
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ethynyl-substituted benzosiloxaboroles: the role of C(π)⋯B interactions in their crystal packing and use in Cu(I)-catalyzed 1,3-dipolar cycloaddition†
P. Pacholakab,
K. Durka *a,
K. Woźniakb,
J. Krajewskac,
A. E. Laudy*c and
S. Luliński*a
*a,
K. Woźniakb,
J. Krajewskac,
A. E. Laudy*c and
S. Luliński*a
aFaculty of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warsaw, Poland. E-mail: krzyysztof.durka@pw.edu.pl; sergiusz.lulinski@pw.edu.pl
bUniversity of Warsaw, Faculty of Chemistry, Pasteura 1, 02-093 Warsaw, Poland
cDepartment of Pharmaceutical Microbiology and Bioanalysis, Medical University of Warsaw, Banacha 1b, 02-097 Warsaw, Poland. E-mail: alaudy@wp.pl
First published on 17th May 2024
Abstract
The synthesis and characterization of two novel 6-ethynyl-7-halogen substituted benzosiloxaboroles (Hal = F, Cl) is reported. The crystal structures of these compounds show a unique type of supramolecular assembly dictated by distinctive C(π)⋯B interactions resulting in the formation of columnar networks involving alternating ethynyl groups and boron atoms. The QTAIM, NBO and NCI analyses were performed in order to obtain a deeper quantitative insight into the nature of these interactions including energy and charge density distribution. The fluoro derivative 1c was used as a starting material in Cu-catalyzed 1,3-dipolar cycloaddition reactions with substituted benzenesulfonyl azides giving rise to benzosiloxaboroles with pendant 1-(arylsulfonyl)-1,2,3-triazole-4-yl functionalities or analogous ionic species, i.e., 1,2,3-triazolium arylsulfonates. Screening of antimicrobial activity of obtained derivatives against a wide selection of Gram-positive and Gram-negative bacteria as well as fungi strains was performed and the obtained results were compared with the data obtained previously for related benzosiloxaborole derivatives.
1 Introduction
A specific class of cyclic boron hemiesters called benzoxaboroles have been known for 70 years1 but they have attracted increased attention only during the last 15 years in the field of medicinal chemistry.2 Extensive studies revealed that functionalized benzoxaboroles are promising small-molecule therapeutic agents possessing antibacterial, antifungal, anti-inflammatory and anticancer activity.3 For example, fluorinated benzoxaborole called tavaborole (trade name Kerydin) exhibits high antifungal activity and was commercialized to treat onychomycosis – a fungal infection of the nail and nail bed.4 5-(4-Cyanophenoxy)benzoxaborole called crisaborole (trade name Eucrisa) was approved for the treatment of mild-to-moderate atopic dermatitis (eczema).5 The activity of benzoxaboroles relies primarily on the binding of the Lewis acid boron centre to the biological target via the formation of a strong covalent bond. The mechanism of action of benzoxaboroles relies on their physicochemical specificity based on the enhanced character of the boron atom. The examples of approved benzoxaborole therapeutic agents as well as other derivatives which are currently under investigation demonstrate that the mode of bioactivity depends on structural modification of the benzoxaborole core. In 2015, our group proposed benzosiloxaboroles as silicon congeners of benzoxaboroles and demonstrated high antifungal activity of simple fluorinated derivatives.6 Further research revealed the potency of selected derivatives including oxaborole–benzosiloxaborole hybrid II as KPC β-lactamase inhibitors.7 Recently, we found that the antibacterial activity of benzosiloxaboroles involves a different mechanism than for related benzoxaboroles pointing to the importance of the introduced structural change.8 In fact, replacement of the CH2 with the SiMe2 group in the oxaborole ring results in increased Lewis acidity and lipophilicity which may be responsible for specific biological activity. Recently, we have also studied crystal structures of functionalized benzosiloxaboroles showing that there is a strong effect of substitution pattern on their supramolecular assembly, mostly manifested by formation of various hydrogen-bonded motifs.9 Those results revealed that benzosiloxaboroles are interesting also from the perspective of crystal engineering.In medicinal chemistry, the conjugation a core of a molecule with different pharmacophores is one of key synthetic concepts as resulting hybrid structures may show higher affinity to a biological target. Herein, we turned our attention to triazole scaffold as it is present in many compounds showing diverse biological properties. For example, vicinal diaryl triazoles were found to be promising tubulin polymerization inhibitors, COX-2 inhibitors, and CB1 receptor antagonists.10 Essramycin (Scheme 2), is (1,2,4-triazolo)pyrimidine derivative active against several Gram-positive and Gram-negative bacteria such as Bacillus subtilis, Staphylococcus aureus, Micrococcus luteus, E. coli, and Pseudomonas aeruginosa.11 Triazole-containing compounds are widely used as topical and systemic antifungal agents. Fluconazole, the first-generation 1,2,4-triazole antifungal agent, is used for fungal infections, including candidiasis, blastomycosis, cryptococcosis, histoplasmosis, coccidioidomycosis, dermatophyteosis and pityriasis versicolor.12 1,2,3-Triazole moiety is a part of tazobactam – inhibitor of β-lactamases from class A according to Ambler classification (especially SHV, TEM and CTX-M enzymes).13 Tazobactam combined with the β-lactam antibiotic such as piperacillin is used to treat infections caused by Pseudomonas aeruginosa. Moreover, tazobactam with the new cephalosporin ceftolozane is dedicated for treatment of the extended-spectrum β-lactamase (ESBL)-producing Enterobacterales and multidrug-resistant (MDR) P. aeruginosa infections.13b Rufinamide is a new anti-epileptic drug used in combination with other medication and therapy to treat Lennox–Gastaut syndrome and various other seizure disorders.14 In the context of this work, a recent synthesis of a novel boronic acid (MB076) with a pendant 1,2,3-triazole-4-carboxylic acid moiety as a highly effective inhibitor of class C Acinetobacter-derived cephalosporinases is interesting.15 6-Substituted triazolyl benzoxaboroles were very recently identified as selective carbonic anhydrase inhibitors.16
As a continuation of our studies on functionalized benzosiloxaboroles, we present herein the synthesis and in-depth structural characterization of ethynyl-substitited derivatives which highlights the effect of the π-hole on the boron atom on crystal packing. These relatively simple compounds were converted to derivatives bearing 1-arylsulfonyl-1,2,3-triazole-4-yl moiety. Such a structural design was inspired by our previous results showing that arylsulfonamido17 arylsulfonato,18 and (β-phenylsulfonyl)ethylthio8 substituted benzosiloxaboroles (Scheme 1, general structures III, IV, and V, respectively) exhibit high activity against the Gram-positive cocci such as methicillin-sensitive Staphylococcus aureus (MSSA), methicillin-resistant S. aureus (MRSA), Enterococcus faecalis and Enterococcus faecium. Based on the presented literature background,10–16 we assumed that the introduction of 1,2,3-triazole spacer between arylsulfonyl group and benzosiloxaborole might have further positive impact on the antimicrobial potency.
