
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConvenient synthesis and X-ray determination of 2-amino-6H-1,3,4-thiadiazin-3-ium bromides endowed with antiproliferative activity†
Hendawy N. Tawfeekab,
Alshaimaa Abdelmoezcd,
Kholood A. Dahlouse,
Bahaa G. M. Youssif *c,
Stefan Bräse*f,
Kari Rissaneng,
Martin Niegerh and
Essmat M. El-Sherefa
*c,
Stefan Bräse*f,
Kari Rissaneng,
Martin Niegerh and
Essmat M. El-Sherefa
aChemistry Department, Faculty of Science, Minia University, El Minia, 61519 Egypt
bUnit of Occupational of Safety and Health, Administration Office of Minia University, El-Minia 61519, Egypt
cPharmaceutical Organic Chemistry Department, Faculty of Pharmacy, Assiut University, Assiut 71526, Egypt. E-mail: bgyoussif2@gmail.com; bahaa.youssif@pharm.aun.edu.eg; Tel: +20-1098294419
dDepartment of Neurology, Ulm University, Ulm, Germany
eDepartment of Chemistry, College of Science, King Saud University, Riyadh 11451, Saudi Arabia
fInstitute of Biological and Chemical Systems, IBCS-FMS, Karlsruhe Institute of Technology, 76131 Karlsruhe, Germany. E-mail: braese@kit.edu
gDepartment of Chemistry, University of Jyväskylä, PO Box 35, 40014 Jyväskylä, Finland
hDepartment of Chemistry, University of Helsinki, PO Box 55, A. I. Virtasen Aukio 1, 00014 Helsinki, Finland
First published on 27th June 2024
Abstract
A new series of 1,3,4-thiadiazin-3-ium bromide derivatives 9a–g were prepared as a six-member ring by interactions between 4-substituted thiosemicarbazides 8a–e and α-halo ketones 2a,b. The reaction was conducted using hydrazine-NH2 and yielded a hexagonal shape. The structures of all obtained compounds have been verified using IR, NMR spectra, mass spectrometry, elemental analysis, and X-ray crystallography. The X-ray crystallographic analysis of compounds 9a and 9b has revealed that the salt is formed with the nitrogen atom N3 when the aromatic substituents 9a and 9d are present, but in the case of compounds 9b, 9c, 9e, 9f, and 9g with the aliphatic substituent, the salt is formed outside the ring. Compounds 9a–g were evaluated for antiproliferative activity as multitargeted inhibitors. Results revealed that targets 9a–g displayed good antiproliferative activity, with GI50 ranging from 38 nM to 66 nM against a panel of four cancer cell lines compared to the reference Erlotinib (GI50 = 33 nM). Compounds 9a, 9c, and 9d were the most potent antiproliferative derivatives, with GI50 values of 43, 38, and 47 nM, respectively. Compounds 9a, 9c, and 9d were evaluated for their inhibitory activity against EGFR, BRAFV600E, and VEGFR-2. The in vitro experiments demonstrated that the compounds being examined exhibit potent antiproliferative properties and have the potential to function as multitargeted inhibitors. In addition, the western blotting investigation demonstrated the inhibitory effects of 9c on EGFR, BRAFV600E, and VEGFR-2.
1. Introduction
Cancer remains a significant global problem. It is the second leading cause of death after heart problems.1 There are currently more than 100 different forms of cancer, each necessitating a distinct diagnosis and therapy.2 Many effective anti-cancer medicines, including standard chemotherapy drugs, suppress cell division and DNA replication. Nevertheless, many medications on the market are generic and often have common adverse effects.3 Thus, searching for new lead structures and chemical components to develop effective, innovative anti-cancer drugs has become a top priority in medicinal chemistry, considering current cancer treatments' limitations and adverse effects.4–6Anti-cancer medication discovery has heavily focused on developing treatments targeting a specific site with good efficacy and specificity. Clinical observations, such as identifying drug resistance in cancer treatment, have shown that focusing on one target may not always provide the intended biological outcome, even if the target is deactivated or suppressed.7–9 The resistance develops due to the target's self-modification through mutation or the cancer cell's adoption of new routes for growth and multiplication.10 Targeting a single oncoprotein has not led to successful treatment and may not be enough to establish long-lasting remission in patients.11 Thus, adjusting the biological network is acknowledged to be advantageous.
There are currently two opposing methodologies for designing multi-targeting medicines. Combination medication therapy involves using numerous drugs that act on different targets to provide an additive or synergistic impact. Combination therapies have shown success in treating metastatic melanoma with BRAF mutations.12,13 The FDA has approved the combination of dabrafenib (BRAF inhibitor) and trametinib (MEK inhibitor) based on preclinical evidence of increased apoptosis and delayed resistance to serine/threonine-protein kinase B-Raf.14,15 Phase III clinical trials have shown good results for combination therapy using RAF inhibitor (vemurafenib) and MEK inhibitor (cobimetinib) in treating BRAF mutant melanoma.16 Another successful example of combination therapy involves utilizing palbociclib and letrozole to treat advanced breast cancer.17
The second approach is to identify and develop medicines that target numerous oncogenic pathways simultaneously. Multi-targeting therapies entail the discovery of a single agent capable of simultaneously acting on two or more targets.18–20 The US Food and Drug Administration (FDA) has approved Lenvima (Lenvatinib) as a receptor tyrosine kinase inhibitor that blocks the kinase activities of vascular endothelial growth factor (VEGF) receptors VEGFR1, VEGFR2, and VEGFR3.21 Cabozantinib, known as cabometyx, is an FDA-approved small molecule inhibitor targeting the tyrosine kinases c-Met and VEGFR-2. It has effectively reduced tumor growth, metastasis, and angiogenesis.21
Nitrogen and sulfur-containing compounds are commonly used as the basis for various biological components in medicinal chemistry.22–24 Thiadiazine-based compounds have garnered considerable attention for their broad applicability as physiologically active molecules.25,26 Thiadiazine derivatives have shown significant cytotoxic effects on cancer cell lines by disrupting DNA synthesis, causing cell cycle arrest, inhibiting tumor cell invasion and migration, triggering apoptosis through the mitochondrial pathway, and blocking various nuclear enzymes.27–31
Ragab et al.27 described the design, synthesis, and biological testing of novel 1,3,4-thiadiazine-based derivatives as potential anti-cancer agents against non-small cell lung cancer (NSCLC) cells. The most effective compounds were chosen for further study as multitargeted inhibitors of VEGFR-2, BRAFV600E, and matrix metalloproteinase 9 (MMP9). Compound I (Fig. 1) showed the most promising results among the compounds tested. It demonstrated inhibitory activity against VEGFR-2, similar to Sorafenib, with an IC50 value of 0.11 ± 0.01 μM. It also exhibited the most potent suppression of BRAFV600E activity, with an IC50 value of 0.178 ± 0.004 μM, and MMP9 inhibition, with an IC50 value of 0.08 ± 0.004 μM.
