with complex C1, indicating cell cycle arrest at the S phase. This arrest may be attributed to the binding of complex C1 with the DNA, which hindered the replication of DNA and the production of new cells.95 On the other hand, the cell cycle analysis for HeLa cells treated with complex C2 showed no significant changes in either the G0/G1 or S phases compared to the untreated HeLa cells. Conversely, there was a significant reduction in the G2/M phase (20.3% compared to the control cells' 15.96%, p = 0.042). These data suggest the cells were prevented from entering mitotic cell division.
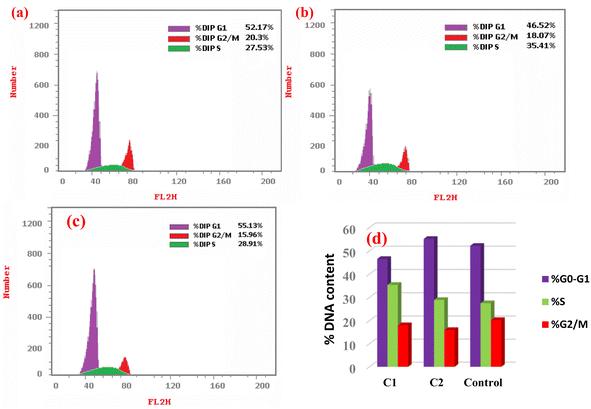 |
| | Fig. 12 Cell cycle phases (%) in (a) control (untreated HeLa cells), (b) HeLa treated with complex C1, (c) HeLa treated with complex C2 at IC50 concentrations for 24 h. (d) Plot presenting the cell cycle distribution (%). | |
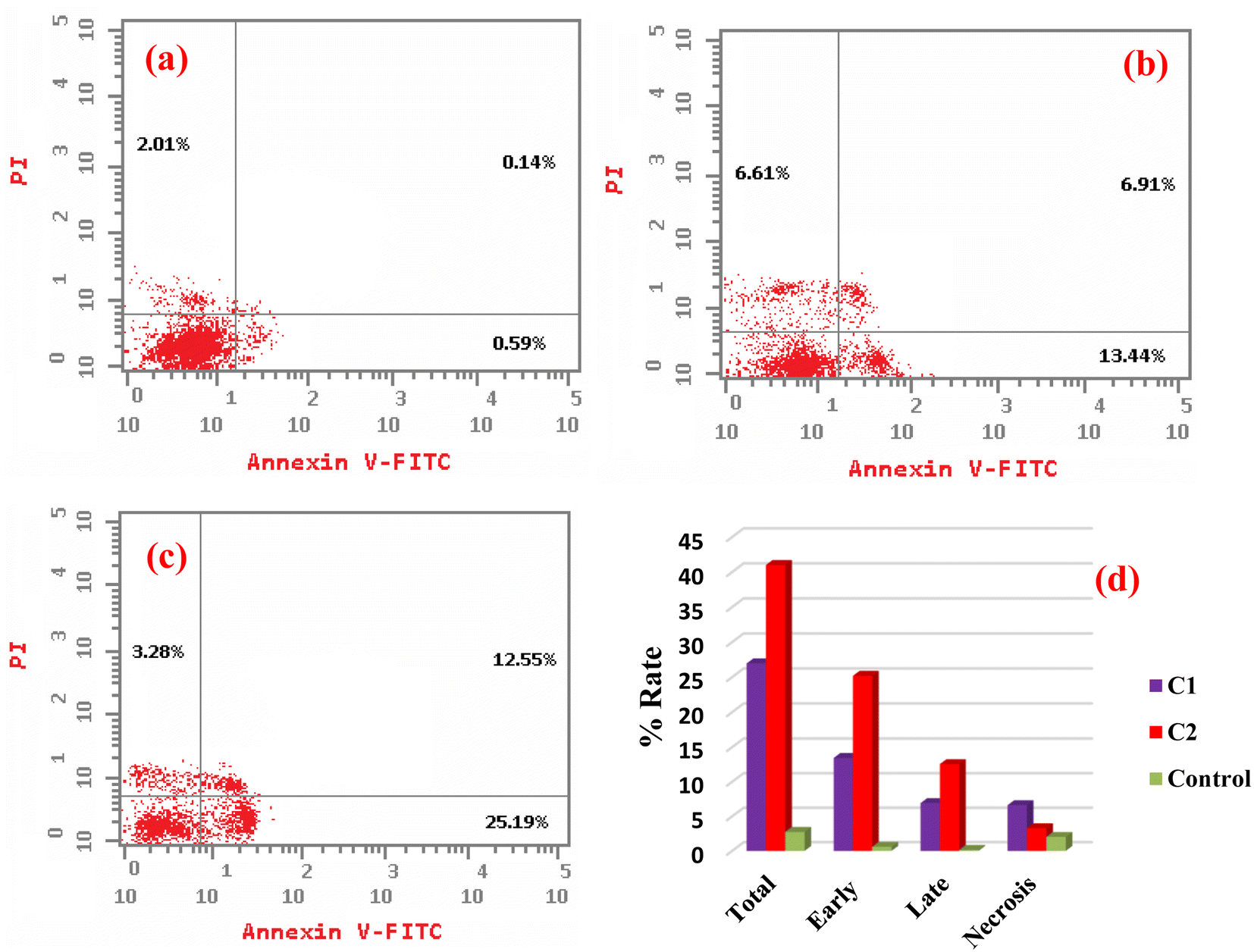
4.3.2. Induction of cell death mode by complexes C1 and C2. The induction of cell death was studied by flow cytometry using the Annexin V/propidium iodide (PI) method to distinguish between live and dead cells in four quadrants, namely Q1 (viable cells, lower left), Q2 (early apoptotic cells, lower right), Q3 (late apoptotic cells, upper right), and Q4 (necrotic cells, upper left), as shown in Fig. 13. After 24 h incubation of HeLa cells with either complexes C1 or C2 and by investigating their IC50, it was observed that the proportion of both early and late apoptotic cells was significantly increased for both complexes compared to untreated HeLa cells as the control, i.e., 23-fold and 49-fold for complex C1, and 42.7-fold and 89.6-fold for complex C1. Compared to the necrotic cells, there were no significant changes (3.3-fold for C1, and 1.6-fold for C2). The data suggest that these metal complexes demonstrating the cell death via early and late apoptosis.96
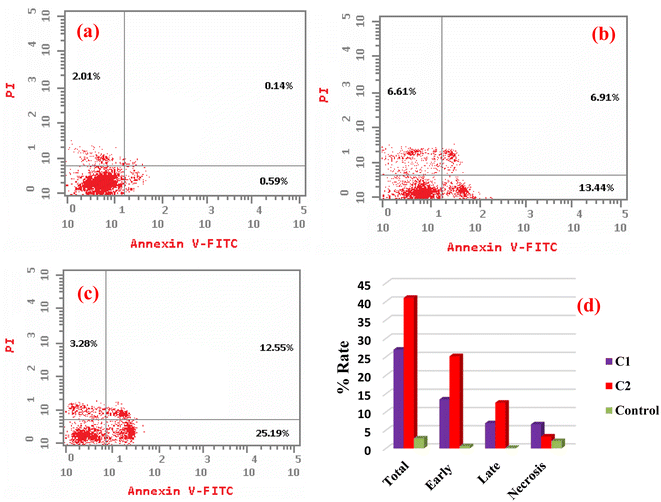 |
