
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-yield synthesis of hydroxylated boron nitride nanosheets and their utilization in thermally conductive polymeric nanocomposites†
Feng Yuan‡
a,
Qinhan Guan‡a,
Xuehan Doua,
Han Yanga,
Yiming Honga,
Yawen Xuea,
Zhenxing Caoa,
Haiyan Li b,
Zexiao Xuc and
Yuyang Qin*abc
b,
Zexiao Xuc and
Yuyang Qin*abc
aSchool of Materials Engineering, Changshu Institute of Technology, Changshu, 215500, China. E-mail: yuyang_qin@outlook.com
bSchool of Chemistry and Chemical Engineering, Northeast Petroleum University, Daqing, 163318, China
cSuzhou Jiren High-Tech Materials Co., Ltd, Suzhou, China
First published on 5th July 2024
Abstract
Hexagonal boron nitride nanosheets (BNNSs) possess remarkable potential for various applications due to their unprecedented properties. However, the scalable production of BNNSs with both expansive surface and high solubility continues to present a significant challenge. Herein, we propose an innovative and efficient two-step method for manufacturing hydroxyl-functionalized BNNSs (OH-BNNSs). Initially, hydroxyl groups are covalently attached to bulk hexagonal boron nitride (h-BN) surfaces through H2O2 treatment. Then, the hydroxyl-functionalized h-BN undergoes exfoliation on account of a sudden increase in interlayer gas pressure generated by the vigorous decomposition of H2O2 in alkali solutions, resulting in the creation of OH-BNNSs. This approach produces relatively large flakes with an average dimension of 1.65 μm and a high yield of 45.2%. The resultant OH-BNNSs exhibit remarkable stability and dispersibility in a range of solvents. Their integration into thermoplastic polyurethane (TPU) significantly enhances both thermal conductivity and stability, attributed to the excellent compatibility with the resin matrix. This study represents a significant advancement in the functionalization and exfoliation of h-BN, opening new avenues for its promising applications in polymer composites.
Hexagonal boron nitride nanosheets (BNNSs), akin in structure to graphene, stand out as exceptional two-dimensional (2D) ordered crystals due to their unparalleled and diverse properties. Leveraging the highly polar B–N bonds, BNNSs intrinsically possess excellent resistance to corrosion and oxidation (sustaining up to 800 °C).1,2 Moreover, their defect-free 2D honeycomb arrangement endows BNNSs with high thermal conductivity and impressive mechanical strength.3,4 Remarkably, the wide energy band gap inherent in BNNSs positions them as exceptional insulating thermal conductivity fillers for the thermal management of high-power electronics. These intriguing properties designate BNNSs as promising candidates for a multitude of specialized applications, such as nanocomposite materials,5–8 catalyst supports,9,10 optical devices,11,12 drug deliveries,13,14 protective coatings15,16 and electronic devices.17,18 To maximize their potential in diverse applications, it is imperative to develop a facile and scalable method for the preparation of high-quality BNNSs.
Considering the layered nature of hexagonal boron nitride (h-BN), exfoliation from its bulk form has been a prevailing way to obtain single- or few-layered BNNSs. Chemical exfoliation strategy appears to be a facile technique for the production of BNNSs, typically involving oxidization, decomposition, or intercalation of bulk h-BN powder. However, these various chemical fabrication routes generally exhibit limited exfoliation efficiency, primarily due to the strong “lip–lip” interactions between adjacent BN layers.19–21 Furthermore, the predominant highly reactive chemicals utilized in these exfoliation routes often consist of specialized compounds, known for their toxicity and challenging handling procedures. In contrast to these chemical approaches, a multitude of physical strategies have emerged for the massive production of BNNSs by peeling or shearing forces, such as mechanical cleavage22–24 and ultrasonic-assisted liquid exfoliation.25–28 Nevertheless, the mechanism behind these physical methods presents a significant challenge in producing functional groups on the BNNSs, consequently restricting their dispersion in a wide range of solvents and polymer matrices, especially at higher concentrations. Meanwhile, the as-prepared BNNSs unavoidably suffer from plenty of structural defects during the violent processes of most mechanical treatments.29
Comparatively speaking, the functionalization of BNNSs emerges as a promising technique to introduce novel features and applications to nanoparticles, thereby enhancing the mechanical, thermal, and electrochemical performance of BNNSs composites.30–32 Noteworthy efforts have been devoted to the efficient and reliable fabrication of hydroxyl-functionalized BNNSs (OH-BNNSs). Sainsbury et al. employed a multistep strategy, involving oxygen radical functionalization followed by hydrolytic defunctionalization with H2O2/H2SO4, to produce OH-BNNSs.33 Xiao and co-workers achieved simultaneous exfoliation and hydroxylation of h-BN at a high yield through the steam treatment of bulk h-BN powders.34 Based on the pioneering hydroxide-assisted ball milling technique, Jeon and colleagues successfully fabricated OH-BNNSs in a one-step process with a yield of 18%.29 Recently, a peroxynitrous acid-assisted hydrolysis method has been developed, which was a novel and effective technique to functionalize h-BN to obtain a few-layered h-BN-OH.35 Notably, the incorporation of hydroxyl (–OH) groups can enhance the dispersion of BNNSs in diverse solvents by tailoring the surface energy. Furthermore, the –OH groups exhibit the capability to interact with numerous inorganic or organic substances, making BNNSs highly desirable for potential applications.14,34 Despite the promising attributes offered by OH-BNNSs, the fabrication process involves sophisticated multiple-step procedures, extra-high temperature or relatively low yield. Consequently, it still remains a technical challenge to achieving large-scale and efficient preparation of OH-BNNSs, necessitating a more facile, economic and environmentally-friendly approach.