 | ||
| Fig. 1 Structures of some reported 1,3,4-thiadiazine-based multitargeted anti-cancer agents and target compounds 9a–g. | ||
Another study explored the synthesis of some novel anti-cancer agents derived from triazolothiadiazine. When tested against a panel of NCI-60 cancer cell lines.29 Compound II (Fig. 1) showed a potential antiproliferative effect. Compound II was evaluated against 16 kinases to determine its antiproliferative mechanism. The study significantly suppressed EGFR, VEGFR-2, CDK-2, and GSK-3β. Furthermore, compound II increased the amount of active caspase-3, causing cell cycle arrest at the G2-M phase with apoptotic action.
On the other hand, phenacyl bromides, also known as α-halo ketones and primary alkyl halides, play a crucial role in chemical synthesis. They serve as crucial building blocks in heterocyclic constructions as a vital intermediate for synthesizing various bioactive chemicals and natural products.32 Also, thiosemicarbazides are a vital class of organic compounds that operate as the backbone of a range of heterocyclic compounds of considerable interest in medical and industrial uses.33,34 Additionally, interactions with phenacyl bromides revealed that thiosemicarbazides have several active centers.35 Busby and Domincy were the first to investigate the behavior of 4-substituted thiosemicarbazides toward haloketones, obtaining 2-amino-1,3,4-thiadiazines.36 Pfeiffer et al. synthesized 1,3,4-thiadiazines by combining α-halo ketones and 1,2,4-trialkyl thiosemicarbazides. The structure of the thiadiazines was confirmed through desulfurization with acetic acid.37 Several methods synthesized 1,3,4-thiadiazine, including interactions between thiosemicarbazides and α-halo ketones in different solvents.38,39 However, Aly et al.40 demonstrated the one-pot synthesis of 2-ylidenehydrazonothiazoles from the reaction of 4-substituted thiosemicarbazides, ketones, and phenacyl bromide, and they proposed that the product was obtained through the formation of the hydrazinylthiazole intermediate, which contradicted the findings of Pfeiffer et al.41
In light of the previous data on the multitargeted inhibitory action of 1,3,4-thiadiazine derivatives and our ongoing efforts to develop multitargeted anti-cancer agents,6,7,19,42–44 this investigation focuses on the synthesis of new 1,3,4-thiadiazine derivatives (9a–g, Fig. 1) as multitargeted antiproliferative agents.
2. Experiments
2.1. Chemistry
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 1594 (CC) cm−1. 1H NMR (300 MHz, CDCl3): δH = 3.91 (s, 2H, CH2), 7.35–7.48 (m, 6H, Ar-H), 7.88–7.94 (m, 2H, Ar-H), 8.01–8.04 (m, 2H, Ar-H), 9.45 (bs, 1H, exocyclic-NH), 12.85 (bs, 1H, cyclic-NH+) ppm. 13C NMR (75 MHz, DMSO-d6): δc = 23.26 (CH2), 123.84, 126.01, 127.67, 128.80, 129.46, 130.03 (Ar-CH), 132.94, 139.15 (Ar–C), 144.32 (thiadiazine-C5), 150.71 (thiadiazine-C2) ppm. MS (70 eV) m/z (%): 347 (M+, 9), 267 (100), 155 (10), 119 (5). Anal. calcd for C15H14BrN3S (348.26) calcd: C, 51.73; H, 4.05; N, 12.07; S, 9.21. Found: C, 51.81; H, 3.99; N, 12.13; S, 9.28.), 5.72–5.83 (m, 1H, Allyl-CH), 7.33–7.44 (m, 3H, Ar-H), 7.72–7.79 (m, 2H, Ar-H), 10.62 (bs, 1H, exocyclic-NH+), 12.98 (bs, 1H, cyclic-NH) ppm. 13C NMR (75 MHz, CDCl3): δc = 22.99 (CH2), 48.01 (CH2-Allyl), 119.91 (Allyl-CH2), 127.01, 129.02, 130.17, 131.71 (Ar-CH), 132.18 (Ar-C), 149.36 (C5), 166.75 (C2) ppm. MS (70 eV) m/z (%): 311 (M+, 15), 283 (28), 199 (54), 189 (10), 119 (33), 102 (100). Anal. calcd for C12H14BrN3S (311.23) calcd: C, 46.16; H, 4.52; N, 13.46; S, 10.27. Found: C, 46.23; H, 4.59; N, 13.39; S, 10.30.N), 1538, 1445 (CC) cm−1. 1H NMR (300 MHz, CDCl3): δH = 125–1.33, 1.36–1.69, 2.01–2.04 (m, 10H, cyclohexyl-CH2), 3.02–3.09 (m, 1H, cyclohexyl-CH), 3.55 (s, 2H, CH2), 7.33–7.37 (m, 3H, Ar-H), 7.80–7.82 (m, 2H, Ar-H), 10.20 (bs, 1H, exocyclic-NH+), 11.84 (bs, 1H, cyclic-NH) ppm. 13C NMR (75 MHz, CDCl3): δc = 22.46 (CH2), 24.54, 25.23, 33.30 (cyclohexyl-CH2), 52.98 (cyclohexyl-CH), 126.34, 128.42, 129.50 (Ar-CH), 135.31 (Ar-C), 147.38 (C5), 166.85 (C2) ppm. MS (70 eV) m/z (%): 353 (M+, 3), 274 (M+-HBr, 100), 192 (7), 133 (3), 104 ([PhCN]+, 4). Anal. calcd for C15H20BrN3S (354.31) calcd: C, 50.85; H, 5.69; N, 11.86; S, 9.05. Found: C, 50.78; H, 5.63; N, 11.90; S, 9.11.
