
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvent accessible surface area-assessed molecular basis of osmolyte-induced protein stability†
Shampa Raghunathan *
*
École Centrale School of Engineering, Mahindra University, Hyderabad 500043, India. E-mail: shampa.raghunathan@mahindrauniversity.edu.in
First published on 9th August 2024
Abstract
In solvent-modulated protein folding, under certain physiological conditions, an equilibrium exists between the unfolded and folded states of the protein without any need to break or make a covalent bond. In this process, interactions between various protein groups (peptides) and solvent molecules are known to play a major role in determining the directionality of the chemical reaction. However, an understanding of the mechanism of action of the co(solvent) by a generic theoretical underpinning is lacking. In this study, a generic solvation model is developed based on statistical mechanics and the thermodynamic transfer free energy model by considering the microenvironment polarity of the interacting co(solvent)–protein system. According to this model, polarity and the fractional solvent-accessible surface areas contribute to the interaction energies. The present model includes various orientations of participating interactant solvent surfaces of suitable areas. As model systems, besides the backbone we consider naturally occurring amino acid residues solvated in ten different osmolytes, small organic compounds known to modulate protein stability. The present model is able to predict the correct trend of the osmolyte–peptide interactions ranging from stabilizing to destabilizing not only for the backbone but also for side chains. Our model predicts Asn, Gln, Asp, Glu, Arg and Pro to be highly stable in most of the protecting osmolytes while Ala, Val, Ile, Leu, Thr, Met, Lys, Phe, Trp and Tyr are predicted to be moderately stable, and Ser, Cys and Histidine are predicted to be least stable. However, in denaturing solvents, both backbone and side chain models show similar stabilities in urea and guanidine. One of the important aspects of this model is that it is essentially parameter-free and consistent with the electrostatics of the interaction partners that make this model suitable for estimating any solute–solvent interaction energies.
Introduction
Biochemical reactions in living organisms take place in aqueous environments, often containing various small organic molecules.1,2 In the equilibrium protein folding reaction, unfolded (U) ⇌ native (N), no covalent bonds are broken or formed, instead an equilibrium exists with solvent interactions causing certain changes to the folded or unfolded states of the protein.3 Osmolytes are small organic compounds known to modulate protein stability, and are ubiquitous in living organisms mainly to counter the osmotic stress,1,4,5 hence they play a role in pharmaceuticals.6 Denaturing osmolytes push the equilibrium toward U, whereas protecting osmolytes push the equilibrium toward N.7 Urea, naturally found in bacteria8 and mammals,9 has been extensively used as a chemical denaturant in solvent-assisted protein denaturation studies.10–12 A favorable protein–osmolyte interaction enhances the preference for protein conformations with greater solvent-exposed surface area, i.e., the unfolded state.13–16 Alternatively, protecting osmolytes play a central role in stabilizing intracellular proteins in order to control osmotic stress in marine organisms.1 Trimethylamine N-oxide (TMAO) – a protecting osmolyte – favors the native structure of a protein and its stabilization has often been discussed by means of its exclusion from the vicinity of the protein surface.17,18 However, in the case of mixed-component solutions the effect of protein-denaturing osmolytes can be counteracted by protecting ones.19 In a recent experimental study, both types of osmolytes, stabilizing (betaine) and destabilizing (urea), were shown to exert an enthalpy effect on the hydration sphere of the protein in both folded and unfolded forms.20 To date, such osmolyte-induced protein stability has been characterized by the transfer free energy (TFE)21–23 model which reveals that denaturing/protecting osmolytes are preferentially accumulated/excluded around the protein backbone in single-component osmolyte solutions.7 Nevertheless, there is no universal molecular theory that can explain the mechanism of osmolyte-induced protein stability. Mechanistic inferences from the past studies suggest that the dominant driving force could be the interaction of osmolyte with either the backbone,7 or the side chain,24 or even both.14,25Existing conventional methods for calculating solvation free energies with the atomistic molecular dynamics (MD) simulations are classified into two types: (1) implicit and (2) explicit solvation free energy methods.26 Both come with advantages and disadvantages considering the cost of computing solute–solvent interactions. Implicit solvation models give a quick qualitative estimate of the free energies whereas explicit solvation models calculate quantitative free energies. Due to high computational cost, explicit models are often limited to a solute consisting of few tens of atoms interacting with a few hundreds of small solvent molecules.27,28 Computed free energies are shown to be force field dependent12,29 especially in case of urea and TMAO which are the most widely studied solvents to investigate co-solvent effects on protein stability.30 Nevertheless, to obtain a generic model for a range of stabilizing to destabilizing solvents31,32 consistency measures would play a factor33 while choosing a force field that might have been parameterized to predict a particular physical phenomenon. Further complications may arise for multi-component solvent mixtures34 where each type of solute–solvent interaction differs from another.35 In a TFE model, the solvation free energy of a solute in pure solvent is subtracted from the solvation free energy of the same in solvent mixtures.36 Many theoretical and experimental studies of two-component37 and three-component solvent30 mixtures have been shown to work successfully with the TFEs applying conventional thermodynamics integration methods.26 While the development of solvation models has benefited from solvation free energy calculations using conventional methods, a generic solvation model for solute in multi-component solvent mixtures is lacking.
Above all else is the undeniable fact that solvent interactions38 play a very important role in the protein denaturation/renaturation process. Having identified the problem at hand, in the present study a statistical mechanics model is developed based on the polarity and the fractional solvent accessible surface areas (SASA) of the interacting molecule, inspired by TFE model of Street et al.7 This model was shown to reproduce transfer free energies of peptide backbones for a range of stabilizing to destabilizing osmolytes. The interaction energy is a function of the peptide polarity and the interaction degeneracy, the latter being a function of the SASA of osmolytes. One of the main aspects of this work is that the model proposed here is fairly generic, and it considers the fundamental electrostatic interactions between a pair of interaction partners while explicitly considering the accessible surface areas instead of just point-charges.
Recent studies have shown how microenvironment polarity modulates many-fold protein stability in the presence of solvent molecules. A single-atom substitution induces significant stability without altering the amino acid sequence and structure of a protein.39,40 Therefore it is important to include all sub-sets of interactions (between solute and solvent) for quantitative prediction. The existing backbone model7 was lacking (i) a generic framework for the backbone as well as side chains, (ii) a suitable method for choosing interaction sites, (iii) an n-site model beyond 3-site, (iv) a regular protocol for electrostatics, and (v) a method for microstate formation by considering all possible permutations of solute charge sites interacting with a solvent surface of a given polarity.
In the current work a generic model is developed. In case of a 3-site peptide backbone model C, N, O atoms are chosen as charge sites instead of choosing N, O, O7 arbitrarily. The current model is flexible about choosing interaction sites. In the regular protocol for electrostatics, for example, atomic partial charges are used for computing interaction energies. A method is introduced for generating sub-sets of microstates.
