
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visible light mediated iron-catalyzed addition of oxamic acids to imines†
Margaux Badufle,
Frédéric Robert and
Yannick Landais*
and
Yannick Landais*
Univ. Bordeaux, CNRS, Bordeaux INP, ISM, UMR 5255, F-33400 Talence, France. E-mail: yannick.landais@u-bordeaux.fr
First published on 18th April 2024
Abstract
Oxamic acids where shown to add to imines, providing a broad range of α-aminoacid amides in generally good yields. The process is efficient on pre-formed imines but may also be conducted using a 3-component strategy by simply mixing aldehydes, amines and oxamic acids in the presence of ferrocene, acting both as a photocatalyst under visible light and as a Lewis acid. The reaction proceeds through the addition onto the imine of a carbamoyl radical intermediate generated through a charge transfer from the carboxylate ligand to a Fe(III) species (LMCT).
Carbamoyl radicals add efficiently to a wide range of unsaturated systems, including alkenes,1 alkynes,2 arenes3 and heteroarenes (Minisci reaction),4 allowing the straightforward incorporation of the amide functional group onto carbon skeletons (Fig. 1A). The generation and use of this class of radicals has recently undergone a major boom.5 Carbamoyl radicals can be generated from a fairly wide variety of precursors, including formamides, oxamic acids and their corresponding potassium salts or activated esters, 4-substituted-1,4-dihydropyridines (DHPs) esters and carbamoyl chlorides.5 Photo-induced and photocatalyzed processes have profoundly modified recently the way to generate carbamoyl radicals and extended their scope of application.5b,c Amongst the above precursors, formamides appear as relatively ideal and their conversion into carbamoyl radicals has been carried out through hydrogen-atom transfer process using either Fenton-type methods6 or direct or indirect HAT using TBADT or alkoxy radicals.7 Although quite efficient, this approach suffers from the potential concurrent C–H abstraction at CHO and α to nitrogen. In turn, carbamoyl chlorides have the disadvantage of being accessible using particularly toxic phosgene derivatives,8 while DHP ester derivatives suffer from their low attractiveness in terms of atom-economy.9 In contrast, oxamic acids are readily available by combination of amines with oxalic acid derivatives5b and only produce CO2 as by-product upon oxidation, explaining their attractiveness as precursors of carbamoyl radicals.1k,3,4c,5b
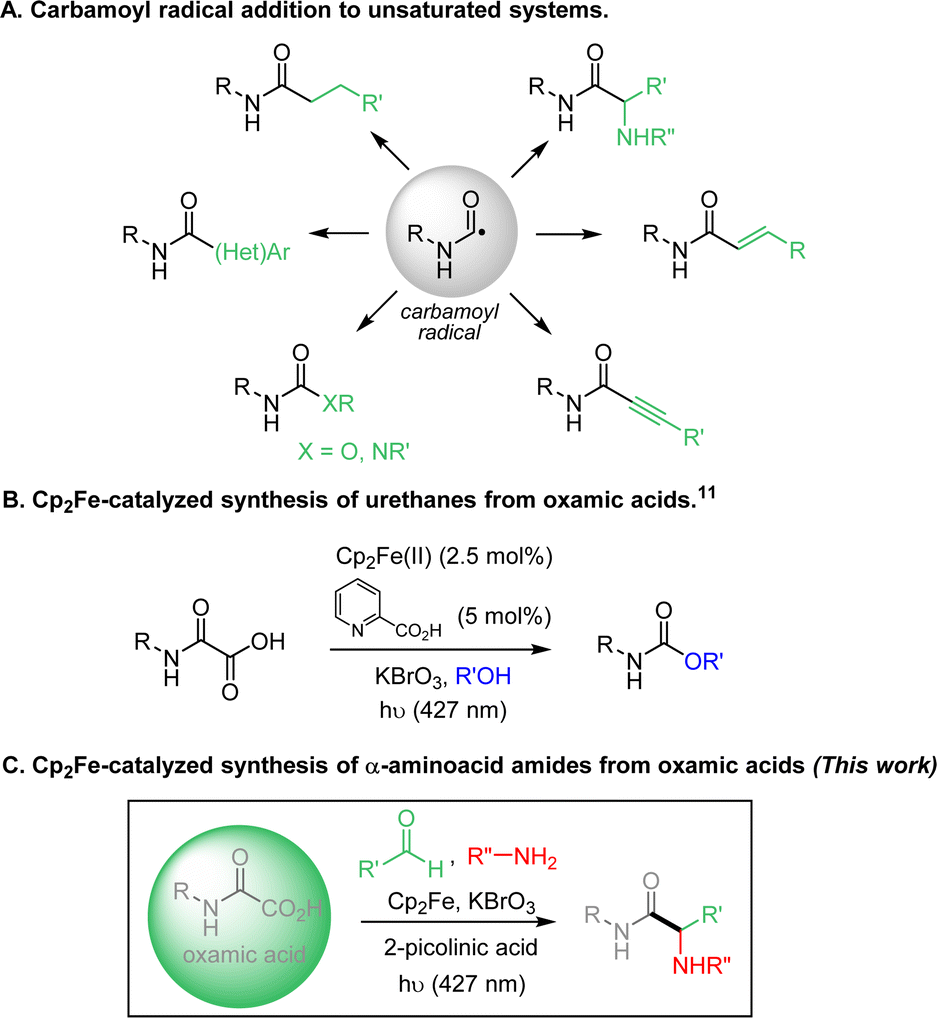 | ||
| Fig. 1 Generation of carbamoyl radicals from oxamic acids and their additions to unsaturated systems. | ||