N), 1594 (CC) cm−1. 1H NMR (300 MHz, CDCl3): δH = 3.91 (s, 2H, CH2), 7.35–7.48 (m, 6H, Ar-H), 7.88–7.94 (m, 2H, Ar-H), 8.01–8.04 (m, 2H, Ar-H), 9.45 (bs, 1H, exocyclic-NH), 12.85 (bs, 1H, cyclic-NH+) ppm. 13C NMR (75 MHz, DMSO-d6): δc = 23.26 (CH2), 123.84, 126.01, 127.67, 128.80, 129.46, 130.03 (Ar-CH), 132.94, 139.15 (Ar–C), 144.32 (thiadiazine-C5), 150.71 (thiadiazine-C2) ppm. MS (70 eV) m/z (%): 347 (M+, 9), 267 (100), 155 (10), 119 (5). Anal. calcd for C15H14BrN3S (348.26) calcd: C, 51.73; H, 4.05; N, 12.07; S, 9.21. Found: C, 51.81; H, 3.99; N, 12.13; S, 9.28.), 5.72–5.83 (m, 1H, Allyl-CH), 7.33–7.44 (m, 3H, Ar-H), 7.72–7.79 (m, 2H, Ar-H), 10.62 (bs, 1H, exocyclic-NH+), 12.98 (bs, 1H, cyclic-NH) ppm. 13C NMR (75 MHz, CDCl3): δc = 22.99 (CH2), 48.01 (CH2-Allyl), 119.91 (Allyl-CH2), 127.01, 129.02, 130.17, 131.71 (Ar-CH), 132.18 (Ar-C), 149.36 (C5), 166.75 (C2) ppm. MS (70 eV) m/z (%): 311 (M+, 15), 283 (28), 199 (54), 189 (10), 119 (33), 102 (100). Anal. calcd for C12H14BrN3S (311.23) calcd: C, 46.16; H, 4.52; N, 13.46; S, 10.27. Found: C, 46.23; H, 4.59; N, 13.39; S, 10.30.N), 1538, 1445 (CC) cm−1. 1H NMR (300 MHz, CDCl3): δH = 125–1.33, 1.36–1.69, 2.01–2.04 (m, 10H, cyclohexyl-CH2), 3.02–3.09 (m, 1H, cyclohexyl-CH), 3.55 (s, 2H, CH2), 7.33–7.37 (m, 3H, Ar-H), 7.80–7.82 (m, 2H, Ar-H), 10.20 (bs, 1H, exocyclic-NH+), 11.84 (bs, 1H, cyclic-NH) ppm. 13C NMR (75 MHz, CDCl3): δc = 22.46 (CH2), 24.54, 25.23, 33.30 (cyclohexyl-CH2), 52.98 (cyclohexyl-CH), 126.34, 128.42, 129.50 (Ar-CH), 135.31 (Ar-C), 147.38 (C5), 166.85 (C2) ppm. MS (70 eV) m/z (%): 353 (M+, 3), 274 (M+-HBr, 100), 192 (7), 133 (3), 104 ([PhCN]+, 4). Anal. calcd for C15H20BrN3S (354.31) calcd: C, 50.85; H, 5.69; N, 11.86; S, 9.05. Found: C, 50.78; H, 5.63; N, 11.90; S, 9.11.2.2. Crystal X-ray structure determination of 9a and 9b
The single-crystal X-ray diffraction studies were carried out on a Rigaku XtaLAB Synergy R diffractometer with HyPix-Arc 100 detector at 133(2) K using Cu-Kα radiation (9a, λ = 1.54178 Å, PhotonJet R rotating anode generator) or a Bruker D8 Venture diffractometer with a PhotonII detector at 173(2) K using Cu-Kα radiation (9b, λ = 1.54178 Å). Dual space methods (SHELXT) [G. M. Sheldrick, Acta Crystallogr., 2015, A71, 3–8] were used for structure solution, and refinement was carried out using SHELXL-2014 (full-matrix least-squares on F2) [G. M. Sheldrick, Acta Crystallogr., 2015, C71, 3–8]. Hydrogen atoms were localized by difference electron density determination and refined using a riding model. An extinction correction and a semi-empirical absorption correction were applied.9a: colourless crystals, C15H14N3S⋯Br, Mr = 348.26, crystal size 0.18 × 0.14 × 0.13 mm, monoclinic, space group P21/n (no. 14), a = 8.9077(1) Å, b = 9.1847(1) Å, c = 18.1367(2) Å, β = 93.364(1)°, V = 1481.29(3) Å3, Z = 4, ρ = 1.562 mg m3, μ(Cu-Kα) = 5.04 mm−1, F(000) = 704, T = 133 K, 2θmax = 158.6°, 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 484 reflections, of which 3177 were independent (Rint = 0.016), 188 parameters, 2 restraints, R1 = 0.020 (for 3161 I > 2σ(I)), wR2 = 0.051 (all data), S = 1.09, largest diff. peak/hole = 0.33/−0.31 e Å−3.
484 reflections, of which 3177 were independent (Rint = 0.016), 188 parameters, 2 restraints, R1 = 0.020 (for 3161 I > 2σ(I)), wR2 = 0.051 (all data), S = 1.09, largest diff. peak/hole = 0.33/−0.31 e Å−3.
9b: yellow crystals, C12H14N3S⋯Br, Mr = 312.23, crystal size 0.14 × 0.25 × 0.20 mm, orthorhombic, space group Pbca (no. 61), a = 7.4414(3) Å, b = 18.2272(7) Å, c = 19.6573(7) Å, V = 2666.24(18) Å3, Z = 8, ρ = 1.556 mg m3, μ(Cu-Kα) = 5.51 mm−1, F(000) = 1264, T = 173 K, 2θmax = 144.2°, 19426 reflections, of which 2613 were independent (Rint = 0.033), 161 parameters, 2 restraints, R1 = 0.022 (for 2558 I > 2σ(I)), wR2 = 0.056 (all data), S = 1.05, largest diff. peak/hole = 0.28/−0.25 e Å−3.