The 3-site (N+O−O−) backbone unit with a single backbone configuration7 does not include any sub-sets (see Fig. 1a). In the current study, multiple configurations of solute interaction sites are considered based on the presence of the charge sites. For example, in case of a 3-site (C N O) model (see Fig. 1b), the number of solute interaction sites are as follows: one configuration with 3 charges, three configurations with 2 charges each, and three configurations with 1 charge each. Now, each of the configurations incorporates all possible orientations following the same principle stated in ref. 7 (described in detail in Section 2). Presently, partial charges of atoms are used from the popular force fields CHARMM,41 and OPLS42 for proteins. We further extend the evaluation of our model by applying it to nineteen naturally occurring amino acids (glycine excluded) besides the backbone. The current study explores the osmolyte–protein interactions via their constituent side chains’/backbone’s impact on the protein stability. We show that our generic model is able to produce reasonable free energies of amino acids for a diverse set of stabilizing and destabilizing osmolytes in a mixed solvent environment.
 | ||
| Fig. 1 Schematic view of an interaction. (a) A 3-site backbone model (N+O−O−) and solvent accessible surface areas with partially positive (SA+), negative (SA−), and neutral (SA0) charges as was originally proposed.7 (b) A 3-site model with solute atoms O, N, and C and solvent accessible surface areas with partially positive (SA+), negative (SA−), and neutral (SA0) charges as proposed in the current work. Consequently, a 3-site model will have now 7 sub-sets. See ESI, Fig. S1† for more on microstate counting. | ||
Methods and models
SASA-based solvation model
In the model proposed here, polar interactions are of prime concern, and it relies on fundamental principles of chemistry, e.g., the oxygen atom is the most electronegative, followed by nitrogen, then carbon and sulfur (O > N > C ≈ S). In this model, peptides are divided into three groups: (1) polar positive (the amide nitrogen, bearing a partial positive charge), (2) polar negative (the carbonyl oxygen, bearing a partial negative charge), and (3) nonpolar (neutral atoms), resulting in three different interaction sites (i, j, k). This model treats all nitrogen and oxygen atoms as equally polar. Solvents are divided into three groups based on their polar surface areas, e.g., surface areas (SAs) with partially positive (SA+), negative (SA−), and neutral (SA0) charges. Thus, three types of peptide–solvent interactions are defined for these three groups: favorable, unfavorable, and neutral, having energies of −1, 1, and 0 kcal mol−1, respectively. Favorable interactions occur between polar groups with opposite charges (polar positive/SA− or polar negative/SA+), unfavorable interactions are between polar groups with like charges (polar positive/SA+ or polar negative/SA−), and neutral interactions involve nonpolar groups (nonpolar/SA0). Therefore, i, j, k can vary between +, −, 0 resulting in a total of 33 = 27 possible microstates, with degeneracies (energetically equivalent ways of making an interaction).n-Site model
Fig. 1b demonstrates the model for a 3-site case including all possible sub-sets accounting for microenvironments. For an interaction site containing nP number of sites taken k at a time, there can be many combinations excluding the empty set,
 | (1) |
For example, for an interaction site containing 3 sites (nP = 3) will give to 7 unique configurations. Each configuration will follow eqn (1)–(7) (ESI, Section 1†). The Boltzmann distribution over different configurations gives the final transfer free energy.
