
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The influence of bicarbonate concentration and ionic strength on peroxide speciation and overall reactivity towards UO2†
Daniel Olsson *,
Hazal Aydogan and
Mats Jonsson
*,
Hazal Aydogan and
Mats Jonsson
Department of Chemistry, KTH Royal Institute of Technology, Stockholm, SE-100 44, Sweden. E-mail: daniols@kth.se
First published on 20th May 2024
Abstract
H2O2 produced from water radiolysis is expected to play a significant role in radiation induced oxidative dissolution of spent nuclear fuel under the anoxic conditions of a deep geological repository if the safety-barriers fail and ground water reaches the fuel. It was recently found that the coordination chemistry between U(VI), HCO32− and H2O2 can significantly suppress H2O2 induced dissolution of UO2 in 10 mM bicarbonate. This was attributed to the much lower reactivity of the U(VI)O22+-coordinated O22− as compared to free H2O2. We have extended the study to lower bicarbonate concentrations and explored the impact of ionic strength to elucidate the rationale for the low reactivity of complexed H2O2. The experimental results clearly show that dissolution of U(VI) becomes suppressed at [HCO3−] < 10 mM. Furthermore, we found that the reactivity of the peroxide in solutions containing U(VI) becomes increasingly more suppressed at lower carbonate concentration. The suppression is not influenced by the ionic strength, which implies that the low reactivity of O22− in ternary uranyl-peroxo-carbonato complexes is not caused by electrostatic repulsion between the negatively charged complex and the negatively charged UO2-surface as we previously hypothesized. Instead, the suppressed reactivity is suggested to be attributed to inherently higher stability of the peroxide functionality as a ligand to UO22+ compared to as free H2O2.
1. Introduction
Uranium dioxide (UO2) is used as the nuclear fuel in most commercial Light Water Reactors (LWR). After use in the reactor, the fuel matrix still consists of ∼95 percent UO2 and a small percentage of fission products and heavier actinides responsible for the increased radioactivity that persists long after the fuel has been removed from the reactor.1 Confinement of the spent fuel in the geosphere has been internationally accepted as the safest option to handle nuclear waste.1a,2 This solution is not without challenges as both natural and engineered barriers will have to withstand corrosion and mechanical stress for at least 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 years to be considered safe.3 If the barriers are breached, the nuclear fuel will be in contact with groundwater.
000 years to be considered safe.3 If the barriers are breached, the nuclear fuel will be in contact with groundwater.
Predictive modeling of the release of long-lived and radiotoxic actinides and fission products upon groundwater intrusion largely relies on accurate prediction of the dissolution rate of the fuel matrix (mainly UO2).4 UO2 has low solubility under the reducing conditions expected at potential repository sites.5 However, oxidation of U(IV) occurs as a result of water radiolysis as the groundwater is exposed to ionizing radiation leading to the formation of strong oxidants (as well as strong reductants).6 It has been shown that U(VI) dissolution under typical repository conditions can mainly be attributed to oxidation induced by the aqueous radiolysis product H2O2.7 The solubility of U(VI) increases significantly by the presence of bicarbonate (expected in concentrations 1–10 mM in groundwater).8