In this context, we recently reported several methods to generate carbamoyl radicals from simple oxamic acids, using photoredox conditions (including visible and NIR light) and electrochemistry.4b,10 More recently, our laboratory developed a new oxamic acid decarboxylation procedure using Cp2Fe as a catalyst and KBrO3 as an oxidant, which relied on a photocatalyzed LMCT process (Fig. 1B).11 This modified procedure has the advantage to be practically simple, does not require strict anhydrous and deaerated conditions and uses cheap iron catalysts and oxidant. We report here an application of this visible-light mediated LMCT process to the addition of oxamic acids onto imines (Fig. 1C). The addition of the carbamoyl motif to imines has been very little developed to date,12,13 even though it gives access to the α-aminoacid amide fragment, the polypeptides basic unit. This iron-based procedure enables the carbamoylation of imines in generally good yields under mild conditions. A three-component process, through the simple mixing of oxamic acids, anilines and aldehydes under standard catalytic conditions was also developed, which showed broad scope and robustness.
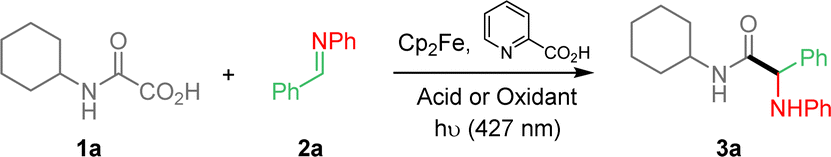
Reaction conditions were first optimized with the preparation of α-aminoacid amide 3a, as summarized in Table 1, using pre-formed imine 2a and oxamic acid 1a as models. Cp2Fe (2.5 mol%) was used as the iron catalyst in the presence of 2-picolinic acid as the ligand in DCE. Without any acid or oxidant additives, the coupling led to only 2% of the desired amide 3a (Table 1, entry 1). Acids such as trifluoroacetic acid and BF3–Et2O were then added to activate the imine function, which led to improved yields of 3a as shown in entries 2 and 3, in good agreement with studies of Jacobi von Wangelin et al.12b This result thus shows that the catalytic cycle is operative in the absence of a terminal oxidant (vide infra). However, all our efforts to further improve the conversion under these “acidic conditions” met with failure. We then turned our attention to the use of KBrO3, which proved to be the best terminal oxidant in our previous studies.11 This modification led to much improved yield (entry 4). Solvents were also varied, indicating that DCE is superior (entries 5 and 6). Various amount of KBrO3 was then tested, showing that optimal yield could be reached using only 0.5 eq. of oxidant (entries 8 and 9). Varying the amount of oxamic acid (entry 10) or imine (11) slightly improved the yield. However, we also observed that under these conditions, the excess of 1a or 2a made the final purification of 3a more tedious. The amount of ligand was also varied (entries 