
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of novel phthalazine-based derivatives with potent cytotoxicity against HCT-116 cells through apoptosis and VEGFR2 inhibition†
Donia El Sayeda,
Samir M. El Rayes *a,
Hamdy A. Solimana,
Imad Eddin AlBalaab,
Mansour S. Alturkic,
Abdulaziz Hassan Al Khzemc,
Mohammed Abdullah Alsharifd and
Mohamed S. Nafie*ae
*a,
Hamdy A. Solimana,
Imad Eddin AlBalaab,
Mansour S. Alturkic,
Abdulaziz Hassan Al Khzemc,
Mohammed Abdullah Alsharifd and
Mohamed S. Nafie*ae
aChemistry Department, Faculty of Science, Suez Canal University, P.O. 41522, Ismailia, Egypt. E-mail: mohamed_nafie@science.suez.edu.eg; samir_elrayes@science.suez.edu.eg
bScience Department, Faculty of Basic Educations, PAAET, Kuwait, Safat 22081, Kuwait
cDepartment of Pharmaceutical Chemistry, College of Clinical Pharmacy, Imam Abdulrahman Bin Faisal University, P.O. Box 1982, Dammam 31441, Eastern Province, Kingdom of Saudi Arabia
dKing Fahad Armed Forces Hospital, Al Kurnaysh Rd, Al Andalus, Jeddah 23311, Kingdom of Saudi Arabia
eDepartment of Chemistry, College of Sciences, University of Sharjah, P.O. 27272, Sharjah, United Arab Emirates. E-mail: mohamed.elsayed@sharjah.ac.ae
First published on 24th April 2024
Abstract
The parent ethyl 3-(4-benzyl-1-oxophthalazin-2(1H)-yl) propanoate (3) has 25 compounds. Their respective mono, dipeptides and hydrazones derivatives were produced by chemoselective N-alkylation via addition reaction of 4-benzylphthalazin-1(2H)-one (2) with ethyl acrylate and anhydrous potassium carbonate to give ethyl 3-(4-benzyl-1-oxophthalazin-2(1H)-yl) propanoate (3). The ester 3 was hydrazinolyzed to give the corresponding hydrazide 3-(4-benzyl-1-oxophthalazin-2(1H)-yl) propanehydrazide (5), then azide 6 coupled with amino acid ester hydrochloride and/or amines to afford several parent esters 8a–c, then a series of hydrazinolyzed reactions occurred to give corresponding hydrazides 9a–c. The hydrazide 9a was subjected to the azide coupling procedure, which resulted in the formation of various dipeptides. Subsequently, it was condensed with various aldehydes to yield hydrazone derivatives 13a–d. Interestingly, compounds 9c, 12b, and 13c exhibited potent cytotoxicity with IC50 values of 1.58, 0.32 and 0.64 μM compared to sorafenib (IC50 = 2.93 μM). Compound 12b exhibited potent VEGFR2 inhibition by 95.2% with an IC50 value of 17.8 μM compared to sorafenib (94.7% and IC50 of 32.1 μM). For apoptosis activity, 12b-treatment induced apoptosis in HCT-116 cells by 21.7-fold, arresting the cell proliferation at S-phase. Finally, it formed a good binding affinity towards VEGFR2 protein with a binding energy of −10.66 kcal mol−1, and it formed binding interactions with the key interactive amino acids.
Introduction
Generally, cancer seems to be the leading cause of death in high- and upper-middle-income countries1 and the second most common cause of death after cardiovascular disease.2 According to previous studies, around 18.1 million new cases of cancer were detected, with lung cancer accounting for 18.4%, followed by breast (11.6%), prostate (7.1%), colorectal (6.1%), stomach, and liver cancer.3,4 About 10 million people died of cancer in 2020, while 19.3 million new cases were identified.5 Cancer occurs when abnormal cells divide rapidly and spread to other parts of the body and tissues, finally forming a tumor.4 The toxicity and the side effects of present antineoplastic drugs, along with the appearance of drug resistance, are the Major drawbacks of chemotherapy.6 Despite advances in our understanding of the biochemical processes involved in carcinogenesis and fifty years of chemotherapy research, there are still many obstacles to overcome before cancer treatments can be considered effective. These include diversity in tumor cells, drug resistance, therapy-related side effects, and the limitations of animal models.7 Cancer chemotherapy has been developed for molecular treatments that are more selective and do not have the toxicity of typical cytotoxic drugs.8 Heterocyclic compounds have been applied to treat a variety of diseases, including cancer. Biological molecules in our body, such as DNA, RNA, and vitamins, contain heterocyclic core rings, which make heterocyclic compounds advance significantly in the medicinal field.4,9 Hydrazine-containing compounds have attracted much attention due to their pharmacological properties and clinical uses.10,11 Hydrazides are an essential class of chemicals for novel medication development because they contain H-bond donors/acceptors that can create H-bonds with their recipients within the target protein active sites.12 Phthalazin-1(2H)-one derivatives are a class of diaza heterobicycles known for their potential medical applications. Thus, this class of compounds has been shown to have a wide range of biological properties, such as anti-diabetic and anti-cancer.13 Over the last two decades, there has been a significant focus on producing many phthalazines as promising drug targets for cancer treatment14 and other biological activity, as shown in (Fig. 1). The phthalazine derivative azelastine 1 is an antihistamine used to treat allergic rhinitis.15 Zopolrestat 5 is a phthalazinone derivative that has been examined in clinical studies. It inhibits aldose reductase and has the potential to prevent retinopathy, neuropathy, and cataract formation in diabetes.16 The aminophthalazine and hydrazinylphthalazine moiety can also be found in the core of many commercial drugs, such as hydralazine 2,17 carbazeran 6,18 and budralazine, which are used for the treatment of heart failure, as well as in the structure of the effective anti-cancer drugs.19 Moreover, in recent years, there has been interest in using several VEGFR-2 inhibitors for targeted cancer therapy, which contain phthalazinone derivative such as vatalanib 3, ZD 6474 4,20 and other compounds 1-(4-chlorophenyl)-3-(4-((4-chlorophthalazin-1-yl)amino)phenyl)urea (7), and 1-(4-chloro-3-(trifluoromethyl)phenyl)-3-(4-((4-chlorophthalazin-1-yl)oxy)phenyl)urea (8) which showed the significant inhibitory effects.21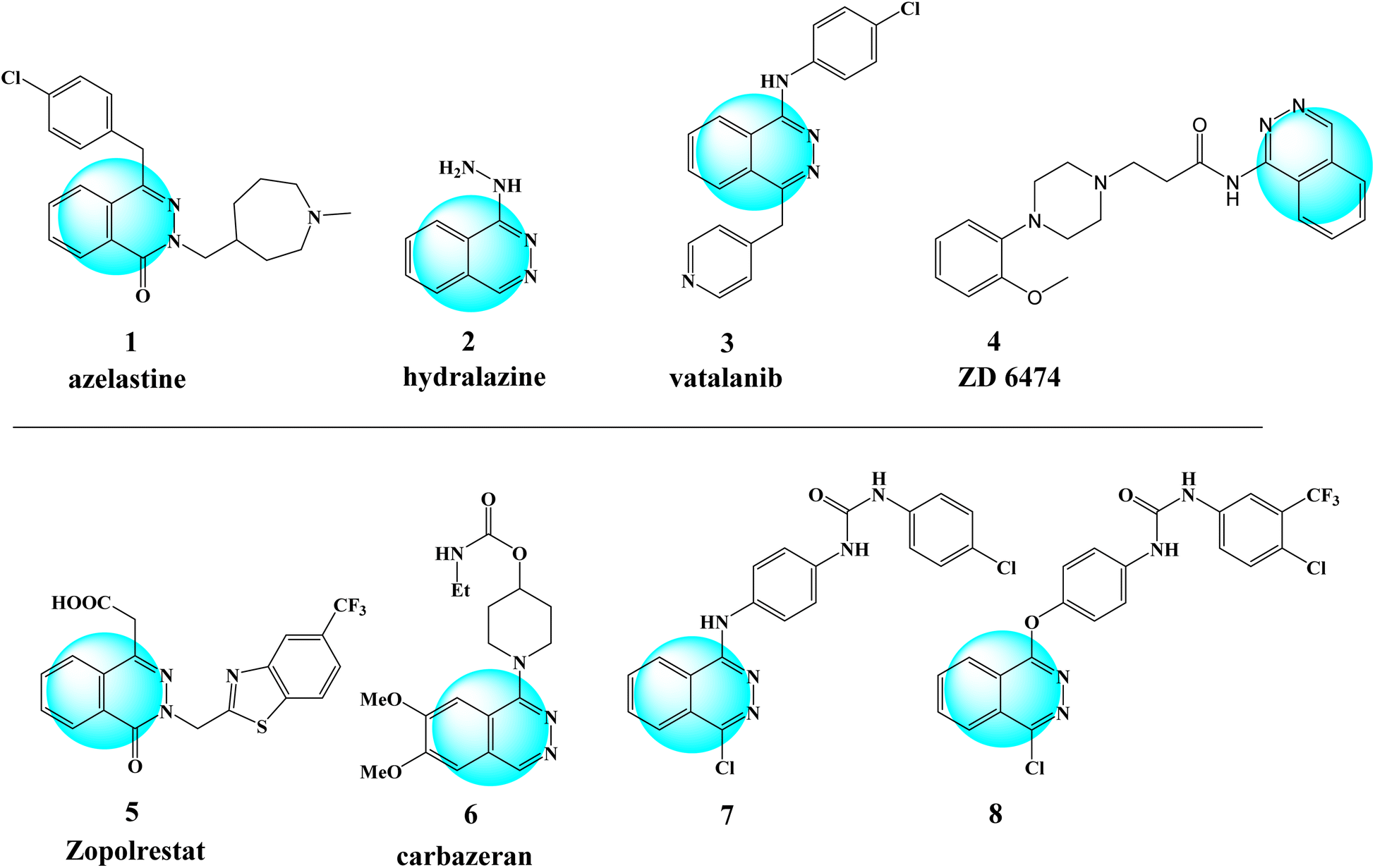 | ||
| Fig. 1 Structure of biologically active phthalazine derivatives. | ||
Accordingly, we aimed to design and synthesize novel phthalazine-based amine and amino acid derivatives with characterization and purity, and to investigate their cytotoxicity against HCT-116 cells along with investigating both molecular target; vascular endothelial growth factor receptor 2 (VEGFR2) with apoptosis-induction as the cell death mechanism.
Results & discussion
Recent studies15,22 revealed how to control chemoselective alkylation of amides and thioamides separately. As a follow-up to these results, we chose to apply them to the structure modification of 4-benzylphthalazin-1(2H)-one (2), our model heterocyclic amide. The addition reaction of the model nucleophile 2 with ethyl acrylate gave ethyl 3-(4-benzyl-1-oxophthalazin-2(1H)-yl) propanoate (3). According to Pearson's hard soft acid base principle, reaction control points like basicity and nucleophilicity of both N and O atoms determine how the addition reaction behaves toward electrophiles. Instead of occurring on the O atom or even at both atoms in a competitive reaction, this reaction only happens on the N atom. The resulting chemoselective N-alkylation reaction can be effectively interpreted as the result of the interaction between the high-energy HOMO at the nitrogen atom of the nucleophile and the low-energy LUMO of the electrophile, which creates a narrow energy gap and high reactivity that ultimately results in N-alkylation.29 The ester 3 interacted with either sodium hydroxide or hydrazine hydrate to form 3-((4-benzyl-1-oxophthalaz)by-1H)-yl) propanoic acid (4) and 3-(4-benzyl-1-oxophthalazin-2(1H)-yl) propanehydrazide (5). The acetic acid derivative 4 and hydrazide 5 are very interesting precursors for modifying the structure of 4-benzylphthalazin-1(2H)-one (2) by attaching amines or amino acids via peptide bond using either N,N′-dicyclohexylcarbodiimide (DCC) or azide coupling conditions. At ambient temperature, 3-(4-benzyl-1-oxophthalazin-2(1H)-yl) propanoic acid (4) reacted with various amines under DCC. Conditions, and produced 2-(4-benzyl-1-oxophthalazin-2(1H)-yl)-N-alkyl-propanamide 7a–g (Scheme 1).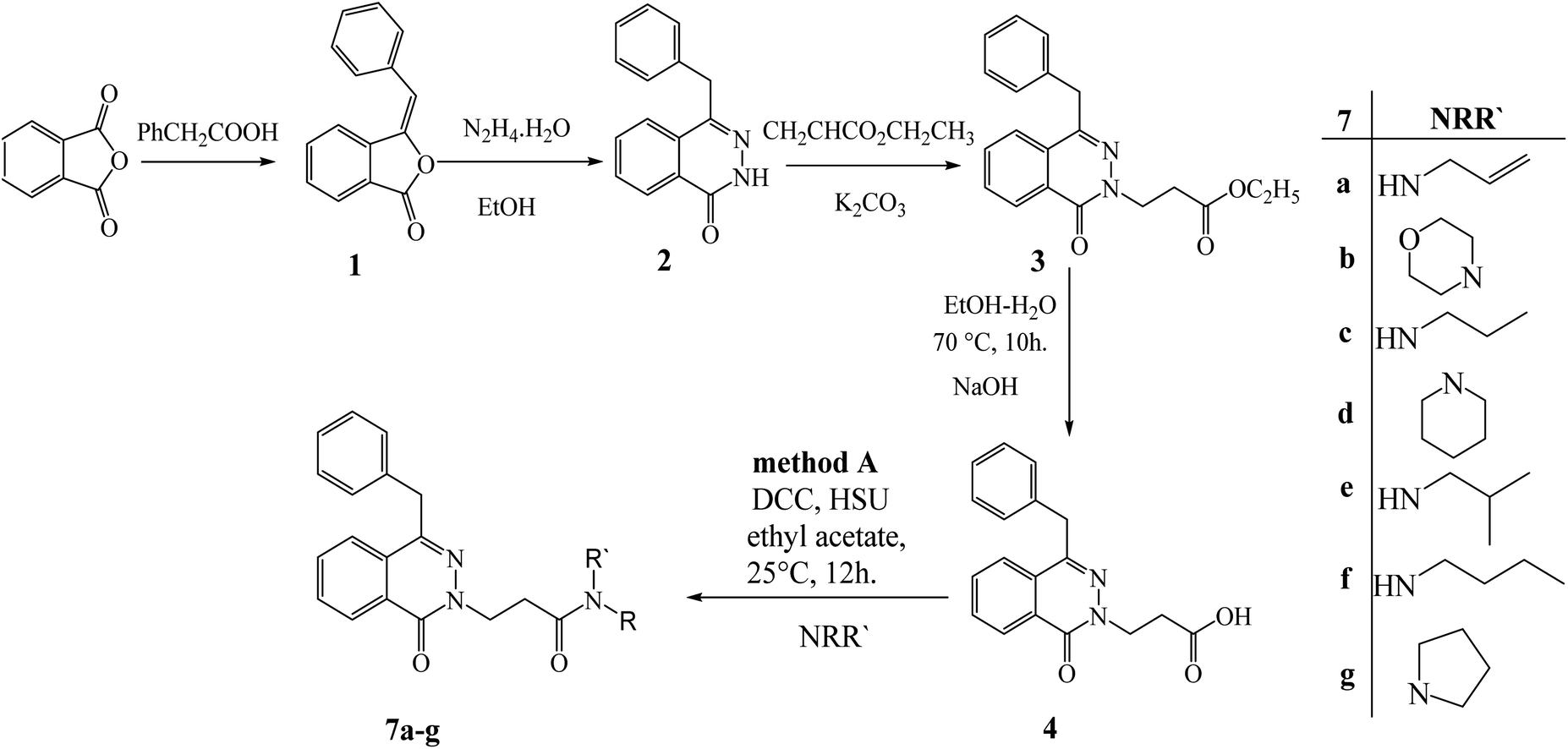 | ||
| Scheme 1 Preparation of 2-(4-benzyl-1-oxophthalazin-2(1H)-yl)-N-alkyl-propanamide 7a–g by method (A). | ||
Methyl (3-[4-benzyl-1-oxophthalazin-2(1H)-yl) propanoyl amino] alkanoates 8a–c were obtained via reaction of 3-(4-benzyl-1-oxophthalazin-2(1H)-yl) propanoic acid (4) with different amino acid ester hydrochloride under DCC conditions (Scheme 2).
