
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNickel-catalyzed γ-alkylation of cyclopropyl ketones with unactivated primary alkyl chlorides: balancing reactivity and selectivity via halide exchange†
Zheng-Ying Wang,
Shi-Zheng Liu,
Cong Guo,
Yi-Zheng Cheng,
Qiang Li *,
Jianmin Dou* and
Dacheng Li*
*,
Jianmin Dou* and
Dacheng Li*
Shandong Provincial Key Laboratory of Chemical Energy Storage and Novel Cell Technology, School of Chemistry and Chemical Engineering, Liaocheng University, Liaocheng 252000, P. R. China. E-mail: liqiang9@lcu.edu.cn; jmdou@lcu.edu.cn; lidacheng62@163.com
First published on 22nd April 2024
Abstract
A novel method was developed for synthesizing γ-alkyl ketones via nickel-catalyzed cross-electrophile coupling of cyclopropyl ketones and non-activated primary alkyl chlorides. High reactivity and selectivity can be achieved with sodium iodide as a crucial cocatalyst that generates a low concentration of alkyl iodide via halide exchange, thus avoiding the formation of alkyl dimers. This reaction possessed excellent regioselectivity and high step economy circumventing in situ or pregenerated organometallics.
Ketones are among the most versatile and fundamental functional groups in organic synthesis, widely present in natural products, pharmaceuticals, and materials.1 More efforts have been committed to developing novel and efficient synthetic ketone methods over the past decades.2 The emergence of cross-electrophile coupling reactions provided a turning point for synthesizing ketones.3 A large number of commercially available coupling reagents were exploited, significantly increasing the substrate range of the ketones and avoiding the use of low abundance starting materials. While progress in the synthesis of ketones has been obtained to a certain extent, modifying the remote sites of ketones, specifically those bearing γ-substitutions, remains challenging. Only limited examples of the highly stereoselective synthesis for γ-functionalized ketones have been reported. For example, Studer et al.4 developed a photoredox-catalyzed γ-C(sp3)–H functionalization of ketones using the stoichiometric α-aminoxy propionic acid as an auxiliary (Scheme 1A). Thus, developing new coupling partners without auxiliaries directly to prepare the γ-functionalized ketones is still highly desirable.