12 and 13) as well as that of the iron catalyst (entry 14), which did not modify the yield to a large extent. Control experiments were performed, showing that, Cp2Fe and light were both essential for the process to occur (entries 15 and 16), while the absence of ligand had a minor effect on the conversion (entry 17). Finally, the process was repeated using air and O2 atmosphere as oxidants (entries 18 and 19). Air led to a moderate yield, while pure oxygen provided 3a in trace amount.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entrya | Oxamic acid (eq.) | Imine (eq.) | Acid or oxidant (eq.) | Fe cat. (mol%) | Ligand (mol%) | Solvent | Yieldb (%) |
| a Unless otherwise mentioned, all reactions were performed with Cp2Fe (2.5 mol%) and ligand (5 mol%) in the indicated solvent (0.1 M), in a sealed tube.b Isolated yields of 3a.c Absence of blue LED.d Absence of Cp2Fe.e Absence of ligand.f Yields of 3a determined by 1H NMR with 1,3,5-trimethylbenzene as an external standard. | |||||||
| 1 | 1.0 | 1.0 | — | 2.5 | 5.0 | DCE | 2 |
| 2 | 1.0 | 1.0 | TFA (1.0) | 2.5 | 5.0 | DCE | 48 |
| 3 | 1.0 | 1.0 | BF3–Et2O (1.0) | 2.5 | 5.0 | DCE | 20 |
| 4 | 1.0 | 1.0 | KBrO3 (1.0) | 2.5 | 5.0 | DCE | 71 |
| 5 | 1.0 | 1.0 | KBrO3 (1.0) | 2.5 | 5.0 | MeCN | 45 |
| 6 | 1.0 | 1.0 | KBrO3 (1.0) | 2.5 | 5.0 | PhCl | 25 |
| 7 | 1.0 | 1.0 | KBrO3 (0.5) | 2.5 | 5.0 | DCE | 70 |
| 8 | 1.0 | 1.0 | KBrO3 (0.2) | 2.5 | 5.0 | DCE | 60 |
| 9 | 1.0 | 1.0 | KBrO3 (2.0) | 2.5 | 5.0 | DCE | 71 |
| 10 | 1.3 | 1.0 | KBrO3 (0.5) | 2.5 | 5.0 | DCE | 77 |
| 11 | 1.0 | 1.3 | KBrO3 (0.5) | 2.5 | 5.0 | DCE | 75 |
| 12 | 1.3 | 1.0 | KBrO3 (0.5) | 2.5 | 2.5 | DCE | 59 |
| 13 | 1.3 | 1.0 | KBrO3 (0.5) | 2.5 | 7.5 | DCE | 76 |
| 14 | 1.3 | 1.0 | KBrO3 (0.5) | 5.0 | 10 | DCE | 70 |
| 15c | 1.3 | 1.0 | KBrO3 (0.5) | 2.5 | 5.0 | DCE | NA |
| 16d | 1.3 | 1.0 | KBrO3 (0.5) | — | 5.0 | DCE | NA |
| 17e | 1.3 | 1.0 | KBrO3 (0.5) | 2.5 | — | DCE | 69 |
| 18f | 1.3 | 1.0 | Air | 2.5 | 5.0 | DCE | 45 |
| 19f | 1.3 | 1.0 | O2 | 2.5 | 5.0 | DCE | 5 |
From these results (Table 1, entry 7), the substrate scope was extended, varying the nature of oxamic acids 1 using pre-formed imines 2a (Scheme 1). The mild reaction conditions allowed the formation of various α-aminoacid amides 3a–r in moderate to high yields. Reaction conditions are compatible with the presence on the oxamic acid structure of electron-rich arenes such as thiophene (3f) or alkoxyarenes as in 3m–o, and electron-poor arenes (3p–r). Substrates having benzylic hydrogens as in 3d–e, 3k or 3l led to high isolated yields, suggesting that competitive 1,5-HAT from a putative aminyl radical does not operate. Ortho-substituted