The reaction between H2O2 and UO2 is known to occur as a competition between oxidation of UO2 (leading to oxidative dissolution) and UO2 catalyzed decomposition of H2O2 leading to the formation of oxygen and water but leaving the UO2 surface unaffected.9 It was previously found that the rate of the reaction between H2O2 and UO2, but not the oxidative efficiency (the dissolution yield) is influenced by the U(VI) concentration in 10 mM bicarbonate.10 This was attributed to varying fractions of stable uranyl-peroxo-carbonato complexes limiting the amount of reactive H2O2 available to the UO2 surface. In such a system the overall reaction scheme can be described as:
| pUO22+ + qH2O2 + rCO32− ⇆ [(UO2)p(O2)q(CO3)r]2p−2q−2r+2qH+ (ref. 11) | (1) |
 | (2) |
 | (3) |
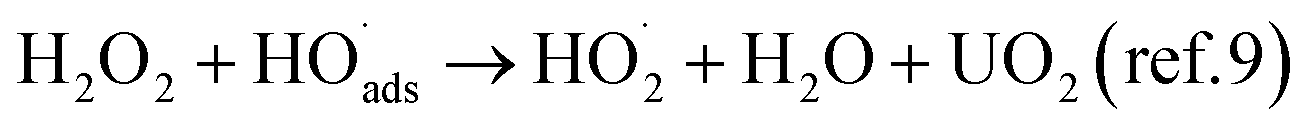 | (4) |
 | (5) |
Both H2O2-induced oxidation of UO2 (reaction (3)) and UO2-catalyzed decomposition of H2O2 (reactions (4) and (5)) have the surface bound hydroxyl radical as a common intermediate. The U(V) produced in reaction (3) is further oxidized into U(VI) in a subsequent step. Direct oxidation of U(V) by OH˙ and disproportionation of two U(V) into U(IV) and U(VI) have been proposed as two possible mechanisms.12,13 Dissolution of U(VI) follows the second oxidation step as a result of complexation reaction(s) between U(VI) and carbonate:
| UO22+(s) + nCO32− → UO2(CO3)n2−2n(aq) (ref. 1b) | (6) |
The equilibria described by reaction (1) indicates that dissolved U(VI) could theoretically suppress the dissolution rate (reaction (6)) by binding carbonate ligands as well as the rate of oxidation (reaction (3)) by binding peroxo-ligands. The affinity of U(VI) towards peroxo- and carbonato ligands follows the order CO32− < O22−.14 Groundwaters present at different potential repository sites can vary in terms of composition. As mentioned above, the concentration of HCO3−/CO32− can vary by one order of magnitude. The ionic strength also varies between sites. In our previous work, we explored the impact of peroxide speciation on the overall peroxide reactivity at 10 mM HCO3− and constant ionic strength (mainly determined by NaHCO3). Hence, the impact of HCO3−/CO32− concentration as well as ionic strength on this system must be explored before peroxide speciation can be correctly accounted for in safety assessments of geological repositories for spent nuclear fuel.
In this work the impact of uranyl-peroxo-carbonato speciation on the overall kinetics of H2O2 induced dissolution of hyper-stoichiometric UO2 has been studied at various carbonate concentrations within the range 1–10 mM. In addition, the ionic strength dependence on the peroxide reactivity was studied as a means of testing if the lower reactivity of the peroxo-ligands in the complexes is due to electrostatic repulsion between the negatively charged U(VI)-peroxo-carbonato complexes and the negatively charged UO2 surface as was previously hypothesized.10,15
2. Materials and methods
The chemicals used throughout the experiments were of reagent grade or higher. Stock solutions of UO2(NO3)2 × 6H2O (Westinghouse AB), H2O2, KI, Arsenazo-III, NaClO4 × H2O (Sigma Aldrich) and NaHCO3 (Merck) where prepared using ultra-pure water (18.2 MΩ cm, Merck MilliQ).The UO2 powder (Westinghouse AB) used throughout these experiments has the specific surface area (BET) 5.4 ± 0.2 m2 g−1 and oxidation state UO2.34, as determined in a previous work.9b The powder was washed prior to exposure to remove U(VI) (i.e., pre-oxidized UO2). The washing procedure has been described in detail elsewhere.10
The speciation was varied by an initial addition of uranyl nitrate. Speciation was simulated in SPANA,16 using stability constants published by Zanonato et al. and ionic strength correction based on the Specific Ion-Interaction Theory (SIT) model.11,17 For convenience, the stability constants used for our speciation calculations are listed in Table 1.