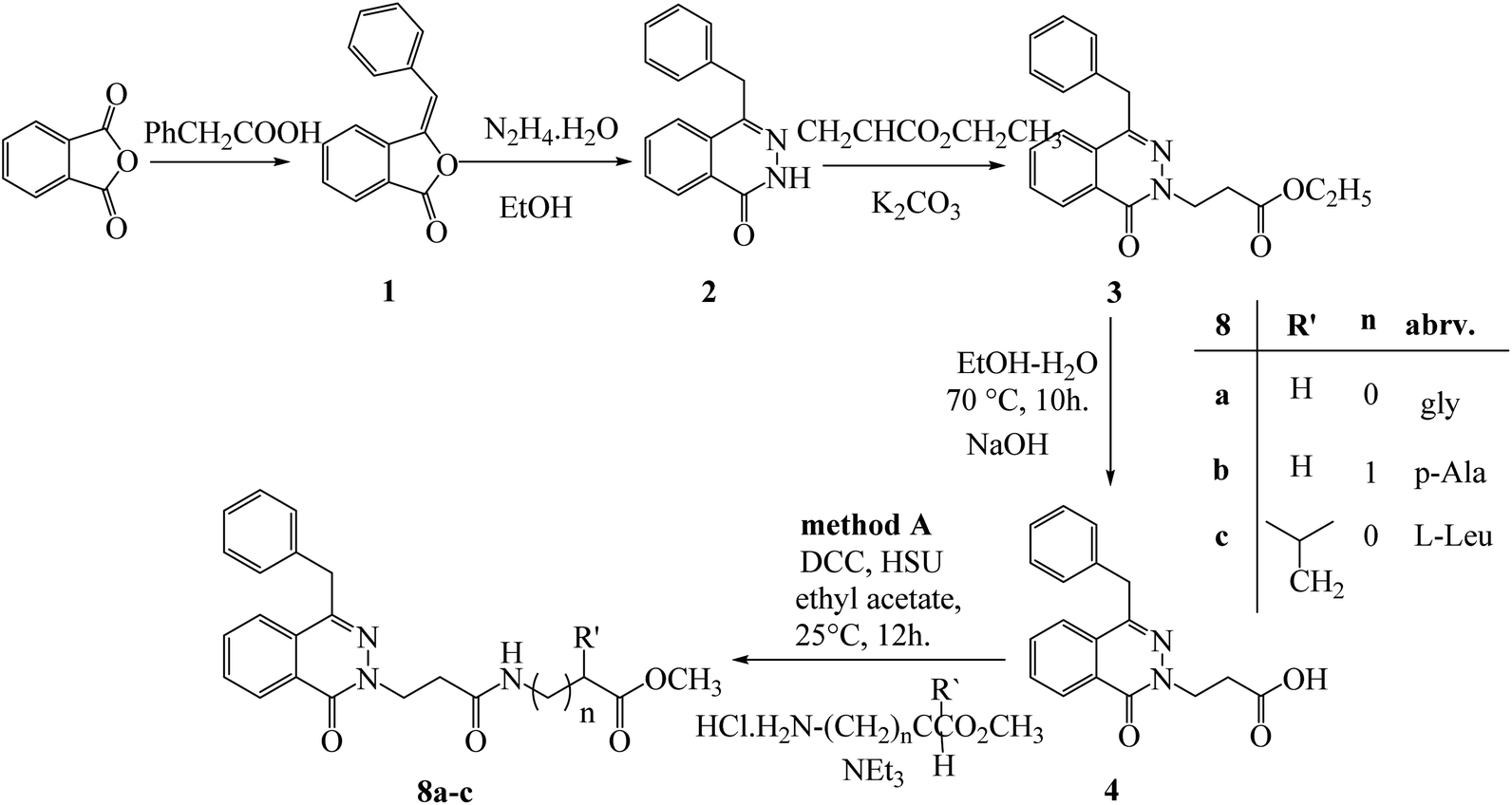 | ||
| Scheme 2 Preparation of methyl (3-[4-benzyl-1-oxophthalazin-2(1H)-yl) propanoyl amino] alkanoates 8a–c by method (A). | ||
3-(4-Benzyl-1-oxophthalazin-2(1H)-yl) propanehydrazide (5) was obtained via reaction of ester 3 with hydrazine hydrate in ethanol under reflux for 6 h. Under azide coupling condition, hydrazide 5 was reacted with different amines in presence of NaNO2/HCl; the amide derivatives 7a–g were obtained (Scheme 3).The 1H NMR spectra of all compounds displayed a sharp singlet signal around 4.30 ppm for (CH2-Ph), a multiplet peaks around 4.62–4.36 ppm for (CH2CH2CO), a multiplet peaks around 2.95–2.85 ppm for (CH2CH2CO) and the aromatic protons appeared between 8.46 and 7.15 ppm. 13C NMR spectra revealed the methylene carbon of the (CH2-Ph) group at 38 ppm. While (NCH2CH2CO) (attached to N-2) appeared between 47.0 and 46.0 ppm, (CH2CH2CO) appeared between 30.0 and 35.0 ppm, all the aromatic carbons were found between 145.85 and 125.00 ppm. The carbonyl group of the phthalazinone ring was observed around 158.68 ppm. The additional significant data could be discussed as follows: the 1H NMR spectrum of ester 3 gave additional signals at 4.05–3.99 ppm multiplet peaks for OCH2CH3, and 1.09 ppm triplet peaks for CH3. The 13C-NMR spectrum has signals at 171.35 for the carbonyl group of esters, peaks at 60.53, and 14.35 ppm for CH2CH3 & CH3 respectively. The IR showed the presence of 2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bands at 1720 and 1639 cm−1. The 1H NMR of hydrazide 5 showed the hydarzino (NHNH2) group protons at 4.18 ppm for NH2 and 9.11 for NH. The IR showed the presence of NHNH2 bands at 3300 and 3194 cm−1. 1H NMR and 13C NMR were used to elucidate the synthetic construction of N-allyl-3-(4-benzyl-1-oxophthalazin-2(1H)-yl) propanamide (7a), which yielded the following signals: the olefinic methylene protons (CHCH2) appeared as two doublet at 5.06 ppm for the cis proton with coupling constant values Jcis = 10.4, while the trans proton appeared at 5.14 ppm with coupling constant Jtrans = 17.4. The olefinic CH (CHCH2) appeared as multiplet at 5.85–5.75 ppm and NHCH2 appeared as triplet at 4.59 ppm; the 13C-NMR spectrum has signals at 134.17, 116.32, 42.01 ppm for (CHCH2), (CHCH2) and (CH2CHCH2) respectively. The NMR spectrum of compound 7b showed multiplet signals at 3.67–3.65 ppm for 2OCH2 and 3.53–3.50 ppm for 2NCH2 in morpholine moiety. And the corresponding carbons appeared at 66.85, 66.65 ppm for (2CH2O), and 46.03, 41.93 ppm for (2CH2N).
O bands at 1720 and 1639 cm−1. The 1H NMR of hydrazide 5 showed the hydarzino (NHNH2) group protons at 4.18 ppm for NH2 and 9.11 for NH. The IR showed the presence of NHNH2 bands at 3300 and 3194 cm−1. 1H NMR and 13C NMR were used to elucidate the synthetic construction of N-allyl-3-(4-benzyl-1-oxophthalazin-2(1H)-yl) propanamide (7a), which yielded the following signals: the olefinic methylene protons (CHCH2) appeared as two doublet at 5.06 ppm for the cis proton with coupling constant values Jcis = 10.4, while the trans proton appeared at 5.14 ppm with coupling constant Jtrans = 17.4. The olefinic CH (CHCH2) appeared as multiplet at 5.85–5.75 ppm and NHCH2 appeared as triplet at 4.59 ppm; the 13C-NMR spectrum has signals at 134.17, 116.32, 42.01 ppm for (CHCH2), (CHCH2) and (CH2CHCH2) respectively. The NMR spectrum of compound 7b showed multiplet signals at 3.67–3.65 ppm for 2OCH2 and 3.53–3.50 ppm for 2NCH2 in morpholine moiety. And the corresponding carbons appeared at 66.85, 66.65 ppm for (2CH2O), and 46.03, 41.93 ppm for (2CH2N).
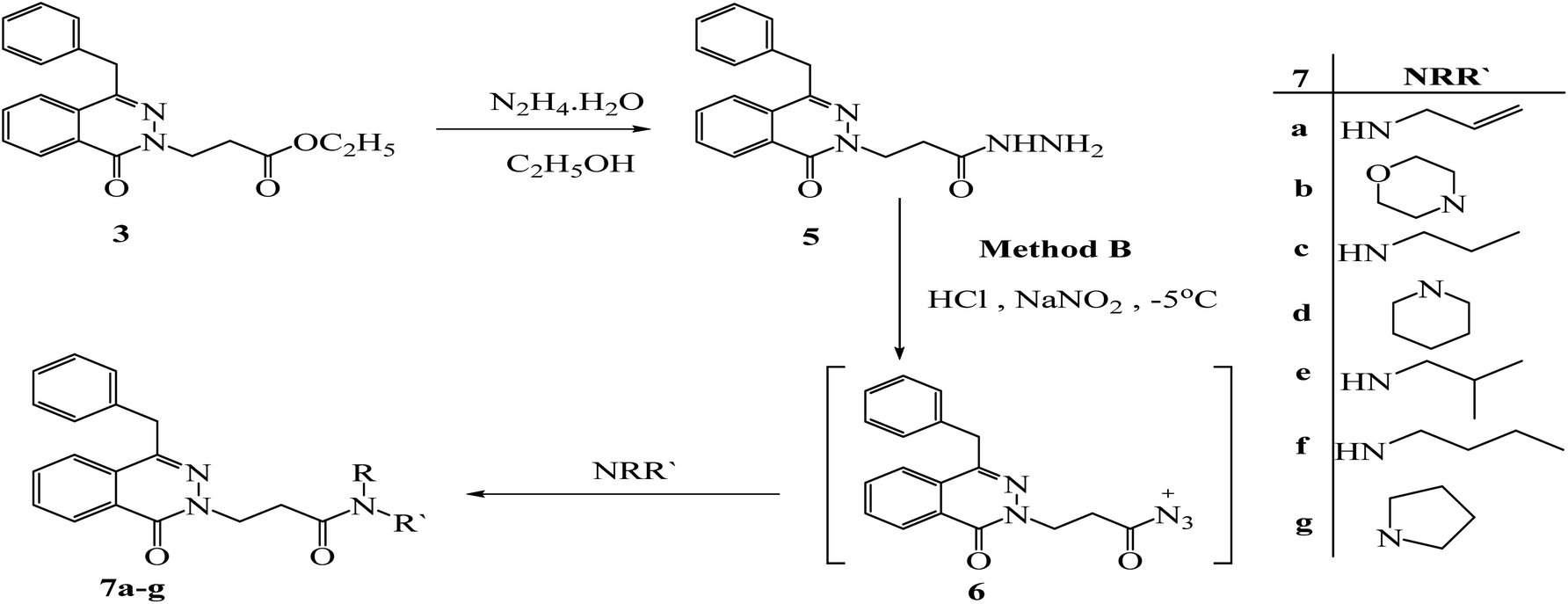 | ||
| Scheme 3 Preparation of 2-(4-benzyl-1-oxophthalazin-2(1H)-yl)-N-alkyl-propanamide 7a–g by method (B). | ||
Hydrazide 5 was reacted with different amino acid hydrochloride in presence of NaNO2/HCl; methyl (3-[4-benzyl-1-oxophthalazin-2(1H)-yl) propanoyl amino] alkanoates 8a–c were obtained (Scheme 4).
 | ||
| Scheme 4 Preparation of methyl (3-[4-benzyl-1-oxophthalazin-2(1H)-yl) propanoyl amino] alkanoates 8a–c by method (B). | ||
The azide method gave the same product as DCC-method but with higher yield 60–78%. So, hydrazide 5 is used as a starting point to create new phthalazinone compounds with significant biological activity. By attaching another amino acid via a peptide bond applying an azide condition. The azide technique is a well-known peptide synthesis technique that minimizes racemization while avoiding interferometer byproduct.20 The esters 8a–c was believed to be a major stage in the chemical structure modification of the phthalazinone nucleus. Hydrazides 9a–c were obtained via reaction of esters 8a–c with hydrazine hydrate in ethanol under reflux for 6 h (Scheme 5). The compound 8a has the 1H-NMR spectrum of characteristic following signals: a multiplet signals at 4.04–4.01 ppm of NHCH2CO and a singlet peak at 3.67 ppm of OCH3. The 13C-NMR spectrum has signals at 170.69, 52.13, and 41.28 ppm for (CO) ester, (OCH3), and (NHCH2), respectively. Compared with compound 9a, the signal of OCH3 disappeared, and new signals developed as broad signals at 9.07 and 3.40 ppm for (NHNH2) and (NH2), respectively. Also, The IR showed the presence of NHNH2 bands at 3292 and 3200 cm−1 which confirm the formation of new hydrazide.
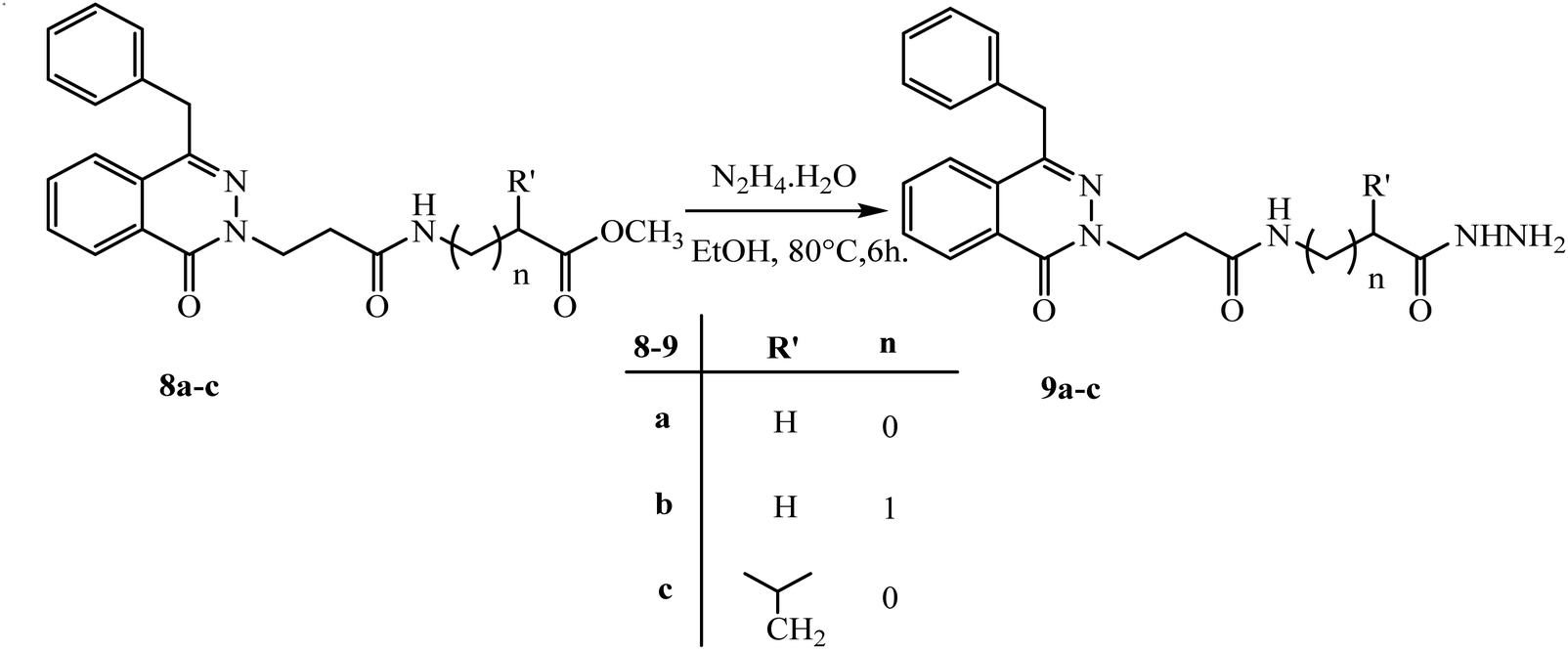 | ||
| Scheme 5 Preparation of various 3-(4-benzyl-1-oxophthalazin-2(1H)-yl)-N-(2-hydrazinyl-2-oxoethyl)alkanamide 9a–c. | ||
Under azide coupling conditions, 3-(4-benzyl-1-oxophthalazin-2(1H)-yl)-N-(2-hydrazinyl-2-oxoethyl)propanamide (9a) was reacted with different amines such as allyl, n-butyl, piperidin, and morpholine and obtained N-substituted-3-((4-benzyl-1-oxophthalazin-2(1H)-yl)-2-oxoethyl) propanamides 11a–d (Scheme 6). In respect to compound 7a, compound 11a showed additional peaks for NHCH2CO as multiplet at 3.97–3.95 in 1H NMR, and signals at 169.00 and 43.59 ppm for (CO) and (NHCH2CO) in 13C NMR. The IR showed the addition of new carbonyl at 1630 cm−1. The structure of compound 11c was confirmed from 1H NMR which showed additional signals as following a multiplet at 3.55–3.52 ppm for NCH2, a multiplet at 3.31–3.28 ppm for NCH2, a quartet signals at 1.63 ppm for CH2CH2CH2, and a triplet signal at 1.53 ppm for CH2CH2CH2) which confirmed the presence of piperidine ring, and the corresponding carbons showed signals at 45.42, 43.11, 24.31, 26.11 and 25.36 ppm respectively. Compound 11d exhibited additional peaks for NHCH2CO as multiplet at 4.03–4.02 ppm and corresponding carbon at 41.17 ppm compared to compound 7b.