 | ||
| Scheme 1 Examples of biologically active benzosiloxaboroles. | ||
 | ||
| Scheme 2 Biologically active compounds comprising 1,2,4- and 1,2,3-triazole rings including recent examples of organoboron compounds. | ||
2 Results and discussion
2.1 Synthesis and crystal structure of 6-ethynyl-7-halogen substituted benzosiloxaboroles 1c and 2c
In a three-step procedure, 4-bromo-2-fluoro-1-iodobenzene 1 (Scheme 3) was converted to the respective TMS-protected ethynyl derivative 1a via Sonogashira coupling as reported previously19 followed by deprotonation with LDA at −78 °C and trapping of corresponding aryllithium intermediates with Me2Si(H)Cl to give arylsilane 1b in high yield. The lithiation occurred regioselectively at the 3-position flanked by two halogens which is activated by a strong cumulated ortho-acidifying effect of those two substituents.20 Compound 1b was subjected to Br/Li exchange with t-BuLi in Et2O at −100 °C followed by trapping with B(OMe)3. The hydrolysis effected with 1 M NaOH/H2O mixture resulted in a cleavage of the Si–H bond which occurs rapidly due to ortho-assistance of the anionic boronate group.21 The strongly alkaline conditions result also in removal of the trimethylsilyl group leading directly to the targeted derivative 1c which is the first example of the ethynyl-substituted benzosiloxaborole (Scheme 3). 7-Chloro-6-ethynyl derivative 2c was obtained using a similar three-step protocol starting with 4-bromo-2-chloro-1-iodobenzene 2. The overall yields of 1c and 2c were 77 and 62%, respectively. Both products were isolated as powders well soluble in most organic solvents. The 1H and 13C NMR spectra of 1c and 2c are in agreement with their structures whilst 11B NMR resonances at ca. 30 ppm are typical of other benzosiloxaboroles.6 It is worth noting that according to our best knowledge there are no published examples of ethynyl-substituted benzoxaboroles, i.e., direct analogues of 1c and 2c. | ||
| Scheme 3 Synthesis of 6-ethynyl-7-halogen substituted benzosiloxaboroles 1c and 2c. | ||
2.2 Crystal structures 1c and 2c: the role of C(π)⋯B interactions
The geometry of benzosiloxaborole core in 1c is essentially the same as in related compounds reported by us previously.9 The supramolecular assembly of 1c shows centrosymmetric hydrogen-bonded (HB) dimers (O⋯O distance of 2.784(1) Å, motif D1, Fig. 1a) typical of boronic acid derivatives22 including most of benzoxaboroles and many benzosiloxaboroles.6,22 However, the molecules within a dimer are not coplanar but they are significantly displaced one to another so the angle between mean planes defined by the four O atoms of the dimeric motif and the atoms of the siloxaborole ring is 26.7°. Such geometrical deformations are also reflected in decreased interaction energy between molecules. According to DFT calculation performed at M06-2X/6-311++G(d,p) level of theory, the D1 dimer interaction energy in 1c is equal to 53.4 kJ mol−1, i.e., lower compared to typical HB dimers in benzoxa- and benzosiloxaboroles (∼58 kJ mol−1). In addition, boron atoms show characteristic interactions with the π-density of two ethynyl groups of neighboured molecules (motifs D2a and D2b, Fig. 1c) characterized by respective intermolecular B⋯C(terminal) contacts of 3.394(2) (motif D2a) and 3.626(2) Å (motif D2b). The C(π)⋯B interactions, which can be considered as a type of π-hole triel bonds,23 are well visible on the Hirshfeld surfaces mapped over the dnorm property as shown in Fig. 2a. Specifically, the two major red spots are associated to O–H⋯O hydrogen bonds, while the two other red spots visible above the boron atom and ethynyl group correspond to C(π)⋯B interactions. Overall, the propagation of D2 dimeric motifs along the [100] direction leads to the formation of a 1D columnar assembly (Fig. 1c). The important contribution from π-stacking interaction between parallel aromatic rings should be also encountered thus giving rise to quite significant D2a and D2b dimer interaction energies of 34.5 and 37.3 kJ mol−1, respectively (Table 1).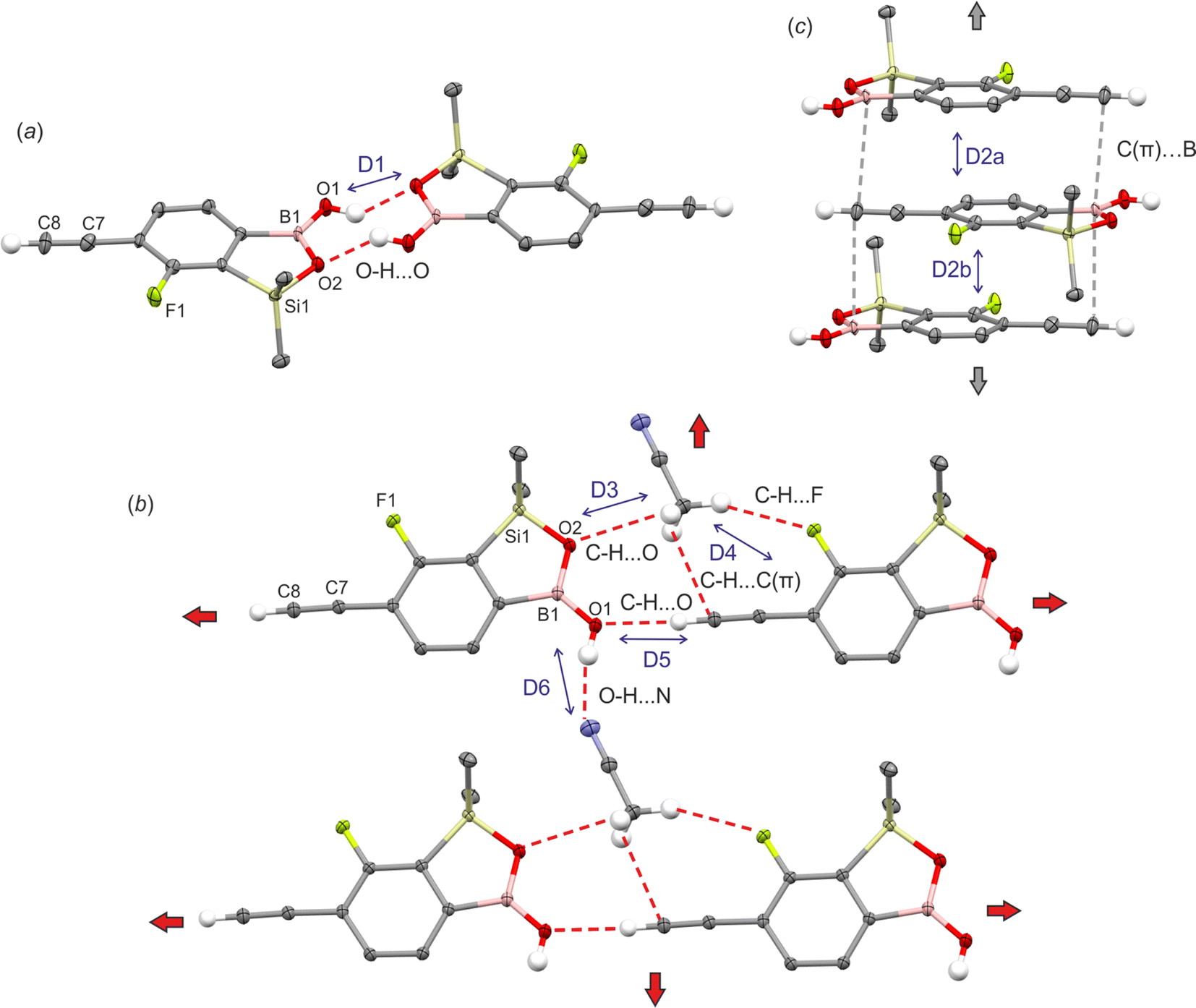 | ||
| Fig. 1 Fragments of crystal structures of 1c and 1c·MeCN showing the formation of (a) centrosymmetric hydrogen-bonded dimer in 1c, (b) molecular layer in 1c·MeCN and (c) columnar motifs (1c, 2c and 1c·MeCN) held by C(π)⋯B interactions. Note that two different D2 motifs (D2a and D2b) are distinguished in case of 1c and 2c. For 1c·MeCN the symmetry center appears between each pair of molecules within D2 motif. | ||
 | ||
| Fig. 2 (a) Hirshfeld surfaces generated for 1c molecule with mapped dnorm property value over the range −0.50 to 1.30. (b) NBO orbitals participating in the intermolecular interaction with ethynyl group. (c) Combined QTAIM and NCI analysis of 1c. Bond critical points are represented by yellow spheres, BCP corresponding to C(π)⋯B interaction is represented by blue sphere. The color scale for the NCI plot is −0.01 < sign(λ2)ρ < 0.01; Reduced gradient density (RGD) isosurface of 0.3. The green color of RGD surface represents weak attractive interaction. | ||
| Structure | Motif | Interaction | dX⋯Ya/Å | Eintb/kJ mol−1 |
|---|---|---|---|---|
| a The intermolecular distance between heavy atoms.b Interaction energies calculated at M06-2X/6-311++G(d,p) level of theory, positions of non-hydrogen atoms were retained from the crystal structure, and positions of hydrogen atoms were fully optimized. | ||||