CCDC 2329624 (9a) and 2329625 (9b) contain the supplementary crystallographic data for this paper.
2.3. Biology
2.4. Western blotting assay
The western blotting analysis54 aids in elucidating the probable mechanisms behind the observed antiproliferative activity of 9c. Appendix A contain all details related to the assay methodology.3. Results and discussion
3.1. Chemistry
According to prior research and our own,35,41,55 the interaction between thiosemicarbazides and phenacyl bromides is principally determined by the reaction conditions, substituted position, and solvent employed, as seen in Scheme 1. All obtained compounds were confirmed by X-ray crystallographic determination. | ||
| Scheme 1 Reactivity of substituted thiosemicarbazides toward phenacyl bromide. | ||
Based on previous research and our interest in phenacyl bromide reactions, we found that the reaction between phenacyl bromide and 1/or 2-substituted thiosemicarbazides results in the formation of a five-membered ring (thiazole-ring). In the current study, the reaction of phenacyl bromides (2a,b) with 4-substituted thiosemicarbazides (8a–e) in absolute ethanol with no catalyst resulted in the synthesis of 2-substituted amino-1,3,4-thiadiazinium bromide derivatives (9a–g). This reaction formed six-membered rings, as shown in Scheme 2. In this context, the differential behavior of 4-substituted thiosemicarbazides 8a–e versus 1/or 2-substituted thiosemicarbazides influences hexagonal ring formation. This mismatch explains the observed differential in nucleophilicity of the NH2 group, which first reacts with the carbonyl group before cycling through the SH group. This effect compelled the reaction to yield thiadiazine as six-membered ring, rather than the five-membered ring characteristic of thiazole.
 | ||
| Scheme 2 Synthesis of 1,3,4-thiadiazin-3-ium bromide derivatives 9a–g. | ||
All obtained compounds are recrystallized from ethanol, and their structures were confirmed with advanced spectroscopic analysis tools such as IR, mass, and NMR spectrum (1H and 13C). Also, the structures are unambiguously confirmed with X-ray crystallographic analysis.
To illustrate our obtained products, we chose compound 9a (Fig. 2) as an example, which was assigned as 5-phenyl-2-(phenylamino)-6H-1,3,4-thiadiazin-3-ium bromide. In the IR spectrum of compound 9a, broad bands were observed at 3164 cm−1 due to NH 3015 cm−1 because of aromatic-CH—the other two peaks at 1617 and 1594 cm−1 due to CN and CC, respectively. Also, the 1H NMR spectra of 9a showed three broad singlet signals at δH = 3.91, 9.45, and 12.85 ppm, which were assigned as 1,3,4-thiadizinium-CH2,56 exocyclic-NH and 1,3,4-thiadizinium-NH+, respectively. The two NH-protons show a downfield shift, attributed to the high deshielding caused by the positive charge on 1,3,4-thiadizinium-ringas observed from the X-ray structure analysis of 9a (Fig. 2); furthermore, in 13C NMR spectrum of 9a signals at 23.26, 144.32, and 150.71 ppm, which were assigned as CH2, thiadiazinium-C5 and thiadiazinium-C2, respectively, in addition to aromatic carbons. Through the preceding data and X-rays displayed in Fig. 2 and 3, we do not doubt that the structures for our obtained products are confirmed.
 | ||
| Fig. 2 X-ray crystal structure of compound 9a (5-phenyl-2-(phenylamino)-6H-1,3,4-thiadiazin-3-ium bromide) (displacement parameters are drawn at 50% probability level). | ||
 | ||
| Fig. 3 X-ray crystal structure of compound 9b ((Z)-N-(5-phenyl-3,6-dihydro-2H-1,3,4-thiadiazin-2-ylidene)prop-2-en-1-aminium bromide) (displacement parameters are drawn at 50% probability level). | ||
Furthermore, the NMR of compound 9b, also known as (Z)-N-(5-phenyl-3,6-dihydro-2H-1,3,4-thiadiazin-2-ylidene)prop-2-en-1-aminium bromide. The 1H NMR spectrum revealed two downfield shifts at δ 12.98 and 10.62 ppm, corresponding to cyclic-NH and exocyclic-NH+. The allylic protons appeared at δH = 4.01–4.04 ppm as triplet with two protons for allyl-CH2, at 5.23–5.34 ppm, as doublet of doublet (dd) with two protons for allyl-CH2 and multiplet signal with one proton at δH = 5.72–5.83 ppm, for the allylic-CH, in addition to singlet signal with upfield chemical shift at δH = 4.10 ppm, which assigned as CH2-group. The inverted values of the cyclic and exocyclic-NH's in both compounds 9a and 9b are owing to the delocalization of the positive charge over the cyclic-N and amino-nitrogen (exocyclic-N), as well as the hydrogen bond between the nitrogen atoms-H and bromine.
The mass spectra of both 9a and 9b compounds revealed molecular ion peaks (M+) at 347 and 311 m/z, confirming that the products are formed as a result of the reaction between phenacyl bromide and 4-phenyl/allyl thiosemicarbazide, with the elimination of an H2O molecule and another HBr molecule. Moreover, the structure of 9b was confirmed by X-ray crystallography, as illustrated in Fig. 3.