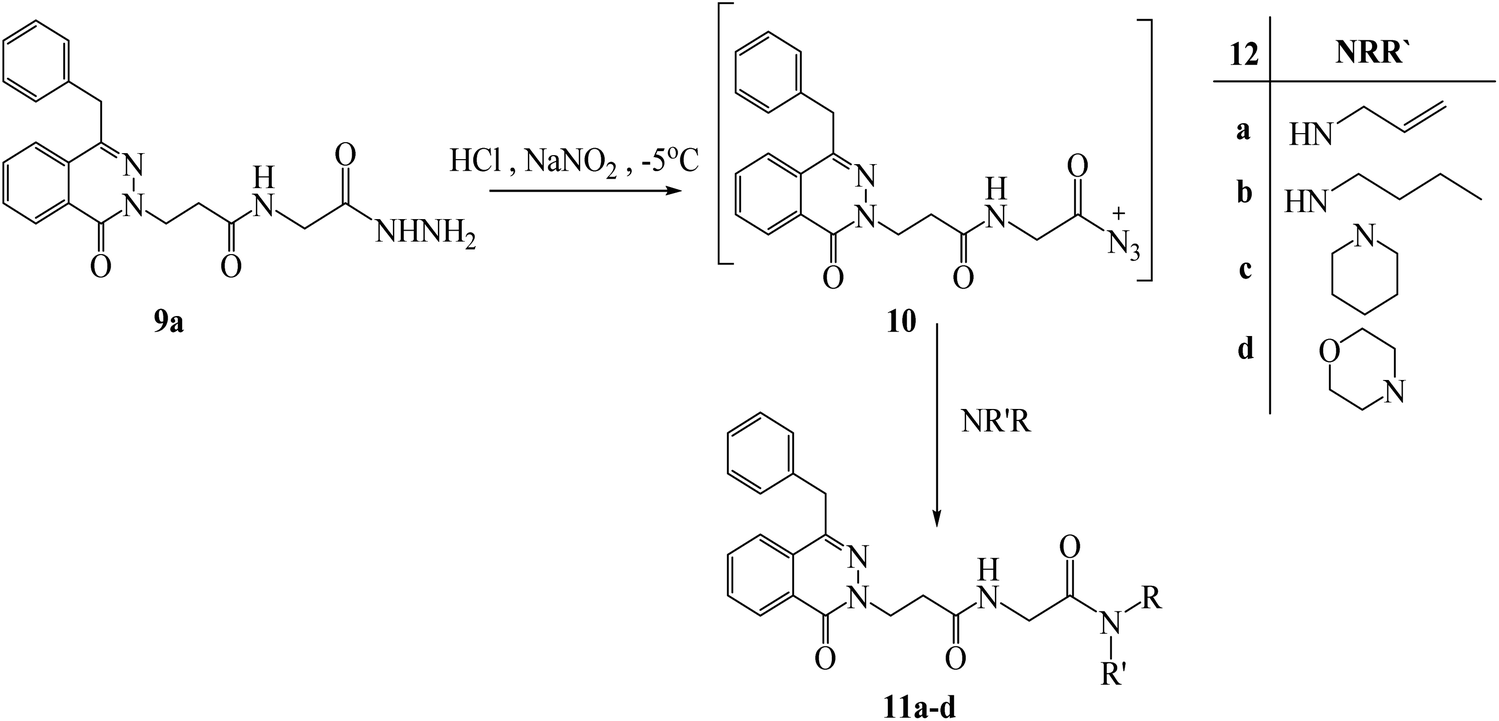 | ||
| Scheme 6 Preparation of N-substituted-3-((4-benzyl-1-oxophthalazin-2(1H)-yl)-2-oxoethyl) propanamides 11a–d. | ||
Similarly, hydrazide 9a was reacted with various amino acid methyl esters such as glycine, β-alanine, and leucine via azide coupling condition and produced dipeptide compounds 12a–c with an appropriate yield (Scheme 7). The formation of dipeptide was confirmed by using different analysis such as 1H NMR and 13C NMR. The compound 12c showed two multiplet at 7.44–7.42 and 7.18–7.15 for 2 NH, a multiplet at 4.12–3.93 ppm for NHCHCO, a singlet peak at 3.66 ppm for OCH3, a doublet at 1.61 ppm for CH2CH, a triplet at 1.24 ppm for CH2CH, and a multiplet at 0.92–0.87 ppm for 2CH3. The 13C-NMR spectrum has signals at 173.20, 171.36, and 169.22 ppm for three carbonyl groups, also showed signals at 52.16, 50.92, 41.01, 35.13, 24.80, 22.74, and 21.78 ppm for (NHCHCO), (OCH3), (CH2CH(CH3)2), (CH2CH2CO), (CH(CH3)2), and (2CH3) respectively. The IR showed the presence of 2 (CO) of dipeptide at 1645, and 1628 cm−1.
 | ||
| Scheme 7 Preparation of methyl-[3-(4-benzyl-1-oxo-1H-phthalazin-2-yl)-acetylamino] alkanoates 12a–c. | ||
Condensation of the hydrazide 3-(4-benzyl-1-oxophthalazin-2(1H)-yl)-N-(2-hydrazinyl-2-oxoethyl)propenamide (9a) with different aldehydes such as 4-nitro benzaldehyde, p-chloro benzaldehyde, anisaldehyde and 5-bromo-2-hydroxybenzaldehyde in ethanol under reflux 24 h; new hydrazone derivatives 13a–d were obtained respectively in acceptable yields (Scheme 8). Finally, the formation of some hydrazones was confirmed by using different analysis such as 1H NMR and IR. The compound 13a show Z/E isomers mixture in 78/22 ratio. The NMR spectrum of compound 13a showed a two singlet peaks at 11.71 and 11.68 ppm for CONHN, a two singlet peaks at 8.09 and 8.41 ppm for NCH, and new aromatic at 7.96–7.81 ppm as a multiplet and 7.36 ppm as doublet. The IR showed the presence of new H aromatic at 3208 and 3114 cm−1, (NO2) at 1524 cm−1, and (CN) at 1597 cm−1.
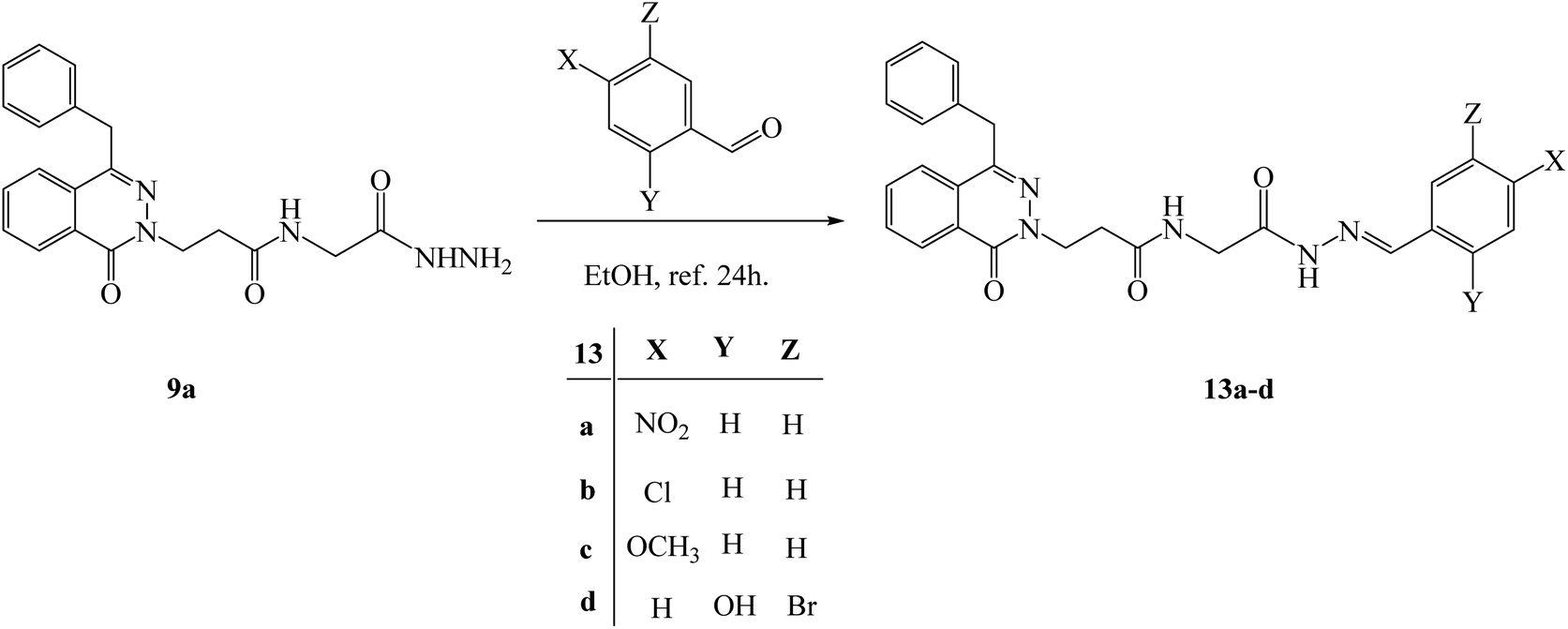 | ||
| Scheme 8 Synthesis of some hydrazone derivatives 13a–d. | ||
Biological investigation
Cytotoxicity against HCT-116 cells
The MTT test was used to measure cytotoxic activity. This assay relies on metabolically active cells reducing a yellow tetrazolium salt, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, or MTT, to purple formazan crystals. The tested compounds were investigated for their cytotoxic activity against colon (HCT-116) cancer cells using the MTT assay (Fig. 2). As summarized in Table 1 with the IC50 values. Interestingly, compounds 9c, 12b, and 13c exhibited potent cytotoxicity with IC50 values of 1.58, 0.32 and 0.64 μM compared to sorafenib (IC50 = 3.23 μM). Compounds 5, 11a, and 7f exhibited moderate cytotoxicity with IC50 values range of 7.98–18.8 μM, while other compounds showed poor cytotoxicity with higher IC50 values. Additionally, the most promising cytotoxic compounds weren't cytotoxic against normal WI-38 cells with higher IC50 values than 50 μM. Hence, these compounds were worth testing for effective molecular target and apoptosis-induction activity.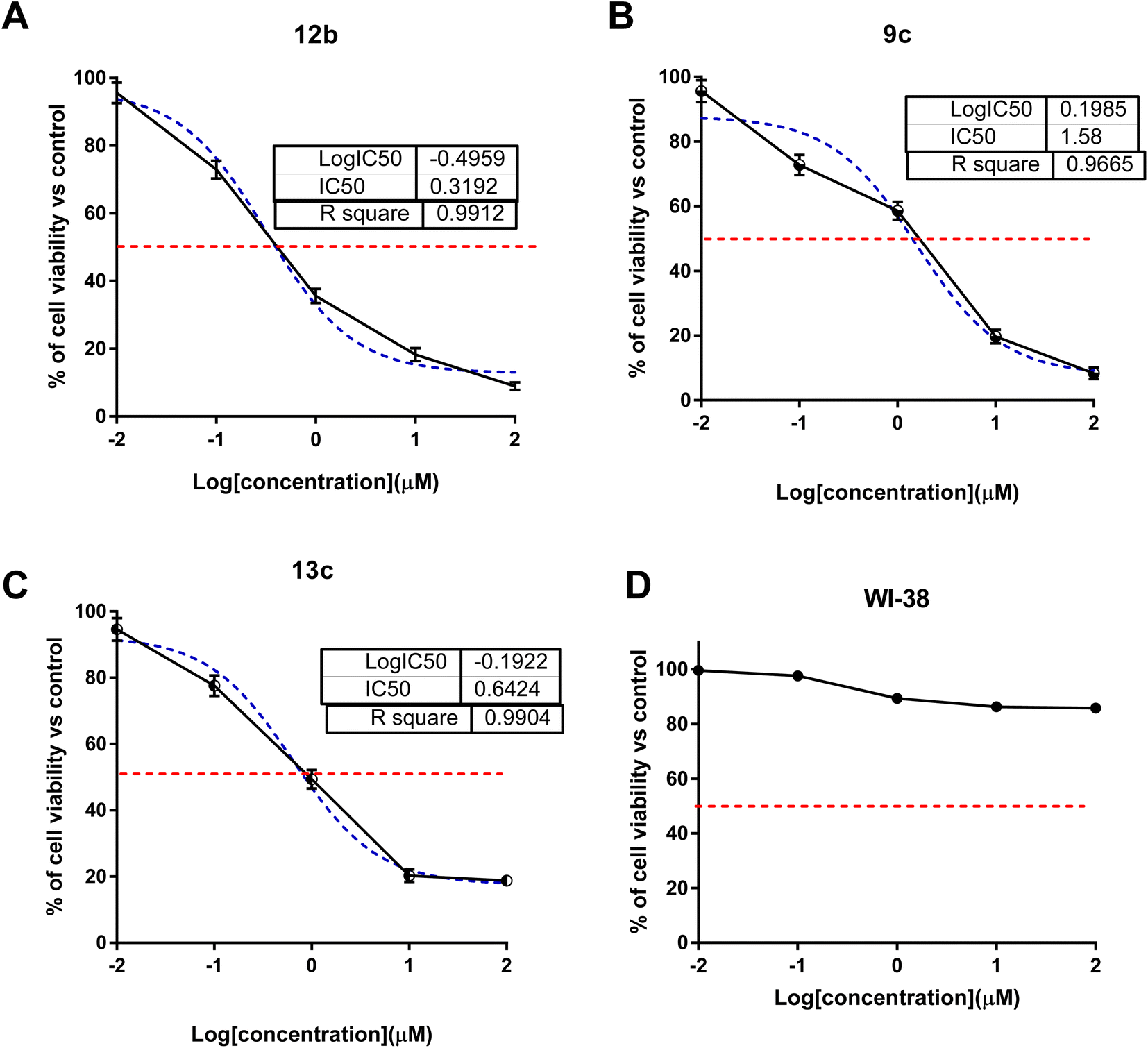 | ||
| Fig. 2 Cell viability versus log concentrations of compounds 9c, 12b, and 13c (A–C) against cancer HCT-116 cells, while (D) is the cytotoxicity against 12b against WI-38 normal cells using MTT assay. Values are expressed as mean ± SD of three independent values. | ||
| Compounds | IC50 (μM) ± SDa | Compounds | IC50 (μM) ± SDa |
|---|---|---|---|
| a IC50 values were calculated as the average of three independent trials using a dose–response curve in GraphPad prism. NT = not tested. | |||
| 5 | 13.8 ± 0.2 | 9c | 1.58 ± 0.21 |
| 7a | 34.8 ± 1.9 | 11a | 7.98 ± 0.7 |
| 7b | 24.3 ± 1.5 | 11d | ≥50 |
| 7d | 39.5 ± 0.9 | 12b | 0.32 ± 0.01 |
| 7f | 18.8 ± 0.6 | 12c | 48.7 ± 1.6 |
| 8a | 40.8 ± 2.1 | 13a | 45.7 ± 1.8 |
| 8c | ≥50 | 13c | 0.64 ± 0.05 |
| 9a | 16.4 ± 0.4 | 13d | |
| Sorafenib | 3.23 ± 0.03 | ||
VEGFR enzyme inhibition
VEGFR is one type of tyrosine kinase (TK) receptor, it was conducted through measuring the percentage of enzyme inhibition at different concentrations using Luminescent assay kit. Compounds 9c, 13c, and 12b were tested for VEGFR2 inhibition, as seen in Table 2. They had promising VEGFR2 inhibition percentages of 92.4, 95.2, and 96.4 with IC50 values of 21.8, 17.8, 19.8 nM compared to sorafenib with 94.7% and IC50 value of 32.1 nM. Hence, compound 12b exhibited potent VEGFR2 inhibition compared to sorafenib.| Compound | VEGFR2 | |
|---|---|---|
| % Of inhibition at [10 μM] | IC50 [nM] ± SDa | |
| a Values are expressed as an average of three independent replicates. IC50 values were calculated using sigmoidal non-linear regression curve fit of percentage inhibition against five concentrations of each compound. | ||
| 9c | 92.4 ± 1.9 | 21.8 ± 1.8 |
| 12b | 95.2 ± 2.1 | 17.8 ± 1.6 |
| 13c | 96.4 ± 2.8 | 19.8 ± 0.6 |
| Sorafenib | 94.68 ± 3.4 | 32.1 ± 0.9 |
Apoptosis-induction activity
To investigate cells with apoptotic cell death, Annexin V/PI procedure was commonly utilized. Combination of propidium iodide (PI) with Annexin V, can distinguish between viable, apoptotic, and necrotic cells by measuring changes in plasma membrane permeability and integrity. Additionally, cell cycle analysis was conducted to measure the percentage of cells population at each stage. Compound 12b was investigated regarding the apoptosis-induction activity in HCT-116 cells (Fig. 3). It induced total apoptosis in HCT-116 cells by 27.57% compared to untreated cells (0.9%). It caused late apoptosis by 7.67% and early apoptosis by 19.9%. So, 12b-treatment induced apoptosis by 21.7-fold. Regarding the cell phase at which cell proliferation was arrested, cell cycle analysis was performed; 12b treatment caused cell cycle arrest at S-phase, increasing the cell population by 38.3% compared to 27.8% in the untreated cells. Cells in G2-phase were non-significantly increased from 15.6% to 20.4%. At the same time, cell population at G1-phase decreased from 56.5% to 41.27%.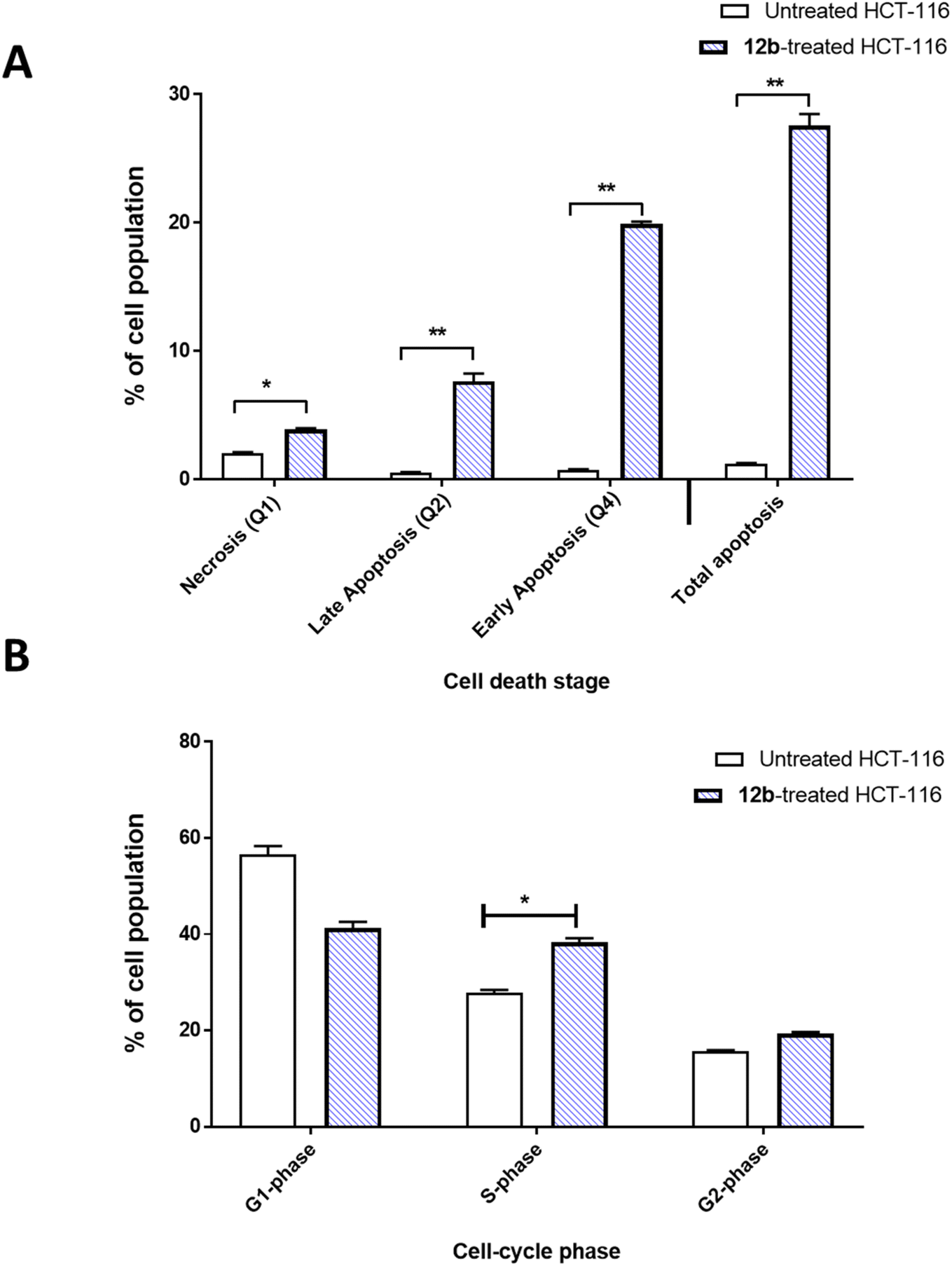 | ||
| Fig. 3 Flow cytometry analysis for apoptosis/necrosis assessment in the untreated and 12b-treated HCT-116 cells with the IC50 value of 0.32 μM for 48 h. (A) Bar representation with cell percentage at each stage. (B) Bar representation for the cell cycle analysis reflecting the cell population in each phase “G1, S, and G” phases. Values are expressed as mean ± SD of three independent trials “*(P ≤ 0.05), and **(P ≤ 0.001) are significantly different using the un-paired test in GraphPad prism”. | ||