Eight stabilizing osmolytes (trimethylamine N-oxide (TMAO), betaine, sarcosine, proline, trehalose, sucrose, glycerol and sorbitol), a destabilizing osmolyte (urea) and a related denaturant (guanidine) (see Fig. 2) are considered in the present study. They are further categorized into: (1) methylamines: TMAO, betaine, and sarcosine; (2) sugars: sucrose, trehalose; (3) polyols: glycerol, sorbitol. All solvent accessible polar and nonpolar SAs were taken from an earlier study.7 A probe radius of 1.4 Å was used. Twenty one amino acids including two neutral and one positively charged histidine residues Hsd, Hse, and Hsp, respectively, constitute a set of model systems studied here. Glycine was not considered (see Fig. 3). Plots were made using the Python toolkit RDKit.43 At first model structures were converted into SMILES44,45 strings, then similarity maps were constructed with partial charges extracted from CHARMM topology files. Different solvent environments were used including pure water as well as aqueous-osmolytes solutions.
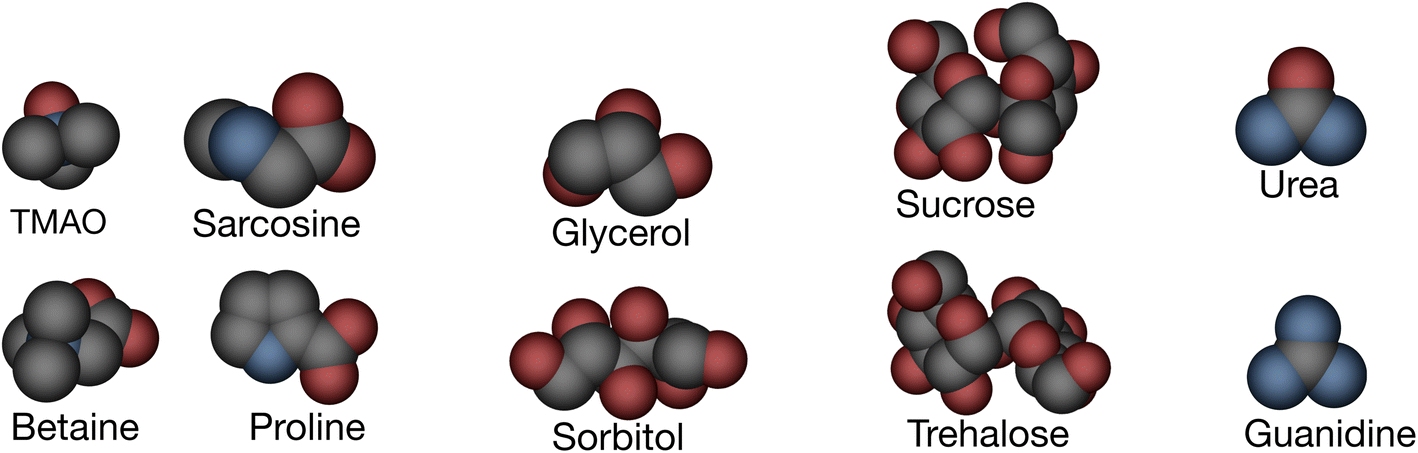 | ||
| Fig. 2 Molecular structures of various osmolytes, shown in space-filling representations and color-coded by atom type. Oxygen (red), nitrogen (blue), and carbon (grey); protecting osmolytes are TMAO, sarcosine, betaine, proline, sucrose, trehalose, glycerol, sorbitol, and denaturants are urea and guanidine. | ||
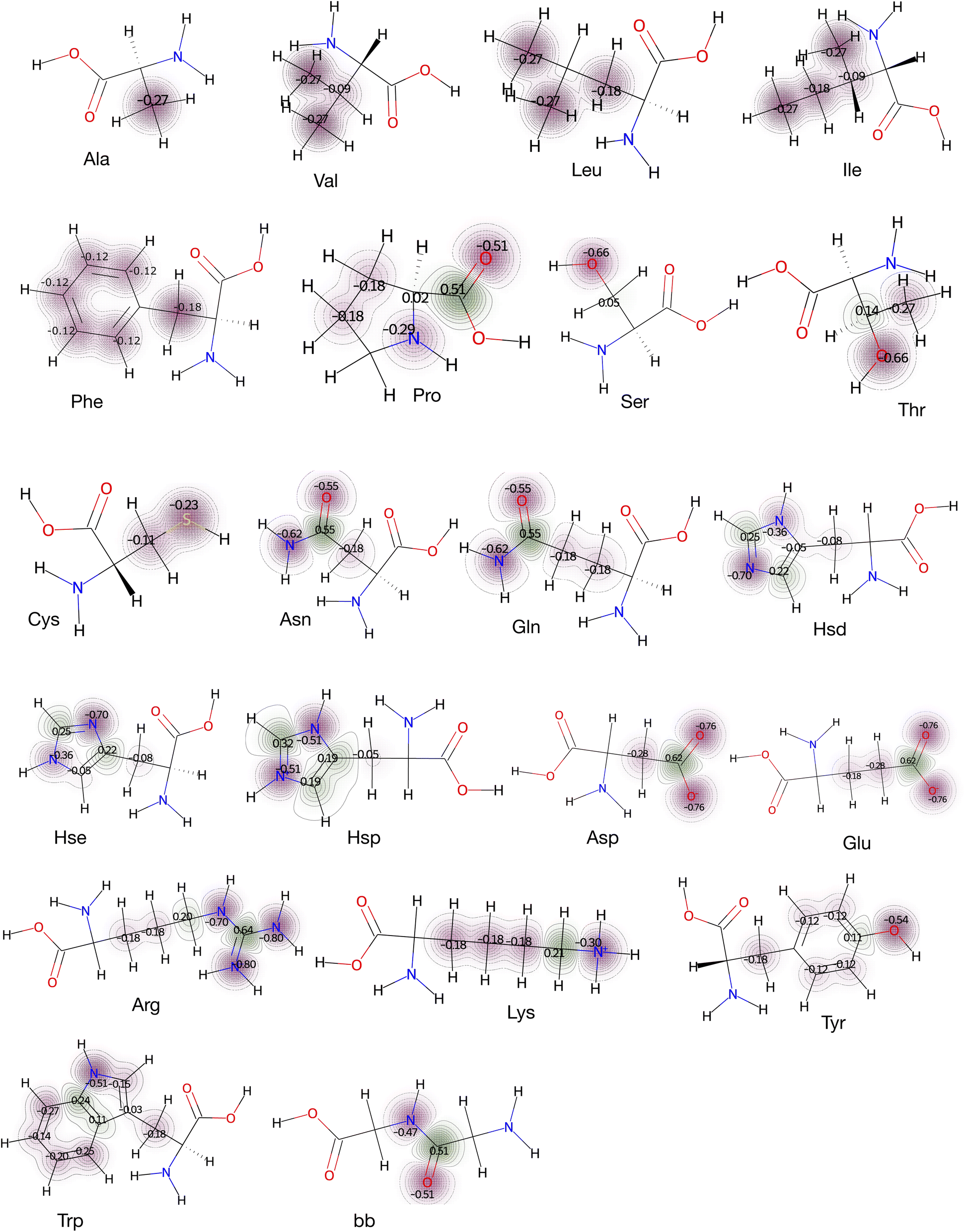 | ||
| Fig. 3 Molecular electrostatics of amino acid side chains and backbone (bb). Atomic partial charges assigned in the CHARMM force field to 21 amino acids (Gly was excluded, and three protonated states of histidine, Hsd, Hse and Hsp were included). n-Number of atomic charges define the n-site model. For example, amino acids Ala and Phe use 1-site and 6-site models, respectively. For Leu, the γ-carbon which is buried, was not included as a charge site. | ||
Results
Transfer free energies of the N+O−O− model
Street et al. proposed a quantitative model for solvent (water and osmolyte) interactions with backbone polar groups (the amide nitrogen bearing a partial positive charge, and the carbonyl oxygen bearing a partial negative charge).7 The backbone amide nitrogen was assigned one solvent interaction site, whereas the larger carbonyl oxygen, containing two lone pair electrons, is assigned two such sites. Together, they make a 3-site (N+O−O−) model, Fig. 1a. This 3-site model with a single backbone configuration during interaction with the osmolyte surface of a given polarity gave identical interaction energies to what was reported earlier.7 In Fig. 4, the blue triangles represent those values. | ||
| Fig. 4 Transfer free energies of various 3-site models. The N+O−O− configuration is highlighted by blue triangles. Red circles represent all possible 33 = 27 arrangements between the solute’s three charge sites (+1, 0, −1) and the solvent’s polarity-specific surfaces (SA+,SA0,SA−). | ||
Now, the question is how to realize a 3-site (N+O−O−) model from interactions between an arbitrary solute with three charge sites (+1, 0, −1) and polarity-specific solvent surfaces (SA+, SA0, SA−) irrespective of whether water or osmolyte is used. The answer is by considering all possible permutations of solute charge sites and solvent surfaces of specific polarity. This gives rise to 33 = 27 possible arrangements. They are as follows: 111, 110, 11−1, 101, 100, 10−1, 1−11, 1−10, 1−1−1, 011, 010, 01−1, 001, 000, 00−1, 0−11, 0−10, 0−1−1, −111, −110, −11−1, −101, −100, −10−1, −1−11, −1−10, −1−1−1. Each one represents a 3-site model. The 9th one (1−1−1) can be taken as (N+O−O−), and their corresponding transfer free energies are highlighted by blue triangles in Fig. 4. The red circles are for those above mentioned 27 arrangements. One can easily understand that there will be redundancy, for example, 21st (−11−1) and 25th (−1−11) will give same transfer free energies as the 9th one (1−1−1), because there are no other parameters to distinguish them. This whole exercise gives a pretty good test for identifying the arrangement that gives transfer free energies close to experimental values. Conformational energies shown in red circles in Fig. 4 vary over a wider range in the case of sugars and polyols, and guanidine, than in methylamines, proline, and urea. Those conformations describe poorly the real scenario, for example, where all three charge sites have the same charges; this is especially apparent in case of sorbitol, sucrose and trehalose. Essentially these three osmolytes have larger surface areas than other osmolytes too.