The X-ray structure of compound 9b confirms the loss of one molecule of H2O, and the HBr molecule adheres to the crystallite, as indicated in the mass fragmentation pattern of compound 9b in Fig. 4.
 | ||
| Fig. 4 Fragmentation pattern of compound 9b. | ||
Furthermore, X-ray crystallographic studies validated the structure of all obtained products, as clarified by the crystallographic data of compounds 9a and 9b (note that the crystallographic numbering does not mirror the systematic IUPAC numbering) (Fig. 2 and 3). The X-ray data of 9a revealed that its molecular formula is C15H14N3S·Br with a weight of m/z = 348.26, a colorless crystal, and a monoclinic shape with space group P21/n. The measured bond lengths of S1–C2 and S1–C6 bond length are 1.7304 (14) and 1.8148 (19) Å, respectively, are slightly different from the S–C single bond of 1.737 Å, and this is attributed to the sp2 character of C2 and the sp3 character of C6. The bond lengths of C2–N3 and C2–N7 are equal to 1.3346 (18) Å, and 1.3259 (19) Å showed some double bond character as they are comparable to the C–N σ-bond length of 1.47 Å and this confirmed the delocalization of the double bond as well as the positive charge. In contrast, the X-ray data of compound 9b showed that the crystal has an orthorhombic shape with space group pbca. It is observed from the bond length table of compound 9b that the S1–C2 and S1–C6 bond lengths are 1.7290 (15) and 1.8034 (16) Å, respectively, are slightly different from the S–C single bond of 1.737 Å and this is attributed to the sp2 character of C2 and the sp3 character of C6. The bond lengths of C2–N3 (1.331 (2)) Å and C2–N7 (1.316 (2)) Å suggest that these bonds have some double bond character as they are comparable to the C–N σ-bond length of 1.47 Å, the N3–N4 bond length is 1.3954(18) Å is closed to the N–N single bond 1.401 Å. Observing the mentioned values of bond lengths confirms the presence of a positive charge, which makes the bond length longer.
The suggested mechanism for forming thiadiazin-3-ium bromides 9a–g derivatives is as depicted in Scheme 3. Initially, a nucleophilic attack from the NH2 group to the carbonyl group and condensation occurs with a loss of H2O molecule. After that, inter-nucleophilic occurs via S-atom (SN2 reaction S-alkylation step) on the methylene group, and cyclization takes place, resulting in the formation of the salt 12 which undergoes rearrangement and forming 2-amino-1,3,4-thiadiazinium bromide derivatives 8a–g.
 | ||
| Scheme 3 Suggested mechanism for the formation of 2-amino-1,3,4-thiadiazinium bromide derivatives 9a–g. | ||
3.2. Biology
| Comp. | Cell viability% | Antiproliferative activity IC50 ± SEM (nM) | ||||
|---|---|---|---|---|---|---|
| A-549 | MCF-7 | Panc-1 | HT-29 | Average (GI50) | ||
| 9a | 91 | 43 ± 4 | 40 ± 4 | 44 ± 4 | 44 ± 4 | 43 |
| 9b | 90 | 56 ± 5 | 52 ± 5 | 58 ± 5 | 58 ± 5 | 56 |
| 9c | 92 | 37 ± 3 | 35 ± 3 | 38 ± 3 | 40 ± 4 | 38 |
| 9d | 89 | 48 ± 4 | 44 ± 4 | 48 ± 4 | 47 ± 4 | 47 |
| 9e | 92 | 62 ± 6 | 58 ± 5 | 64 ± 6 | 64 ± 6 | 62 |
| 9f | 89 | 65 ± 6 | 64 ± 6 | 66 ± 6 | 66 ± 6 | 66 |
| 9g | 88 | 54 ± 5 | 51 ± 5 | 56 ± 5 | 56 ± 5 | 54 |
| Erlotinib | ND | 30 ± 3 | 40 ± 3 | 30 ± 3 | 30 ± 3 | 33 |
In general, targets 9a–g displayed good antiproliferative activity, with GI50 ranging from 38 nM to 66 nM against the four cancer cell lines, when compared to the reference Erlotinib (GI50 = 33 nM), and all examined compounds demonstrated less potent efficacy than Erlotinib. Compounds 9a, 9c, and 9d were the most potent antiproliferative derivatives, with GI50 values of 43, 38, and 47 nM, respectively.
Compound 9c (Ar = cyclohexyl, Y = H) was the most potent derivative of all developed compounds, with a GI50 value of 38 nM, and was more effective than Erlotinib against the MCF-7 cancer cell line, with an IC50 value of 35 nM versus Erlotinib's IC50 value of 40 nM. The replacement of the cyclohexyl group in the thiadiazine moiety with a phenyl group, as in compound 9a (Ar = Ph, Y = H), or with an allyl group, as in compound 9b (Ar = Allyl, Y = H), resulting in a reported decrease in antiproliferative activity. Compounds 9a and 9b had GI50 values of 43 and 56 nM, respectively, which were 1.2- and 1.5-fold lower than 9c, suggesting that the cyclohexyl group at position 2 of the thiadiazine moiety is better tolerated for antiproliferative action than the phenyl and allyl groups.
Moreover, the substitution pattern of the phenyl group at position 5 of the thiadiazine moiety might greatly influence the antiproliferative activity of these molecules. For instance, the 4-bromo derivative 9g (Ar = cyclohexyl, Y = Br) was less effective than the unsubstituted derivative 9c (Ar = cyclohexyl, Y = H). Compound 9g exhibited a GI50 value of 54 nM, suggesting that substituting a bromine atom in the para-position of the phenyl group is not beneficial for its activity. The same holds when comparing compound 9a (Ar = Ph, Y = H) with compound 9d (Ar = Ph, Y = Br). It was found that compound 9a had a GI50 value of 43 nM, but compound 9d had a GI50 value of 47 nM.
Finally, compounds 9e (Ar = Me, Y = Br) and 9f (Ar = Et, Y = Br) were the least effective, with GI50 values of 62 and 66 nM, respectively. This suggests that having methyl and ethyl groups at position 2 of the thiadiazine moiety is not beneficial for activity.