Molecular docking studies
One of the structural bioinformatics tools that can be used to highlight the binding mode disposition of compounds towards the protein active site, molecular docking study, was utilized. Compound 12b was subjected to a molecular docking study to highlight the virtual mechanism of binding towards the VEGFR2 protein (Fig. 4); it maintained the biding mode disposition of the co-crystallized ligand; it was docked inside the VEGFR2 binding site with biding energy of −10.66 kcal mol−1, and it formed binding interactions with Cys 919 with bond length of 1.77 Å, and it formed arene–arene interactions with Lys 838.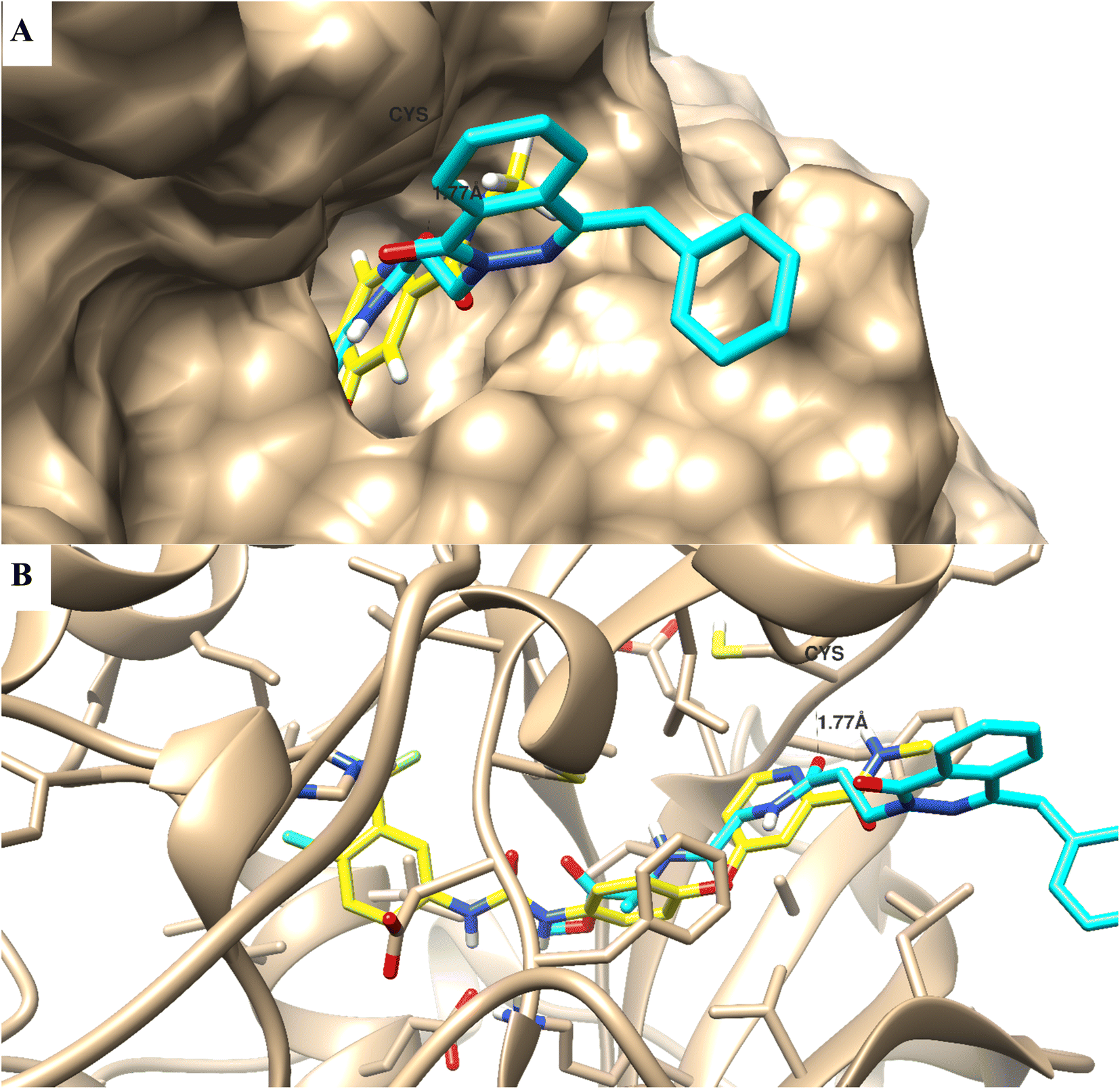 | ||
| Fig. 4 Binding mode and ligand–receptor interactions of the co-crystallized ligand (yellow-colored) and compound 12b (cyan-colored) inside the receptor binding site of VEGFR2 protein. (A) Surface presentation, and (B) interactive binding mode. | ||
Experimental part
1-Chemistry
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) & ethyl acetate/petroleum ether (3:1)”, the spots on thin layer plates were detected by UV lamp. The melting points were determined using a Buchi 510 melting-point system and are uncorrected. IR spectra were recorded in KBr on FTIR Mattson Spectrometers. Nuclear Magnetic Resonance (1H-NMR & 13C-NMR) spectra were measured on Bruker spectrophotometer operating at (400 MHZ) using the appropriate deuterated solvents with chemical shift (δ) expressed in ppm downfield from TMS as internal standard at “nuclear magnetic resonance laboratory, Faculty of Science, Sohag University”. Elemental analyses were performed on a Flash EA-1112 instrument at the “Micro Analytical Laboratory, Faculty of Science, Cairo University, Egypt”. Compounds 1 and 2 were prepared according to the literature procedure.14,23
:5) & ethyl acetate/petroleum ether (3:1)”, the spots on thin layer plates were detected by UV lamp. The melting points were determined using a Buchi 510 melting-point system and are uncorrected. IR spectra were recorded in KBr on FTIR Mattson Spectrometers. Nuclear Magnetic Resonance (1H-NMR & 13C-NMR) spectra were measured on Bruker spectrophotometer operating at (400 MHZ) using the appropriate deuterated solvents with chemical shift (δ) expressed in ppm downfield from TMS as internal standard at “nuclear magnetic resonance laboratory, Faculty of Science, Sohag University”. Elemental analyses were performed on a Flash EA-1112 instrument at the “Micro Analytical Laboratory, Faculty of Science, Cairo University, Egypt”. Compounds 1 and 2 were prepared according to the literature procedure.14,23
Preparation of ethyl 3-(4-benzyl-1-oxophthalazin-2(1H)-yl) propanoate (3). A reaction of compound 2 (2.36 g, 0.01 mol), ethyl acrylate (2.0024 g, 0.02 mol) and anhydrous potassium carbonate (0.05 g, 0.01 mol) was refluxed for 48 h, cooled in ice and the white precipitate filtered.
White crystals; yield (1.62 g, 68%); mp: 88–90 °C; 1H NMR (400 MHz, DMSO-d6) (δ, ppm), (J, Hz): 8.28–8.25 (m, 1H, ArH), 7.89–7.86 (m, 1H, ArH), 7.82–7.75 (m, 2H, ArH), 7.33–7.30 (m, 2H, ArH), 7.27–7.23 (m, 2H, ArH), 7.18–7.15 (m, 1H, ArH), 4.42–4.38 (m, 2H, CH2CH2CO), 4.27 (s, 2H, CH2-ph), 4.05–3.99 (m, 2H, CH2CH3), 2.84 (q, J = 6.4, 2H, CH2CH2CO), 1.09 (t, J = 6.8, 3H, CH3). 13C NMR (101 MHz, DMSO) δ 171.35 (CO) ester, 158.68 (CO) ring, 145.53 (C-Ar), 138.45 (C-Ar), 133.73 (CH-Ar), 132.11 (CH-Ar), 128.95 (C-Ar & 2CH-Ar), 128.79 (2CH-Ar), 127.88 (C-Ar), 126.96 (CH-Ar), 126.82 (CH-Ar), 126.07 (CH-Ar), 60.53 (CH2CH3), 46.39 (CH2CH2CO), 38.22 (CH2ph), 33.19 (CH2CH2CO), 14.35 (CH3). IR (KBr) (cm−1) 3082 (H-Ar), 2978 (H-Al), 1720 (CO) ring, 1639 (CO) ester, 1579 (CC). MS (MALDI, positive mode, matrix DHB) m/z: 359.41 (M + Na)+. Elemental Analysis calculated for C20H20N2O3 (336.4) C, 71.41; H, 5.99; N, 8.33 found: C, 71.45; H, 5.94; N, 8.37.
Procedure for preparation of 3-(4-benzyl-1-oxophthalazin-2(1H)-yl) propanoic acid (4). The procedure used for hydrolysis of ester 3 was reported in previous work,24 to a solution of ethyl 3-(4-benzyl-1-oxophthalazin-2(1H)-yl) propanoate (3) (3.3615 g, 1.0 mmol) in 70% ethyl alcohol (10 ml), NaOH (0.6 g, 1.5 mmol) and 10 ml H2O were added, and the reaction mixture was heated under reflux for 10 h. The reaction mixture was cooled and acidification by dil. HCl. The precipitated residue was crystallized from ethyl alcohol.
White crystals; yield (74%); mp: 154–156 °C; 1H NMR (400 MHz, DMSO-d6) (δ, ppm), (J, Hz): 8.28–8.25 (m, 1H, ArH), 7.89–7.86 (m, 1H, ArH), 7.82–7.75 (m, 2H, ArH), 7.33–7.30 (m, 2H, ArH), 7.27–7.23 (m, 2H, ArH), 7.18–7.15 (m, 1H, ArH), 4.42–4.38 (m, 2H, CH2CH2CO), 4.27 (s, 2H, CH2-ph), 2.84 (q, J = 6.4, 2H, CH2CH2CO).
Procedure for preparation of 3-(4-benzyl-1-oxophthalazin-2(1H)-yl) propanehydrazide (5). A mixture of 3 (3.3615 g, 0.01 mol) and hydrazine hydrate (0.5 ml, 0. 01 mol) in ethanol (30 ml) was refluxed for 6 h. The separated solid was filtered off and recrystallized from ethanol to give compound 5.
Off-white crystals; yield (2.15 g, 63.97%); mp: 170 °C; 1H NMR (400 MHz, DMSO-d6) (δ, ppm), (J, Hz): 9.11 (bs, 1H, D2O exchangeable, NH), 8.29–8.26 (m, 1H, ArH), 7.92–7.90 (m, 1H, ArH), 7.84–7.79 (m, 2H, ArH), 7.36–7.34 (m, 2H, ArH), 7.29–7.26 (m, 2H, ArH), 7.21–7.18 (m, 1H, ArH), 4.40–4.35 (m, 2H, CH2CH2CO), 4.30 (s, 2H, CH2-ph), 4.18 (bs, 2H, D2O exchangeable, NH2), 2.62–2.58 (m, 2H, CH2CH2CO). 13C NMR (101 MHz, DMSO) δ 169.75 (CO), 158.59 (CO) ring, 145.53 (C-Ar), 138.56 (C-Ar), 133.72 (C-Ar), 132.11 (CH-Ar), 129.04 (C-Ar & 2CH-Ar), 128.85 (2CH-Ar), 127.99 (C-Ar), 126.96 (CH-Ar), 126.84 (CH-Ar), 126.13 (CH-Ar), 47.33 (CH2CH2), 38.22 (CH2ph), 32.91 (CH2CH2). IR (KBr) (cm−1): 3022, 2965, 3300 (N-H), 3194 (NH2), 1739. 1643 (CO), 1583. MS (MALDI, positive mode, matrix DHB) m/z: 345.39 (M + Na)+. Elemental Analysis calculated for C18H18N4O2 (322.4) C, 67.07; H, 5.63; N, 17.38 found: C, 67.02; H, 5.67; N, 17.35.
Method A. DCC coupling. The procedure used for DCC-HSU (dicyclohexyl carbodiimide-hydroxysuccinimide) coupling was reported in previous work24 using 3-(4-benzyl-1-oxophthalazin-2(1H)-yl) propanoic acid (4) (3.08 g, 10.0 mmol) and the same molar equivalents of DCC, HSU and amines. The pure product of 2-(4-benzyl-1-oxophthalazin-2(1H)-yl)-N-alkyl-propanamide 7a–g were obtained by column separation using petroleum ether/ethyl acetate 3
:1 as eluent.