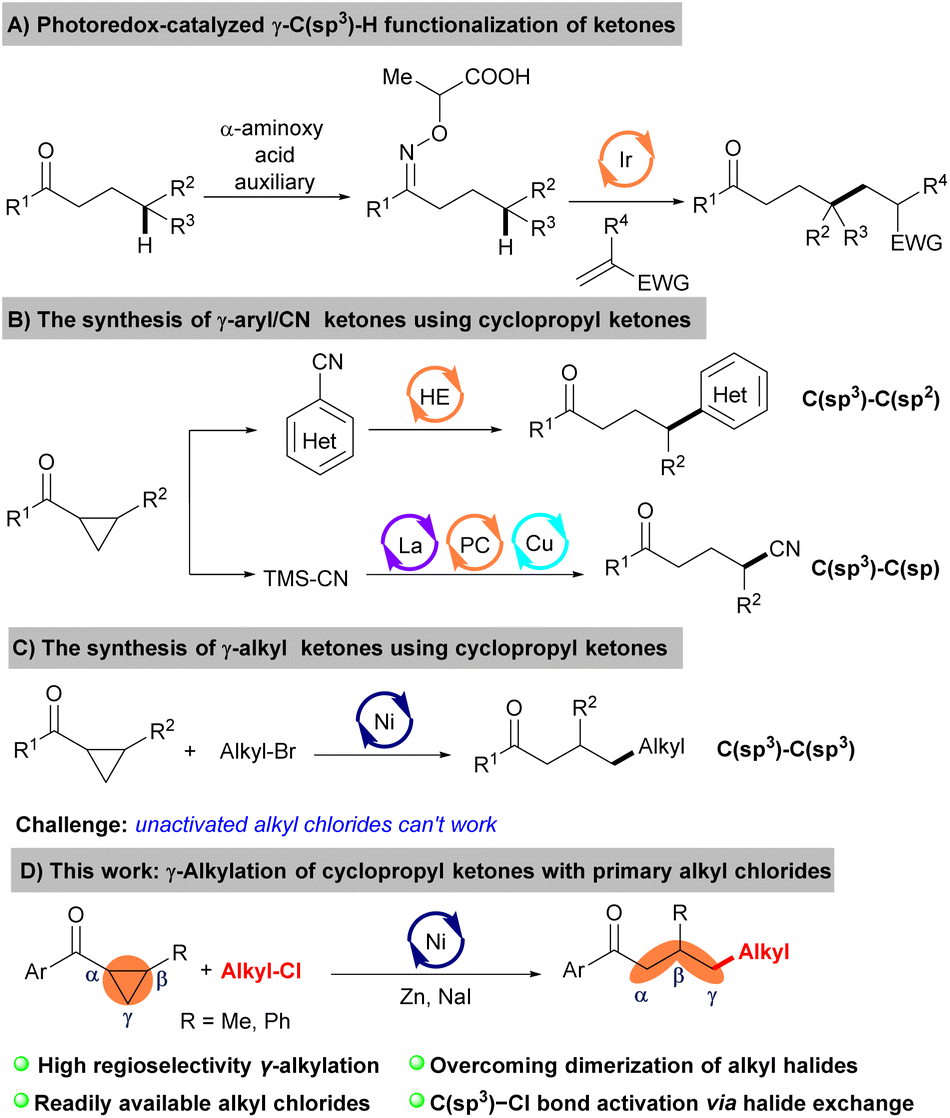 | ||
| Scheme 1 Synthesis of the γ-substituted ketones. | ||
Cyclopropyl ketones serve as valuable building blocks, readily attainable from diverse initial substrates,5 facilitating the synthesis of γ-functionalized ketone molecules via ring-opening reactions.6
In this vein, Opatz and co-workers developed a Hantzsch ester-mediated synthesis of γ-hetarylketones starting from cyclopropyl ketones.7 Furthermore, Xiao groups found a triple catalytic system merging photoredox catalysis with Lewis acid and copper catalysis, which fulfilled the ring-opening cyanation reaction of cyclopropyl ketones with TMSCN (Scheme 1B).8 However, all these reactions are constrained to affording the C(sp3)–C(sp2) or C(sp3)–C(sp) bond formation. Therefore, a general and efficient procedure for C(sp3)–C(sp3) bond formation by the ring opening reactions of cyclopropyl ketones is still highly demanded.
Recently, nickel-catalyzed reductive ring opening reaction of cyclopropyl ketones with alkyl bromides was independently reported by Wang and Mao groups.9 The above two works elucidate the activation mechanism of alkyl bromide through a two-electron process or a single electron transfer (SET) process, wherein the alkyl chloride moieties exhibit notable tolerance (Scheme 1C). Considering alkyl chlorides as versatile building blocks in organic synthesis due to their commercial availability and chemical stability,10 we wish to develop the ring opening reactions of cyclopropyl ketones with non-activated alkyl chlorides to synthesize γ-alkyl ketones. The main challenge to realizing this transformation was balancing alkyl–Cl bond reactivity and cross-selectivity.11 There is a pressing need to develop a highly efficient catalytic system with mild reaction conditions to overcome the dual reactivity-selectivity challenge of unactivated alkyl chlorides. Herein, we show that this can be accomplished by halide exchange to generate a low concentration of more reactive alkyl iodides from the alkyl–Cl starting material, thereby avoiding the formation of alkyl dimers (Scheme 1D).