| 1c | D1 | O–H⋯O | 2.784(1) | 53.4 |
| D2a | C(π)⋯B | 3.394(2) | 34.5 | |
| D2b | C(π)⋯B | 3.626(2) | 37.3 | |
| 2c | D1 | O–H⋯O | 2.793(2) | 50.7 |
| D2a | C(π)⋯B | 3.362(2) | 41.4 | |
| D2b | C(π)⋯B | 3.640(2) | 42.2 | |
| 1c·MeCN | D2 | C(π)⋯B | 3.528(2) | 45.3 |
| D3 | C–H⋯O | 3.189(2) | 24.0 | |
| D4 | C–H⋯F | 3.542(2) | 8.3 | |
| C–H⋯C(π) | 3.267(2) | |||
| D5 | C–H⋯O | 3.155(1) | 10.0 | |
| D6 | O–H⋯N | 2.853(2) | 28.9 | |
In order to get deeper insight into the nature of C(π)⋯B interaction we have performed natural bond orbital (NBO) analysis and, in parallel, topological analysis of electron density within quantum theory of atoms in molecules (QTAIM) approach. The energetic contributions of intermolecular donor–acceptor interactions was estimated by 2nd-order perturbation theory (Table S1, ESI†). It comes out that the formation of C(π)⋯B interaction mostly originates from electron donation from ethynyl πCC orbital to empty pB orbital (orbital interaction energy E = 5.1 kJ mol−1 in D2a and E = 2.6 kJ mol−1 in D2b dimers, Fig. 2b). It is supported by the back-donation from phenyl π-electron density (represented by localized πCC orbital) to antibonding π*CC of ethynyl group (E ≈ 1.0 kJ mol−1). In addition, the non-covalent interaction index (NCI) was calculated for 2Da dimer. It revealed the typical pattern of π-stacking interactions reflected by the green area covering almost the whole interface between two adjacent aromatic rings (Fig. 2c). The reduced gradient density (RGD) isosurface is extended on the regions corresponding to the C(π)⋯B contact confirming the weak attractive character of this interaction. The QTAIM analysis revealed the appearance of bond critical point (BCP) located in-between the ethynyl group and boron atom with the electron density (ρ) of 0.036 e Å−3 and negative Laplacian (∇2ρ) of 0.34 e Å−5. The amount of electron density at BCP is slightly lower compared to the value found at BCP in model BCl3–ethynyl complex (ρ = 0.045 e Å−3) studied by Grabowski,24 and lies in the range of typical density values for two stacking aromatic rings (0.03–0.07 e Å−3).25 At this point, it should be noted that the bond path does not directly connect the ethynyl triple bond and boron atom but it ends at BCP of the B–C bond. Similar effect was already described for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(π)⋯B interaction in the crystal structure of (E)-(4-phenylbut-1-en-1-yl)boronic acid.26 The bond path might not be revealed from QTAIM analysis despite an attractive character of the C(π)⋯B interaction. Moreover, the presence of electron rich oxygen atoms at the boron atom may hinder the formation of the mentioned bond path.
C(π)⋯B interaction in the crystal structure of (E)-(4-phenylbut-1-en-1-yl)boronic acid.26 The bond path might not be revealed from QTAIM analysis despite an attractive character of the C(π)⋯B interaction. Moreover, the presence of electron rich oxygen atoms at the boron atom may hinder the formation of the mentioned bond path.
The crystallization of 1c in acetonitrile afforded monosolvate 1c·MeCN. Unlike crystal structures of most benzoxaboroles including the case of non-solvated 1c, in 1c·MeCN the BOH group adopts anti conformation. The O atom of this group serves as a hydrogen bond acceptor for the ethynyl H atom of a neighbored molecule (dO⋯H = 2.21 Å, dC⋯O = 3.155(1) Å, motif D5) which results in the formation of a chain running along the c axis (Fig. 1b). The chains are cross-linked through interactions with MeCN molecules. First, the H atom of the BOH group is involved in O–H⋯N hydrogen bond (dN⋯H = 2.03 Å, dO⋯N = 2.853(2) Å, motif D6). In addition, the methyl group of the solvent guest molecule is anchored to two molecules of 1c through weaker C–H⋯F (dC⋯F = 3.542(2) Å, motif D4), C–H⋯O (dC⋯O = 3.189(2) Å, motif D3) and C–H⋯C(π-ethynyl) (dC⋯C = 3.267(2) Å, as a part of motif D4) interactions. This results in a creation of a layer parallel to the (010) plane. The layers are generally assembled through π-stacking interactions but again characteristic intermolecular B⋯C(terminal) symmetrical contacts of 3.528(2) Å with the π-density of two ethynyl groups of neighboured molecules from both adjacent layers are observed. The interaction energy between molecules within D2 dimer is quite significant and equal to 45.3 kJ mol−1.
Compound 2c forms non-solvated crystals both from CHCl3 and MeCN solution. It is isostructural with 1c which means that the replacement of fluorine with chlorine has a marginal effect on the supramolecular assembly. As for 1c, centrosymmetric dimers are formed (O⋯O) distance of 2.793(2) Å, i.e., by only 0.005 Å longer compared to (1c) and molecules within a dimer are significantly displaced one to another at the same angle of 25.9° as for 1c. Owing to the longer O⋯O distance, the interaction energy between molecules is slightly lower compared to 1c (50.7 kJ mol−1). The C(π)⋯B interactions are characterized by respective intermolecular B⋯C(terminal) contacts of 3.362(2) and 3.640(2) Å, i.e., distance alternation is slightly smaller than for 1c. This resulted in increased intermolecular interaction energy for D2a (41.4 kJ mol−1) and D2b (42.2 kJ mol−1) dimers. Finally, C(π)⋯B orbital interaction energies and AIM parameters at corresponding BCPs are comparable for all studied systems (Tables S1.3 and S1.4, ESI†).