aryloxamic acids were converted into amides 3k and 3o in satisfying yields. Oxamic acids issued from α-aminoacid 4b led to the desired product (3g) in moderate yield as a mixture of two diastereomers in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. Surprisingly, secondary oxamic acid provided the desired product 3h albeit in modest yield, while the same precursor was shown to fail to deliver the desired isocyanate under similar conditions.11 N-Acetylphenylhydrazone, known for its high reactivity toward C-centered radicals also failed to react under our conditions (ESI†).14 The scope of the methodology was further extended varying the nature of all partners as summarized in Scheme 2.
:1 ratio. Surprisingly, secondary oxamic acid provided the desired product 3h albeit in modest yield, while the same precursor was shown to fail to deliver the desired isocyanate under similar conditions.11 N-Acetylphenylhydrazone, known for its high reactivity toward C-centered radicals also failed to react under our conditions (ESI†).14 The scope of the methodology was further extended varying the nature of all partners as summarized in Scheme 2.
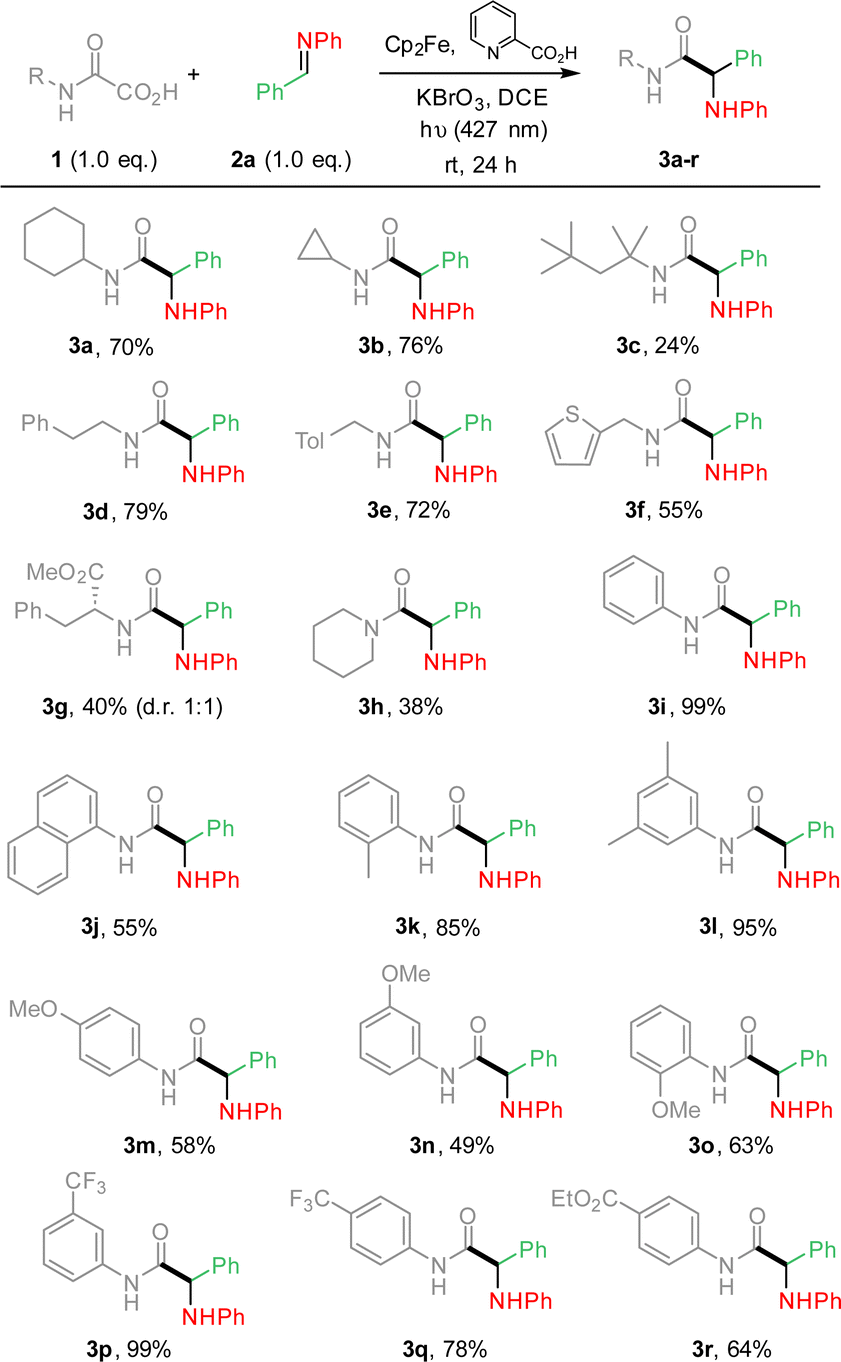 | ||
| Scheme 1 Photocatalyzed Cp2Fe-mediated addition of oxamic acids onto imines. Oxamic acid scope. | ||
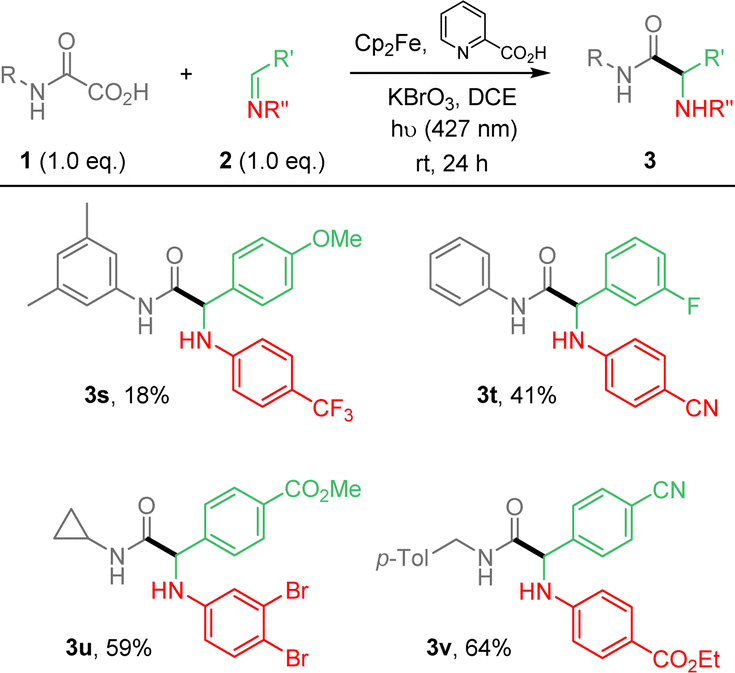 | ||
| Scheme 2 Photocatalyzed Cp2Fe-mediated addition of oxamic acids onto imines. 2-component strategy. | ||
We then decided to implement the methodology by developing a more practical 3-component approach to extend the scope of application and potentially enable future automation of this reaction. Optimization of the process was carried out, using oxamic acid 1a, benzaldehyde 4a and aniline 5a to afford α-aminoacid amide 3a (ESI†). The best yield was obtained using a 1.0:1.3:1.3 ratio of 1a/4a/5a (Table S1, ESI†). However, due to purification issues, conditions using 1.0/1.0/1.0 ratio were finally retained and applied to the 3-component process as summarized in Scheme 3. Compounds 3 were thus generally accessible in moderate to good yields. The 3-component approach compares well with the 2-component version in terms of yields (Scheme 2 vs. 3). As above, reaction conditions are compatible with various substituents and functional groups on arene moieties, including free OH, halogens, esters, nitriles and fluorine-containing substituents. 3y having an amino substituent on the aldehyde fragment was observed in 1H NMR but could not be isolated pure. Finally aliphatic aldehydes and aliphatic or benzylamines as well as sulfonylamines (not shown) were tested and did not provide the desired addition products (ESI†).
