
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLED-induced Ru-photoredox Pd-catalyzed C–H arylation of (6-phenylpyridin-2-yl)pyrimidines and heteroaryl counterparts†
Ioakeim M. Antoniou ,
Natalia Ioannou,
Nikos Panagiotou‡
and
Savvas N. Georgiades*
,
Natalia Ioannou,
Nikos Panagiotou‡
and
Savvas N. Georgiades*
Department of Chemistry, University of Cyprus, 1 Panepistimiou Avenue, Aglandjia, 2109, Nicosia, Cyprus. E-mail: georgiades.savvas@ucy.ac.cy
First published on 16th April 2024
Abstract
N-heterocycles are essential building blocks and scaffolds in medicinal chemistry. A Pd-catalyzed, Ru-photoredox-mediated C–H arylation is applied herein, for converting a series of functionality-inclusive (6-phenylpyridin-2-yl)pyrimidines to single arylated derivatives, using phenyldiazonium tetrafluoroborate as aryl source. This green chemistry-compliant transformation is induced by LED light. The drug-like modular substrates are constructed via combination of Biginelli multi-component condensation and Suzuki C–C cross-coupling, in order to strategically install, adjacent to the Ph-ring intended to undergo C–H arylation, a (6-pyridin-2-yl)pyrimidine that plays the role of a chelating directing moiety for the C–H arylation catalyst. The scope has been demonstrated on a series of 26 substrates, comprising diverse Ph-ring substituents and substitution patterns, as well as with 13 different aryl donors. Substrates in which the Ph-ring (arylation acceptor) was replaced by an electron-rich heteroaryl counterpart (2-/3-thiophene or -benzofuran) have also been examined and found to undergo arylation regioselectively. End-product conformations afford interesting motifs for occupying 3D chemical space, as implied by single-crystal X-ray diffraction, which has allowed the elucidation of six structures of aryl derivatives and one of an unprecedented pyrimidine-pyridine-benzofuran carbopalladated complex, believed to be a C–H activation derivative.
Introduction
C–H activation approaches are gaining ground in recent years as innovative enabling technologies for the controlled functionalization of substrates considered to be either unreactive or unselective towards conventional organic chemistry transformations.1 By means of C–H bond activation, useful chemical moieties can be introduced in a single stage, thus by-passing the need for multi-step, laborious and costly synthetic processes. Transformations of this type, often developed within the context of sustainability, have already yielded diverse derivatives of bioactive and medicinal compounds, fine chemicals and various classes of added-value materials.2The inclusion of an efficient directing moiety in the substrate structure is an essential strategy for controlling site-selectivity and enhancing conversion in a C–H activation reaction. This may, in fact, even enable accessing products that are not attainable by alternative methods, by means of overriding intrinsic substrate reactivity.3 Nitrogen heterocycles are entities often selected as directing moieties,4 owing both to their Lewis basicity and ability to coordinate to metal catalysts, as well as other properties (e.g., pharmacological) that they bring to end-products, should they be retained in the final structure.
Pyridine is a frequently employed directing group, especially in cases of catalytic C–H arylation on adjacent phenyl rings (Scheme 1A), with a significant body of research on this topic having emerged over the last few years. Metal complex catalysts for C–H arylation of phenylpyridines are predominantly based on ruthenium,5 rhodium6 or palladium,7 and to a lesser extent iron,8 cobalt9 or nickel.10 Aryl donors have been diverse, including aryl halides,5o,p,6i benzoyl halides,6g aryl acylperoxides,7i arylboronic acids,7k Grignard reagents,8b triarylindium reagents,6a arylstannanes,6h arylsilanes,5e aryltrifluoroborate salts,7j diaryliodonium salts7f or alternative hypervalent iodine reagents7b and phenyldiazonium salts.7h Some of the described methods are also compatible with the use of (the less basic) pyrimidine as directing moiety in structurally related 2-/4-phenylpyrimidine substrates (Scheme 1B).11,6a,d,i,7h,8b Despite a significant number of highly efficient, N-directed, catalytic C–H arylation protocols, applications have been largely limited to relatively simple and unfunctionalized phenylpyridine and phenylpyrimidine substrates (Scheme 1A and B). This may be indicative of the various restrictions imposed by protocols that entail high temperature requirement, costly and/or extravagant catalysts or additives and overly reactive or sacrificial fragment-containing aryl donors, in relation to substrate tolerance or process practicality and viability considerations.
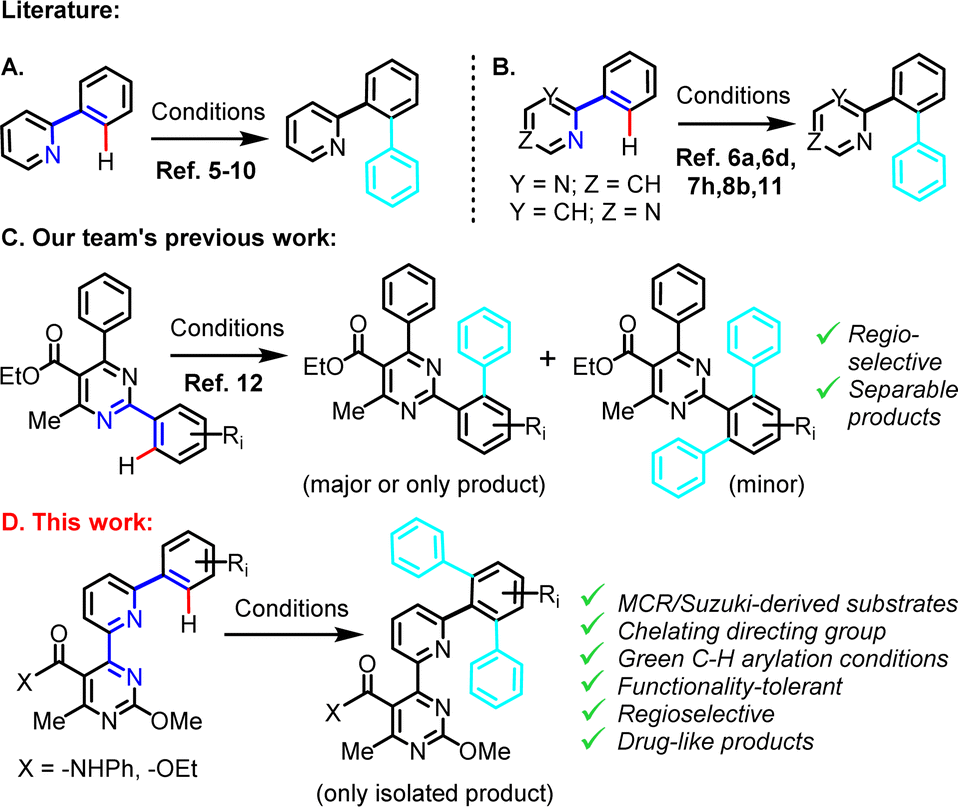 | ||
| Scheme 1 Previously reported N-directed C–H arylations, demonstrated on simple 2-phenylpyridines (A), simple 2-/4-phenylpyrimidines (B) or 2,6-diphenylpyrimidines (C) vs. the current work (D), which exploits a MCR/Suzuki-derived chelating directing group and drug-like multi-substituted (6-phenylpyridin-2-yl)pyrimidine substrates. The scissile C–H bond is shown in red, the added phenyl rings in cyan, while the connectivity of the atoms that engage in metal catalyst coordination is highlighted in blue. | ||
Having identified an emerging need to address the issue of C–H arylation in structurally complex substrates of medicinal relevance, while taking into account chemical functionality tolerance and applicability considerations, we have recently investigated a 2,6-diphenylpyrimidine series of polysubstituted substrates under mild (r.t., light-induced) C–H phenylation conditions (Scheme 1C).12 These involved Pd(II)-catalysis combined with Ru(II)- or Ir(III)-photoactivation to generate phenyl radicals from a readily accessible phenyldiazonium precursor. This methodology, which afforded C–H phenylation on challenging substrates regioselectively, was inspired from pre-existing work by Sanford and co-workers7f,h and was adapted/extended by our team, for application on the system at hand.
In the current work (Scheme 1D), we describe an advancement of this C–H arylation methodology, as a late-stage derivatization tool, in conjunction with a (medicinally validated) multi-component reaction (Biginelli condensation), whose aim herein is to enable early-stage construction of a 4-(pyridin-2-yl)pyrimidine chelator precursor. The assembled chelator is intended to play the role of the directing moiety in the final C–H arylation stage of a linear synthesis. This is advantageous, since the proposed synthetic process exploits a previously unexplored for C–H arylation directing moiety (i.e., pyrimidine-pyridine-phenyl), which is far superior compared to the simple N-atoms typically used in phenylpyridine or phenylpyrimidine substrates. Overall, the modular substrates employed for C–H arylation in the current study are structurally more complex and functionality-inclusive compared to simple 2-phenylpyridines or 2-/4-phenylpyrimidines typically encountered in previous N-directed C–H arylation studies. Nonetheless, the C–H arylation proceeds efficiently, yielding a single isolated product. Additionally, this study provides a molecular library of new drug-like compounds with potentially interesting 3D conformations, in which the medicinally relevant 4-(pyridin-2-yl)pyrimidine fragment constitutes an integral component. The fact that this green chemistry-compliant method is amenable to parallel synthesis format, renders it a valuable modern tool in the context of lead compound derivatization.