We first conducted the optimized conditions for the γ-alkylation of cyclopropyl phenyl ketone 1a with Phenylpropyl chloride 2a (Table 1). As a result, 10 mol% NiBr2(dme), and 10 mol% 4,4′-dimethyl-2,2′-bipyridine (L4) were employed in the presence of 3 equiv. of Zn powder and 1.5 equiv. of sodium iodide (NaI) in N,N-dimethylacetamide (DMA) at 50 °C for 24 h. Under these conditions, 1,7-diphenylheptan-1-one (3aa) was obtained in 72% isolated yield. Meanwhile, we detected the byproduct of alkyl homo-coupling (4a) in 7% of GC yield (entry 1). Initially, the 6,6′-dimethyl-2,2′-bipyridine ligand L1 was ineffective for this reaction (entry 2). When electron-rich bipyridine ligand L2 was employed instead of L4, the yield of 3aa was only slightly decreased (entry 3). Furthermore, the electron-poor bipyridine ligand L3 also turned out to be inappropriate (entry 4). Compared to bipyridine ligands, phenanthroline ligands L5 and L6 exhibited a propensity for greater alkyl dimerization. The enhanced yield observed with the L6 was attributed to its electronic effect (entries 5 and 6). When using terpyridine L7 as a ligand, no cross-coupling product was observed, but it was accompanied by a significant dimer byproduct (entry 7). Overall, the choice of the ligand had an unneglectable impact on the outcome distribution (See Table S1 in the ESI† for full details). When switching the catalyst from NiBr2(dme) to NiCl2(dme) or NiCl2, the desired product was afforded a lower yield (entries 8–10). The reaction using NiBr2 gained a terrible result (entry 9) (See Table S2 in the ESI†). Reducing the amount of NaI to 50 mol% resulted in a sharply decreased yield (entry 11). Notably, the target coupling product 3aa was not generated when this reaction was conducted without NaI (entry 12). The catalyst loading can be reduced by 5 mol%, diminishing the reaction yield (entry 13). Employing Mn instead of Zn gave an inferior outcome (entry 14). Lessening the temperature from 50 °C to r.t. produced 3aa only in 29% yield (entry 15). Raising the temperature to 80 °C resulted in a trace amount of the desired product (entry 16).
|
|||
|---|---|---|---|
| Entry | Variations from standard conditions | 3aab (%) | 4ab (%) |
| a Unless otherwise specified, the reactions were carried out in a Schlenk tube under nitrogen in the presence of 1a (0.2 mmol), 2a (0.4 mmol), NiBr2(dme) (10 mol%), L4 (10 mol%), NaI (1.5 equiv.), and Zn (3.0 equiv.) in DMA (0.5 mL) at 50 °C for 24 h.b Yields are corrected GC yields vs. hexadecane internal standard.c Yields of the isolated product through column chromatography. N.D. = not detected. | |||
| 1 | None | 78 (72)c | 7 |
| 2 | L1 instead of L4 | N.D. | N.D. |
| 3 | L2 instead of L4 | 74 | 5 |
| 4 | L3 instead of L4 | 21 | 18 |
| 5 | L5 instead of L4 | 23 | 19 |
| 6 | L6 instead of L4 | 61 | 15 |
| 7 | L7 instead of L4 | N.D. | 36 |
| 8 | NiCl2(dme) instead of NiBr2(dme) | 67 | 8 |
| 9 | NiBr2 instead of NiBr2(dme) | 20 | 13 |
| 10 | NiCl2 instead of NiBr2(dme) | 61 | 11 |
| 11 | NaI (50 mol%) was used | 33 | 2 |
| 12 | W/O NaI | 0 | 0 |
| 13 | 5 mol% catalyst loading | 30 | 26 |
| 14 | Mn instead of Zn | 31 | 22 |
| 15 | At r.t. | 29 | 3 |
| 16 | At 80 °C | Trace | 29 |
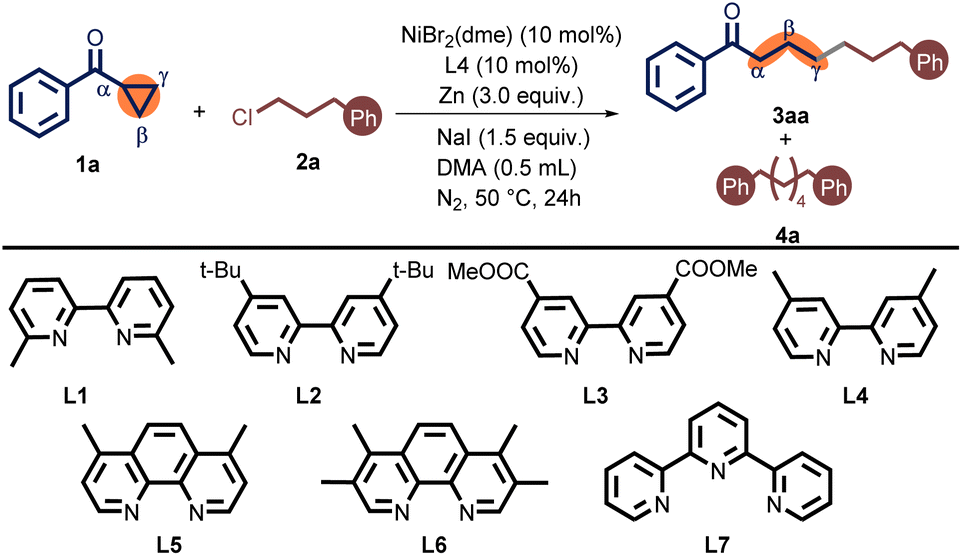
We also tested the influence of other reaction parameters, including the use of different additives, solvents and concentrations, but these studies did not lead to an improvement on the conditions in Table 1, entry 1 (see the ESI† for full details).
