
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceKeto–enol equilibrium: stable tautomers of ortho-, meta-, and para-hydroquinones in large aromatics†
Vincent Silber ,
Christophe Gourlaouen* and
Romain Ruppert*
,
Christophe Gourlaouen* and
Romain Ruppert*
Institut de Chimie, UMR CNRS 7177, Université de Strasbourg, 4 Rue Blaise Pascal, 67000 Strasbourg, France. E-mail: rruppert@unistra.fr; gourlaouen@unistra.fr
First published on 15th April 2024
Abstract
The synthesis of [6]helicene para-quinone starting from the 1,4-dimethoxy-[6]helicene derivative is presented. The demethylation reaction with boron tribromide led to unexpected results. Instead of the expected para-hydroquinone, the diketone tautomeric form was isolated. In contrast to the 1,4-hydroquinone and 1,4-dihydroxynaphthalene, the stable tautomers for the [4] and [6]helicenes were the aromatic diketones. These experimental results were corroborated by calculations. Additional calculations showed that these tautomeric species were indeed the stable forms of 1,4 and 1,3-hydroquinones when present in larger aromatics, in drastic contrast with 1,2-dihydroxy-aromatics (known as catechol).
Introduction
[n]Helicenes are well-known molecules with the smallest one being first synthesised at the beginning of the twentieth century,1 and the seminal articles of Newman gave a new impulse to the field.2 The helicenes were prepared by various synthetic methodologies, with photochemical dehydrogenative cyclisations of stilbene derivatives or metal-catalysed cyclisations being widely used reactions. Very long helicenes and sophisticated structural modifications of helicenes have been described in the last twenty years.3 More recently, with the aim of varying the redox or photochemical properties of these molecules, various functionalities were introduced. In particular, metal-based organometallic derivatives were prepared and studied for their circularly polarised luminescence properties.4 Other redox-active compounds containing pyridinium units have also been recently described.5 The quinone–hydroquinone redox system is widely studied because it is present in many biological redox mediators or cellular cofactors. The naphthoquinones or anthraquinones are generally functionalized and in some case the unexpected 1,4-diketo tautomeric form of the corresponding reduced 1,4-hydronaphthoquinones or 1,4-hydroanthraquinones were isolated and characterised.6 This unexpected tautomeric 1,4-diketo form was also found during oxidative coupling of 2-hydroxyanthracene to form BINOL derivatives by the group of Shinokubo.7 Generally BINOL derivatives do not exhibit keto–enol tautomerism due to instability of the diketones. However, steric hindrance and a large conversion barrier was invoked to explain the stability of the diketo tautomer. More than twenty years ago, many helicenic quinone compounds have been prepared by the group of T. J. Katz.8 In these seminal contributions, several large π-conjugated helicene-1,4-quinones were obtained and some of them aggregated to form even larger helical entities.9 These helicenes were generally obtained by Diels–Alder cyclisation in good yields.10 These quinones were initially prepared to study, after monoelectronic reduction, the electronic delocalisation through the fused aromatics of the helicene. In several reports, the generation of the 1,4-hydroquinone moiety was proposed as intermediate, but not fully characterised. In addition, notably during the alkylation of these hydroquinones with alcohols in acidic media, the presence of the dione tautomers as intermediates was suggested. Later, the group of Diederich described the synthesis and study of functionalised [6]helicenes containing an ortho-quinone moiety at one extremity of the π-conjugated molecules (see Scheme 1).11 These compounds were prepared to check their optical switching properties upon reduction of the quinones to their radical-anions, or complexation of the catechol units by metal ions. The last steps during the synthesis of these ortho-quinone helicenes consisted of demethylation of the 1,2-dimethoxy derivative to the 1,2-dihydroxy derivative (the catechol) and oxidation to the ortho-quinone. In these reports, the 1,2-hydroquinones were fully characterised by 1H and 13C NMR as the dihydroxy tautomers.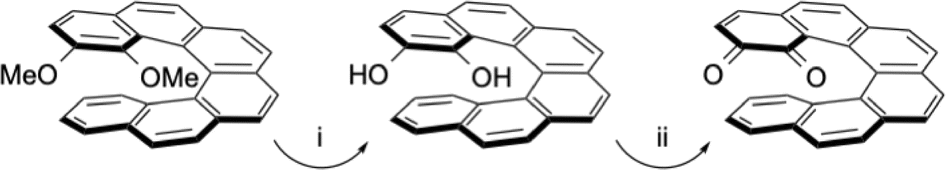 | ||
| Scheme 1 Synthetic route to the ortho-quinone described by the group of F. Diederich: (i) BBr3, CH2Cl2 (ii) AgO, CH3CN. | ||
Results and discussion
During the course of our work on helicene–porphyrin conjugates, we prepared helicenic compounds containing electron rich methoxy substituents located at one extremity of the conjugated aromatic.12 The desired [6]helicenic starting materials were prepared by usual synthetic procedures described for these types of compounds.13 The presence of the two methoxy substituents led to a small decrease of the photochemical cyclisation yield in the last step (52% instead of 76% for unsubstituted aromatics in general). The synthesis of 1 was reported earlier,12 but is shown again here, because the lithiation of 1 led also to 2 as side-product after reaction with electrophiles (Scheme 2).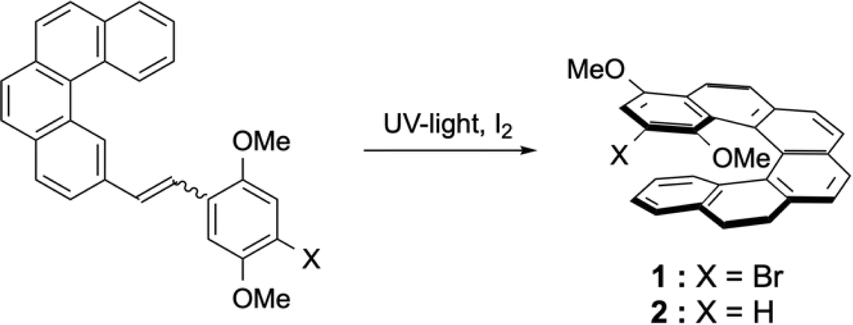 | ||
| Scheme 2 Syntheses of [6]helicenes 1 and 2. | ||
We were not able to obtain single crystals of helicene 2, but could grow single crystals of 1 suitable for X-ray analysis. The structure was solved (see Fig. 1) and the exact positions of the two methoxy groups was therefore unambiguously confirmed. Compound 1 crystallised in the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and both enantiomers were present in the unit cell. All molecules were “isolated” and no particular π–π stacking was observed in the structure.
and both enantiomers were present in the unit cell. All molecules were “isolated” and no particular π–π stacking was observed in the structure.