| | Fig. 13 Apoptotic/necrotic dot plots of (a) untreated HeLa cells, (b) cells treated with complex C1, (c) cells treated with complex C2 for 24 h. (d) Plot presenting the total, early, and late apoptosis, and necrosis percentage. | |
5. Conclusions
In this study, three new metal complexes of Pd(II), Pt(II), and Ag(I) (C1–C3) derived from 3-acetylcoumarin benzoylhydrazone (HL) were prepared and characterized by elemental analysis, spectroscopic techniques, and DFT calculations. The analytical data revealed that the ligand acted as an ONO-donor forming square planar geometries for the Pd(II) and Pt(II) complexes. The HL was coordinated to Pd(II) centers in its deprotonated form, while in the case of Pt(II) it was coordinated in its neutral form. However, in the Ag(I) complex it was coordinated as a neutral ON-donor. The binding affinity of all the compounds toward ctDNA, yeast tRNA, and BSA was assessed by electronic and fluorescence spectra. All the compounds showed the intercalative mode of binding, and the data revealed that the Pd(II) and Pt(II) complexes showed the highest binding strength (Kb) to these molecules. This latter observation was further emphasized by our molecular docking calculations, which provided a higher binding score for the Pt(II) complex than Ag(I). The key interaction responsible for the tight interaction between the targeted BSA and the two complexes was mainly a cation–pi interaction, where the cationic residues arginine and lysine represent the cationic motifs. Furthermore, the cytotoxic activity (in vitro) of the compounds was estimated against a non-cancerous WI38 and cancerous HeLa and MCF7 cell lines using the MTT assay for 24 and 48 h. The data showed that the Pd(II) and Pt(II) complexes had the highest toxicity based on the IC50 and selective indices (SI) values, consistent with the biomolecular interaction studies. Furthermore, the results from flow cytometry demonstrated that complexes C1 and C2 caused cell cycle arrest at S and G1/S, respectively.