With optimized reaction conditions, a range of non-activated primary alkyl chlorides were examined for the coupling reaction with 1a or 1f, furnishing a series of γ-alkylated ketones 3aa–3ah (Table 2). In the synthesis of the relatively longer-chain product 3aa, it was found that the coupling product was obtained with a satisfactory yield. In contrast, a shorter-chain alkyl substrate was converted to 3ab in a moderate yield. Simple unsubstituted alkyl chlorides turned out to be an appropriate substrate. n-Alkyl chlorides containing alkyl chains with seven or eight carbon atoms, 1-chloroheptane (2c) and 1-chlorooctane (2d), the relevant products 3ac and 3ad were delivered in 68% and 72% yield, respectively. Alkyl chloride with branching, chloro-iso-octane (2e), also provided the target yield, albeit in a 27% output. Other functional groups, such as ester (3af, 3ag) and trimethylsilyl (3ah), were well tolerated.
| a Unless otherwise specified, the reactions were carried out in a Schlenk tube under nitrogen in the presence of 1a (0.2 mmol), 2a (0.4 mmol), NiBr2(dme) (10 mol%), L4 (10 mol%), NaI (1.5 equiv.), and Zn (3.0 equiv.) in DMA (0.5 mL) at 50 °C for 24 h. |
|---|
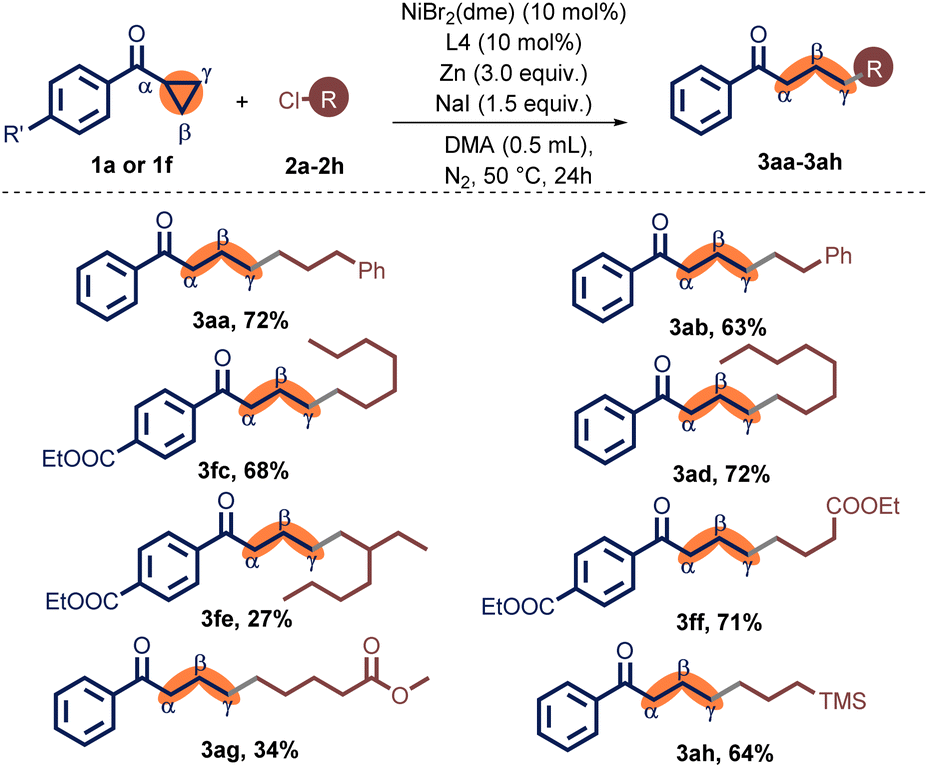 |