Herein, we demonstrate an efficient and simple two-step method for the functionalization and exfoliation of commercial h-BN by H2O2-assisted hydroxylation treatment. This strategy involves a hydrothermal reaction with H2O2 (30%) to yield hydroxyl-functionalized h-BN (OH-BN). Subsequently, scalable exfoliation of OH-BN is accomplished based on the violent exothermic decomposition of H2O2 in alkaline solutions. Noteworthy, the exfoliation process differs from conventional thermal exfoliation methods, as it can be carried out at room temperature. The –OH groups on the surfaces of OH-BN can be effectively reserved in the exfoliated products, directly yielding OH-BNNSs without any additional post-processing steps. This procedure is environmentally friendly, easily manageable and cost-effective. Moreover, the synthetic route demonstrates high efficiency with a yield of up to 45.2%. A mechanism based on the H2O2-assisted hydroxylation and exfoliation technique has been proposed for the efficient functionalization of bulk h-BN powders. The as-prepared OH-BNNSs demonstrate an average size of 1.65 μm and exhibit outstanding dispersion stability in various solutions. In addition, the OH-BNNSs/thermoplastic polyurethane (TPU) composites exhibit excellent forward thermal conductivity and thermal stability characteristics.
Results and discussion
The schematic illustration in Fig. 1 depicts a high yielding synthesis procedure of OH-BNNSs. The powdered h-BN undergoes hydrothermal reaction in aqueous H2O2 solution (Fig. 1a). Under the impact of strong oxidation property of H2O2, B sites along the edges and near the defects were effectively activated, facilitating an effective interaction with hydroxide ions. This results in the introduction of a large number of –OH groups to h-BN surfaces, achieving hydroxyl-functionalized h-BN. Under alkaline conditions, the ionization balance of H2O2 moves towards the direction of generating peroxy groups, resulting in an increase in the concentration of the peroxy groups, thereby accelerating the decomposition process. Specifically, the decomposition of H2O2 under alkaline conditions can be expressed as 2H2O2 → 2H2O + O2, where O2 is one of the products. This process can not only occur at room temperature, but the decomposition speed is slow, and it is significantly accelerated under alkaline conditions. With the violent exothermic decomposition of H2O2 under concentrated alkaline conditions (Fig. 1b), the dynamically formed O2 can intercalate into OH-BN layers, facilitating large-scale exfoliation. It should be noted that the well preservation of –OH groups on OH-BN surfaces play a pivotal role in preventing the agglomeration of exfoliated layers during this procedure. | ||
| Fig. 1 Schematic illustration of the hydroxylation and exfoliation of h-BN powders. The whole process involves the following two steps: (a) the hydroxylation of h-BN by H2O2 treatment and (b) the exfoliation of OH-BN by the violent exothermic decomposition of H2O2. | ||
Preparation of OH-BN by H2O2 treatment
The successful hydroxylation of h-BN can be verified by XPS analysis. In Fig. 2a, the comparative XPS surveys of OH-BN and h-BN powders reveal four peaks corresponding to O, N, C and B elements, respectively. The presence of C elements is expected, considering the common occurrence of carbon contamination in BN nanomaterials.29,36 Notably, the intensity of O 1s in OH-BN is unusually intensive, leading to a decrease in the B![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O atomic ratio from 35.54 in h-BN to 7.15. This ratio is comparable to the value of our previous report, in which OH-BN was successfully prepared via hydrothermal reactions with NaOH.37 As is evident from Fig. 2b and c, the B and N atoms of h-BN are bound only to each other, indicating that the additional O element is associated with the physically absorbed moisture.29,38
:O atomic ratio from 35.54 in h-BN to 7.15. This ratio is comparable to the value of our previous report, in which OH-BN was successfully prepared via hydrothermal reactions with NaOH.37 As is evident from Fig. 2b and c, the B and N atoms of h-BN are bound only to each other, indicating that the additional O element is associated with the physically absorbed moisture.29,38
 | ||
| Fig. 2 (a) XPS survey scans of the h-BN and OH-BN. (b) High-resolution B 1s XPS scans of the h-BN and OH-BN. (c) High-resolution N 1s XPS scans of the h-BN and OH-BN. | ||
In the case of OH-BN, the B 1s spectrum is observed to be fitted with two peaks (Fig. 2b). The prominent peak centered at 190.2 eV is ascribed to the B–N bond, while the small one around 191.1 eV corresponds to the B–O bond.29,39 Conversely, the N 1s peak of OH-BN remains symmetric with respect to its center located at 397.7 eV (Fig. 2c), implies that the oxidation primarily occurs in B atoms rather than N atoms. This observation aligns with previous reports, further emphasizing the oxidation sensitivity of B sites.22,29,34,40 XPS observation of OH-BN was further corroborated by solid-state 1H NMR information (Fig. S1†). A chemical shift (δ) of 2.02 ppm clearly appears in OH-BN, which is a distinct peak for the –OH group. The –OH content can be determined through TGA analysis, as depicted in Fig. 3. The raw h-BN powders possess high thermal stability, with only 0.28% water evaporation until 700 °C. However, due to the removal of –OH groups, a significant weight loss has occurred in OH-BN. In combination with the weight loss data of h-BN, the content of –OH on OH-BN surface is calculated to be 9.09 wt%. Additionally, the Raman spectra of the parent h-BN and OH-BN both exhibit the E2g phonon mode at 1367.6 cm−1 (Fig. S2†), indicating that the intrinsic hexagonal structure of h-BN remains undamaged during the oxidation process.
