
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal free synthesis of thermally stable blue fluorescent m-terphenyls by ring transformation of 2H-pyran-2-ones: chemical synthesis, spectroscopy and computational studies†
Priyanka B. Kolea,
Kokila Sakthivela,
Sanja J. Armaković bd,
Stevan Armakovićcd,
Muzaffar Iqbale,
Fateh V. Singh*a and
Shiva Prasad Kollur*f
bd,
Stevan Armakovićcd,
Muzaffar Iqbale,
Fateh V. Singh*a and
Shiva Prasad Kollur*f
aChemistry Division, School of Advanced Sciences (SAS), VIT Chennai, Vandalur-Kelambakkam Road, Chennai-600 127, Tamil Nadu, Chennai, India. E-mail: fatehveer.singh@vit.ac.in
bUniversity of Novi Sad, Faculty of Sciences, Department of Chemistry, Biochemistry and Environmental Protection, 21000 Novi Sad, Serbia
cUniversity of Novi Sad, Faculty of Sciences, Department of Physics, 21000 Novi Sad, Serbia
dAssociation for the International Development of Academic and Scientific Collaboration (AIDASCO), 21000 Novi Sad, Serbia
eDepartment of Pharmaceutical Chemistry, College of Pharmacy, King Saud University, Riyadh 11451, Saudi Arabia
fSchool of Physical Sciences, Amrita Vishwa Vidyapeetham, Mysuru Campus, Mysuru – 570 026, Karnataka, India. E-mail: shivachemist@gmail.com
First published on 24th May 2024
Abstract
A simple yet convenient nucleophile-induced synthetic route for the construction of thermally stable fluorescent active functionalized stilbenes has been delineated. The nucleophile-induced base encouraged synthetic protocol was performed under mild conditions without harming the environment and products were achieved in good yields. The synthesized stilbenes showed amazing emission properties and good thermal stability. Synthesized products displayed interesting positive solvatochromism in different solvents based on variation in polarity. Further, we present a comprehensive analysis of the eight molecules, leveraging a combination of Density Functional Tight Binding (DFTB), Density Functional Theory (DFT) calculations, and Molecular Dynamics (MD) simulations. This integrated approach allowed for an in-depth exploration of the electronic structures, reactivity profiles, and dynamic behaviors of these complex molecular systems. Our findings reveal significant insights into the physicochemical properties of the synthesized molecules, contributing to a deeper understanding of their potential applications in various fields.
Introduction
The naturally occurring substituted aromatic compounds that have m-terphenyl scaffolds are enhanced with remarkable medicinal properties.1 The photophysical and optical properties of these molecules have garnered a lot of interest in the field of materials chemistry.2,3 More significantly though, these substances are necessary building blocks for the production of liquid crystals, dendrimers, ligands needed for organometallic synthesis, cyclic ketones, and optically active cyclophanes, among other chemicals.4–8 Additionally, a crucial component of their ability to function as chemical reagents is the presence of different substituents in the terphenyl moiety.9 Because of their amazing electronic potential, they are sought-after substrates for the development of host and electron-transporting materials for organic light-emitting devices (OLEDs).10 The naturally occurring m-terphenyl-cored scaffolds are depicted in Fig. 1. Trifucol (1), a member of the Fucaceae family, was isolated from brown algae and possesses significant antioxidant activity.11 A natural supplement called macranthol (2) is made from Illicium dunnianum Tutch and has been shown to have antidepressant properties in rats.12 Dictyoterphenyl A (3) has specific antiproliferative properties against cancer cells and is isolated from Dictyostelium cellular slime moulds.13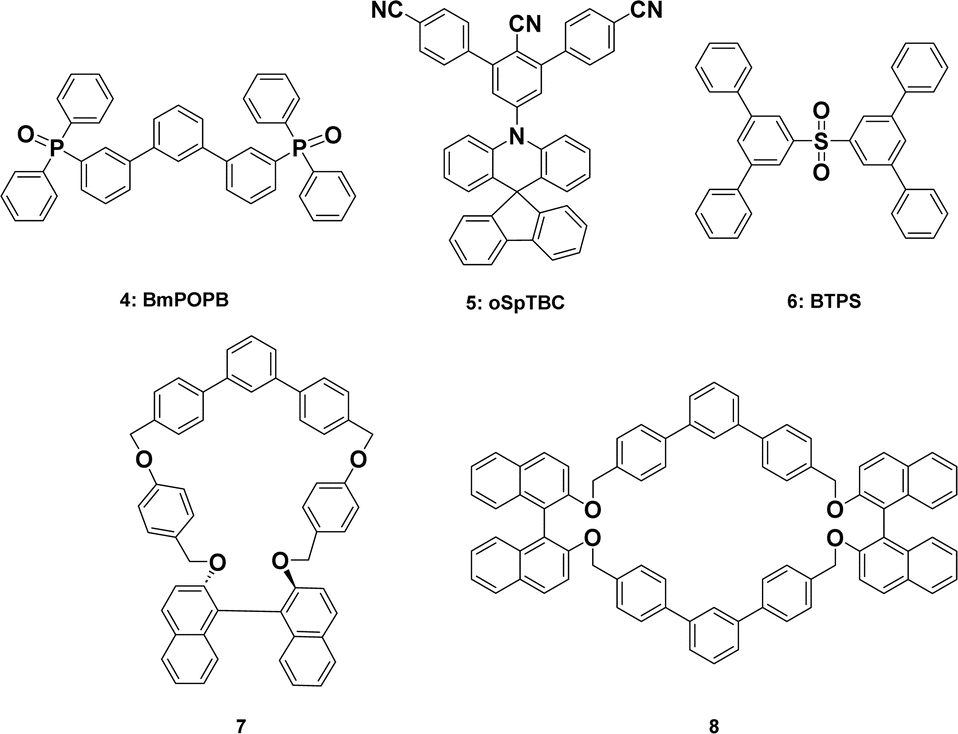 | ||
| Fig. 1 Naturally occurring biologically active m-terphenyl-cored compounds (1–3) and photo-physically active m-terphenyl-cored compounds (4–8). | ||
Since m-terphenyls take a long time to conjugate, the molecules implanted in these scaffolds exhibit a wide range of photophysical properties in addition to their biological ones.14,15 Fig. 1 shows the photophysically active m-terphenyl skeletons, which are represented as [1,1′:3′,1′′-terphenyl]. Organic light emitting devices (OLEDs) might greatly benefit from the very effective electron-transporting substance known as 3,3′′-diylbis(diphenylphosphine oxide) (BmPOPB, 4).16 The compound designated oSpTBC (5) is referred to as a delayed fluorescence emitter.17 BTPS (6) denotes OLED use.18,19 The remaining two cyclophanes, 7 and 8, are employed as metal trapping agents due to the hollow hole that exists inside the structural framework.20,21 Furthermore, polyfunctionalized derivatives of m-terphenyl are employed as strong UV filters.22 Furthermore, it was observed that the poly aromatic compounds with donor–acceptor cores have exceptional fluorescence.23–29 To highlight the photophysical characteristics of extended conjugation systems, we have synthesized phenyl substituted m-terphenyl scaffolds.
A number of disadvantages are linked to the majority of existing techniques, including the need for a metal catalyst, laborious reaction conditions such high temperatures and high pressures, and lengthy reaction times. Notwithstanding the abundance of available tactics, a novel style that is compatible with present methodologies and provides flexibility in incorporating a broad range of functional groups into designed phenyl substituted m-terphenyl scaffolds is still necessary. Also, the electrical and steric properties of the biphenyl/terphenyl backbone are easily manipulated using standard organic synthesis methods. m-Terphenyl-derived ligands are particularly important in coordination and organometallic chemistry because they have a larger steric bulk than corresponding biphenyls and p-terphenyls. Yet, the m-terphenyl backbone allows the insertion of three donating moieties in close physical proximity to one another by functionalizing the locations ortho to the CAr–CAr bonds (Fig. 2). Though this latter aspect has been explored in detail for m-terpyridine complexes, relatively little work has been done on tri-ortho-substituted m-terphenyl ligands.30
 | ||
| Fig. 2 (a) Absorbance and (b) fluorescent emission spectra of V-shaped biphenyl-flanked m-terphenyls 11a–h. | ||
In contemporary research, sophisticated atomic-level computational techniques are indispensable for pinpointing optimal candidates in pharmaceutical and material science domains and for understanding their functional mechanisms.31,32 Quantum-mechanical approaches, particularly those grounded in density functional theory (DFT), have demonstrated exemplary efficiency in balancing precision and resource expenditure, especially for assessing localized reactivity.33,34 Alongside quantum mechanics (QM) methods, Molecular Dynamics (MD) simulations have been notably effective in scenarios demanding the explicit incorporation of solvent molecules.35–37 This technique is also beneficial for examining larger molecular assemblies comprising numerous atoms. To thoroughly evaluate the reactivity of newly synthesized molecular structures, employing a variety of computational approaches and analysing diverse reactivity indicators is recommended. Thus, our research involves extensive computational analysis to deepen our understanding of the reactive characteristics of the newly synthesized molecules from the synthesized series of molecules.