Next, we explored the substituents on the phenyl ring of the aryl cyclopropyl ketones (Table 3). Initially, we discovered that electron-donating tertiary butyl and methoxy groups on the para position of the benzene ring provided the products 3ba–3ca in low yields. Gratifyingly, electron-withdrawing CF3, F, and ester substituents on the para position of the benzene ring were well accommodated, and the products 3da–3fa were delivered in 73–85% yields. In contrast, when the aryl group was substituted with 3-methyl, the target product 3ha was obtained in a moderate yield. Remarkably, the 2-substituted phenyl cyclopropyl ketone (1g) provided a medium yield compared to the 4-substituted one (1d). To our delight, the pyridinyl cyclopropyl ketone was also well accommodated, producing the coupling products 3ia in 67% yield. In addition, trans-disubstituted cyclopropanes (1j, 1k) were pertinent precursors, albeit the more significant steric hindrance can cut the yield. Unfortunately, alkyl-substituted cyclopropyl ketones were unreactive in this reaction.
| a Unless otherwise specified, the reactions were carried out in a Schlenk tube under nitrogen in the presence of 1a (0.2 mmol), 2a (0.4 mmol), NiBr2(dme) (10 mol%), L4 (10 mol%), NaI (1.5 equiv.), and Zn (3.0 equiv.) in DMA (0.5 mL) at 50 °C for 24 h. |
|---|
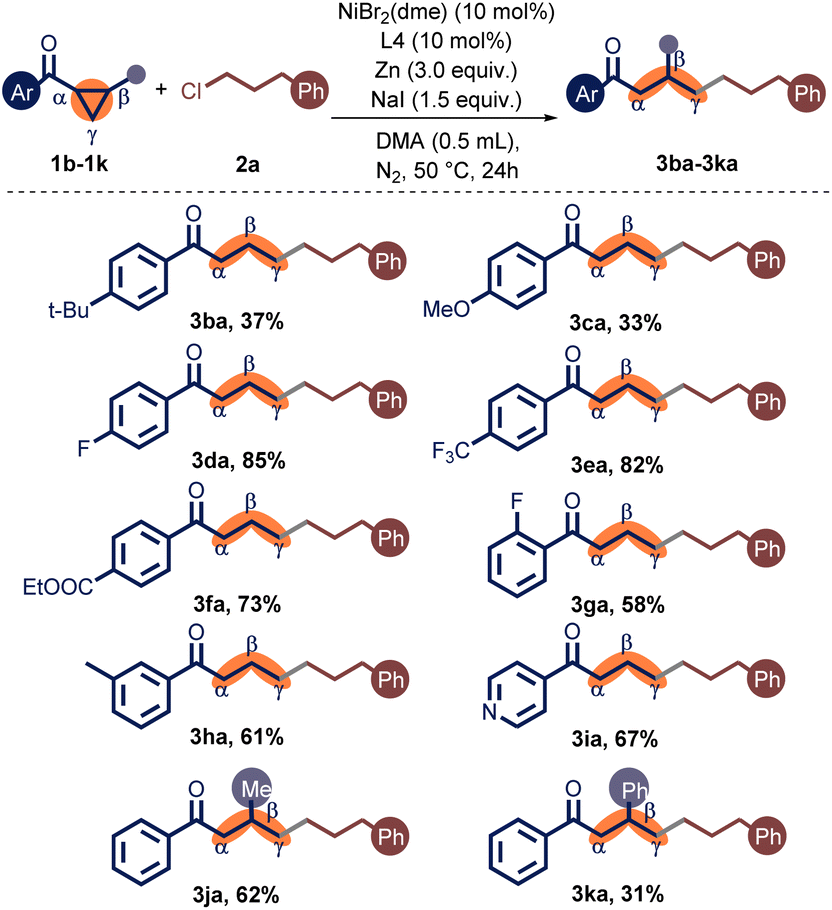 |
Moreover, the reaction was performed on a 10 mmol scale of 1a under standard conditions (Scheme 2). The product 3aa was also successfully isolated in 70% yield (1.86 g) that did not necessitate an extended reaction time, demonstrating the scalability of this transformation.