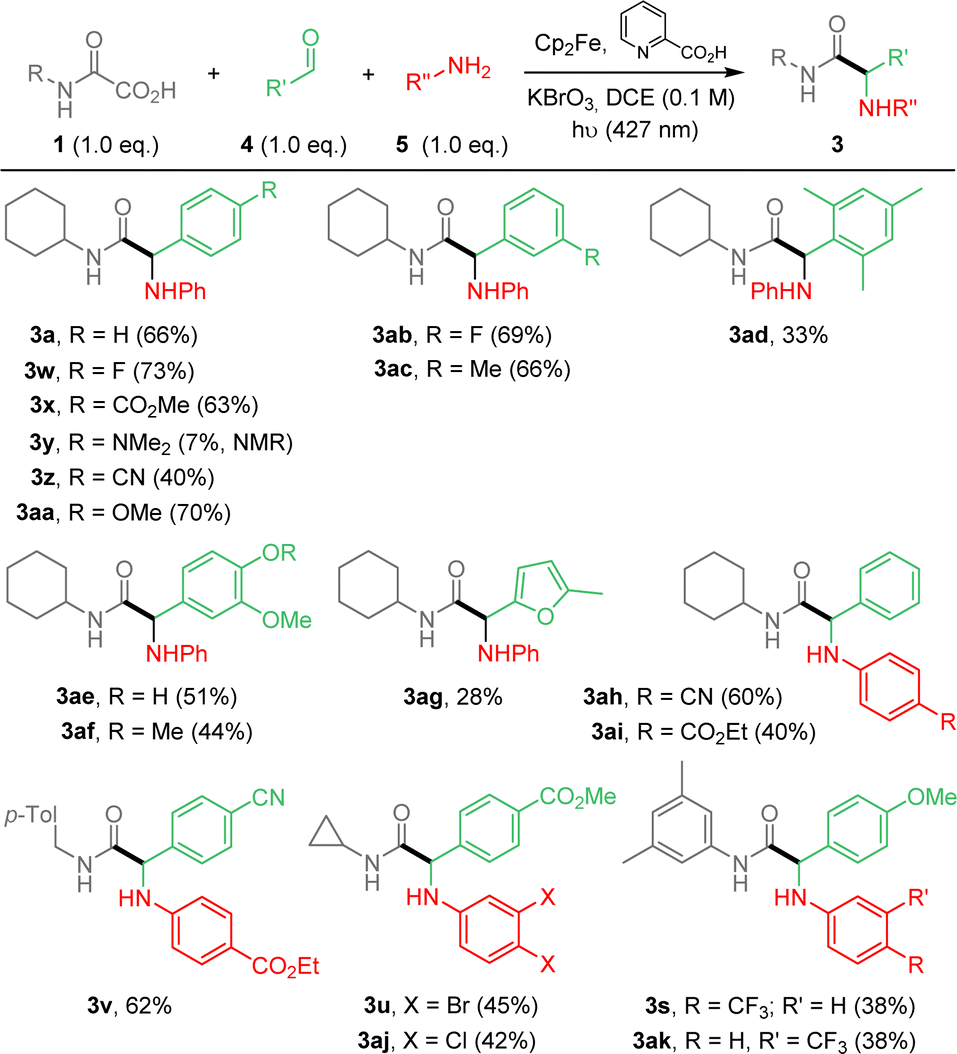 | ||
| Scheme 3 Photocatalyzed Cp2Fe-mediated addition of oxamic acids onto imines. 3-component strategy. | ||
Several control experiments were finally carried out to get mechanistic insights (ESI†). For instance, reaction between 1a and 2a under standard conditions, but in the presence of TEMPO, did not provide 3a, suggesting that the process follows a radical pathway. Finally, addition of 1,1-diphenylethylene to 1a and 2a under conditions above, led to a mixture in which the product resulting from the carbamoyl addition onto the olefin was isolated in 33% yield, indicating that a carbamoyl radical was generated during the process. On the basis of the above results and previous reports,11,12b a tentative mechanism is finally proposed in Fig. 2. Cp2Fe and 2-picolinic acid likely provides mixed Cp2Fe–picolinate complexes, which upon oxidation with KBrO3 generate the catalytically active species, i.e. Fe(III)Ln.15 The latter then combines with oxamic acid 1 to afford the iron–carboxylate I, which suffers a photoactivated Ligand to Metal Charge Transfer (LMCT) leading to carboxyl radical II and Fe(II)Ln.16–18 Decarboxylation of II then forms the carbamoyl radical III, which can add to the imine IV, activated by a Brønsted (TFA, picolinic acid, oxamic acid) or a Lewis acid (BF3 or Fe(III)Ln) to provide the cation-radical V. The latter may finally be reduced by Fe(II)Ln (path a), to give upon protonation the final α-aminoacid amide 3, regenerating Fe(III)Ln.19 Oxidation potentials of +0.44 V (vs. SCE)20 and in the range +0.6 to +0.9 V (vs. SCE)21 in CH3CN respectively for Cp2Fe/Cp2Fe+ and ArNH2+˙/ArNH2 indicate that V may effectively be reduced to provide 3 and regenerate Fe(III)Ln. However the very close potential values may also explain the recourse to an external source of oxidant to maintain the catalytic cycle. It is worth mentioning that additional experiments using Cp2Fe+ as a catalyst for the reaction between 1a and 2a, in the absence of KBrO3, led to 3a in 52% yield, further supporting the catalytic cycle proposed in Fig. 2. In the absence of a Brønsted acid, Fe(III)Ln and Fe(II)Ln species likely play a dual role in the catalytic cycle, the former as an oxidant of the oxamate I and both as Lewis acids, to activate the imine partner,14 which does not react in the absence of acid activation (vide supra).12 The need for 0.5 eq. of KBrO3 to reoxidize Fe(II)Ln into Fe(III)Ln may also be required as to maintain sufficient Fe(III) in the catalytic cycle (path b), as Fe(III) is known to bind strongly to nitrogen,14 preventing the restoration of the catalytic cycle. Further studies are ongoing in our laboratory to clarify this point.