Results and discussion
Here in, we delineate the metal-free and convenient strategy for the synthesis of bulky biphenyl flanked V-shaped m-terphenyls scaffolds with maximum yields by the ring transformation of 4-amino-6-biphenyl-2H-pyran-2-ones using biphenyl ketone as a source of carbanion. The primary precursor 2H-pyran-2-one was synthesized by the reaction of 4-acetyl biphenyl with methyl-2-cyano-3,3-dimethylsulfanylacrylate in DMSO under alkaline conditions at ambient temperature.38,39 Besides, the 2H-pyran-2-one was reacted with various secondary amines in methanol at reflux temperature to achieve the synthesis of 4-amino-6-biphenyl-2H-pyran-2-ones.40,41Our earliest efforts were dealt with the screening of suitable base for the ring transformation of 6-([1,1′-biphenyl]-4-yl)-2-oxo-4-(piperidin-1-yl)-2H-pyran-3-carbonitrile 9a. The bases of different pKb in DMF have been used for the ring transformation of substrate 9a to corresponding biphenyl flanked V-shaped m-terphenyls derivatives 11a–h using ketone 10 as a source of carbanion and results are outlined below (Table 1). Initially, the reaction was performed with hard base NaOH (pKb value: 0.2) and the reaction produced up to 57% of ring transform product (Table 1, entry 1). To our delight, the formation of ring transformed product was improved up to 77% and 86% respectively by using other hard bases like LiOH (pKb value: −0.36) and KOH (pKb value: 0.5) (Table 1, entries 2 and 3). Similarly, this reaction was tested with comparatively soft bases such as the Cs2CO3, K2CO3 (pKb value: 3.75) and NaHCO3 (pKb value: 8.0) under same reaction condition but the yield of ring transformation product was reduced significantly 11a (Table 1, entries 4–6). Surprisingly, it was difficult to corelate the product yields with the hardness or softness and pKb of the bases used during these studies.
|
||||
|---|---|---|---|---|
| Entry | Solvent | Base | Time (h) | Yield 11a (%) |
| 1 | DMF | NaOH | 12 | 57 |
| 2 | DMF | LiOH | 10 | 77 |
| 3 | DMF | KOH | 10 | 86 |
| 4 | DMF | Cs2CO3 | 12 | 46 |
| 5 | DMF | K2CO3 | 12 | 41 |
| 6 | DMF | NaHCO3 | 12 | 33 |
| 7 | DMSO | KOH | 10 | 84 |
| 8 | MeCN | KOH | 10 | 78 |
| 9 | EtOAc | KOH | 10 | 70 |
| 10 | 1,4-Dioxane | KOH | 14 | — |
| 11 | MeOH | KOH | 14 | 21 |
| 12 | EtOH | KOH | 14 | 13 |
| 13 | Toluene | KOH | 12 | 29 |
| 14 | THF | KOH | 14 | — |
| 15 | AcOH | KOH | 14 | — |
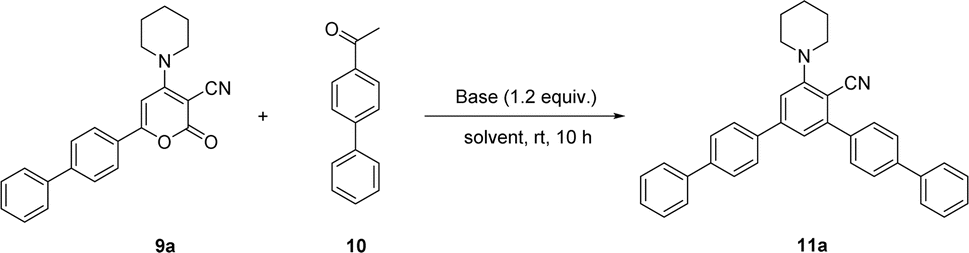
After getting the suitable base, our target was shifted towards screening of the appropriate solvents for ring transformation reaction. Different solvents were examined for the ring transformation of lactone 9a to corresponding biphenyl flanked V-shaped m-terphenyls derivative 11a and results are listed below (Table 1). Initially, the ring transformation reaction of lactone 9a was performed in polar aprotic solvents like, DMSO and MeCN, hence the reaction product 11a was obtained in 84 and 78% yields respectively (Table 1, entries 7 and 8). The occurrence of desired product was reduced up to 70% in EtOAc (Table 1, entry 9). In order to check the performance of polar protic solvents, MeOH and EtOH were used for the transformation reaction, but the yields of products were reduced into 21% and 13% respectively (Table 1, entries 11 and 12). The course of reaction was investigated in nonpolar solvent such as toluene and ring transformed product was obtained in low yield (Table 1, entry 13). Finally, reaction was also performed in solvents like THF and 1,4-dioxane but reaction failed in these solvents (Table 1, entries 10 and 14). This transformation reaction could not proceed in acetic acid due to salt formation and starting material was fully recovered (Table 1, entry 15).
Considering the above optimized conditions in hand: 1.2 equivalent of KOH as base and DMF as favourable solvent, we synthesized bulky biphenyl flanked V-shaped m-terphenyls 11a–h from 6-biphenyl-2H-pyran-2-ones 9a–h (Table 1, entries 1–14) via ring transformation strategy. All reactions were carried out at ambient temperature for 10–14 h under optimized conditions. The desired products were obtained in good yields.
Furthermore, 4-amino-6-biphenyl-2H-pyran-2-ones 9a–h were synthesized and further reacted with acetyl biphenyl 10 under optimized conditions (Table 2, entries 1–8). All the starting materials were easily transformed in to biphenyl-flanked V-shaped m-terphenyl 11a–h. The ring transformation of substrates 9d and 9g containing acyclic tert-amino groups gave reaction products 11d and 11g in comparatively low yields (Table 2, entries 4 and 7). Whereas other substrates embedded with cyclic tert-amino groups 9a–c, e, f, h delivered high yields of expected products 11a–c, e, f, h (Table 2, entries 1–3, 5, 6 and 8).
|
|||
|---|---|---|---|
| Entry | 9 | Time (h) | Yield (%) |
| 1 | 9a: Piperidin-1-yl | 10 | 11a: 96 |
| 2 | 9b: N-Phenylpiperazin-1-yl | 10 | 11b: 90 |
| 3 | 9c: Pyrrolidin-1-yl | 12 | 11c: 87 |
| 4 | 9d: Diethyl amine | 14 | 11d: 70 |
| 5 | 9e: N-Methylpiperazin-1-yl | 10 | 11e: 86 |
| 6 | 9f: Morpholin-1-yl | 12 | 11f: 88 |
| 7 | 9g: Dimethylamine | 14 | 11g: 65 |
| 8 | 9h: N-Ethylpiperazin-1-yl | 10 | 11h: 84 |
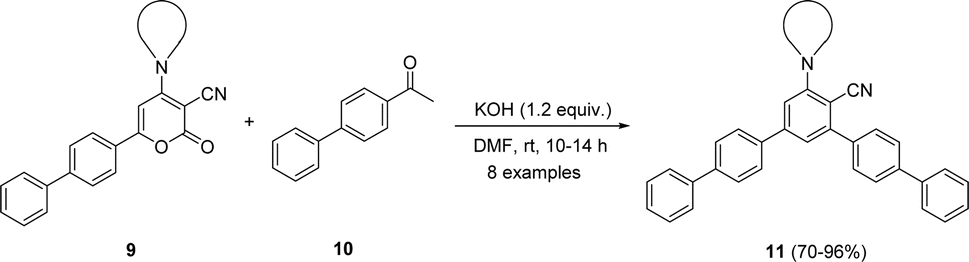
The mechanistic pathway for the synthesis of biphenyl-flanked m-terphenyls 11 from functionalized 2H-pyranone 9 is exemplified in Scheme 1. Upon reaction of ketone with KOH generates carbanion at alpha carbon, which further attack at C-6 position of 2H-pyranone 9 lead the formation of intermediate [A]. Promptly intermediate [A] gets intramolecular cyclization involves carbonyl group and C-3 position of pyran ring caused intermediate [B]. The intermediate [C] was formed by decarboxylation followed by dehydration of intermediate [B]. Further, intermediate [C] aromatized to affords the 5′′-amino-[1,1′:4′,1′′:3′′,1′′′:4′′′,1′′′′-quinquephenyl]-4′′-carbonitrile 11.