| Compound | EGFR inhibition IC50 ± SEM (nM) | BRAFV600E inhibition IC50 ± SEM (nM) | VEGFR-2 inhibition IC50 ± SEM (nM) |
|---|---|---|---|
| 9a | 92 ± 7 | 83 ± 6 | 2.80 ± 0.02 |
| 9c | 86 ± 6 | 74 ± 5 | 2.20 ± 0.02 |
| 9d | 96 ± 7 | 88 ± 6 | 3.25 ± 0.03 |
| Erlotinib | 80 ± 5 | 60 ± 5 | ND |
| Vemurafenib | ND | 30 ± 3 | ND |
| Sorafenib | ND | ND | 0.17 ± 0.01 |
3.3. Western blot analysis
Based on previous results, compound 9c is highly effective at inhibiting EGFR, BRAFV600E, and VEGFR-2, significantly suppressing cell proliferation. The western blotting analysis54 aids in elucidating the probable mechanisms behind the observed antiproliferative activity of 9c. The findings of the western blotting analysis agreed with the data obtained from the in vitro assays for EGFR, BRAFV600E, and VEGFR-2 inhibition. Compound 9c exhibited a significant decrease in the levels of the specific enzymes, with inhibition percentages ranging from 47% to 58% when compared to the positive control A549 untreated cells (as shown in Table 3 and Fig. 5). The findings of this study confirm the outcomes of the in vitro experiment, demonstrating that compound 9c exerts its antiproliferative effects via multitargeted inhibitory action.| S | Compound | Western blotting | β-Actin | ||||
|---|---|---|---|---|---|---|---|
| Code | MW | Cells | OD | ||||
| EGFR | VEGFR-2 | BRAFV600E | |||||
| 1 | 9c | 354.31 | A549 | 0.288 | 0.346 | 0.219 | √ |
| 2 | Control | — | A549 | 0.695 | 0.662 | 0.507 | √ |
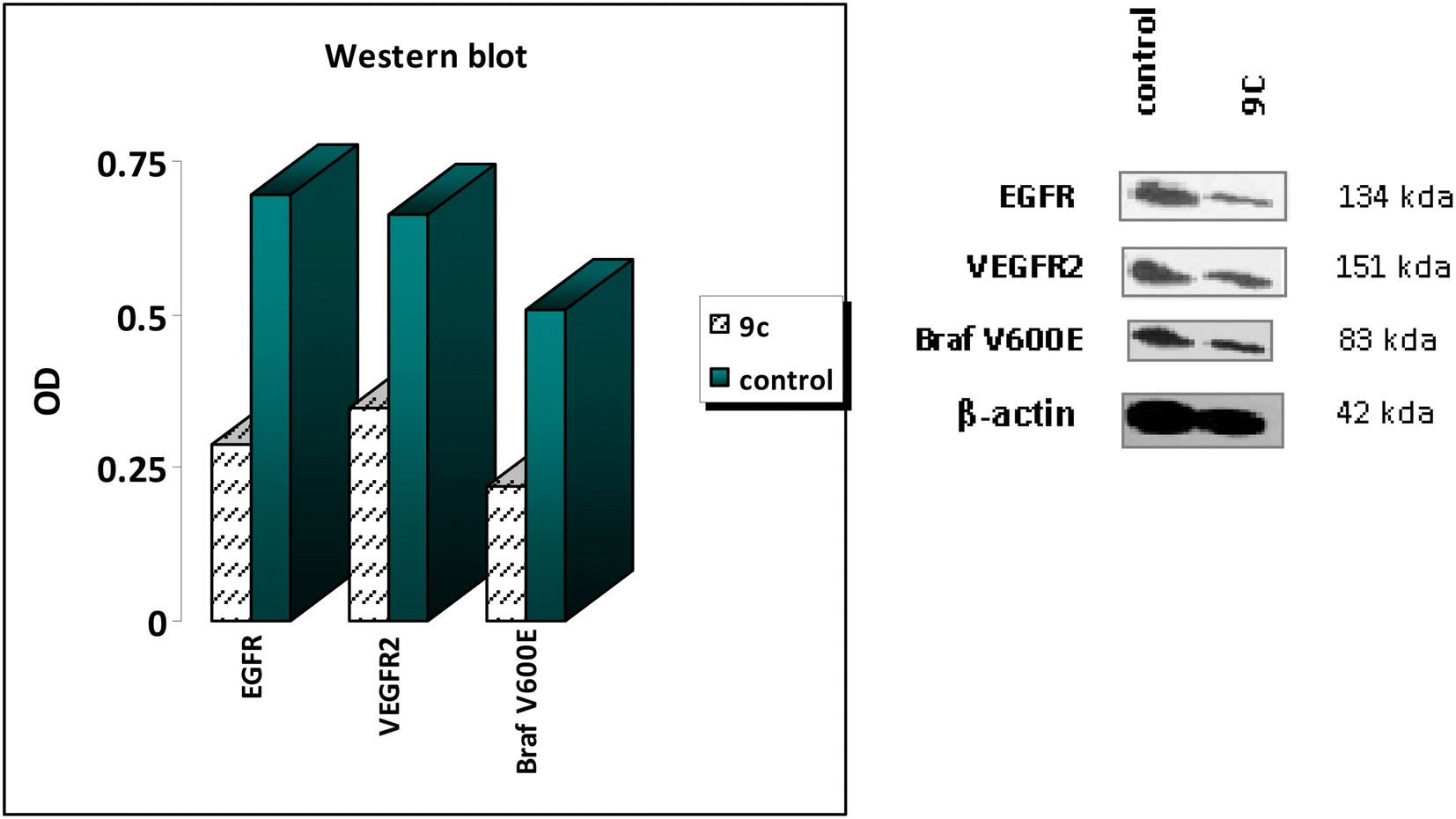 | ||
| Fig. 5 Results of western blotting of compound 9c against EGFR, VEGFR-2, and BRAFV600E. | ||
4. Conclusion
This study discusses the preparation of thiadiazines heterocycle 9a–g through the reaction between 4-substituted thiosemicarbazides and α-halo ketones. The accurate structure was established and verified by X-ray diffraction analysis. A comparison was conducted, revealing contradictory findings in other scholarly works. Compounds 9a–g were investigated as EGFR, BRAFV600E, and VEGFR-2 inhibitors in quest of a multitargeted antiproliferative scaffold. The results showed that compound 9c is a promising antiproliferative agent inhibiting EGFR, BRAFV600E, and VEGFR-2. On the other hand, further structural modifications may be required to successfully acquire more potent lead compounds to develop future cancer medicines.Sample availability
Samples of compounds 9a–g are available from the authors.Author contributions
Bahaa G. M. Youssif, Essmat M. El-Sheref, and Hendawy N. Tawfeek: conceptualization, methodology, writing, editing and revision. Alshaimaa Abdelmoez and Kholood Dahlous: writing, editing and revision. S. Bräse: writing and editing. Kari Rissanen, M. Nieger: X-ray analysis.Conflicts of interest
The authors state that they do not have any known competing financial interests or personal links that could appear to have influenced the work disclosed in this study.Acknowledgements
This work was funded by the Researchers Supporting Project Number (RSP2024R388) at King Saud University, Riyadh, Saudi Arabia. The authors also acknowledge support from the KIT-Publication Fund of the Karlsruhe Institute of Technology.References
- W. Cao, H.-D. Chen, Y.-W. Yu, N. Li and W.-Q. Chen, Chinese Med. J., 2021, 134, 783–791 CrossRef PubMed.