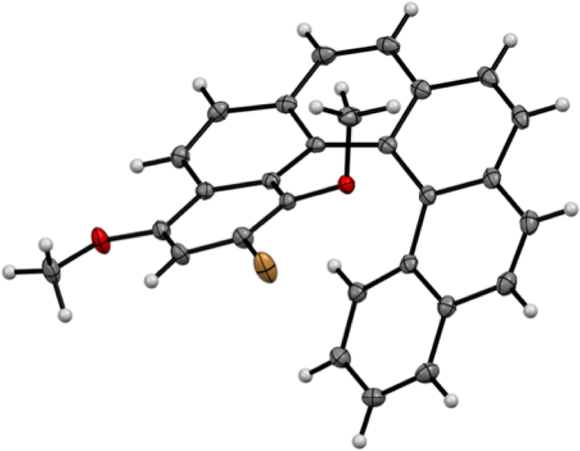 | ||
| Fig. 1 X-ray structure of the starting bromo-dimethoxy-[6]helicene 1, only one enantiomer shown (CCDC no. 2092869†). | ||
The two enantiomers of helicene 1 were separated by preparative chiral HPLC and their properties determined. The ECD spectra of the enantiomers are typical of [6]helicenes (see Fig. 2) and the measured [α]25λ were also similar to reported values (±2850, 3050, 3800, and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 at 589, 578, 546, and 436 nm respectively, in dichloromethane, c = 0.21).
000 at 589, 578, 546, and 436 nm respectively, in dichloromethane, c = 0.21).
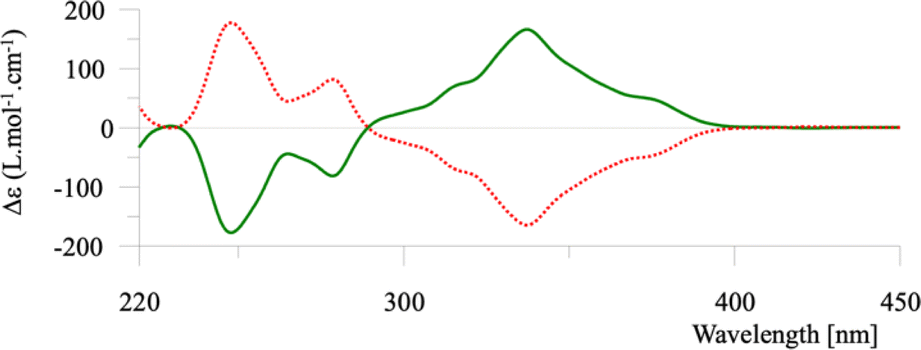 | ||
| Fig. 2 ECD spectra of the two enantiomers of compound 1. | ||
The bromo-dimethoxy derivative 1 of the [6]helicene was the starting compound for further functionalization. After bromine–lithium exchange, addition of the electrophiles always afforded the desired products, together with variable amounts of debrominated dimethoxy[6]helicene 2 as side-product. Compound 2 could alternatively be prepared by following the same procedure as for the synthesis of 1, but without the bromine substituent in the starting material. Compound 2 was then reacted with BBr3, the classical reagent used to demethylate methoxyaromatics. To our surprise, the expected para-hydroquinone 3 was never isolated after this reaction. Instead, the isomeric reduced form 4 and the oxidized form of the hydroquinone (the para-quinone 6) were isolated as sole products (see Scheme 3). The mono-ethoxylated product 5 was formed in our initial attempt, because non-distilled dichloromethane (containing less than 1% of ethanol as stabilising agent) was used for the work-up and chromatographic separations. The addition of alcohols in acidic media at room temperature was described earlier for phenolic helicenic compounds (the so-called Russig–Laatsch reaction).14 Using distilled dichloromethane the reaction gave only two products, the reduced form 4 and the quinone 6, and again no hydroquinone tautomer 3. The presence of quinone 6 was due to oxidation of 4 by air during work-up and chromatography.
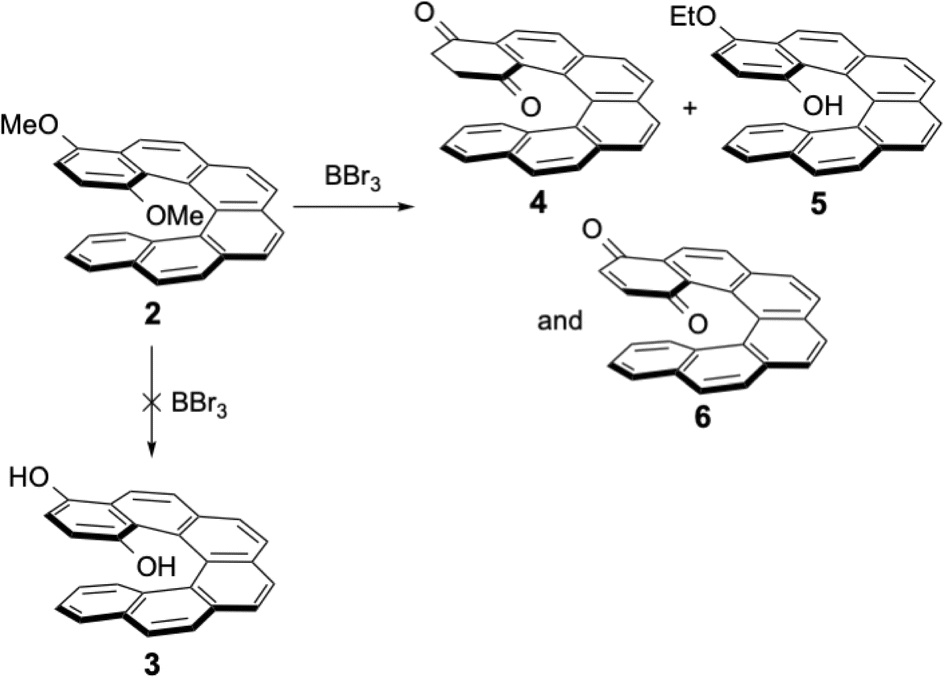 | ||
| Scheme 3 Reaction of the 1,4-dimethoxy-[6]helicene 2 with boron tribromide. | ||
The structures of compounds 4 and 6 could be easily deduced from the 1H and 13C NMR spectra. For para-quinone 6, only aromatic protons were observed, no sp3 carbons were found in the 13C spectrum, and the two 13C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O signals were located at 185.2 and 185.6 ppm (the chemical shift of the 13CO signal of 1,4-benzoquinone is 187 ppm). For the second compound 4, the four CH2 protons next to the two carbonyl functions were non-equivalent because this [6]helicene is chiral, and four well-separated ddd signals were observed between 3 and 1.7 ppm in the 1H NMR spectrum (see Fig. 3, insert). In the 13C NMR spectrum, two CH2 carbons (DEPT 135) were found at 35.8 and 38.3 ppm and the two 13CO signals were found at 195.9 and 196.7 ppm. These are values characteristic of ketones (acetophenone 13CO signal at 195.7 ppm, or tetralone 13CO signal at 197.8 ppm) and very different from the measured values for the two 13CO carbons of quinone 6.