| Species | Log10β11 |
|---|---|
| HO2− | −11.29 |
| OH− | −13.72 |
| HCO3− | 9.69 |
| H2CO3(g) | 15.60 |
| (UO2)2(OH)22+ | −6.07 |
| UO2(OH)3− | −19.69 |
| UO2(CO3)34− | 21.76 |
| UO2(CO3)22− | 14.93 |
| UO2CO3 | 8.57 |
| (UO2)3(CO3)66− | 53.82 |
| UO2(O2)(OH)− | −14.16 |
| (UO2)2(O2)2(OH)− | −15.82 |
| (UO2)(O2)(CO3)24− | 4.37 |
| UO2(O2)(CO3)2− | 1.47 |
| (UO2)2(O2)2(CO3)2− | 1.98 |
| (UO2)2(O2)(CO3)22− | 18.31 |
| (UO2)2(O2)(CO3)46− | 27.4 |
| (UO2)2(CO3)(OH)3− | −1.89 |
| UO2(O2)(OH)− | −2.67 |
| (UO2)2(O2)(OH)− | 7.16 |
The interactions between UO22+ and NO3− were not included in the speciation calculations as they are too weak to significantly affect the speciation under the conditions used in this study. The apparent equilibrium constants (β) found in literature for the UO2NO3+ complex at 25 °C are within the range 0.15 to 0.65,18 considerably weaker interactions than for the dominant uranyl-carbonato- and uranyl-peroxo-carbonato-complexes (see Table 1).
The concentration of U(VI) was measured by the absorbance of U(VI)-(1,8-dihydroxynaphthalene-3,6-disulphonic acid-2,7-bis[(azo-2)-phenylarsonic acid]) complex at 653 nm (Arsenazo III method)19 using calibrations determined through titration (available as ESI† to our previous work10). The total peroxide concentration (free H2O2 and U(VI)-coordinated O22−) was measured indirectly by fully converting the peroxide into I3− in the presence of a molybdenum catalyst (see Ghormley triiodide method20). I3− strongly absorbs light at 360 nm where an overlap with the absorbance of U(VI) is avoided. The extinction coefficient for I3− at 360 nm is 2.16 × 104 dm3 mol−1 cm−1. Control experiments have shown that the results of the methods used to quantify uranium in solution Taand total peroxide in solution are not affected by speciation within the variation in this work.21
The pH was allowed to vary but was measured at several points during each experiment using a Thermo Scientific™ Orion Star™ A111 Benchtop pH Meter. Observed changes in pH were accounted for when performing speciation calculations. pH data for all exposures are included as ESI (Tables S1–S3).†
No blanks were used in this study to control for peroxide decomposition in the aqueous phase. As demonstrated in our previous work,21 decomposition in the aqueous phase has been found to be orders of magnitude slower than the decomposition of H2O2 on UO2 under conditions similar to those studied in this work.
3. Results
A series of experiments were performed using an initial H2O2 concentration of 0.2 mM and bicarbonate concentrations of 1, 2, 5 and 10 mM in 30 ml solutions containing 50 mg UO2 powder. Total peroxide (H2O2 + peroxo-ligands) and U(VI) (UO22+ + uranyl coordination centers) concentrations as function of time are presented in Fig. 1a and b, respectively. | ||
| Fig. 1 Peroxide concentration (a) and U(VI) concentration (b) as a function of exposure time in 30 ml bicarbonate solutions with 50 mg UO2 powder (SA/V = 9000 m−1). | ||
Error bars represents standard deviations based on three replicate exposures. Note that while all other aliquot samples where both filtered and centrifugated, samples from the exposures using 2 mM bicarbonate were only filtered, resulting in larger uncertainty (likely caused by small UO2 particles remaining in the sample).