Results and discussion
To produce the desired modular substrates for C–H arylation, we have employed the Biginelli reaction, a 3-component MCR, which affords the medicinally-relevant 3,4-dihydropyrimidin-2(1H)-one heterocycle in a first, atom-economic step, from three simple and readily available components. It is noted that the Biginelli-derived multisubstituted 3,4-dihydropyrimidin-2(1H)-one scaffold has been linked to diverse biological activities, such as anti-inflammatory, antimicrobial, antimitotic, antifungal, antioxidant, antihypertensive, adrenoreceptor antagonistic and calcium channel-blocking.13 In this case, Sn(II)-catalyzed Biginelli condensation conditions were applied,14 with 2-methoxyethanol as solvent, using a combination of 6-bromo-2-pyridinecarboxaldehyde (1), urea (2) and either 3-oxo-N-phenylbutanamide (3) or methyl 3-oxobutanoate (4), to construct phenylamide- or ethyl ester-substituted 3,4-dihydropyrimidinones 5 and 6, respectively (Scheme 2). While Biginelli condensations employing simple picolyl aldehyde components often suffer from low isolated yields,15 likely due to difficulties in isolation of these polar products, in our case the opposite was true, as a result of using a bromo-substituted picolyl aldehyde (1). Presence of bromide both reduced solubility and facilitated product handling and isolation after reaction completion, by means of filtration and recrystallization, thus affording satisfactory to high yields (5 in 95%; 6 in 68%).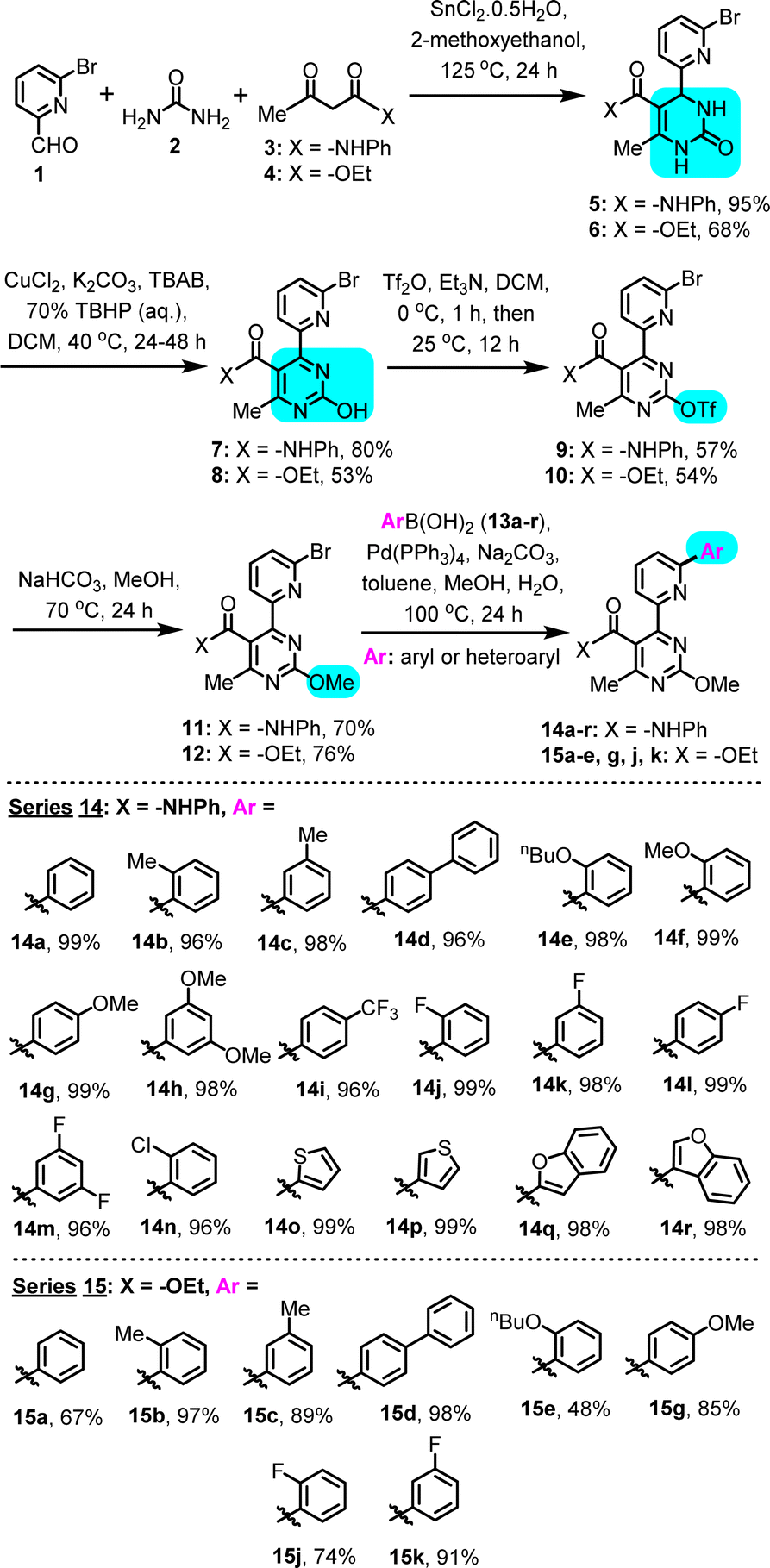 | ||
| Scheme 2 Biginelli/Suzuki synthetic route for generation of the (6-(hetero)arylpyridin-2-yl)pyrimidine substrates. | ||
Compounds 5 and 6 where subsequently converted to the corresponding pyrimidin-2-ols (7 and 8, respectively), via a Cu(II)-catalyzed oxidation in the presence of Luperox 70 wt%, in a mixed solvent (water–DCM), under basic conditions.16 In this biphasic system, the substrates demonstrated low solubility, therefore, we introduced a phase-transfer catalyst (tetrabutylammonium bromide, TBAB), which led to a significant improvement in yields (7 in 80%; 8 in 53%). The hydroxypyrimidine scaffold obtained in this step is also considered medicinally-relevant, as it can correlate to nucleobase analogues, vitamins, unnatural amino-acids, natural products and known pharmaceuticals.17 The chemistry described herein provides a way to functionalize it in a way not previously explored, in order to access unprecedented derivatives for biological evaluation.
Pyrimidin-2-ols 7 and 8 could be further elaborated into activated derivatives, namely trifluoromethanesulfonic esters 9 (57%) and 10 (54%), respectively, to allow the introduction of a ‘cap’ on the –OH and avoid interference in subsequent synthetic steps. Introduction of the triflate was achieved with trifluoromethane-sulfonic anhydride in DCM, in the presence of excess TEA.12,18 Triflate derivatives 9 and 10 were submitted to treatment with NaHCO3 in MeOH, under reflux, to achieve conversion to the corresponding methyl ethers, 11 (70%) and 12 (76%), respectively, by means of addition–elimination. Each of the desired methyl ethers was accompanied by a percentage (<30%) of the triflate methanolysis product – pyrimidin-2-ol 7 or 8. Nonetheless, this 2-stage conversion of pyrimidin-2-ols 7/8 to methoxy-‘capped’ derivatives 11/12 was found to be superior compared to a direct (single-stage) methylation of the pyrimidin-2-ols with MeI, since the latter afforded complex mixtures of constitutional isomers, resulting from non-selective Me addition on various heteroatoms of the substrate, under all examined conditions.
The synthesis of the 4-(6-(hetero)arylpyridin-2-yl)pyrimidine substrates was completed by means of a series of parallel Suzuki–Miyaura C–C cross-couplings with boronic acids 13a–r, employing Pd(PPh3)4 as catalyst in a mixed toluene–methanol–water solvent system,19 which took place on the bromo-substituted position of the pyridine in compounds 11 and 12 in high yields, to introduce a new (unsubstituted or substituted) phenyl or heteroaryl (2-/3-thiophene or 2-/3-benzofuran) ring system. The synthesized C–C coupling derivatives (Scheme 2, inset: series 14 from precursor 11; series 15 from precursor 12) encompass a representative range of substitution patterns and substituent electronic characteristics, while exhibiting unoccupied C–H sites, available for the ensuing C–H arylation step. The extended oligoaryl scaffold constructed via the Suzuki–Miyaura step combines the powerful (6-pyridin-2-yl)pyrimidine chelating moiety with the intended C–H arylation locus – the phenyl/heteroaryl ring – thus affording the possibility for N-directed C–H activation. As substrates for C–H arylation, compounds of series 14 and 15 (Scheme 2) are more functionality-inclusive, compared to ones investigated in prior C–H arylation studies.