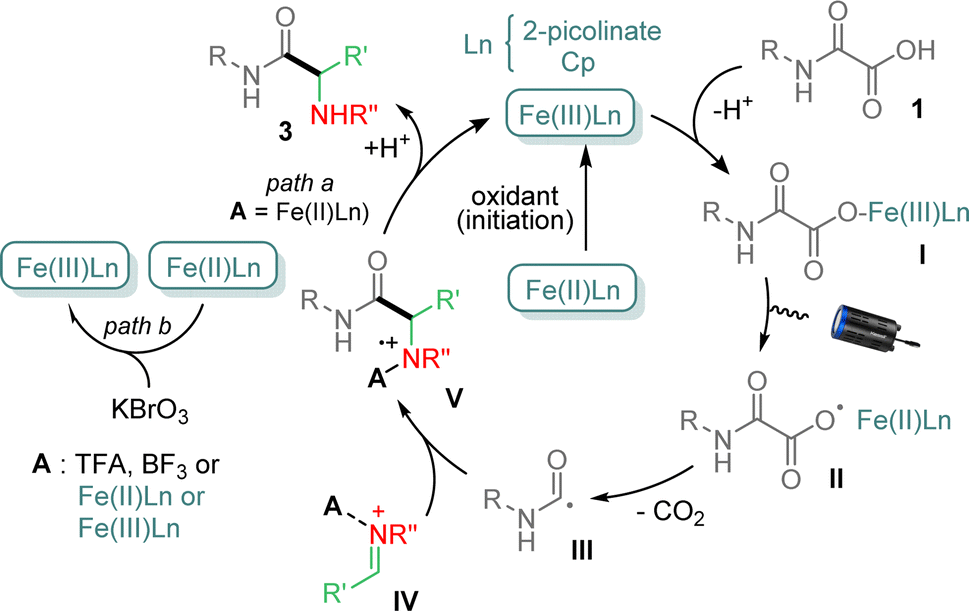 | ||
| Fig. 2 Mechanism of the photocatalyzed Cp2Fe-mediated addition of oxamic acids onto imines. | ||
In summary, we reported a straightforward photoactivated ferrocene-mediated addition of oxamic acids onto imines, which provides a broad range of α-aminoacid amides in good yields. The process may be carried out on pre-formed imines or by simply mixing aldehydes, amines and oxamic acids, in the presence of the iron-complex catalyst. The reaction proceeds through the formation of a nucleophilic carbamoyl radical species generated through a LMCT from an oxamate–Fe(III) intermediate. Iron complexes are believed to play a dual role, both as oxidant of the oxamic acid and as Lewis acid to activate the imine partner. The methodology, which uses readily available starting materials, catalyst and oxidant proceeds under mild conditions and should thus find useful for synthetic applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the ANR (NCO-INNOV, No. 20-CE07-0015-01), the University of Bordeaux and the CNRS for financial support. We thank the analytical facilities CESAMO for NMR, and mass spectrometry studies.Notes and references
- (a) L. Friedman and H. Shechter, Tetrahedron Lett., 1961, 2, 238 CrossRef; (b) D. Elad and J. Rokach, J. Org. Chem., 1964, 29, 1855 CrossRef CAS; (c) W.-P. Mai, G.-C. Sun, J.-T. Wang, G. Song, P. Mao, L.-R. Yang, J.-W. Yuan, Y.-M. Xiao and L.-B. Qu, J. Org. Chem., 2014, 79, 8094 CrossRef CAS PubMed; (d) Q. Jiang, J. Jia, B. Xu, A. Zhao and C.-C. Guo, J. Org. Chem., 2015, 80, 3586 CrossRef CAS PubMed; (e) S. Fujiwara, Y. Shimizu, T. Shin-ike and N. Kambe, Org. Lett., 2001, 3, 2085 CrossRef CAS PubMed; (f) H. Wang, L.-N. Guo, S. Wang and X.-H. Duan, Org. Lett., 2001, 17, 3054 CrossRef PubMed; (g) S. B. Herzon and A. G. Myers, J. Am. Chem. Soc., 2005, 127, 5342 CrossRef CAS PubMed; (h) A. F. Bella, L. V. Jackson and J. C. Walton, Org. Biomol. Chem., 2004, 2, 421 RSC; (i) R. S. Grainger, M. Betou, L. Male, M. B. Pitak and S. J. Coles, Org. Lett., 2012, 14, 2234 CrossRef CAS PubMed; (j) R. S. Grainger and E. J. Welsh, Angew. Chem., Int. Ed., 2007, 46, 5377 CrossRef CAS PubMed; (k) M. Betou, L. Male, J. W. Steed and R. S. Grainger, Chem.