Method B. Azide coupling. A cold solution of propanehydrazide 5 (3.22 g, 10 mmol) at (−5 °C) in acetic acid (60 ml) and hydrochloric acid (5N, 30 ml) was added portion wise under stirring to a cold solution (0 °C) of sodium nitrite (0.7 g, 0.01 mol) in water (30 ml). After 30 minutes of stirring at the same temperature, the azide that was produced in situ was extracted using cold ethyl acetate. It was then washed several times with cold water and 5% Na2CO3. The azide 6 was utilized in the following stage without additional purification after drying over anhydrous sodium sulphate. After making the cold-dried azide solution beforehand, 12 mmol of various amines were added to it. Afterwards, the mixture was kept 24 h in the refrigerator and then at room temperature for another 24 h. The reaction mixture was filtered, and the filtrated solution washed with “0.1N HCl, 5% Na2CO3” and water then dried over anhydrous sodium sulphate, the solvent was evaporated in vacuum to give amides 7a–g.
Synthesis of N-allyl-3-(4-benzyl-1-oxophthalazin-2(1H)-yl)propanamide (7a). White crystals; yield (Method A 35%, Method B 60%); mp: 171 °C; 1H NMR (400 MHz, chloroform-d) (δ, ppm), (J, Hz): 8.43–8.41 (m, 1H, ArH), 7.75–7.68 (m, 3H, ArH), 7.32–7.30 (m, 4H, ArH), 7.24–7.20 (m, 1H, ArH), 6.56 (brs, 1H, D2O exchangeable, NH), 5.85–5.75 (m, 1H, CHCH2), 5.14 (d, Jtrans = 17.2, 1H, CHCH2), 5.06 (d, Jcis = 10.4, 1H, CHCH2),4.59 (t, J = 7.2, 2H, NHCH2), 4.30 (s, 2H, CH2-ph), 3.89–3.86 (t, J = 6.4, 2H, CH2CH2CO), 2.89–2.86 (t, J = 7.8, 2H, CH2CH2CO). 13C NMR (101 MHz, CDCl3) δ 170.11 (C
O), 159.48 (CO) ring, 145.72 (C-Ar), 137.68 (C-Ar), 134.17 (CHCH2), 132.99 (CH-Ar), 131.31 (CH-Ar), 129.20 (C-Ar), 128.73 (2CH-Ar), 128.40 (2CH-Ar), 128.15 (C-Ar), 127.20 (CH-Ar), 126.78 (CH-Ar), 125.22 (CH-Ar), 116.32 (CHCH2), 47.31 (CH2CH2CO), 42.01 (CH2CHCH2), 38.90 (CH2ph), 35.61 (CH2CH2CO). IR (KBr) cm−1: 3063, 2916, 3300, 2851 (H-Al), 3237 (H-ole), 1724, 1651, 1579. MS (MALDI, positive mode, matrix DHB) m/z: 370.43 (M + Na)+. Elemental analysis calculated for C21H21N3O2 (347.4) C, 72.60; H, 6.09; N, 12.10 found: C, 72.62; H, 6.05; N, 12.15.
Synthesis of 4-benzyl-2-(3-morpholino-3-oxopropyl) phthalazin-1(2H)-one (7b). Off-white crystals; yield (Method A 40%, Method B 62%); mp: 90 °C; 1H NMR (400 MHz, chloroform-d) (δ, ppm), (J, Hz): 8.45–8.43 (m, 1H, ArH), 7.75–7.69 (m, 3H, ArH), 7.31 (d, J = 8, 4H, ArH), 7.23–7.22 (m, 1H, ArH), 4.61–4.57 (m, 2H, CH2CH2CO), 4.30 (s, 2H, CH2-ph), 3.67–3.65 (m, 6H, 2CH2O & CH2N), 3.53–3.50 (m, 2H, CH2N), 2.96–2.92 (m, 2H, CH2CH2CO). 13C NMR (101 MHz, CDCl3) δ 169.14 (C
O), 159.33 (CO), 145.46 (C-Ar), 137.79 (C-Ar), 132.90 (CH-Ar), 131.24 (CH-Ar), 129.29 (C-Ar), 128.70 (2CH-Ar), 128.42 (2CH-Ar), 128.21 (C-Ar), 127.10 (CH-Ar), 126.74 (CH-Ar), 125.18 (CH-Ar), 66.85 (CH2O), 66.65 (CH2O), 48.00 (CH2CH2CO), 46.03 (CH2N), 41.93 (CH2N), 38.91 (CH2ph), 31.73 (CH2CH2CO). IR (KBr) cm−1: 3025, 2965, (2924 & 2851) H-Al, 1661, 1630, 1579. MS (MALDI, positive mode, matrix DHB) m/z: 400.46 (M + Na)+. Elemental analysis calculated for C22H23N3O3 (377.4) C, 70.01; H, 6.14; N, 11.13 found: C, 70.06; H, 6.18; N, 11.18.
Synthesis of 3-(4-benzyl-1-oxophthalazin-2(1H)-yl)-N-propylpropanamide (7c). White crystals; yield (Method A 50%, Method B 69%); mp: 184 °C; 1H NMR (400 MHz, chloroform-d) (δ, ppm), (J, Hz): 8.43–8.40 (m, 1H, ArH), 7.75–7.67 (m, 3H, ArH), 7.30 (d, J = 7.6, 4H, ArH), 7.23–7.21 (m, 1H, ArH), 6.57 (brs, 1H, D2O exchangeable, NH), 4.57 (t, J = 7.2, 2H, CH2CH2CO), 4.30 (s, 2H, CH2-ph), 3.22–3.16 (m, 2H, NHCH2), 2.85 (t, J = 7.2, 2H, CH2CH2CO), 1.53–1.43 (m, 2H, CH2CH3), 0.87 (t, J = 7.7, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 170.19 (C
O), 159.52 (CO) ring, 145.77 (C-Ar), 137.68 (C-Ar), 137.55 (C-Ar), 133.00 (CH-Ar), 131.30 (CH-Ar), 129.19 (C-Ar), 128.71 (2CH-Ar), 128.39 (2CH-Ar), 128.12 (CH-Ar), 127.15 (CH-Ar), 126.77 (CH-Ar), 125.22 (CH-Ar), 47.36 (CH2CH2CO), 41.34 (CH2CH2CH3), 38.90 (CH2ph), 35.73 (CH2CH2CO), 22.69 (CH2CH2CH3), 11.31 (CH3). IR (KBr) cm−1: 3296 (N-H), 3088, 2949, 2880 (H-Al), 1726, 1637, 1583. MS (MALDI, positive mode, matrix DHB) m/z: 372.45 (M + Na)+. Elemental analysis calculated for C21H23N3O2 (349.4) C, 72.18; H, 6.63; N, 12.03 found: C, 72.12; H, 6.68; N, 12.08.
Synthesis of 4-benzyl-2-(3-oxo-3-(piperidin-1-yl) propyl) phthalazin-1(2H)-one (7d). Off-white crystals; yield (Method A 55%, Method B 70%); mp: 80 °C; 1H NMR (400 MHz, chloroform-d) (δ, ppm), (J, Hz): 8.44–8.42 (m, 1H, ArH), 7.73–7.67 (m, 3H, ArH), 7.28–7.27 (m, 4H, ArH), 7.21–7.19 (m, 1H), 4.60–4.56 (m, 2H, CH2CH2CO), 4.29 (s, 2H, CH2-ph), 3.58–3.55 (m, 2H, NCH2), 3.44–3.42 (m, 2H, NCH2), 2.95–2.85 (m, 2H, CH2CH2CO), 1.63–1.54 (m, 2H, 3CH2). 13C NMR (101 MHz, CDCl3) δ 168.68 (C
O), 159.30 (CO)ring, 145.32 (C-Ar), 137.87 (C-Ar), 132.79 (CH-Ar), 131.12 (CH-Ar), 129.28 (C-Ar), 128.67 (2CH-Ar), 128.41 (2CH-Ar), 128.26 (C-Ar), 127.09 (CH-Ar), 126.69 (CH-Ar), 125.13 (CH-Ar), 48.14 (CH2CH2CO), 46.63 (NCH2), 42.63 (NCH2), 38.92 (CH2ph), 31.97 (CH2CH2CO), 26.48 (CH2CH2CH2), 25.53 (CH2CH2CH2), 24.52 (CH2CH2CH2). IR (KBr) cm−1: 3057, 2927, 2851 (H-Al), 1734, 1657, 1579. MS (MALDI, positive mode, matrix DHB) m/z: 398.48 (M + Na)+. Elemental analysis calculated for C23H25N3O2 (375.5) C, 73.57; H, 6.71; N, 11.19 found: C, 73.52; H, 6.75; N, 11.14.
Synthesis of 3-(4-benzyl-1-oxophthalazin-2(1H)-yl)-N-isobutylpropanamide (7e). White crystals; yield (Method A 45%, Method B 65%); mp: 164 °C 1H NMR (400 MHz, chloroform-d) (δ, ppm), (J, Hz): 8.46–8.43 (m, 1H, ArH), 7.78–7.70 (m, 3H, ArH), 7.31(d, J = 7.2, 4H, ArH), 7.24–7.23 (m, 1H, ArH), 6.48 (brs, 1H, D2O exchangeable, NH), 4.60–4.41 (m, 2H, CH2CH2CO), 4.30 (s, 2H, CH2-ph), 3.09–3.06 (m, 2H, CH2CH), 2.96–2.86 (m, 2H, CH2CH2CO), 1.79–1.71 (m, 1H, CH2CH), 1.27–0.86 (m, 6H, 2CH3). 13C NMR (101 MHz, CDCl3) δ 170.18 (C
O), 159.56 (CO) ring, 145.81 (C-Ar), 137.67 (C-Ar), 133.02 (CH-Ar), 131.32 (CH-Ar), 129.21 (C-Ar), 128.73 (2CH-Ar), 128.53 (CH-Ar), 128.39 (CH-Ar), 128.14 (C-Ar), 127.20 (CH-Ar), 126.79 (CH-Ar), 125.23 (CH-Ar), 47.38 (CH2CH2CO), 47.01 (CH2CH), 38.93 (CH2ph), 35.80 (CH2CH2CO), 29.66 (CH3), 28.36 (CH3), 20.06 (CH(CH3)2). IR (KBr) cm−1: 3296 (N-H), 3090, 2957, 2865 (H-Al), 1730, 1639, 1583. MS (MALDI, positive mode, matrix DHB) m/z: 386.47 (M + Na)+. Elemental analysis calculated for C22H25N3O2 (363.5) C, 72.70; H, 6.93; N, 11.56 found: C, 72.73; H, 6.95; N, 11.59.
Synthesis of 3-(4-benzyl-1-oxophthalazin-2(1H)-yl)-N-butylpropanamide (7f). White crystals; yield (Method A 55%, Method B 67%); mp: 140 °C; 1H NMR (400 MHz, chloroform-d) (δ, ppm), (J, Hz): 8.46–8.43 (m, 1H, ArH), 7.76–7.71 (m, 3H, ArH), 7.31 (d, J = 7.2, 4H, ArH), 7.24–7.21 (m, 3H, ArH), 6.47 (brs, 1H, D2O exchangeable, NH), 4.59–4.55 (m, 2H, CH2CH2CO), 4.31 (s, 2H, CH2-ph), 3.28–3.20 (m, 2H, NHCH2), 2.90–2.83 (m, 2H, CH2CH2CO), 1.30–1.26 (m, 2H, CH2 CH2CH3), 0.95–0.91 (m, 2H, CH2CH3), 0.85 (t, J = 7, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 170.13 (C
O), 159.58 (CO) ring, 145.82 (C-Ar), 137.66 (C-Ar), 133.02 (CH-Ar), 131.33 (CH-Ar), 129.20 (C-Ar), 128.73 (2CH-Ar), 128.39 (2CH-Ar), 128.13 (C-Ar), 127.20 (CH-Ar), 126.78 (CH-Ar), 125.23 (CH-Ar), 47.31 (CH2CH2CO), 39.36 (NHCH2), 38.92 (CH2ph), 35.77 (CH2CH2CO), 29.66 (CH2CH2CH3), 20.03 (CH2CH3), 13.64 (CH3). IR (KBr) cm−1: 3300, 3080, 2937, 2863 (H-Al), 1732, 1639, 1567. MS (MALDI, positive mode, matrix DHB) m/z: 386.47 (M + Na)+. Elemental analysis calculated for C22H25N3O2 (363.5) C, 72.70; H, 6.93; N, 11.56 found: C, 72.74; H, 6.96; N, 11.51.
Synthesis of 4-benzyl-2-(3-oxo-3-(pyrrolidin-1-yl) propyl) phthalazin-1(2H)-one (7g). Off-white crystals; yield (Method A 50%, Method B 64%); mp: 86 °C; 1H NMR (400 MHz, chloroform-d) (δ, ppm), (J, Hz): 8.43–8.41 (m, 1H, ArH), 7.72–7.65 (m, 3H, ArH), 7.27–7.26 (m, 4H, ArH), 7.20–7.18 (m, 1H, ArH), 4.61–4.53 (m, 2H, CH2CH2CO), 4.27 (s, 2H, CH2-ph), 3.48–3.41 (m, 4H, CH2NCH2), 2.88–2.85 (m, 2H, CH2CH2CO), 1.94–1.79 (m, 4H, CH2CH2). 13C NMR (101 MHz, CDCl3) δ 169.02 (C
O), 159.25 (CO), 145.23 (C-Ar), 137.88 (C-Ar), 132.78 (CH-Ar), 131.10 (CH-Ar), 129.24 (C-Ar), 128.66 (2CH-Ar), 128.40 (2CH-Ar), 128.25 (C-Ar), 127.10 (CH-Ar), 126.67 (CH-Ar), 125.11 (CH-Ar), 47.54 (CH2CH2CO), 46.62 (NCH2CH2), 45.60 (NCH2CH2), 38.90 (CH2ph), 33.31 (CH2CH2CO), 26.04 (NCH2CH2), 24.37 (NCH2CH2). IR (KBr) cm−1: 3025, 2965, 2873 (H-Al), 1661, 1643, 1579. MS (MALDI, positive mode, matrix DHB) m/z: 384.46 (M + Na)+. Elemental analysis calculated for C22H23N3O2 (361.4) C, 73.11; H, 6.41; N, 11.63 found: C, 73.16; H, 6.44; N, 11.68.
Method A. DCC coupling. The procedure used for DCC-HSU (dicyclohexyl carbodiimide-hydroxysuccinimide) coupling was reported in previous work24 using 3-(4-benzyl-1-oxophthalazin-2(1H)-yl) propanoic acid (4) (3.08 g, 10.0 mmol) and the same molar equivalents of DCC, HSU and amino acids methyl ester hydrochloride. The pure product of methyl (3-[4-benzyl-1-oxophthalazin-2(1H)-yl) propanoyl amino]alkanoates 8a–c were obtained by column separation using petroleum ether/ethyl acetate 3
:1 as eluent.
Method B. Azide coupling. An ice-cold solution of sodium nitrite (0.7 g, 0.01 mol) in water (30 ml) was gradually added to a cold solution of propane hydrazide (5, 3.22 g, 10 mmol) in acetic acid (60 ml) and hydrochloric acid (5N, 30 ml) while stirring. The temperature of the mixture was kept at −5 °C. After 30 minutes of stirring at the same temperature, the azide that was produced in situ was extracted using cold ethyl acetate. It was then washed several times with cold water and 5% Na2CO3. After drying over anhydrous sodium sulphate, the azide (6) was used without further in the next step. Amino acids methyl ester hydrochloride (15 mmol); “glycine, β-Alanine, and L-Leucine” which were placed with triethyl amine (1 g, 10 mmol) in ethyl acetate solution at (−5 °C) for 15 minutes. Then the amino acid methyl ester hydrochloride solution was added to the previously prepared cold dried solution of the azide. Afterwards, the mixture was kept 24 h in the refrigerator and then at room temperature for another 24 h. After filtering the reaction mixture, the resulting solution was rinsed with 0.1N HCl, 5% Na2CO3, and water. It was then dried over anhydrous sodium sulphate. The solvent was subsequently evaporated under vacuum, and the remaining ethyl acetate-petroleum ether substance was crystallized to produce esters 8a–c.