Optimization of conditions for the C–H arylation stage was required, and results for C–H arylation attempts on the simplest substrate (14a), as a model system, are summarized in Table 1. Phenyldiazonium tetrafluoroborate (16) was employed as a readily available Ph-source, and the effects of a Pd(II)-catalyst, a Ru(II)-photoinitiator and a Ag(I)- or Ag(II)-oxidant were evaluated in MeOH, under photoactivation conditions induced by a household LED lamp. The optimization revealed the following: (a) phenyldiazonium tetrafluoroborate has to be present in excess (4 equiv.) relative to the substrate for maximum conversion, thus eliminating possibility of mono-arylation; (b) light, as well as a Pd(II)-catalyst and a Ru(II)-photoinitiator are essential for the reaction to occur; (c) Pd(II)-catalysts with chelating, non-exchangeable ligands (e.g., Pd(dppf)Cl2) are unsuitable, likely due to blocking coordination sites and excluding the substrate from forming a stable complex; and (d) presence of Ag(I) or Ag(II), albeit not essential, significantly enhances yields, especially AgOAc, which suggests a potential role for silver salts in maintaining the catalytic activity of palladium, in agreement with previous reports by others.20
|
||||||
|---|---|---|---|---|---|---|
| Entry | PhN2·BF4a equiv. | Cat. equiv. | Ru(II) equiv.b | Oxid. equiv. | LED lightc | % |
| a Phenyl donor.b [Ru(bpy)3]Cl2 photoinitiator.c Phillips, 12.5 W, 1521 lumen, emission wavelength range 400–700 nm. | ||||||
| 1 | 1 | Pd(OAc)2 10 mol% | 5 mol% | AgOAc 2 equiv. | Yes | 11 |
| 2 | 2 | Pd(OAc)2 10 mol% | 5 mol% | AgOAc 2 equiv. | Yes | 27 |
| 3 | 3 | Pd(OAc)2 10 mol% | 5 mol% | AgOAc 2 equiv. | Yes | 36 |
| 4 | 4 | Pd(OAc)2 10 mol% | 5 mol% | AgOAc 2 equiv. | Yes | 61 |
| 5 | 5 | Pd(OAc)2 10 mol% | 5 mol% | AgOAc 2 equiv. | Yes | 60 |
| 6 | 4 | 0 mol% | 5 mol% | AgOAc 2 equiv. | Yes | 0 |
| 7 | 4 | Pd(OAc)2 10 mol% | 0 mol% | AgOAc 2 equiv. | Yes | 0 |
| 8 | 4 | Pd(OAc)2 10 mol% | 5 mol% | AgOAc 2 equiv. | No | 0 |
| 9 | 4 | Pd(OAc)2 5 mol% | 5 mol% | AgOAc 2 equiv. | Yes | 42 |
| 10 | 4 | Pd(OAc)2 10 mol% | 5 mol% | AgOAc 1 equiv. | Yes | 39 |
| 11 | 4 | PdCl2 10 mol% | 5 mol% | AgOAc 2 equiv. | Yes | 60 |
| 12 | 4 | Pd(dppf)Cl2 10 mol% | 5 mol% | AgOAc 2 equiv. | Yes | 0 |
| 13 | 4 | Pd(OAc)2 10 mol% | 5 mol% | 0 equiv. | Yes | 25 |
| 14 | 4 | Pd(OAc)2 10 mol% | 5 mol% | AgO 2 equiv. | Yes | 32 |
| 15 | 4 | Pd(OAc)2 10 mol% | 5 mol% | AgNO3 2 equiv. | Yes | 28 |
| 16 | 4 | Pd(OAc)2 10 mol% | 5 mol% | AgOAc 3 equiv. | Yes | 61 |
| 17 | 4 | Pd(OAc)2 10 mol% | 5 mol% | AgOAc 0.5 equiv. | Yes | 32 |
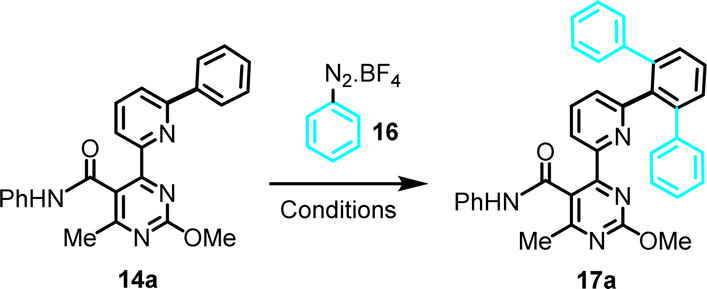
Further optimization was not attempted, after recognizing the synthetic challenge posed by this functionality-dense substrate. A yield >60% in this model reaction was deemed to be of synthetic utility. It is noteworthy that the remaining percentage mostly accounts for unreacted starting material. Based on the above, condition set #4 (Table 1) was selected as optimal working conditions, for application on all substrates shown in Scheme 2, both from the amide and ester set (14a–r and 15a–e, g, j, k, respectively).
This catalytic C–H arylation protocol is green chemistry-compliant and readily amenable to parallel synthesis. A non-toxic solvent is employed (MeOH), the aryl donor only generates benign N2 gas and (easily separable) homocoupling by-product (1,1′-biphenyl), apart from the only arylation derivative, while the transformation occurs at room temperature, activated simply by a commercial white LED lamp (Phillips, 12.5 W, 1521 lumen, emission wavelength 400–700 nm).
All substrates included in this study behaved similarly under the selected conditions. The C–H phenylation results for the amide- and ester-containing substrates are shown in Schemes 3 and 4, respectively. C–H phenylation occurred exclusively on unoccupied ortho-positions of the aryl moiety connected to the pyridine's C2. Under the selected conditions, substrates exhibiting a single unoccupied ortho-site (pre-existing 2-substituent) afforded the expected mono-aryl derivatives, while all substrates with two such sites, either equivalent (i.e., unsubstituted, 4-substituted or 3,5-disubstituted) or non-equivalent (3-substituted), afforded the bis-aryl derivative as the only observed product, in yields which in some cases surpassed 70%. All isolated C–H arylation derivatives were systematically characterized by means of 1H and 13C NMR, FT-IR, LC-MS(API) and melting point determination (for solids), while ones for which crystals were obtained (17b, d, f, h, j, r) were also characterized by single-crystal X-ray diffraction (XRD) (for characterization and selected crystallographic data, see the ESI†).
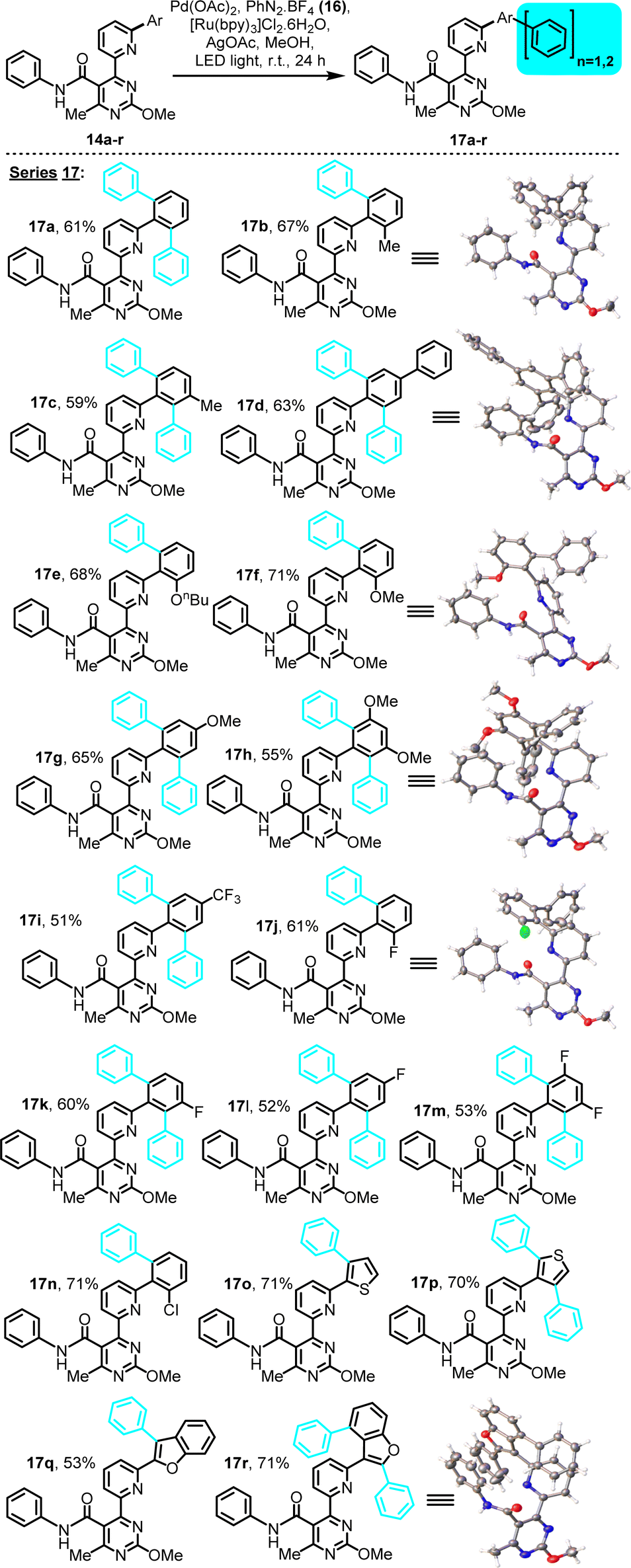 | ||
| Scheme 3 LED-activated, Pd-catalyzed, Ru-photoredox-mediated C–H phenylation on 4-(6-(hetero)arylpyridin-2-yl)pyrimidine substrates bearing a phenylamide substituent. | ||
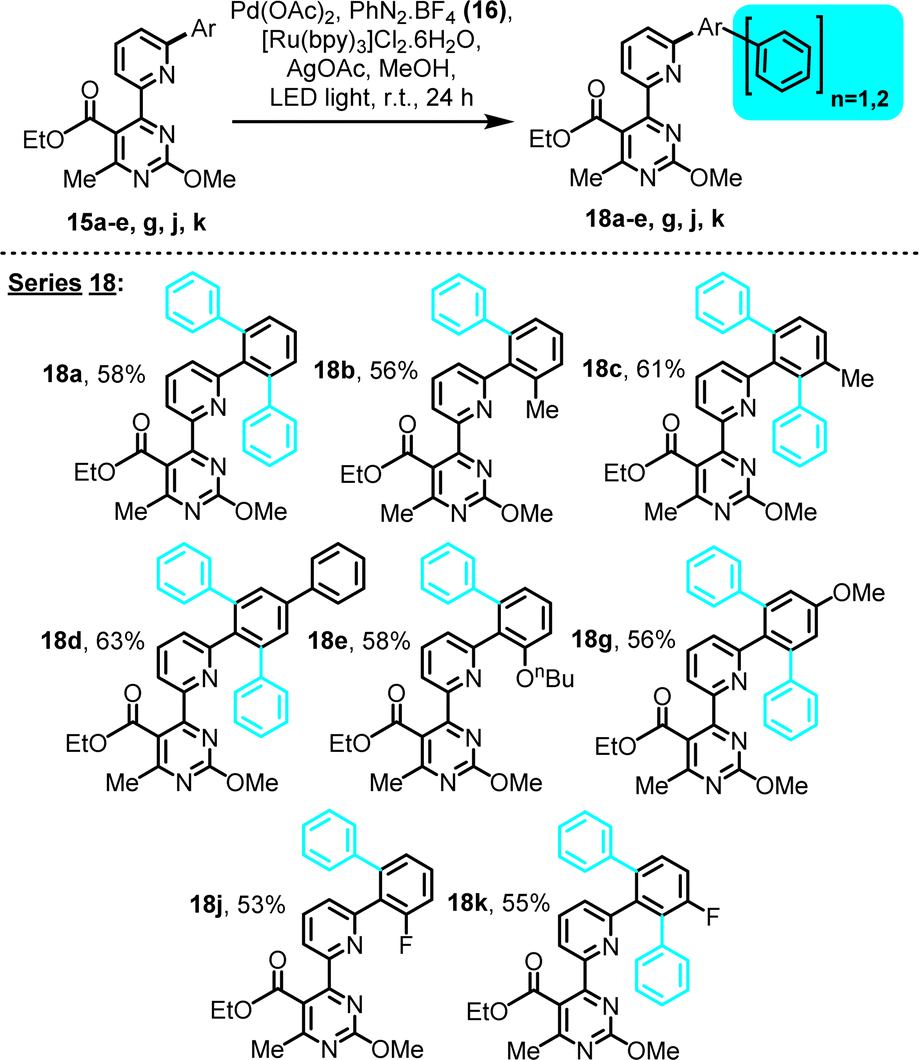 | ||
| Scheme 4 LED-activated, Pd-catalyzed, Ru-photoredox-mediated C–H phenylation on 4-(6-phenylpyridin-2-yl)pyrimidine substrates bearing an ethyl ester substituent. | ||
In the amide series, substrates exhibiting either electron-donating or electron-withdrawing substituents and diverse substitution patterns (i.e., 2-, 3- or 4-monosubstitution, 3,5-disubstitution) on the arylation-receiving phenyl ring performed comparably (Scheme 3), indicating that electronic effects of pre-existing substituent(s) do not significantly impact this arylation, unlike the case of a pyrimidine-directed arylation in our previous study, where the substrates' electronic features proved significant.12 This difference could be indicative of a strong influence of the chelating (6-pyridin-2-yl)pyrimidine directing moiety employed herein on the arylation mechanism, especially on facilitating the initial C–H palladation step. The current C–H arylation occurs independently of pre-existing substituent effects, thus broadening the synthetic scope with regard to substrate diversity.