2.3 The use of 1c in Cu(I)-catalyzed 1,3-dipolar cycloaddition
Compound 1c was utilized in Cu(I)-catalyzed azide–alkyne 1,3-dipolar cycloaddition (CuAAC) reactions with selected organic azides with a special emphasis on arylsulfonyl azides (easily accessible from the nucleophilic substitution of respective chlorides with NaN3 in acetone27). Previously, thermally induced azide–alkyne cycloaddition was reported for the preparation of a some boronated 1,2,3-triazoles.28 There are only a few reports on arylboronic-triazole conjugates up-to-date. For the first time, such compounds synthesized via CuAAC were reported by Hall et al.29 Kumar et al. synthesized a library of 1H-1,2,3-triazole-tethered 4-aminoquinoline-benzoxaborole hybrids and aryl-substituted benzoxaborole analogues. Obtained products were screened for their anti-plasmodial efficacy against both chloroquine-susceptibility 3D7 and chloroquine-resistant W2 strains of P. falciparum.30 The Passerini three component selective synthesis of benzoxaboroles with pendant tetrazole substituents shows also some resemblance to the present work due to the use of boronic acid and azide precursors.31 The standard CuSO4/sodium ascorbate catalytic system32 proved not effective in our CuAAC-based protocol utilizing 1c and various arylsulfonyl azides as starting materials. In contrast, Cu(I) thiophene-2-carboxylate (CuTC, 0.1 equiv.) proved more effective as a catalyst while the reactions were conducted in a biphasic water/toluene mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).33 However, the use of simple benzenesulfonyl azide did not afford the expected cycloaddition product. Instead, the respective 4-(7-fluoro-3-hydroxy-1,1-dimethyl-1,3-dihydrobenzo[c][1,2,5]oxasilaborol-6-yl)-1H-1,2,3-triazol-3-ium salt with the benzenesulfonate counterion 3a, resulting from the hydrolytic abstraction of benzenesulfonyl group, was isolated. An analogous result was obtained with 4-chlorophenylsulfonyl azide. Treatment of 3a with aq. NaOH followed by careful acidification with aq. HCl to pH = 5 afforded the neutral product 3c. However, in selected cases it was possible to isolate intermediates, i.e., the elaborated protocol allowed for the synthesis of a series of functionalized benzosiloxaboroles 4a–4c (Scheme 4). The products of studied CuAAC reactions were obtained in moderate yields (33–68%). Overall, the cycloaddition reaction proceeds smoothly but it is difficult to control the subsequent potential abstraction of arylsulfonyl moiety.
:1).33 However, the use of simple benzenesulfonyl azide did not afford the expected cycloaddition product. Instead, the respective 4-(7-fluoro-3-hydroxy-1,1-dimethyl-1,3-dihydrobenzo[c][1,2,5]oxasilaborol-6-yl)-1H-1,2,3-triazol-3-ium salt with the benzenesulfonate counterion 3a, resulting from the hydrolytic abstraction of benzenesulfonyl group, was isolated. An analogous result was obtained with 4-chlorophenylsulfonyl azide. Treatment of 3a with aq. NaOH followed by careful acidification with aq. HCl to pH = 5 afforded the neutral product 3c. However, in selected cases it was possible to isolate intermediates, i.e., the elaborated protocol allowed for the synthesis of a series of functionalized benzosiloxaboroles 4a–4c (Scheme 4). The products of studied CuAAC reactions were obtained in moderate yields (33–68%). Overall, the cycloaddition reaction proceeds smoothly but it is difficult to control the subsequent potential abstraction of arylsulfonyl moiety.
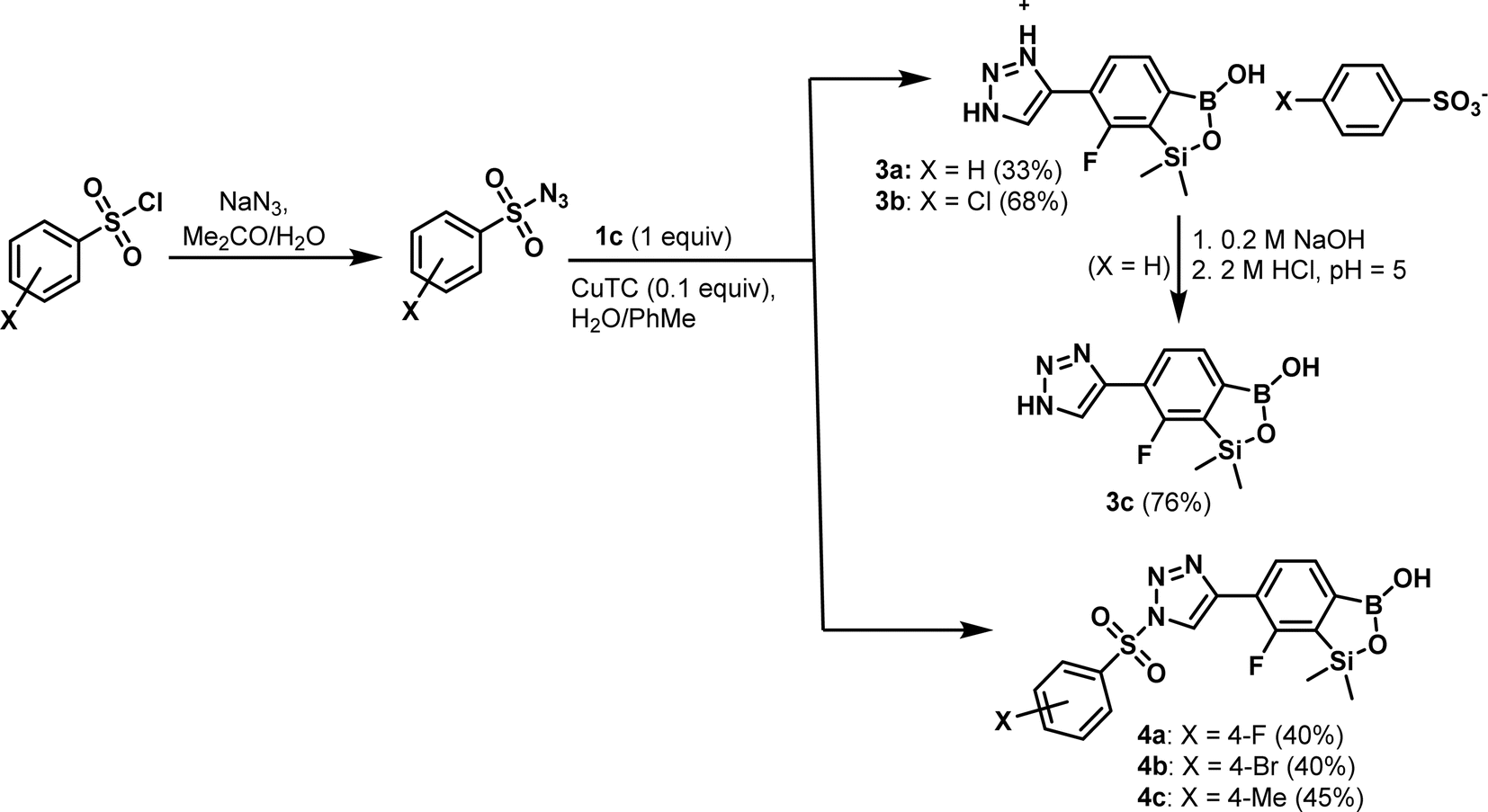 | ||
| Scheme 4 Application of Cu(I)-catalyzed azide–alkyne 1,3-dipolar cycloaddition for the synthesis of 1,2,3-triazole-4-yl functionalized benzosiloxaboroles 3a–3c and 4a–4c. | ||
All products were characterized by 1H, 13C and 19F NMR spectroscopy as well as HRMS. In the NMR spectra of 4a–4c the signal of the proton at the 5-position of 1,2,3-triazole ring at ca. 8.8–9.1 ppm appears always as a doublet with a coupling constant of ca. 3.5 Hz which may be attributed the existence of a through-space 1H–19F coupling with the fluorine atom. In addition, compound 4d bearing the pendant 2-fluorophenylsulfonyl moiety was characterized by single crystal X-diffraction (Fig. 3a) as a monosolvate 4d CHCl3. It should be noted that we were unable to obtain pure bulk 4d and the crystal was grown from the mixture containing mainly a respective ionic species analogous to 3a–3b. In the molecule 4d the triazole ring is essentially coplanar with the benzosiloxaborole core (the interplanar angle of benzene and triazole ring is 10.5°) as that conformation apparently gains stabilization owing to the short intramolecular C10(H)⋯F1 contact of 2.37 Å which is in agreement with 1H NMR data. Compound 4d also forms dimers but the angle of displacement of molecules within a dimer (defined above for 1c) is only 10.3° (Fig. 4a). The packing also shows stacking of the adjacent molecules characterized by interactions of the boron atom with the π-density of the triazole rings. The distance between the B atom and triazole ring centroid is 3.61 Å. The partially disordered CHCl3 molecules are closely paired34 in the crystal structure cavities and show H-bonding interactions with triazole rings as the C19(H)⋯N3 distance is relatively short (3.193 Å).