While the rate of peroxide consumption (decomposition via reactions (3) and (4)) was found to be similar at all four carbonate concentrations (Fig. 1a), there is a clear trend in the amount of dissolved U(VI), where the rate of dissolution as well as the final concentration (after 5 h) increases with increasing carbonate concentration. The ratio between dissolved UO22+ and carbonate is probably crucial here. This possibility was further explored by adding 0.3 mM U(VI) prior to the experiment. In this series of experiments, the initial bicarbonate concentrations were the same as in the experiments without initially added U(VI) but towards the end of each exposure the concentration was increased to ∼40 mM. The late addition of carbonate was done to elucidate the reasons behind the limitation in uranium dissolution. The resulting peroxide concentrations and U(VI) concentrations as a function of time are presented in Fig. 2a and b, respectively.
 | ||
| Fig. 2 Peroxide concentration (a) and U(VI) concentration (b) as a function of exposure time in 20 ml solutions containing 1–10 mM bicarbonate, 0.2 mM initial peroxide and 0.3 mM initial U(VI), with 30 mg UO2 powder suspension (SA/V = 8100 m−1). | ||
As can be seen, the carbonate concentration dependence is even more pronounced in the systems initially containing 0.3 mM U(VI). In this case, also the peroxide consumption kinetics is significantly affected. Note that a lower SA/V was used when compared to the uranyl free systems in Fig. 1, which is expected to reduce the rate of a given surface reaction by 11 percent. To allow further analysis of the system, speciation calculations were performed. The calculated free peroxide (H2O2 + HO2−) – and free carbonate (HCO3− + CO32−) – as functions of time evolution and total carbonate concentration are presented in Fig. 3a and b respectively. The uranyl speciation as a function of total carbonate concentration at the start of the exposures and at the time of the carbonate additions are presented in Fig. 4a and b respectively. The speciation calculations were based on the data presented in Fig. 2a and b and the measured pH (see ESI, Fig. S2†).
 | ||
| Fig. 3 Calculated fractions of free H2O2 (a) and free carbonate species (HCO3− + CO32−) (b) as functions of time evolution and total carbonate concentration. Time points; 1 = start of reaction, 2 = after initial drop (5 h) and 3 = prior to bicarbonate addition (20 or 26 h), as indicated in Fig. 2. | ||
 | ||
| Fig. 4 Calculated fractions of uranyl species as functions of total carbonate concentration at (a) the start of the exposures and (b) the time of NaHCO3 addition. *The 40 mM bicarbonate speciation was calculated for the 1 mM bicarbonate system after raising the total carbonate concentration. | ||
Based on the calculated fractions of free H2O2 (Fig. 3a) and our previous conclusion that coordinated peroxides (O22− ligands) are passive towards the UO2 surface,10 kinetic suppression due to O22− coordination is expected to have a greater effect at lower bicarbonate concentrations, as a result of less competition from UO2(CO3)34− in favor of uranyl-peroxo-carbonato complexes (see Fig. 4a). The suppressing effect is also expected to increase with the progression of time due to the increase in uranyl concentration from oxidative dissolution of the UO2 powder. These predictions are in line with the observed trends for the peroxide concentrations as functions of time (Fig. 2a). Raising the total carbonate concentration from 1 to 40 mM is expected to increase the fraction of free H2O2 to ∼70 percent due to the formation of UO2(CO3)34− at the expense of uranyl-peroxo-carbonato complexes, while a small fraction of ternary complexes (∼20 and 10 percent respectively) is expected to exist in the form of (UO2)2(O2)(CO3)46− and (UO2)(O2)(CO3)24− as seen in Fig. 4b. The high increase in the fraction of free H2O2 at the moment of carbonate addition explains the rapid increase in reactivity towards the UO2 powder which is observed as the carbonate concentrations are raised.
Another possibility is that the suppression of the peroxide reactivity and subsequent uranyl dissolution is partly caused by a blockage of reactive UO2 sites by U(VI) due to a low fraction of free carbonate species (HCO3− + CO32−). This could potentially suppress dissolution and the regeneration of the reactive sites. The fraction of free carbonate is expected to decrease with carbonate concentration due to the lower excess of total carbonate relative to U(VI) coordination centers (see Fig. 3b). This fraction is also expected to change slightly as time progresses due to changes in U(VI) and peroxide concentration. Even in 1 mM bicarbonate, more than half of the total carbonate (>0.5 mM) is expected to remain as free carbonate species. Prevented dissolution of U(VI) due to limitations in free carbonate species is thus not a likely explanation for the observed suppression of peroxide reactivity and subsequent uranyl dissolution.