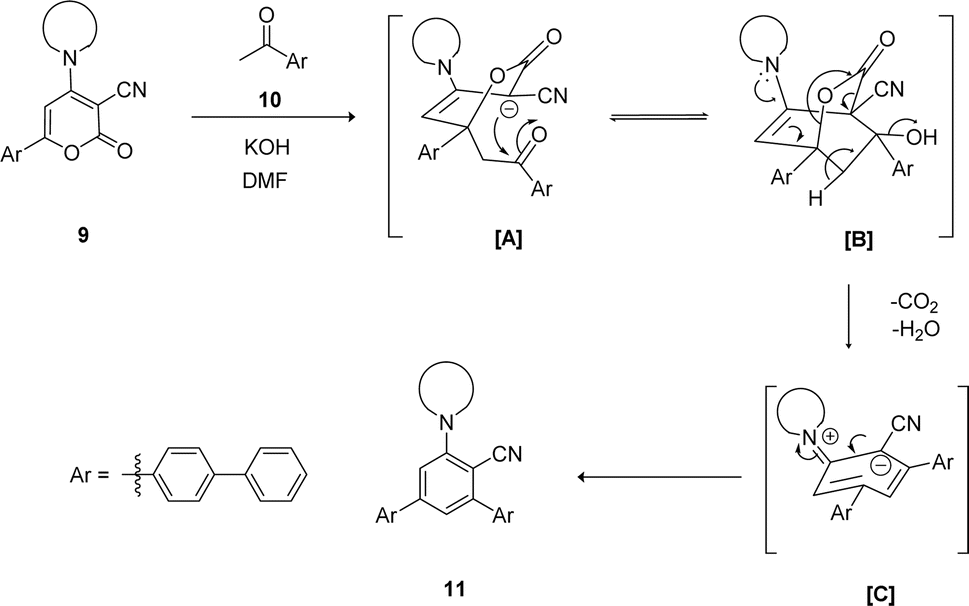 | ||
| Scheme 1 Plausible mechanism for synthesis of biphenyl flanked V-shaped m-terphenyls 11a–h from 6-biphenyl-2H-pyran-2-ones 9a–h. | ||
Photophysical properties of synthesized V-shaped biphenyl-flanked m-terphenyls
The photophysical activity of synthesized V-shaped biphenyl-flanked m-terphenyls enhanced due to the presence of donor and acceptor moieties. The photophysical properties for V-shaped biphenyl-flanked m-terphenyls was investigated in MeCN (1 × 10−5 M). The compounds 11a–h indicated short-range absorbance maxima from 272–285 nm and long-range absorbance maxima from 365–372 nm (Fig. 2(a)). The compounds embedded with N-methylpiperazine 11e and N-ethylpiperazine 11h found to improves fluorescence emitting at 434 nm whereas, compounds with N-phenylpiperazine 11b was observed to quench the fluorescence emitting at 435 nm. This depends on the ability towards stabilization of delocalization and maintenance of aromaticity within the structural architecture. Similarly, compounds with pyrrolidine 11c and piperidine 11a exhibited good emission at 459 and 463 nm, respectively. The acyclic amines attached to the main structural skeletons of m-terphenyls 11d and 11g disclosed better emissions at respectable λmax 460 and 458 nm. Morpholine holding molecule 11f displayed less intense emission peak at 459 nm (Fig. 2(b)).Next, we delved the solvatochromic properties of compound 11c, and the results showed modest bathochromic shifts in favour of more polar solvents from less polar, non-polar solvents (Fig. 3). The compounds 11c revealed absorbance in the range of 281–294 nm and red shift emissions from λmax 455–475 nm.
 | ||
| Fig. 3 Solvatochromic effect of biphenyl-flanked m-terphenyls derivatives 11c. | ||
The better solvatochromic shift was witnessed in DMSO then any other solvents due to greater dipole moment of 11c in the excited state than that in ground state during photo-induced electronic transition, which connects the intramolecular charge transfer features of particular compounds.
Further, the concentration based fluorescent nature of compound 11c was studied (Fig. 4). On dilution of compound increases the fluorescent intensity with the same wavelength evidenced that, the molecule exist as a monomer. The maximum intensity is observed at 7.5 × 10−5 M concentration, here after the fluorescent intensity started to decrease.
 | ||
| Fig. 4 Concentration study of biphenyl-flanked m-terphenyls derivatives 11c. | ||
Thermal analysis for synthesized V-shaped biphenyl-flanked m-terphenyls derivatives 11c
The thermogravimetric analysis (TGA) and differential thermal analysis (DTA) were carried out of m-terphenyl derivative 11c to evaluate their thermal behaviour (Fig. 5) using alumina as reference. The TG curve for compound 11c exposed that this compound stay thermally stable up to 293 °C and underwent single step weight loss and ensuing decomposition in the temperature range of 293–475 °C. Further, DTA curve has not pictured any sharp peak revealed that; the substance 11c may exist as an amorphous solid.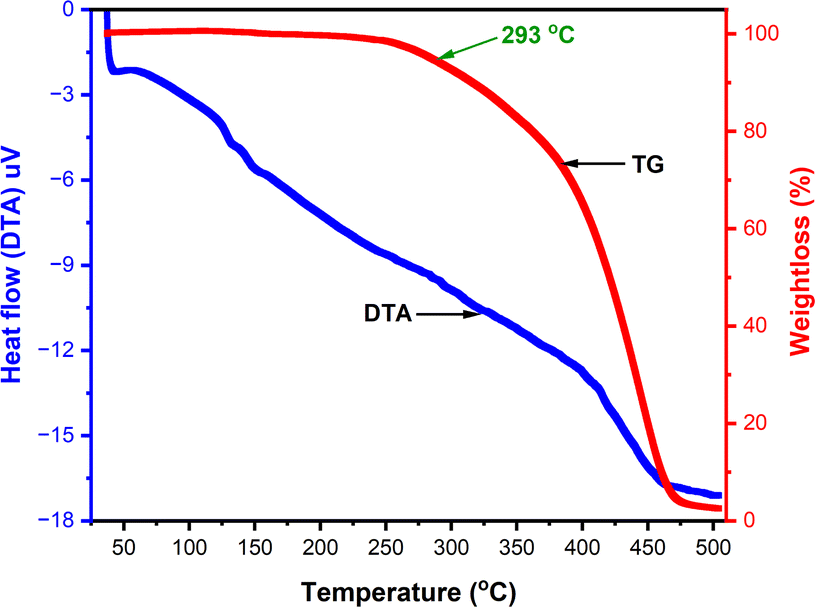 | ||
| Fig. 5 The thermogravimetric analysis and differential thermal analysis of 11c. | ||
Local reactivity of molecules
The study considered the local reactive characteristics of molecules by employing two prominent quantum molecular indicators: the MEP and ALIE. These two descriptors are rooted in the electron density concept, a cornerstone in the quantum mechanical analysis of molecular structures. These descriptors give information on two critical molecular attributes. High or low MEP values pinpoint areas within the molecule likely to interact due to electrostatic forces with oppositely charged entities. In contrast, ALIE highlights regions within the molecule that are more inclined to electron loss, thus indicating susceptibility to electrophilic attacks.A particularly effective method for visualizing these properties involves overlaying the calculated values of these descriptors onto the electron density surface of the molecule. This approach facilitates the identification of areas with extreme descriptor values, thereby aiding in the recognition of molecular regions that are particularly reactive or interactive with other molecules. In Fig. 6, we have illustrated this concept by presenting the MEP and ALIE surfaces of molecules 11g and 11h.
 | ||
| Fig. 6 (a) MEP surface of 11g and (b) ALIE surface of 11h. | ||
For the sake of simplicity, in Fig. 6, we provided MEP and ALIE surfaces of the derivative with the lowest MEP value (11g) and the derivative with the lowest ALIE value (11h). All MEP and ALIE values are jointly presented in Fig. S2 and S3 of the ESI.†
A study of extreme MEP values indicated that the minimal values ranged in the interval of around just 2 kcal mol−1. A similar situation was observed in the case of maximal MEP values, with the exception of derivative 11d. According to these two descriptors, the two derivatives mentioned might be the most reactive. With the lowest MEP value of −41.43 kcal mol−1, the 11g derivative might be the most reactive derivative with respect to electrostatic interactions. From the topological standpoint, in the case of MEP quantum molecular descriptor, the CN group appears to be most significant in all cases since extreme values are found in its vicinity, as presented in Fig. 7(a) and S1 and S2 of the ESI.† The reason for this is that in a cyano (CN) group, the nitrogen atom has a significant electron-withdrawing effect due to its electronegativity. This creates a region of high electron density around the nitrogen atom. It is also known that the carbon–nitrogen triple bond in the CN group is highly polar. The nitrogen, being more electronegative than carbon, pulls electron density towards itself. This results in a partial negative charge on the nitrogen and a partial positive charge on the carbon. Consequently, the nitrogen atom in the CN group exhibits a low electrostatic potential, as it is a region where electrons are more densely localized.
On the other side, with the lowest ALIE value of 196.07 kcal mol−1, derivative 11h might be the most reactive molecule among studied from the aspect of sensitivity towards electrophilic attacks. From the topological standpoint, in the case of ALIE quantum-molecular descriptor, aside from the benzene rings, the lowest ALIE values have been identified in the near vicinity of the nitrogen atom of pyrazine ring. This could be explained by the fact that the pyrazine fragment has a lone pair of electrons, contributing to the increased electron density in this region of the molecule and thus making it easier for electrons to be removed.