O signals were located at 185.2 and 185.6 ppm (the chemical shift of the 13CO signal of 1,4-benzoquinone is 187 ppm). For the second compound 4, the four CH2 protons next to the two carbonyl functions were non-equivalent because this [6]helicene is chiral, and four well-separated ddd signals were observed between 3 and 1.7 ppm in the 1H NMR spectrum (see Fig. 3, insert). In the 13C NMR spectrum, two CH2 carbons (DEPT 135) were found at 35.8 and 38.3 ppm and the two 13CO signals were found at 195.9 and 196.7 ppm. These are values characteristic of ketones (acetophenone 13CO signal at 195.7 ppm, or tetralone 13CO signal at 197.8 ppm) and very different from the measured values for the two 13CO carbons of quinone 6.
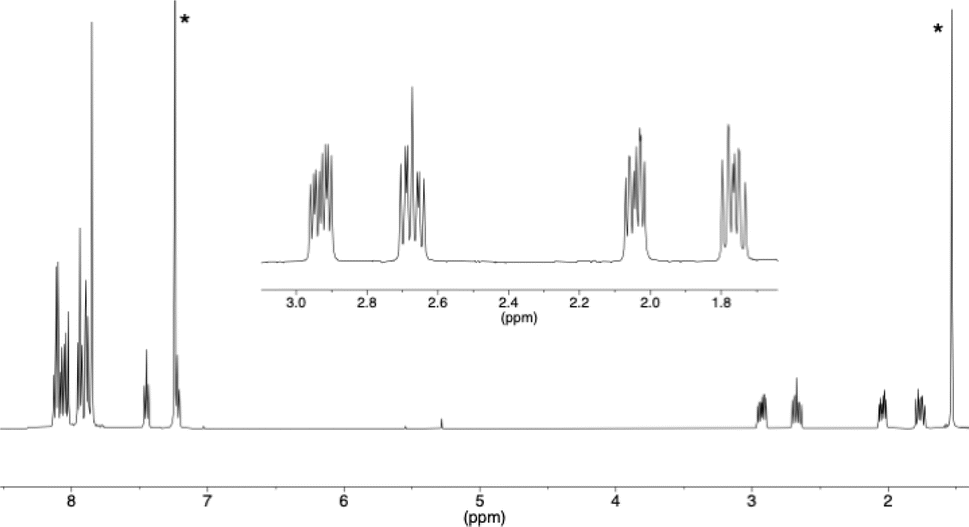 | ||
| Fig. 3 1H NMR spectrum of compound 4 with an expansion of the 1.7 to 3 ppm area (* residual CHCl3 and H2O). | ||
Wondering if the stability of this tautomer was specific to these large [6]helicenes, we started to check the products obtained after the demethylation reaction of other 1,4-dimethoxy aromatics, notably smaller fused aromatics. Under the same experimental conditions, the 1,4-dimethoxynaphthalene 7 was treated with boron tribromide and as expected the 1,4-dihydroxynaphthalene 8 was isolated together with the 1,4-naphthoquinone 10 (see Scheme 4). The diketone 9 was known because the 1,4-dihydroxynaphthalene 8 and the diketone 9 are two tautomers which are in equilibrium. However, at room temperature and under neutral conditions, the phenolic form is the stable form and the diketone 9 is not present.
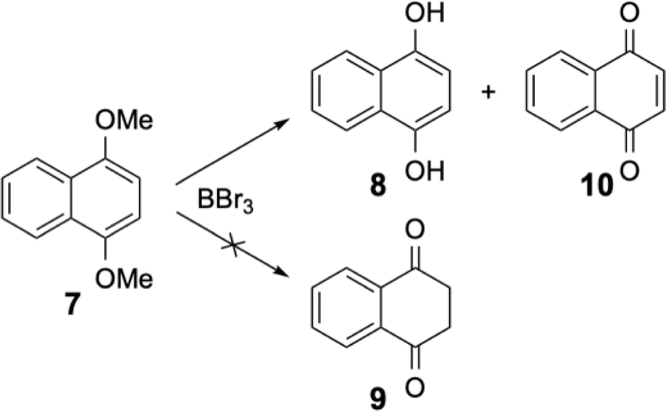 | ||
| Scheme 4 Demethylation of 1,4-dimethoxynaphthalene 7 with boron tribromide. | ||
The diketone 9 can be isolated from very acidic media15 or by melting the solid at high temperature.16 Both conditions (acidic medium or elevated temperatures) were very different from the mild experimental procedures used for our demethylation and the slightly basic treatment to neutralise boron tribromide should highly favor the phenolic form. This was true for the naphthalene derivative, but the opposite was found for the [6]helicenic aromatic for which only the diketone form 4 was isolated. Helicenic quinones were often described in the literature and when the authors had to formulate the reduced forms (as an intermediate in various reactions), they always described them as the phenolic form.14 Puzzled by our observations, we also checked the dimethoxy derivative 11 of a compound containing four fused aromatic rings. Again, under the same experimental conditions, only two products were isolated from this reaction, i.e. the diketone 12 and the oxidised form, quinone 14 (see Scheme 5). It should be emphasised that compound 12 was quite rapidly oxidised to quinone 14 in solution. Consequently, the NMR spectra of 12 were always polluted by small amounts of quinone 14 (see ESI†).