As we lack the thermodynamic data to simulate the surface speciation, another possibility to consider is that the strong affinity of U(VI) towards O22− ligands could result in the formation of adsorbed uranyl-peroxo/uranyl-peroxo-carbonato complexes with low solubility in favor of readily soluble uranyl-carbonato complexes at the reactive sites. In this case, the addition of 40 mM would favor formation of soluble uranyl-carbonato complexes and increase the surface reactivity by removing the uranyl, thus removing the blockage of the reactive sites. However, as the passivating effect of uranyl on the peroxide reactivity towards the metal oxide surface has been observed for H2O2 degradation on ZrO2 (where no oxidized sites are formed)10 this is likely not the main reason for the observed peroxide passivation towards UO2.
To further explore the systems with high fractions of complexed peroxide, experiments with initial additions of 0.3 mM U(VI) in 1 and 10 mM bicarbonate were performed at different ionic strengths controlled by addition of NaClO4. The ionic strength was varied to explore the influence of electrostatic repulsion on the surface reactivity of complexed peroxide. Peroxide and U(VI) concentrations as functions of exposure time in 10 mM bicarbonate with an initial U(VI) concentration of 0.3 mM are presented in Fig. 6a and b, respectively.
In 10 mM carbonate, the ionic strength does not appear to significantly affect the rate of peroxide consumption (Fig. 5a) or the subsequent U(VI) dissolution (Fig. 5b). The absence of an ionic strength effect contradicts the previous hypothesis, that suppression of the peroxide consumption rate in systems with a high fraction of complexed peroxide is caused by electrostatic repulsion.10 Instead, we conclude that the complexed peroxide is inherently more stable than free H2O2 and therefore less reactive toward UO2.
 | ||
| Fig. 5 Peroxide concentration (a) and U(VI) concentration (b) as functions of exposure time in 20 ml solutions containing 10 mM bicarbonate, 0.3 mM initial U(VI) and 0–1 M NaClO4, with 30 mg UO2 powder suspension (SA/V = 8100 m−1). | ||
Peroxide- and U(VI) concentrations as function of time for the corresponding exposures in 1 mM bicarbonate are presented in Fig. 6a and b, respectively.
 | ||
| Fig. 6 Peroxide concentration (a) and U(VI) concentration (b) as a function of exposure time in 20 ml solutions containing 1 mM bicarbonate, 0.3 mM initial U(VI) and 0–1 M NaClO4, with 30 mg UO2 powder suspension (SA/V = 8100 m−1). | ||
Regardless of ionic strength, a fast initial drop in peroxide concentration (Fig. 6a) as well as in U(VI) concentration (Fig. 6b) can be observed. Increasing the ionic strength results in more substantial initial drops in the two concentrations. A similar effect has previously been observed in bicarbonate-free water.15 At the highest ionic strengths the U(VI) and peroxide concentrations keep decreasing after the initial fast drop and reach values close to the detection limit after 20 h. The apparent 1:1 ratio between the peroxide and uranyl disappearance from the aqueous phase in these two systems is likely caused by the formation of a secondary phase (such as studtite) on the UO2 surface. Upon increasing the bicarbonate concentration to 40 mM, a rapid increase in U(VI) concentration is observed at all ionic strengths. Interestingly, the peroxide concentration is also observed to initially increase with the bicarbonate addition at the two highest ionic strengths, followed by the type of decrease expected for reactive H2O2 (towards UO2). This initial, partial recovery of peroxide supports the idea of a peroxide decrease as a result of the formation of a secondary phase rather than a decrease caused by the typical H2O2 consumption on UO2 where the peroxide would be permanently lost.
At lower ionic strength where a significant amount of peroxide remains in solution, the peroxide concentration immediately starts to drop upon addition of bicarbonate. This indicates increased peroxide reactivity towards UO2 by the release of H2O2, as expected due to the formation of UO2(CO3)34− in favor of uranyl-peroxo-carbonato complexes (as was observed in NaClO4-free solutions (Fig. 2a)). The initial fast drop in U(VI) and peroxide concentrations in combination with the rapid increase in concentrations upon bicarbonate addition at the two highest ionic strengths, indicate that increasing the ionic strength favors adsorption of the uranyl peroxo-carbonato complex(es).