Intramolecular non-covalent interactions
Non-covalent intramolecular interactions play a crucial role in determining the behaviour, structure, and reactivity of various chemical entities. While strong covalent bonds primarily hold molecules together, it is the more subtle non-covalent interactions that are equally vital in providing molecular stability and functionality. These interactions, which include hydrogen bonds, van der Waals forces, π–π stacking, and electrostatic forces, are key in shaping a molecule's conformational stability, electronic structure, and its ability to bind with other entities like ligands, substrates, or solvents. Gaining insights into these non-covalent intramolecular forces is essential across various disciplines, especially in materials science and pharmaceutical development.DFT calculations play a fundamental role in understanding non-covalent interactions at an atomic level. This computational approach helps in pinpointing atoms involved in non-covalent interactions and categorizing them based on their strength. Building on foundational research.42,43 Our study investigates intramolecular non-covalent interactions within the synthesized molecules. We apply techniques such as the reduced density gradient (RDG) and atoms in molecules (AIM) analysis. Using the powerful Multiwfn software on the atomistica.online platform, we employ RDG scatter plots and corresponding surfaces to visualize these interactions.44,45 All RDG scatter plots are presented in Fig. S4 of the ESI,† while here in the main text we present the RDG scatter plot of the 11e molecule, in Fig. 7, which is the only derivative with two identified noncovalent interactions, compared to all other derivatives that have only one noncovalent interaction, or neither one (such as derivative 11d).
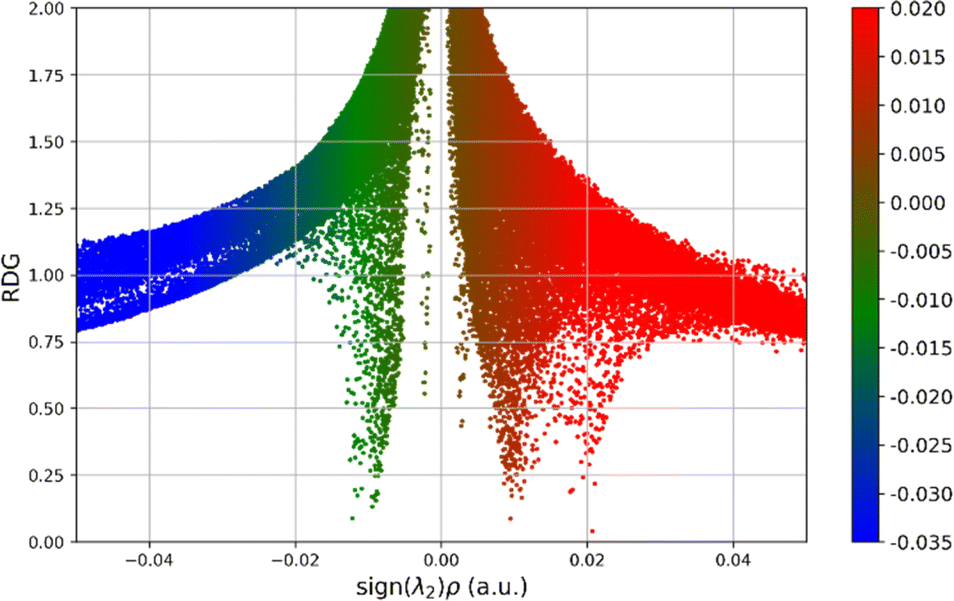 | ||
| Fig. 7 RDG scatter plots of 11e molecule. | ||
Within the provided RDG scatter plot, red dots correspond to steric repulsion, for example, when two electron clouds are in close proximity and repel each other, indicating destabilizing interactions. This is characterized by low RDG values and positive sign (λ)2ρ. Green dots represent van der Waals interactions, characterized by a low RDG and sign (λ)2ρ values closer to zero. Blue dots indicate areas where there are strong, attractive interactions, such as hydrogen bonds, characterized by low RDG and a negative sign (λ)2ρ. The results presented in Fig. S4 of the ESI† suggest that the dominant attractive interaction is the van der Waals interaction, as indicated by the majority of green points. In addition to the dominant van der Waals interaction, strong attraction through hydrogen bonding is hardly evidenced by the rare presence of blue points.
The analysis of RDG scatter plots reveals that all derivatives are characterized by a relatively similar distribution of RDG points. The only notable difference among RDG scatter plots is the presence of a vertical line of green-coloured points close to zero values of the sign (λ)2ρ, as can be seen in Fig. S4 of the ESI.† This specificity corresponds to van der Waals interactions and can be seen also in Fig. 7.
The strengths of noncovalent interactions indicated in Fig. S5 of the ESI† are summarized in Table 3. In Fig. 8, we present the RDG surfaces with corresponding noncovalent interactions of the 11e molecule, the only case where two intramolecular noncovalent interactions were identified.
| Molecule | NCIs | NCI #2 | NCIs |
|---|---|---|---|
| 11a | −0.0124 | 11e | −0.0124 & −0.0099 |
| 11b | −0.0125 | 11f | −0.0124 |
| 11c | −0.0135 | 11g | −0.0129 |
| 11d | — | 11h | −0.0123 |
 | ||
| Fig. 8 RDG surfaces and noncovalent interactions with numerated noncovalent interactions of 11e molecule (blue colour corresponds to repealing interaction, while red colour RDG surfaces correspond to attractive interaction). | ||
In all cases, intramolecular noncovalent interactions were formed between the carbon atom of the CN group and the nearby hydrogen atom of the pyrazine fragment. In the case of the 11e derivative, the second noncovalent interaction also involves the aforementioned carbon atom of the CN group, and the hydrogen atom of the nearby benzene ring. Analysis of the strengths of noncovalent interactions shows that all noncovalent interactions are of very similar strength, except for the second noncovalent interaction in the 11e derivative, which is somewhat weaker, as summarized in Table 3.
Interactions of 11a–h molecules with water
Understanding the interactions between molecules with pharmaceutical potential and water is essential for comprehending their behaviour in biological systems. To investigate this, we carried out Molecular Dynamics (MD) simulations using a force field approach, focusing on the synthesized molecule immersed in a medium of approximately 2000 water molecules. This procedure was replicated for all molecules, meaning that 8 MD simulations were conducted to understand the interactions of these molecules with water. After generating trajectories over a simulation period of 10 nanoseconds, we calculated two key parameters: the interaction energies between the molecules and water (Ewint), and the radial distribution functions (RDFs) for the systems where the highest interaction energies between a synthesized and water were observed. We present our findings on the interaction energies with water, as shown in Fig. 9. | ||
| Fig. 9 Magnitudes of interaction energies between synthesized molecules and water. | ||
The results shown in Fig. 9 reveal significant differences in the interaction energies (Ewint) among the synthesized molecules, 11a–h. The 11g molecule exhibits the lowest magnitude of Ewint, with a value of 67.56 kcal mol−1. The highest Ewint is observed for the 11b molecule, suggesting it has the strongest interactions with water.
A more detailed examination of this molecule in this study involved the analysis of radial distribution functions (RDFs), which revealed the atoms of the 11b with the most significant interactions with water, Fig. 10. Namely, the RDFs showcased the importance of only three atoms of the 11b molecule, all three of them being the nitrogen atoms (N1, N9 and N12). Further analysis of RDFs indicated that peaks of maximal g(r) values are located at distances higher than 3 Å, while the maximal g(r) values of N9 and N12 are very low. These insights indicate that within 11b there are no atoms with very strong interactions with water, and that the overall strongest Ewint value is a result of additive interactions of the large number of atoms constituting the 11b molecule.