Preferential osmolyte interactions with the N+O−O− model
Preferential interaction helps to characterize the concentration of the solvent near the solute compared to bulk. Here preferential interactions were computed as average osmolyte occupancy on an interaction site and in the bulk (see ESI, Section 1†). For this, 1 M osmolyte was considered, and the relative difference between 〈Opref〉 and 〈Obulk〉 was scaled.7 Fig. 5 shows the correlation between the local concentration of the osmolyte around the model and measured ΔGtr (transfer free energy) values. From TMAO to guanidine, a good negative correlation is seen indicating stabilizing osmolytes have lower local concentration compared to destabilizing osmolytes around the backbone. Interestingly, from this graph one can see the result of a particular conformation is very different than the others. This arises from the fact that conformation with solute charges q+, q0, q0 interacts less favorably with the fractional polar surfaces of the osmolytes, hence its poor contribution. In fact, local osmolyte concentration around each conformation may tell about its accumulation/depletion ability.46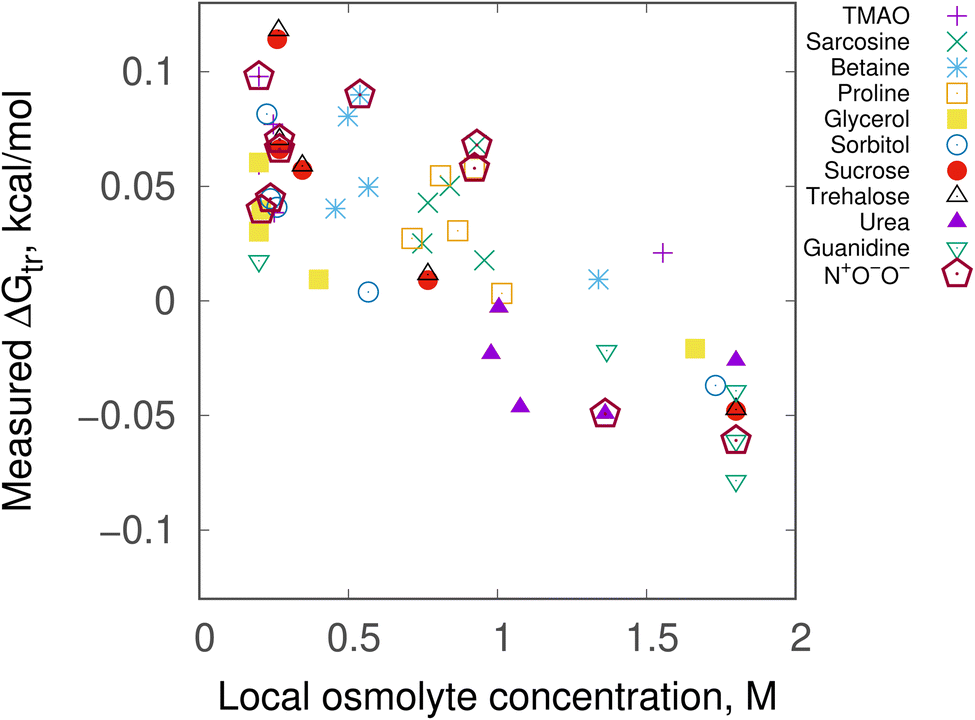 | ||
| Fig. 5 Local osmolyte concentration vs. measured transfer free energies, ΔGtr of the 3-site backbone model. ΔGtr values are given for all seven conformations of all ten osmolytes. Values for the lone N+O−O− configuration which was originally proposed7 are shown in maroon open pentagons for all ten osmolytes. In all cases, 1 M osmolyte solution was taken. Local concentration is plotted as scaled deviation between average occupancy of osmolyte on an interaction site (〈Opref〉) and in the bulk (〈Obulk〉). | ||
Additionally, the ΔGtr values were computed beyond 1 M osmolyte (aqueous) solution. Experimental transfer free energies along with the results from the present model are plotted in Fig. 6. Sarcosine, urea and guanidine are considered for this test as experimental data for these osmolytes are available.47,48 We can find a certain degree of similarities between experimental and computational results—this implies that our model is flexible enough to compute ΔGtr beyond 1 M concentration. In order to further assess the quality of the results shown in Fig. 6, two different standard charge parameters, CHARMM and OPLS were chosen while varying the number of interaction sites present in the peptide backbone. In ESI, Fig. S2† the results are plotted. Sarcosine and urea were shown to give consistent results for 1 M, 2 M, 4 M and 6 M aqueous osmolyte solutions by using each of the 3-site, 4-site and 5-site models, whereas guanidine seemed to vary more at higher concentrations among three different models. The present model is based on solute-polarity and factional surface areas (SAs) of solvents used. Consequently, values of fractional SAs play an important role. Further tests are needed for better realization.
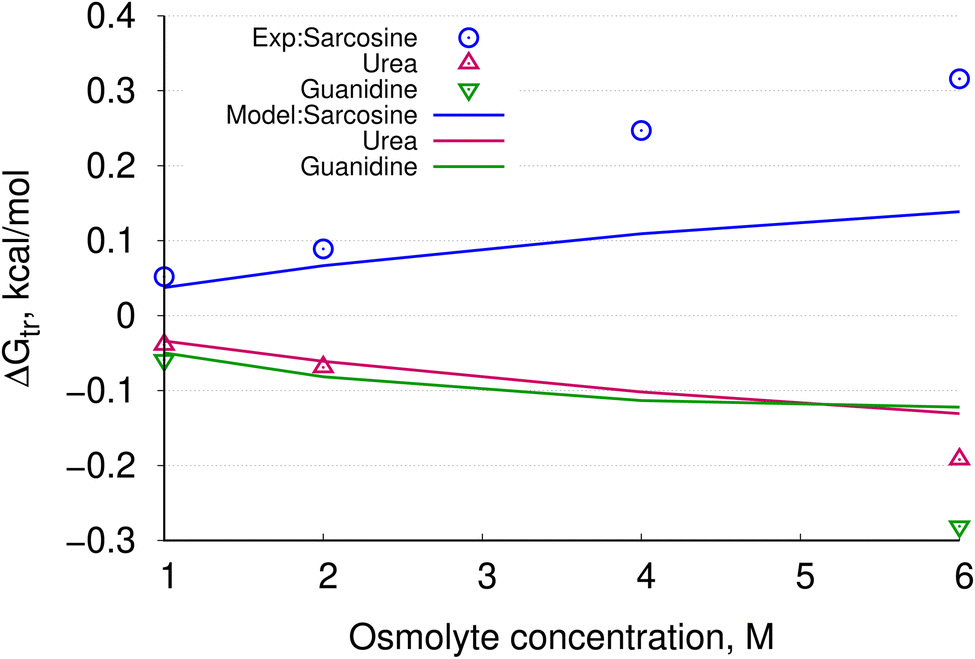 | ||
| Fig. 6 Calculated ΔGtr values for the backbone model as osmolyte concentration is increased from >1 M. Available experimental data are plotted using blue circles (sarcosine), magenta triangles (urea), and green inverted triangles (guanidine). Solid lines represent values calculated in this study. | ||
For calculating the interaction energies for the side chain model (see Fig. 3), explicit atomic charges from the CHARMM36 (ref. 41) force fields were used. Charges were assigned for a particular non-hydrogen atom of a molecule except the α-carbon (Cα). In case of proline, nitrogen and Cα atoms are given charges as they are fused to form a ring. Additionally, here C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O is assigned charges to consider Pro as a whole amino acid residue with side chain and backbone contributions. Hydroxylic (–OH) and thiol (–SH) hydrogens (of Ser, Thr, Tyr, and Cys) were not considered as charge sites, unless stated otherwise. The number of charge sites is shown in Fig. 3. The presence of n charge sites defines an n-site model. For amino acid residues Ala, Val, and Phe, the models are 1-site, 3-site, and 6-site, respectively. By including all combinations (eqn (1)) for these model systems, average interaction energies were calculated followed by transfer free energies.