Calculated fractions of free H2O2 and free carbonate at the start of the exposures (t = 0) are presented in Fig. 5a and b respectively.
The fractions of free H2O2 (Fig. 7a) and free carbonate (Fig. 7b) change slightly with ionic strength but are mainly determined by the total carbonate concentration. At 10 mM bicarbonate, the fraction of free H2O2 is expected to decrease from 35 to 21 percent upon increasing the ionic strength. This is not reflected in the seemingly unaffected rates observed in Fig. 5a and b. Instead, the unaffected rates suggests that the stability of the coordinated O22− is not significantly affected by ionic strength (at least not at these concentrations of uranyl, peroxide and carbonate).
 | ||
| Fig. 7 Calculated fractions (at t = 0) of free H2O2 (a) and free carbonate species (HCO3− + CO32−) (b) as functions of NaClO4- and total carbonate-concentration. | ||
In the 1 mM bicarbonate system, the initial fraction of free H2O2 is expected to decrease from 5 percent to considerably less than 1 percent with increasing ionic strength. Hence, we would expect the consumption of peroxide and the release of U(VI) to be very slow and to become even slower with increasing ionic strength, provided that the screening of electrostatic repulsion between the uranyl-peroxo-carbonato complexes and the UO2 surface does not catalyze degradation of coordinated O22− on UO2. Any kinetic effect on the surface reaction between H2O2 and UO2 (if present) is masked by the continuous adsorption of uranyl and peroxide. However, the 1:1 ratio between loss of uranyl and peroxide in the aqueous phase is consistent with a low fraction of H2O2 to a point where loss of peroxide due to degradation of H2O2 on the UO2 is negligible.
4. Conclusions
The experiments performed in this work show that the bicarbonate concentration has little impact on the rate of H2O2 consumption on UO2 but a significant effect on the release of U(VI) in systems initially free from dissolved U(VI).In systems initially containing U(VI), the concentration of bicarbonate has a significant effect on both the consumption of peroxide and the release of U(VI), likely by influencing the ratio between reactive, free H2O2, and non-reactive U(VI)-coordinated O22−.
In 10 mM bicarbonate the consumption of peroxide as well as the release of U(VI) are unaffected by changes in the ionic strength while at lower bicarbonate concentration (1 mM), ionic strength is observed to influence the solubility of uranyl-peroxo-carbonato complexes. The combination of high ionic strength and peroxide predominantly in the form of U(VI)-coordinated O22− appear to favor adsorption of uranyl-peroxo-/uranyl-peroxo-carbonato species in bicarbonate deficient systems.
Speciation calculations based on the stability constants for uranyl-carbonato and uranyl-peroxo-carbonato complexes shed some light on the experimental observations. A stronger suppression of the peroxide reactivity towards UO2 is expected due to a lower fraction of uranyl-carbonato complex favoring higher fractions of passive peroxide in uranyl-peroxo-carbonate complexes. The absence of an ionic strength effect at high bicarbonate concentration shows that the reason for the low surface reactivity of the ternary peroxo complexes is not electrostatic repulsion, but likely due to an inherently higher thermodynamic stability compared to that of free H2O2.
Conflicts of interest
There are no conflicts to declare.References
- (a) V. K. Verma and K. Katovsky, in Spent Nuclear Fuel and Accelerator-Driven Subcritical Systems, Springer Singapore, 2019, pp. 1–19 CrossRef; (b) D. W. Shoesmith, J. Nucl. Mater., 2000, 282, 1–31 CrossRef CAS; (c) S. Sunder, Nucl. Technol., 1998, 122, 211–221 CrossRef CAS.
- J. C. S. Long and R. C. Ewing, Annu. Rev. Earth Planet. Sci., 2004, 32, 363–401 CrossRef CAS.