 | ||
| Fig. 3 TGA patterns of h-BN and OH-BN. | ||
Preparation of OH-BNNSs by the decomposition of H2O2
After oxidation, the substantial residual H2O2 can serve as decomposing agents, which contribute to the disruption of interlayer interactions in OH-BN. Rapid decomposition in concentrated alkaline environments leads to a sudden increase in interlayer pressure. The successful exfoliation of OH-BN is verified by the morphology characterization using SEM and TEM. As illustrated in Fig. 4a and b, the SEM images of h-BN powders exhibit typical laminar structures with smooth surfaces as well as clear edges. After H2O2 treatment, the resultant OH-BN still remains a thick flake structure, but the edges become rough (Fig. 4c). This is primarily attributed to the fact that oxidation predominantly occurs in the region near the edges.34 In contrast to the opaque surfaces of h-BN and OH-BN, the obtained OH-BNNSs appear highly transparent to the electron beam, as revealed in low magnification TEM image (Fig. 4d). Also, the structural features of OH-BN can be more clearly observed in the high-magnification TEM. Note that numerous thin ripples can be seen on the film-like surfaces of OH-BNNSs, indicating their ultrathin nature (Fig. 4e). Moreover, the exfoliation process preserves the hexagonal lattice structure, as confirmed by the typical hexagonal BN result in the XRD spectrum (Fig. S3†). Moreover, taking the (100) peak as a reference, the intensity of the (004) plane from OH-BNNSs become unusually intensive in relation to that of their 3D structures, providing further evidence for the efficient exfoliation. Due to their ultrathin nature, some curled edges are visible at certain locations (as marked by a blue rectangle in Fig. 4e), allowing for the acquisition of a cross-sectional view of the nanosheet. As shown in Fig. 4f, three parallel fringes are clearly observable, signifying the presence of three stacked layers in this sample. In addition, the six-fold symmetrical characteristic of OH-BNNSs is further supported by the corresponding selected area electron diffraction (SAED) pattern (Fig. 4g), which is consistent with the analysis of XRD pattern and previous reports.37,41 In order to further investigate the thickness and lateral size of the OH-BNNSs, AFM characterizations were performed. A mass of ultrathin nanosheets can be seen in the AFM image (Fig. 5a). AFM characterizations were performed to investigate the thickness and lateral size of the OH-BNNSs. Numerous ultrathin nanosheets can be observed in the AFM image (Fig. 5a). It has been previously demonstrated that the AFM height of a monolayer BNNS increases to 1 nm on account of the trapped solvent and underlying substrate.42 The measured heights correspond to 3 and 4 atomic layers, respectively (Fig. 5b). The thickness and lateral size distributions were statistically evaluated on 50 sheets and presented in Fig. 5c and d, respectively. Notably, a significant majority of the measured nanosheets consist of less than 10 atomic layers (3 ≤ nm), with an average value of 3–4 layers (Fig. 5c). | ||
| Fig. 4 SEM images of the pristine h-BN (a and b), and OH-BN powders (c). TEM images of the OH-BNNSs at different magnifications (d and e). High-resolution TEM image of the folded edge (wherein the interplanar spacing has been marked) marked in (e) with a blue rectangle (f). The selected area electron diffraction (SAED) pattern of OH-BNNSs (g). | ||
 | ||
| Fig. 5 (a) AFM topography image of the OH-BNNSs, scale bar = 1 μm and (b) the corresponding height profile along the line. Statistical analyses on the number of layers per sheet (c) and the lateral size (d) over a sample set of 50 pieces of OH-BNNSs. | ||
The successful preparation of few-layered nanosheets was further verified by Raman analysis, as shown in Fig. S4.† Owing to the slightly elongated B–N bonds and reduced interlayer interactions, the Raman E2g mode of OH-BNNSs shows a slight redshift of 3 cm−1 compared to that of the bulk OH-BN. This is a typical result of the exfoliation of bulk h-BN into bilayers or few-layers nanosheets. Meanwhile, the as-produced OH-BNNSs are relatively large flakes with an average lateral size of 1.65 μm (Fig. 5d), significantly larger than those reported by many other exfoliation methods, as presented in Table 1. In the case of OH-BN, a remarkably broadened peak around 3200 cm−1 together with a small peak centered at approximately 1200 cm−1 differs from the spectra of the initial h-BN flakes (Fig. 6a). These additional peaks are attributed to the stretching vibration of B–OH and in-plane bending of B–O, respectively. The results further confirm the successful hydroxylation of h-BN, consistent with previous reports on the FT-IR spectra of hydroxyl-functionalized h-BN.29,36,40,45 Notably, the two absorptions originating from the B–O–H vibrations remain visible after exfoliation. Further evidence for the existence of –OH groups on the exfoliated products comes from XPS analysis. As shown in Fig. S5,† the OH-BNNSs exhibit essentially the same peaks as OH-BN, with a negligible increase in the B:O atomic ratio (from 7.15 of OH-BN to 7.2). Moreover, apart from the B–N bonds, additional peaks assigned to the B–O bonds also clearly appear in OH-BNNSs (Fig. 6b).