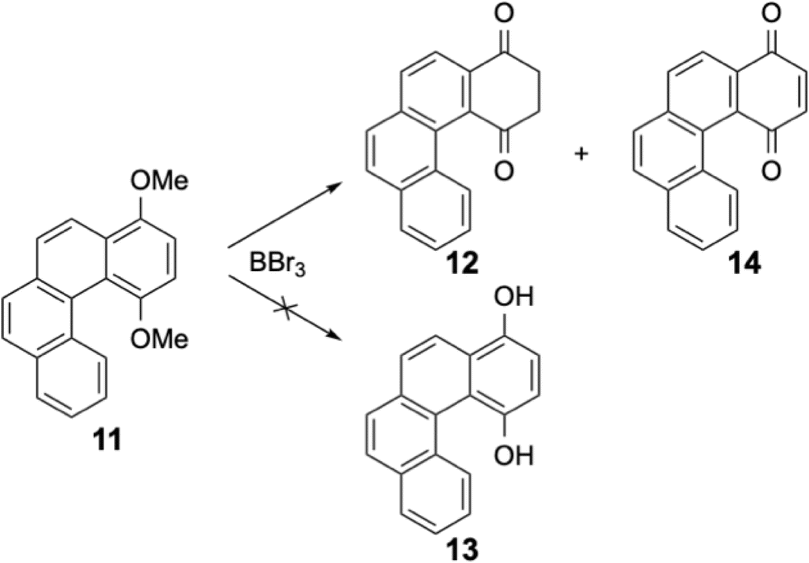 | ||
| Scheme 5 Demethylation of compound 11 (1,4-dimethoxy-[4]helicene). | ||
The 1H NMR spectrum of the compound 12 confirmed the diketone structure because two CH2 resonances were observed at 3.31 and 3.52 ppm; the molecule racemizes too rapidly and the two protons of each CH2 group were now equivalent. The 13CO signals were found at 195.1 and 202.1 ppm for diketone 12 and at 185.3 and 188.5 ppm for quinone 14.
Clearly, the tautomeric equilibrium varied with the length of the aromatic 1,4-hydroquinones. The phenolic form was present for small aromatics whereas in the case of larger fused aromatics like these helicenes, the diketone was the major (and sole) tautomer found for the reduced forms of the para-quinones. These experimental findings were somehow counterintuitive for synthetic chemists because a loss of aromaticity is involved. This prompted us to examine the energy differences involved in these tautomeric equilibria. According to DFT calculations, the tautomeric equilibrium is displaced towards the diketone derivative for all compounds presenting a number of fused aromatic rings equal or greater than two (see Table 1).
| Number of fused aromatic cycles [n] | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| ΔG (kcal mol−1) | |||||||
| Dichloromethane | +6.4 | −6.7 | −5.0 | −4.0 | −3.4 | −4.0 | −3.9 |
| Gas phase | +6.6 | −6.3 | −5.0 | −2.2 | −1.7 | −3.2 | −3.6 |
This observation is consistent with experimental results, as shown by Thomson,16 even if harsh conditions were needed to convert the phenol tautomer to the diketone in the case of the naphthohydroquinone. All C–C bond lengths in 1,4-hydroquinone (one aromatic ring) are almost identical. In contrast, the bond lengths in all other 1,4-hydroxy tautomers show alternating values for the bond lengths in the terminal aromatic rings (see ESI†). Furthermore, the diketone tautomers might allow a greater flexibility of the terminal ring, which can then better adapt to the constraints induced by the helical structure (see ESI† for the calculated dihedral angles). Both electronic and geometric effects seem to favor the diketone tautomers for all polyaromatics of this series up to the [6]helicene. From the results of the F. Diederich group, it was clear that 1,2-dihydroxy forms are the stable tautomers for [6]helicenes and this study demonstrated that the 1,4-diones were the stable tautomers for [6]helicenes. Calculations to determine the difference in energy between the tautomers with all possible hydroquinone isomers of [6]helicenes were performed and corroborated these experimental results. Fig. 4 represents the values of ΔG to undergo the transformations from the dihydroxy forms to the dione tautomers (ortho, meta and para structures).