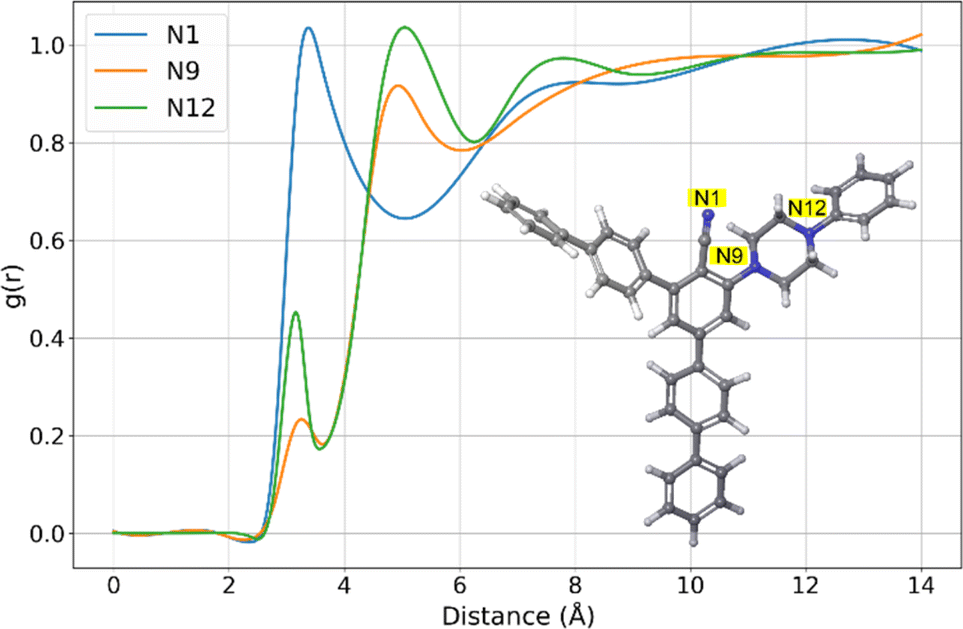 | ||
| Fig. 10 Representative RDFs of 11b together with enumerated atoms with significant interactions with water. | ||
Identification of excipients
A key step in creating innovative pharmaceuticals is the selection of appropriate excipient materials to complement new active ingredients. These excipients are crucial for improving the physical properties of pharmaceutical products, such as their stability, solubility, and bioavailability. It is essential to understand that there isn't a universal excipient suitable for all active ingredients. The challenge lies in identifying the most appropriate excipient for each distinct active ingredient. Fortunately, a range of established excipient options is known for their beneficial properties, often considered in developing new active ingredients.An important factor in determining the compatibility between an active ingredient and an excipient is their solubility parameter. Generally, the closer these parameters are between the two substances, the more compatible they are likely to be. Additionally, finding the right excipient is aided by the capability to compute or estimate solubility parameters. This can be done through MD simulations, using the equation:
 | (1) |
To assess solubility parameters, we carried out three MD simulations. In each, 32 molecules of a specific derivative were placed in a cubic simulation box. These simulations followed the same parameters as our previous water interaction studies, except the simulation time was extended to 20 nanoseconds. Concurrently, we performed similar simulations for established excipient substances. In particular, we have focused on excipients with antioxidant properties commonly used as stabilizers in the pharmaceutical industry. These include polyvinylpyrrolidone (PVP), butylated hydroxytoluene (BHT), butylated hydroxyanisole (BHA), and ascorbic acid (AA).
We then proceeded to evaluate and contrast the solubility parameters of the synthesized derivatives with those of the well-known excipients. This analysis aimed to identify the most compatible excipient for the molecules under study. A summary of findings on solubility parameters can be found in Table 4.
| Molecule | δ [MPa1/2] |
|---|---|
| 11a | 18.832 |
| 11b | 19.304 |
| 11c | 19.515 |
| 11d | 18.682 |
| 11e | 18.833 |
| 11f | 19.259 |
| 11g | 18.979 |
| 11h | 18.938 |
| PVP | 18.515 |
| BHT | 17.544 |
| BHA | 21.486 |
| AA | 32.695 |
According to the results in Table 4, all synthesized derivatives have very similar values of δ, ranging from 18.832 to 19.515. The lowest δ has been calculated for the 11a molecule, while the highest δ was calculated for the 11g molecule. An average value of δ for the molecules is around 19.042 MPa1/2, which almost perfectly fits the δ value of the PVP and BHT excipient. Although the BHA excipient exhibits a somewhat higher value of δ, it remains sufficiently close to the values of the studied molecules to be considered compatible. However, in contrast, the δ value for AA is notably higher, suggesting incompatibility between the studied molecules and AA in terms of excipient stabilization.
Materials and methods
The chemicals used for synthesis and characterization studies were purchased from Alfa Aesar, Thermo Fisher Scientific India Private Limited and Avra Synthesis Pvt. Ltd. All the chemicals were reagent grade and employed without further purification. The reaction progress was predicted with the help of thin-layer chromatography (TLC) on Merck pre-coated sheets of silica gel (60 mesh) and column chromatography was performed with silica gel (100–200 mesh). The 1H and 13C NMR spectra were got using deuterated chloroform (CDCl3, δ 77.00 ppm for 13C NMR) as the solvent and Me4Si (δ = 0 ppm) as the internal standard on a Bruker AV-400 at 400 and 100 MHz, respectively. The chemical shifts (δ) are given in ppm and the coupling constants (J) are mentioned in Hertz. Signal peaks are designated as s, singlet; d, doublet; t, triplet; q, quartet and m, multiplet. Melting points were measured by using REMI DDMS 2545 melting point equipment. The molecular weights of the compounds (m/z) were obtained using Shimadzu GC-mass spectrometer-QP2020 and also on WATERSXEVO G2-XS-QToF High-Resolution Mass Spectrometer. FT-IR spectral analysis was carried out on a PerkinElmer Spectrum.Experimental section
General procedure I: for the synthesis of functionalized 2H-pyranones
A mixture of ethyl 2-cyano-3,3-bis(methylthio)acrylate (1.0 mmol, 1.0 equiv.) and 4-acetyl biphenyl (0.8 mmol, 0.8 equiv.) in DMSO (7 mL) and powdered KOH (1.5 mmol, 1.5 equiv.) were stirred for 12 h. The reaction progress monitored using TLC. After the completion of reaction, crushed ice was added to the reaction mixture and obtained precipitate 6-([1,1′-biphenyl]-4-yl)-4-(methylthio)-2-oxo-2H-pyran-3-carbonitrile was filtered and washed with water and MeOH (3 × 5 mL), then air dried. The mixture of 6-([1,1′-biphenyl]-4-yl)-4-(methylthio)-2-oxo-2H-pyran-3-carbonitrile (1.0 mmol, 1 equiv.) and secondary amine (1.5 mmol, 1.5 equiv.) in absolute MeOH (5 mL) warmed at 50 °C and stirred for 8 h. The reaction progress monitored using TLC. Once the reaction was over, the obtained precipitate 6-([1,1′-biphenyl]-4-yl)-4-(methylthio)-2-oxo-2H-pyran-3-carbonitrile 9 was filtered and washed with absolute MeOH (3 × 4 mL) and dried.General procedure II: for the synthesis of V-shaped biphenyl-flanked m-terphenyls derivatives
A mixture of 6-([1,1′-biphenyl]-4-yl)-2-oxo-4-(amino)-2H-pyran-3-carbonitrile 9 (1.0 mmol, 1.0 equiv.) and 4-acetyl biphenyl 10 (1.0 mmol, 1.0 equiv.) in dry DMF (5 mL) taken in cleaned round bottom flask. A powdered KOH (1.2 mmol, 1.2 equiv.) was added to the reaction mixture and allowed to stir. Up on completion of reaction, reaction mixture was poured in the beaked containing ice-cold water, further neutralized with 10 M HCl. Then extracted using EtOAc (3 × 5 mL) and the combined organic layer was dried with Na2SO4, solvent was evaporated. The crude further purified by column chromatography on silica gel (100–200 mesh) using hexane ethyl acetate as an eluent to afford the product in good amount. The obtained solid was characterized by spectroscopic and spectrometric analysis.