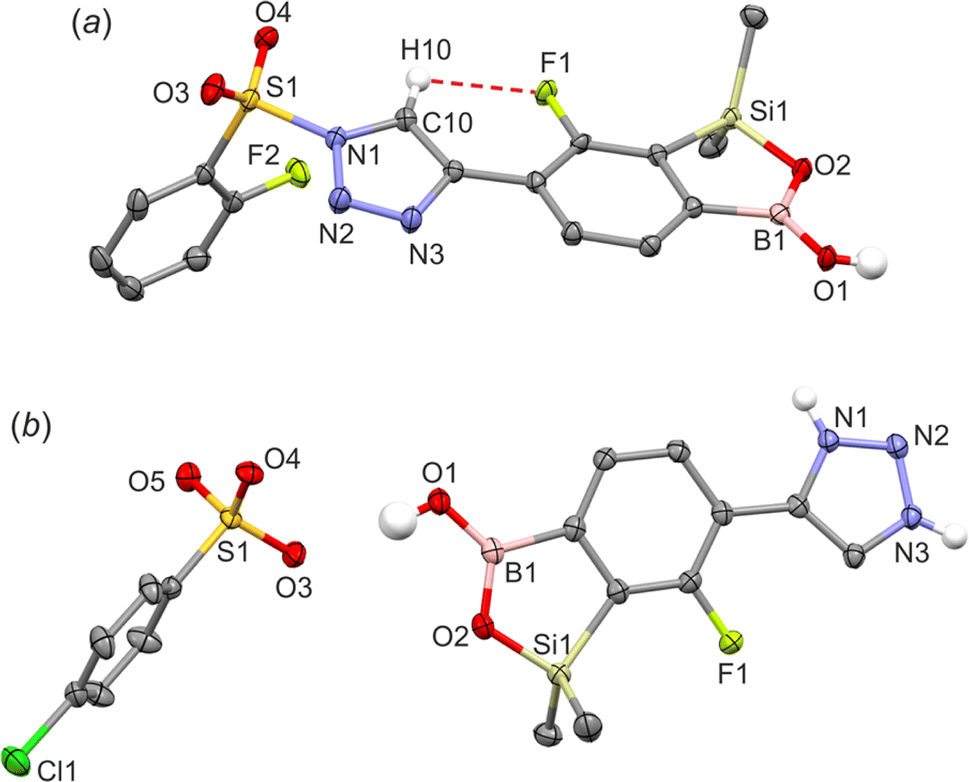 | ||
| Fig. 3 The molecular structures of (a) 4d and (b) 3b. | ||
 | ||
| Fig. 4 Fragments of the supramolecular structures of (a) 4d and (b) 3b. | ||
The geometry of the cationic part of compound 3b is similar to that of the analogous fragment in 4d (Fig. 3b). Unlike 4d, the cations do not assemble through the formation of centrosymmetric dimeric motifs involving B(O)OH groups. Instead, two types of charge-assisted NH⋯O hydrogen bonds (dN⋯O = 2.684 Å, dN⋯O = 2.669 Å) link cationic triazolium moieties with two neighbored arylsulfonate anions (Fig. 4b). This is complemented by another hydrogen bond formed between the BOH group and the third oxygen atom of the sulfonate group (dO⋯O = 2.882 Å). Overall, this results in a layer network located symmetrically with respect to the (100) plane. The layers interact by means of weak C–H⋯Cl interactions of methyl groups with chloro substituents. In addition, we attempted to grow single crystals of the neutral compound 3c. However, we obtained invariably an amorphous material which can be attributed to dynamic equilibria between various forms of 3c, presumably involving proton exchange at the triazole ring and/or aggregation processes involving formation of N–B coordination bonds. This is indicated by strongly broadened signals in the 1H NMR spectrum of 3c (Fig. S3.17†).
We have also studied the reactions of 1c with functionalized alkyl azides XCH2CH2N3 (X = OH, NHSO2Ph). Since CuTC was not effective as a catalyst in both cases, we decided to use the classical system CuSO4/sodium ascorbate in H2O/THF mixture. This approach did not result in expected benzosiloxaboroles as cycloaddition reactions were accompanied by extensive protodeboronation, presumably catalyzed by Cu2+ cations. Thus, the respective structurally extended 1,3-diaryl-1,1,3,3-tetramethyldisiloxanes 5a–5b were isolated in reasonable yields as isolable products of a dehydrative condensation of initially formed aryldimethylsilanols (Scheme 5). This was confirmed for 5b by X-ray crystallography showing a specific curled conformation of the molecule stabilized through two intramolecular NH⋯N hydrogen bonds (dN⋯N = 2.922 Å, dN⋯N = 2.929 Å) linking sulfonamide groups and triazole rings (Fig. 5). In addition, π–π stacking interactions of internal benzene and triazole rings play also some role (in the former case, it would be probably more precise to invoke mutual C–F dipole–dipole or C–F⋯π interactions).