 | ||
| Scheme 2 Gram-scale experiment. | ||
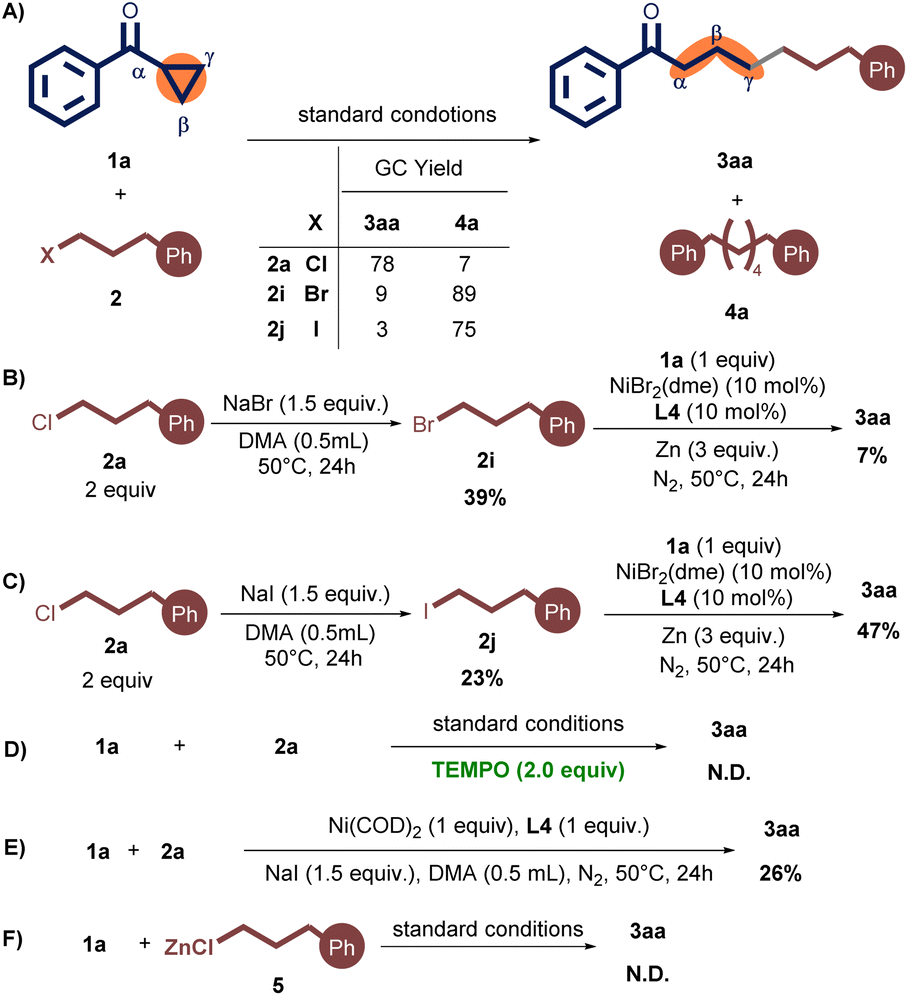
To further understand the reaction mechanism, several control experiments were performed (Scheme 3). First, when an alkyl bromide 2i or alkyl iodide 2j was employed instead of 2a, 3aa was obtained in only 9% or 3% yield, along with dimer product 4a in 89% or 75% yield during the standard reaction conditions (Scheme 3A).12
 | ||
| Scheme 3 Control experiments. | ||
Next, the reaction between 2a with NaBr (1.5 equiv.) produced 2i in 39% yield, which upon reaction with 1a under standard conditions produced 3aa in a yield of only 7% (Scheme 3B). Analogously, employing NaI (1.5 equiv.) in the reaction of 2a afforded 2j in 23% yield. The ensuing reaction of 2j with 1a under standard conditions resulted in a 47% yield of 3aa (Scheme 3C).13 More importantly, the cross-coupled product 3aa was not observed without NaI (Table 1, entry 12). These experiments showed that NaI as a crucial cocatalyst in this reaction likely serves a dual role: (1) generating a low concentration of more reactive alkyl iodides through halide exchange, thereby effectively suppressing the formation of alkyl dimers,11,14 and (2) enhancing the reductive coupling process may be attributed to its potential facilitation of either the reduction of the nickel catalyst or the formation of a more reactive nickelate species.14,15