O is assigned charges to consider Pro as a whole amino acid residue with side chain and backbone contributions. Hydroxylic (–OH) and thiol (–SH) hydrogens (of Ser, Thr, Tyr, and Cys) were not considered as charge sites, unless stated otherwise. The number of charge sites is shown in Fig. 3. The presence of n charge sites defines an n-site model. For amino acid residues Ala, Val, and Phe, the models are 1-site, 3-site, and 6-site, respectively. By including all combinations (eqn (1)) for these model systems, average interaction energies were calculated followed by transfer free energies.
TFEs using CHARMM and OPLS partial charges
The model presented here is based on SASA, and solvent polarity. Naturally the current model is dependent on the number of charge sites present in a solute (here, amino acids). We have performed this test by increasing the number of charge sites on the peptide backbone unit from three → four → five. Besides CHARMM force fields we also performed this test using OPLS42 which is widely used for amino acids. Fig. 7a and b present results for CHARMM and OPLS force fields, respectively. Clearly, varying the number of charge sites affects ΔGtr values (for more examples, see Fig. S4–S6 in the ESI†). TMAO, sarcosine, betaine, and proline are the least affected, and the rest (for example, glycerol, sorbitol, sucrose, and trehalose) among the stabilizing osmolytes are most affected in both CHARMM and OPLS force fields. In the case of the destabilizing osmolytes, ΔGtr of urea is less dependent on number of charge sites. However, guanidine seems to show more influence over varying charge sites. In fact, similar behavior in conformational energies is manifested in Fig. 4. Overall CHARMM and OPLS have similar results irrespective of the number of charge sites. | ||
| Fig. 7 Computed ΔGtr values for peptide backbone (a single unit) utilizing (a) CHARMM and (b) OPLS partial charges. For partial charges of 3-site, 4-site and 5-site models, see ESI, Fig. S3.† | ||
Osmolyte specific interactions
Calculated ΔGtr values of all models (Fig. 3) are plotted as a heatmap in Fig. 8. Among the stabilizing osmolytes the model predicts the sugars sucrose and trehalose to have the least solubility (with +ve sign) across all amino acids. That is, amino acids are more soluble in water than in trehalose and sucrose. ΔGtr values are significantly higher than other stabilizing osmolytes. Therefore, we can predict sucrose and trehalose to be better stabilizers of amino acids. Cys becomes slightly more soluble in sarcosine than in any other stabilizing osmolytes. Proline and sarcosine show moderate solubility of amino acids compared to other stabilizing osmolytes, whereas TMAO and betaine show slightly less solubility towards amino acids than them. By looking at their consistent poor solubility throughout all amino acids, we can infer TMAO and betaine to be more general stabilizers for proteins. In the case of two destabilizing osmolytes, guanidine seems to be a stronger destabilizer of amino acids than urea. | ||
| Fig. 8 Transfer free energies (ΔGtr) of side chain models calculated using explicit charges, and considering all k-combinations in all ten osmolytes. The color bar runs from red (−0.13) to blue (0.15) representing ΔGtr values in kcal mol−1. The red color represents better solubility of amino acids in osmolyte than those in blue color. The size of the square displays counting of occurrence of the value. For example, Arg has the greatest solubility in guanidine, and the corresponding ΔGtr value has lower distribution indicated by the smallest red square. Arg has the lowest solubility in trehalose, and corresponding ΔGtr value has higher distribution indicated by a large, dark blue square. | ||
Charged amino acids contribute predominantly to electrostatics
Amino acid specific contributions are as follows: (1) hydrophobic residues: Ala, Val, Leu, Ile, Phe and Pro consistently show lower solubility in stabilizing osmolytes, and higher solubility in urea and guanidine. (2) Hydrophilic neutral residues with –O/S–H group: Ser, Thr and Cys show the same trend as hydrophobic residues. However, ΔGtr values vary within a lower range (see, light blue squares) as compared to hydrophobic residues. (3) Hydrophilic neutral residues, amide containing: Asn, Gln, and neutral histidines Hsd and Hse have ΔGtr values varying over a slightly wider range although the same trend of solubility in various osmolytes persists. (4) Hydrophilic charged residues: Asp, Glu, and Arg have the highest range of ΔGtr values with almost similar distribution (similar square sizes) except for guanidine, which shows the most favorable ΔGtr with least distribution/population. Positively charged histidine Hsp is the least stable in polyols and sugars. In contrast, all other amino acids show high stability in sugars. (5) Amphipathic residues: Lys, Tyr, Met and Trp, like other amino acids are the least soluble in sugars and moderately soluble in the rest of the osmolytes; apparently they show the same trend of solubility, nearly like hydrophobic residues.48,49 Backbone: bb behaves very similarly to Thr. Given its simplicity, the present model reproduces the trend in ΔGtr values in all cases.Discussion
The present model provides free energy estimates of solutes while transferring from pure aqueous solvent to aqueous osmolyte solutions. The peptide backbone model7 is extended beyond three interaction sites with atomic partial charges and a new strategy to include sub-sets of microstates to predict transfer free energies of solutes in general. Amino acid side chains and backbone are used as model systems. The model predicts the general trend of the behavior of the protein–osmolyte solutions rather well. The nature of various types of stabilizing osmolytes, like methylamines, proline, polyols, and sugars seem to be captured well by the model. For example, from earlier studies we know that TMAO, sorbitol, sucrose and trehalose fall into the category which stabilizes proteins well in both their native and denatured states.31 Our model predicts this trend. One exception is that amino acids are predicted to be slightly less stable in TMAO compared to sugars. Betaine seems to be comparable to TMAO which was reported earlier50 though their mechanism of action was not the same. Besides, our model shows proline and glycerol to be responsible for the moderate change in protein stability as reported earlier.31 Sarcosine is the milder one to modulate protein stability amongst the stabilizing osmolytes. Denaturing osmolytes are well predicted by our model. In order to reproduce the experimental transfer free energy data as closely as possible, several groups have performed extensive MD simulations,17,50,51 and developed new models52–55 for amino acids as well as for a whole protein. Recently, the interaction between RNase A protein and sorbitol/sucrose was studied56 using a model which is based on MD simulations’ data that are able to describe preferential exclusion of sorbitol/sucrose from protein better than using standard CHARMM36 (ref. 57) and KBP58 force fields for sorbitol and sucrose. That model provides a better estimation of preferential exclusion parameters for both high (1.0 M) and low (0.5 M) concentrations of co-solutes. Here, the current model shows that free energy changes due to folding/unfolding of a protein is linearly dependent on osmolyte concentration for both the stabilizing and destabilizing osmolytes. In fact this was earlier predicted by the parent backbone model,7 and here, we show this with a more general model. The present