Synthesis of methyl (3-(4-benzyl-1-oxophthalazin-2(1H)-yl) propanoyl) glycinate (8a). White crystals; yield (Method A 50%, Method B 70%); mp: 152 °C; 1H NMR (400 MHz, chloroform-d) (δ, ppm), (J, Hz): 8.44–8.41 (m, 1H, ArH), 7.74–7.66 (m, 3H, ArH), 7.29–7.26 (m, 4H, ArH), 7.22–7.20 (m, 1H, ArH), 6.92 (brs, 1H, D2O exchangeable, NH), 4.62–4.57 (m, 2H, CH2CH2CO), 4.30 (s, 2H, CH2-ph), 4.04–4.01 (m, 2H, NHCH2CO), 3.67 (s, 3H, OCH3), 2.94–2.89 (m, 2H, CH2CH2CO). 13C NMR (101 MHz, CDCl3) δ 170.69 (C
O), 170.23 (CO), 159.43 (CO) ring, 145.69 (C-Ar), 137.74 (C-Ar), 132.95 (CH-Ar), 131.27 (CH-Ar), 129.21 (C-Ar), 128.70 (2CH-Ar), 128.40 (2CH-Ar), 128.17 (C-Ar), 127.19 (CH-Ar), 126.74 (CH-Ar), 125.20 (CH-Ar), 52.13 (OCH3), 47.13 (CH2CH2CO), 41.28 (NHCH2), 38.84 (CH2ph), 35.15 (CH2CH2CO).IR (KBr) cm−1: 3308 (N-H), 3067, 2957, 2845 (H-Al), 1749, 1643, 1630 (CO) ester, 1581. MS (MALDI, positive mode, matrix DHB) m/z: 402.43 (M + Na)+. Elemental analysis calculated for C21H21N3O4 (379.4) C, 66.48; H, 5.58; N, 11.08 found: C, 66.44; H, 5.53; N, 11.13.
Synthesis of methyl 3-(3-(4-benzyl-1-oxophthalazin-2(1H)-yl) propanamido) propanoate (8b). White crystals; yield (Method A 55%, Method B 69%); mp: 150 °C; 1H NMR (400 MHz, chloroform-d) (δ, ppm), (J, Hz): 8.36–8.34 (m, 1H, ArH), 7.69 (d, J = 7.6, 1H, ArH), 7.64–7.60 (m, 2H, ArH), 7.25 (t, J = 8.4, 4H, ArH), 7.18–7.16 (m, 1H, ArH), 6.92–6.88 (m, 1H, D2O exchangeable, NH), 4.55–4.51 (m, 2H, CH2CH2CO), 4.26 (s, 2H, CH2-ph), 3.56 (s, 3H, CH3), 3.51–3.46 (m, 2H, NHCH2CH2), 2.82–2.78 (m, 2H, NHCH2CH2), 2.51–2.47 (m, 2H, CH2CH2CO). 13C NMR (101 MHz, CDCl3) δ 172.71 (C
O), 170.51 (CO), 159.33 (CO)ring, 145.64 (C-Ar), 137.73 (C-Ar), 132.95 (CH-Ar), 131.25 (CH-Ar), 129.10 (C-Ar), 128.70 (2CH-Ar), 128.37 (2CH-Ar), 128.04 (C-Ar), 127.05 (CH-Ar), 126.74 (CH-Ar), 125.23 (CH-Ar), 51.65 (OCH3), 47.37 (CH2CH2CO), 38.86 (CH2ph), 35.34 (NHCH2CH2CO), 34.97 (NHCH2CH2CO), 33.81 (CH2CH2CO). IR (KBr) cm−1: 3408 (NH), 3065, 2965, (2916 & 2857) H-Al, 1720, 1663, 1655 (CO) ester, 1585. MS (MALDI, positive mode, matrix DHB) m/z: 416.46 (M + Na)+. Elemental analysis calculated for C22H23N3O4 (393.4) C, 67.16; H, 5.89; N, 10.68 found: C, 67.14; H, 5.84; N, 10.64.
Synthesis of methyl (3-(4-benzyl-1-oxophthalazin-2(1H)-yl) propanoyl) leucinate (8c). White crystals; yield (Method A 60%, Method B 78%); mp: 100 °C; 1H NMR (400 MHz, chloroform-d) (δ, ppm), (J, Hz): 8.45–8.42 (m, 1H, ArH), 7.74–7.68 (m, 3H, ArH), 7.29 (d, J = 7.2, 4H, ArH), 7.20 (d, J = 7.6, 1H, ArH), 6.79 (d, J = 8.4, 1H, D2O exchangeable, NH), 4.65–4.58 (m, 3H, CH2CH2CO & NHCHCO), 4.30 (s, 2H, CH2-ph), 3.63 (s, 3H, OCH3), 2.92 (t, J = 7.2, 2H, CH2CH2CO), 1.62–1.50 (m, 3H, CH2CH(CH3)2), 0.87–0.83 (m, 6H, 2CH3). 13C NMR (101 MHz, CDCl3) δ 173.43 (C
O), 170.21 (CO), 159.53 (CO) ring, 145.85 (C-Ar), 137.69 (C-Ar), 133.03 (CH-Ar), 131.33 (CH-Ar), 129.13 (C-Ar), 128.75 (2CH-Ar), 128.37 (2CH-Ar), 128.11 (C-Ar), 127.19 (CH-Ar), 126.78 (CH-Ar), 125.29 (CH-Ar), 52.14 (NHCH2CO), 50.76 (OCH3), 46.99 (CH2CH2CO), 41.33 (CH2CH), 38.94 (CH2ph), 35.33 (CH2CH2CO), 24.81 (CH(CH3)2), 22.74 (CH3), 21.79 (CH3). IR (KBr) cm−1: 3306, 3065, 2973, 2920 (H-Al), 1749, 1737, 1641 (CO) ester, 1581. MS (MALDI, positive mode, matrix DHB) m/z: 458.54 (M + Na)+. Elemental analysis calculated for C25H29N3O4 (436.0) C, 68.95; H, 6.71; N, 9.65 found: C, 68.90; H, 6.76; N, 9.60.
Synthesis of hydrazides 9a–c. To a solution of ester 8a (3.49 g, 0.01 mol) in ethyl alcohol (30 ml) was added hydrazine hydrate (0.5 ml, 0.01 mol). The reaction mixture was refluxed for 6 h, cooled and the white precipitate filtered and recrystallized from ethanol to obtain the corresponding hydrazide 3-(4-benzyl-1-oxophthalazin-2(1H)-yl)-N-(2-hydrazineyl-2-oxoethyl) propenamide (9a). By the same method, 3-(4-benzyl-1-oxophthalazin-2(1H)-yl)-N-(3-hydrazineyl-3-oxopropyl) propanamide (9b) and 3-(4-benzyl-1-oxophthalazin-2(1H)-yl)-N-(1-hydrazineyl-4-methyl-1-oxopentan-2-yl)propanamide (9c) can be prepared from reflux of the ester 8b and 8c (3.8042 g, 0.01 mol) and (4.3552 g, 0.01 mol) in ethyl alcohol (30 ml) with hydrazine hydrate (0.5 ml, 0.01 mol)for 6 h and then recrystallized from boiling ethanol.
Synthesis of 3-(4-benzyl-1-oxophthalazin-2(1H)-yl)-N-(2-hydrazinyl-2-oxoethyl) propanamide (9a). White crystals; yield (90%); mp: 170 °C; 1H NMR (400 MHz, DMSO-d6) (δ, ppm), (J, Hz): 9.07 (brs, 1H, D2O exchangeable, NHNH2), 8.28 (d, J = 8.4, 2H, ArH), 7.90 (d, J = 8, 1H, ArH), 7.83–7.76 (m, 2H, ArH), 7.34–7.25 (m, 4H, ArH), 7.19–7.15 (m, 1H, D2O exchangeable, NH), 4.40–4.36 (m, 2H, CH2CH2CO), 4.30 (s, 2H, CH2-ph), 3.69–3.66 (m, 2H, NHCH2CO), 3.40 (brs, 2H, D2O exchangeable, NH2), 2.72 (t, J = 7.6, 2H, CH2CH2CO). 13C NMR (101 MHz, DMSO) δ 170.79 (C
O), 168.75 (CO), 158.64 (CO) ring, 145.58 (C-Ar), 138.58 (CH-Ar), 133.71 (CH-Ar), 132.11 (CH-Ar), 129.02 (C-Ar & 2CH-Ar), 128.84 (2CH-Ar), 127.98 (C-Ar), 126.96 (CH-Ar), 126.85 (C-Ar), 126.14 (CH-Ar), 47.28 (CH2CH2CO), 41.42 (NHCH2CO), 38.22 (CH2ph), 34.57 (CH2CH2CO). IR (KBr) cm−1: 3445, 3292 (NH-NH2), 3200 (NH-NH2), 3059, 2947, 2935, 1720, 1634, 1620, 1581. MS (MALDI, positive mode, matrix DHB) m/z: 402.43 (M + Na)+. Elemental analysis calculated for C20H21N5O3 (379.4) C, 63.31; H, 5.58; N, 18.46 found: C, 63.34; H, 5.53; N, 18.41.
Synthesis of 3-(4-benzyl-1-oxophthalazin-2(1H)-yl)-N-(3-hydrazineyl-3-oxopropyl) propanamide (9b). White crystals; yield (88%); mp: 222 °C; 1H NMR (400 MHz, DMSO-d6) (δ, ppm), (J, Hz): 9.09–9.03 (m, 1H, D2O exchangeable, NHNH2), 8.26–8.10 (m, 2H, ArH), 7.88–7.77 (m, 3H, ArH), 7.33–7.23 (m, 4H, ArH), 7.17 (brs, 1H, D2O exchangeable, NH), 4.35–4.33 (m, 2H, CH2CH2CO), 4.28 (s, 2H, CH2-ph), 3.43 (brs, 2H, D2O exchangeable, NH2), 2.63 (s, 2H), 2.63–2.60 (m, 2H, NHCH2CH2), 1.84–1.76 (m, 4H, 2CH2CH2CO). 13C NMR (101 MHz, DMSO) δ 170.26 (C
O), 170.20 (CO), 158.56 (CO) ring, 145.42 (C-Ar), 138.61 (C-Ar), 133.68 (CH-Ar), 132.08 (CH-Ar), 129.02 (C-Ar& 2CH-Ar), 128.83 (2CH-Ar), 128.03 (C-Ar), 126.97 (CH-Ar), 126.84 (CH-Ar), 126.14 (CH-Ar), 47.43 (CH2CH2CO), 38.24 (CH2ph), 35.84 (NHCH2CH2CO), 34.70 (NHCH2CH2CO), 34.07 (CH2CH2CO). IR (KBr) cm−1: 3427, 3300 (NH-NH2), 3210 (NH-NH2), 3086, 2931, 2863, 1655, 1649, 1608, 1581. MS (MALDI, positive mode, matrix DHB) m/z: 416.46 (M + Na)+. Elemental analysis calculated for C21H23N5O3 (393.4) C, 64.11; H, 5.89; N, 17.80 found: C, 64.14; H, 5.85; N, 17.84.
Synthesis of 3-(4-benzyl-1-oxophthalazin-2(1H)-yl)-N-(1-hydrazinyl-4-methyl-1-oxopentan-2-yl) propanamide (9c). White crystals; yield (89%); mp: 220 °C; 1H NMR (400 MHz, DMSO-d6) (δ, ppm), (J, Hz): 9.13 (brs, 1H, D2O exchangeable, NHNH2), 8.29–8.25 (m, 1H, ArH), 8.08–8.05 (m, 1H, ArH), 7.90–7.77 (m, 3H, ArH), 7.35–7.16 (m, 4H, ArH), 7.20–7.16 (m, 1H, D2O exchangeable, NH), 4.45–4.37 (m, 3H, CH2CH2CO & NHCHCO), 4.30 (s, 2H, CH2-ph), 3.43 (brs, 2H, D2O exchangeable, NH2), 2.77–2.65 (m, 2H, CH2CH2CO), 1.43–1.39 (m, 3H, CH2CH), 0.76 (d, J = 6.8, 6H, 2CH3). 13C NMR (101 MHz, DMSO) δ 171.66 (C
O), 170.04 (CO), 158.58 (CO)ring, 145.45 (C-Ar), 138.59 (C-Ar), 133.64 (CH-Ar), 132.03 (CH-Ar), 129.01 (C-Ar & 2CH-Ar), 128.83 (2CH-Ar), 128.04 (C-Ar), 126.93 (CH-Ar), 126.85 (CH-Ar), 126.11 (CH-Ar), 50.17 (NHCHCO), 47.23 (CH2CH2CO), 41.57 (CH2CH(CH3)2), 38.27 (CH2ph), 34.58 (CH2CH2CO), 24.62 (CH2CH(CH3)2), 23.25 (CH3), 22.11 (CH3). IR (KBr) cm−1: 3414, 3292 (NH-NH2), 3116 (NH-NH2), 3065, 2959, 2922, 2867, 1732, 1645, 1604, 1585. MS (MALDI, positive mode, matrix DHB) m/z: 458.54 (M + Na)+. Elemental analysis calculated for C24H29N5O3 (435.5) C, 66.19; H, 6.71; N, 16.08 found: C, 66.14; H, 6.76; N, 16.03.
Synthesis of N-(2-(allylamino)-2-oxoethyl)-3-(4-benzyl-1-oxophthalazin-2(1H)-yl) propanamide (11a). Off-white crystals; yield (68%); mp: 165 °C; 1H NMR (400 MHz, chloroform-d) (δ, ppm), (J, Hz): 8.38–8.35 (m, 1H, ArH), 7.71–7.64 (m, 3H, ArH), 7.39–7.36 (m, 1H, ArH), 7.29 (d, J = 7.2, 3H, ArH), 7.21–7.18 (m, 3H, D2O exchangeable, 2NH & ArH), 5.83–5.74 (m, 1H, CHCH2), 5.15 (d, Jtrans = 17.1, 1H, CHCH2), 5.05 (d, Jcis = 10.4, 1H, CHCH2), 4.58 (t, J = 6.7, 2H, CH2CH2CO), 4.28 (s, 2H, CH2-ph), 3.97–3.95 (m, 2H, NHCH2CO), 3.86–3.83 (m, 2H, NHCH2CH), 2.88 (t, J = 7.6, 2H, CH2CH2CO). 13C NMR (101 MHz, CDCl3) δ 171.31 (C
O), 169.00 (CO), 159.45 (CO) ring, 145.82 (C-Ar), 137.69 (C-Ar), 133.96 (CHCH2), 133.00 (CH-Ar), 131.30 (CH-Ar), 129.22 (C-Ar), 128.73 (2CH-Ar), 128.39 (2CH-Ar), 128.08 (C-Ar), 127.17 (CH-Ar), 126.77 (CH-Ar), 125.25 (CH-Ar), 116.20 (CHCH2), 47.29 (CH2CH2CO), 43.59 (NHCH2CO), 41.86 (CH2CHCH2), 38.88 (CH2ph), 35.34 (CH2CH2CO). IR (KBr) cm−1: 3300, 3086, 2969, (2920 & 2853) H-Al, 1720, 1645, 1630, 1579. MS (MALDI, positive mode, matrix DHB) m/z: 427.48 (M + Na)+. Elemental analysis calculated for C23H24N4O3 (404.5) C, 68.30; H, 5.98; N, 13.85 found: C, 68.35; H, 5.93; N, 13.80.
Synthesis of 3-(4-benzyl-1-oxophthalazin-2(1H)-yl)-N-(2-(butylamino)-2-oxoethyl) propanamide (11b). Off-white crystals; yield (66%); mp: 161 °C; 1H NMR (400 MHz, chloroform-d) (δ, ppm), (J, Hz): 8.41–8.36 (m, 1H, ArH), 7.72–7.66 (m, 3H, ArH), 7.29 (d, J = 8.0, 4H, ArH), 7.20–7.18 (m, 1H, ArH), 7.12–7.10 (m, 1H, D2O exchangeable, NHCH2), 6.92–6.88 (m, 1H, D2O exchangeable, NHCH2CH2), 4.60–4.54 (m, 2H, CH2CH2CO), 4.28 (s, 2H, CH2-ph), 3.90 (s, 2H, NHCH2CO), 3.25–3.17 (m, 2H, NHCH2CH2), 2.87 (t, J = 7.4, 2H, CH2CH2CO), 1.49–1.41 (m, 2H, CH2CH2CH3), 1.36–1.27 (m, 2H, CH2CH3), 0.89 (t, J = 7.6, 3H, CH3). 13C NMR (101 MHz, CDCl3) δ 171.26 (C
O), 170.14 (CO), 169.01 (CO) ring, 145.83 (C-Ar), 137.68 (C-Ar), 133.02 (CH-Ar), 131.31 (CH-Ar), 129.22 (C-Ar), 128.72 (2CH-Ar), 128.38 (2CH-Ar), 128.08 (C-Ar), 127.13 (CH-Ar), 126.76 (CH-Ar), 125.26 (CH-Ar), 47.30 (CH2CH2CO), 43.56 (NHCH2CO), 39.39 (NHCH2), 38.88 (CH2ph), 35.32 (CH2CH2CO), 31.64 (CH2CH2CH3), 20.03 (CH2CH2CH3), 13.67 (CH3). IR (KBr) cm−1: 3288, 3090, 2985, 2931, 2873 (H-Al), 1728, 1645, 1626, 1583. MS (MALDI, positive mode, matrix DHB) m/z: 443.52 (M + Na)+. Elemental analysis calculated for C24H28N4O3 (420.5) C, 68.55; H, 6.71; N, 13.32 found: C, 68.50; H, 6.76; N, 13.37.