| Method | Operation | Lateral size | Yield (%) | Ref. |
|---|---|---|---|---|
| Ultrasonication | Sonication in DMF | — | 0.1 | 25 |
| Sonication in ethanol/water | — | 2.6 | 26 | |
| Sonication in methanesulfonic acid | ≤500 nm | 15 | 27 | |
| Sonication in green solvent | 77 nm | 23 | 28 | |
| Expansion-assisted sonication | 1.6 μm | 26 | 37 | |
| Mechanical cleavage | Urea-assisted ball milling | 200–300 nm | 85 | 22 |
| Hydroxide-assisted ball milling | 1.5 μm | 18 | 29 | |
| Vortex fluidic exfoliation | — | 5 | 23 | |
| Ball-milled in organic solvent | ≤300 nm | — | 24 | |
| Chemical exfoliation | Peroxynitrous acid-assisted hydrolysis | — | 95 | 35 |
| A modified Hummers' method | 6.5 | 43 | ||
| Exfoliation by molten hydroxides | — | 0.191 | 38 | |
| Thermal exfoliation | Exfoliation by heating in air | 100–200 nm | 65 | 40 |
| Exfoliation by the steam treatment | — | 31.6 | 34 | |
| Novel exfoliation | Water freezing expansion exfoliation | — | 0.12 | 44 |
| Exfoliation using supercritical fluid | — | 10 | 41 | |
| Exfoliation by decomposition of H2O2 | 1.65 μm | 45.2 | This work |
 | ||
| Fig. 6 (a) FT-IR spectra of the pristine h-BN powders, the as-produced OH-BN and OH-BNNSs. (b) High-resolution B1s XPS scan of the OH-BNNSs. | ||
The introduction of –OH groups effectively improves the dispersibility of OH-BNNSs in various solvents. For comparative purpose, unfunctionalized BNNSs were also fabricated through the direct treatment of h-BN powders in H2O2 under concentrated alkaline solutions. The violent exothermic decomposition of H2O2 could provide sufficient energy to overcome the interlayer force of h-BN, resulting in successful exfoliation, as confirmed by the TEM image (Fig. S6a†). Whereas, the FT-IR result reveals the absence of –OH groups on the surfaces of the as-obtained BNNSs, as observed in Fig. S6b.† It is known that BNNSs are typical superhydrophobic materials with highly limited solubility in water.46 Therefore, the dispersibility of OH-BNNSs in water can serve as a representative example to demonstrate the improved dispersion properties of functionalized BNNSs.16,47
UV-vis spectroscopy was adopted to probe the change in dispersibility of OH-BNNSs in water with respect to the pure BNNSs. For UV-vis analysis, nanosheets were dispersed in water at a nominal concentration of 1 mg mL−1 through bath sonication, followed by standing for a week. As illustrated in Fig. 7, OH-BNNSs exhibit significantly higher absorbance than that of the unfunctionalized BNNSs, indicating a substantial increase in water solubility due to the surface –OH groups. The inset in Fig. 7 further visually demonstrates the remarkably enhanced solubility of OH-BNNSs in water. To determine the precise concentration, the dispersions were firstly filtered, dried, and then weighed. The concentration of BNNSs is only 0.032 mg mL−1, while that of the OH-BNNSs is found to be as high as 0.337 mg mL−1, corresponding to a 9-fold increase in concentration. Moreover, the dispersibility of OH-BNNS in various organic solvents, such as ethanol, IPA and DMF, also shows a drastic increment relative to the BNNSs, as shown in Fig. S7.†
 | ||
| Fig. 7 UV-vis spectra of the BNNSs and OH-BNNSs in water, with the inset showing photographs of the BNNSs and OH-BNNSs in water for 1 week after sonication. | ||
To demonstrate the high efficiency of the present synthesis method, the yield of OH-BNNSs was determined. The output per single production run was measured to be 0.904 g, resulting in a high yield of up to 45.2%. Whereas, the direct treatment of h-BN in H2O2 under concentrated alkaline solutions led to a decreased yield of 36.6%. Hence, it is reasonable to suggest that the achieved high yield is partially attributed to the existence of –OH groups on h-BN surfaces. As discussed above, the grafting of –OH groups can prevent agglomeration during exfoliation, thus greatly promoting the exfoliation of OH-BN. The yield of our method is also significantly higher than that found in most reported exfoliation methods, as shown in Table 1. It is noteworthy that although certain reported methods have achieved ultrahigh yields of 85%22 or 65%,40 the resultant BNNSs typically demonstrate relatively small sizes ranging from 100 to 300 nm.
Thermal performance of the OH-BNNSs/TPU composites