model correctly predicts the m values (the change in transfer free energies w.r.t. the change in osmolyte concentration), implying proteins will be forced to fold in the presence of TMAO at lower concentrations whereas they will unfold in the presence of urea at relatively higher concentrations.Diehl et al. recently reported α-value and group transfer free energy (GTFE) predictions using vapor pressure osmometry (VPO) data of functional groups of proteins and solubility data of amino acids and dipeptides for osmolyte glycine betaine, proline and urea. These two methods yielded significantly different predictions. On the one hand, the GTFE analysis needed further scrutiny as it used glycine as a reference to obtain side chain contributions. On the other hand, α-value analysis used more extensive data sets.59 Finally, we note that Diehl et al. compared their VPO results with those from solubility data. While many amino acids preferred to interact with betaine instead of water as concluded by both two techniques, they also showed some differences. For example, solubility-data estimates showed a weak preference of valine and leucine for betaine (and proline) compared to water while VPO-data estimates showed a weak preference of valine and leucine for water than for betaine.59 In an earlier study by Auton and Bolen,53 valine and leucine were shown to have weak favorable interactions for water too. Additionally GTFE experiments found sodium salts of glutamate and aspartate to interact highly favorably with betaine while VPO results indicate the largest unfavorable interaction with betaine.59 Another study showed a pH dependent preference of folate (vitamin B9) with betaine using solubility and VPO experiments.60 At pH 7, solubility-data estimates found folate to interact with both water and betaine with a TFE of about 0.089 ± 0.03 kcal mol−1.60 Specifically, Bhojane et al. reported that the glutamate tail of folate prefers to interact with water, while the aromatic rings of folate prefer betaine. The ambiguity toward predicting the right protein stability in osmolyte was not explored further. One of the reasons is the scarcity of data for a range of stabilizing to destabilizing osmolytes.
Atomic type contribution differs in various solvent environments: a case study of Arg
The present model predicts the overall trend of both stabilizing and destabilizing osmolytes satisfactorily well. However it fails in the cases of aromatic and charged cationic side chains. In case of aromatic residues the model predicts positive ΔGtr values (about 0.13 kcal mol−1) contradictory to the original TFE model of Liu and Bolen.48 The maximum discrepancy is seen in the case of Arg; here ΔGtr is 0.15 kcal mol−1. Therefore, Arg was chosen for further analysis. The charge surface of Arg is divided into aliphatic C and cationic N groups as Diehl et al. suggested recently.59 Further, their contributions towards ΔGtr were computed individually. It was found that for polyols, sugars and even for urea, the aliphatic C group’s ΔGtr values are negative (Fig. 9). However, the cationic N groups’ ΔGtr values are positive and larger in magnitude. As a result, net ΔGtr values are positive. Considering the simplicity of the present model it is apparent that the magnitude of the net ΔGtr value reported currently agrees with the original transfer free energy data. The present model might predict better TFE values for aromatic and charged cationic residues if the number of charge sites and charge values are optimized. This can be further explored in future studies. | ||
| Fig. 9 Transfer free energies (ΔGtr) of aliphatic C and cationic N groups of Arg. They are calculated using explicit charges, and considering all k-combinations in all ten osmolytes. | ||
Many earlier models used SASA to compute solvation free energies, and were able to predict ΔGtr values satisfactorily.52,61 However, none considered the polar fraction of osmolytes’ surface areas, interaction degeneracies (between solute and solvent) and microenvironment polarity in a unified model for predicting transfer free energies of a solute in the presence of a cosolvent. Fig. 10 presents a comprehensive view of the outcomes of our present model. This figure shows a comparative stability of various side chain models in protecting osmolytes. For example, Asn, Gln, Asp, Glu, Arg and Pro are highly stable in most of the protecting osmolytes; Ala, Val, Ile, Leu, Thr, Met, Lys, Phe, Trp and Tyr show moderate stability; Ser, Cys and histidine show poor stability. However, in denaturing solvents no such clear distinction can be made between side chain models as most of them show similar stabilities in urea and guanidine. Our model uses statistical mechanics to predict solvation thermodynamics, and is essentially parameter-free, and while consistent with electrostatic considerations is flexible about n-site choices. The proposed model is devoid of any rigorous conventional force field based molecular simulations. Considering its mathematical simplicity, and usage of model systems’ electrostatics primarily, the current model predicts satisfactory transfer free energies of amino acids/peptides from pure aqueous to aqueous osmolyte solutions for a range of stabilizing to destabilizing osmolytes.
 | ||
| Fig. 10 Wordcloud representation of stabilities of side chain models in all ten osmolytes. In each sub-set, the larger the size of the word the higher the ΔGtr value. | ||
Conclusions
Solvent effects play the primary role in modulating protein stability. Peptide backbone and side chains are known to contribute diversely to this stability. However their mechanisms of action are yet to be fully understood/described by means of a generic theoretical model. In this study, solvent induced protein stability was quantified at the molecular level in terms of the interactions between various osmolytes (ranging from the stabilizing to destabilizing) and model amino acid side chains and the backbone. In particular, we presented transfer free energies of model compounds from water to osmolyte. The average energy of each model in various osmolyte solutions was calculated using statistical mechanics, where the number of interaction sites of a model is the number of heavy atoms present. Force field assigned charges were considered for determining the interaction energies, while solvent polarity and fractional solvent accessible surface areas were used to estimate the binding-efficiency fraction between solvent and solute interaction. The model presented in this work predicts the correct trend of osmolyte’s behavior in a parameter-insensitive fashion. The current model has the potential to be developed further by including the optimized number of charge sites and charge values. Hence, we believe that the model can be of general use for estimating solute–solvent interaction energies of biomolecules in solvent environments which is of great interest in drug discovery and therapeutics.Data availability
The data supporting the findings of this study are available within the article and ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author thanks HPC facilities of Department of Computer Science and Engineering, Mahindra University.References
- P. H. Yancey, M. E. Clark, S. C. Hand, R. D. Bowlus and G. N. Somero, Living with water stress: evolution of osmolyte systems, Science, 1982, 217, 1214–1222 CrossRef CAS.
- P. H. Yancey, Organic osmolytes as compatible, metabolic and counteracting cytoprotectants in high osmolarity and other stresses, J. Exp. Biol., 2005, 208, 2819–2830 CrossRef CAS PubMed.