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :49), IR (ATR): 2209 cm−1 (CN); 1H NMR (400 MHz, CDCl3): δ = 1.53–1.57 (m, 2H, CH2), 1.73–1.78 (m, 4H, 2CH2), 3.17–3.23 (m, 4H, 2NCH2), 7.14 (s, 1H, ArH), 7.22 (s, 1H, ArH), 7.27 (t, J = 7.2 Hz, 2H, ArH), 7.37 (t, J = 7.2 Hz, 4H, ArH), 7.51–7.65 (m, 12H, ArH); 13C NMR (100 MHz, CDCl3): δ = 24.1, 26.2 (2C), 53.7 (2C), 104.3, 116.1, 118.2, 121.4, 127.1 (2C), 127.2 (2C), 127.3 (2C), 127.6 (2C), 127.7 (4C), 128.8 (2C), 128.9 (2C), 129.4 (3C), 137.9, 138.8, 140.4, 140.5, 141.5, 145.6, 147.6, 158.9; GC-MS: m/z = 491 [M + 1]+; HRMS (ESI): calcd for C36H30N2 490.6500 [M+]; found 490.6526.:49), IR (ATR): 2217 cm−1 (CN); 1H NMR (400 MHz, CDCl3): δ = 3.34–3.42 (m, 8H, 4NCH2), 6.80 (t, J = 7.2 Hz, 1H, ArH), 6.91 (d, J = 8.0 Hz, 2H, ArH), 7.10–7.24 (m, 3H, ArH), 7.26–7.32 (m, 3H, ArH), 7.37 (t, J = 7.2 Hz, 4H, ArH), 7.52–7.66 (m, 12H, ArH); 13C NMR (100 MHz, CDCl3): δ = 49.7 (2C), 52.1 (2C), 104.4, 116.1, 116.5 (2C), 117.9, 120.2, 122.3, 127.1 (2C), 127.2 (2C), 127.4 (2C), 127.6, 127.7 (3C), 127.8 (2C), 128.8 (2C), 128.9 (2C), 129.2 (2C), 129.4 (2C), 137.7, 138.5, 140.3, 140.5, 140.6, 140.7, 145.8, 147.8, 151.2, 157.4; GC-MS: m/z = 568 [M + 1]+; HRMS (ESI): calcd for C41H33N3 567.2674 [M+]; found 567.2698.:49), IR (ATR): 2200 cm−1 (CN); 1H NMR (400 MHz, CDCl3): δ = 1.93–1.98 (m, 4H, 2CH2), 3.62–3.68 (m, 4H, 2NCH2), 6.84 (s, 1H, ArH), 6.94 (s, 1H, ArH), 7.24–7.30 (m, 2H, ArH), 7.37 (t, J = 7.2 Hz, 4H, ArH), 7.50–7.65 (m, 12H, ArH); 13C NMR (100 MHz, CDCl3): δ = 25.9 (2C), 50.7 (2C), 93.6, 111.5, 117.2, 126.8, 127.0 (2C), 127.1 (2C), 127.2 (2C), 127.4, 127.6 (2C), 127.7 (2C), 128.8 (2C), 128.9 (2C), 129.6 (2C), 129.9, 138.7, 139.2, 140.4, 140.7, 141.2, 141.3, 145.2, 148.6, 152.5; GC-MS: m/z = 477 [M + 1]+.:49), IR (ATR): 2215 cm−1 (CN); 1H NMR (400 MHz, CDCl3): δ = 1.16 (t, J = 6.8 Hz, 6H, 2CH3), 3.39 (q, J = 7.2 Hz, 4H, 2CH2), 7.15 (s, 1H, ArH), 7.18 (s, 1H, ArH), 7.26–7.30 (9m, 2H, ArH), 7.37 (t, J = 7.2 Hz, 4H, ArH), 7.52–7.65 (m, 12H, ArH); 13C NMR (100 MHz, CDCl3): δ = 12.8 (2C), 46.9 (2C), 103.6, 117.3, 118.7, 127.1 (2C), 127.2 (2C), 127.3 (2C), 127.6, 127.7 (3C), 128.8 (2C), 128.9 (2C), 129.5 (2C), 138.1, 138.9, 140.4, 140.6, 141.4, 141.5, 145.0, 148.0, 156.0; GC-MS: m/z = 479 [M + 1]+.:49), IR (ATR): 2219 cm−1 (CN); 1H NMR (400 MHz, CDCl3): δ = 2.30 (s, 3H, Me), 2.59–2.65 (m, 4H, 2NCH2), 3.27–3.31 (m, 4H, 2NCH2), 7.13–7.17 (m, 1H, ArH), 7.25–7.32 (m, 3H, ArH), 7.37 (t, J = 7.6 Hz, 4H, ArH), 7.51–7.65 (m, 12H, ArH); 13C NMR (100 MHz, CDCl3): δ = 46.1, 52.0 (2C), 55.2 (2C), 104.2, 116.0, 118.1, 122.0, 127.1 (2C), 127.2 (2C), 127.4 (2C), 127.6, 127.8 (3C), 128.8 (3C), 128.9 (3C), 129.4 (2C), 137.7, 138.6, 140.3, 140.5, 141.5, 141.6, 145.8, 147.7, 157.6; GC-MS: m/z = 506 [M + 1]+.:49), IR (ATR): 2210 cm−1 (CN); 1H NMR (400 MHz, CDCl3): δ = 3.25 (t, J = 4.4 Hz, 4H, 2NCH2), 3.87 (t, J = 4.4 Hz, 4H, 2OCH2), 7.13–7.18 (m, 1H, ArH), 7.25–7.33 (m, 3H, ArH), 7.38 (t, J = 7.2 Hz, 4H, ArH), 7.52–7.66 (m, 12H, ArH); 13C NMR (100 MHz, CDCl3): δ = 52.4 (2C), 67.1 (2C), 104.4, 115.9, 117.9, 122.4, 127.1 (2C), 127.2 (2C), 127.4 (2C), 127.6, 127.7 (2C), 127.8 (2C), 128.8 (3C), 128.9 (2C), 129.4 (2C), 137.6, 138.4, 140.2, 140.4, 141.6, 141.7, 145.9, 147.8, 157.4; GC-MS: m/z = 493 [M + 1]+; HRMS (ESI): calcd for C35H28N2O 492.2202 [M+]; found 492.2150.:49), IR (ATR): 2210 cm−1 (CN); 1H NMR (400 MHz, CDCl3): δ = 3.05 (s, 6H, 2Me), 7.09 (ds, J = 1.2 Hz, 1H, ArH), 7.16 (s, 1H, ArH), 7.24–7.33 (m, 2H, ArH), 7.38 (t, J = 7.2 Hz, 4H, ArH), 7.53–7.66 (m, 12H, ArH); 13C NMR (100 MHz, CDCl3): δ = 43.8 (2C), 100.9, 114.6, 118.9, 120.2, 127.1 (3C), 127.2 (2C), 127.3 (2C), 127.6, 127.7 (3C), 127.8 (2C), 128.8 (2C), 128.9 (2C), 129.5 (2C), 138.0, 138.8, 140.3, 140.6, 141.5, 145.5, 148.0, 157.9; GC-MS: m/z = 451 [M + 1]+.:49), IR (ATR): 2210 cm−1 (CN); 1H NMR (400 MHz, CDCl3): δ = 1.05 (t, J = 6.8 Hz, 3H, Me), 2.44 (q, J = 7.2 Hz, 2H, CH2), 2.62–2.67 (m, 4H, 2NCH2), 3.28–3.33 (m, 4H, 2NCH2), 7.15 (ds, J = 1.2 Hz, 1H, ArH), 7.24–7.33 (m, 3H, ArH), 7.37 (t, J = 7.6 Hz, 4H, ArH), 7.51–7.65 (m, 12H, ArH); 13C NMR (100 MHz, CDCl3): δ = 12.1, 52.0 (2C), 52.3, 52.9 (2C), 104.1, 115.9, 118.1, 122.0, 127.1 (2C), 127.2 (2C), 127.4 (2C), 127.6, 127.7 (3C), 127.8 (2C), 128.8 (2C), 128.9 (2C), 129.1 (2C), 137.7, 138.6, 140.3, 140.5, 141.5, 141.6, 145.8, 147.7, 157.6; GC-MS: m/z = 520 [M + 1]+.
:49), IR (ATR): 2209 cm−1 (CN); 1H NMR (400 MHz, CDCl3): δ = 1.53–1.57 (m, 2H, CH2), 1.73–1.78 (m, 4H, 2CH2), 3.17–3.23 (m, 4H, 2NCH2), 7.14 (s, 1H, ArH), 7.22 (s, 1H, ArH), 7.27 (t, J = 7.2 Hz, 2H, ArH), 7.37 (t, J = 7.2 Hz, 4H, ArH), 7.51–7.65 (m, 12H, ArH); 13C NMR (100 MHz, CDCl3): δ = 24.1, 26.2 (2C), 53.7 (2C), 104.3, 116.1, 118.2, 121.4, 127.1 (2C), 127.2 (2C), 127.3 (2C), 127.6 (2C), 127.7 (4C), 128.8 (2C), 128.9 (2C), 129.4 (3C), 137.9, 138.8, 140.4, 140.5, 141.5, 145.6, 147.6, 158.9; GC-MS: m/z = 491 [M + 1]+; HRMS (ESI): calcd for C36H30N2 490.6500 [M+]; found 490.6526.:49), IR (ATR): 2217 cm−1 (CN); 1H NMR (400 MHz, CDCl3): δ = 3.34–3.42 (m, 8H, 4NCH2), 6.80 (t, J = 7.2 Hz, 1H, ArH), 6.91 (d, J = 8.0 Hz, 2H, ArH), 7.10–7.24 (m, 3H, ArH), 7.26–7.32 (m, 3H, ArH), 7.37 (t, J = 7.2 Hz, 4H, ArH), 7.52–7.66 (m, 12H, ArH); 13C NMR (100 MHz, CDCl3): δ = 49.7 (2C), 52.1 (2C), 104.4, 116.1, 116.5 (2C), 117.9, 120.2, 122.3, 127.1 (2C), 127.2 (2C), 127.4 (2C), 127.6, 127.7 (3C), 127.8 (2C), 128.8 (2C), 128.9 (2C), 129.2 (2C), 129.4 (2C), 137.7, 138.5, 140.3, 140.5, 140.6, 140.7, 145.8, 147.8, 151.2, 157.4; GC-MS: m/z = 568 [M + 1]+; HRMS (ESI): calcd for C41H33N3 567.2674 [M+]; found 567.2698.:49), IR (ATR): 2200 cm−1 (CN); 1H NMR (400 MHz, CDCl3): δ = 1.93–1.98 (m, 4H, 2CH2), 3.62–3.68 (m, 4H, 2NCH2), 6.84 (s, 1H, ArH), 6.94 (s, 1H, ArH), 7.24–7.30 (m, 2H, ArH), 7.37 (t, J = 7.2 Hz, 4H, ArH), 7.50–7.65 (m, 12H, ArH); 13C NMR (100 MHz, CDCl3): δ = 25.9 (2C), 50.7 (2C), 93.6, 111.5, 117.2, 126.8, 127.0 (2C), 127.1 (2C), 127.2 (2C), 127.4, 127.6 (2C), 127.7 (2C), 128.8 (2C), 128.9 (2C), 129.6 (2C), 129.9, 138.7, 139.2, 140.4, 140.7, 141.2, 141.3, 145.2, 148.6, 152.5; GC-MS: m/z = 477 [M + 1]+.:49), IR (ATR): 2215 cm−1 (CN); 1H NMR (400 MHz, CDCl3): δ = 1.16 (t, J = 6.8 Hz, 6H, 2CH3), 3.39 (q, J = 7.2 Hz, 4H, 2CH2), 7.15 (s, 1H, ArH), 7.18 (s, 1H, ArH), 7.26–7.30 (9m, 2H, ArH), 7.37 (t, J = 7.2 Hz, 4H, ArH), 7.52–7.65 (m, 12H, ArH); 13C NMR (100 MHz, CDCl3): δ = 12.8 (2C), 46.9 (2C), 103.6, 117.3, 118.7, 127.1 (2C), 127.2 (2C), 127.3 (2C), 127.6, 127.7 (3C), 128.8 (2C), 128.9 (2C), 129.5 (2C), 138.1, 138.9, 140.4, 140.6, 141.4, 141.5, 145.0, 148.0, 156.0; GC-MS: m/z = 479 [M + 1]+.:49), IR (ATR): 2219 cm−1 (CN); 1H NMR (400 MHz, CDCl3): δ = 2.30 (s, 3H, Me), 2.59–2.65 (m, 4H, 2NCH2), 3.27–3.31 (m, 4H, 2NCH2), 7.13–7.17 (m, 1H, ArH), 7.25–7.32 (m, 3H, ArH), 7.37 (t, J = 7.6 Hz, 4H, ArH), 7.51–7.65 (m, 12H, ArH); 13C NMR (100 MHz, CDCl3): δ = 46.1, 52.0 (2C), 55.2 (2C), 104.2, 116.0, 118.1, 122.0, 127.1 (2C), 127.2 (2C), 127.4 (2C), 127.6, 127.8 (3C), 128.8 (3C), 128.9 (3C), 129.4 (2C), 137.7, 138.6, 