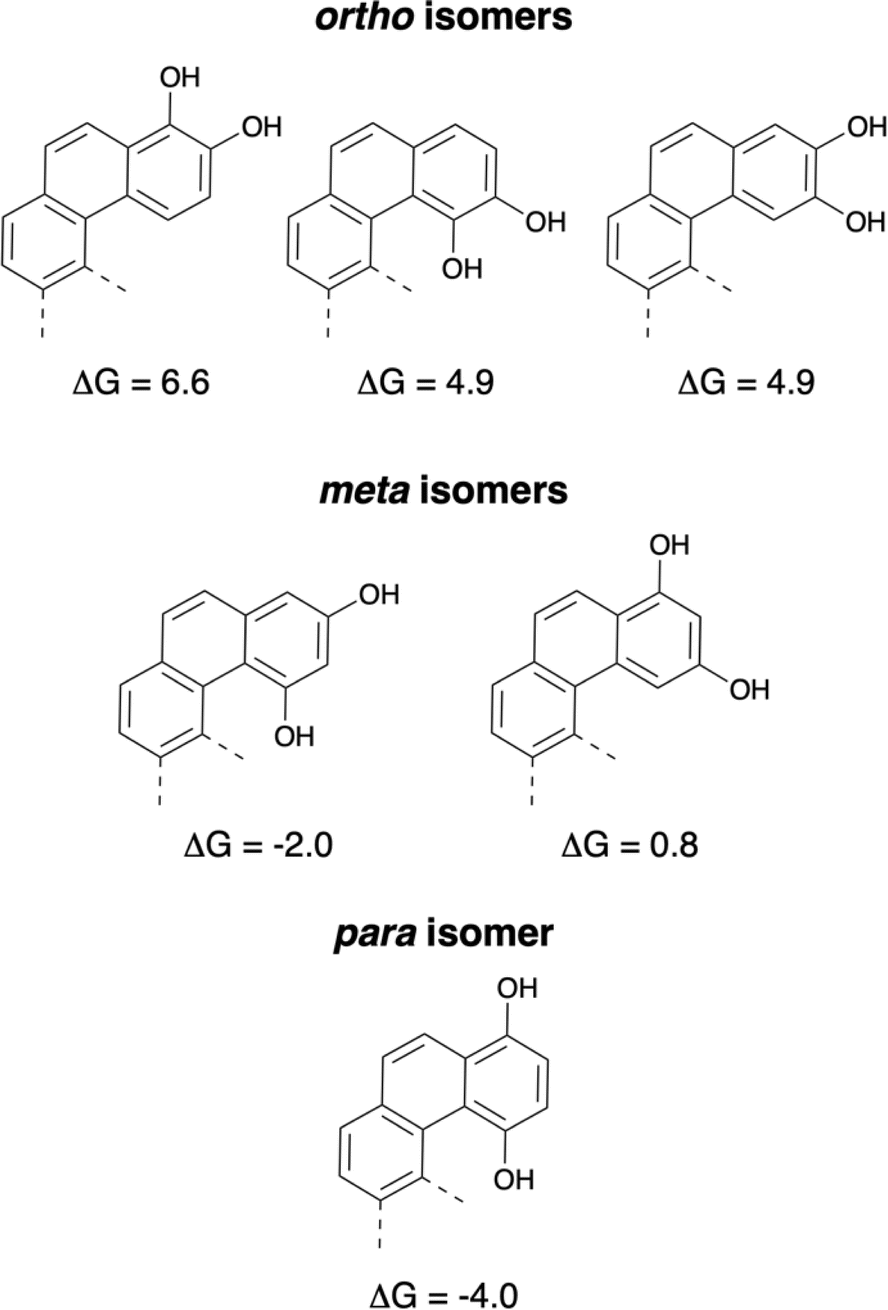 | ||
| Fig. 4 Energy differences to obtain the dione isomers from the dihydroxy tautomers (values in kcal mol−1). | ||
For the three ortho isomers, the stable tautomers are the dihydroxy or catechols derivatives (ΔG = 6.6, 4.9, and 4.9 kcal mol−1). In the case of the para isomer, the stable tautomer is clearly the dione (ΔG = −4 kcal mol−1). These results are in good agreement with the values obtained earlier for the stable tautomers of naphthohydroquinones (see ESI, Fig. S45†). For the meta-naphthohydroquinone, the dione is the stable tautomer (see ESI†), but for the two possible meta isomers of [6]helicenes, the results were different. In the case of the 1,3-meta isomer the dione is the stable tautomer (ΔG = −2 kcal mol−1), whereas for the 2,4-meta isomer the dihydroxy tautomer seems to be favored (ΔG = 0.8 kcal mol−1).
In an attempt to understand the origin of the different stability of the tautomers, Electron Localisation Function (ELF) analysis17 was performed on the reference benzene and naphthalene rings as well as on compounds 8 and 9 (Scheme 4). The aromaticity of naphthalene is smaller than that of benzene18 as can be seen on the ELF function (see ESI, Table S2†): the C–C ELF basin are identical in benzene whereas they are closer to an alternation of single and double bond in naphthalene. The ELF C–C basins in the naphthohydroquinone 8 are very close to that of naphthalene: the binding scheme is essentially the same. On the contrary in the dione 9, the population of the ELF C–C basins of the remaining aromatic ring is very similar to that of benzene. We can then propose an hypothesis for the difference in the diol/dione equilibrium which is a balance between aromaticity, internal H-bond and geometry distortion induced by the presence of the C–C single bond in the dione form. For the para structures 8–9 and 3–4, the equilibrium is displaced towards the dione form as the loss of the aromaticity of the terminal cycle is compensated by the reinforcement of the aromaticity of the other rings and furthermore the carbonyle function are also conjugated with the π-system. On the contrary, for the ortho structures, the equilibrium is displaced towards the diol form because the loss of the internal H-bond existing between the two O–H group is not compensated by the greater aromaticity of the dione form. In the meta structures, only one carbonyle group is conjugated with the π-system and the dione form is less stable than in the para isomer, furthermore the out-of-plane distortion due to the presence of the C–C single bond may further disfavour the dione form. This explains the smaller equilibrium displacement for the meta structures.