 | ||
| Scheme 5 Formation of functionalized 1,3-diaryl-1,1,3,3-tetramethyldisiloxanes 5a–5b. | ||
 | ||
| Fig. 5 The molecular structure of 5b viewed from two different perspectives. | ||
2.4 Antimicrobial activity
Taking into account the direct antimicrobial activity of various benzosiloxaborole derivatives demonstrated previously6,7,17,18,35 in the current research we determined the activity of the newly synthesized compounds against a wide panel of Gram-positive, Gram-negative bacteria and yeasts strains. All obtained results were summarized in Tables S2.1 and S2.3 in the ESI.† 8 of 10 tested compounds displayed moderate to weak activity against Gram-positive cocci, with MICs ranging from 12.5 to 200 mg L−1 (Tables 2 and S2.1†). Among these active compounds, derivatives 2c and 3c were highly active against standard staphylococci, including MRSA, with MIC 12.5 mg L−1. Despite the approval of new drugs and actions taken around the world to limit the spread of drug-resistant strains, MRSA strains are still one of the most common pathogens causing nosocomial infections. MRSA strains have been entered on the WHO list of the most dangerous pathogens for humans.36 It is worth emphasizing that MRSA strains are resistant to almost all β-lactams (except ceftaroline and ceftobiprole) and are often resistant to many other groups of antibiotics as macrolides, tetracyclines, aminoglycosides, and fluoroquinolones.37,38 However, the most promising compounds 2c and 3c, showed from 6- to 12-fold less activity than linezolid as a reference agent. Linezolid is a representative of a relatively new group of drugs active against cocci, including MRSA strains.38 In our previous publication, we reported that among benzenesulfonato benzosiloxaborole derivatives display the highest potency (MICs 0.39–3.12 mg L−1) against Staphylococcus spp.18 However, expanding the structure of compounds tested in this study with arylsulfonyl group resulted in a reduction in activity against staphylococci and enterococci (from 2- to 4-fold reduction in the MIC values of compounds 3a, 3b, 4b, and 4c, compared to 3c) (Table 2). It seems that this can be attributed to the facile abstraction of the arylsufonyl group under aqueous conditions resulting in the formation of respective 1,2,3-triazolium arylsulfonates where only the cationic part comprising the siloxaborole ring exhibits antibacterial acitivity. For most compounds, the minimal bactericidal concentration (MBC) values were high (>400 mg L−1). Notably, the so-called Eagle effect (also known as the paradoxical growth reported for various antibiotics)39 was observed in the case of S. aureus ATCC 6538P (for 1c, 2c and 3c) and S. epidermidis ATCC 12228 (for 1c and 3c). It implies occurrence of two MBC values as determined by us previously for other substitued benzosiloxaboroles.17,18 The first MBC values were 2-fold higher than the MIC values. Furthermore, a progressive increase in the number of surviving bacteria was observed at higher concentrations followed by a subsequent decrease. If the bacterial population was reduced again to the MBC threshold, a second MBC value (at 200 ≥ 400 mg L−1) could be determined.| Agent tested | MICb in mg L−1 [MBC in mg L−1] | ||||
|---|---|---|---|---|---|
| S. aureus ATCC 6538P | S. aureus ATCC 43300 MRSA | S. epidermidis ATCC 12228 | E. faecalis ATCC 29212 | E. faecium ATCC 6057 | |
| a The highest activity against Gram-positive bacteria indicated by the low MIC values (≤12.5 mg L−1) is shown in boldface. (—) the inhibition zone was not observed in the disc-diffusion method. The diameter of the paper discs was 9 mm.b Only the MBC values ≤400 mg L−1 are presented.c The Eagle effect39 was observed during the determination of the MBC value of the same tested agents against Staphylococcus spp. strains. The Eagle effect is shown in the italic face.d LIN, linezolid was used as a reference agent active against Gram-positive bacteria. The diameter of a commercial disc containing 0.03 mg of linezolid was 6 mm; the MIC of linezolid was determined according to the CLSI recommendations.40 | |||||
| 1c/147 | 25 [50/400]c | 25 [50] | 12.5 [25/>400]c | 100 | 100 |
| 2c/253 | 12.5 [25/200]c | 12.5 [25] | 12.5 [400] | 50 | 50 |
| 3a/166 | 25 | 50 | 50 | 200 | 200 |
| 3b/170 | 50 | 50 | 50 | 200 | 200 |
| 3c/254 | 12.5 [25/>400]c | 12.5 [25] | 12.5 [25/>400]c | 100 | 50 |
| 4a/167 | 25 | 50 | 50 | 200 | 200 |
| 4b/169 | 25 | 50 | 50 | 200 | 200 |
| 4c/171 | 25 | 50 | 50 | 200 | 200 |
| LINd | 1 [>128] | 2 [>128] | 1 [>128] | 2 [>128] | 2 [>128] |
Only 1c and 2c displayed weak activity against Gram-negative rods with MICs ranging from 200 to >400 mg L−1 (Table S2.2†). Considering that resistance of Gram-negative rods is frequently associated with efflux pump activity,41 we also determined the MICs of newly compounds in the presence of efflux pump inhibitor, i.e., phenylalanine-arginine-β-naphthylamide (PAβN).42 In the case of Escherichia coli, Klebsiella pneumoniae, Serratia marcescens and Stenotrophomonas maltophilis strains the MICs of compounds 1c, 2c and 3c are reduced at least 4-fold in the presence of PAβN. This means that these compounds are actively removed by efflux pumps of bacterial cells.
Considering previously reported antifungal activity of some benzosiloxaboroles (with MICs ranging from 0.78–12.5 mg L−1)6,7 we investigated the activity of new derivatives against 7 standard yeast strains. The moderate antifungal activity was observed only for 1c and 2c with MICs ranging from 12.5 to 400 mg L−1 (Table S2.3†).
3 Conclusions
In conclusion, a convenient protocol for the synthesis of halogenated benzosiloxaboroles 1c and 2c comprising reactive ethynyl substituents was elaborated. It should be noted that analogous functionalized benzoxaboroles are still unknown which strengthens the importance of presented results. Intriguingly, C(π)⋯B interactions seem to play a decivise in role in supramolecular organization of these compounds which is confirmed by the fact that it is retained in the crystal structure of the solvate 1c MeCN. This observation indicates a high potential of the ethynyl group in crystal engineering of boracyclic compounds. CuAAC reaction of 1c using arylsulfonyl azides gave rise to respective products 4a–4c bearing 1-(arylsulfonyl)-1H-1,2,3-triazol-4-yl functionalities. However, compounds of this type are rather prone to hydrolysis as evidenced by the isolation of 1,2,3-triazolium arylsulfonate salts 3a–3b and the neutral benzosiloxaborole 3c with attached 1,2,3-triazol-4-yl moiety. In turn, the analogous cycloaddition reactions with functionalized alkyl azides XCH2CH2N3 (X = OH, NHSO2Ph) were accompanied by extensive protodeboronation and thus furnished respective 1,3-diaryl-1,1,3,3-tetramethyldisiloxanes 5a–5b as isolable products. Structural studies of final products were complemented by evaluation of antimicrobial actitivity which revealed that the ethynyl-substituted derivatives 1c and 2c are more potent antibacterial agents than structurally expanded 1,2,3-triazolyl derivatives. Based on previous findings, we assumed initially that the presence of arylsulfonyl end groups could be beneficial but it was definitely not the case. This might be rationalized by the tendency of these compounds to hydrolysis which can occur under physiological conditions. Further studies on the use of ethynyl-substituted benzosiloxaboroles as a platform for the construction of structurally extended systems including nucleoside conjugates are currently in progress and the results will be reported in due course.4 Experimental section