140.3, 140.5, 141.5, 141.6, 145.8, 147.7, 157.6; GC-MS: m/z = 506 [M + 1]+.:49), IR (ATR): 2210 cm−1 (CN); 1H NMR (400 MHz, CDCl3): δ = 3.25 (t, J = 4.4 Hz, 4H, 2NCH2), 3.87 (t, J = 4.4 Hz, 4H, 2OCH2), 7.13–7.18 (m, 1H, ArH), 7.25–7.33 (m, 3H, ArH), 7.38 (t, J = 7.2 Hz, 4H, ArH), 7.52–7.66 (m, 12H, ArH); 13C NMR (100 MHz, CDCl3): δ = 52.4 (2C), 67.1 (2C), 104.4, 115.9, 117.9, 122.4, 127.1 (2C), 127.2 (2C), 127.4 (2C), 127.6, 127.7 (2C), 127.8 (2C), 128.8 (3C), 128.9 (2C), 129.4 (2C), 137.6, 138.4, 140.2, 140.4, 141.6, 141.7, 145.9, 147.8, 157.4; GC-MS: m/z = 493 [M + 1]+; HRMS (ESI): calcd for C35H28N2O 492.2202 [M+]; found 492.2150.:49), IR (ATR): 2210 cm−1 (CN); 1H NMR (400 MHz, CDCl3): δ = 3.05 (s, 6H, 2Me), 7.09 (ds, J = 1.2 Hz, 1H, ArH), 7.16 (s, 1H, ArH), 7.24–7.33 (m, 2H, ArH), 7.38 (t, J = 7.2 Hz, 4H, ArH), 7.53–7.66 (m, 12H, ArH); 13C NMR (100 MHz, CDCl3): δ = 43.8 (2C), 100.9, 114.6, 118.9, 120.2, 127.1 (3C), 127.2 (2C), 127.3 (2C), 127.6, 127.7 (3C), 127.8 (2C), 128.8 (2C), 128.9 (2C), 129.5 (2C), 138.0, 138.8, 140.3, 140.6, 141.5, 145.5, 148.0, 157.9; GC-MS: m/z = 451 [M + 1]+.:49), IR (ATR): 2210 cm−1 (CN); 1H NMR (400 MHz, CDCl3): δ = 1.05 (t, J = 6.8 Hz, 3H, Me), 2.44 (q, J = 7.2 Hz, 2H, CH2), 2.62–2.67 (m, 4H, 2NCH2), 3.28–3.33 (m, 4H, 2NCH2), 7.15 (ds, J = 1.2 Hz, 1H, ArH), 7.24–7.33 (m, 3H, ArH), 7.37 (t, J = 7.6 Hz, 4H, ArH), 7.51–7.65 (m, 12H, ArH); 13C NMR (100 MHz, CDCl3): δ = 12.1, 52.0 (2C), 52.3, 52.9 (2C), 104.1, 115.9, 118.1, 122.0, 127.1 (2C), 127.2 (2C), 127.4 (2C), 127.6, 127.7 (3C), 127.8 (2C), 128.8 (2C), 128.9 (2C), 129.1 (2C), 137.7, 138.6, 140.3, 140.5, 141.5, 141.6, 145.8, 147.7, 157.6; GC-MS: m/z = 520 [M + 1]+.Computational details
This research utilized multiple theoretical approaches in its computational analysis.46 Initially, the molecular structure was pre-optimized using the GFN2-xTB method, a technique developed by Prof. Stefan Grimme and his team.47,48 These GFN2-xTB calculations were executed using version 6.6.1 of the xTB program, accessible through the atomistica.online platform, a publicly available molecular modelling resource at https://atomistica.online/. DFT calculations involved the B3LYP density functional and Pople-type basis sets,49 using the 6-31G(d,p) basis set for re-optimization of the focal molecule. Vibrational frequency analyses, confirming the molecules' true ground states, resulted in positive values only. For determining specific properties like molecular electrostatic potential (MEP) and average local ionization energy (ALIE), an M06-2X functional with 6-311++G(d,p) basis set was employed.50,51 Reduced density gradient (RDG) scatter plots and RDG surfaces were obtained by using the Multiwfn program via the online tools of atomistica.online for automatization. Necessary files (.wfn) for application of the Multiwfn program were obtained by single point energy calculations at the same level of theory as for MEP and ALIE using the ORCA 5.0.4 molecular modelling package. Input files for ORCA were created by the Online ORCA input generator of atomistica.online.MD simulations were pivotal in exploring the interactions between the title molecules and water and in calculating its solubility parameter, which was essential for selecting appropriate excipients. The MD simulations were conducted in two scenarios. The first one involved a single molecule from the synthesized series of molecules surrounded by roughly 2000 water molecules in a cubic box. The second involved 32 molecules of each of the molecules from the synthesized molecules alone in the cubic simulation box. These simulations employed the OPLS4 force field with simulation time set to 10 ns, with an NPT ensemble and a 9 Å cut-off radius. DFT calculations were performed using the Jaguar program, while MD simulations were performed with the Desmond program of the Schrödinger Materials Science Suite 2023-2.52
Conclusions
In conclusion, we have established an efficient synthetic route towards donor–acceptor stilbenes by utilizing base promoted ring transformation of 6-styrenyl-2H-pyran-2-ones with different acyclic nucleophiles and we could afford altered derivatives under mild reaction conditions. Our protocol is operationally convenient and free from toxic transition metals. This reaction proceeds in good yields without inert atmosphere. Our procedure is alternate route over numerous metals catalysed cross coupling reactions. Moreover, synthesised stilbenes exhibited interesting fluorescence properties and displayed positive solvatochromic behaviour with the solvents of different polarities. Infect, to our delight the studied compounds revealed good thermal stability. Furthermore, the scope of similar ring transformation methodology is under investigation in our laboratory. Computational part of this study provides a comprehensive analysis of eight molecules, using DFTB, DFT calculations, and MD simulations. The analysis of MEP and ALIE descriptors revealed that the 11e molecule showed a distinct susceptibility to electrophilic attacks. DFT calculations also provided insights into intramolecular non-covalent interactions, with molecule 11e being the only molecule with two non-covalent interactions. On the other hand, MD simulations revealed significant differences in the interaction energies between synthesized molecules and water. The findings suggest that 11b has the strongest interaction with water, however, due to the additive contribution of constituting atoms. The close match in solubility parameters between PVP and the synthesized molecules suggests high compatibility, which is crucial for pharmaceutical development.Conflicts of interest
There are no conflicts to declare.Acknowledgements
FVS is thankful to CSIR, New Delhi [Grant No. 02/(0330)/17-EMR] for the financial support. Authors are thankful to SAIF, VIT Vellore for providing spectroscopic analysis data. PBK is thankful to CSIR, New Delhi for Senior Research Fellowship. SA and SJA acknowledge the financial support of the Ministry of Education, Science and Technological Development of the Republic of Serbia (Grants No. 451-03-66/2024-03/200125 & 451-03-65/2024-03/200125) computing resources of the National AI platform of the Republic of Serbia (https://www.ai.gov.rs/). SPK extend acknowledgements to the Director, Amrita Vishwa Vidyapeetham, Mysuru, for infrastructural support. The authors extend their appreciation to “Researchers Supporting Project number (RSPD2024R734), King Saud University, Riyadh, Saudi Arabia” for financial support.Notes and references
- B. Rashidzadeh, F. Jafarpour and A. Saednya, ARKIVOC, 2008, 17, 167–172 Search PubMed.