The functionality-rich products obtained through this effort are conformationally appealing oligo-aryl systems, as suggested by the elucidated crystal structures of six members of the amide series (Scheme 3, inset, series 17), affording unusual architectures and conformations for occupying 3D chemical space. Hence, they should be of interest to future screening efforts for identifying either bioactive compounds or useful ligands for metal coordination and catalysis.
Within the amide subset of C–H phenylation derivatives (Scheme 3, series 17), of particular importance are examples 17o–r, involving electron-rich heteroaryl rings of 2- and 3-thiophene, as well as 2- and 3-benzofuran, as the phenyl-receiving moieties. In these examples, phenyl transfer was observed exclusively on positions dictated by the directing effect of the (6-pyridin-2-yl)pyrimidine moiety, but not on other (electron-rich) positions as dictated by the heteroaryl's intrinsic reactivity. This observed regioselectivity could find application in medicinal and materials chemistry, for site-selective functionalization of these valuable heteroaryl ring systems. For substrates 14p and 14r, which exhibit two non-equivalent positions for C–H phenylation, the bis-phenyl adduct is the only isolated product. In particular, bis-phenylated derivative 17r, that originates from 3-benzofuranyl substrate 14r, suggests that a 6-membered carbopalladated intermediate, in addition to a 5-membered one, may be in play, en route to the observed product, as will be discussed below.
The series of ester substrates afforded similar type products (Scheme 4, series 18), at yields comparable to the amide series, indicating a tolerance of the selected reaction conditions to ester functionalities.
We subsequently explored the scope of this transformation with regard to the aryl donor component. In particular 4-iPr, 4-tBu, 4-Me2N, 4-Et2N, 3-NO2, 4-NO2, 4-F, 4-Cl, 2-Br, 4-Br and 4-I substituted phenyldiazonium tetrafluoroborates, as well as the 1-naphthyl counterpart, were successfully employed in combination with substrate 14b (2-Me pre-existing substituent), to yield the corresponding C–H arylation derivatives (Scheme 5, series 20). All diazonium tetrafluoroborates were in-house prepared, immediately prior to use (see ESI†). This study revealed a general compatibility of the reaction with various types of diazonium salts that contain diverse functionalities. A sensitivity to the steric bulk of the arylating agent was suggested by the 1-naphthyl case (20c), as reflected by the moderate yield.
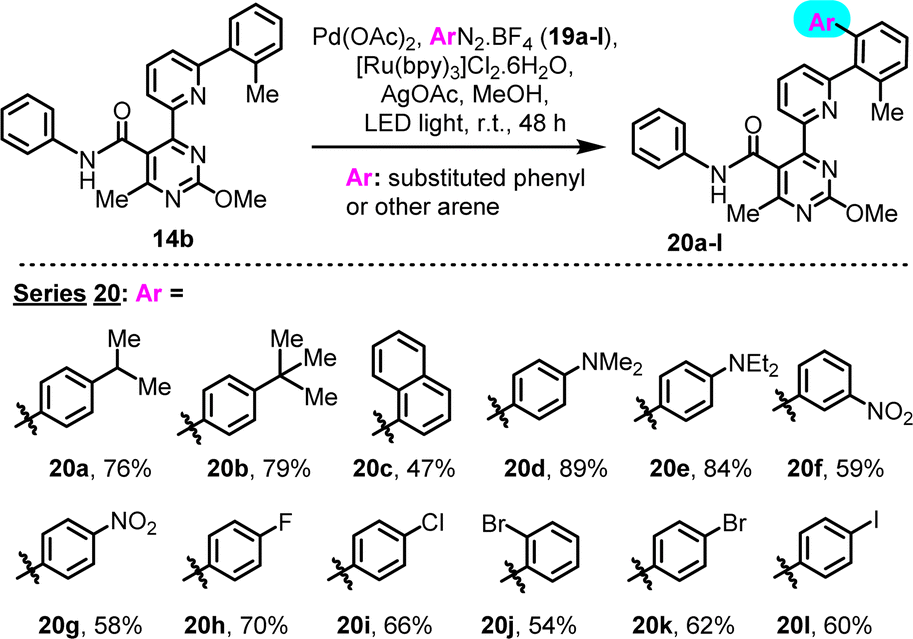 | ||
| Scheme 5 Investigation of scope of LED-activated C–H arylation, with regard to structures of diazonium salt aryl donors. | ||
Mechanistically, we propose the early generation of a square planar Pd(II)-species via C–H palladation, with participation of the Biginelli/Suzuki-assembled tridentate (6-phenylpyridin-2-yl)pyrimidine motif (Scheme 6). Chelation is believed to be key in this initial activation event, based on the superiority of this system relative to a phenylpyrimidine counterpart.12 The Ru(II)/(III)-photoredox cycle, triggered by light, is well-established in the literature7h and in our described system photoactivation is brought about by a commercial LED light. Single-electron transfer from the photoexcited Ru(II)*-species to the diazonium salt results in dissociation of the latter to generate an aryl radical. We have verified the intermediacy of a radical species, by examining a model reaction (14a + 16) under the selected reaction conditions (Table 1, entry 4) and time duration (24 h), in the presence of a known radical scavenger, diphenyl-1-picrylhydrazyl (DPPH).21 The efficient suppression of conversion, and the 90% recovery of starting material observed after workup and column chromatography, was attributed to the compromise imposed by DPPH to radical reactivity, as has been reported in other studies employing radical scavengers.22 A combination of the aryl radical with the organopalladium(II)-species may lead to a transient Pd(III)-complex. Such intermediates have been frequently invoked in palladium catalytic cycles,7h but rarely isolated. Ritter has shown that certain Pd(III)-complexes with N^N-bidentate and two acetate ligands are bimetallic,23 however no information is available on tridentate-monoacetate ligand combinations, in cases of complexes generated via radical uptake. Transfer of a single electron from the Pd(III) to the Ru(III) affords a Pd(IV)-hexacoordinated species and regenerates the Ru(II)-photosensitizer. Finally, reductive elimination results in the release of the C–H arylated derivative and regeneration of the Pd(II)-catalyst. Substrates with a second available ortho-site (e.g., 14r) may enter a second catalytic round to install an additional aryl ring.
 | ||
| Scheme 6 Proposed mechanism for the LED-activated, Pd-catalyzed, Ru-photoredox-mediated C–H arylation on 4-(6-phenylpyridin-2-yl)pyrimidine substrates with (above) stoichiometric AgOAc. The reaction is suppressed in the presence of DPPH (L = general ligand or the solvent; DPPH = diphenyl-1-picrylhydrazyl). | ||
The role of silver acetate is collectively significant and can be attributed to:20 (a) the ability of Ag(I) to serve as a stoichiometric oxidant in the event of formation of Pd(0) degradation species, ensuring return of the catalyst to the Pd(II) oxidation state; (b) the common counterion it shares with Pd(OAc)2, enriching acetate concentration to ensure acetate ligand transfer to palladium in the reductive elimination step; (c) acetate being superior to other counterions tested, which is compliant with the bridging ability of carboxy-ligands to form transient bimetallic complexes between Ag(I) and Pd(II), while also facilitating the Pd-mediated C–H activation step; and (d) the halide scavenging ability of silver(I), that ensures halides pose no competition for acetate. Some of these aspects are shown in Scheme 6.
Evidence of what is proposed to be a stable tetracoordinated, carbopalladated derivative of a mechanistic intermediate was obtained from a partially purified sample of the C–H phenylation reaction that generated product 17r from precursor 14r. Upon silica column purification of an unquenched reaction mixture, 17r was obtained along with traces of an orange impurity. Attempts to crystallize this product from DCM solution, with slow diffusion of pentane, afforded colorless needle-like crystals of 17r mingled with orange block-like crystals of the unknown impurity (Fig. 1). After manually segregating the two types of crystals under a microscope, both structures were solved by XRD. Crystallography unambiguously confirmed the bis-phenyl adduct structure proposed for 17r (see Scheme 3, inset), and revealed that the orange impurity was the distorted square-planar Pd(II)-complex 23 (Fig. 1).