- G. D. Rose, P. J. Fleming, J. R. Banavar and A. Maritan, A backbone-based theory of protein folding, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16623–16633 CrossRef CAS PubMed.
- M. A. Schroer, Y. Zhai, D. F. Wieland, C. J. Sahle, J. Nase, M. Paulus, M. Tolan and R. Winter, Exploring the piezophilic behavior of natural cosolvent mixtures, Angew. Chem., Int. Ed., 2011, 50, 11413–11416 CrossRef CAS.
- A. Rani and P. Venkatesu, Changing relations between proteins and osmolytes: a choice of nature, Phys. Chem. Chem. Phys., 2018, 20, 20315–20333 RSC.
- S. R. Wlodarczyk, D. Custódio, A. Pessoa Jr and G. Monteiro, Influence and effect of osmolytes in biopharmaceutical formulations, Eur. J. Pharm. Biopharm., 2018, 131, 92–98 CrossRef CAS.
- T. O. Street, D. W. Bolen and G. D. Rose, A molecular mechanism for osmolyte-induced protein stability, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13997–14002 CrossRef CAS.
- G. Godara, C. Smith, J. Bosse, M. Zeidel and J. Mathai, Functional characterization of Actinobacillus pleuropneumoniae urea transport protein, ApUT, Am. J. Physiol.: Regul., Integr. Comp. Physiol., 2009, 296, R1268–R1273 CrossRef CAS PubMed.
- C. M. R. LeMoine and P. J. Walsh, Evolution of urea transporters in vertebrates: adaptation to urea’s multiple roles and metabolic sources, J. Exp. Biol., 2015, 218, 1936–1945 CrossRef.
- W. Kauzmann, Advances in Protein Chemistry, Elsevier, 1959, vol. 14, pp. 1–63 Search PubMed.
- J. L. England, V. S. Pande and G. Haran, Chemical denaturants inhibit the onset of dewetting, J. Am. Chem. Soc., 2008, 130, 11854–11855 CrossRef CAS PubMed.
- D. R. Canchi and A. E. García, Cosolvent effects on protein stability, Annu. Rev. Phys. Chem., 2013, 64, 273–293 CrossRef CAS PubMed.
- G. S. Jas, E. C. Rentchler, A. M. Słowicka, J. R. Hermansen, C. K. Johnson, C. R. Middaugh and K. Kuczera, Reorientation motion and preferential interactions of a peptide in denaturants and osmolyte, J. Phys. Chem. B, 2016, 120, 3089–3099 CrossRef CAS PubMed.
- A. S. Holehouse, K. Garai, N. Lyle, A. Vitalis and R. V. Pappu, Quantitative assessments of the distinct contributions of polypeptide backbone amides versus side chain groups to chain expansion via chemical denaturation, J. Am. Chem. Soc., 2015, 137, 2984–2995 CrossRef CAS PubMed.
- R. Zangi, R. Zhou and B. Berne, Urea’s action on hydrophobic interactions, J. Am. Chem. Soc., 2009, 131, 1535–1541 CrossRef CAS PubMed.
- S. Raghunathan, T. Jaganade and U. Priyakumar, Urea-aromatic interactions in biology, Biophys. Rev., 2020, 12, 65–84 CrossRef CAS PubMed.
- B. J. Bennion and V. Daggett, Counteraction of urea-induced protein denaturation by trimethylamine N-oxide: a chemical chaperone at atomic resolution, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 6433–6438 CrossRef CAS PubMed.
- Z. Su, F. Mahmoudinobar and C. L. Dias, Effects of trimethylamine-N-oxide on the conformation of peptides and its implications for proteins, Phys. Rev. Lett., 2017, 119, 108102 CrossRef PubMed.
- Q. Zou, B. J. Bennion, V. Daggett and K. P. Murphy, The molecular mechanism of stabilization of proteins by TMAO and its ability to counteract the effects of urea, J. Am. Chem. Soc., 2002, 124, 1192–1202 CrossRef CAS PubMed.
- M. Stasiulewicz, A. Panuszko, P. Bruździak and J. Stangret, Mechanism of Osmolyte Stabilization–Destabilizationof Proteins: Experimental Evidence, J. Phys. Chem. B, 2022, 126, 2990–2999 CrossRef CAS.
- C. Tanford, Protein denaturation: Part C. Theoretical models for the mechanism of denaturation, Adv. Protein Chem., 1970, 24, 1–95 CrossRef CAS.
- A. Arsiccio, P. Ganguly and J.-E. Shea, A Transfer Free Energy Based Implicit Solvent Model for Protein Simulations in Solvent Mixtures: Urea-Induced Denaturation as a Case Study, J. Phys. Chem. B, 2022, 4472–4482 CrossRef CAS.
- E. A. Oprzeska-Zingrebe, M. Kohagen, J. Kästner and J. Smiatek, Unfolding of DNA by co-solutes: insights from Kirkwood–Buff integrals and transfer free energies, Eur. Phys. J.: Spec. Top., 2019, 227, 1665–1679 CAS.
- D. R. Canchi and A. E. García, Backbone and side-chain contributions in protein denaturation by urea, Biophys. J., 2011, 100, 1526–1533 CrossRef CAS PubMed.
- B. Moeser and D. Horinek, Unified description of urea denaturation: backbone and side chains contribute equally in the transfer model, J. Phys. Chem. B, 2014, 118, 107–114 CrossRef CAS PubMed.
- C. Chipot and A. Pohorille, Free Energy Calculations: Theory and Applications in Chemistry and Biology, Springer, 2007, pp. 33–75 Search PubMed.
- C. J. D. Foumthuim, M. Carrer, M. Houvet, T. Škrbić, G. Graziano and A. Giacometti, Can the roles of polar and non-polar moieties be reversed in non-polar solvents?, Phys. Chem. Chem. Phys., 2020, 22, 25848–25858 RSC.
- C. J. D. Foumthuim and A. Giacometti, Solvent quality and solvent polarity in polypeptides, Phys. Chem. Chem. Phys., 2023, 25, 4839–4853 RSC.
- M. R. Shirts, J. W. Pitera, W. C. Swope and V. S. Pande, Extremely precise free energy calculations of amino acid side chain analogs: comparison of common molecular mechanics force fields for proteins, J. Chem. Phys., 2003, 119, 5740–5761 CrossRef CAS.
- P. Ganguly, N. F. van der Vegt and J.-E. Shea, Hydrophobic association in mixed urea–TMAO solutions, J. Phys. Chem. Lett., 2016, 7, 3052–3059 CrossRef CAS PubMed.
- M. Auton, J. Rösgen, M. Sinev, L. M. F. Holthauzen and D. W. Bolen, Osmolyte effects on protein stability and solubility: a balancing act between backbone and side-chains, Biophys. Chem., 2011, 159, 90–99 CrossRef CAS.