–Eur. J., 2014, 20, 6505 CrossRef CAS PubMed; (l) A. Millan-Ortiz, G. Lopez-Valdez, F. Cortez-Guzman and L. D. Miranda, Chem. Commun., 2015, 51, 8345 RSC; (m) D. M. Kitcatt, K. A. Scott, E. Rongione, S. Nicolle and A.-L. Lee, Chem. Sci., 2023, 14, 9806 RSC.
- J.-J. Wu, Y. Li, H.-Y. Zhou, A.-H. Wen, C.-C. Lun, S.-Y. Yao, Z. Ke and B.-H. Ye, ACS Catal., 2016, 6, 1263 CrossRef CAS.
- (a) M. Yuan, L. Chen, J. Wang, S. Chen, K. Wang, Y. Xue, G. Yao, Z. Luo and Y. Zhang, Org. Lett., 2015, 17, 346 CrossRef CAS PubMed; (b) W. F. Petersen, R. J. K. Taylor and J. R. Donald, Org. Biomol. Chem., 2017, 15, 5831 RSC; (c) S. Maiti, S. Roy, P. Ghosh, A. Kasera and D. Maiti, Angew. Chem., Int. Ed., 2022, 61, e202207472 CrossRef CAS PubMed; (d) V. Hutskalova, F. Bou Hamdan and C. Sparr, Org. Lett., 2024, 26, 2768 CrossRef CAS PubMed; (e) D. Duan and L. Song, Org. Chem. Front., 2024, 11, 47 RSC.
- (a) F. Minisci, A. Citterio, E. Vismara and C. Giordano, Tetrahedron, 1985, 41, 4157 CrossRef CAS; (b) A. Hussain Jatoi, G. G. Pawar, F. Robert and Y. Landais, Chem. Commun., 2019, 55, 466 RSC; (c) D. T. Mooney, P. R. Moore and A.-L. Lee, Org. Lett., 2022, 24, 8008 CrossRef CAS PubMed.
- (a) C. Chatgilialoglu, D. Crich, M. Komatsu and I. Ryu, Chem. Rev., 1999, 99, 1991 CrossRef CAS PubMed; (b) M. Ogbu, G. Kurtay, F. Robert and Y. Landais, Chem. Commun., 2022, 58, 7593 RSC; (c) B. T. Matsuo, P. H. R. Oliveira, E. F. Pissinati, K. B. Vega, I. S. de Jesus, J. T. M. Correia and M. Paixao, Chem. Commun., 2022, 58, 8322 RSC and references therein..
- (a) V. G. Correia, J. C. Abreu, C. A. E. Barata and L. H. Andrade, Org. Lett., 2017, 19, 1060 CrossRef CAS PubMed; (b) M. N. Sanabria, M. M. Hornink, V. G. Correia and L. H. Andrade, Org. Process Res. Dev., 2020, 24, 2288 CrossRef CAS.
- (a) M. C. Quattrini, S. Fujii, K. Yamada, T. Fukuyama, D. Ravelli, M. Fagnoni and I. Ryu, Chem. Commun., 2017, 53, 2335 RSC; (b) I. Kim, G. Kang, K. Lee, B. Park, D. Kang, H. Jung, Y.-T. He, M.-H. Baik and S. Hong, J. Am. Chem. Soc., 2019, 141, 9239 CrossRef CAS PubMed.
- E. P. Beato, D. Mazzarella, M. Balletti and P. Melchiorre, Chem. Sci., 2020, 11, 6312 RSC.