Abbreviations
| B3LYP | Becke, 3-parameter, Lee–Yang–Parr |
| BSA | Bovine serum albumin |
| ctDNA | Calf thymus deoxyribonucleic acid |
| DFT | Density functional theory |
| DMSO | Dimethylsulfoxide |
| EB | Ethidium bromide |
| FTIR | Fourier-transform infrared |
| HeLa | Cervical carcinoma cell line |
| HOMO | Highest unoccupied molecular orbital |
| Kb | Intrinsic binding constant |
| Ksv | Stern–Volmer constant |
| Kq | Quenching binding constant |
| LANL2DZ | Los Alamos National Laboratory 2 double-zeta |
| LMCT | Ligand-to-metal charge transfer |
| LUMO | Lowest unoccupied molecular orbital |
| MCF7 | Mammary gland breast cancer |
| MGL | Machine graded lumber |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide |
| NA | Nucleic acid |
| NMR | Nuclear magnetic resonance |
| PBS | Phosphate buffered saline |
| PI | Propidium iodide |
| SI | Selective index |
| tRNA | Transfer ribonucleic acid |
| Tris–HCl | Tris(hydroxymethyl)aminomethane hydrochloride |
| UV-vis | Ultraviolet-visible spectroscopy |
| WI38 | Human lung fibroblast cell line |
| εa | Apparent extinction coefficients |
| εb | Extinction coefficients bound compound |
| εf | Extinction coefficients of the free complex |
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Author contributions
Shadia A. Elsayed: supervision, conceptualization, formal analysis, software, resources, visualization, methodology, software, validation, writing – original draft. Islam M. Elnabky: software, validation, methodology. Ahmed M. El-Hendawy: supervision, conceptualization, visualization, writing – review & editing. Mohamed M. Aboelnga: supervision, visualization, data curation, software, formal analysis, calculation, writing – original draft.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
We acknowledge the Ministry of Higher Education of Egypt for CIQAP (CP3-016-MAN) project, and the Academy of Scientific Research and Technology (ASRT), Egypt, under initiatives of science up capacity building (Grant No. 6389) for research equipment funds.
References
- T. Pereira, D. Franco, F. Vitorio and A. Kummerle, Curr. Top. Med. Chem., 2018, 18, 124–148 CrossRef CAS PubMed.
- O. Halter and H. Plenio, Eur. J. Inorg. Chem., 2018, 2018, 2935–2943 CrossRef CAS.
- P. Manojkumar, T. K. Ravi and G. Gopalakrishnan, Acta Pharm., 2009, 59, 159–168 CAS.
- M. P. Sathisha, U. N. Shetti, V. K. Revankar and K. S. R. Pai, Eur. J. Med. Chem., 2008, 43, 2338–2346 CrossRef CAS PubMed.
- M. Mladenović, N. Vuković, S. Sukdolak and S. Solujić, Molecules, 2010, 15, 4294–4308 CrossRef PubMed.
- M. P. Sathisha, U. N. Shetti, V. K. Revankar and K. S. R. Pai, Eur. J. Inorg. Chem., 2008, 43, 2338–2346 CAS.