- M. I. Ojovan, W. E. Lee and S. N. Kalmykov, in An Introduction to Nuclear Waste Immobilisation, Elsevier, 3rd edn, 2019, pp. 433–461 Search PubMed.
- T. E. Eriksen, D. W. Shoesmith and M. Jonsson, J. Nucl. Mater., 2012, 420, 409–423 CrossRef CAS.
- A. S. Kertes and R. Guillaumont, Nucl. Chem. Waste Manage., 1985, 5, 215–219 CrossRef CAS.
- S. Le Caër, Water, 2011, 3, 235–253 CrossRef.
- E. Ekeroth, O. Roth and M. Jonsson, J. Nucl. Mater., 2006, 355, 38–46 CrossRef CAS.
- (a) F. King, in Geological Repository Systems for Safe Disposal of Spent Nuclear Fuels and Radioactive Waste, ed. M. J. Apted and J. Ahn, Woodhead Publishing, 2nd edn, 2017, pp. 365–408 Search PubMed; (b) M. Somervuori, E. Isotahdon, M. Nuppunen-Puputti, M. Bomberg, L. Carpén and P. Rajala, Corros. Mater. Degrad., 2021, 603–624 CrossRef.
- (a) A. Hiroki and J. A. Laverne, J. Phys. Chem. B, 2005, 109, 3364–3370 CrossRef CAS PubMed; (b) A. Barreiro Fidalgo, Y. Kumagai and M. Jonsson, J. Coord. Chem., 2018, 71, 1799–1807 CrossRef CAS.
- D. Olsson, J. Li and M. Jonsson, ACS Omega, 2022, 7, 15929–15935 CrossRef CAS PubMed.
- P. L. Zanonato, P. Di Bernardo, Z. Szabó and I. Grenthe, Dalton Trans., 2012, 41, 11635 RSC.
- E. Ekeroth and M. Jonsson, J. Nucl. Mater., 2003, 322, 242–248 CrossRef CAS.
- (a) K.-U. Ulrich, E. S. Ilton, H. Veeramani, J. O. Sharp, R. Bernier-Latmani, E. J. Schofield, J. R. Bargar and D. E. Giammar, Geochim. Cosmochim. Acta, 2009, 73, 6065–6083 CrossRef CAS; (b) J. Li, X. Liu and M. Jonsson, Inorg. Chem., 2023, 62, 7413–7423 CrossRef CAS PubMed.
- S. O. Odoh and G. Schreckenbach, Inorg. Chem., 2013, 52, 5590–5602 CrossRef CAS PubMed.
- G. El Jamal, J. Li and M. Jonsson, Eur. J. Inorg. Chem., 2021, 2021, 4175–4182 CrossRef CAS.
- I. Puigdomènech, E. Colàs, M. Grivé, I. Campos and D. G. Cobos, Mater. Res. Soc. Symp. Proc., 2014, 1665, 111–116 CrossRef.
- J. N. Brönsted, J. Am. Chem. Soc., 1922, 44, 877–898 CrossRef.
- (a) M. H. Brooker, C.-B. Huang and J. Sylwestrowicz, J. Inorg. Nucl. Chem., 1980, 42, 1431–1440 CrossRef CAS; (b) M. D. Marcantonatos, M. Deschaux and F. Celardin, Chem. Phys. Lett., 1980, 69, 144–150 CrossRef CAS; (c) O. M. Suleimenov, T. M. Seward and J. K. Hovey, J. Solution Chem., 2007, 36, 1093–1102 CrossRef CAS.
- S. Savvin, Talanta, 1961, 8, 673–685 CrossRef CAS.
- J. A. Ghormley and A. C. Stewart, J. Am. Chem. Soc., 1956, 78, 2934–2939 CrossRef CAS.
- J. Li, L. Li and M. Jonsson, Ecotoxicol. Environ. Saf., 2023, 263, 115297 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra02281e |
| This journal is © The Royal Society of Chemistry 2024 |