The reactive OH-BNNSs prepared by this method are particularly valuable in many applications, especially as thermal conducting fillers for polymer composites.48 FTIR analyses for the pure TPU resin and TPU composites containing 10 wt% BNNSs and 10 wt% OH-BNNSs were firstly performed to check for –OH functionality, as shown in Fig. 8. In the case of pure TPU resin, the existence of two important absorption peaks, including the stretching vibrations of –NCO, –NHCOO–, confirm that isocyanate-terminated TPU was successfully synthesized. For BNNSs/TPU composites, as expected, in addition to the absorption peaks assigned to BNNSs, the absorption peak attributed to –NCO valence bond vibration is also observed. This indicates that there is no chemical binding between –NCO groups of TPU and BNNSs. Regarding of OH-BNNSs/TPU, the peak assigned to the –NCO groups disappears. These results confirm that the terminal –NCO groups in TPU completely react with –OH groups in OH-BNNSs fillers.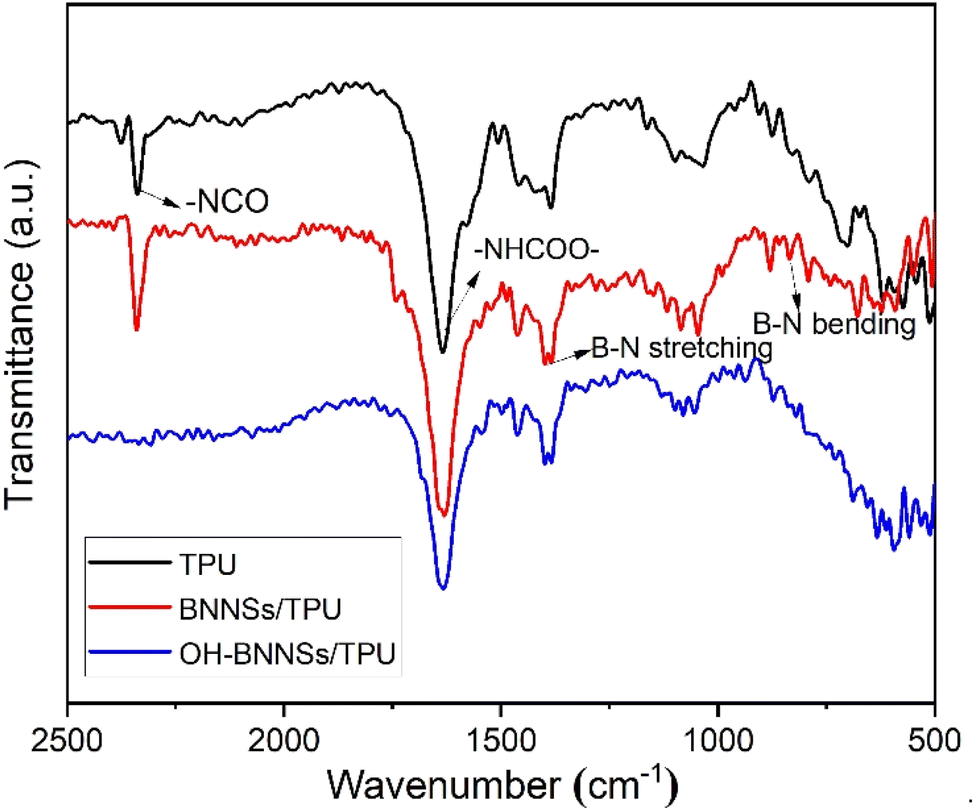 | ||
| Fig. 8 FT-IR spectra of the TPU and TPU composites with 10 wt% BNNSs and 10 wt% OH-BNNSs. | ||
The insufficient bonding sites between BNNSs and TPU severely limit the homogeneous dispersion of BNNSs, as revealed in the cross-sectional SEM image (Fig. 9a). The observation shows the presence of numerous prominent large flakes and aggregates in the BNNSs/TPU composites. The strong chemical bonding between –OH and –NCO groups can facilitate the uniform dispersion of OH-BNNSs and the strong interfacial adhesion between OH-BNNSs and TPU, as indicated in the SEM characterization (Fig. 9b). The OH-BNNSs are evenly embedded in TPU matrices, without interfacial debonding between fillers and matrices. Note that, the “disappearance” of most nanosheets in the OH-BNNSs/TPU composites provides further support for the uniform dispersion and the strong interfacial adhesion.27 Such homogeneous distribution and strong interface can effectively facilitate the utilization of the ultrahigh thermal conductivity of the BNNSs while simultaneously decreasing the phonon scattering at the interfaces. Consequently, OH-BNNSs exhibit much higher effectiveness in improving the thermal conductivity of TPU compared to BNNSs. As shown in Fig. 10a, the thermal conductivity can reach up to 3.097 Wm−1 K−1 in OH-BNNSs/TPU composite with a 30 wt% loading, while the corresponding thermal conductivity of BNNSs/TPU composite is only 1.672 Wm−1 K−1. It is also worth noting that the difference in thermal conductivity between OH-BNNSs/TPU and BNNSs/TPU composites becomes even more remarkable at high filler contents. As presented in Fig. 10b, for OH-BNNSs/TPU composites, the thermal improvement (defined as the percentage increase in thermal conductivity compared to that of BNNSs/TPU composite) quickly increases from 29.5 at 5 wt% loading to 85.2 at 30 wt% loading. For BN-based composites, the relationship between thermal conductivity and filler content can be modeled as follows:49
| logλc = (1 − V)log(Cmλm) + VCflogλf |
 | ||
| Fig. 9 The SEM images of the fractured surfaces of TPU composites with 10 wt% BNNSs (a) and 10 wt% OH-BNNSs (b). | ||
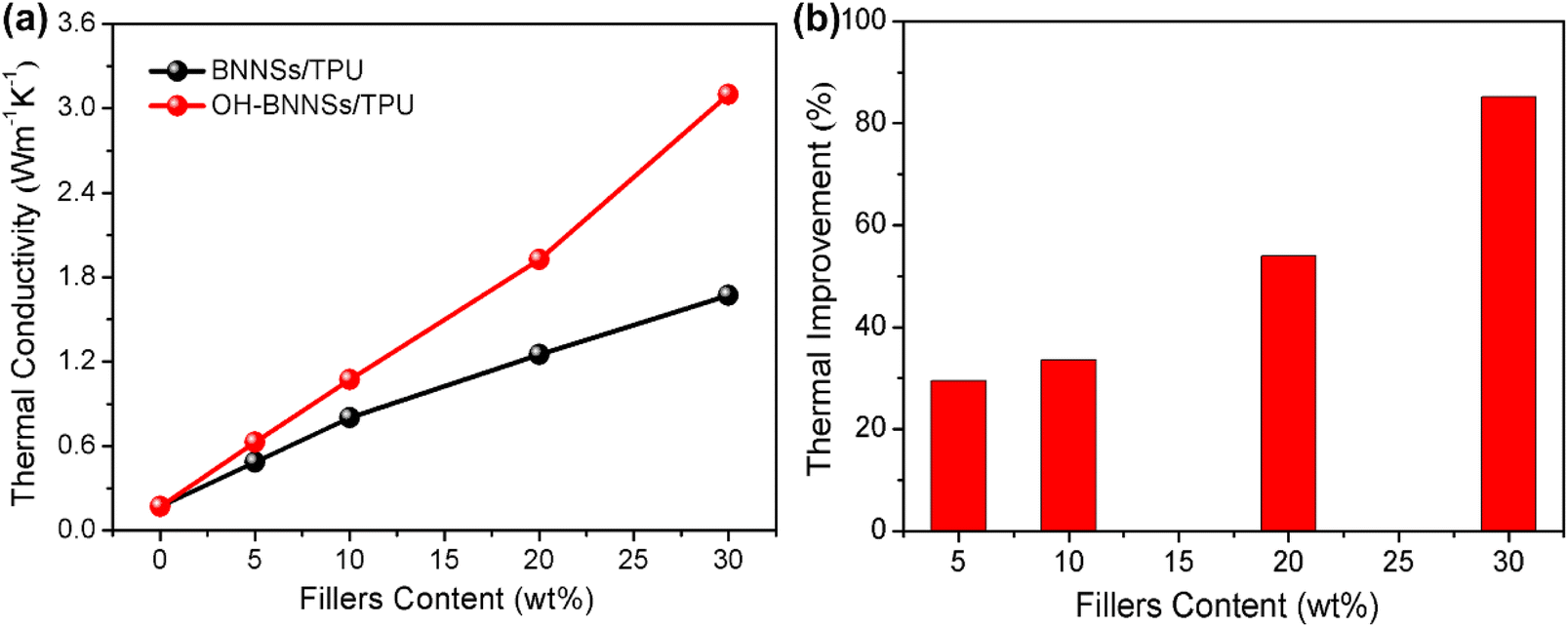 | ||
| Fig. 10 (a) Thermal conductivity of the BNNSs/TPU and OH-BNNSs/TPU composites. (b) The thermal conductivity improvement of OH-BNNSs/TPU composites in comparison with the BNNSs/TPU composites. | ||
The thermal stability of OH-BNNSs/TPU composites was also evaluated by TGA analysis, and the corresponding curves are shown in Fig. 11. It is observed that all OH-BNNSs/TPU composites display similar thermal degradation profiles irrespective of the OH-BNNSs content. This indicates that the incorporation of OH-BNNSs has no significant effect on the degradation mechanism of the TPU matrix. However, the onset degradation temperature, defined as that corresponding to 5 wt% weight loss, increases monotonously as the loading of OH-BNNSs increases, as illustrated in an amplified pattern in the inset of Fig. 11. Also, a comparative study was conducted on TGA-DTG plots of the pure TPU resin and the composite containing 30 wt% OH-BNNSs. As illustrated in Fig. S8,† for the composite with 30 wt% fillers, the onset degradation temperature increases up to 342.8 °C, improving by 37.4 °C compared to that of the neat TPU resin. Moreover, the pure TPU resin exhibits the highest mass loss rate at around 377 °C, while that of the composite containing 30 wt% fillers appears at a higher temperature (436 °C). The maximum mass loss rate of pure TPU is also significantly higher than that of the composite containing 30 wt% filler. The enhancement in thermal stability is primarily attributed to the high thermal conductivity of OH-BNNSs/TPU composites.