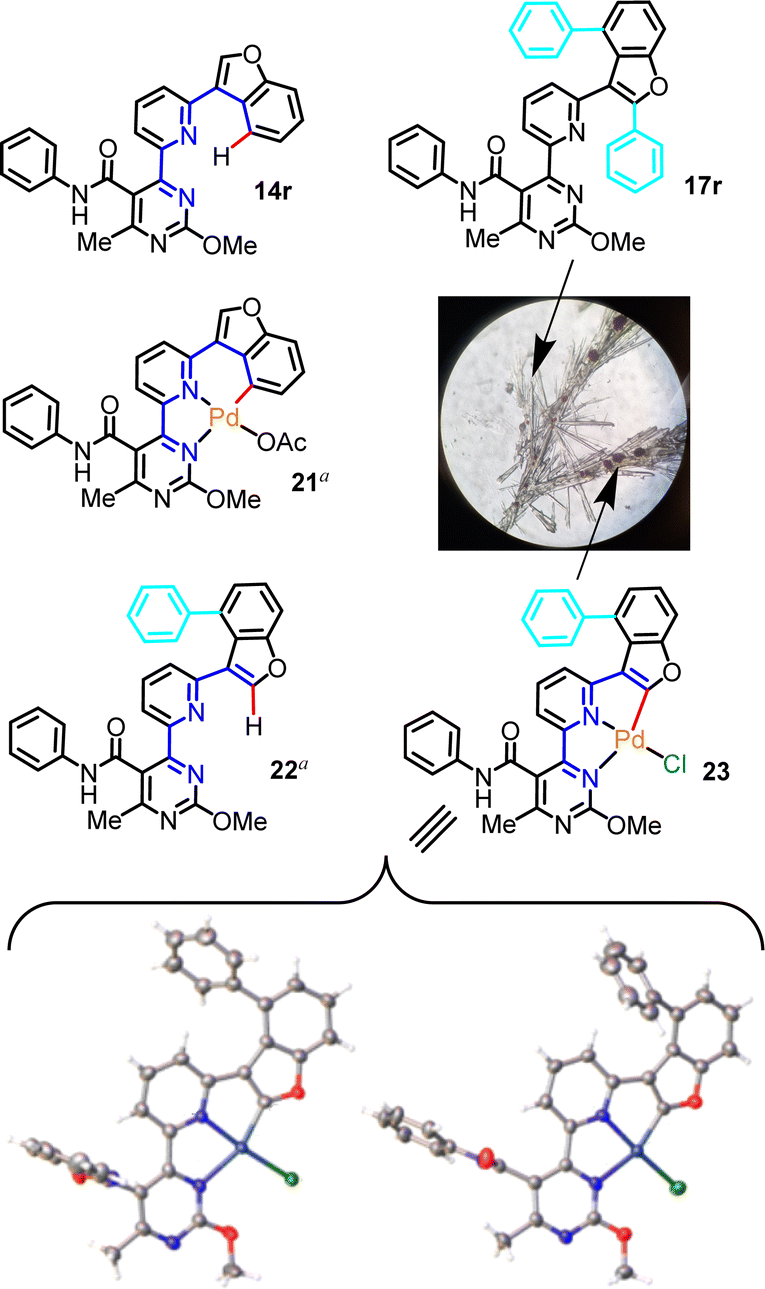 | ||
| Fig. 1 Structures of substrate 14r, isolated bis-phenyl end-product 17r and presumptive mechanistic intermediates (21, 22) or isolated organometallic derivatives (23) in the conversion of 14r to 17r. Colorless needle crystals of 17r and orange block crystals of 23 were obtained from a recrystallization, thus allowing elucidation of the corresponding structures. The X-ray crystal structure of carbopalladated derivative 23 reveals two alternative conformations. Color code in crystal structures: C: grey; H: white; O: red; N: blue; Cl: green; Pd: dark blue (a = not isolated). | ||
Interestingly, 23 contains the retron of one C–H phenylation event (post-functionalization adduct), on the C4 of the benzofuran ring, while the C2 of the benzofuran is involved in organometallic bond with Pd(II), as would be expected prior to the C–H phenylation of position C2 (pre-functionalization complex with regard to the second C–H phenylation that eventually leads to 17r). Additionally, both the pyridine and pyrimidine N-atoms are coordinated on the Pd(II) center, while the fourth coordination site is occupied by a terminally-ligated chloride, likely originating from the [Ru(bpy)3]Cl2 photoinitiator, which has replaced the acetate. To the best of our knowledge, this is the first report of a 4-(6-(benzofuran-3-yl)pyridin-2-yl)pyrimidine motif in the role of a tridentate ligand in palladium chemistry. It is noteworthy that two alternative conformations of complex 23 were detected in the crystallographic cell, which differ in the angles formed between the phenylamide and phenyl substituents relative to the metal-promoted hexacyclic central scaffold (Fig. 1). The tridentate ligand's bite angle was found to be 158.3° and 158.5° in these conformations, in agreement with other tridentate ligand types reported in the literature.24
We believe that Pd(II)-complex 23 represents a derivative of a [5,5]-membered organometallic precursor (the acetate counterpart) en route to product 17r, and a similar type of precursor would be expected for all other substrates discussed in this study. Moreover, the occurrence of C–H phenylation on the benzofuran C4, prior to the formation of 23, implies that another organopalladated [5,6]-ring species, the hypothetical 21, is a possible intermediate in this particular reaction and a direct precursor to presumptive mono-aryl derivative 22. Formation of complex 21 as intermediate is deemed possible, since various other types of stable [5,6]-ring complexes of Pd(II) are known.25
Experimental
Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 to 1:10) and then ethyl acetate–methanol (10:1), to afford 0.62 g (1.85 mmol, 53%) of compound 8 as a sub-yellow powder.:1 to 5:1), to afford 0.65 g (1.25 mmol, 57%) of compound 9 as a white solid.:1), to afford 0.28 g (0.6 mmol, 54%) of compound 10 as a sub-yellow powder.:1 to 1:1) to afford 0.105 g (0.26 mmol, 70%) of compound 11 as a white foamy solid.:1 to 2:1) to afford 0.12 g (0.34 mmol, 85%) of compound 12 as a white powder.:1 mixture (8 mL), which had nitrogen bubbled through it, was added to the reaction. Finally, Na2CO3 (0.275 g, 2.6 mmol, 6.5 equiv.) dissolved in water (2.5 mL), which also had nitrogen bubbled through it, was added to the flask. The mixture was heated at 100 °C with vigorous stirring for 12 h. It was subsequently cooled down to r.t. and ethyl acetate was added. The mixture was transferred to a separatory funnel and washed (×3) with aqueous Na2CO3 10 wt% solution and water. The combined organic phase was dried over Na2SO4. After filtering out the drying agent, the solvent was removed under vacuum. The crude sample was re-dissolved in DCM and applied to a silica column for flash chromatography. Elution took place using a hexane–ethyl acetate step gradient (different in each case depending on product polarity) to afford products of type 14.:1 mixture (8 mL), which had nitrogen bubbled through it, was added to the reaction. Finally, Na2CO3 (0.275 g, 2.6 mmol, 6.5 equiv.) dissolved in water (2.5 mL), which also had nitrogen bubbled through it, was added to the mixture. The mixture was heated at 100 °C with vigorous stirring for 12 h. It was subsequently cooled down to r.t. Aqueous NH4Cl 25 wt% solution was added to the flask and the mixture was transferred to a separatory funnel and extracted with ethyl acetate (×3). The combined organic extract was dried over Na2SO4. After filtering out the drying agent, the solvent was removed under vacuum. The crude sample was redissolved in DCM and applied to a silica column for flash chromatography. Elution took place using a hexane–ethyl acetate step gradient (from 10:1 to 4:1) to afford products of type 15.:0 to 3:1), to afford C–H arylated derivative of type 18.