- D. Kim, V. Coropceanu and J. L. Bredas, J. Am. Chem. Soc., 2011, 133, 17895–17900 CrossRef CAS.
- J. Ortyl, J. Wilamowski, P. Milart, M. Galek and R. Popielarz, Polym. Test., 2015, 48, 151–159 CrossRef CAS.
- H. Hart and P. Rajakumar, Tetrahedron, 1995, 51, 1313–1336 CrossRef CAS.
- P. Rajakumar and M. Srisailas, Tetrahedron Lett., 1997, 38, 5323–5326 CrossRef CAS.
- M. Olaru, J. Beckmann and C. I. Rat, Organometallics, 2014, 33, 3012–3020 CrossRef CAS.
- R. S. Grewal, H. Hart and T. K. Vinod, J. Org. Chem., 1992, 57, 2721–2726 CrossRef CAS.
- K. Goto, G. Yamamoto, B. Tan and R. Okazaki, Tetrahedron Lett., 2001, 42, 4875–4877 CrossRef CAS.
- M. Abbass, C. Kuhl, C. Manthey, A. Muller and U. Luning, Collect. Czech. Chem. Commun., 2004, 69, 1325–1344 CrossRef CAS.
- M. Romain, S. Thiery, A. Shirinskaya, C. Declairieux, D. Tondelier, B. Geffroy, O. Jeannin, J. Rault-Berthelot, R. Metivier and C. Poriel, Angew. Chem., Int. Ed., 2014, 54, 1–6 Search PubMed.
- E. Gopi and I. N. N. Namboothiri, J. Org. Chem., 2014, 79, 7468–7476 CrossRef CAS PubMed.
- J. Li, D. Geng, J. Xu, L. J. Weng, Q. Liu and L. T. Yi, Eur. J. Pharmacol., 2013, 707, 112–119 CrossRef CAS.
- H. Kikuchi, Y. Matsuo, Y. Katou, Y. Kubohara and Y. Oshima, Tetrahedron, 2012, 68, 8884–8889 CrossRef CAS.
- K. Huang, X. Ke, H. Wang, J. Wang, C. Zhou, X. Xu, L. Liu and J. Li, Org. Biomol. Chem., 2015, 13, 4486–4493 RSC.
- E. Hola, J. Ortyl, M. Jankowska, M. Pilch, M. Galek, M. F. Savary, B. Graff, C. Dietlin and J. Lalevee, Polym. Chem., 2020, 11, 922–935 RSC.
- Q. Zhang, B. Wang, J. Tan, G. Mu, W. Yi, X. Lv, S. Zhuang, W. Liu and L. Wang, J. Mater. Chem. C, 2017, 5, 8516–8526 RSC.
- Z.-P. Chen, D.-Q. Wang, M. Zhang, K. Wang, Y.-Z. Shi, J.-X. Chen, W.-W. Tao, C.-J. Zheng, S.-L. Tao and X.-H. Zhang, Adv. Opt. Mater., 2018, 6, 1800935 CrossRef.
- D. C. M. Albanese, S. Brunialti and D. Destro, Catalysts, 2016, 6, 142–150 CrossRef.
- J. Y. Lee, J. Inf. Disp., 2014, 15, 139–144 CrossRef CAS.
- P. Rajakumar and M. Srisailas, Tetrahedron, 2001, 57, 9749–9754 CrossRef CAS.
- P. Rajakumar and M. Srisailas, Tetrahedron, 2003, 59, 5373–5376 CrossRef CAS.
- R. J. I. Tamargo, Y. R. Lee and T. N. Poudel, RSC Adv., 2016, 6, 70311–70319 RSC.
- S. E. Shetgaonkar, S. P. Kollur, R. R. Pillai, K. Thangavel, S. J. Armaković, S. Armaković, C. Shivamallu, R. G. Amachawadi, A. Syed, A. M. Elgorban and A. H. Bahkali, Symmetry, 2021, 13, 1619 CrossRef CAS.
- P. B. Kole and F. V. Singh, J. Mol. Struc., 2022, 1250, 131622 CrossRef CAS.
- M. Krishnan and F. V. Singh, J. Mol. Struc., 2022, 1256, 132544 CrossRef CAS.
- M. Krishnan and F. V. Singh, Synthesis, 2022, 54, 5273–5280 CrossRef CAS.
- M. Krishnan and F. V. Singh, New J. Chem., 2023, 47, 15827–15846 RSC.
- P. B. Kole, S. P. Kollur, H. D. Revanasiddappa, C. Shivamallu, R. A. Costa, E. S. Junior, L. M. Anselmo, J. N. da Silva, C. Srinivasa, A. Syed and F. V. Singh, Polycyclic Aromat. Compd., 2023, 43, 2177–2195 CrossRef CAS.
- R. Mamgain, R. J. Gana, A. Malik and F. V. Singh, New J. Chem., 2024, 48, 342–350 RSC.
- R. C. Guadagnin, C. A. Suganuma, F. V. Singh, A. S. Vieira, R. Cella and H. A. Stefani, Tetrahedron Lett., 2008, 49, 4713–4716 CrossRef CAS.
- R. Thomas, M. Hossain, Y. S. Mary, K. S. Resmi, S. Armaković, S. J. Armaković, A. K. Nanda, V. K. Ranjan, G. Vijayakumar and C. Van Alsenoy, J. Mol. Struc., 2018, 1158, 156–175 CrossRef CAS.
- J. S. Al-Otaibi, Y. S. Mary, Y. S. Mary and R. Thomas, Comput. Theor. Chem., 2022, 1208, 113569 CrossRef CAS.
- J. N. C. Mishma, V. B. Jothy, A. Irfan, B. Narayana, S. N. Kodlady and S. Muthu, J. Mol. Liq., 2023, 376, 121439 CrossRef CAS.
- M. Sumithra, N. Sundaraganesan, R. Rajesh, V. Vetrivelan, V. Ilangovan, S. Javed and S. Muthu, Chem. Phys. Impt., 2023, 6, 100145 CrossRef.
- K. Haruna, V. S. Kumar, S. J. Armaković, S. Armaković, Y. S. Mary, R. Thomas, S. A. Popoola, A. R. Almohammedi, M. S. Roxy and A. A. Al-Saadi, Spectrochim. Acta, Part A, 2020, 228, 117580 CrossRef CAS PubMed.
- A. Goel and F. V. Singh, Tetrahedron Lett., 2005, 46, 5585–5587 CrossRef CAS.
- A. Goel and V. J. Ram, Tetrahedron, 2009, 65, 7865–7913 CrossRef CAS.
- A. Goel, F. V. Singh, M. Dixit, D. Verma, R. Raghunandan and P. R. Maulik, Chem.–Asian J., 2007, 2, 239–247 CrossRef CAS PubMed.
- F. V. Singh, V. Kumar, B. Kumar and A. Goel, Tetrahedron, 2007, 63, 10971–10978 CrossRef CAS.
- S. Chandrasekar and F. V. Singh, Chemistry Select, 2020, 5, 7452–7459 CAS.
- C. Subashini, L. J. Kennedy and F. V. Singh, J. Mol. Struc., 2021, 1226, 129365 CrossRef.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- A. Otero-de-la-Roza, E. R. Johnson and J. Contreras-Garcia, Phys. Chem. Chem. Phys., 2012, 14, 12165–12172 RSC.
- S. Armaković and S. J. Armaković, Mol. Simul., 2023, 49, 117–123 CrossRef.
- S. Armaković and S. J. Armaković, Mol. Simul., 2024, 50, 560–570 CrossRef.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- T. Lu and Q. Chen, J. Mol. Model., 2020, 26, 315 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS.
- F. Neese, J. Comput. Chem., 2023, 44, 381–396 CrossRef CAS.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS.
- K. J. Bowers, E. Chow, H. Xu, R. O. Dror, M. P. Eastwood, B. A. Gregersen, J. L. Klepeis, I. Kolossvary, M. A. Moraes, F. D. Sacerdoti, J. K. Salmon, Y. Shan and D. E. Shaw, Scalable algorithms for molecular dynamics simulations on commodity clusters, in Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Association for Computing Machinery, New York, NY, USA, 2006, p. 84 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C NMR and mass spectra. MEP, ALIE surfaces, RDG scatter plots and RDG surfaces. See DOI: https://doi.org/10.1039/d4ra02375g |
| This journal is © The Royal Society of Chemistry 2024 |