Experimental
Materials and methods
Commercially available reagent grade materials such as 2-naphthaldehyde and 2,6-dimethoxy-4-bromobenzaldehyde were used as obtained. The synthesis of 2-bromo-benzo[c]phenanthrene as well as its subsequent functionalisation were carried out following methodology described in the literature12,13 except for the photocyclisation steps which were adapted and are described below. THF was distilled from sodium/benzophenone ketyl. Column chromatography was performed with silica gel from Merck (Kieselgel 60; 63–200 μm or 40–63 μm). 1H NMR spectra were recorded on Bruker Avance 400 (400 MHz) or 500 (500 MHz) spectrometers. Chemical shifts are given in parts per million (ppm) by taking the solvent as a reference δCHCl3 = 7.26 ppm for 1H NMR and δCHCl3 = 77.16 ppm for 13C NMR. The coupling constants (J) are given in Hertz (Hz) and the multiplicity of the signals are expressed as: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet. Mass spectrometry (MS and HRMS) experiments were performed on a Bruker Daltonics microTOF spectrometer (Bruker Daltonik GmbH, Bremen, Germany) by the Service de Spectrométrie de Masse de la Fédération de Chimie “Le Bel” (FR 2010). Irradiation of stilbene derivatives was carried out with a Heraeus TQ 150 mercury vapor lamp in a 400 mL photoreactor. The protons of 1H spectra were assigned according to Martin's proposed nomenclature.1aSynthetic procedures
Compounds 4, 5, and 6
With ethanol stabilised dichloromethane. Under argon, 4 mL of BBr3 (1 M in CH2Cl2) were added to the solid 1,4-dimethoxyhexahelicene (160 mg, 0.41 mmol, 1 eq.) and the resulting solution was stirred at room temperature for 1 h. The solution was poured in 50 mL of water. The organic layer was washed with a saturated aqueous solution of NH4Cl (2 × 20 mL), dried over MgSO4 and filtered. Column chromatography (SiO2, CH2Cl2(EtOH stabilised)/cyclohexane 40/60) allowed to isolate 4 (m = 68 mg, 0.42 mmol, 43%), 5 (33 mg, 0.09 mmol, 22%) and 6 (51 mg, 0.14 mmol, 34%).With freshly distilled dichloromethane. Under argon, 4 mL of BBr3 (1 M in CH2Cl2) were added to the solid 1,4-dimethoxyhexahelicene (160 mg, 0.41 mmol, 1 eq.) and the resulting solution was stirred at room temperature for 1 h. The solution was poured in 50 mL of water. The organic layer was washed with a saturated aqueous solution of NH4Cl (2 × 20 mL), dried over MgSO4 and filtered. Column chromatography (SiO2, CH2Cl2(distilled)/cyclohexane 40/60) allowed to isolate 5 (77 mg, 0.21 mmol, 51%) and 6 (71 mg, 0.20 mmol, 48%). Compound 4: 1H NMR (CDCl3, 500 MHz, 25 °C): δH (ppm) = 8.14 (d, J = 8.3 Hz, 1H, H5), 8.11 (d, J = 8.3 Hz, 1H, H6), 8.08 (d, J = 8.4 Hz, 1H, H13), 8.05 (d, J = 8.3 Hz, 1H, H7–12), 7.99–7.93 (m, 2H, H7–12 + H16), 7.91 (m, 2H, 2*H7–12), 7.87 (bs, 2H, 2*H7–12), 7.47 (ddd, J = 8.0, 7.0, 1.2 Hz, 1H, H15), 7.29–7.19 (m, 1H, H14), 2.95 (ddd, J = 16.8, 8.0, 4.8 Hz, 1H, H2–3), 2.69 (ddd, J = 16.6, 9.6, 6.7 Hz, 1H, H2–3), 2.06 (ddd, J = 14.8, 6.7, 4.8 Hz, 1H, H2–3), 1.78 (ddd, J = 14.8, 9.6, 8.0 Hz, 1H, H2–3). 13C NMR (126 MHz, CDCl3): δC (ppm) = 196.7, 195.9, 139.2, 135.9, 133.5, 133.0, 133.0, 132.6 (CH), 131.1, 130.3 (CH), 130.1, 129.9, 129.1 (CH), 129.0 (CH), 127.9 (CH), 127.9 (CH), 126.8 (CH), 126.6 (CH), 126.4 (CH), 126.2 (CH), 125.4 (CH), 125.2, 123.5 (CH), 38.3 (CH2), 35.8 (CH2). ESI-TOF-HR-MS (m/z): calcd for ([M + K+]) 399.0782; found 399.0793. Compound 5: 1H NMR (CDCl3, 500 MHz, 25 °C): δH (ppm) = 8.51 (d, J = 8.6 Hz, 1H, H5–12), 8.16–8.05 (m, 3H, 3*H5–12), 8.03 (d, J = 8.1 Hz, 1H, H5–12), 7.96 (bs, 2H, 2*H5–12), 7.94 (d, J = 8.6 Hz, 1H, H5–12), 7.83 (dd, J = 7.9, 1.5 Hz, 1H, H13), 7.21 (ddd, J = 8.0, 7.0, 1.3 Hz, 1H, H14), 7.03 (d, J = 8.6 Hz, 1H, H2–3), 6.71 (d, J = 8.4 Hz, 1H, H16), 6.63 (ddd, J = 8.5, 6.9, 1.5 Hz, 1H, H15), 6.20 (d, J = 8.4 Hz, 1H, H2–3), 4.28 (dq, J = 8.5, 6.9 Hz, 1H, CH2), 4.13 (dq, J = 8.7, 6.9 Hz, 1H, CH2), 3.86 (s, 1H, OH), 1.59 (t, J = 6.9 Hz, 3H, CH3). 13C NMR (126 MHz, CDCl3): δC (ppm) = 148.9, 146.1, 132.5, 132.4, 132.0, 130.6, 128.7 (CH), 128.5, 127.5 (CH), 127.5 (CH), 127.3 (CH), 127.2 (CH), 127.2 (CH), 126.5 (CH), 126.2 (CH), 125.7 (CH), 125.6 (CH), 124.4, 123.9 (CH), 123.4, 123.0, 122.4 (CH), 121.6, 111.8 (CH), 108.7 (CH), 65.1 (CH2), 15.2 (CH3). Compound 6: 1H NMR (CDCl3, 500 MHz, 25 °C): δH (ppm) = 8.22 (d, J = 8.2 Hz, 1H, H5), 8.19 (d, J = 8.2 Hz, 1H, H6), 8.14 (d, J = 8.4 Hz, 1H, H13), 8.04–7.83 (6d + m, 7H, 6*H7–12 + H16), 7.40 (ddd, J = 7.9, 6.8, 1.2 Hz, 1H, H15), 7.14 (ddd, J = 8.4, 6.9, 1.4 Hz, 1H, H14), 6.54 (d, J = 10.1 Hz, 1H, H2), 6.00 (d, J = 10.1 Hz, 1H, H3). 13C NMR (126 MHz, CDCl3): δC (ppm) = 185.6, 185.2, 140.0 (CH), 135.9, 134.9 (CH), 134.4, 133.6, 133.0 (CH), 132.5, 131.3, 130.5 (CH), 130.0, 129.7, 129.4 (CH), 128.9 (CH), 127.9 (CH), 126.8 (CH), 126.6 (CH), 126.4 (CH), 126.4 (CH), 126.3 (CH), 126.2 (CH), 126.1, 125.2, 123.0 (CH). ESI-TOF-HR-MS (m/z): calcd for ([M + H+]) 359.1067; found 359.1063.