- S. A. Patil, V. Kandathil, A. Sobha, S. B. Somappa, M. R. Feldman, A. Bugarin and S. A. Patil, Molecules, 2022, 27, 5220 CrossRef CAS PubMed.
- S. Balcioğlu, M. Karataş, B. Ates, B. Alici and I. Ozdemir, Bioorg. Med. Chem. Lett., 2020, 30, 126805 CrossRef PubMed.
- C. Ranjan Sahoo, J. Sahoo, M. Mahapatra, D. Lenka, P. Kumar Sahu, B. Dehury, R. Nath Padhy and S. Kumar Paidesetty, Arab. J. Chem., 2021, 14, 102922 CrossRef CAS.
- B. S. Creaven, E. Czeglédi, M. Devereux, É. A. Enyedy, A. Foltyn-Arfa Kia, D. Karcz, A. Kellett, S. McClean, N. V. Nagy, A. Noble, A. Rockenbauer, T. Szabó-Plánka and M. Walsh, Dalton Trans., 2010, 39, 10854–10865 RSC.
- A. Pangal, J. Shaikh and E. Khan, Int. J. Pharmaceut. Sci. Rev. Res., 2017, 42, 161–168 CAS.
- S. Aslkhademi, N. Noshiranzadeh, M. S. Sadjadi, K. Mehrani and N. Farhadyar, Polyhedron, 2019, 160, 115–122 CrossRef CAS.
- R. Kaplánek, M. Havlík, B. Dolenský, J. Rak, P. Džubák, P. Konečný, M. Hajdúch, J. Králová and V. Král, Bioorg. Med. Chem., 2015, 23, 1651–1659 CrossRef PubMed.
- S. Omidi and A. Kakanejadifard, RSC Adv., 2020, 10, 30186–30202 RSC.
- G. Le Goff and J. Ouazzani, Bioorg. Med. Chem., 2014, 22, 6529–6544 CrossRef CAS PubMed.
- Y. Li, Z. Yang, M. Zhou, J. He, X. Wang, Y. Wu and Z. Wang, J. Mol. Struct., 2017, 1130, 818–828 CrossRef CAS.
- P. K. Suganthy, R. N. Prabhu and V. S. Sridevi, Polyhedron, 2015, 88, 57–62 CrossRef CAS.
- R. N. Prabhu and R. Ramesh, J. Organomet. Chem., 2012, 718, 43–51 CrossRef CAS.
- H. H. Monfared, M. Vahedpour, M. M. Yeganeh, M. Ghorbanloo, P. Mayer and C. Janiak, Dalton Trans., 2011, 40, 1286–1294 RSC.
- S. A. Elsayed, I. M. Elnabky, A. di Biase and A. M. El-Hendawy, Appl. Organomet. Chem., 2022, 36, e6481 CrossRef CAS.
- T. Nasr, S. Bondock and M. Youns, Eur. J. Med. Chem., 2014, 76, 539–548 CrossRef CAS PubMed.
- M. Mishra, K. Tiwari, S. Shukla, R. Mishra and V. P. Singh, Spectrochim. Acta, Part A, 2014, 132, 452–464 CrossRef CAS PubMed.
- I. Ali, K. Saleem, D. Wesselinova and A. Haque, Med. Chem. Res., 2013, 22, 1386–1398 CrossRef CAS.
- P. P. Netalkar, A. Kamath, S. P. Netalkar and V. K. Revankar, Spectrochim. Acta, Part A, 2012, 97, 762–770 CrossRef CAS PubMed.
- Y.-h. Li, B.-d. Wang and Z.-y. Yang, Spectrochim. Acta, Part A, 2007, 67, 395–401 CrossRef PubMed.
- Ł. Balewski, S. Szulta, A. Jalińska and A. Kornicka, Front. Chem., 2021, 9, 781779 CrossRef PubMed.
- C. T. G. Retnam, S. V. Rose and B. S. Kumari, J. Mol. Struct., 2023, 1282, 135162 CrossRef CAS.
- L. Dkhar, A. K. Verma, V. Banothu, W. Kaminsky and M. R. Kollipara, Appl. Organomet. Chem., 2022, 36, e6589 CrossRef CAS.