:5 to 1:10) and then ethyl acetate–methanol (10:1), to afford 0.62 g (1.85 mmol, 53%) of compound 8 as a sub-yellow powder.:1 to 5:1), to afford 0.65 g (1.25 mmol, 57%) of compound 9 as a white solid.:1), to afford 0.28 g (0.6 mmol, 54%) of compound 10 as a sub-yellow powder.:1 to 1:1) to afford 0.105 g (0.26 mmol, 70%) of compound 11 as a white foamy solid.:1 to 2:1) to afford 0.12 g (0.34 mmol, 85%) of compound 12 as a white powder.:1 mixture (8 mL), which had nitrogen bubbled through it, was added to the reaction. Finally, Na2CO3 (0.275 g, 2.6 mmol, 6.5 equiv.) dissolved in water (2.5 mL), which also had nitrogen bubbled through it, was added to the flask. The mixture was heated at 100 °C with vigorous stirring for 12 h. It was subsequently cooled down to r.t. and ethyl acetate was added. The mixture was transferred to a separatory funnel and washed (×3) with aqueous Na2CO3 10 wt% solution and water. The combined organic phase was dried over Na2SO4. After filtering out the drying agent, the solvent was removed under vacuum. The crude sample was re-dissolved in DCM and applied to a silica column for flash chromatography. Elution took place using a hexane–ethyl acetate step gradient (different in each case depending on product polarity) to afford products of type 14.:1 mixture (8 mL), which had nitrogen bubbled through it, was added to the reaction. Finally, Na2CO3 (0.275 g, 2.6 mmol, 6.5 equiv.) dissolved in water (2.5 mL), which also had nitrogen bubbled through it, was added to the mixture. The mixture was heated at 100 °C with vigorous stirring for 12 h. It was subsequently cooled down to r.t. Aqueous NH4Cl 25 wt% solution was added to the flask and the mixture was transferred to a separatory funnel and extracted with ethyl acetate (×3). The combined organic extract was dried over Na2SO4. After filtering out the drying agent, the solvent was removed under vacuum. The crude sample was redissolved in DCM and applied to a silica column for flash chromatography. Elution took place using a hexane–ethyl acetate step gradient (from 10:1 to 4:1) to afford products of type 15.:0 to 3:1), to afford C–H arylated derivative of type 18.X-ray diffraction (XRD)
Single crystal X-ray diffraction data were collected on a Rigaku Synergy S X-ray diffractometer, equipped with a HyPix-6000HE area detector utilizing Cu-Kα (λ = 1.54184 Å) radiation. A suitable crystal was mounted on a Hampton cryoloop with Paratone-N oil and transferred to a goniostat, where it was cooled for data collection. The structures were solved by direct methods using SHELXT and refined on F2 using full-matrix least squares employing SHELXL14.1.26 The software packages used were: CrysAlis CCD for data collection and CrysAlis RED for cell refinement and data reduction (CrysAlis CCD and CrysAlis RED, version p171.38.46, Oxford Diffraction Ltd, Abingdon, Oxford, 2017); WinGX27 or OLEX228 for geometric calculations; and DIAMOND for molecular graphics (K. Brandenburg, DIAMOND, version 3.2k, Crystal Impact GbR, Bonn, Germany, 2014). The non-H atoms were treated anisotropically, whereas the aromatic hydrogen atoms were placed in calculated, ideal positions and refined as riding on their respective carbon atoms. To limit the disorder of functional groups or lattice solvent molecules, various restraints have been applied in the refinement of the crystal structures. Electron density contributions from disordered guest molecules were handled using the SQUEEZE procedure from the PLATON software suite.29 All the crystal structures described in this study were deposited via the joint CCDC/FIZ Karlsruhe deposition service and have been assigned the deposition numbers CCDC 2307358–2307364.†
Conclusions
A short and efficient synthetic route was described for the generation of a collection of 26 modular oligoaryl compounds, belonging to the families of (6-phenylpyridin-2-yl)pyrimidines and (6-heteroarylpyridin-2-yl)pyrimidines. These proved to be ideal substrates for late-stage C–H arylation. For this purpose, a robust and reproducible C–H arylation protocol was applied, which was compatible with sustainable chemistry principles, such as the use of a benign solvent, a Pd(II)-catalyst and a commercial LED lamp as activation source to promote Ru-photoredox chemistry, in the absence of a heating source. The simplicity of this method renders it a useful synthetic tool, amenable to parallel synthesis of molecular libraries. The novelty of our approach stems from the use of a multi-component reaction (Biginelli) in the early stages of the linear synthetic route, to strategically pre-assemble the (N-based) chelating moiety that later serves both as a highly efficient directing group for the catalyst in the C–H arylation, as well as an integral component of the final arylated products, intended to contribute to a potentially unique conformation.The successful application of the C–H arylation on the series of 26 substrates showed a general applicability of the reaction, tolerance to various functionalities of the substrate, such as amides, esters, ethers, halogens (F, Cl), as well as electron-deficient (pyrimidine, pyridine) and electron-rich (thiophene, benzofuran) heteroaromatics. A series of 13 diazonium tetrafluoroborates, employed under the selected C–H arylation conditions in the role of aryl donor, showed compatibility of this transformation with branched alkyl, naphthyl, dialkylamino, nitro and (any) halogen substituent on the aryl donor. Notably, the pyrimidine-pyridine-phenyl chelating moiety present in the substrates enhances reaction conversion, relative to unfunctionalized substrates that employ a simple N-based directing group. Moreover, regioselectivity appears to be predominantly dictated by the directing group, and in certain cases overrides the substrates' intrinsic reactivity. Such processes are highly desirable in sustainable chemistry, to facilitate direct access to specific products, by means by-passing longer alternative routes that employ conventional chemistry and are often prone to limitations due to substrate reactivity predispositions. These features enabled fine-tuning of the reaction conditions, to achieve absence of formation of any competitive product. The bis-adducts, where possible, were consistently the only isolated products, while no mono-adduct from substrates with two available reaction sites was observed. This departs from our own previous findings pertaining to a library of 2,6-diphenylpyrimidine counterparts,12 where the mono-aryl adduct was the major product but bis-adducts were also possible, suggesting that the current type of substrates, unlike our previous series, undergo a second arylation more readily after they have one aryl group already incorporated. This finding suggests a key role of substrate architecture, creating valuable precedent for informed substrate design in future C–H activation efforts.
Finally, the XRD crystal structure of isolated [5,5]-carbopalladated complex 23 provides support for our mechanistic proposal for this C–H arylation and highlights the possibility of manipulating the rather underexplored [5,6]-carbopalladated counterparts for obtaining access to unprecedented C–H arylation derivatives.
Author contributions
IMA: compound synthesis and characterization, crystal growing, data collection and processing, manuscript draft writing; NI: compound synthesis and characterization, data collection and processing; NP: XRD data collection and processing, manuscript draft writing; SNG: project supervision, compound synthesis and characterization, data collection and processing, final manuscript writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SNG gratefully acknowledges the University of Cyprus for funding of this project through allocation of public funds, as well as for the award of a research fellowship to IMA. SNG thanks COST organization and the Action Chair for his participation in COST Action CA15106 (C–H Activation in Organic Synthesis, CHAOS).References
- K. M. Altus and J. A. Love, The continuum of carbon–hydrogen (C–H) activation mechanisms and terminology, Commun. Chem., 2021, 4, 173 CrossRef PubMed.
- (a) T. Dalton, T. Faber and F. Glorius, C−H Activation: Toward Sustainability and Applications, ACS Cent. Sci., 2021, 7, 245 CrossRef CAS PubMed; (b) T. Rogge, N. Kaplaneris, N. Chatani, J. Kim, S. Chang, B. Punji, L. L. Schafer, D. G. Musaev, J. Wencel-Delord, C. A. Roberts, R. Sarpong, Z. E. Wilson, M. A. Brimble, M. J. Johansson and L. Ackermann, C–H activation, Nat. Rev. Methods Primers, 2021, 1, 43 CrossRef CAS; (c) A. Yamashita, H. Nishiyama, S. Inagi and I. Tomita, Synthesis of π-conjugated poly(arylene)s by polycondensation of 1,4-bis(3-methylpyridin-2-yl)benzene and aryl dibromides through regiospecific C-H functionalization process, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 2771 CrossRef CAS.
- J. M. Zakis, T. Smejkal and J. Wencel-Delord, Cyclometallated complexes as catalysts for C-H activation and functionalization, Chem. Commun., 2022, 58, 483 RSC.
- K. Murali, L. A. Machado, R. L. Carvalho, L. F. Pedrosa, R. Mukherjee, E. N. da Silva Jr and D. Maiti, Decoding Directing Groups and their Pivotal Role in C–H Activation, Chem.–Eur. J., 2021, 27, 12453 CrossRef CAS PubMed.
- (a) R. González-Fernández, P. Crochet and V. Cadierno, Hydrophilic (η6-Arene)–Ruthenium(II) Complexes with P–OH Ligands as Catalysts for the Isomerization of Allylbenzenes and C–H Bond Arylation Reactions in Water, Organometallics, 2019, 38, 3696 CrossRef; (b) C. Binnani, S. C. Mandal, B. Pathak and S. K. Singh, Ruthenium-Catalyzed C-H Bond Activation/Arylation Accelerated by Biomass-Derived Ligands, Eur. J. Inorg. Chem., 2019, 2019, 2844 CrossRef CAS; (c) D. Sierra, C. Contreras, J. Francos, J. Gómez and V. Cadierno, Novel ferrocenylphosphino sulfonates: Synthesis, crystal structure and preliminary application as ligands in aqueous catalysis, J. Organomet. Chem., 2018, 854, 106 CrossRef CAS; (d) N. Kaloğlu, İ. Özdemir, N. Gürbüz, H. Arslan and P. H. Dixneuf, Ruthenium(η6,η1-arene-CH2-NHC) Catalysts for Direct Arylation of 2-Phenylpyridine with (Hetero)Aryl Chlorides in Water, Molecules, 2018, 23, 647 CrossRef PubMed; (e) P. Nareddy, F. Jordan and M. Szostak, Ruthenium(II)-catalyzed ortho-C–H arylation of diverse N-heterocycles with aryl silanes by exploiting solvent-controlled N-coordination, Org. Biomol. Chem., 2017, 15, 4783 RSC; (f) L. V. Graux, M. Giorgi, G. Buono and H. Clavier, [RuCl2(η6-p-cymene)] complexes bearing phosphinous acid ligands: preparation, application in C–H bond functionalization and mechanistic investigations, Dalton Trans., 2016, 45, 6491 RSC; (g) G. M. Reddy, N. S. S. Rao, P. Satyanarayana and H. Maheswaran, PhI(OCOCF3)2-mediated ruthenium catalyzed highly site-selective direct ortho-C–H monoarylation of 2-phenylpyridine and 1-phenyl-1H-pyrazole and their derivatives by arylboronic acids, RSC Adv., 2015, 5, 105347 RSC; (h) P. B. Arockiam, C. Fischmeister, C. Bruneau and P. H. Dixneuf, Ruthenium(II)-catalyzed selective monoarylation in water and sequential functionalisations of C–H bonds, Green Chem., 2013, 15, 67 RSC; (i) M. Christakakou, M. Schön, M. Schnürch and M. D. Mihovilovic, Arylation of Pyridines via Suzuki–Miyaura Cross-Coupling and Pyridine-Directed C–H Activation Using a Continuous-Flow Approach, Synlett, 2013, 24, 2411 CrossRef CAS; (j) H. Li, W. Wei, Y. Xu, C. Zhang and X. Wan, Ru-catalyzed aerobic oxidative coupling of arylboronic acids with arenes, Chem. Commun., 2011, 47, 1497 RSC; (k) P. B. Arockiam, C. Fischmeister, C. Bruneau and P. H. Dixneuf, C-H Bond Functionalization in Water Catalyzed by Carboxylato Ruthenium(II) Systems, Angew. Chem., Int. Ed., 2010, 49, 6629 CrossRef CAS; (l) N. Luo and Z. Yu, RuCl3·x H2O-Catalyzed Direct Arylation of Arenes with Aryl Chlorides in the Presence of Triphenylphosphine, Chem.–Eur. J., 2010, 16, 787 CrossRef CAS PubMed; (m) B. Yu, X. Yian, S. Wang, N. Tang and C. Xi, A Highly Efficient Ruthenium(II) Catalyst with(1,2-Diarylvinyl)phosphine Ligands for Direct OrthoArylation of 2-Arylpyridine with Aryl Chlorides, Organometallics, 2010, 29, 3222 CrossRef CAS; (n) A. Prades, M. Poyatos and E. Peris, η6-Arene)ruthenium(N-heterocyclic carbene) Complexes for the Chelation-Assisted Arylation and Deuteration of Arylpyridines: Catalytic Studies and Mechanistic Insights, Adv. Synth. Catal., 2010, 352, 1155 CrossRef CAS; (o) S. Oi, S. Fukita, N. Hirata, N. Watanuki, S. Miyano and Y. Inoue, Ruthenium complex-catalyzed direct ortho arylation and alkenylation of 2-arylpyridines with organic halides, Org. Lett., 2001, 3, 2579 CrossRef CAS PubMed; (p) Y.-Q. He, Z.-Q. Li and Y.-W. Zhong, Synthesis of Dibenzo[f,h]quinolines by Stepwise C–H Arylation of 2-Phenylpyridine and Reductive Cyclodehydrogenation, Synthesis, 2023, 55, 2779 CrossRef CAS.