- (a) N. Alandini, L. Buzzetti, G. Favi, T. Schulte, L. Candish, K. Collins and P. Melchiorre, Angew. Chem., Int. Ed., 2020, 59, 5248 CrossRef CAS PubMed; (b) L. Cardinale, M. O. Konev and A. Jacobi von Wangelin, Chem.–Eur. J., 2020, 26, 8239 CrossRef CAS PubMed.
- (a) G. G. Pawar, F. Robert, E. Grau, H. Cramail and Y. Landais, Chem. Commun., 2018, 54, 9337 RSC; (b) M. Ogbu, D. M. Bassani, F. Robert and Y. Landais, Chem. Commun., 2022, 58, 8802 RSC; (c) I. M. Ogbu, J. Lusseau, G. Kurtay, F. Robert and Y. Landais, Chem. Commun., 2020, 56, 12226 RSC.
- G. Kurtay, J. Lusseau, F. Robert and Y. Landais, Synlett, 2024, 35, 342 CrossRef CAS.
- (a) N. Pastori, C. Greco, A. Clerici, C. Punta and O. Porta, Org. Lett., 2010, 12, 3898 CrossRef CAS PubMed; (b) L. Cardinale, M.-O. W. S. Schmotz, M. O. Konev and A. Jacobi von Wangelin, Org. Lett., 2022, 24, 506 CrossRef CAS PubMed and references cited therein..
- (a) J. Chen and R. F. Cunico, Tetrahedron Lett., 2003, 44, 8025 CrossRef CAS; (b) J. T. Reeves, Z. Tan, M. A. Herbage, Z. S. Han, M. A. Marsini, Z. Li, G. Li, Y. Xu, K. R. Fandrick, N. C. Gonnella, S. Campbell, S. Ma, N. Grinberg, H. Lee, B. Z. Lu and C. H. Senanayake, J. Am. Chem. Soc., 2013, 135, 5565 CrossRef CAS PubMed.
- R. Tang, Z. Shao, J. Wang, Z. Liu, Y.-M. Li and Y. Shen, J. Org. Chem., 2019, 84, 8177 CrossRef CAS PubMed , and references cited therein..
- (a) G. Balavoine, D. H. R. Barton, I. Boivin and A. Gref, Tetrahedron Lett., 1990, 31, 659 CrossRef CAS; (b) S. Tanaka, Y. Kon, A. Ogawa, Y. Uesaka, M. Tamura and K. Sato, ChemCatChem, 2016, 8, 2930 CrossRef CAS.
- (a) A. Sugimori and T. Yamada, Bull. Chem. Soc. Jpn., 1986, 59, 3911 CrossRef CAS; (b) Z. Li, X. Wang, S. Xia and J. Jin, Org. Lett., 2019, 21, 4259 CrossRef CAS; (c) G. Feng, X. Wang and J. Jin, Eur. J. Org Chem., 2019, 6728 CrossRef CAS; (d) Y.-C. Lu and J. G. West, Angew. Chem., Int. Ed., 2023, 62, e2022130 Search PubMed; (e) G. A. Lutovsky, S. N. Gockel, M. W. Bundesmann, S. W. Bagley and T. P. Yoon, Chem, 2023, 9, 1610 CrossRef CAS PubMed; (f) S.-C. Kao, K.-J. Bian, X.-W. Chen, Y. Chen, A. A. Marti and J. G. West, Chem Catal., 2023, 3, 100603 CrossRef CAS PubMed; (g) M. Ding, S. Zhou, S. Yao, C. Zhu, W. Li and J. Xie, Chin. J. Chem., 2024, 42, 351 CrossRef CAS; (h) S. Wang, T. Li, C. Gu, J. Han, C.-G. Zhao, C. Zhu, H. Tan and J. Xie, Nat. Commun., 2022, 13, 2432 CrossRef CAS PubMed.
- (a) J. K. Kochi, J. Am. Chem. Soc., 1962, 84, 2121 CrossRef CAS; (b) M. Treacy and T. Rovis, J. Am. Chem. Soc., 2021, 143, 2729 CrossRef PubMed; (c) F. Julia, ChemCatChem, 2022, 14, e2022009S CrossRef.
- J. Chen and W. R. Browne, Coord. Chem. Rev., 2018, 374, 15 CrossRef CAS.
- L. Zhu, P. Xiong, Z.-Y. Mao, Y.-H. Wang, X. Yan, X. Lu and H.-C. Xu, Angew. Chem., Int. Ed., 2016, 55, 2226 CrossRef CAS PubMed.
- (a) D. Bao, B. Millare, W. Xia, B. G. Steyer, A. A. Gerasimenko, A. Ferreira, A. Contreras and V. I. Vullev, J. Phys. Chem. A, 2009, 113, 1259 CrossRef CAS PubMed; (b) V. V. Pavlishchuk and A. W. Addison, Inorg. Chim. Acta, 2000, 298, 97 CrossRef CAS.
- A. K. V. Mruthunjaya and A. A. J. Torriero, Molecules, 2023, 28, 471 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra02258k |
| This journal is © The Royal Society of Chemistry 2024 |