- S. A. Elsayed, H. M. El-Gharabawy, I. S. Butler and F. M. Atlam, Appl. Organomet. Chem., 2020, 34, e5643 CrossRef CAS.
- S. A. Aboafia, S. A. Elsayed, A. K. El-Sayed and A. M. El-Hendawy, J. Mol. Struct., 2018, 1158, 39–50 CrossRef CAS.
- S. A. Elsayed, I. M. Elnabky, A. di Biase and A. M. El-Hendawy, Appl. Organomet. Chem., 2022, 36, e6481 CrossRef CAS.
- M. M. Aboelnga, RSC Adv., 2022, 12, 15543–15554 RSC.
- M. M. Aboelnga and S. Kalyaanamoorthy, ACS Sustainable Chem. Eng., 2022, 48, 15857–15868 CrossRef.
- R. Kaur, M. M. Aboelnga, D. J. Nikkel and S. D. Wetmore, Phys. Chem. Chem. Phys., 2022, 24, 29130–29140 RSC.
- M. M. Aboelnga and J. W. Gauld, J. Phys. Chem. B, 2017, 25, 6163–6174 CrossRef PubMed.
- M. M. Aboelnga, J. J. Hayward and J. W. Gauld, ACS Catal., 2017, 7, 51805193 Search PubMed.
- S. Forli, R. Huey and M. Pique, et al., Nat. Protoc., 2016, 11, 905–919 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 16, 2785–2791 CrossRef PubMed.
- S. D. Rossella Castagna, P. Colnago, A. Serafini, E. Parisini and C. Bertarelli, ACS Omega, 2019, 8, 13270–13278 CrossRef PubMed.
- T. D. Goddard, E. F. Pettersen, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25(13), 1605–1612 CrossRef PubMed.
- A. Hussain, M. F. AlAjmi, M. T. Rehman, S. Amir, F. M. Husain, A. Alsalme, M. A. Siddiqui, A. A. AlKhedhairy and R. A. Khan, Sci. Rep., 2019, 9, 1–17 CrossRef PubMed.
- M. Carcelli, P. Cozzini, T. Maccagni, C. Pelizzi and L. Righi, Inorg. Chim. Acta, 2000, 303, 238–243 CrossRef CAS.
- F. Hueso-Ureña, N. A. Illán-Cabeza, M. N. Moreno-Carretero, A. L. Peñas-Chamorro and R. Faure, Polyhedron, 2000, 19, 689–693 CrossRef.
- F. B. Tamboura, P. M. Haba, M. Gaye, A. S. Sall, A. H. Barry and T. Jouini, Polyhedron, 2004, 23, 1191–1197 CrossRef.
- R. S. Hunoor, B. R. Patil, D. S. Badiger, R. S. Vadavi, K. B. Gudasi, V. M. Chandrashekhar and I. S. Muchchandi, Spectrochim. Acta, Part A, 2010, 77, 838–844 CrossRef PubMed.
- R. C. Maurya and S. Rajput, J. Mol. Struct., 2007, 833, 133–144 CrossRef CAS.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Part B, Applications in Coordination, Organometallic, and BioInorg. Chem., John Wiley & Sons, Inc., 6th edn, 2009 Search PubMed.
- M. Ghorbanloo, R. Bikas and G. Małecki, Inorg. Chim. Acta, 2016, 445, 8–16 CrossRef CAS.
- V. Kamat, A. Kotian, A. Nevrekar, K. Naik, D. Kokare and V. K. Revankar, Inorg. Chim. Acta, 2017, 466, 625–631 CrossRef CAS.
- G. S. Kurdekar, S. M. Puttanagouda, N. V. Kulkarni, S. Budagumpi and V. K. Revankar, Med. Chem. Res., 2011, 20, 421–429 CrossRef CAS.
- R. S. Hunoor, B. R. Patil, D. S. Badiger, R. S. Vadavi, K. B. Gudasi, V. Chandrashekhar and I. Muchchandi, Spectrochim. Acta, Part A, 2010, 77, 838–844 CrossRef PubMed.
- I. L. Paiva, G. S. G. de Carvalho, A. D. da Silva, P. P. Corbi, F. R. G. Bergamini, A. L. B. Formiga, R. Diniz, W. R. do Carmo, C. Q. F. Leite, F. R. Pavan and A. Cuin, Polyhedron, 2013, 62, 104–109 CrossRef CAS.