- (a) R. Riveiros, R. Tato, J. P. Sestelo and L. A. Sarandeses, Triorganoindium Reagents in Rh-Catalyzed C–H Activation/C–C Cross-Coupling Reactions of 2-Arylpyridines, Molecules, 2018, 23, 1582 CrossRef PubMed; (b) G. Meng and M. Szostak, Site-Selective C–H/C–N Activation by Cooperative Catalysis: Primary Amides as Arylating Reagents in Directed C–H Arylation, ACS Catal., 2017, 7, 7251 CrossRef CAS; (c) G. Meng and M. Szostak, Rhodium-Catalyzed C–H Bond Functionalization with Amides by Double C–H/C–N Bond Activation, Org. Lett., 2016, 18, 796 CrossRef CAS PubMed; (d) M.-Z. Lu, P. Lu, Y.-H. Xu and T.-P. Loh, Mild Rh(III)-Catalyzed Direct C–H Bond Arylation of (Hetero)Arenes with Arylsilanes in Aqueous Media, Org. Lett., 2014, 16, 2614 CrossRef CAS PubMed; (e) Q. Shuai, L. Yang, X. Guo, O. Baslé and C.-J. Li, Rhodium-Catalyzed Oxidative C−H Arylation of 2-Arylpyridine Derivatives via Decarbonylation of Aromatic Aldehydes, J. Am. Chem. Soc., 2010, 132, 12212 CrossRef CAS PubMed; (f) T. Vogler and A. Studer, Oxidative coupling of arylboronic acids with arenes via Rh-catalyzed direct C-H arylation, Org. Lett., 2008, 10, 129 CrossRef CAS PubMed; (g) X. Zhao and Z. Yu, Rhodium-Catalyzed Regioselective C-H Functionalization via Decarbonylation of Acid Chlorides and C-H Bond Activation under Phosphine-Free Conditions, J. Am. Chem. Soc., 2008, 130, 8136 CrossRef CAS PubMed; (h) S. Oi, S. Fukita and Y. Inoue, Rhodium-catalysed direct ortho arylation of 2-arylpyridines with arylstannanes via C–H activation, Chem. Commun., 1998, 2439 RSC; (i) H. Teng, X. Shao, Z.-F. Zhang, K.-F. Yang, M.-D. Su, L.-W. Xu and L. Yang, Directing group assisted rhodium-catalyzed formal C–H arylation and carbonylative arylation of arenes with aryl halides in the presence of CO, J. Catal., 2024, 429, 115234 CrossRef CAS.
- (a) M. V. N. Raju, G. M. Reddy, B. J. Reddy, V. J. Rao and S. P. Parvathaneni, Pd(II) catalyzed regioselective ortho arylation of 2-arylpyridines, 1-phenyl-1H-pyrazoles, and N-pyridinylcarbazoles with diaryliodonium salts, Monatsh. Chem., 2022, 153, 1261 CrossRef CAS; (b) Y. He, L. Huang, L. Xie, P. Liu, Q. Wei, F. Mao, X. Zhang, J. Huang, S. Chen and C. Huang, Palladium-Catalyzed C−H Bond Functionalization Reactions Using Phosphate/Sulfonate Hypervalent Iodine Reagents, J. Org. Chem., 2019, 84, 10088 CrossRef CAS PubMed; (c) J. Yu, W. Lv and G. Cheng, Palladium-Catalyzed Site-Selective C–H Arylation of 2,2′-Bipyridine-6-carboxamides via a Rollover Cyclometalation Pathway, Org. Lett., 2018, 20, 4732 CrossRef CAS PubMed; (d) L. Su, D.-D. Guo, B. Li, S.-H. Guo, G.-F. Pan, Y.-R. Gao and Y.-Q. Wang, Palladium-Catalyzed Direct Monoarylation of Aryl C−H Bonds with Iodoarenes, ChemCatChem, 2017, 9, 2001 CrossRef CAS; (e) J. Feng, G. Lu, M. Lv and C. Cai, Palladium(II)-Catalyzed Oxidative ortho-Arylation of 2-Phenylpyridines, Synlett, 2013, 24, 2153 CrossRef CAS; (f) S. R. Neufeldt and M. S. Sanford, Combining Transition Metal Catalysis with Radical Chemistry: Dramatic Acceleration of Palladium-Catalyzed C-H Arylation with Diaryliodonium Salts, Adv. Synth. Catal., 2012, 354, 3517 CrossRef CAS PubMed; (g) W. Li, Z. Yin, X. Jiang and P. Sun, Palladium-Catalyzed Direct Ortho C-H Arylation of 2-Arylpyridine Derivatives with Aryltrimethoxysilane, J. Org. Chem., 2011, 76, 8543 CrossRef CAS PubMed; (h) D. Kalyani, K. B. McMurtrey, S. R. Neufeldt and M. S. Sanford, Room-Temperature C–H Arylation: Merger of Pd-Catalyzed C–H Functionalization and Visible-Light Photocatalysis, J. Am. Chem. Soc., 2011, 133, 18566 CrossRef CAS; (i) W.-Y. Yu, W. N. Sit, Z. Zhou and A. S.-C. Chan, Palladium-catalyzed decarboxylative arylation of C-H bonds by aryl acylperoxides, Org. Lett., 2009, 11, 3174 CrossRef CAS; (j) J.-H. Chu, S.-L. Tsai and M.-J. Wu, Palladium(II)-Catalyzed ortho Arylation of 2-Phenylpyridines with Potassium Aryltrifluoroborates by C-H Functionalization, Synthesis, 2009, 22, 3757 Search PubMed; (k) S. Kirchberg, T. Vogler and A. Studer, Directed Palladium-Catalyzed Oxidative C-H Arylation of (Hetero)arenes with Arylboronic Acids by Using TEMPO, Synlett, 2008, 2841 CAS.
- (a) N. Yoshikai, S. Asako, T. Yamakawa, L. Ilies and E. Nakamura, Iron-Catalyzed C–H Bond Activation for the ortho-Arylation of Aryl Pyridines and Imines with Grignard Reagents, Chem.–Asian J., 2011, 6, 3059 CrossRef CAS PubMed; (b) J. Norinder, A. Matsumoto, N. Yoshikai and E. Nakamura, Iron-Catalyzed Direct Arylation through Directed C−H Bond Activation, J. Am. Chem. Soc., 2008, 130, 5858 CrossRef CAS PubMed.
- B. Li, Z.-H. Wu, Y.-F. Gu, C.-L. Sun, B.-Q. Wang and Z.-J. Shi, Direct Cross-Coupling of C-H Bonds with Grignard Reagents through Cobalt Catalysis, Angew. Chem., Int. Ed., 2011, 50, 1109 CrossRef CAS PubMed.
- R. A. Ragtap, V. Soni and B. Punji, Expeditious and Solvent-Free Nickel-Catalyzed C−H Arylation of Arenes and Indoles, ChemSusChem, 2017, 10, 2242 CrossRef PubMed.
- T. Mandal, M. Mondal and J. Choudhury, Hypercrosslinked Polymer Platform-Anchored Single-Site Heterogeneous Pd–NHC Catalysts for Diverse C–H Functionalization, Organometallics, 2021, 40, 2443 CrossRef CAS.
- S. N. Georgiades, P. G. Nicolaou and N. Panagiotou, N-Directed Pd-Catalyzed Photoredox-Mediated C–H Arylation for Accessing Phenyl-Extended Analogues of Biginelli/Suzuki-Derived Ethyl 4-Methyl-2,6-diphenylpyrimidine-5-carboxylates, Catalysts, 2021, 11, 1071 CrossRef CAS.
- (a) H. Nagarajaiah, A. Mukhopadhyay and J. N. Moorthy, Biginelli reaction: An overview, Tetrahedron Lett., 2016, 57, 5135 CrossRef CAS; (b) Â. de Fátima, T. C. Braga, L. da Silva Neto, B. S. Terra, B. G. F. Oliveira, D. L. da Silva and L. V. Modolo, A mini-review on Biginelli adducts with notable pharmacological properties, J. Adv. Res., 2015, 6, 363 CrossRef PubMed.
- D. Russowsky, F. A. Lopes, V. S. S. da Silva, K. F. S. Canto, M. G. Montes D'Oca and M. N. Godoi, Multicomponent Biginelli's Synthesis of 3,4-Dihydropyrimidin-2(1H)-ones Promoted by SnCl2.2H2O, J. Braz. Chem. Soc., 2004, 15, 165 CrossRef CAS.