Computational details
All calculations were done with GAUSSIAN 09 (version D01) at DFT level of theory (ωB97XD functional). All atoms were described by 6-31+G** basis set. Solvent corrections of dichloromethane were introduced through PCM model. Frequency calculations were done on the basis of optimised geometry within harmonic approximation. Gibbs free energies were extracted from this frequency analysis. The tautomerism equilibrium were computed from the reaction: ΔG = G(diketo) − G(diol). Topological analyses were performed on the optimized structure wavefunction using the TOPMOD package.19Crystallography
A specimen of C28H19BrO2, approximate dimensions 0.150 mm × 0.200 mm × 0.200 mm, was used for the X-ray crystallographic analysis. The X-ray intensity data were measured (λ = 0.71073 Å). The integration of the data using a monoclinic unit cell yielded a total of 91304 reflections to a maximum θ angle of 27.92° (0.76 Å resolution), of which 9892 were independent (average redundancy 9.230, completeness = 99.9%, Rint = 4.68%, Rsig = 2.15%) and 8445 (85.37%) were greater than 2σ(F2). The final cell constants of a = 14.6897(5) Å, b = 14.0457(6) Å, c = 20.6528(7) Å, β = 104.4410(10)°, volume = 4126.6(3) Å3, are based upon the refinement of the XYZ-centroids of reflections above 20 σ(I). The calculated minimum and maximum transmission coefficients (based on crystal size) are 0.6577 and 0.7456. The structure was solved and refined using the Bruker SHELXTL Software Package, using the space group P21/c (14), with Z = 8 for the formula unit, C28H19BrO2. The final anisotropic full-matrix least-squares refinement on F2 with 563 variables converged at R1 = 2.95%, for the observed data and wR2 = 7.64% for all data. The goodness-of-fit was 1.006. The largest peak in the final difference electron density synthesis was 0.749 e− Å−3 and the largest hole was −0.733 e− Å−3 with an RMS deviation of 0.061 e− Å−3. On the basis of the final model, the calculated density was 1.504 g cm−3 and F(000), 1904 e−. CCDC no. 2092869.†
Conclusions
In conclusion, the well-known redox system quinone–hydroquinone led to unexpected results for the parent [4]helicene and [6]helicene “hydroquinones”. The 1,4-hydroquinone (or phenolic form) which was expected and which is indeed present after reduction of benzoquinone or 1,4-naphthoquinone, was not observed after demethylation of the 1,4-dimethoxy derivatives of slightly larger π-conjugated fused aromatics. These experimental results were then corroborated by DFT calculations, which clearly indicated that the diketone tautomers were the more stable forms for larger fused aromatic “hydroquinones”.Conflicts of interest
There are no conflicts to declare.Acknowledgements
VS thanks the French Ministry of Research for his PhD fellowship. We thank the HPC computer center of the University of Strasbourg for the computation facilities. We thank Nathalie Gruber for solving the structure, Marion Jean and Nicolas Vanthuyne (Chiropole, Aix-Marseille University) for chiral HPLC separations.Notes and references
- (a) R. H. Martin, Angew Chem. Int. Ed. Engl., 1974, 13, 649–660 CrossRef; (b) M. Gingras, Chem. Soc. Rev., 2013, 42, 968–1006 RSC; (c) M. Gingras, G. Félix and R. Peresutti, Chem. Soc. Rev., 2013, 42, 1007–1050 RSC; (d) M. Gingras, Chem. Soc. Rev., 2013, 42, 1051–1095 RSC; (e) Y. Shen and C.-F. Chen, Chem. Rev., 2012, 112, 1463–1535 CrossRef CAS PubMed.
- (a) M. S. Newman and D. Lednicer, J. Am. Chem. Soc., 1955, 77, 3420–3421 CrossRef CAS; (b) M. S. Newman and W. B. Lutz, J. Am. Chem. Soc., 1956, 78, 2469–2473 CrossRef CAS.
- Selected examples: (a) R. H. Martin and M. Baes, Tetrahedron, 1975, 31, 2135–2137 CrossRef CAS; (b) J. Roose, S. Achermann, O. Dumele and F. Diederich, Eur. J. Org Chem., 2013, 3223–3231 CrossRef CAS; (c) K. Mori, T. Murase and M. Fujita, Angew. Chem., Int. Ed., 2015, 54, 6847–6851 CrossRef CAS PubMed; (d) H. A. Staab, M. Diehm and C. Krieger, Tetrahedron Lett., 1994, 35, 8357–8360 CrossRef CAS; (e) I. G. Stara, I. Stary, A. Kollarovic, F. Teply, S. Vyskocil and D. Saman, Tetrahedron Lett., 1999, 40, 1993–1996 CrossRef CAS; (f) A. Jancarik, J. Rybacek, K. Cocq, J. V. Chocholousova, J. Vacek, R. Pohl, L. Bednerova, P. Fiedler, I. Cisarova, I. G. Stara and I. Stary, Angew. Chem., Int. Ed., 2013, 52, 9970–9975 CrossRef CAS PubMed.
- (a) N. Saleh, C. Shen and J. Crassous, Chem. Sci., 2014, 5, 3680–3694 RSC; (b) E. S. Gauthier, R. Rodriguez and J. Crassous, Angew. Chem., Int. Ed., 2020, 59, 22840–22856 CrossRef CAS PubMed.
- (a) J. R. Brandt, L. Pospisil, L. Bednerova, R. Correa da Costa, A. J. P. White, T. Mori, F. Teply and M. J. Fuchter, Chem. Commun., 2017, 53, 9059–9062 RSC; (b) L. E. R. Buckley, B. J. Coe, D. Rusanova, S. Sanchez, M. Jirasek, V. D. Joshi, J. Vavra, D. Khobragade, L. Pospisil, S. Ramesova, I. Cisarova, D. Saman, R. Pohl, K. Clays, N. Van Steerteghem, B. S. Brunschwig and F. Teply, Dalton Trans., 2017, 46, 1052–1064 RSC.