 | ||
| Fig. 11 TGA patterns of OH-BNNSs/TPU composites with various loadings. The inset shows the dependence of the onset degradation temperature on the filler content. | ||
Conclusions
In summary, we have demonstrated a novel and facile two-step route to exfoliate and functionalize hydrophobic h-BN into hydrophilic OH-BNNSs. This method boasts the advantages such as easy operation, low cost, and the avoidance of high temperature and vacuum systems. Furthermore, the strategy is particularly efficient, yielding a high percentage of 45.2%. The as-obtained products exhibit a few-layered and large-area morphology, along with excellent dispersion stability in various solutions. This method is easily scalable and holds promise for the large-scale preparation of other layered nanomaterials. Furthermore, OH-BNNSs/TPU composites show significantly higher thermal conductivity compared to the counterpart of BNNSs/TPU composites, with an 85.2% improvement at 30% loading. Simultaneously, the OH-BNNSs/TPU composite demonstrates a remarkable improvement in thermal stability. Owing to the robust interfacial affinity between OH-BNNSs and polymer matrices, the OH-BNNSs are predestined to be promising fillers in highly thermoconductive polymeric composites.Materials and methods
Materials
The bulk h-BN (purity > 99.5%) was purchased from Dandong Rijin Science and Technology Co., Ltd, China. Aqueous solution of hydrogen peroxide (H2O2, 30%), ammonia water (25%), 4,4′-phenyl methane diisocyanate (MDI), 1,4-butanediol (BDO), isopropyl alcohol (IPA), ethanol and N,N-dimethylformamide (DMF) were purchased from J&K Chemical in Shanghai. Poly (butylenes adipate) glycol (PBA) was kindly supplied by Qingdao Xinyutian Chemical Co., Ltd, China. The OH value of PBA is 53–59 mg KOH per g. The molecular Weight of PBA is 2000. All reagents were of analytical grade and used directly without further purification. Deionized water was used throughout all experiments.Hydroxylation and exfoliation of h-BN
In a typical synthesis, 2 g h-BN powders were dispersed in 500 mL aqueous solution of H2O2 by sonication for 10 min. Under vigorous stirring, the mixture was refluxed for 5 h at 120 °C. After the hydrothermal treatment, the required volume of ammonia water was added to the dispersion above, and then the mixture was kept vigorous stirring for 15 min at room temperature. Subsequently, the suspension was centrifuged at 3000 rpm for 30 min to exclude the remaining large particles. The OH-BNNSs, dispersed in the supernatant fraction, were collected by filtration, washed thrice with deionized water, and dried under vacuum.To obtain the mid-products, a fraction of the resultant dispersion after the hydrothermal reaction was filtered and repeatedly rinsed with deionized water. Finally, the mid-product, denoted as OH-BN, was dried at 80 °C for 8 h. For comparison, unfunctionalized BNNSs were also fabricated through direct treatment of the h-BN in H2O2 under concentrated alkaline conditions, without the hydrothermal reactions between h-BN with H2O2.
Fabrication of OH-BNNSs/TPU composites
PBA, MDI and BDO, with the weight ratio of 200:52:9, were used to prepare isocyanate-terminated thermoplastic polyurethane elastomer (TPU) resins. The OH-BNNSs/TPU composites were prepared by an in situ polymerization process as follows: Firstly, PBA was vacuum dehydrated at 100–120 °C for 2 h. After cooling to 70–80 °C, the required quantity of MDI, dissolved in DMF, was added to initiate prepolymerization until the value of –NCO % reached the predetermined content. The chain extension process was then initiated by the addition of BDO. The process BJO continued until a viscous and transparent liquid was observed. Subsequently, OH-BNNSs, dispersed homogeneously in DMF via vigorous ultrasonic dispersion, were slowly dropped into the above liquid under rapid stirring. After that, the mixture was further vigorously stirred for 2 h. Finally, the mixture was poured into a Teflon mold and the solvent was evaporated at 80 °C for 12 h, followed by curing at 120 °C for 24 h. The blank TPU and BNNSs/TPU composites were also prepared by the same procedure above, except that no OH-BNNSs was added and BNNSs substituted for OH-BNNS, respectively.