- L. M. Fostiak, I. García, J. K. Swearingen, E. Bermejo, A. Castiñeiras and D. X. West, Polyhedron, 2003, 22, 83–92 CrossRef CAS.
- N. K. Ngan, K. M. Lo and C. S. R. Wong, Polyhedron, 2012, 33, 235–251 CrossRef CAS.
- S. A. Elsayed, E. E. Saleh, M. M. Aboelnga and E. A. Toson, J. Inorg. Biochem., 2023, 241, 112132 CrossRef CAS PubMed.
- A. M. Ismail, S. A. El Sayed, I. S. Butler and S. I. Mostafa, J. Mol. Struct., 2020, 1200, 127088 CrossRef CAS.
- M. Muralisankar, J. Haribabu, N. S. Bhuvanesh, R. Karvembu and A. Sreekanth, Inorg. Chim. Acta, 2016, 449, 82–95 CrossRef CAS.
- G. Ayyannan, M. Mohanraj, M. Gopiraman, R. Uthayamalar, G. Raja, N. Bhuvanesh, R. Nandhakumar and C. Jayabalakrishnan, Inorg. Chim. Acta, 2020, 119868 CrossRef CAS.
- V. Censi, A. B. Caballero, M. Perez-Hernandez, V. Soto-Cerrato, L. Korrodi-Gregorio, R. Perez-Tomas, M. M. Dell'Anna, P. Mastrorilli and P. Gamez, J. Inorg. Biochem., 2019, 198, 110749 CrossRef CAS PubMed.
- S. Delaney, M. Pascaly, P. K. Bhattacharya, K. Han and J. K. Barton, Inorg. Chem., 2002, 41, 1966–1974 CrossRef CAS PubMed.
- K. L. Bergeron, E. L. Murphy, O. Majofodun, L. D. Muñoz, J. C. Williams and K. H. Almeida, Mutat. Res. Genet. Toxicol. Environ. Mutagen, 2009, 673, 141–148 CrossRef CAS PubMed.
- K. Sudeepa, N. Narsimha, B. Aparna, S. Sreekanth, A. V. Aparna, M. Ravi, J. Mohmed and C. S. Devi, J. Chem. Sci., 2018, 130, 52 CrossRef.
- Y. Jung and S. J. Lippard, Chem. Rev., 2007, 107, 1387–1407 CrossRef CAS PubMed.
- H.-K. Liu and P. J. Sadler, Acc. Chem. Res., 2011, 44, 349–359 CrossRef CAS PubMed.
- E. Movahedi, A. R. Rezvani and H. Razmazma, Int. J. Biol. Macromol., 2019, 126, 1244–1254 CrossRef CAS PubMed.
- A. Wolfe, G. H. Shimer Jr and T. Meehan, Biochemistry, 1987, 26, 6392–6396 CrossRef CAS PubMed.
- Y. Sun, S. Bi, D. Song, C. Qiao, D. Mu and H. Zhang, Sens. Actuators, B, 2008, 129, 799–810 CrossRef CAS PubMed.
- M. V. Babak, S. M. Meier, A. A. Legin, M. S. Adib Razavi, A. Roller, M. A. Jakupec, B. K. Keppler and C. G. Hartinger, Chem.–Eur. J., 2013, 19, 4308–4318 CrossRef CAS PubMed.
- V. D. Suryawanshi, L. S. Walekar, A. H. Gore, P. V. Anbhule and G. B. Kolekar, J. Biol. Chem., 2016, 6, 56–63 Search PubMed.
- S. Kathiresan, S. Mugesh, M. Murugan, F. Ahamed and J. Annaraj, RSC Adv., 2016, 6, 1810–1825 RSC.
- M. Idowu, E. Lamprecht and T. Nyokong, J. Photochem. Photobiol. Chem., 2008, 198, 7–12 CrossRef CAS.
- K. Sakthikumar, R. V. Solomon and J. D. Raja, RSC Adv., 2019, 9, 14220–14241 RSC.
- M. Anjomshoa, S. J. Fatemi, M. Torkzadeh-Mahani and H. Hadadzadeh, Spectrochim. Acta, Part A, 2014, 127, 511–520 CrossRef CAS PubMed.