4.1 General comments
Solvents used for reactions were dried by heating to reflux with sodium/benzophenone and distilled under argon. Starting materials including halogenated benzenes, ethynyltrimethylsilane, Pd(PPh3)4, CuI, chlorodimethylsilylane, alkyllithiums, diisopropylamine, trimethyl borate, sodium azide, CuTC, arylsulfonyl chlorides, as well as other reagents were used as received without further purification. In the 13C NMR spectra, the resonances of boron-bound carbon atoms were not observed in some cases as a result of their broadening by a quadrupolar boron nucleus. 1H and 13C NMR chemical shifts are given relative to TMS using residual solvent resonances. 11B and 19F NMR chemical shifts are given relative to BF3·Et2O and CFCl3, respectively.4.2 Synthesis
:3) and collected by filtration to afford the product. It was obtained as a white powder, mp: 183–186 °C (decomposition). Yield 0.20 g (33%). 1H NMR (400 MHz, DMSO-d6) δ 8.25 (d, J = 3.8 Hz, 1H), 8.17 (t, J = 7.4 Hz, 1H), 7.75 (dd, J = 7.4, 2.0 Hz, 1H), 7.61–7.58 (m, 2H), 7.33–7.30 (m, 3H), 0.45 (s, 6H) ppm. 13C NMR (101 MHz, DMSO-d6) δ 160.4 (d, J = 246.7 Hz), 148.0, 144.2 (broad), 139.5, 135.5 (d, J = 31.9 Hz), 130.5 (J = 2.1 Hz), 128.6, 128.3 (J = 3.0 Hz), 127.8, 125.5, 119.5 (d, J = 16.0 Hz), −0.5 ppm. 11B NMR (96 MHz, DMSO-d6) δ 29.9 ppm. 19F NMR (376 MHz, DMSO-d6) δ −105.71 ppm. HRMS (ESI, positive ion mode): calcd for C10H12BFN3O2Si+ [M]+ 264.0770; found 264.0771.4.3 Single crystal X-ray diffraction and structural analysis
Single crystals were grown by a solvent–evaporation method under air from the CHCl3 or acetone solutions. Selected crystals were measured at low temperature (100 K) with the use of Oxford Cryosystems nitrogen gas-flow device. The crystal structures were established in a conventional way via X-ray data refinement employing the Independent Atom Model (IAM). Data reduction and analysis were carried out with the CrysAlisPro suites of programs.44 All structures were solved by direct methods using SHELXS-97 (ref. 45) and refined using SHELXL-2016.46 All non-H atoms were refined anisotropically. All C–H atoms were placed in calculated positions with C–H distances of 0.95 Å and Uiso(H) = 1.2Ueq(C). The position H atoms of hydroxy groups were located from a difference electron density maps. The O–H distances were fixed to 0.84 Å with a standard deviation of 0.01 Å and the directionality of O–H was refined freely. The Uiso parameter was set to 1.5Ueq with respect to oxygen atoms. The crystal structure 1c contains large residual density peak (3.17 e Å−3), which cannot be reasonably explained by the mode. All-important crystallographic data including measurement, reduction, structure solution and refinement details are placed in Table S1 in ESI† or in the associated CIF files or can be retrieved from the Cambridge Crystallographic Data Centre as supplementary deposition no. 2304134 (1c), 2304135 (1c·MeCN), 2304136 (2c), 2304137 (3b), 2304138 (4d) and 2304139 (5b).4.4 Antimicrobial activity
Author contributions
S. L. and A. E. L.: conceptualization of the paper and supervision of the research; S. L.: funding acquisition, P. P.: synthesis and compound characterization, K. D. and K. W.: single-crystal X-ray diffraction; K. D.: theoretical calculations; J. K.: antimicrobial activity studies; K. D., A. E. L., and S. L.: data analysis; P. P., K. D., J. K., A. E. L. and S. L.: writing the original draft. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Centre (Poland) within the framework of the project DEC-UMO-2018/31/B/ST5/00210. Work implemented as a part of Operational Project Knowledge Education Development 2014–2020 cofinanced by European Social Fund (the TRIBIOCHEM interdisciplinary PhD programme (P.P.)). We also acknowledge the support by the Warsaw University of Technology. The authors are grateful to Dr P. H. Marek-Urban for the measurement of the crystal structure of 1c.References
- K. Torssell, Ark. Kemi, 1957, 10, 507–511 CAS.
- S. J. Baker, Y.-K. Zhang, T. Akama, A. Lau, H. Zhou, V. Hernandez, W. Mao, M. R. K. Alley, V. Sanders and J. J. Plattner, J. Med. Chem., 2006, 49, 4447–4450 CrossRef CAS PubMed.
- (a) C. T. Liu, J. W. Tomsho and S. J. Benkovic, Bioorg. Med. Chem., 2014, 22, 4462–4473 CrossRef CAS PubMed; (b) J. Zhang, M. Zhu, Y. Lin and H. Zhou, Sci. China: Chem, 2013, 56, 1372–1381 CrossRef CAS; (c) A. Adamczyk-Woźniak, K. M. Borys and A. Sporzyński, Chem. Rev., 2015, 115, 5224–5247 CrossRef PubMed; (d) P. S. Coghi, Y. Zhu, H. Xie, N. S. Hosmane and Y. Zhang, Molecules, 2021, 26, 3309 CrossRef CAS PubMed; (e) D.-C. Huang, Z. He, D. Guo, F. Deng, Q. Bian, H. Zhang, A. S. Ali, M.-Z. Zhang, W.-H. Zhang and Y.-C. Gu, J. Agric. Food Chem., 2023, 71, 6226–6235 CrossRef CAS PubMed.
- N. Sharma and D. Sharma, J. Pharmacol. Pharmacother., 2015, 6, 236–239 CrossRef PubMed.
- Anacor Pharmaceuticals, FDA Approves Anacor Pharmaceuticals' Crisaborole, 2016, https://www.investor.anacor.com/ Search PubMed.
- A. Brzozowska, P. Ćwik, K. Durka, T. Kliś, A. E. Laudy, S. Luliński, J. Serwatowski, S. Tyski, M. Urban and W. Wróblewski, Organometallics, 2015, 34, 2924–2932 CrossRef CAS.
- K. Durka, A. E. Laudy, Ł. Charzewski, M. Urban, K. Stępień, S. Tyski, K. A. Krzyśko and S. Luliński, Eur. J. Med. Chem., 2019, 171, 11–24 CrossRef CAS PubMed.
- K. Nowicki, J. Krajewska, T. M. Stępniewski, M. Wielechowska, P. Wińska, A. Kaczmarczyk, J. Korpowska, J. Selent, P. H. Marek-Urban, K. Durka, K. Woźniak, A. E. Laudy and S. Luliński, RSC Med. Chem., 2024 10.1039/d4md00061g.
- K. Durka, A. Zuba, P. H. Marek-Urban, K. Nowicki, J. Drapała, K. Woźniak and S. Luliński, CrystEngComm, 2023, 25, 6329–6342 RSC.
- L. Todorov and I. Kostova, Front. Chem., 2023, 11, 1247805 CrossRef CAS PubMed.
- U. Battaglia and C. J. Moody, J. Nat. Prod., 2010, 73, 1938–1939 CrossRef CAS PubMed.
- (a) S. M. Grant and S. P. Clissold, Drugs, 1990, 39, 877–916 CrossRef CAS PubMed; (b) S. G. Agalave, S. R. Maujan and V. S. Pore, Chem.–Asian J., 2011, 6, 2696–2718 CrossRef CAS PubMed.
- (a) D. Yahav, C. G. Giske, A. Gramatniece, H. Abodakpi, V. H. Tam and L. Leibovicib, Clin. Microbiol. Rev, 2021, 34, e00115–e00120 CrossRef CAS PubMed; (b) H. S Sader, R. K Flamm, C. G. Carvalhaes and M. Castanheira, Antimicrob. Agents Chemother., 2018, 62, 015877 Search PubMed.
- S. Hakimian, A. Cheng-Hakimian, G. D. Anderson and J. W. Miller, Expert Opin. Pharmacother., 2007, 8, 1931–1940 CrossRef CAS PubMed.
- R. A. Powers, C. M. June, M. C. Fernando, E. R. Fish, O. L. Maurer, R. M. Baumann, T. J. Beardsley, M. A. Taracila, S. D. Rudin, K. M. Hujer, A. M. Hujer, N. Santi, V. Villamil, M. L. Introvigne, F. Prati, E. Caselli, R. A. Bonomo and B. J. Wallar, J. Med. Chem., 2023, 66, 8510–8525 CrossRef CAS PubMed.
- A. Nocentini, A. Bonardi, C. Bazzicalupi, V. Alterio, D. Esposito, S. M. Monti, M. Smietana, G. De Simone, C. T. Supuran, P. Gratteri and J.-Y. Winum, J. Med. Chem., 2023, 66(12), 8118–8129 CrossRef CAS PubMed.
- J. Krajewska, K. Nowicki, K. Durka, P. H. Marek-Urban, P. Wińska, T. Stępniewski, K. Woźniak, A. E. Laudy and S. Luliński, RSC Adv., 2022, 12, 23099–23117 RSC.
- P. Pacholak, J. Krajewska, P. Wińska, J. Dunikowska, U. Gogowska, J. Mierzejewska, K. Durka, K. Woźniak, A. E. Laudy and S. Luliński, RSC Adv., 2021, 11, 25104–25121 RSC.