- A. Arsiccio and R. Pisano, Water entrapment and structure ordering as protection mechanisms for protein structural preservation, J. Chem. Phys., 2018, 148, 055102 CrossRef CAS.
- J. Rösgen, Synergy in protein–osmolyte mixtures, J. Phys. Chem. B, 2015, 119, 150–157 CrossRef.
- A. C. M. Ferreon, M. M. Moosa, Y. Gambin and A. A. Deniz, Counteracting chemical chaperone effects on the single-molecule α-synuclein structural landscape, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17826–17831 CrossRef CAS.
- J. M. Schurr, D. P. Rangel and S. R. Aragon, A contribution to the theory of preferential interaction coefficients, Biophys. J., 2005, 89, 2258–2276 CrossRef CAS.
- Y. Nozaki and C. Tanford, The solubility of amino acids and related compounds in aqueous urea solutions, J. Biol. Chem., 1963, 238, 4074–4081 CrossRef CAS.
- T. Jaganade, A. Chattopadhyay, S. Raghunathan and U. D. Priyakumar, Urea-water solvation of protein side chain models, J. Mol. Liq., 2020, 311, 113191 CrossRef CAS.
- J. A. Schellman, Fifty years of solvent denaturation, Biophys. Chem., 2002, 96, 91–101 CrossRef CAS.
- B. Khatri, S. Raghunathan, S. Chakraborti, R. Rahisuddin, S. Kumaran, R. Tadala, P. Wagh, U. D. Priyakumar and J. Chatterjee, Desolvation of peptide bond by O to S substitution impacts protein stability, Angew. Chem., Int. Ed., 2021, 60, 24870–24874 CrossRef CAS.
- E. M. Adams, S. Pezzotti, J. Ahlers, M. Rüttermann, M. Levin, A. Goldenzweig, Y. Peleg, S. J. Fleishman, I. Sagi and M. Havenith, Local mutations can serve as a game changer for global protein solvent interaction, JACS Au, 2021, 1, 1076–1085 CrossRef CAS.
- R. B. Best, X. Zhu, J. Shim, P. E. M. Lopes, J. Mittal, M. Feig and A. D. MacKerell, Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ1 and χ2 Dihedral Angles, J. Chem. Theory Comput., 2012, 8, 3257–3273 CrossRef CAS.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- G. Landrum, et al., Rdkit: Open-Source Cheminformatics Software, 2016, vol. 149, p. 150, https://www.rdkit.org/, https://github.com/rdkit/rdkit Search PubMed.
- D. Weininger, SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules, J. Chem. Inf. Comput. Sci., 1988, 28, 31–36 CrossRef CAS.
- D. Weininger, A. Weininger and J. L. Weininger, SMILES. 2. Algorithm for generation of unique SMILES notation, J. Chem. Inf. Comput. Sci., 1989, 29, 97–101 CrossRef CAS.
- V. Vagenende and B. L. Trout, Quantitative characterization of local protein solvation to predict solvent effects on protein structure, Biophys. J., 2012, 103, 1354–1362 CrossRef CAS PubMed.
- M. Auton and D. W. Bolen, Additive transfer free energies of the peptide backbone unit that are independent of the model compound and the choice of concentration scale, Biochemistry, 2004, 43, 1329–1342 CrossRef CAS PubMed.
- Y. Liu and D. Bolen, The peptide plays a dominant role in protein stabilization by naturally occurring osmolytes, Biochemistry, 1995, 34, 12884–12891 CrossRef CAS PubMed.
- Y. Qu, C. Bolen and D. Bolen, Osmolyte-driven contraction of a random coil protein, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 9268–9273 CrossRef CAS.
- Y.-T. Liao, A. C. Manson, M. R. DeLyser, W. G. Noid and P. S. Cremer, Trimethylamine N-oxide stabilizes proteins via a distinct mechanism compared with betaine and glycine, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2479–2484 CrossRef CAS.
- P. Ganguly, P. Boserman, N. F. van der Vegt and J.-E. Shea, Trimethylamine N-oxide Counteracts Urea Denaturation by Inhibiting Protein–Urea Preferential Interaction, J. Am. Chem. Soc., 2018, 140, 483–492 CrossRef CAS.
- M. Auton, L. M. F. Holthauzen and D. W. Bolen, Anatomy of energetic changes accompanying urea-induced protein denaturation, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 15317–15322 CrossRef CAS.
- M. Auton and D. W. Bolen, Predicting the energetics of osmolyte-induced protein folding/unfolding, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 15065–15068 CrossRef CAS.
- J. Rösgen and R. Jackson-Atogi, Volume exclusion and H-bonding dominate the thermodynamics and solvation of trimethylamine-N-oxide in aqueous urea, J. Am. Chem. Soc., 2012, 134, 3590–3597 CrossRef.
- P. Dullinger and D. Horinek, Solvation of Nanoions in Aqueous Solutions, J. Am. Chem. Soc., 2023, 145, 24922–24930 CAS.
- A. Arsiccio, P. Ganguly, L. La Cortiglia, J.-E. Shea and R. Pisano, ADD Force Field for Sugars and Polyols: Predicting the Additivity of Protein–Osmolyte Interaction, J. Phys. Chem. B, 2020, 124, 7779–7790 CrossRef CAS.
- O. Guvench, E. Hatcher, R. M. Venable, R. W. Pastor and A. D. MacKerell Jr, CHARMM additive all-atom force field for glycosidic linkages between hexopyranoses, J. Chem. Theory Comput., 2009, 5, 2353–2370 CrossRef CAS PubMed.
- T. Cloutier, C. Sudrik, H. A. Sathish and B. L. Trout, Kirkwood–Buff-derived alcohol parameters for aqueous carbohydrates and their application to preferential interaction coefficient calculations of proteins, J. Phys. Chem. B, 2018, 122, 9350–9360 CrossRef CAS PubMed.
- R. C. Diehl, E. J. Guinn, M. W. Capp, O. V. Tsodikov and M. T. Record Jr, Quantifying additive interactions of the osmolyte proline with individual functional groups of proteins: comparisons with urea and glycine betaine, interpretation of m-values, Biochemistry, 2013, 52, 5997–6010 CrossRef CAS PubMed.
- P. P. Bhojane, M. R. Duff Jr, K. Bafna, G. P. Rimmer, P. K. Agarwal and E. E. Howell, Aspects of weak interactions between folate and glycine betaine, Biochemistry, 2016, 55, 6282–6294 CrossRef CAS PubMed.
- B. M. Baynes and B. L. Trout, Proteins in mixed solvents: a molecular-level perspective, J. Phys. Chem. B, 2003, 107, 14058–14067 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Methods and models; microstate counting; ΔGtr vs. solvent molarity; partial charges; computed ΔGtr values for varying n-site model. See DOI: https://doi.org/10.1039/d4ra02576h |
| This journal is © The Royal Society of Chemistry 2024 |