Synthesis of 3-(4-benzyl-1-oxophthalazin-2(1H)-yl)-N-(2-oxo-2-(piperidin-1-yl) ethyl) propanamide (11c). Off-white crystals; yield (61%); mp: 110 °C; 1H NMR (400 MHz, chloroform-d) (δ, ppm), (J, Hz): 8.44–8.42 (m, 1H, ArH), 7.71–7.65 (m, 3H, ArH), 7.29–7.23 (m, 3H, ArH), 7.21–7.17 (m, 1H, ArH), 7.05 (brs, 1H, D2O exchangeable, NH), 4.60 (t, J = 7.6, 2H, CH2CH2CO), 4.30 (s, 2H, CH2-ph), 4.03 (s, 2H, NHCH2CO), 3.55–3.52 (m, 2H, NCH2), 3.31–3.28 (m, 2H, NCH2), 2.87 (t, J = 7.4, 2H, CH2CH2CO), 1.63 (q, J = 5.6, 2H, CH2CH2CH2), 1.53 (t, J = 5.6, 4H, CH2CH2CH2). 13C NMR (101 MHz, CDCl3) δ 170.46 (C
O), 165.99 (CO), 159.23 (CO) ring, 145.36 (C-Ar), 137.94 (CH-Ar), 132.79 (CH-Ar), 131.12 (CH-Ar), 129.23 (C-Ar), 128.66 (2CH-Ar), 128.36 (2CH-Ar), 128.35 (C-Ar), 127.24 (CH-Ar), 126.63 (CH-Ar), 125.15 (CH-Ar), 47.22 (CH2CH2CO), 45.42 (NCH2CH2), 43.11 (NCH2CH2), 41.31 (NHCH2CO), 38.88 (CH2ph), 35.04 (CH2CH2CO), 26.11 (CH2CH2CH2), 25.36 (CH2CH2CH2), 24.31 (CH2CH2CH2). IR (KBr) cm−1: 3308, 3059, 2933, 2853 (H-Al), 1730, 1657, 1630, 1581. MS (MALDI, positive mode, matrix DHB) m/z: 455.53 (M + Na)+. Elemental analysis calculated for C25H28N4O3 (432.5) C, 69.42; H, 6.53; N, 12.95 found: C, 69.47; H, 6.58; N, 12.90.
Synthesis of 3-(4-benzyl-1-oxophthalazin-2(1H)-yl)-N-(2-morpholino-2-oxoethyl) propanamide (11d). Off-white crystals; yield (62%); mp: 123 °C; 1H NMR (400 MHz, chloroform-d) (δ, ppm), (J, Hz): 8.39–8.37 (m, 1H, ArH), 7.69–7.62 (m, 3H, ArH), 7.26–7.22 (m, 5H, ArH), 7.18–7.14 (m, 1H, D2O exchangeable, NH), 4.56 (t, J = 7.2, 2H, CH2CH2CO), 4.26 (s, 2H, CH2-ph), 4.03–4.02 (m, 2H, NHCH2CO), 3.61–3.59 (m, 4H, 2CH2O), 3.55–3.52 (m, 2H, NCH2), 3.35–3.33 (m, 2H, NCH2), 2.87 (t, J = 7.6, 2H, CH2CH2CO). 13C NMR (101 MHz, CDCl3) δ 170.71 (C
O), 166.79 (CO), 159.24 (CO) ring, 145.45 (C-Ar), 137.90 (C-Ar), 132.85 (CH-Ar), 131.19 (CH-Ar), 129.18 (C-Ar), 128.67 (2CH-Ar), 128.35 (2CH-Ar), 127.72 (C-Ar), 127.17 (CH-Ar), 126.65 (CH-Ar), 125.18 (CH-Ar), 66.58 (CH2O), 66.27 (CH2O), 47.25 (CH2CH2CO), 44.85 (NCH2CH2O), 42.24 (NCH2CH2O), 41.17 (NHCH2CO), 38.82 (CH2ph), 34.99 (CH2CH2CO). IR (KBr) cm−1: 3308, 3057, 2963, (2931 & 2855) H-Al, 1732, 1663, 1636, 1581. MS (MALDI, positive mode, matrix DHB) m/z: 457.52 (M + Na)+. Elemental analysis calculated for C24H26N4O4 (434.5) C, 66.34; H, 6.03; N, 12.89 found: C, 66.39; H, 6.08; N, 12.86.
After combining “glycine, β-Alanine, and L-Leucine” with triethyl amine (1 g, 10 mmol) in an ethyl acetate solution at −5 °C for 15 minutes, the amino acid methyl ester hydrochloride solution was added to the azide cold dried solution that had been previously made. The next step was to chill the combination for 24 hours before letting it sit at room temperature for another hour. Products 12a–c were obtained by filtering the reaction mixture, washing the resulting solution with 0.1N HCl, 5% Na2CO3, and water, and finally drying it over anhydrous sodium sulfate. The solvent was subsequently evaporated under vacuum.
Synthesis of methyl (3-(4-benzyl-1-oxophthalazin-2(1H)-yl) propanoyl) glycylglycinate (12a). White crystals; yield (72%); mp: 178 °C; 1H NMR (400 MHz, chloroform-d) (δ, ppm), (J, Hz): 8.35–8.33 (m, 1H, ArH), 7.71–7.64 (m, 3H, ArH), 7.59–7.58 (m, 1H, ArH), 7.45 (brs, 1H, D2O exchangeable, NH), 7.26 (d, J = 7.2, 4H, ArH), 7.19 (brs, 1H, D2O exchangeable, NH), 4.58 (t, J = 6.8, 2H, CH2CH2CO), 4.28 (s, 2H, CH2-ph), 4.02–3.99 (m, 4H, 2NHCH2CO), 3.67 (s, 3H, OCH3), 2.89 (t, J = 7.2, 2H, CH2CH2CO). 13C NMR (101 MHz, CDCl3) δ 171.41 (C
O), 170.21 (CO), 169.73 (CO), 159.48 (CO) ring, 145.87 (C-Ar), 137.70 (C-Ar), 132.97 (CH-Ar), 131.29 (CH-Ar), 129.22 (C-Ar), 128.71 (2CH-Ar), 128.39 (2CH-Ar), 128.06 (C-Ar), 127.09 (CH-Ar), 126.75 (CH-Ar), 125.25 (CH-Ar), 52.17 (OCH3), 47.36 (CH2CH2CO), 43.26 (NHCH2CO), 41.14 (NHCH2COO), 38.85 (CH2ph), 35.33 (CH2CH2CO). IR (KBr) cm−1: 3388, 3314, 3071, 2945, 2861 (H-Al), 1761, 1744, 1665, 1637 (CO) ester. MS (MALDI, positive mode, matrix DHB) m/z: 459.48 (M + Na)+. Elemental analysis calculated for C23H24N4O5 (436.5) C, 63.29; H, 5.54; N, 12.84 found: C, 63.25; H, 5.59; N, 12.89.
Synthesis of methyl 3-(2-(3-(4-benzyl-1-oxophthalazin-2(1H)-yl) propanamido) acetamido) propanoate(12b). Off-white crystals; yield (70%) mp; 120 °C; 1H NMR (400 MHz, chloroform-d) (δ, ppm), (J, Hz): 8.40–8.37 (m, 1H, ArH), 7.74–7.65 (m, 4H, ArH), 7.27 (d, J = 8, 4H, ArH), 7.21–7.17 (m, 2H, D2O exchangeable, 2NH), 4.58 (t, J = 7.6, 2H, CH2CH2CO), 4.29 (s, 2H, CH2-ph), 3.91–3.89 (m, 2H, NHCH2CO), 3.65 (s, 3H, OCH3), 3.53–3.47 (m, 2H, NHCH2CH2), 2.88 (t, J = 7.6, 2H, CH2CH2CO), 2.55–2.51 (m, 2H, NHCH2CH2). 13C NMR (101 MHz, CDCl3) δ 172.58 (C
O), 171.24 (CO), 169.21 (CO), 159.45 (CO) ring, 145.87 (C-Ar), 137.70 (C-Ar), 133.04 (CH-Ar), 131.37 (CH-Ar), 129.18 (C-Ar), 128.74 (2CH-Ar), 128.38 (2CH-Ar), 128.04 (C-Ar), 127.11 (CH-Ar), 126.78 (CH-Ar), 125.29 (CH-Ar), 51.79 (OCH3), 47.34 (CH2CH2CO), 43.37 (NHCH2CO), 38.88 (CH2ph), 35.30 (CH2CH2CO), 35.09 (NHCH2CH2CO), 33.78 (NHCH2CH2CO), 29.03 (NHCH2CH2). Dept 135 (101 MHz, CDCl3) δ 132.97 (CH-Ar), 131.31 (CH-Ar), 128.71 (2CH-Ar), 128.38 (2CH-Ar), 127.13 (CH-Ar), 126.75 (CH-Ar), 125.24 (CH-Ar), 51.70 (OCH3), 47.33 (CH2CH2CO), 43.46 (NHCH2CO), 38.93 (CH2-ph), 35.34 (CH2CH2CO), 35.12 (NHCH2CH2CO), 33.82 (NHCH2CH2CO). IR (KBr) cm−1: 3296, 3086, 3065, 2955, 2849 (H-Al), 1724, 1734, 1643, 1624 (CO) ester, 1583. MS (MALDI, positive mode, matrix DHB) m/z: 473.51 (M + Na)+. Elemental analysis calculated for C24H26N4O5 (450.5) C, 63.99; H, 5.82; N, 12.44 found: C, 63.95; H, 5.87; N, 12.40.
Synthesis of methyl (3-(4-benzyl-1-oxophthalazin-2(1H)-yl) propanoyl) glycylleucinate (12c). Off-white crystals; yield (73%); mp: 125 °C; 1H NMR (400 MHz, chloroform-d) (δ, ppm), (J, Hz): 8.37–8.35 (m, 1H, ArH), 7.70–7.63 (m, 3H, ArH), 7.44–7.42 (m, 2H, D2O exchangeable, ArH & NH), 7.28–7.24 (m, 4H, ArH), 7.18–7.15 (m, 1H, D2O exchangeable, NH), 4.56 (t, J = 7.1, 2H, CH2CH2CO), 4.26 (s, 2H, CH2-ph), 4.12–3.93 (m, 3H, NHCH2CO & NHCHCO), 3.66 (s, 3H, OCH3), 2.87 (t, J = 7.2, 2H, CH2CH2CO), 1.61 (d, J = 6.8, 2H, CH2CH), 1.24 (t, J = 6.4, 1H, CH2CH), 0.92–0.87 (m, 6H, 2CH3). 13C NMR (101 MHz, CDCl3) δ 173.20 (C
O), 171.36 (CO), 169.22 (CO), 159.35 (CO) ring, 145.78 (C-Ar), 137.72 (C-Ar), 132.94 (CH-Ar), 131.25 (CH-Ar), 129.20 (C-Ar), 128.69 (2CH-Ar), 128.38 (2CH-Ar), 128.09 (C-Ar), 127.15 (CH-Ar), 126.72 (CH-Ar), 125.21 (CH-Ar), 52.16 (NHCHCO), 50.92 (OCH3), 47.38 (CH2CH2CO), 43.27 (NHCH2CO), 41.01 (CH2CH(CH3)2), 38.83 (CH2ph), 35.13 (CH2CH2CO), 24.80 (CH(CH3)2), 22.74 (CH3), 21.78 (CH3). IR (KBr) cm−1: 3300, 3220, 3063, 2961, 2867 (H-Al), 1741, 1730, 1645, 1628 (CO) ester, 1583. MS (MALDI, positive mode, matrix DHB) m/z: 515.59 (M + Na)+. Elemental analysis calculated for C27H32N4O5 (492.6) C, 65.84; H, 6.55; N, 11.37 found: C, 65.84; H, 6.55; N, 11.37.
Synthesis of (Z)-3-(4-benzyl-1-oxophthalazin-2(1H)-yl)-N-(2-(2-(4-nitrobenzylidene) hydrazinyl)-2-oxoethyl)propanamide (13a). A mixture of hydrazide 9a (3.53 g, 0.01 mol) and 4-nitrobenzaldehyde (3.02 g, 0.02 mol) in ethanol (30 ml) was refluxed for 24 h. By cooling the solid product formed, filtered off and recrystallized from ethanol solvent gave compound 13a.
Off-white crystals; yield (80%); mp: 240 °C; 1H NMR (400 MHz, chloroform-d) (δ, ppm), (J, Hz): δ = (Z/E isomers mixture 78/22) 11.71& 11.68 (2 s, 1H, D2O exchangeable, CONHN), 8.09 & 8.41 (2 s, 1H, NCH), 8.34–8.25 (m, 4H, ArH), 7.96–7.81 (m, 5H, ArH), 7.36 (d, J = 7.7, 2H, ArH), 7.28 (t, J = 8, 3H, ArH), 7.18 (s, 1H, D2O exchangeable, CONHCH2), 4.42–4.38 (m, 2H, CH2CH2CO), 4.32 (s, 2H, CH2-ph), 4.29 (s, 1H, NHCH2CO), 3.86 (s, 1H, NHCH2CO), 2.77 (s, 2H, CH2CH2CO). IR (KBr) cm−1: 3431, 3355, 3208 (H-Ar), 3114 (H-Ar), 3087, 2957, 2925, 2851, 1739, 1694, 1622, 1524 (NO2), 1597 (CN). MS (MALDI, positive mode, matrix DHB) m/z: 535.55 (M + Na)+. Elemental analysis calculated for C27H24N6O5 (512.5) C, 63.27; H, 4.72; N, 16.40 found: C, 63.22; H, 4.77; N, 16.45.
Synthesis of (Z)-3-(4-benzyl-1-oxophthalazin-2(1H)-yl)-N-(2-(2-(4-chlorobenzylidene) hydrazinyl)-2-oxoethyl)propanamide (13b). A mixture of hydrazide 9a (3.53 g, 0.01 mol) and p-chlorobenzaldehyde (2.81 g, 0.02 mol) in ethanol (30 ml) was refluxed for 24 h. By cooling the solid product formed, filtered off and recrystallized from ethanol solvent gave compound 13b.
Off-white crystals; yield (85%); mp: 190 °C; 1H NMR (400 MHz, chloroform-d) (δ, ppm), (J, Hz): δ = (Z/E isomers mixture 80/20) 10.17 &10.55 (2 s, 1H, D2O exchangeable, CONHN), 8.44–8.34 (m, 1H, ArH), 7.94 & 8.18 (2 s, 1H, NCH), 7.76–7.70 (m, 3H, ArH), 7.64–7.61 (m, 2H, ArH), 7.40–7.37 (m, 3H, ArH), 7.28–7.26 (m, 4H, ArH), 7.19 & 7.45 (2 s, 1H, D2O exchangeable, CONHCH2), 4.69–4.64 (m, 2H, CH2CH2CO), 4.54–4.52 (m, 2H, NHCH2CO), 4.33 & 4.05 (2 s, 2H, CH2-ph), 3.01–2.90 (m, 2H, CH2CH2CO).IR (KBr) cm−1: 3406, 3296, (3214 & 3132) H-Ar, 3064, 2962, 2927, 1688, 1643, 1632, 1581. MS (MALDI, positive mode, matrix DHB) m/z: 524.99 (M + Na)+. Elemental analysis calculated for C27H24ClN5O3 (502.0) C, 64.60; H, 4.82; Cl, 7.06; N, 13.95 found: C, 64.65; H, 4.87; Cl, 7.04; N, 13.90.
Synthesis of (Z)-3-(4-benzyl-1-oxophthalazin-2(1H)-yl)-N-(2-(2-(4-methoxybenzylidene) hydrazinyl)-2-oxoethyl)propanamide (13c). A mixture of hydrazide 9a (3.53 g, 0.01 mol) and 4-methoxybenzaldehyde (2.72 g, 0.02 mol) in ethanol (30 ml) was refluxed for 24 h. By cooling the solid product formed, filtered off and recrystallized from ethanol solvent gave compound 13c.
Off-white crystals; yield (89%); mp: 191 °C; 1H NMR (400 MHz, DMSO-d6) (δ, ppm), (J, Hz): δ = (Z/E isomers mixture 75/25) 11.29& 11.23 (2 s, 1H, D2O exchangeable, CONHN), 8.29 & 8.38 (2 s, 1H, NCH), 8.21–8.18 (m, 1H, ArH), 7.94–7.80 (m, 2H, ArH), 7.86–7.80 (m, 2H, ArH), 7.64–7.59 (m, 2H, ArH), 7.37–7.27 (m, 4H, ArH), 7.18 & 7.09 (2 s, 1H, D2O exchangeable, CONHCH2), 4.43–4.39 (m, 2H, CH2CH2CO), 4.31 (s, 2H, CH2-ph), 4.24 (s, 2H, NHCH2CO), 3.80 (s, 3H, OCH3), 2.79–2.75 (m, 2H, CH2CH2CO). MS (MALDI, positive mode, matrix DHB) m/z: 520.58 (M + Na)+. Elemental analysis calculated for C28H27N5O4 (497.6) C, 67.59; H, 5.47; N, 14.08 found: C, 67.54; H, 5.45; N, 14.04.