- See for example: (a) D. M. Bird, M. Boldt and T. H. Koch, J. Am. Chem. Soc., 1989, 111, 1148–1150 CrossRef CAS; (b) M. A. Schätzle, S. M. Husain, S. Ferlaino and M. Müller, J. Am. Chem. Soc., 2012, 134, 14742–14745 CrossRef PubMed; (c) S. M. Husain, M. A. Schätzle, S. Lüdeke and M. Müller, Angew. Chem., Int. Ed., 2014, 53, 9806–9811 CrossRef CAS PubMed; (d) A. T. Dharmaraja, C. Jain and H. Chakrapani, J. Org. Chem., 2014, 79, 9413–9417 CrossRef CAS PubMed.
- T. Matsuno, Y. Koyama, S. Hiroto, J. Kumar, T. Kawai and H. Shinokubo, Chem. Commun., 2015, 51, 4607–4610 RSC.
- (a) S. D. Dreher, D. Weix and T. J. Katz, J. Org. Chem., 1999, 64, 3671–3678 CrossRef CAS PubMed; (b) C. Nuckolls, T. J. Katz, G. Katz, P. J. Collings and L. Castellanos, J. Am. Chem. Soc., 1999, 121, 79–88 CrossRef CAS; (c) T. Thongpanchang, K. Paruch, T. J. Katz, A. L. Rheingold, K.-C. Lam and L. Liable-Sands, J. Org. Chem., 2000, 65, 1850–1856 CrossRef CAS PubMed.
- (a) L. Vyklicky, S. H. Eichhorn and T. J. Katz, Chem. Mater., 2003, 15, 3594–3601 CrossRef CAS; (b) M. A. Shcherbina, X.-B. Zeng, T. Tadjiev, G. Ungar, S. H. Eichhorn, K. E. S. Phillips and T. J. Katz, Angew. Chem., Int. Ed., 2009, 48, 7837–7840 CrossRef CAS PubMed; (c) C. Nuckolls and T. J. Katz, J. Am. Chem. Soc., 1998, 120, 9541–9544 CrossRef CAS.
- (a) B. Yang, L. Liu, T. J. Katz, C. A. Liberko and L. L. Miller, J. Am. Chem. Soc., 1991, 113, 8993–8994 CrossRef CAS; (b) C. A. Liberko, L. L. Miller, T. J. Katz and L. Liu, J. Am. Chem. Soc., 1993, 115, 2478–2482 CrossRef CAS.
- (a) D. Schweinfurth, M. Zalibera, M. Kathan, C. Shen, M. Mazzolini, N. Trapp, J. Crassous, G. Gescheidt and F. Diederich, J. Am. Chem. Soc., 2014, 136, 13045–13052 CrossRef CAS PubMed; (b) D. Schweinfurth, M. Mazzolini, D. Neshchadin, C. Hoyer, R. Geier, K. Gatterer, N. Trapp, G. Gescheidt and F. Diederich, Chem.–Eur. J., 2016, 22, 7152–7157 CrossRef CAS PubMed.
- (a) V. Silber, N. Gruber, M. Jean, N. Vanthuyne and R. Ruppert, Chem. Commun., 2022, 58, 6012–6015 RSC; (b) V. Silber, M. Jean, N. Vanthuyne, N. Del Rio, P. Matozzo, J. Crassous and R. Ruppert, Org. Biomol. Chem., 2023, 21, 8924–8935 RSC.
- M. Jakubec, T. Beranek, P. Jakubik, J. Sykora, J. Zadny, W. Cirkva and J. Storch, J. Org. Chem., 2018, 83, 3607–3616 CrossRef CAS PubMed.
- (a) S. D. Dreher, K. Paruch and T. J. Katz, J. Org. Chem., 2000, 65, 806–814 CrossRef CAS; (b) S. D. Dreher, T. J. Katz, K. -C Lam and A. L. Rheingold, J. Org. Chem., 2000, 65, 815–822 CrossRef CAS; (c) F. Russig and J. Prakt, Chem, 1900, 62, 30–60 CAS; (d) H. Laatsch, Liebigs Ann. Chem., 1980, 140–157 CrossRef CAS.
- (a) E. P. Kündig, A. Enriquez Garcia, T. Lomberget and G. Bernadinelli, Angew. Chem., Int. Ed., 2006, 45, 98–101 CrossRef PubMed; (b) E. P. Kündig and A. Enriquez Garcia, Beilstein J. Org. Chem., 2008, 4, 37 Search PubMed.
- (a) R. H. Thomson, J. Chem. Soc., 1950, 1737–1742 RSC; (b) D. B. Bruce and R. H. Thomson, J. Chem. Soc., 1952, 2759–2766 RSC; (c) R. H. Thomson, Q. Rev., Chem. Soc., 1956, 10, 27–43 RSC; (d) M. S. Pearson, B. J. Jensky, F. H. Greer, J. P. Hagstrom and N. M. Wells, J. Org. Chem., 1978, 43, 4617–4622 CrossRef CAS.
- (a) A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397–5403 CrossRef CAS; (b) A. Savin, R. Nesper, S. Wengert and T. F. Fässler, Angew Chem. Int. Ed. Engl., 1997, 36, 1808–1832 CrossRef CAS; (c) B. Silvi and A. Savin, Nature, 1994, 371, 683–686 CrossRef CAS; (d) A. Savin, B. Silvi and F. Colonna, Can. J. Chem., 1996, 74, 1088–1096 CrossRef CAS.
- S. W. Slayden and J. F. Liebman, Chem. Rev., 2001, 101, 1541–1566 CrossRef CAS PubMed.
- (a) S. Noury, X. Krokidis, F. FusterandB. Silvi, TopMod Package, 1997, this package is available on the web site of the Laboratoire de Chimie Théorique, Université Pierre et Marie Curie, UMR CNRS 7616, https://www.lct.jussieu.fr Search PubMed; (b) S. Noury, X. Krokidis, F. Fuster and B. Silvi, Comput. Chem., 1999, 23, 597–604 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2092869. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra02202e |
| This journal is © The Royal Society of Chemistry 2024 |