- K. El-Adl, H. M. Sakr, R. G. Yousef, A. B. Mehany, A. M. Metwaly, M. A. Elhendawy, M. M. Radwan, M. A. ElSohly, H. S. Abulkhair and I. H. Eissa, Bioorg. Chem., 2021, 114, 105105 CrossRef CAS PubMed.
- A. S. Correia, F. Gärtner and N. Vale, Heliyon, 2021, 7(1), e05948 CrossRef CAS PubMed.
- R. A. Mekheimer, S. M. Allam, M. A. Al-Sheikh, M. S. Moustafa, S. M. Al-Mousawi, Y. A. Mostafa, B. G. Youssif, H. A. Gomaa, A. M. Hayallah and M. Abdelaziz, Bioorg. Chem., 2022, 121, 105693 CrossRef CAS PubMed.
- M. A. Mahmoud, A. F. Mohammed, O. I. Salem, S. M. Rabea and B. G. Youssif, J. Mol. Struct., 2023, 1282, 135165 CrossRef CAS.
- F. O. A. Frejat, H. Zhai, Y. Cao, L. Wang, Y. A. Mostafa, H. A. Gomaa, B. G. Youssif and C. Wu, Bioorg. Chem., 2022, 126, 105922 CrossRef PubMed.
- L. H. Al-Wahaibi, M. A. Mahmoud, Y. A. Mostafa, A. E. Raslan and B. G. Youssif, J. Enzyme Inhib. Med. Chem., 2023, 38, 376–386 CrossRef CAS PubMed.
- L. N. Puls, M. Eadens and W. Messersmith, Oncologist, 2011, 16, 566–578 CrossRef CAS PubMed.
- A. D. Boran and R. Iyengar, Current Opinion in Drug Discovery & Development, 2010, vol. 13, p. 297 Search PubMed.
- J. J. Frost, Int. J. Unconv. Comput., 2021, 16, 41–78 Search PubMed.
- D. C. Altieri, Future Oncol., 2010, 6, 487–489 CrossRef CAS PubMed.
- K. Paraiso, I. Fedorenko, L. Cantini, A. Munko, M. Hall, V. Sondak, J. Messina, K. Flaherty and K. Smalley, Br. J. Cancer, 2010, 102, 1724–1730 CrossRef CAS PubMed.
- K. T. Flaherty, J. R. Infante, A. Daud, R. Gonzalez, R. F. Kefford, J. Sosman, O. Hamid, L. Schuchter, J. Cebon and N. Ibrahim, N. Engl. J. Med., 2012, 367, 1694–1703 CrossRef CAS PubMed.
- G. V. Long, D. Stroyakovskiy, H. Gogas, E. Levchenko, F. de Braud, J. Larkin, C. Garbe, T. Jouary, A. Hauschild and J. J. Grob, N. Engl. J. Med., 2014, 371, 1877–1888 CrossRef PubMed.
- C. Robert, B. Karaszewska, J. Schachter, P. Rutkowski, A. Mackiewicz, D. Stroiakovski, M. Lichinitser, R. Dummer, F. Grange and L. Mortier, N. Engl. J. Med., 2015, 372, 30–39 CrossRef PubMed.
- A. Ribas, R. Gonzalez, A. Pavlick, O. Hamid, T. F. Gajewski, A. Daud, L. Flaherty, T. Logan, B. Chmielowski and K. Lewis, Lancet Oncol., 2014, 15, 954–965 CrossRef CAS PubMed.
- R. S. Finn, M. Martin, H. S. Rugo, S. Jones, S.-A. Im, K. Gelmon, N. Harbeck, O. N. Lipatov, J. M. Walshe and S. Moulder, N. Engl. J. Med., 2016, 375, 1925–1936 CrossRef CAS PubMed.
- C. T. Keith, A. A. Borisy and B. R. Stockwell, Nat. Rev. Drug Discov., 2005, 4, 71–78 CrossRef CAS PubMed.
- L. H. Al-Wahaibi, A. F. Mohammed, F. E.-Z. S. Abdel Rahman, M. H. Abdelrahman, X. Gu, L. Trembleau and B. G. Youssif, J. Enzym. Inhib. Med. Chem., 2023, 38, 2218602 CrossRef PubMed.
- L. H. Al-Wahaibi, A. F. Mohammed, M. H. Abdelrahman, L. Trembleau and B. G. Youssif, Pharmaceuticals, 2023, 16, 1039 CrossRef CAS PubMed.
- US Department of Health and Human Services, Nutr. Today, 1990, 25(6), 29–39 Search PubMed.
- A. A. Mourad, Y. W. Rizzk, I. Zaki, F. Z. Mohammed and M. El Behery, J. Mol. Struct., 2021, 1242, 130722 CrossRef CAS.
- A. Ahmad, H. Varshney, A. Rauf, A. Sherwani and M. Owais, Arab. J. Chem., 2017, 10, S3347–S3357 CrossRef CAS.
- A. Anant, A. Ali, A. Ali, G. Gupta and V. Asati, J. Mol. Struct., 2021, 1245, 131079 CrossRef CAS.
- S. H. Ali and A. R. Sayed, Synth. Commun., 2021, 51, 670–700 CrossRef CAS.
- A. A. Abu-Hashem, A.-R. El-Gazzar, H. N. Hafez, A. A. M. Abdelgawad and M. A. Gouda, Mini-Rev. Org. Chem., 2024, 21, 151–171 CrossRef CAS.
- F. A. Ragab, S. A. Abdel-Aziz, M. Kamel, A. M. A. Ouf and H. A. Allam, Bioorg. Chem., 2019, 93, 103323 CrossRef PubMed.
- B. Zhang, Y.-H. Li, Y. Liu, Y.-R. Chen, E.-S. Pan, W.-W. You and P.-L. Zhao, Eur. J. Med. Chem., 2015, 103, 335–342 CrossRef CAS PubMed.