- N. Li, Z. Li, X. Zhang and R. Hua, Int. J. Mol. Sci., 2013, 14, 23257–23273 CrossRef PubMed.
- F. Mongin and M. Schlosser, Tetrahedron Lett., 1996, 37, 6551–6554 CrossRef CAS.
- K. Durka, M. Urban, M. Czub, M. Dąbrowski, P. Tomaszewski and S. Luliński, Dalton Trans., 2018, 47, 3705–3716 RSC.
- K. Durka, K. N. Jarzembska, R. Kamiński, S. Luliński, J. Serwatowski and K. Woźniak, Cryst. Growth Des., 2012, 12, 3720–3734 CrossRef CAS.
- (a) J. Zhang, W. Li, J. Cheng, Z. Liu and Q. Li, RSC Adv., 2018, 8, 26580–26588 RSC; (b) M. A. A. Ibrahim, A. S. M. Rady, J. H. Al-Fahemi, E. M. Z Telb, S. A. Ahmed, A. M. Shawky and N. A. M. Moussa, ChemistrySelect, 2020, 5, 13223–13231 CrossRef CAS; (c) R. Oshimizu, N. Ando and S. Yamaguchi, Angew. Chem., Int. Ed., 2022, 61, e20220939 CrossRef PubMed.
- S. J. Grabowski, Molecules, 2015, 20, 11297–11316 CrossRef CAS PubMed.
- (a) C. Trujillo and G. Sánchez-Sanz, ChemPhysChem, 2016, 17, 395–405 CrossRef CAS PubMed; (b) B. Kelly, G. Sánchez-Sanz, F. Blanco and I. Rozas, Comput. Theor. Chem., 2012, 998, 64–73 CrossRef CAS.
- E. C. Escudero-Adán, A. Bauzá, C. Lecomte, A. Frontera and P. Ballester, Phys. Chem. Chem. Phys., 2018, 20, 24192–24200 RSC.
- (a) T. Jiang, Z.-Y. Gu, L. Yin, S.-Y. Wang and S.-J. Ji, J. Org. Chem., 2017, 82, 7913–7919, DOI:10.1021/acs.joc.7b01127; (b) J. Gui, H. Xie, H. Jiang and W. Zeng, Org. Lett., 2019, 21, 2804–2807 CrossRef CAS PubMed.
- (a) J. Huang, S. J. F. Macdonald and J. P. A. Harrity, Chem. Commun., 2009, 436–438 RSC; (b) J. Huang, S. J. F. Macdonald, A. W. J. Cooper, G. Fisher and J. P. A. Harrity, Tetrahedron Lett., 2009, 50, 5539–5541 CrossRef CAS.
- S. Mothana, J.-M. Grassot and D. G. Hall, Angew. Chem., Int. Ed., 2010, 49, 2883–2887 CrossRef CAS PubMed.
- A. Saini, S. Kumar, R. Raj, S. Chowdhary, M. Gendrot, J. Mosnier, I. Fonta, B. Pradines and V. Kumar, Bioorg. Chem., 2021, 109, 104733 CrossRef CAS PubMed.
- A. Singh and R. Kumar, Chem. Commun., 2021, 57, 9708–9711 RSC.
- V. Hong, S. Presolski, C. Ma and M. G. Finn, Angew. Chem., Int. Ed., 2009, 48, 9879–9883 CrossRef CAS PubMed.
- J. Raushel and V. V. Fokin, Org. Lett., 2010, 12, 4952–4955 CrossRef CAS PubMed.
- K. Biradha and M. Fujita, Chem. Lett., 2000, 29, 350–351 CrossRef.
- J. Krajewska, S. Tyski and A. E. Laudy, Antimicrob. Agents Chemother., 2023, 67, 1–14, DOI:10.1128/aac.01373-22.
- Prioritization of pathogens to guide discovery, research and development of new antibiotics for drug-resistant bacterial infections, including tuberculosis, World Health Organization, 2017, https://www.apps.who.int/iris/handle/10665/311820, accessed 1 February 2024 Search PubMed.
- T. J. Foster, FEMS Microbiol. Rev., 2017, 41, 430–449 CrossRef CAS PubMed.
- Y. Guo, G. Song, M. Sun, J. Wang and Y. Wang, Front. Cell. Infect. Microbiol., 2020, 10, 107 CrossRef PubMed.
- (a) H. Eagle and A. D. Musselman, J. Exp. Med., 1948, 88, 99–131 CrossRef CAS PubMed; (b) A. Prasetyoputri, A. M. Jarrad, M. A. Cooper and M. A. T. Blaskovich, Trends Microbiol., 2019, 27, 339–354 CrossRef CAS PubMed.
- Clinical and Laboratory Standards Institute, Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically. Approved Standard, CLSI guideline M07-A9, Wayne, PA, USA, 9th edn, 2012 Search PubMed.
- (a) A. E. Laudy, Pol. J. Microbiol., 2018, 67, 129–135 CrossRef PubMed; (b) A. Słoczyńska, M. E. Wand, L. J. Bock, S. Tyski and A. E. Laudy, Int. J. Mol. Sci., 2023, 24, 9525 CrossRef PubMed.
- (a) O. M. Zając, S. Tyski and A. E. Laudy, Biology, 2022, 11, 1044 CrossRef PubMed; (b) H. Nikaido and J.-M. Pagès, FEMS Microbiol. Rev., 2012, 36, 340–363 CrossRef CAS PubMed; (c) T. J. Opperman and S. T. Nguyen, Front. Microbiol., 2015, 6, 421 Search PubMed.
- K. Lee, V. C. Pham, M. J. Choi, K. J. Kim, K.-T. Lee, S.-G. Han, Y. G. Yu and J. Y. Lee, Bioorg. Med. Chem. Lett., 2013, 23, 75–80 CrossRef CAS PubMed.
- Rigaku Oxford Diffraction, Crysalis Pro v. 1.171.38.46, 2018 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory; International Series of Monographs on Chemistry; Oxford University Press, Oxford, New York, 1994 Search PubMed.
- K. A. Todd, AIMAll(Version 19.10.12), TK Gristmill Software, Overland Park K. S., USA, 2019 Search PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- E. D. Glendening, D. M. Hiatt and F. Weinhold, Comprehensive Computational Chemistry, 2024, vol. 2, pp. 406–421 Search PubMed.
- Avogadro: An Open-Source Molecular Builder and Visualization Tool, Version 1.2.0., https://www.avogadro.cc/.
- (a) European Committee on Antimicrobial Susceptibility Testing, EUCAST disk diffusion method for antimicrobial susceptibility testing, 2023, Document version 11.0., https://www.eucast.org/ Search PubMed; (b) European Committee on Antimicrobial Susceptibility Testing, Method for the determination of broth dilution MIC of antifungal agents for yeasts, 2020, Document E.DEF 7.3.2., https://www.eucast.org/ Search PubMed.
- (a) Clinical and Laboratory Standards Institute, Method for Antifungal Disk Diffusion Susceptibility Testing of Yeasts. Approved Standard, CLSI guideline M44-A2, Wayne, PA, USA, 2nd edn, 2009 Search PubMed; (b) Clinical and Laboratory Standards Institute, Methods for Determining Bactericidal Activity of Antimicrobial Agents, CLSI guideline M26-A, Wayne, PA, USA, 1999 Search PubMed.
- R. P. Lamers, J. F. Cavallari and L. L. Burrows, PLoS One, 2013, 8, e60666 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2304134–2304139. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra02137a |
| This journal is © The Royal Society of Chemistry 2024 |