Furthermore, the radical scavenger 2,2,6,6-Tetramethylpiperidine 1-oxyl (TEMPO) was added under the standard reaction conditions, and the desired product 3aa was not detected (Scheme 3D). This result suggests that the radical pathway may be involved in this protocol. In addition, in the presence of 1.0 equiv. of Ni(COD)2, the corresponding product 3aa could be yielded in 26%, indicating the coupling started with Ni(0) species (Scheme 3E).16 Finally, phenethylzinc chloride 5 (0.25 M)17 was synthesized and subjected to the reaction with 1a in the absence of zinc powder (Scheme 3F). These experimental results support that excludes the possibility of an organozinc reagent in the reaction system.
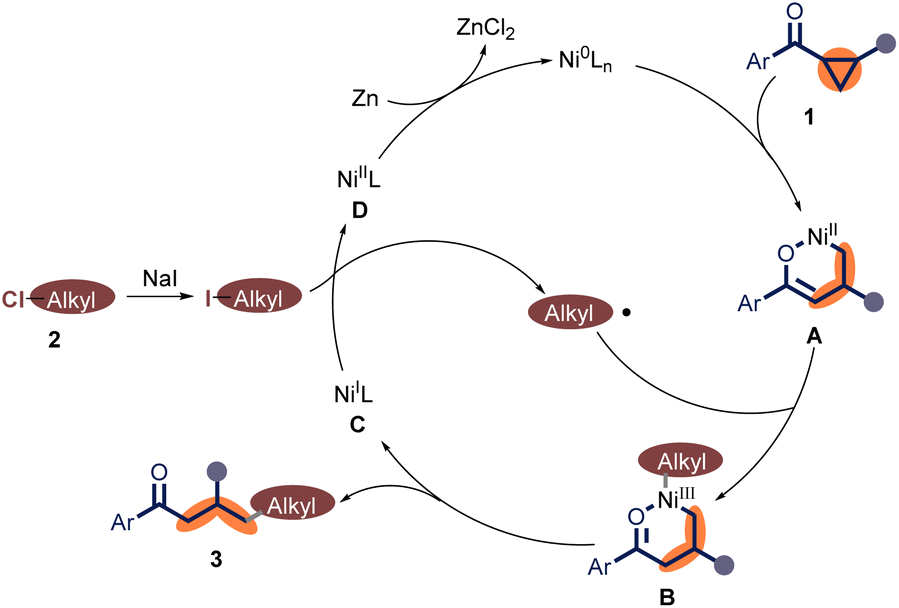
Based on previous reports9,13,18 and these control experiments, a proposed mechanism was shown in Scheme 4. Initially, the oxidative addition of aryl cyclopropyl ketone 1 to Ni(0) produces Ni(II) species A, which reacts with an alkyl radical to form Ni(III) species B. In the next step, intermediate B undergoes reductive elimination to generate the target product γ-alkyl ketones and the Ni(I) species C. Subsequently, the reaction of Ni(I) species C with alkyl iodide (formed via the halide exchange between the alkyl chloride 2 with NaI) affords the alkyl radical and the Ni(II) intermediate D. Finally, Ni(0) species is regenerated for the next catalytic cycle through reduction.