- M. I. Ismail, S. Mohamady, N. Samir and K. A. Abouzid, ACS Omega, 2020, 5, 20170–20186 CrossRef CAS PubMed.
- Q. Xu, K. Bao, M. Sun, J. Xu, Y. Wang, H. Tian, D. Zuo, Q. Guan, Y. Wu and W. Zhang, Sci. Rep., 2017, 7, 11997 CrossRef PubMed.
- L. Ji, Y. Zhou, Q. Yu, Y. Fang, Y. Jiang, Y. Zhao, C. Yuan and W. Xie, J. Mol. Struct., 2021, 1227, 129406 CrossRef CAS.
- R. H. Vekariya, K. D. Patel, N. P. Prajapati and H. D. Patel, Synth. Commun., 2018, 48, 1505–1533 CrossRef CAS.
- A. A. Hassan, N. K. Mohamed, A. A. Aly, H. N. Tawfeek, H. Hopf, S. Bräse and M. Nieger, Mol. Diversity, 2019, 23, 821–828 CrossRef CAS PubMed.
- A. A. Hassan, A. A. Aly, M. Ramadan, N. K. Mohamed, H. N. Tawfeek, S. Bräse and M. Nieger, Monatsh. Chem., 2020, 151, 1453–1466 CrossRef CAS.
- A. A. Hassan, N. K. Mohamed, A. A. Aly, H. N. Tawfeek, S. Bräse and M. Nieger, J. Mol. Struct., 2019, 1176, 346–356 CrossRef CAS.
- H. BoBooth, D. V. Griffiths and M. L. Jozefowicz, J. Chem. Soc., Perkin Trans. 2, 1980, 255 RSC.
- W.-D. Pfeiffer, E. Dilk, H. Rossberg and P. Langer, Synlett, 2003, 2003, 2392–2394 CrossRef.
- W. W. Wardakhan, Arch. Pharmazie, 2006, 339, 608–615 CrossRef CAS PubMed.
- S. Rostamizadeh, A. M. Amani and N. Shadjou, Phosphorus, Sulfur, Silicon Relat. Elem.s, 2012, 187, 238–244 CrossRef CAS.
- A. A. Aly, E. M. El-Sheref, A. B. Brown, S. Bräse, M. Nieger and E.-S. M. Abdelhafez, J. Sulfur Chem., 2019, 40, 641–647 CrossRef CAS.
- W. D. Pfeiffer, D. Junghans, A. S. Saghyan and P. Langer, J. Heterocycl. Chem., 2014, 51, 1063–1067 CrossRef CAS.
- B. G. Youssif, A. M. Gouda, A. H. Moustafa, A. A. Abdelhamid, H. A. Gomaa, I. Kamal and A. A. Marzouk, J. Mol. Struct., 2022, 1253, 132218 CrossRef CAS.
- H. A. El-Sherief, B. G. Youssif, A. H. Abdelazeem, M. Abdel-Aziz and H. M. Abdel-Rahman, Anti Cancer Agents Med. Chem., 2019, 19, 697–706 CrossRef CAS PubMed.
- M. B. Alshammari, A. A. Aly, B. G. Youssif, S. Bräse, A. Ahmad, A. B. Brown, M. A. Ibrahim and A. H. Mohamed, Front. Chem., 2022, 10, 1076383 CrossRef CAS PubMed.
- M. Tišler, Croat. Chem. Acta, 1956, 28, 147–154 Search PubMed.
- A. A. Hassan, N. K. Mohamed, A. A. Aly, M. Ramadan, H. A. Gomaa, A. T. Abdel-Aziz, B. G. Youssif, S. Bräse and O. Fuhr, Molecules, 2023, 28, 7951 CrossRef CAS PubMed.
- A. M. Mohassab, H. A. Hassan, H. A. Abou-Zied, M. Fujita, M. Otsuka, H. A. Gomaa, B. G. Youssif and M. Abdel-Aziz, J. Mol. Struct., 2024, 1297, 136953 CrossRef CAS.
- M. A. Mahmoud, A. F. Mohammed, O. I. Salem, T. M. Almutairi, S. Bräse and B. G. Youssif, J. Enzyme Inhib. Med. Chem., 2024, 39, 2305856 CrossRef PubMed.
- A. A. Aly, M. B. Alshammari, A. Ahmad, H. A. Gomaa, B. G. Youssif, S. Bräse, M. A. Ibrahim and A. H. Mohamed, Arab. J. Chem., 2023, 16, 104612 CrossRef CAS.
- L. H. Al-Wahaibi, A. F. Mohammed, M. H. Abdelrahman, L. Trembleau and B. G. Youssif, Molecules, 2023, 28, 1269 CrossRef CAS PubMed.
- L. H. Al-Wahaibi, M. Hisham, H. A. Abou-Zied, H. A. Hassan, B. G. Youssif, S. Bräse, A. M. Hayallah and M. Abdel-Aziz, Pharmaceuticals, 2023, 16, 1522 CrossRef CAS PubMed.
- A. M. Mohassab, H. A. Hassan, D. Abdelhamid, A. M. Gouda, B. G. Youssif, H. Tateishi, M. Fujita, M. Otsuka and M. Abdel-Aziz, Bioorg. Chem., 2021, 106, 104510 CrossRef CAS PubMed.
- A. A. Marzouk, S. A. Abdel-Aziz, K. S. Abdelrahman, A. S. Wanas, A. M. Gouda, B. G. Youssif and M. Abdel-Aziz, Bioorg. Chem., 2020, 102, 104090 CrossRef CAS PubMed.
- W. N. Burnette, Anal. Biochem., 1981, 112, 195–203 CrossRef CAS PubMed.
- A. A. Hassan, N. K. Mohamed, A. A. Aly, H. N. Tawfeek, S. Bräse and M. Nieger, Monatsh. Chem., 2020, 151, 1143–1152 CrossRef CAS.
- W. D. Jones Jr, J. M. Kane and A. D. Sill, J. Heterocycl. Chem., 1983, 20, 1359–1361 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2329624 (9a) and 2329625 (9b). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra02531h |
| This journal is © The Royal Society of Chemistry 2024 |