- (a) V. Singh, K. Rajput, P. Verma, S. Singh and V. Srivastava, A green approach for the synthesis of 2-oxo-1,2,3,4-tetrahydropyrimidines through oxidative functionalization of methyl arenes/benzyl derivatives via in situ generated urea, Res. Chem. Intermed., 2023, 49, 2969 CrossRef CAS; (b) A. N. Dadhania, V. K. Patel and D. K. Raval, A facile approach for the synthesis of 3,4-dihydropyrimidin-2-(1H)-ones using a microwave promoted Biginelli protocol in ionic liquid, J. Chem. Sci., 2012, 124, 921 CrossRef CAS; (c) H. Biklarian, F. K. Dehbahani and Z. Fakhroueian, 22% Co/CeO2-ZrO2-catalyzed Synthesis of 1,2,3,4-tetrahydro-2-pyrimidinones and -thiones, Lett. Org. Chem., 2012, 9, 580 CrossRef CAS; (d) H. Valizadeh and A. Shockravi, Imidazolium-Based Phosphinite Ionic Liquid as Reusable Catalyst and Solvent for One-Pot Synthesis of 3,4-Dihydropyrimidin-2(1H)-(thio)ones, Heteroat. Chem., 2009, 20, 284 CrossRef CAS; (e) K. Kaur and E. E. Knaus, Synthesis of alkyl 6-methyl-4-(2-pyridyl)-1,2,3,4-tetrahydro-2H-pyrimidine-2-one-5-carboxylates for evaluation as calcium channel antagonists, J. Heterocycl. Chem., 2007, 44, 745 CrossRef CAS; (f) R. Varala, M. M. Alam and S. R. Adapa, Bismuth Triflate Catalyzed One-Pot Synthesis of 3,4-Dihydropyrimidin-2(1H)-ones: An Improved Protocol for the Biginelli Reaction, Synlett, 2003, 67 CAS.
- (a) K. Yamamoto, Y. G. Chen and F. G. Buono, Oxidative Dehydrogenation of Dihydropyrimidinones and Dihydro-pyrimidines, Org. Lett., 2005, 7, 4673 CrossRef CAS PubMed; (b) Z.-J. Quan, F.-Q. Jing, Z. Zhang, Y.-X. Da and X.-C. Wang, Palladium(II) Catalyzed Suzuki/Sonogashira Cross-Coupling Reactions of Sulfonates: An Efficient Approach to C2-Functionalized Pyrimidines and Pyridines, Eur. J. Org Chem., 2013, 2013, 7175 CrossRef CAS.
- (a) I. M. Lagoja, Pyrimidine as constituent of natural biologically active compounds, Chem. Biodiversity, 2005, 2, 1 CrossRef CAS PubMed; (b) S. Prachayasittikul, R. Pingaew, A. Worachartcheewan, N. Sinthupoom, V. Prachayasittikul, S. Ruchirawat and V. Prachayasittikul, Roles of Pyridine and Pyrimidine Derivatives as Privileged Scaffolds in Anticancer Agents, Mini-Rev. Med. Chem., 2017, 17, 869 CAS; (c) R. Kaur, P. Kaur, S. Sharma, G. Singh, S. Mehndiratta, P. M. S. Bedi and K. Nepali, Anti-cancer pyrimidines in diverse scaffolds: A review of patent literature, Recent Pat. Anti-Cancer Drug Discovery, 2015, 10, 23 CrossRef CAS PubMed; (d) V. Sharma, N. Chitranshi and A. K. Agarwal, Significance and Biological Importance of Pyrimidine in the Microbial World, Int. J. Med. Chem., 2014, 2014, 202784 Search PubMed.
- J. Sandosham and K. Undheim, Synthesis of Pyrimidinyl Triflates and Palladium-catalyzed Coupling with Organotin and Organozic Reagents, Heterocycles, 1994, 37, 501 CrossRef CAS.
- C. Sicre, A. A. C. Braga, F. Maseras and M. M. Cid, Mechanistic insights into the transmetalation step of a Suzuki–Miyaura reaction of 2(4)-bromopyridines: characterization of an intermediate, Tetrahedron, 2008, 64, 7437 CrossRef CAS.
- (a) M. D. Lotz, N. M. Camasso, A. J. Canty and M. S. Sanford, Role of Silver Salts in Palladium-Catalyzed Arene and Heteroarene C–H Functionalization Reactions, Organometallics, 2017, 36, 165 CrossRef CAS; (b) K. L. Bay, Y.-F. Yang and K. N. Houk, Multiple roles of silver salts in palladium-catalyzed C-H activations, J. Organomet. Chem., 2018, 864, 19 CrossRef CAS.
- P. Ionita, The Chemistry of DPPH˙ Free Radical and Congeners, Int. J. Mol. Sci., 2021, 22, 1545 CrossRef CAS PubMed.
- Y. Zhang, W. Li, Z. Hu, X. Jing and L. Yu, Mo@PANI-catalyzed oxidative deoximation reaction, Chin. Chem. Lett., 2024, 35, 108938 CrossRef CAS.
- (a) D. C. Powers and T. Ritter, Bimetallic Redox Synergy in Oxidative Palladium Catalysis, Acc. Chem. Res., 2012, 45, 840 CrossRef CAS PubMed; (b) D. C. Powers, D. Benitez, E. Tkatchouk, W. A. Goddard III and T. Ritter, Bimetallic Reductive Elimination from Dinuclear Pd(III) Complexes, J. Am. Chem. Soc., 2010, 132, 14092 CrossRef CAS; (c) D. C. Powers, D. Y. Xiao, M. A. L. Geibel and T. Ritter, On the Mechanism of Palladium-Catalyzed Aromatic C-H Oxidation, J. Am. Chem. Soc., 2010, 132, 14530 CrossRef CAS PubMed; (d) D. C. Powers and T. Ritter, Bimetallic Pd(III) complexes in palladium-catalysed carbon–heteroatom bond formation, Nat. Chem., 2009, 1, 302 CrossRef CAS PubMed; (e) D. C. Powers, M. A. L. Geibel, J. E. M. N. Klein and T. Ritter, Bimetallic Palladium Catalysis: Direct Observation of Pd(III)−Pd(III) Intermediates, J. Am. Chem. Soc., 2009, 131, 17050 CrossRef CAS PubMed.
- (a) J. L. Appleton, V. Silber, L. Karmazin, C. Bailly, J.-C. Chambron, J. Weiss and R. Ruppert, A New Phenanthroline Ligand and the Spontaneous Resolution of its Homoleptic Copper(I) Complex, Eur. J. Org Chem., 2020, 2020, 7320 CrossRef CAS; (b) E. E. Galenko, M. S. Novikov, F. M. Shakirova, J. R. Shakirova, I. V. Kornyakov, V. A. Bodunov and A. F. Khlebnikov, Isoxazole Strategy for the Synthesis of 2,2′-Bipyridine Ligands: Symmetrical and Unsymmetrical 6,6′-Binicotinates, 2,2′-Bipyridine-5-carboxylates, and their Metal Complexes, J. Org. Chem., 2019, 84, 3524 CrossRef CAS PubMed; (c) M. Kuritani, S. Tashiro and M. Shionoya, Organic and Organometallic Nanofibers Formed by Supramolecular Assembly of Diamond-Shaped Macrocyclic Ligands and PdII Complexes, Chem.–Asian J., 2013, 8, 1368 CrossRef CAS PubMed; (d) A. Zucca, G. L. Petretto, M. L. Cabras, S. Stoccoro, M. A. Cinellu, M. Manassero and G. Minghetti, N^N^C platinum(II) and palladium(II) cyclometallates of 6,6’-diphenyl-2,2’-bipyridine, L: Crystal and molecular structure of [Pd(L–H)Cl], J. Organomet. Chem., 2009, 694, 3753 CrossRef CAS.
- (a) Q. Luo, J. Zhang and J. Xia, Developing strong NIR absorption materials through linear planar π-conjugated cyclopalladated complex dimers, Dalton Trans., 2021, 50, 1344 RSC; (b) X.-Q. Zhou, M. Xiao, V. Ramu, J. Hilgendorf, X. Li, P. Papadopoulou, M. A. Siegler, A. Kros, W. Sun and S. Bonnet, The Self-Assembly of a Cyclometalated Palladium Photosensitizer into Protein-Stabilized Nanorods Triggers Drug Uptake In Vitro and In Vivo, J. Am. Chem. Soc., 2020, 142, 10383 CrossRef CAS PubMed; (c) A. Zucca, M. A. Cinellu, M. V. Pinna, S. Stoccoro, G. Minghetti, M. Manassero and M. Sansoni, Cyclopalladation of 6-Substituted-2,2′-bipyridines. Metalation of Unactivated Methyl Groups vs Aromatic C-H Activation, Organometallics, 2000, 19, 4295 CrossRef CAS; (d) G. Minghetti, M. A. Cinellu, G. Chelucci and S. Gladiali, Palladium(II) and platinum(II) derivatives with chiral 2,2′-bipyridines. X-ray structure of C18H15ClN2Pd; C- and N-intramolecular coordination in a six-membered metallacycle, J. Organomet. Chem., 1986, 307, 107 CrossRef CAS.
- G. M. Sheldrick, A short history of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS.
- L. J. Farrugia, Synthesis, X-Ray Crystallography and DFT Studies of Ni(II) Complex with Tetradentate, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis pro-gram, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- A. L. Spek, Single-crystal structure validation with the program PLATON, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary synthetic methods and characterization data; 1H and 13C NMR spectra of synthesized compounds; crystal structures and single-crystal X-ray diffraction data. CCDC 2307358–2307364. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra02173h |
| ‡ Current address: Department of Chemistry, University of Liverpool, Crown Street, Liverpool L69 7ZD, UK. |
| This journal is © The Royal Society of Chemistry 2024 |