 | ||
| Scheme 4 Proposed possible mechanism. | ||
Conclusions
In conclusion, we have developed a nickel-catalyzed γ-alkylation reaction of aryl cyclopropyl ketones with non-activated preliminary alkyl chlorides, providing efficient chemo- and regioselectivity for cross-coupling products using halide exchange. Further efforts are currently under investigation in our labs regarding to the role of sodium iodide in the reaction process.Author contributions
Zheng-Ying Wang: experimental design and optimization, data curation, writing-original draft; Zheng-Ying Wang, Shi-Zheng Liu and Qiang Li: subject selection and writing-review & editing; Cong Guo and Yi-Zheng Cheng: validation; Qiang Li: supervision; Qiang Li, Jianmin Dou, and Dacheng Li: funding acquisition, project administration. All authors read and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the financial support of the Natural Science Foundation of China (grant no. 21802062) and the Natural Science Foundation of Shandong Province (grant no. ZR2016BM18).Notes and references
- For selected reviews, see:
(a) E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed
; (b) N. Stephanopoulos and M. B. Francis, Nat. Chem. Biol., 2011, 7, 876–884 CrossRef CAS PubMed
-
(a) N. J. Lawrence, J. Chem. Soc., Perkin Trans. 1, 1998, 1739–1750 RSC
- For selected reviews, see:
(a) A. K. Pandey, ChemCatChem, 2022, 14, e202101982 CrossRef CAS
- H. Jiang and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 1692–1696 CrossRef CAS PubMed
- For reviews on synthesis of cyclopropanes, see:
(a) L. Dian and I. Marek, Chem. Rev., 2018, 118, 8415–8434 CrossRef CAS PubMed
- For selected reviews, see:
(a) J. Liu, R. Liu, Y. Wei and M. Shi, Trends Chem., 2019, 1, 779–793 CrossRef CAS
- J. Paternoga, J. Kühlborn, N. O. Rossdam and T. Opatz, J. Org. Chem., 2021, 86, 3232–3248 CrossRef CAS PubMed
- J. Liu, X.-P. Liu, H. Wu, Y. Wei, F.-D. Lu, K.-R. Guo, Y. Cheng and W.-J. Xiao, Chem. Commun., 2020, 56, 11508–11511 RSC
-
(a) B. Yuan, D. Ding and C. Wang, ACS Catal., 2022, 12, 4261–4267 CrossRef CAS
-
(a) G. W. Gribble, Acc. Chem. Res., 1998, 31, 141–152 CrossRef CAS
- S. Kim, M. J. Goldfogel, M. M. Gilbert and D. J. Weix, J. Am. Chem. Soc., 2020, 142, 9902–9907 CrossRef CAS PubMed
- Q. Chen, J. You, T. Tian, Z. Li, M. Kashihara, H. Mori and Y. Nishihara, Org. Lett., 2022, 24, 9259–9263 CrossRef CAS PubMed
-
(a) H. Yu and Z.-X. Wang, Org. Biomol. Chem., 2023, 21, 3423–3431 RSC
-
(a) M. R. Prinsell, D. A. Everson and D. J. Weix, Chem. Commun., 2010, 46, 5743–5745 RSC
-
(a) I. Colon and D. R. Kelsey, J. Org. Chem., 1986, 51, 2627–2637 CrossRef CAS
- L. Le, M. Yin, H. Zeng, W. Xie, W. Zhou, Y. Chen, B. Xiong, S.-F. Yin, N. Kambe and R. Qiu, Org. Lett., 2024, 26, 344–349 CrossRef CAS PubMed
- S. Graßl and P. Knochel, Org. Lett., 2020, 22, 1947–1950 CrossRef PubMed
-
(a) S. Ogoshi, M. Nagata and H. Kurosawa, J. Am. Chem. Soc., 2006, 128, 5350–5351 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra02616k |
| This journal is © The Royal Society of Chemistry 2024 |