Synthesis of (Z)-3-(4-benzyl-1-oxophthalazin-2(1H)-yl)-N-(2-(2-(5-bromo-2-hydroxybenzylidene) hydrazineyl)-2-oxoethyl) propanamide (13d). A solution containing 3.53 g of hydrazide 9a and 0.01 mol of 5-bromo-2-hydroxybenzaldehyde in 30 ml of ethanol was refluxed for 24 hours. Compound 13d was obtained by chilling the solid product, filtering it out, and then recrystallizing it from the ethanol solvent.
Off-white crystals; yield (88%); mp: 130 °C; 1H NMR (400 MHz, DMSO-d6) (δ, ppm), (J, Hz): δ = (Z/E isomers mixture 50/50) 11.69 & 11.44 (2 s, 1H, D2O exchangeable, CONHN), 8.41 & 8.35 (2 s, 1H, NCH), 8.30–8.25 (m, 2H, ArH), 8.22 (s, 2H, ArH), 7.91 (d, J = 7.8, 2H, ArH), 7.85–7.76 (m, 4H, ArH), 7.35 (d, J = 7.8, 2H, ArH), 7.30–7.26 (m, 3H, ArH), 7.20–7.17 (m, 1H, D2O exchangeable, NHCH2CO), 4.43–4.36 (m, 2H, CH2CH2CO), 4.31 (s, 2H, CH2-ph), 4.31 (s, 2H, NHCH2CO), 4.24 (s, 1H, D2O exchangeable, OH), 2.77–3.70 (m, 2H, CH2CH2CO). IR (KBr) cm−1: 3433, 3286, 3212 (H-Ar), 3067, 2963, 2920, 1677, 1649, 1624, 1577. MS (MALDI, positive mode, matrix DHB) m/z: 585.44 (M + Na)+. Elemental analysis calculated for C27H24BrN5O4 (562.4) C, 57.66; H, 4.30; Br, 14.21; N, 12.45 found: C, 57.63; H, 4.35; Br, 14.22; N, 12.40.
Biological assays
Cytotoxicity of the synthesized compounds using MTT assay
HCT-116 cancer and normal liver WI-38 cell lines were cultured in complete media of “DMEM at 5% carbon dioxide and 37 °C” following standard tissue culture work. The cells were grown in “10% fetal bovine serum (FBS) and 1% penicillin-streptomycin” in 96-multiwell plate. The synthesized compounds were screened for their cytotoxicity using 20 μL of MTT solution (Promega, USA) for 48 hours25,26 with concentrations of “0.01, 0.1, 1, 10, and 100 μM” for 48 h. The plate was cultured for 3 hours. Percentage of cell viability was calculated following this equation (100 − (Asample)/(Acontrol)) × 100.27VEGFR inhibition
The most promising cytotoxic compounds were subjected to VEGFR2 Kinase Assay Kit Catalog #40325 using ELISA kit ELISA Assay following manufacturer information.28 The luminescence was measured with a microplate reader at 450 nm by ELISA Reader (PerkinElmer). The inhibition percentage was calculated following this equation: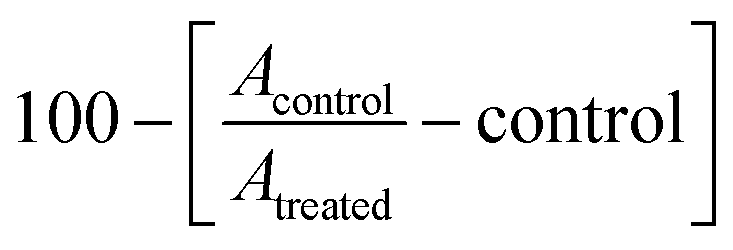 , IC50 was determined using GraphPad prism7.
, IC50 was determined using GraphPad prism7.
Flow cytometry using annexin V/PI staining
After a night of incubation in 6-well culture plates with 3–5 × 105 cells per well, compound 7d was added to the cells and left to treat for 48 hours according to the IC50 values. After that, the cells were incubated in a 100 μL solution of Annexin binding buffer “25 mM CaCl2, 1.4 M NaCl, and 0.1 M Hepes/NaOH, pH 7.4” in the dark for 30 minutes with “Annexin V-FITC solution (1:100) and propidium iodide (PI) at a concentration equivalent to 10 g ml−1”. The labeled cells were then extracted using the Cytoflex FACS machine.29,30
Molecular docking study
Utilizing Maestro, protein, and compound structures were created and optimized. Binding sites inside proteins were then identified using the grid-box dimensions surrounding the co-crystallized ligands. Compounds were docked against the protein structures of VEGFR2 (PDB = 4ASD) using AutoDock Vina software following routine work.31,32 Maestro was utilized to optimize protein and ligand structures. In terms of binding energy and ligand–receptor interactions, binding activities evaluated the results of molecular docking. Chimera-UCSF was then used to complete the visualization.Conclusion
In conclusion, twenty-six new phthalazine derivatives were designed and synthesized beginning with 2-(4-benzyl-1-oxophthalazin-2(1H)-yl)-N-(3-hydrazineyl-3-oxo propyl) acetamide (2) by chemoselective N-alkylation via Michael addition reaction and their structures were interpreted by several analytical and spectroscopic techniques. Interestingly, compound 7d exhibited potent cytotoxicity with an IC50 value 0.38 μM compared to Sorafenib (IC50 = 2.93 μM). Compounds 7d exhibited potent VEGFR2 inhibition by 97.6% with an IC50 value 21.9 μM compared to Sorafenib (94.7% and IC50 of 30.1 μM). For apoptosis activity, 7d-treatment induced apoptosis by 23.6-fold, arresting the cell proliferation at G1-phase. Finally, it formed a good binding affinity towards VEGFR2 protein with a biding energy of −26.8 kcal mol−1, and it formed binding interactions with the key interactive amino acids. Hence, compound 7d was worthy of studying as a target-oriented anti-liver agent with a good selectivity profile.Author contributions
D. E. S., S. M. R., H. A. S. synthesized the entire series of derivatives with the characterization of structure elucidation. At the same time, I. E. A., M. S. A., A. H.·K., and M. A. A. participated in characterization, data analysis, resources, and revision, while M. S. Nafie initiated the idea and design of the biology part by carrying out in vitro cytotoxic screening, flow cytometry, and in silico studies with the linguistic revision and manuscript finalizing. D. E. S., S. M. R., and M. S. Nafie wrote the original draft with the literature review in their corresponding parts. All authors agreed on the manuscript in the final submitted form.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research received no specific grant from funding agencies in the public, commercial, or not-for-profit sectors.References
- G. R. Dagenais, et al., Variations in common diseases, hospital admissions, and deaths in middle-aged adults in 21 countries from five continents (PURE): a prospective cohort study, Lancet, 2020, 395(10226), 785–794, DOI:10.1016/S0140-6736(19)32007-0.
- R. L. Siegel, K. D. Miller and A. Jemal, Cancer statistics, Ca-Cancer J. Clin., 2018, 68(1), 7–30, DOI:10.3322/caac.21442.
- F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jemal, Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries, Ca-Cancer J. Clin., 2018, 68(6), 394–424, DOI:10.3322/caac.21492.
- N. Dhiman, K. Kaur and V. Jaitak, Tetrazoles as anticancer agents: A review on synthetic strategies, mechanism of action and SAR studies, Bioorg. Med. Chem., 2020, 28(15), 115599, DOI:10.1016/j.bmc.2020.115599.
- H. Sung, et al., Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries, Ca-Cancer J. Clin., 2021, 71(3), 209–249, DOI:10.3322/caac.21660.
- N. Vila, P. Besada, J. Brea, M. I. Loza and C. Terán, Novel Phthalazin-1(2H)-One Derivatives Displaying a Dithiocarbamate Moiety as Potential Anticancer Agents, Molecules, 2022, 27(23), 8115, DOI:10.3390/molecules27238115.
- S. Gali, D. Raghu, V. Mallikanti, V. Thumma and N. Vaddiraju, Design, synthesis of benzimidazole tethered 3,4-dihydro-2H-benzo[e] [1, 3] oxazines as anticancer agents, Mol. Diversity, 2023 DOI:10.1007/s11030-023-10661-3.
- B. Yang, Y. S. Yang, N. Yang, G. Li and H.-L. Zhu, Design, biological evaluation and 3D QSAR studies of novel dioxin-containing pyrazoline derivatives with thiourea skeleton as selective HER-2 inhibitors, Sci. Rep., 2016, 6, 27571, DOI:10.1038/srep27571.
- A. Al-Mulla, A Review: Biological Importance of Heterocyclic Compounds, Pharma Chem., 2017, 9(13), 141–147 CAS.
- M. Barge, G. Rashinkar, D. Kanase, S. Mohite and T. Lohar, One-drop organocatalyzed multicomponent synthesis of pyrazolo[1,2-b]phthalazine-diones and pyrazolophthalazinyl quinolines, Res. Chem. Intermed., 2022, 48(12), 5045–5058, DOI:10.1007/s11164-022-04848-w.
- M. F. H. Naglaa and A. E. Galal, Molecular docking and biological assessment of substituted phthalazin-1(2H)-one derivatives, J. Heterocycl. Chem., 2020, 57(4), 1845–1862, DOI:10.1002/jhet.3913.
- R. Narang, B. Narasimhan and S. Sharma, A review on biological activities and chemical synthesis of hydrazide derivatives, Curr. Med. Chem., 2012, 19(4), 569–612, DOI:10.2174/092986712798918789.
- D. C. Izuogu, J. N. Asegbeloyin, M. M. Jotani and E. R. T. Tiekink, 2-[(2,4,6-Tri-methyl-benzene)-sulfon-yl]phthalazin-1(2H)-one: crystal structure, Hirshfeld surface analysis and computational study, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2020, 76(5), 697–702, DOI:10.1107/S2056989020005101.
- M. I. Marzouk, S. A. Shaker, A. A. Abdel Hafiz and K. Z. El-Baghdady, Design and Synthesis of New Phthalazinone Derivatives Containing Benzyl Moiety with Anticipated Antitumor Activity, Biol. Pharm. Bull., 2016, 39(2), 239–251, DOI:10.1248/bpb.b15-00656.
- S. M. Emam, S. M. E. Rayes, I. A. I. Ali, H. A. Soliman and M. S. Nafie, Synthesis of phthalazine-based derivatives as selective anti-breast cancer agents through EGFR-mediated apoptosis: in vitro and in silico studies, BMC Chem., 2023, 17(1), 90, DOI:10.1186/s13065-023-00995-2.
- M. Napoletano, et al., Phthalazine PDE4 inhibitors. Part 2: the synthesis and biological evaluation of 6-methoxy-1,4-disubstituted derivatives, Bioorg. Med. Chem. Lett., 2001, 11(1), 33–37, DOI:10.1016/s0960-894x(00)00587-4.
- I. Graça, E. J. Sousa, P. C. Pinheiro and F. Q. VieiraI, Anti-neoplastic properties of hydralazine in prostate cancer, Oncotarget, 2014, 5(15), 5950–5964, DOI:10.18632/oncotarget.1909.
- Z. Malinowski, E. Fornal, A. Sumara, R. Kontek, K. Bukowski, B. Pasternak, D. Sroczyński, J. Kusz, M. Małecka and M. Nowak, Amino- and polyaminophthalazin-1(2H)-ones: synthesis, coordination properties, and biological activity, Beilstein J. Org. Chem., 2021, 17, 558–568, DOI:10.3762/bjoc.17.50.
- S. Tanaka, M. Tanaka and A. Akashi, Influence of antihypertensive treatment with budralazine on autoregulation of cerebral blood flow in spontaneously hypertensive rats, Stroke, 1989, 20(12), 1724–1729, DOI:10.1161/01.STR.20.12.1724.
- S. M. El Rayes, G. El Enany, I. A. I. Ali, W. Ibrahim and M. S. Nafie, Synthesis of Novel Phthalazinedione-Based Derivatives with Promising Cytotoxic, Anti-bacterial, and Molecular Docking Studies as VEGFR2 Inhibitors, ACS Omega, 2022, 7(30), 26800–26811, DOI:10.1021/acsomega.2c03182.
- S. Elmeligie, A. M. Aboul-Magd, D. S. Lasheen, T. M. Ibrahim, T. M. Abdelghany, S. M. Khojah and K. A. M. Abouzid, Design and synthesis of phthalazine-based compounds as potent anticancer agents with potential antiangiogenic activity via VEGFR-2 inhibition, J. Enzyme Inhib. Med. Chem., 2019, 34(1), 1347–1367, DOI:10.1080/14756366.2019.1642883.
- S. M. El Rayes, G. El-Enany, M. S. Gomaa, I. A. I. Ali, W. Fathalla, F. H. Pottoo and F. A. Khan, Convenient Synthesis of N-Alkyl-2-(3-phenyl-quinoxalin-2-ylsulfanyl)acetamides and Methyl-2-[2-(3-phenyl-quinoxalin-2-ylsulfanyl)acetylamino]alkanoates, ACS Omega, 2022, 7(38), 34166–34176, DOI:10.1021/acsomega.2c03522.
- H. S. Ibrahim, W. M. Eldehna, H. A. Abdel-Aziz, M. M. Elaasser and M. M. Abdel-Aziz, Improvement of antibacterial activity of some sulfa drugs through linkage to certain phthalazin-1(2H)-one scaffolds, Eur. J. Med. Chem., 2014, 85, 480–486, DOI:10.1016/j.ejmech.2014.08.016.
- S. M. El Rayes, A. Aboelmagd, M. S. Gomaa, W. Fathalla, I. A. I. Ali, F. H. Pottoo and F. A. Khan, Newly synthesized 3-(4-chloro-phenyl)-3-hydroxy-2,2-dimethyl-propionic acid methyl ester derivatives selectively inhibit the proliferation of colon cancer cells, RSC Adv., 2020, 10(15), 8825–8841, 10.1039/C9RA10950A.
- T. Mosmann, Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays, J. Immunol. Methods, 1983, 65(1–2), 55–63, DOI:10.1016/0022-1759(83)90303-4.
- S. A. Abdulmalek, A. M. Saleh, Y. R. Shahin and E. F. El Azab, Functionalized siRNA-chitosan nanoformulations promote triple-negative breast cancer cell death via blocking the miRNA-21/AKT/ERK signaling axis: in-silico and in vitro studies, Naunyn-Schmiedeberg's Arch. Pharmacol., 2024 DOI:10.1007/s00210-024-03068-w.
- M. S. Nafie and A. T. A. Boraei, Exploration of novel VEGFR2 tyrosine kinase inhibitors via design and synthesis of new alkylated indolyl-triazole Schiff bases for targeting breast cancer, Bioorg. Chem., 2022, 122, 105708, DOI:10.1016/j.bioorg.2022.105708.
- M. S. Nafie, S. M. Kishk, S. Mahgoub and A. M. Amer, Quinoline-based thiazolidinone derivatives as potent cytotoxic and apoptosis-inducing agents through EGFR inhibition, Chem. Biol. Drug Des., 2022, 99(4), 547–560, DOI:10.1111/cbdd.13997.
- K. M. Dawood, M. A. Raslan, A. A. Abbas, B. E. Mohamed, M. H. Abdellattif, M. S. Nafie and M. K. Hassan, Novel Bis-Thiazole Derivatives: Synthesis and Potential Cytotoxic Activity through Apoptosis with Molecular Docking Approaches, Front. Chem., 2021, 9, 694870, DOI:10.3389/fchem.2021.694870.
- M. S. Nafie, K. Arafa, N. K. Sedky, A. A. Alakhdar and R. K. Arafa, Triaryl dicationic DNA minor-groove binders with antioxidant activity display cytotoxicity and induce apoptosis in breast cancer, Chem.-Biol. Interact., 2020, 324, 109087, DOI:10.1016/j.cbi.2020.109087.
- A. T. A. Boraei, E. H. Eltamany, I. A. I. Ali, S. M. Gebriel and M. S. Nafie, Synthesis of new substituted pyridine derivatives as potent anti-liver cancer agents through apoptosis induction: in vitro, in vivo, and in silico integrated approaches, Bioorg. Chem., 2021, 111, 104877, DOI:10.1016/j.bioorg.2021.104877.
- M. S. Nafie, M. A. Tantawy and G. A. Elmgeed, Screening of different drug design tools to predict the mode of action of steroidal derivatives as anti-cancer agents, Steroids, 2019, 152, 108485, DOI:10.1016/j.steroids.2019.108485.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic characterizations of the synthesized compounds. See DOI: https://doi.org/10.1039/d4ra02103g |
| This journal is © The Royal Society of Chemistry 2024 |