- D. Xu, X. Wang, D. Fei and L. Ding, Nucleos Nucleot., 2010, 29, 854–866 CrossRef CAS PubMed.
- A. Ray, B. K. Seth, U. Pal and S. Basu, Spectrochim. Acta, Part A, 2012, 92, 164–174 CrossRef CAS PubMed.
- J. Toneatto and G. A. Argüello, J. Inorg. Biochem., 2011, 105, 645–651 CrossRef CAS PubMed.
- M. H. Chowdhury, K. Aslan, S. N. Malyn, J. R. Lakowicz and C. D. Geddes, J. Fluoresc., 2006, 16, 295–299 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer science & business media, 2013 Search PubMed.
- J. R. Lakowicz and G. Weber, Biochem., 1973, 12, 4161–4170 CrossRef CAS PubMed.
- W. R. Ware, J. Phys. Chem., 1962, 66, 455–458 CrossRef CAS.
- X. Zhang, S. Li, L. Yang and C. Fan, Spectrochim. Acta, Part A, 2007, 68, 763–770 CrossRef PubMed.
- E. Froehlich, J. Mandeville, C. Jennings, R. Sedaghat-Herati and H. Tajmir-Riahi, J. Phys. Chem. B, 2009, 113, 6986–6993 CrossRef CAS PubMed.
- N. Wang, L. Ye, F. Yan and R. Xu, Int. J. Pharm., 2008, 351, 55–60 CrossRef CAS PubMed.
- R. A. Khan, I. I. BinSharfan, S. S. Alterary, H. Alsaeedi, F. A. Qais, A. AlFawaz, A. D. Hadi and A. Alsalme, Appl. Organomet. Chem., 2022, 36, e6550 CrossRef CAS.
- P. Wiji Prasetyaningrum, A. Bahtiar and H. Hayun, Sci. Pharm., 2018, 86, 25 CrossRef PubMed.
- S. A. Elsayed, H. E. Badr, A. di Biase and A. M. El-Hendawy, J. Inorg. Biochem., 2021, 223, 111549 CrossRef CAS PubMed.
- S. Roy, S. Saha, R. Majumdar, R. R. Dighe and A. R. Chakravarty, Polyhedron, 2010, 29, 3251–3256 CrossRef CAS.
- D. A. Guk, O. O. Krasnovskaya and E. K. Beloglazkina, Russ. Chem. Rev., 2021, 90, 1566 CrossRef.
- H. Y. Khan, M. O. Ansari, G. Shadab, S. Tabassum and F. Arjmand, Bioorg. Chem., 2019, 88, 102963 CrossRef CAS PubMed.
- M. Marloye, G. Berger, M. Gelbcke and F. Dufrasne, Future Med. Chem., 2016, 8, 2263–2286 CrossRef CAS PubMed.
- Y. Wang, J. Hu, Y. Cai, S. Xu, B. Weng, K. Peng, X. Wei, T. Wei, H. Zhou, X. Li and G. Liang, J. Med. Chem., 2013, 56, 9601–9611 CrossRef CAS PubMed.
- K. A. Abu-Safieh, A. S. Abu-Surrah, H. D. Tabba, H. A. AlMasri, R. M. Bawadi, F. M. Boudjelal and L. H. Tahtamouni, J. Chem., 2016, 2016, 508724 Search PubMed.
- S. D. Brown, K. D. Trotter, O. B. Sutcliffe, J. A. Plumb, B. Waddell, N. E. B. Briggs and N. J. Wheate, Dalton Trans., 2012, 41, 11330–11339 RSC.
- S. K. Liew, S. Malagobadan, N. M. Arshad and N. H. Nagoor, Biomolecules, 2020, 10, 138 CrossRef CAS PubMed.
- C.-R. Sihn, E.-J. Suh, K.-H. Lee, T.-Y. Kim and S. H. Kim, Cancer Lett., 2003, 201, 203–210 CrossRef CAS PubMed.
- I. Vermes, C. Haanen and C. Reutelingsperger, J. Immunol. Methods, 2000, 243, 167–190 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Islam M. Elnabky
*,
Islam M. Elnabky