Characterization
X-ray photoelectron spectroscopy (XPS) measurement was performed using a PHI 5700 ESCA System with an aluminum K-α source (1486.6 eV). Raman spectra were recorded through a confocal Raman spectroscopic system with a 532 nm excitation laser. Powder X-ray diffraction (XRD) analysis was acquired by a PANalytical diffractometer, working with Cu Kα radiation. Scanning electron microscopy (SEM) observation was carried out on a FEI Helios Nanolab 600i scanning electron microscope. Transmission electron microscope (TEM) images were obtained using a FEI Tecnai G2 F20 transition electron microscope. The sample for TEM analysis was prepared by drying OH-BNNSs/IPA dispersion on a carbon coated copper grid. Atomic force microscope (AFM) observation was performed using a Digital Instruments-Veeco Metrology Group atomic force microscope. The sample for AFM analysis was prepared by dispersing OH-BNNSs into IPA by sonication and spin coated on a Si substrate. Identification of functional groups was carried out using an AVATAR 360 Fourier transform infrared spectrometer (FT-IR). Ultraviolet and visible spectroscopy (UV-vis) measurement was performed on a Hitachi UH5300 spectrophotometer. Thermal conductivity of the composites was measured using a Hot Disk thermal analyzer (Hot Disk AB, Uppsala, Sweden). At least three separate measurements were carried out for each specimen, and the average value was used. Thermal gravimetric analysis (TGA) was performed with a thermoanalyzer (SDT Q600 V20.9 Build 20, TA Instruments) at a heating rate of 10 °C min−1.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Natural Science Foundation of Jiangsu Province, China (Grant No. BK20201053). We are grateful for the constructive comments and valuable advices from all the reviewers for further improvement of our work.References
- M. Yi, Z. Shen, X. Zhao, S. Liang and L. Liu, Appl. Phys. Lett., 2014, 104, 143101 CrossRef.
- X. Tang, H. Wang, C. Liu, X. Zhu, W. Gao and H. Yin, ACS Appl. Nano Mater., 2021, 4, 12024–12033 CrossRef CAS.
- T. Ouyang, Y. Chen, Y. Xie, K. Yang, Z. Bao and J. Zhong, Nanotechnology, 2010, 21, 245701 CrossRef PubMed.
- L. Song, L. Ci, H. Lu, P. B. Sorokin, C. Jin, J. Ni, A. G. Kvashnin, D. G. Kvashnin, J. Lou, B. I. Yakobson and P. M. Ajayan, Nano Lett., 2010, 10, 3209–3215 CrossRef CAS PubMed.
- T. Feng, J. Cui, M. Ou, R. Li, Z. Zhao, Y. Geng, X. Chen and C. Jiao, Compos. Commun., 2023, 41, 101649 CrossRef.
- Y. Zhang, B. Zhu, X. Cai, K. Qiao, H. Du, M. Zhou, S. Yan, N. Sun and X. Liang, Composites, Part A, 2023, 173, 107632 CrossRef CAS.
- P. Li, W. Cheng, Y. Zhou, D. Zhao, J. Liu, L. Li, X. Ouyang, B. Liu, W. Jia, Q. Xu and K. Ostrikov, Adv. Mater., 2023, 35, 2209452 CrossRef CAS PubMed.
- Q. He, L. Ding, L. Wu, Z. Zhou, Y. Wang, T. Xu, N. Wang, K. Zhang, X. Wang, F. Ding, J. Zhang and Y. Yao, Small Struct., 2023, 4, 2200282 CrossRef CAS.
- Y. Xu, Z. An, X. Yu, J. Yao, Q. Lv, H. Yang, Z. Lv, H. Guo, Q. Jiang, W. Liu, L. Wu, L. Tan, Y. Dai and Y. Tang, J. Catal., 2023, 427, 115094 CrossRef CAS.
- Y. Wang, J. Wang, P. Zheng, C. Sun, J. Luo and X. Xie, J. Energy Chem., 2022, 67, 451–457 CrossRef CAS.
- J. Qi, C. Ma, Q. Guo, C. Ma, Z. Zhang, F. Liu, X. Shi, L. Wang, M. Xue, M. Wu, P. Gao, H. Hong, X. Wang, E. Wang, C. Liu and K. Liu, Adv. Mater., 2023, 2303122 Search PubMed.
- A. Biswas, R. Xu, J. Christiansen-Salameh, E. Jeong, G. A. Alvarez, C. Li, A. B. Puthirath, B. Gao, A. Garg, T. Gray, H. Kannan, X. Zhang, J. Elkins, T. S. Pieshkov, R. Vajtai, A. G. Birdwell, M. R. Neupane, B. B. Pate, T. Ivanov, E. J. Garratt, P. Dai, H. Zhu, Z. Tian and P. M. Ajayan, Nano Lett., 2023, 23, 6927–6936 CrossRef CAS PubMed.
- K. Qiu, W. Zou, Z. Fang, Y. Wang, S. Bell, X. Zhang, Z. Tian, X. Xu, B. Ji, D. Li, T. Huang and J. Diao, ACS Nano, 2023, 17, 4716–4728 CrossRef CAS PubMed.
- J. Zhang, N. Neupane, P. R. Dahal, S. Rahimi, Z. Cao, S. Pandit and I. Mijakovic, ACS Appl. Bio Mater., 2023, 6, 3131–3142 CrossRef CAS PubMed.
- R. Lu, F. Xu, Y. Cui, D. Bao, S. Yuan, Y. Sun, L. Pei, Y. Jiang, J. Zhu and H. Wang, Prog. Org. Coat., 2024, 186, 108004 CrossRef CAS.
- F. Xu, J. Zhu, P. Ye, H. Geng, J. Peng, Y. Cui, D. Bao, R. Lu, X. Shen, H. Zhu, Y. Zhu and H. Wang, Chem. Eng. J., 2023, 473, 145163 CrossRef CAS.
- Z. Li, W. Yang, Z. Chen, C. Qi, C. Zhang, S. Du, M. Li, R. Feng, S. Li, B. Sun, L. Wang, Y. Liu and Y. Li, ACS Appl. Nano Mater., 2024, 7, 4264–4276 CrossRef CAS.
- J. Liu, H. Feng, J. Dai, K. Yang, G. Chen, S. Wang, D. Jin and X. Liu, Chem. Eng. J., 2023, 469, 143963 CrossRef CAS.
- D. Golberg, Y. Bando, Y. Huang, T. Terao, M. Mitome, C. Tang and C. Zhi, ACS Nano, 2010, 4, 2979–2993 CrossRef CAS PubMed.
- Y. Lin and J. W. Connell, Nanoscale, 2012, 4, 6908–6939 RSC.
- Z. Wang, Z. Tang, Q. Xue, Y. Huang, Y. Huang, M. Zhu, Z. Pei, H. Li, H. Jiang, C. Fu and C. Zhi, Chem. Rec., 2016, 16, 1204–1215 CrossRef CAS PubMed.
- W. Lei, V. N. Mochalin, D. Liu, S. Qin, Y. Gogotsi and Y. Chen, Nat. Commun., 2015, 6, 8849 CrossRef CAS PubMed.
- X. Chen, J. F. Dobson and C. L. Raston, Chem. Commun., 2012, 48, 3703–3705 RSC.
- Y. Yao, Z. Lin, Z. Li, X. Song, K.-S. Moon and C.-p. Wong, J. Mater. Chem., 2012, 22, 13494–13499 RSC.
- C. Zhi, Y. Bando, C. Tang, H. Kuwahara and D. Golberg, Adv. Mater., 2009, 21, 2889–2893 CrossRef CAS.
- K.-G. Zhou, N.-N. Mao, H.-X. Wang, Y. Peng and H.-L. Zhang, Angew. Chem., Int. Ed., 2011, 50, 10839–10842 CrossRef CAS PubMed.
- Y. Wang, Z. Shi and J. Yin, J. Mater. Chem., 2011, 21, 11371–11377 RSC.
- A. R. Deshmukh, J. W. Jeong, S. J. Lee, G. U. Park and B. S. Kim, ACS Sustain. Chem. Eng., 2019, 7, 17114–17125 CrossRef CAS.
- D. Lee, B. Lee, K. H. Park, H. J. Ryu, S. Jeon and S. H. Hong, Nano Lett., 2015, 15, 1238–1244 CrossRef CAS PubMed.
- Q. Weng, X. Wang, X. Wang, Y. Bando and D. Golberg, Chem. Soc. Rev., 2016, 45, 3989–4012 RSC.
- C.-G. Yin, Y. Ma, Z.-J. Liu, J.-C. Fan, P.-H. Shi, Q.-J. Xu and Y.-L. Min, Polymer, 2019, 162, 100–107 CrossRef CAS.
- S. Kong, H. Seo, H. Shin, J.-H. Baik, J. Oh, Y.-O. Kim and J.-C. Lee, Polymer, 2019, 180, 121714 CrossRef CAS.
- T. Sainsbury, A. Satti, P. May, Z. Wang, I. McGovern, Y. K. Gun'ko and J. Coleman, J. Am. Chem. Soc., 2012, 134, 18758–18771 CrossRef CAS PubMed.
- F. Xiao, S. Naficy, G. Casillas, M. H. Khan, T. Katkus, L. Jiang, H. Liu, H. Li and Z. Huang, Adv. Mater., 2015, 27, 7196–7203 CrossRef CAS PubMed.
- P. K. Samantaray, R. Sen Gupta and S. Bose, Adv. Sustainable Syst., 2023, 7, 2200385 CrossRef CAS.
- A. S. Nazarov, V. N. Demin, E. D. Grayfer, A. I. Bulavchenko, A. T. Arymbaeva, H.-J. Shin, J.-Y. Choi and V. E. Fedorov, Chem.–Asian J., 2012, 7, 554–560 CrossRef CAS PubMed.
- F. Yuan, W. Jiao, F. Yang, W. Liu, J. Liu, Z. Xu and R. Wang, J. Mater. Chem. C, 2017, 5, 6359–6368 RSC.
- X. Li, X. Hao, M. Zhao, Y. Wu, J. Yang, Y. Tian and G. Qian, Adv. Mater., 2013, 25, 2200–2204 CrossRef CAS PubMed.
- D. Schild, S. Ulrich, J. Ye and M. Stüber, Solid State Sci., 2010, 12, 1903–1906 CrossRef CAS.
- Z. Cui, A. J. Oyer, A. J. Glover, H. C. Schniepp and D. H. Adamson, Small, 2014, 10, 2352–2355 CrossRef CAS PubMed.
- P. Thangasamy and M. Sathish, CrystEngComm, 2015, 17, 5895–5899 RSC.
- M. Du, X. Li, A. Wang, Y. Wu, X. Hao and M. Zhao, Angew. Chem., Int. Ed., 2014, 53, 3645–3649 CrossRef CAS PubMed.
- M. Du, Y. Wu and X. Hao, CrystEngComm, 2013, 15, 1782–1786 RSC.
- C. Li, T. Wang, Y. Wu, F. Ma, G. Zhao and X. Hao, Nanotechnology, 2014, 25, 495302 CrossRef PubMed.
- J. Hou, G. Li, N. Yang, L. Qin, M. E. Grami, Q. Zhang, N. Wang and X. Qu, RSC Adv., 2014, 4, 44282–44290 RSC.
- T. Ikuno, T. Sainsbury, D. Okawa, J. M. J. Fréchet and A. Zettl, Solid State Commun., 2007, 142, 643–646 CrossRef CAS.
- A. Pakdel, C. Zhi, Y. Bando, T. Nakayama and D. Golberg, ACS Nano, 2011, 5, 6507–6515 CrossRef CAS PubMed.
- Z. Li, F. Guo, S. Tian, R. Liu, D. Yang, X. Wang, H. Hu, Y. Wang and J. Zhao, ACS Appl. Nano Mater., 2023, 6, 5365–5373 CrossRef CAS.
- Y. Agari, A. Ueda, M. Tanaka and S. Nagai, J. Appl. Polym. Sci., 1990, 40, 929–941 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra02329c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |