
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuinazolines and thiazolidine-2,4-dions as SARS-CoV-2 inhibitors: repurposing, in silico molecular docking and dynamics simulation
Sanadelaslam S. A. El-Hddad a,
Mohamed H. Sobhyb,
Ahmed El-morsyc,
Nabil A. Shomand and
Khaled El-Adl*be
a,
Mohamed H. Sobhyb,
Ahmed El-morsyc,
Nabil A. Shomand and
Khaled El-Adl*be
aPharmaceutical Chemistry Department, Faculty of Pharmacy, Omar Almukhtar University, Al Bayda 991, Libya
bChemistry Department, Faculty of Pharmacy, Heliopolis University for Sustainable Development, Cairo, Egypt. E-mail: khaled.eladl@hu.edu.eg
cPharmaceutical Chemistry Department, College of Pharmacy, The Islamic University, Najaf, Iraq
dDepartment of Pharmaceutics and Pharmaceutical Technology, Faculty of Pharmacy, Ahram Canadian University, Giza, Egypt
ePharmaceutical Medicinal Chemistry & Drug Design Department, Faculty of Pharmacy (Boys), Al-Azhar University, Cairo11884, Egypt. E-mail: eladlkhaled74@yahoo.com; eladlkhaled74@azhar.edu.eg
First published on 23rd April 2024
Abstract
This paper presents an extensive analysis of COVID-19 with a specific focus on VEGFR-2 inhibitors as potential treatments. The investigation includes an overview of computational methodologies employed in drug repurposing and highlights in silico research aimed at developing treatments for SARS-CoV-2. The study explores the possible effects of twenty-eight established VEGFR-2 inhibitors, which include amide and urea linkers, against SARS-CoV-2. Among these, nine inhibitors exhibit highly promising in silico outcomes (designated as 3–6, 11, 24, 26, 27, and sorafenib) and are subjected to extensive molecular dynamics (MD) simulations to evaluate the binding modes and affinities of these inhibitors to the SARS-CoV-2 Mpro across a 100 ns timeframe. Additionally, MD simulations are conducted to ascertain the binding free energy of the most compelling ligand–pocket complexes identified through docking studies. The findings provide valuable understanding regarding the dynamic and thermodynamic properties of the interactions between ligands and pockets, reinforcing the outcomes of the docking studies and presenting promising prospects for the creation of therapeutic treatments targeting COVID-19.
1. Introduction
The global impact of the COVID-19 pandemic and the escalating number of deaths underscore the pressing requirement for scientists to discover new and potent medications. Despite the considerable challenges and time constraints associated with drug discovery, certain candidates have exhibited effectiveness against severe acute respiratory syndrome coronaviruses (SARS-CoV), offering promising prospects in the quest for viable treatments.1 There exist four distinct types of coronaviruses identified as α, β, γ, and δ. Notably, SARS-CoV, MERS-CoV, and SARS-CoV-2 fall within the category of β-coronaviruses.2 A comparative examination comparing the genetic sequences of these three viruses has unveiled a higher degree of similarity between SARS-CoV-2 and SARS-CoV, with an 89.1% match in their nucleotide sequences, compared to MERS-CoV.3 SARS-CoV-2 possesses a single-stranded, positive-sense RNA genome, approximately 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 nucleotides long, and contains a minimum of six open reading frames (ORFs). The genome is enveloped by nucleocapsid proteins, forming ribonucleoprotein complexes. These ORFs are essential for synthesizing a minimum of 16 non-structural proteins and 4 structural proteins. Distinctively, SARS-CoV-2 features unique polybasic cleavage sites within its RNA genome, a trait that enhances the virus's pathogenicity and transmissibility among other betacoronaviruses.4 Specifically, the 229E gene within the SARS-CoV-2 genome is responsible for encoding two polyproteins, vital for producing functional polypeptides necessary for viral transcription and replication. A crucial component in this process is the 3 chymotrypsin-like proteases (3CLpro or Mpro), which is responsible for the cleavage of polyproteins at a minimum of 11 sites within the central and C-terminal regions, facilitating the release of essential proteins necessary for the virus's replication process.5,6 Given its central role, the viral protease (Mpro) has become a promising target for developing inhibitors against coronaviruses.7–11
000 nucleotides long, and contains a minimum of six open reading frames (ORFs). The genome is enveloped by nucleocapsid proteins, forming ribonucleoprotein complexes. These ORFs are essential for synthesizing a minimum of 16 non-structural proteins and 4 structural proteins. Distinctively, SARS-CoV-2 features unique polybasic cleavage sites within its RNA genome, a trait that enhances the virus's pathogenicity and transmissibility among other betacoronaviruses.4 Specifically, the 229E gene within the SARS-CoV-2 genome is responsible for encoding two polyproteins, vital for producing functional polypeptides necessary for viral transcription and replication. A crucial component in this process is the 3 chymotrypsin-like proteases (3CLpro or Mpro), which is responsible for the cleavage of polyproteins at a minimum of 11 sites within the central and C-terminal regions, facilitating the release of essential proteins necessary for the virus's replication process.5,6 Given its central role, the viral protease (Mpro) has become a promising target for developing inhibitors against coronaviruses.7–11
As of now, there is no targeted antiviral treatment for COVID-19, with the primary strategy being supportive care.12 To expedite the search for effective treatments, researchers have adopted the strategy of drug repurposing, which involves exploring existing drugs for new therapeutic purposes in a short timeframe.13 Molecular docking-based assays have emerged as essential tools for discovering potential antiviral agents, allowing researchers to screen and evaluate drug candidates prior to their synthesis.14 This strategy, known as in silico virtual screening, utilizes computational methods to screen and simulate the interactions of potential drug candidates with the target protein, aiming to identify promising inhibitors for SARS-CoV-2 Mpro.
Repurposing existing drugs, which seeks to discover new therapeutic applications beyond their original medical indications, represents an efficient method in treatment development, including for cancer.15,16 By exploring the potential of drugs that are already approved and in clinical use, this strategy provides a rapid and cost-effective route to identify effective treatments for various conditions. In the context of the COVID-19 outbreak, drug repurposing has been proposed for specific drugs.17–22 The strategy of repurposing existing drugs for COVID-19 treatment takes advantage of the extensive clinical history of commercially available medications, which come with well-documented dosages and toxicity profiles. This familiarity potentially paves the way for these drugs to undergo fast-tracked and cost-efficient phase II–III clinical trials, or to be considered for compassionate use. It's worth noting that a notable number of the drugs being reconsidered for combating COVID-19 were originally developed as anticancer treatments. This resemblance is understandable and quite expected, given that both virus-infected cells and cancer cells exhibit similar characteristics, such as enhanced synthesis of nucleic acids, proteins, lipids, and increased energy metabolism to support their respective programs. Therefore, drugs targeting particular pathways in cancer cells might also impede viral replication. Identifying appropriate drugs for repurposing in treating COVID-19 requires a thorough examination of existing data from diverse sources, such as clinical studies, anecdotal evidence, and documented publications. This study provides a comprehensive analysis of VEGFR-2 inhibitors, initially developed for cancer therapy, exploring their potential repurposing for COVID-19 therapy.
1.1. Rational of the work
Nelfinavir, a potent inhibitor of HIV-1 protease, effectively blocks the replication of SARS-CoV.23,24 It also prevents the cytopathic impact of SARS-CoV virus. Ritonavir and lopinavir, which protease inhibitors employed in the treatment of SARS and MERS, work in similar ways as treatments designed for HIV.25 HIV-1 protease inhibitors, such as fosamprenavir, nelfinavir, saquinavir, atazanavir, darunavir, lopinavir, ritonavir, and indinavir, contain urea and amide linkers as depicted in Fig. 1.26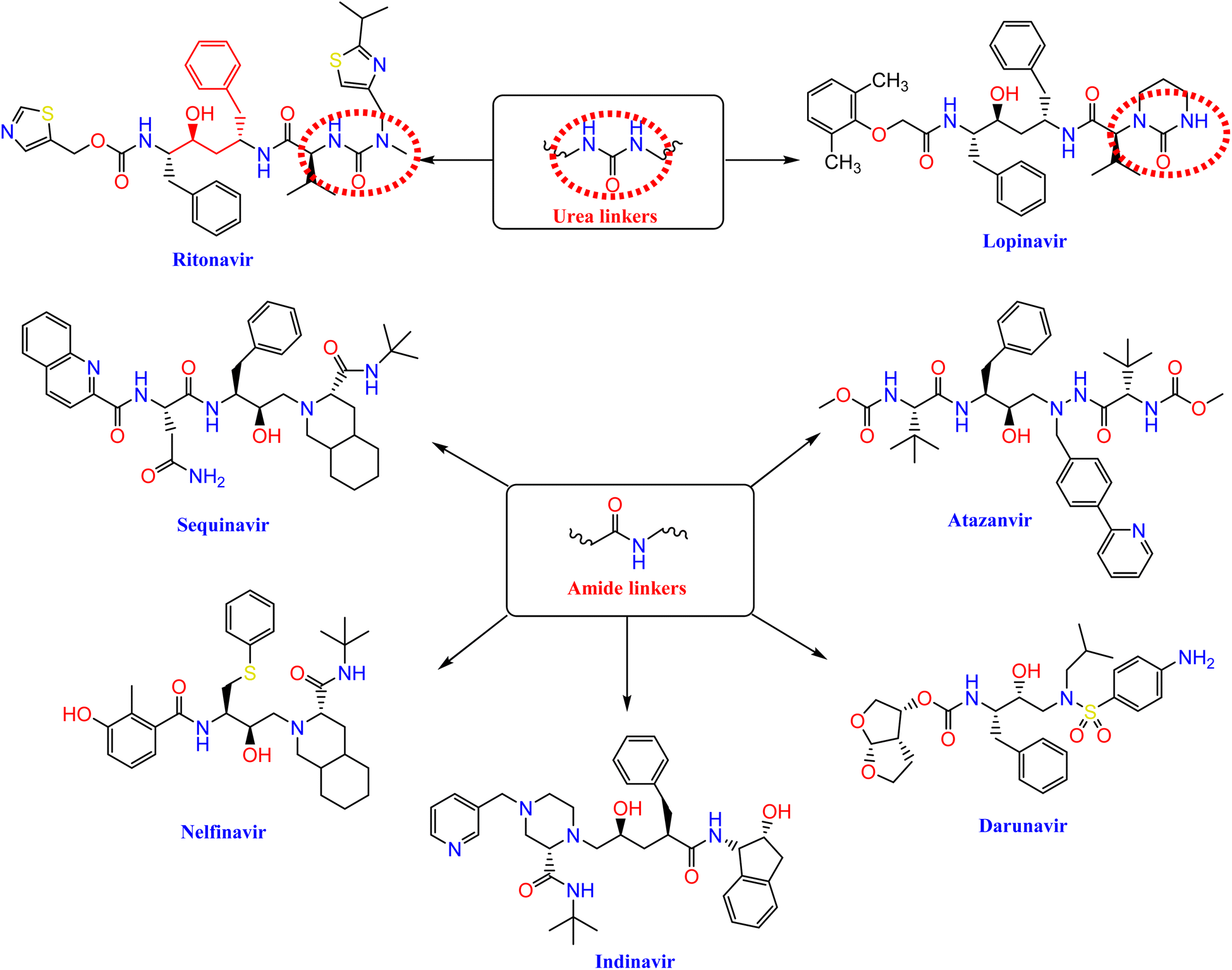 | ||
| Fig. 1 The pharmacophoric amide and urea components of certain HIV-1 protease inhibitors. | ||
Our research is based on the pharmacophoric features of these urea and amide linkers found in VEGFR-2 inhibitors (Fig. 2).
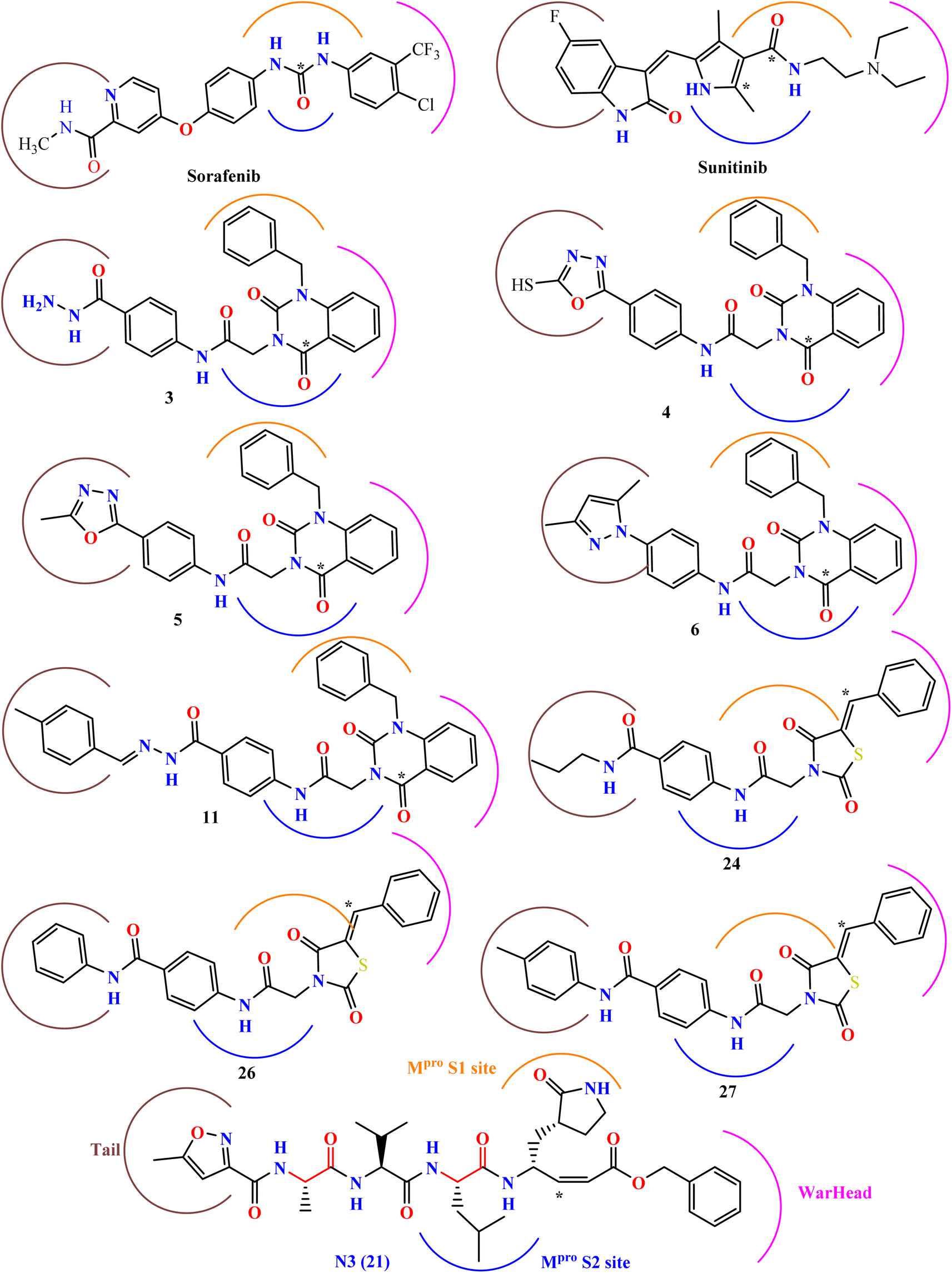 | ||
| Fig. 2 The key pharmacophoric attributes of N3 (Mpro inhibitor) along with noted VEGFR-2 inhibitors incorporating urea and/or amide components. | ||
Additionally, the inhibition activity of SARS-CoV-2 Mpro with N3 is modeled and various N3-analogues are explored for their potential as inhibitors with modifications to the recognition and warhead motifs (Fig. 2 and 3). The use of QM/MM modeling to inhibit Mpro suggests that these compounds might function as irreversible inhibitors or possess the potential to act as reversible inhibitors against Mpro inhibition.27
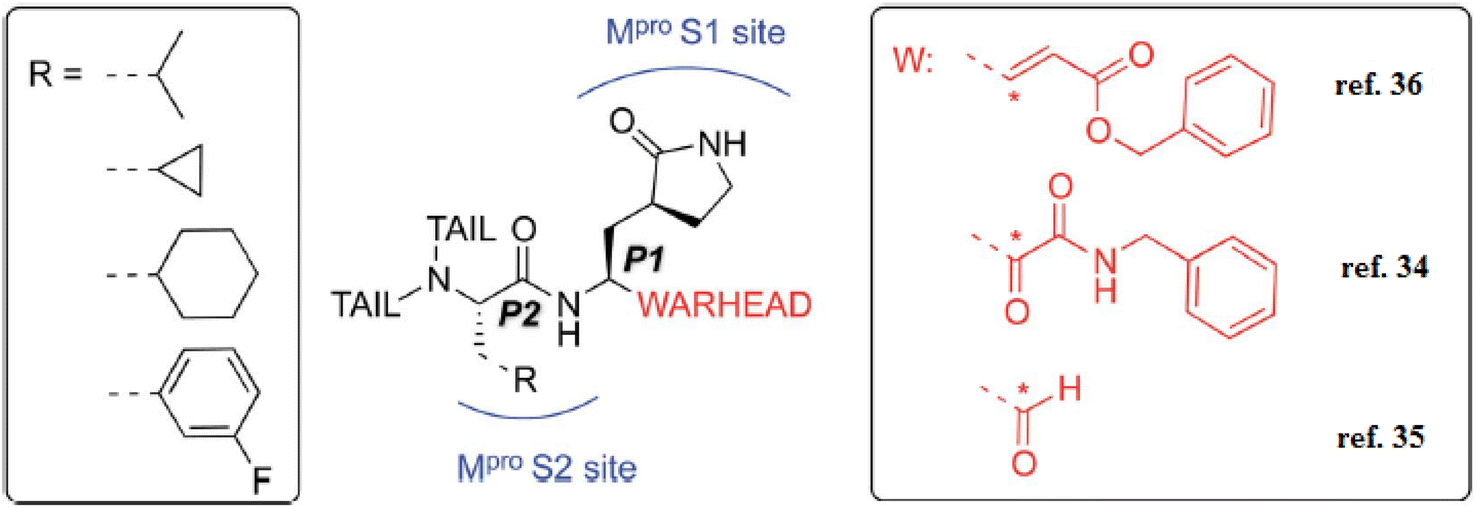 | ||
| Fig. 3 A diagram illustrating the primary categories of covalent inhibitors targeting CoVs Mpro reported to date. The “R” emerging from the P2 residue symbolizes a moderately sized lipophilic group. The tail segment is a significantly variable area concerning both size and configuration and encompasses additional Mpro sub-pockets (i.e. S3 and S4) not depicted in the diagram.27 | ||
The known Mpro predominantly employ a covalent mechanism, featuring a peptidomimetic scaffold at P1 position, typically with a glutamine or an isostere, and a branched lipophilic group at P2, enhancing interaction with the Mpro enzyme.28–30 These inhibitors also incorporate a reactive warhead, as depicted in (Fig. 2 and 3).
In the quest to develop SARS-CoV-2 inhibitors, a diverse array of warheads has been employed, from traditional Michael acceptors (MAs) to activated carbonyl derivatives. MAs are notably favored for targeting cysteine proteases due to their ability to form covalent bonds with the enzyme, ensuring its inhibition. On the other hand, compounds with less reactive warheads, such as those based on carbonyl or nitriles, are known to act as reversible inhibitors. They achieve this by generating metastable adducts, like hemithioketal or thioimidate species, with cysteine residues. The use of MAs is particularly advantageous in achieving sustained inhibition, enhanced target engagement, and greater efficacy compared to other warhead types.31
Our research expects derivatives to act as covalent inhibitors of SARS-CoV-2, akin to N3, including thiazolidine 2,4-dione derivatives and sunitinib, both designed to serve as classical MAs.
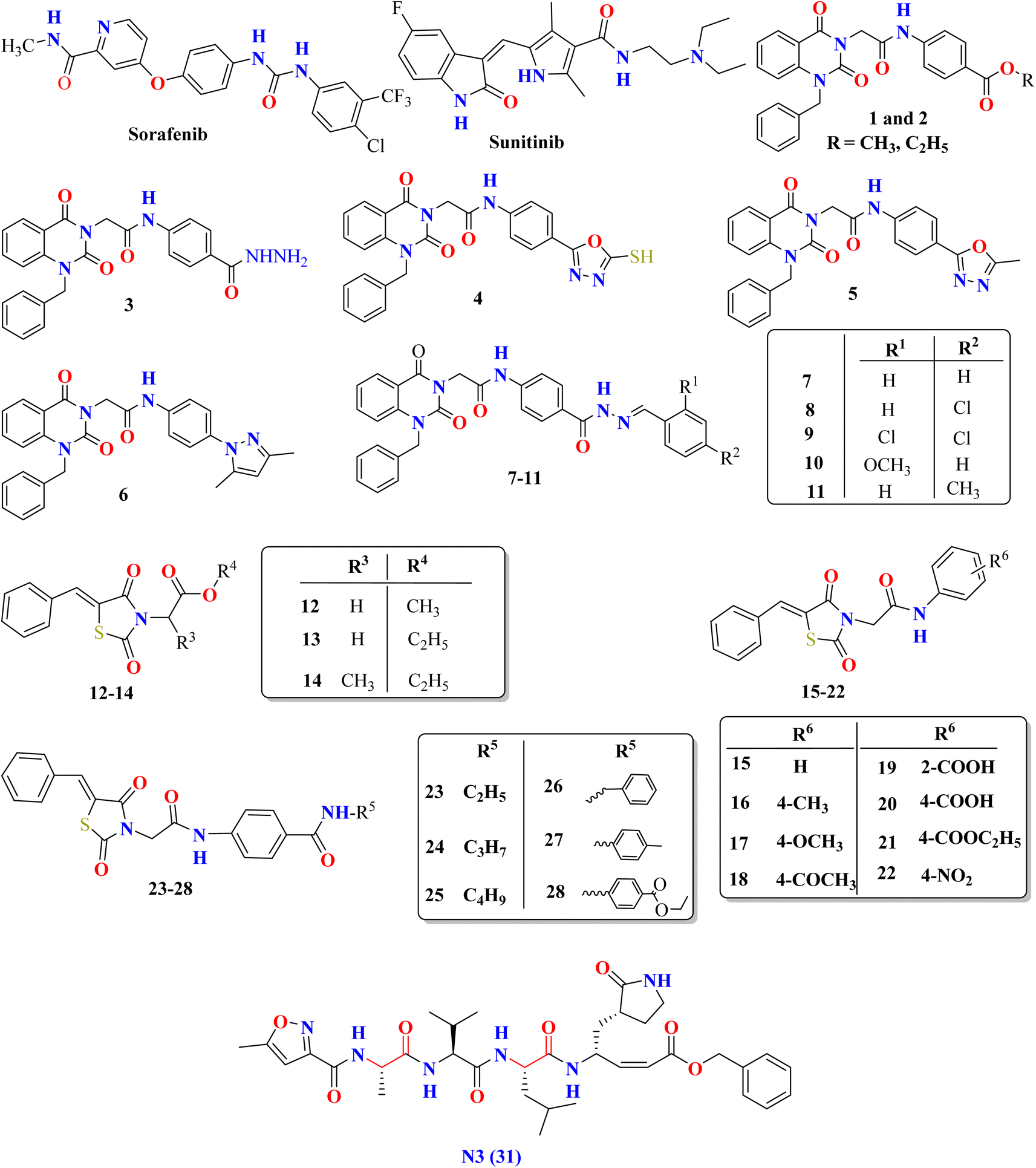
The initial strategy for creating new SARSCoV Mpro inhibitors was to examine their interaction with the N3 ligand, as initially suggested by Yang et al.32 Fig. 4 provides a schematic diagram showing the equilibrated structure of the binding site, highlighting key interactions identified in the molecular dynamics simulations and the X-ray structure reported by Jin et al.,30 represented by dashed lines in blue and red, respectively. In our equilibrated structure, the interaction between the enzyme and the inhibitor closely resembles what was observed through crystallography. This validates our initial structure and allows us to investigate the entire mechanism with confidence. Results from molecular dynamics simulations verify that specific side chains of the N3 residues (P2–P5) do not engage in hydrogen bond interactions. Given the efficacy of this inhibitor, this observation can serve as a reference for designing improved compounds that do not necessitate hydrogen bond interactions at these locations, as noted by Arafet et al.27
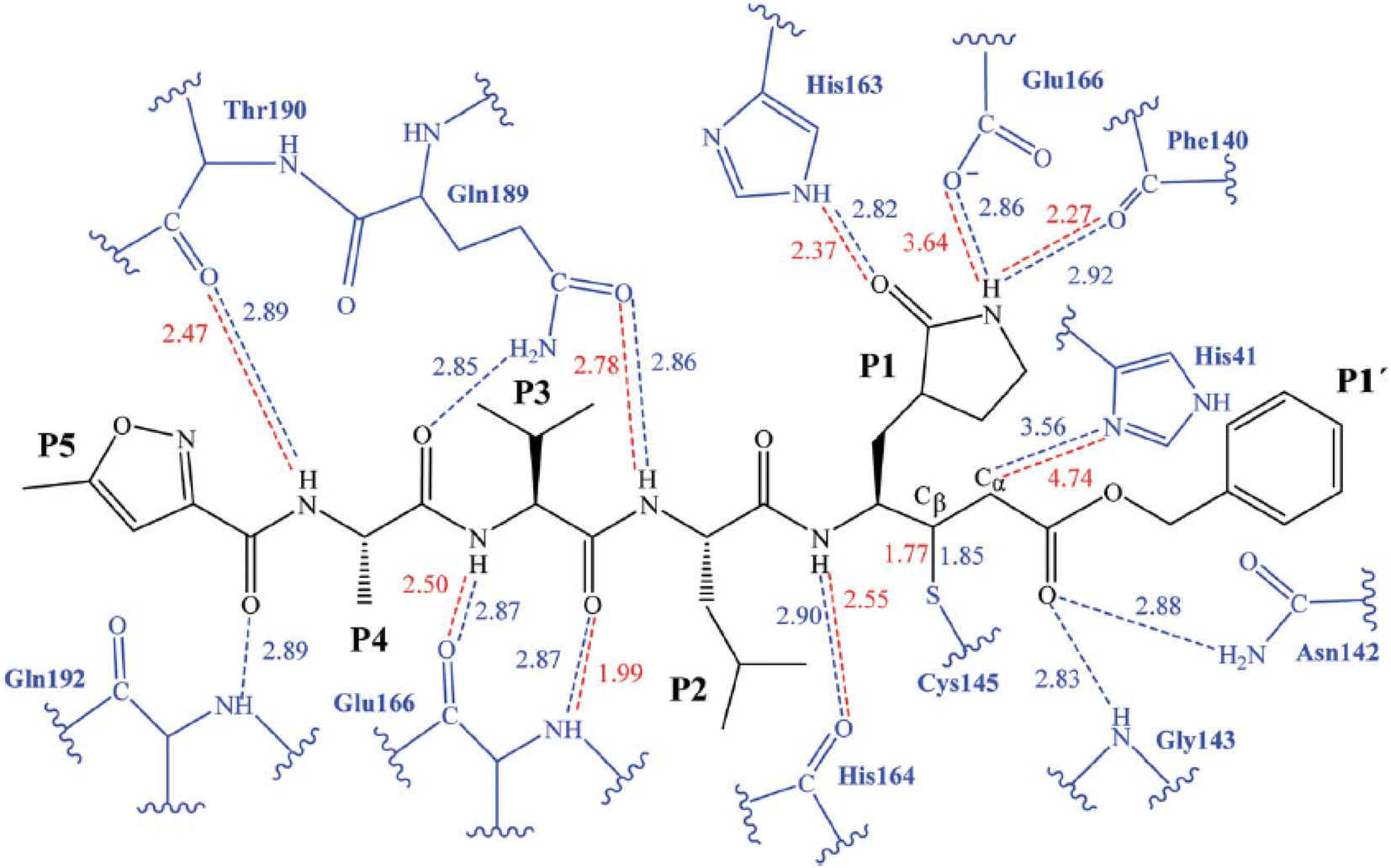 | ||
| Fig. 4 Diagram representing N3 within binding site of SARS-CoV-2 Mpro. Dashed lines indicate hydrogen bonds between the inhibitor and the protein, identified in molecular dynamics simulations (shown in blue) and the X-ray structure (shown in red).30 Average interatomic distances, measured in Ångströms, from both molecular dynamics simulations and X-ray structural data are presented. | ||
In contrast, VEGFR-2 inhibitors,33,34 including sorafenib (with urea linkers) and sunitinib (with amide linkers), as well as our previously reported quinazoline and thiazolidine 2,4-dione derivatives,35,36 display structural resemblances to HIV-1 protease inhibitors. Additionally, they exhibit key pharmacophoric features akin to the co-crystallized inhibitor ligand of SARS-CoV-2 Mpro (N3), as illustrated in Fig. 5. Thus, this research focuses on determining the potential efficacy of various VEGFR-2 inhibitors in combating SARS-CoV-2 through in silico molecular docking and MD simulation methods. Moreover, the VEGFR-2 inhibitors under scrutiny may serve as potential lead compounds for further optimization through structure–activity relationship studies aimed at boosting their effectiveness toward SARS-CoV-2 Mpro.
 | ||
| Fig. 5 The molecular configurations of specific VEGFR-2 inhibitors endorsed by the FDA or under investigation, alongside the co-crystal inhibitor of SARS-CoV-2 Mpro (ligand N3). | ||
2. Materials and methods
In the molecular docking studies, AutoDock Tools software37 and molesoft program35 were used. The protein was handled with the Amber 18 molecular dynamics package38 using the AMBERff14SB force field,39 while the ligands were assigned the GAFF2 force field.40 The reference standard in this research was the crystallized inhibitor ligand (N3).2.1. Design of the evaluated VEGFR-2 inhibitors
Examining of the established compounds and checking the formal charges on atoms through 2D depiction. Energy minimization was applied, and partial charges were automatically calculated. The process involved applying energy minimization and the partial charges were determined. The validated compounds, including N3, were added into same database and stored as an MDB file to carry out docking analyses with the targeted protease.412.2. Preparation of the targeted SARS-CoV-2 Mpro
The structural data for SARS-CoV-2 Mpro was sourced from the Protein Data Bank, identified by the PDB identifier 6LU7 and a resolution of 2.16 Å.30 The crystal structure underwent protonation, and hydrogen atoms were introduced with their conventional three-dimensional geometry. Automatic adjustments were made to correct any errors in atom connections and types, and potential rectifications were applied to the receptor and its atomic structure. The Site Finder tool was employed to identify the identical active site as the co-crystallized inhibitor, and dummy atoms were generated to represent the pocket within that site.422.3. Molecular docking of the evaluated VEGFR-2 inhibitors at active site of Mpro
The database, containing thirty VEGFR-2 inhibitors under investigation (labeled 1–30) alongside the co-crystallized inhibitor N3 (labeled 31), underwent docking analysis. The active site file was loaded, and the overall docking process was started. Adjustments were made to the program's configurations to align with the docking area (using dummy atoms), placement approach (triangle matcher), and evaluation criteria (London dG). A rigid receptor was utilized for refining the poses, and GBVI/WSA dG was used to score and select the top ten poses from a total of one hundred poses for each evaluated compound.43 The database file, containing thirty ligands, was imported, and the system then automatically executed the general docking operations. After completion, resulting poses were analyzed to identify those with optimal ligand–enzyme interactions and satisfactory root mean square deviation (RMSD) values for selection. To validate the target receptor, a docking process was initially performed using only the co-crystallized ligand, and low RMSD values between the docked and crystal conformations indicated a valid performance.44,452.4. Molecular dynamics (MD) simulation
The top eight ranked score complexes (higher binding energies and lower RMSD), along with the sorafenib complex, underwent MD simulation for assessing their affinity for the protease enzyme. Linux 18.04 OS is used for MD simulation and also Amber MD 2018. Additionally, co-crystal ligand (N3) was included as a positive control in the docking and simulation. Force fields for the ligands were derived from GAFF2,40 while the protein utilized the AMBERff14SB force field.39 The complex setup involved extending 15.0 Å in all directions, encapsulating it within an octahedral truncated box filled with TIP3P water, and neutralizing it to achieve a salt concentration equivalent to 150 mM NaCl. The setup underwent several stages of energy minimization and equilibration, progressively reducing positional restraints on both the ligand and protein, until achieving a stable state at a temperature of 310 K and a pressure of 1.0 bar. Subsequently, a 100 ns production run without restraints was conducted, saving the coordinates at 2 ns intervals. The ligand/receptor binding energy has been calculated using the MM/GBSA method, analyzing the trajectory with the MM/PBSA.py script from Amber.46 Calculations were carried out on the final 50 ns segment; utilizing snapshots captured every 1 ns along the simulation trajectory.3. Results and discussion
3.1. VEGF/VEGFR role during COVID-19 progression
VEGF belongs to the family of platelet-derived growth factor supergene and plays an important role in regulation of both pathological and physiological lymphangiogenesis, angiogenesis, and vasculogenesis under different conditions.47 Following SARS-CoV-2 suppression of angiotensin-converting enzyme 2 (ACE2), the VEGF/VEGFR system is also interrupted, and consequently, the modulatory effect on the VEGF activation is hindered.48 It has been documented that some of the VEGF subsets, as primary factors for thrombosis and coagulopathy progression, are up-regulated in COVID-19 positive patients,48 which is supposed to be another reason for the reasonable correlation between VEGF elevated levels and COVID-19 induced ARDS (acute respiratory distress syndrome). In addition, different studies have shown that SARS-CoV-2 leads to accumulating the angiocentric mixed inflammatory cells in post-infection respiratory failure.49VEGF may play a substantial role in the pathogenesis of COVID-19 for multiple reasons: (1) the pulmonary edema, (2) decline oxygen saturation (sO2), and (3) vascular remodeling, in part, due to disturbance of the alveolar-capillary membrane integrity, leading to fibrin deposition and development of the ARDS-related fibro-proliferative phase. Moreover, VEGF can facilitate the virus transmission from the lung to the blood circulation via the impairment of glycocalyx, as a pericellular matrix in the lung tissue.48 Notably, VEGF also has the potential to initiate neuroinflammation in the brain of COVID-19 non-survivors following the induction of inflammatory responses, as well as the disrupting effect on the blood–brain barrier.50 It can be proposed that inhibition of VEGF could lead to vascular normalization and reduce the virus spreading throughout the body fluids. Among various VEGF members, VEGF-A is defined as a key vasodilator and permeability factor involved in angiogenesis, exhibiting a pro/anti-angiogenic property by VEGFR 1/2 activation.51 In detail, VEGFR-1 (sFlt-1) activation appears to be as an endogenous VEGF inhibitor, while VEGFR-2 (KDR/Flk-1) presents an intense tyrosine kinase activity towards pro-angiogenic signals.51 VEGF-C and -D mainly stimulate VEGFR-3 to participate in lymphangiogenesis (Flt-4).51 It has also been well-established that VEGFA-stimulated VEGFR1/2 is considered as one of the critical processes for modulating multiple biological functions, such as ECs (Endothelial cells) proliferation, migration, and vascular permeability.52 To note, VEGFA/VEGFR2 system can also recruit the TSAd (T-cell-specific adapter) protein complex to simultaneously regulate VEGFA induced proto-oncogene tyrosine-protein kinase Src (sarcoma) activation, as well as vascular permeability in ECs. In the setting of COVID-19, VEGFA is also over-expressed in the lung tissue of the non-survivor individuals.51 Beyond the ACE2, other proteins like the neuropilin-1 receptor (NRP-1), as a co-receptor, also participate in SARS-CoV-2's spike protein cell entry. Remarkably, it has been shown that manipulating the VEGF-A165a subtype/b1 domain of NRP-1 signaling, which is up-regulated in the transcriptional levels during COVID-19, can affect disease transmission in asymptomatic subjects.53 Bevacizumab, a humanized anti-VEGF monoclonal antibody, is being used to treat various types of cancer, including metastatic colorectal and renal carcinoma, lung, pancreatic, and breast cancers. Mechanistically, bevacizumab inhibits VEGF-mediated angiogenesis by exclusively targeting VEGF in blood circulation to impede the cancer cells' growth and subsequently confines the blood supply to tumor tissue. In this regard, the therapeutic potential of bevacizumab in COVID-19-induced pneumonia and ARDS is currently under intense investigation. Other classes with secondary anti-VEGF properties refer to sunitinib and sorafenib. These tyrosine kinase inhibitors (TKIs) blockade both cytosolic VEGF and platelet-derived growth factor (PDGF) receptor. However, there are limited data regarding their presumable therapeutic impact against either COVID-19 or non-COVID-19-induced ARDS. Rivoceranib, an oral anti-angiogenesis inhibitor, competitively and selectively can inhibit the VEGFR-2, as well. Cyclosporine, an immunosuppressant agent with a possible anti-VEGF effect, also serves a vascular protective role accompanied by anti-angiogenic and antiapoptotic properties on ECs in low concentrations.51
On the other hand using VEGFA, a physiological ligand for the b1b2 pocket in neuropilin-1 receptor (NRP-1), it was interrogated that whether the Spike protein, the major surface antigen of SARS-CoV-2, could block VEGF-A/NRP-1 signaling to affect pain behaviors. Given parallels between the pro-nociceptive effects of VEGF-A in rodents and humans and clinical findings demonstrating increased VEGF-A levels in bronchial alveolar lavage fluid from COVID-19 patients coupled with substantially lower levels in the sera of asymptomatic individuals compared to symptomatic patients.54
Moreover, World Health Organization (WHO) has labeled several variants, including alpha (B.1.1.7), beta (B.1.351), gamma (P.1), delta (B.1.617.2), and the newly identified omicron (B.1.1.529), as variants of concern because of the potential risks associated with them.55 These variants continue to evolve, leading to different mutations in the Mpro protein that can cause changes in its structure, enzymatic activity, and impact the effectiveness of therapeutic strategies targeting Mpro.56 Recent research has demonstrated that the P108S mutation in SARS-CoV-2 Mpro disrupts the structure around the substrate-binding site, resulting in reduced enzyme activity and less severe disease outcomes in patients infected with the B.1.1.284 sublineage.57 On the other hand, the Omicron variant, known for its high transmissibility, contains a single-point mutation, P132H, in Mpro. However, ongoing mutations in SARS-CoV-2 could lead to changes in Mpro structures, potentially causing resistance to Mpro inhibitors. While the wild-type variant is of lesser interest, we will extend our investigation to include emerging variants. This will ensure the continued applicability of our findings in the dynamic landscape of COVID-19.
Our results augmented the abovementioned data as VEGFR-2 inhibitors can be used in treatment of COVID-19 but more detailed future studies should be carried out to investigate the different mechanisms by which VEGFR-2 inhibitors can inhibit different SARS-CoV-2 variants progression.
3.2. Molecular docking examinations
The Mpro enzyme of the COVID-19 virus possesses a catalytic dyad consisting of Cys and His residues, located within a cleft that bridges domains I and II, serving as the substrate-binding region. The N3 inhibitor is precisely accommodated in the substrate-binding pocket of the COVID-19 virus Mpro, characterized by an asymmetric unit that comprises a single polypeptide. Molecular docking simulations were carried out for VEGFR-2 inhibitors (labeled 1–35) and the N3 inhibitor (labeled 36), targeting the active site of the Mpro enzyme. The results of the docking simulations were benchmarked against the crystal structure of protein and its ligand-binding site showcasing high levels of accuracy and success, as detailed in Table 1. The N3 ligand has been successfully positioned within the active site of Mpro, identified by PDB code 6LU7, achieving an RMSD value of 0.98 Å for the docked ligand. This demonstrates a precise overlay with the original bound ligand, as illustrated in Fig. 6. The findings underscore the high precision of the docking simulation.| Ligands | Binding energy (kcal mol−1) | Amino acids involved | Distance (Å) |
|---|---|---|---|
| Sorafenib | −8.0 | CYS 145, CYS 145, ASN 142 | 4.47, 3.98, 2.98 |
| Sunitinib | −7.5 | CYS 145, GLY 143 | 3.40, 3.11 |
| 1 | −7.9 | GLY 143 | 2.80 |
| 2 | −7.5 | CYS 145, HIP 164, GLU 166 | 3.78, 2.87, 4.89 |
| 3 | −9.2 | SER 144, GLY 143, GLY 143 | 2.99, 2.96, 4.79 |
| 4 | −8.8 | CYS 145, GLY 143 | 3.25, 4.75 |
| 5 | −8.5 | ASN 142, GLY 143 | 3.08, 3.29 |
| 6 | −8.9 | GLN 189, GLY 143, GLY 143, HIE 41 | 3.23, 3.24, 2.82, 3.88 |
| 7 | −8.1 | GLU 166, GLU 166, THR 190, GLU 166 | 3.21, 3.95, 3.53, 4.67 |
| 8 | −8.0 | GLN 189, GLY 143, GLU 166, THR 25 | 3.18, 2.96, 2.80, 4.08 |
| 9 | −7.8 | CYS 145, HIP 164 | 3.67, 3.37 |
| 10 | −7.9 | THR 190, GLU 166 | 3.39, 4.52 |
| 11 | −8.3 | LEU 141, ASN 142, CYS 145, ASN 142 | 3.33, 3.59, 3.17, 4.74 |
| 12 | −6.4 | HIE 41 | 3.85 |
| 13 | −6.7 | GLN 189, GLY 143, SER 144, HIE 41 | 3.53, 2.99, 2.76, 3.87 |
| 14 | −6.1 | CYS 145 | 3.18 |
| 15 | −7.3 | GLN 189, GLY 143, HIE 41 | 3.48, 3.29, 3.92 |
| 16 | −7.8 | HIE 163 | 3.11 |
| 17 | −7.1 | GLY 143, HIE 41 | 3.17, 3.89 |
| 18 | −7.5 | THR 24, GLN 189, GLY 143, THR 25, HIE 41 | 3.11, 3.53, 3.19, 4.09, 3.90 |
| 19 | −7.7 | GLU 166, HIE 41 | 3.10, 3.87 |
| 20 | −7.3 | THR 24, GLN 189, GLY 143, HIE 41 | 3.17, 3.52, 3.22, 3.90 |
| 21 | −7.1 | - - | — |
| 22 | −7.7 | HIE 163 | 3.22 |
| 23 | −7.4 | ASN 142, CYS 145, GLN 189 | 3.56, 3.11, 3.13 |
| 24 | −8.1 | CYS 145, CYS 145, THR 26, MET 165 | 3.46, 3.73, 3.13, 3.88 |
| 25 | −7.4 | CYS 145, CYS 145, ASN 142, THR 26, MET 165 | 3.44, 3.78, 3.35, 3.19, 3.89 |
| 26 | −8.2 | GLY 143, GLU 166, HIE 41, THR 26 | 2.97, 2.95, 4.89, 4.56 |
| 27 | −9.0 | GLN 189, GLY 143, GLU 166, THR 26, GLN 189 | 3.07, 2.84, 2.89, 4.51, 4.28 |
| 28 | −7.3 | SER 144 | 3.05 |
| N3 | −8.1 | MET 165, GLY 143 | 3.40, 2.96 |
 | ||
| Fig. 6 Superimposition of co-crystal ligand (N3) onto the binding pocket of 6LU7 protein structure. | ||
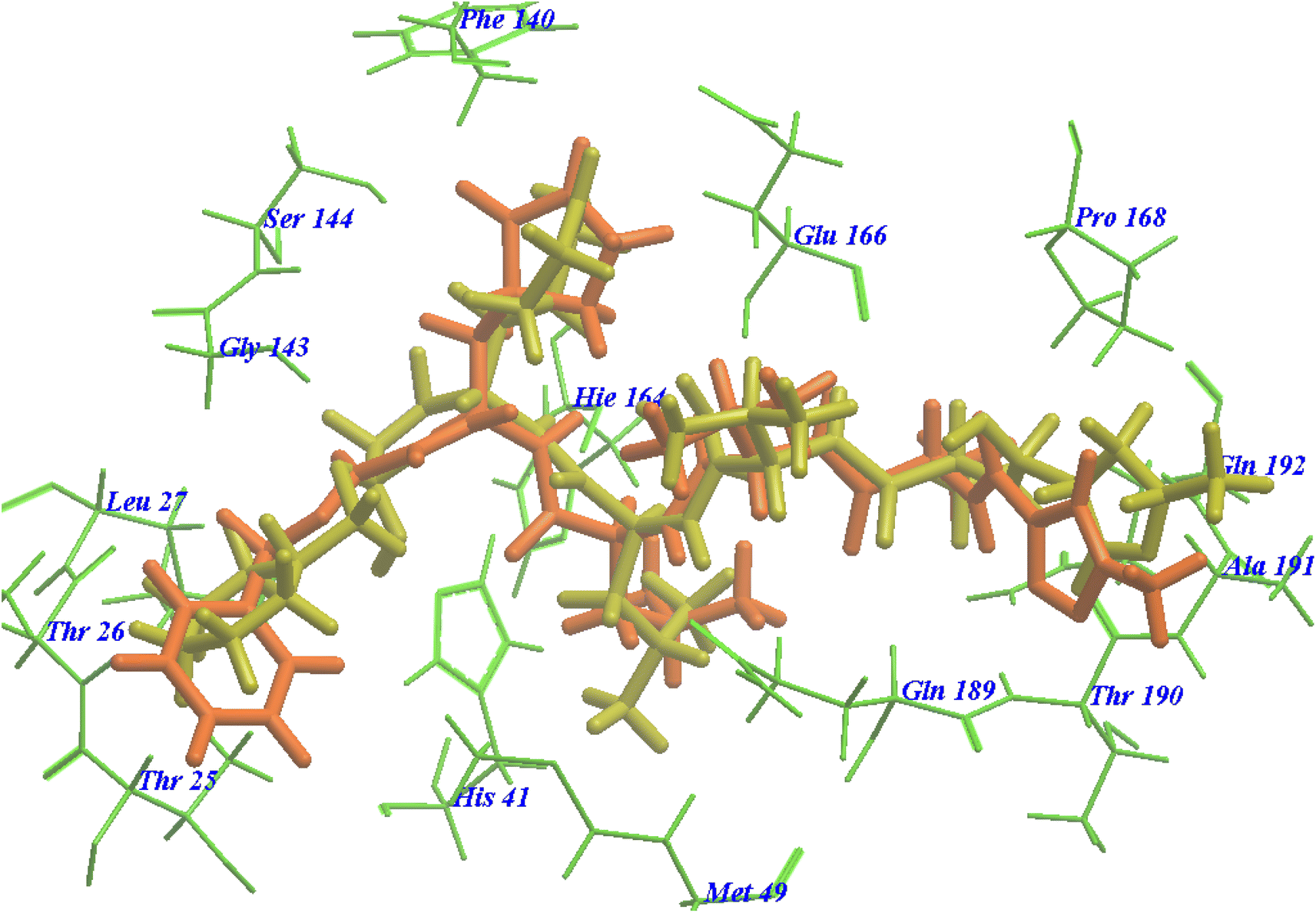
The suggested binding pattern of N3 closely aligns with the findings reported by Jin et al. and demonstrates the formation of eight hydrogen bonds. Specifically, N3 forms two hydrogen bonds with GLU166 (at distances of 1.80 Å and 1.94 Å), one with SER144 (at 1.70 Å), one with GLY143 (at 2.22 Å), one with PHE140 (at 2.43 Å), one with HIE164 (at 2.76 Å), one with GLN189 (at 2.33 Å), and one with THR190 (at 2.22 Å). Additionally, it engages in numerous hydrophobic interactions, occupying various hydrophobic grooves, as depicted in Fig. 7.
 | ||
| Fig. 7 N3 docked within the active site of 6LU7 protein structure. | ||
The majority of compounds exhibited binding patterns similar to that of the N3 inhibitor, with several poses demonstrating improved active modes and strong bonds within the receptor site (Fig. 8).
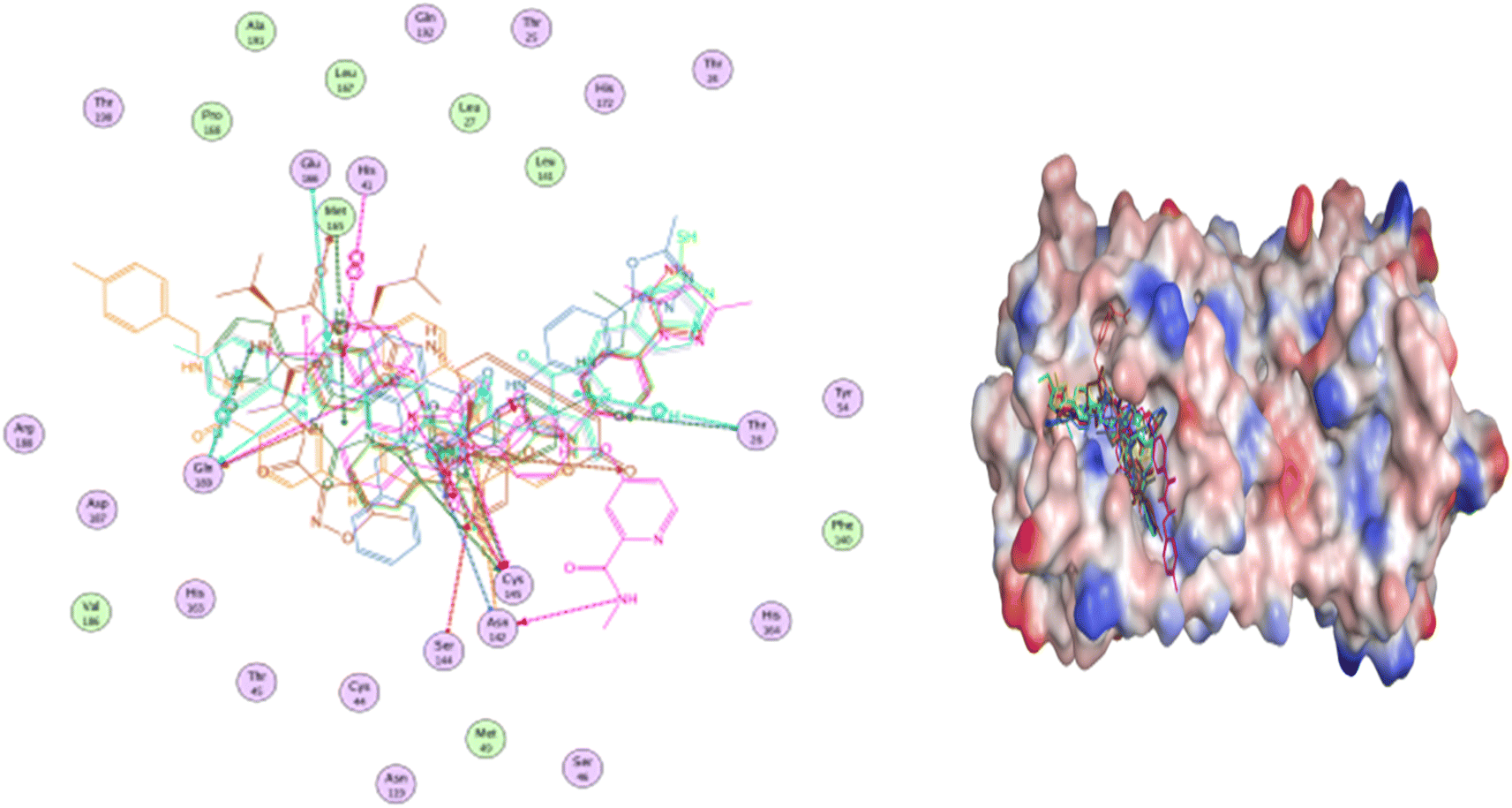 | ||
| Fig. 8 Overlay of certain docked compounds within the active site of 6LU7. | ||
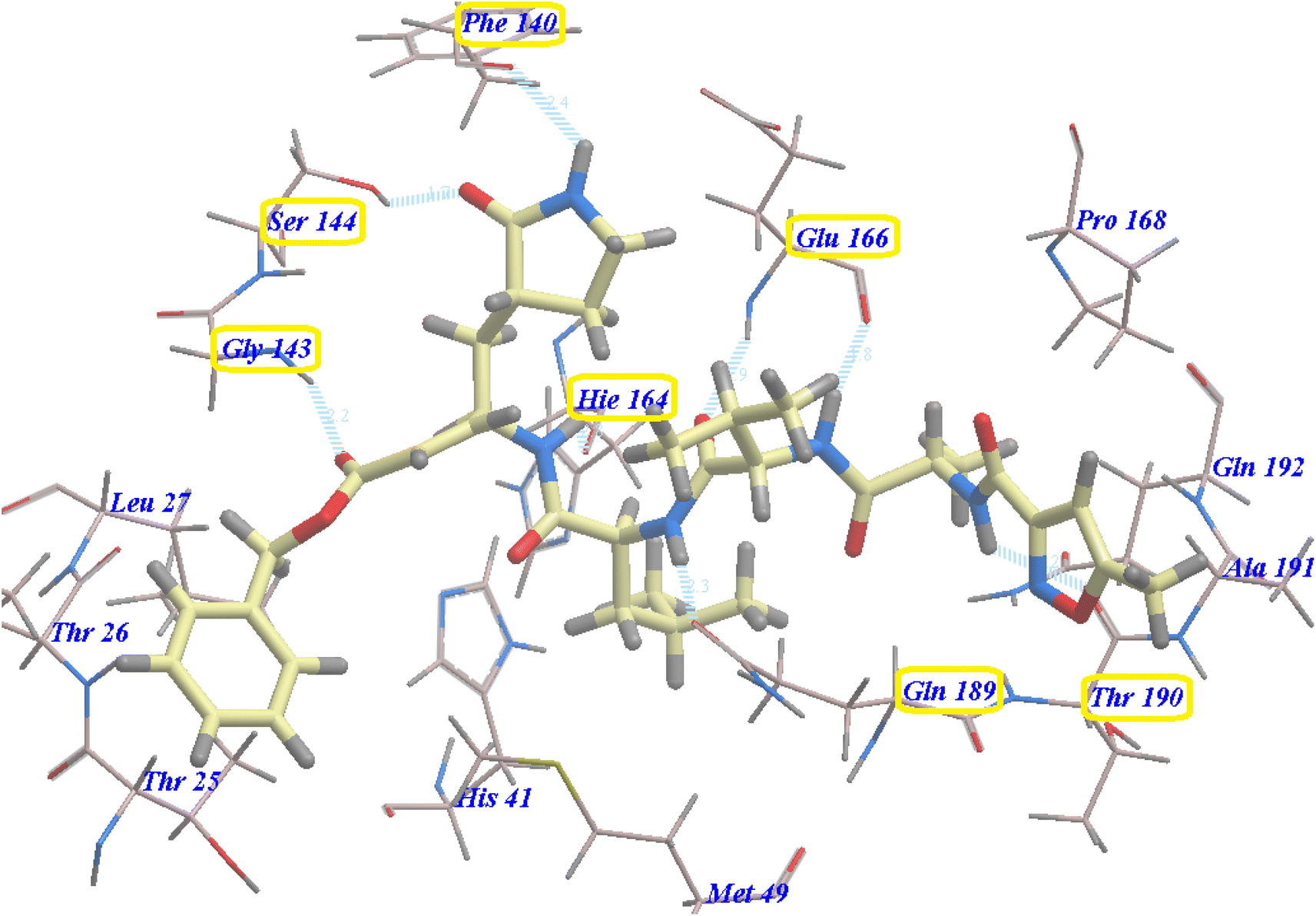
Selected poses were those with the highest scores, reflecting pose stability, and rmsd_refine values, which suggest closeness of the selected pose to the ligand position within the pocket. A variety of electrostatic bonds stabilized the docked compounds within the N3-active site of Mpro (Fig. 9). Energy results and diverse interactions with amino acids within the Mpro protein pocket are detailed in Table 1.
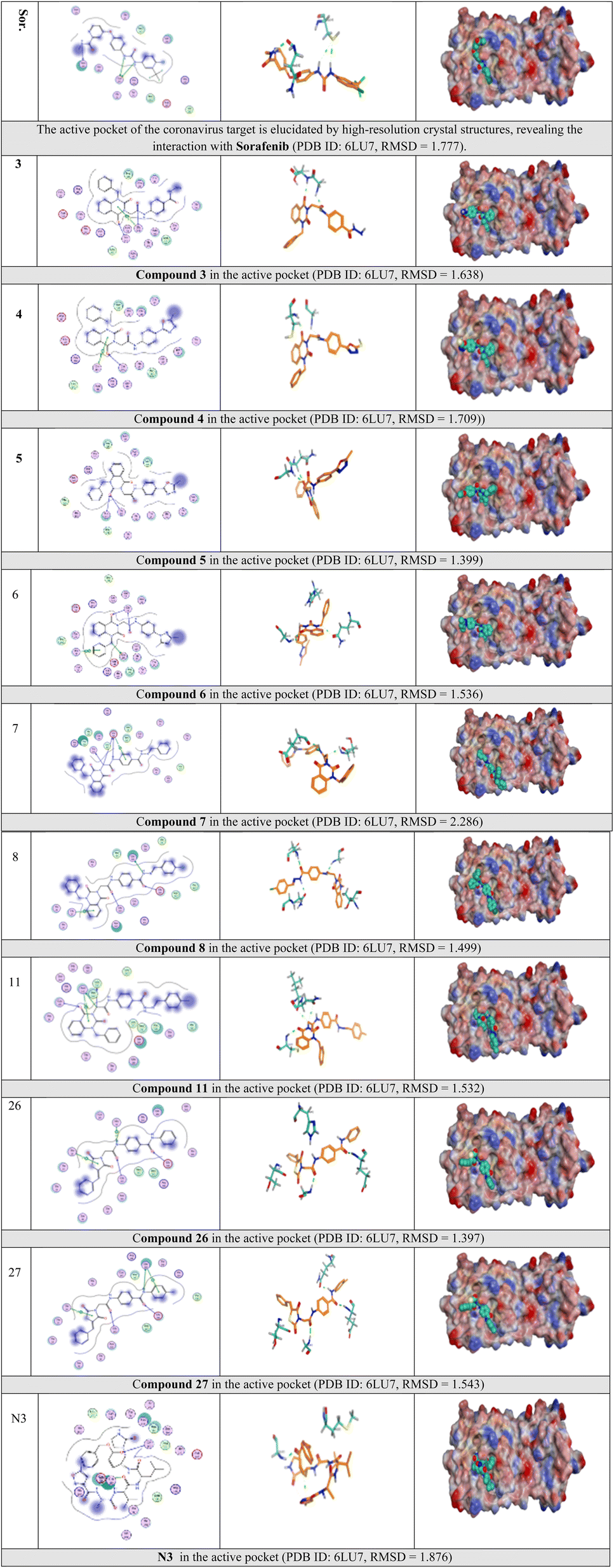 | ||
| Fig. 9 2D, 3D, and surface representations, as well as maps, depicting N3 and VEGFR-2 inhibitors within the Mpro protein, 6LU7. | ||
Lastly, certain VEGFR-2 inhibitors showcased binding scores ranging from −8.0 to −9.2, demonstrating equal or superior potency compared to the co-crystallized ligand N3, which has a score of −8.1.
3.3. Molecular dynamics (MD) simulation
MD simulation studies were employed to explore the relative stability and dynamic behavior of ligand-target complexes, providing insights into their conformational changes over time. Unlike other in silico methods such as molecular docking and mechanics energy minimization, MD simulations allow for a more comprehensive examination of the conformational space of ligand–target interactions, going beyond static image analysis.58The top docked configurations, demonstrating substantial interactions and affinity between the ligand and Mpro, were subjected to an all-atom MD simulation for duration of 100 nanoseconds. This facilitated a detailed investigation of the conformational alterations in the ligand–target complexes of VEGFR-2 inhibitors (3–6, 11, 24, 26, 27, and sorafenib) and N3 within the canonical binding site of SARS-CoV-2 Mpro., shedding light on their dynamic behavior throughout the interaction process.
High RMSD trajectories in molecular dynamics simulations often signal instability within the target protein and substantial conformational shifts. Conversely, elevated complex RMSD values point to a weak affinity between the ligand and its target, highlighting the ligand's struggle to consistently occupy the established binding site.59 In the case of Mpro proteins, the observed RMSD deviations, especially concerning their C-alpha atoms (C-α RMSD), aligned with expected outcomes in MD simulations (Fig. 10). At the onset of the MD simulations, a rise in the RMSD values for the protein's C-α atoms was observed, a phenomenon attributed to the removal of initial restraints. Following this initial phase, the RMSD trajectories for the C-α atoms demonstrated a stable pattern for a significant portion of the simulation duration, specifically more than 50 nanoseconds. Notably, the protein complex involving BIS exhibited a phase of reduced RMSD variability, particularly between 55 and 85 nanoseconds, indicating a period of enhanced stability.
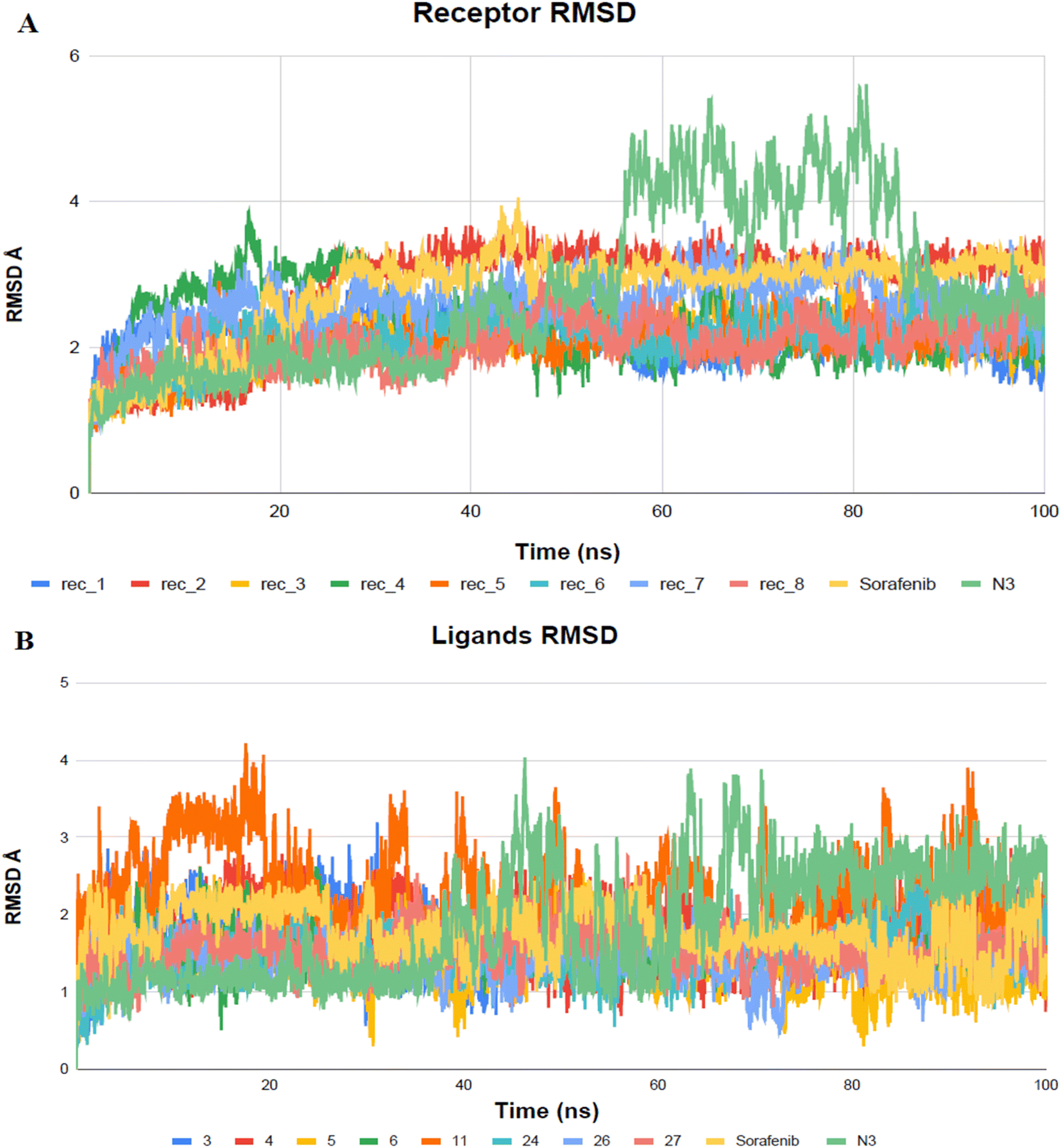 | ||
| Fig. 10 Examination of RMSD trajectories for the complexes formed by the ligand and Mpro protein over MD simulation (100 ns). (A) Represents the backbone RMSD; (B) displays the ligand RMSD vs. time plots for molecular dynamics simulations (nanoseconds). | ||
| Ligands | EEL | VDWAALS | ΔGgas | ΔGsolv | ΔTotal |
|---|---|---|---|---|---|
| 3 | −21.9265 | −45.7109 | −67.6375 | 34.1658 | −33.4717 |
| 4 | −5.1472 | −9.4382 | −14.5854 | 8.4373 | −6.1481 |
| 5 | −13.0207 | −24.5178 | −37.5385 | 21.2820 | −16.2566 |
| 6 | −17.5238 | −36.9243 | −54.4481 | 29.5398 | −24.9083 |
| 11 | −12.3774 | −38.7219 | −51.1008 | 24.5090 | −26.5918 |
| 24 | −12.3596 | −32.1424 | −44.5020 | 23.2094 | −21.2926 |
| 26 | −12.9159 | −33.8802 | −46.7961 | 23.6342 | −23.1619 |
| 27 | −21.8916 | −43.5004 | −65.3920 | 36.9703 | −28.4216 |
| Sorafenib | −39.2863 | −32.4855 | −71.7718 | 50.7580 | −21.0138 |
| N3 | −28.2044 | −47.8873 | −76.0916 | 43.6267 | −32.4650 |
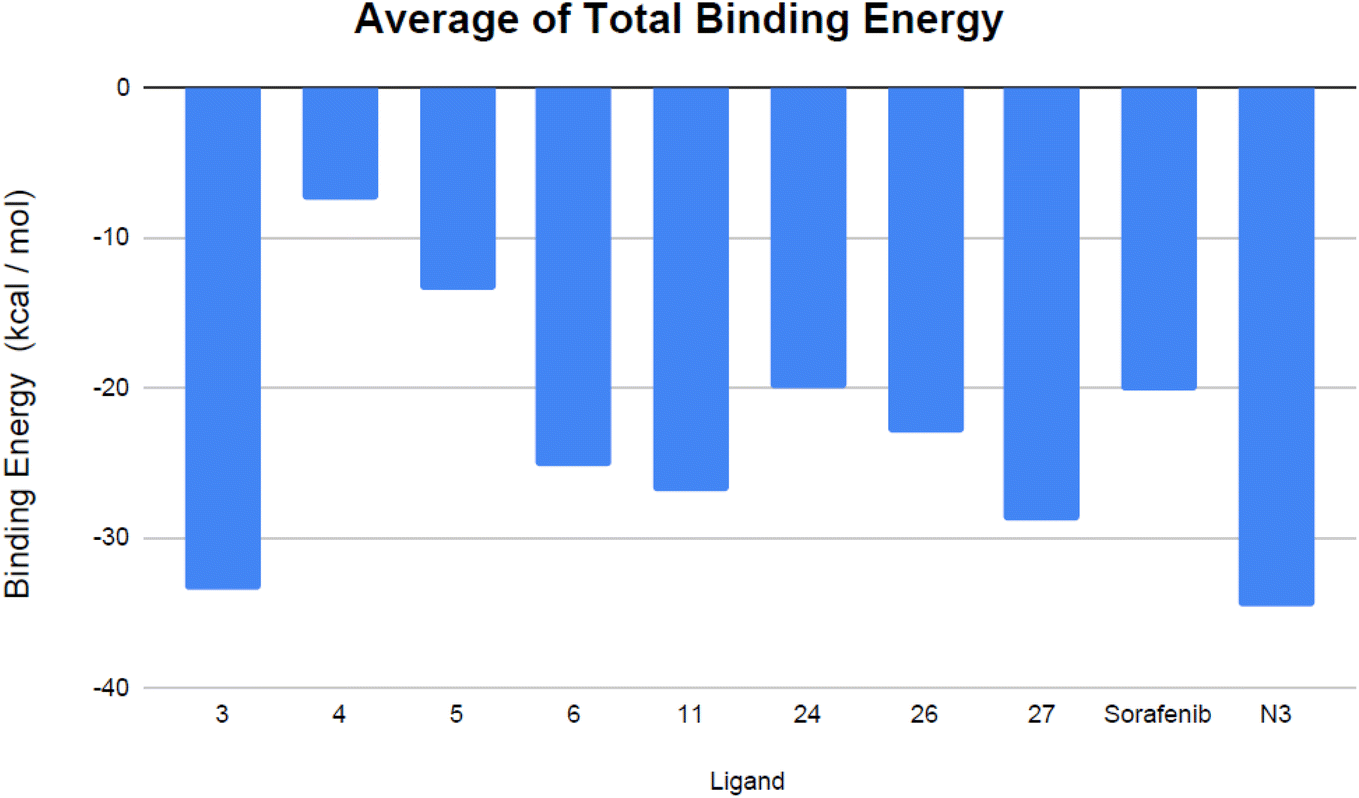 | ||
| Fig. 11 Mean total binding energy (in kcal mol−1) of the evaluated VEGFR-2 inhibitors and the co-crystallized ligand N3. | ||
Analyzing the binding-free energy components, van der Waals interactions emerged as the primary contributor to the ligand-protein complex's free-binding energy, surpassing electrostatic potential energy with the exception of sorafenib. The solute's energy in the gas phase (ΔGgas) encompasses internal energies, along with energies arising from van der Waals interactions and electrostatic forces. Solvation free energy, ΔGsolv, consists of polar and non-polar components, ΔGsolv = ΔGpol + ΔGnonpol. Notably, the N3 inhibitor showed elevated levels of gas-phase energy (ΔGgas) and solvation energy (ΔGsolv) in comparison to the evaluated VEGFR-2 inhibitors, apart from sorafenib, which demonstrated the highest solvation energy. Contemporary studies suggest that the binding site of Mpro is predominantly hydrophobic, attributed to its depth, minimal exposure to solvent, and the presence of conserved residues that line the hydrophobic pocket. Given its hydrophobic characteristics and significant surface area, the Mpro active site may favor robust nonpolar interactions, particularly with N3, due to its extended configuration within the binding site. Furthermore, compounds 3 and 27 exhibited consistent binding behaviors during both the docking studies and the MD simulations. Specifically, compound 3 displayed a more advantageous free-binding energy at the Mpro binding site than the N3 inhibitor, underscoring its heightened affinity for the target as initially indicated by the docking analysis. The remarkable binding energy value observed for the most stable VEGFR-2 inhibitor, compound 3, aligns closely with its leading position in the initial docking score rankings (Table 1).
4. Conclusion
The findings of the study highlight the potential of twenty-eight known VEGFR-2 inhibitors that have amide and urea moieties as possible agents against SARS-CoV-2. Through in silico analysis, nine specific compounds (3–6, 11, 24, 26, 27, and sorafenib) demonstrated promising results and were subjected to extensive MD simulations to assess their inhibitory effects on SARS-CoV-2 Mpro over a duration of 100 ns. By utilizing MM/GBSA calculations to determine binding-free energies, compounds 3 and 27 exhibited significant affinity and energy as VEGFR-2 inhibitors binding to the targeted pocket of SARS-CoV-2 Mpro. These findings substantiate the suggested approach and align with the computational results obtained through molecular docking and dynamics, indicating the potential effectiveness of other studied VEGFR-2 inhibitors. Therefore, we recommend further preclinical and clinical investigations to repurpose FDA-approved VEGFR-2 inhibitors as a strategic response to the Covid-19 crisis. Additionally, these drugs can serve as valuable starting points for structural modifications aimed at enhancing their efficacy against SARS-CoV-2.Conflicts of interest
The authors declare no conflict of interest.References
- J. A. Alara and O. R. Alara, An Overview of the Global Alarming Increase of Multiple Drug Resistant: A Major Challenge in Clinical Diagnosis, Infect. Disord.: Drug Targets, 2024, 24(3), 26–42 Search PubMed
.
- S. R. Khetran and R. Mustafa, Mutations of SARS-CoV-2 structural proteins in the alpha, beta, gamma, and delta variants: bioinformatics analysis, J. Med. Internet Res. Bioinform. Biotechnol., 2023, 4(1), e43906 CrossRef PubMed
- F. Wu, et al., A new coronavirus associated with human respiratory disease in China, Nature, 2020, 579(7798), 265–269 CrossRef CAS PubMed
- Y. Chen, Q. Liu and D. Guo, Emerging coronaviruses: genome structure, replication, and pathogenesis, J. Med. Virol., 2020, 92(4), 418–423 CrossRef CAS PubMed
- K. Anand, et al., Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs, Science, 2003, 300(5626), 1763–1767 CrossRef CAS PubMed
- A. O. Elzupir, Inhibition of SARS-CoV-2 main protease 3CLpro by means of α-ketoamide and pyridone-containing pharmaceuticals using in silico molecular docking, J. Mol. Struct., 2020, 1222, 128878 CrossRef CAS PubMed
- A. A. Elmaaty, et al., In a search for potential drug candidates for combating COVID-19: computational study revealed salvianolic acid B as a potential therapeutic targeting 3CLpro and spike proteins, J. Biomol. Struct. Dyn., 2022, 40(19), 8866–8893 CrossRef CAS PubMed
- R. Soltane, et al., Strong inhibitory activity and action modes of synthetic maslinic acid derivative on highly pathogenic coronaviruses: COVID-19 drug candidate, Pathogens, 2021, 10(5), 623 CrossRef CAS PubMed
- A. A. Zaki, et al., Calendulaglycoside A showing potential activity against SARS-CoV-2 main protease: Molecular docking, molecular dynamics, and SAR studies, J. Tradit. Complementary Med., 2022, 12(1), 16–34 CrossRef CAS PubMed
- Y. Duan, et al., Structural biology of SARS-CoV-2 Mpro and drug discovery, Curr. Opin. Struct. Biol., 2023, 82, 102667 CrossRef CAS PubMed
- X. Jiang, et al., Structure-based development and preclinical evaluation of the SARS-CoV-2 3C-like protease inhibitor simnotrelvir, Nat. Commun., 2023, 14(1), 6463 CrossRef CAS PubMed
- A. Moura, et al., Converging Paths: A Comprehensive Review of the Synergistic Approach between Complementary Medicines and Western Medicine in Addressing COVID-19 in 2020, BioMed, 2023, 3(2), 282–308 CrossRef
- M. Khattab and A. A. Al-Karmalawy, Revisiting activity of some nocodazole analogues as a potential anticancer drugs using molecular docking and DFT calculations, Front. Chem., 2021, 9, 628398 CrossRef CAS PubMed
- C. Milite, et al., Novel 2-substituted-benzimidazole-6-sulfonamides as carbonic anhydrase inhibitors: synthesis, biological evaluation against isoforms I, II, IX and XII and molecular docking studies, J. Enzyme Inhib. Med. Chem., 2019, 34(1), 1697–1710 CrossRef CAS PubMed
- D. Ajmeera and R. Ajumeera, Drug repurposing: A novel strategy to target cancer stem cells and therapeutic resistance, Genes Dis., 2024, 11(1), 148–175 CrossRef CAS PubMed
- V. Kulkarni, et al., Drug repurposing: An effective tool in modern drug discovery, Russ. J. Bioorg. Chem., 2023, 1–10 Search PubMed
- A. A. Elfiky, Anti-HCV, nucleotide inhibitors, repurposing against COVID-19, Life Sci., 2020, 248, 117477 CrossRef CAS PubMed
- H.-H. Fan, et al., Repurposing of clinically approved drugs for treatment of coronavirus disease 2019 in a 2019-novel coronavirus-related coronavirus model, Chin. Med. J., 2020, 133(09), 1051–1056 CrossRef CAS PubMed
- Y. W. Chen, C.-P. B. Yiu and K.-Y. Wong, Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CL pro) structure: virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates, F1000Research, 2020, 9 Search PubMed
- W. Waseem, et al., Drug repurposing of FDA-approved anti-viral drugs via computational screening against novel 6M03 SARS-COVID-19, Ir. J. Med. Sci., 2023, 1–11 Search PubMed
- S. Haid, et al., Repurposing screen identifies novel candidates for broad-spectrum coronavirus antivirals and druggable host targets, Antimicrob. Agents Chmother., 2024, e01210–e01223 Search PubMed
- P. A. Velásquez, et al., Effectiveness of Drug Repurposing and Natural Products Against SARS-CoV-2: A Comprehensive Review, Clin. Pharmacol.: Adv. Appl., 2024, 1–25 Search PubMed
- N. Yamamoto, et al., HIV protease inhibitor nelfinavir inhibits replication of SARS-associated coronavirus, Biochem. Biophys. Res. Commun., 2004, 318(3), 719–725 CrossRef CAS PubMed
- Y. Xue, et al., Repurposing clinically available drugs and therapies for pathogenic targets to combat SARS-CoV-2, MedComm, 2023, 4(3), e254 CrossRef PubMed
- J.-Y. Li, et al., The epidemic of 2019-novel-coronavirus (2019-nCoV) pneumonia and insights for emerging infectious diseases in the future, Microbes Infect., 2020, 22(2), 80–85 CrossRef CAS PubMed
- J. M. Beale and J. H. Block, Organic Medicinal and Pharmaceutical Chemistry, 2011 Search PubMed
- K. Arafet, et al., Mechanism of inhibition of SARS-CoV-2 M pro by N3 peptidyl Michael acceptor explained by QM/MM simulations and design of new derivatives with tunable chemical reactivity, Chem. Sci., 2021, 12(4), 1433–1444 RSC
- L. Zhang, et al., Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors, Science, 2020, 368(6489), 409–412 CrossRef CAS PubMed
- W. Dai, et al., Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease, Science, 2020, 368(6497), 1331–1335 CrossRef CAS PubMed
- Z. Jin, et al., Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors, Nature, 2020, 582(7811), 289–293 CrossRef CAS PubMed
- S. El-Hddad, et al., In silico molecular docking, dynamics simulation and repurposing of some VEGFR-2 inhibitors based on the SARS-CoV-2-main-protease inhibitor N3, J. Biomol. Struct. Dyn., 2023, 41(19), 9267–9281 CrossRef CAS PubMed
- H. Yang, et al., Design of wide-spectrum inhibitors targeting coronavirus main proteases, PLoS Biol., 2005, 3(10), e324 CrossRef PubMed
- S. Takahashi, Vascular endothelial growth factor (VEGF), VEGF receptors and their inhibitors for antiangiogenic tumor therapy, Biol. Pharm. Bull., 2011, 34(12), 1785–1788 CrossRef CAS PubMed
- R. Roskoski Jr, Properties of FDA-approved small molecule protein kinase inhibitors: A 2023 update, Pharmacol. Res., 2023, 187, 106552 CrossRef PubMed
- K. El-Adl, et al., Design, synthesis, docking, ADMET profile, and anticancer evaluations of novel thiazolidine-2, 4-dione derivatives as VEGFR-2 inhibitors, Arch. Pharm., 2021, 354(7), 2000491 CrossRef CAS PubMed
- K. El-Adl, et al., Design, synthesis, molecular docking, anticancer evaluations, and in silico pharmacokinetic studies of novel 5-[(4-chloro/2, 4-dichloro) benzylidene] thiazolidine-2, 4-dione derivatives as VEGFR-2 inhibitors, Arch. Pharm., 2021, 354(2), 2000279 CrossRef CAS PubMed
- G. M. Morris, et al., AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility, J. Comput. Chem., 2009, 30(16), 2785–2791 CrossRef CAS PubMed
- H. Liu, Y. Jin and H. Ding, MDBuilder: a PyMOL plugin for the preparation of molecular dynamics simulations, Briefings Bioinf., 2023, 24(2), bbad057 CrossRef PubMed
- J. A. Maier, et al., ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB, J. Chem. Theory Comput., 2015, 11(8), 3696–3713 CrossRef CAS PubMed
- J. Wang, et al., Development and testing of a general amber force field, J. Comput. Chem., 2004, 25(9), 1157–1174 CrossRef CAS PubMed
- A. Ghanem, et al., Tanshinone IIA synergistically enhances the antitumor activity of doxorubicin by interfering with the PI3K/AKT/mTOR pathway and inhibition of topoisomerase II: in vitro and molecular docking studies, New J. Chem., 2020, 44(40), 17374–17381 RSC
- S. G. Eliaa, et al., Empagliflozin and doxorubicin synergistically inhibit the survival of triple-negative breast cancer cells via interfering with the mTOR pathway and inhibition of calmodulin: in vitro and molecular docking studies, ACS Pharmacol. Transl. Sci., 2020, 3(6), 1330–1338 CrossRef CAS
- R. M. Samra, et al., Bioassay-guided isolation of a new cytotoxic ceramide from Cyperus rotundus L, S. Afr. J. Bot., 2021, 139, 210–216 CrossRef CAS
- I. W. Davis and D. Baker, RosettaLigand docking with full ligand and receptor flexibility, J. Mol. Biol., 2009, 385(2), 381–392 CrossRef CAS PubMed
- B. J. McConkey, V. Sobolev and M. Edelman, The performance of current methods in ligand–protein docking, Curr. Sci., 2002, 845–856 CAS
- B. R. Miller III, et al., MMPBSA. py: an efficient program for end-state free energy calculations, J. Chem. Theory Comput., 2012, 8(9), 3314–3321 CrossRef PubMed
- C. E. F. Pain, S. Cleary, G. Mayell, S. Conrad, K. Harave, S. Duong, P. Sinha, I. Porter and D. Hedrich, C.M. Novel paediatric presentation of COVID-19 with ARDS and cytokine storm syndrome without respiratory symptoms, Lancet Rheumatol., 2020, 2(7), e376–e379 CrossRef PubMed
- Y. Kong, J. Han, X. Wu, H. Zeng, J. Liu and H. Zhang, VEGF-D: a novel biomarker for detection of COVID-19 progression, Crit. Care, 2020, 24(1), 1–4 CrossRef PubMed
- M. Ackermann, S. E. Verleden, M. Kuehnel, A. Haverich, T. Welte, F. Laenger, A. Vanstapel, C. Werlein, H. Stark and A. Tzankov, Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19, N. Engl. J. Med., 2020, 383(2), 120–128 CrossRef CAS PubMed
- X.-X. Yin, X.-R. Zheng, W. Peng, M.-L. Wu and X.-Y. Mao, Vascular Endothelial Growth Factor (VEGF) as a Vital Target for Brain Inflammation during the COVID-19 Outbreak, ACS Chem. Neurosci., 2020 Search PubMed
- A. Sahebnasagh, S. M. Nabavi, H. R. K. Kashani, S. Abdollahian, S. Habtemariam and A. Rezabakhsh, Anti-VEGF agents: As appealing targets in the setting of COVID-19 treatment in critically ill patients, Int. Immunopharmacol., 2021, 101(Pt B), 108257, DOI:10.1016/j.intimp.2021.108257
- Y. Jin, W. Ji, H. Yang, S. Chen, W. Zhang and G. Duan, Endothelial activation and dysfunction in COVID-19: from basic mechanisms to potential therapeutic approaches, Signal Transduction Targeted Ther., 2020, 5(1), 1–13 CrossRef PubMed
- A. Moutal, L. F. Martin, L. Boinon, K. Gomez, D. Ran, Y. Zhou, H. J. Stratton, S. Cai, S. Luo and K. B. Gonzalez, SARS-CoV-2 Spike protein co-opts VEGF-A/Neuropilin-1 receptor signaling to induce analgesia, Pain, 2021, 162(1), 243 CrossRef CAS PubMed
- A. Moutal, L. F. Martin, L. Boinon, K. Gomez, D. Ran, Y. Zhou, H. J. Stratton, S. Cai, S. Luo, K. B. Gonzalez, S. Perez-Miller, A. Patwardhan, M. M. Ibrahim and R. Khanna. SARS-CoV-2 Spike protein co-opts VEGF-A/Neuropilin-1 receptor signaling to induce analgesia, bioRxiv, 2020, preprint:2020.07.17.209288, DOI:10.1101/2020.07.17.209288Update in: Pain, 2021, Jan, 162(1), 243–252.
- S. A. Chen, et al., SARS-CoV-2 Mpro protease variants of concern display altered viral substrate and cell host target Galectin-8 processing but retain sensitivity toward antivirals, ACS Cent. Sci., 2023, 9(4), 696–708 CrossRef CAS PubMed
- N. Yashvardhini, A. Kumar and D. K. Jha, Analysis of SARS-CoV-2 mutations in the main viral protease (NSP5) and its implications on the vaccine designing strategies, Vacunas, 2022, 23, S1–S13 CrossRef CAS PubMed
- K. Abe, et al., Pro108Ser mutation of SARS-CoV-2 3CLpro reduces the enzyme activity and ameliorates the clinical severity of COVID-19, Sci. Rep., 2022, 12(1), 1299 CrossRef CAS PubMed
- M. Karplus and G. A. Petsko, Molecular dynamics simulations in biology, Nature, 1990, 347(6294), 631–639 CrossRef CAS PubMed
- K. Liu, E. Watanabe and H. Kokubo, Exploring the stability of ligand binding modes to proteins by molecular dynamics simulations, J. Comput.-Aided Mol. Des., 2017, 31, 201–211 CrossRef CAS PubMed
- C. N. Cavasotto, Binding free energy calculation using quantum mechanics aimed for drug lead optimization, Quantum mechanics in drug discovery, 2020, p. 257–268 Search PubMed
- T. Hou, et al., Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations, J. Chem. Inf. Model., 2011, 51(1), 69–82 CrossRef CAS PubMed
- N. Forouzesh and N. Mishra, An effective MM/GBSA protocol for absolute binding free energy calculations: A case study on SARS-CoV-2 spike protein and the human ACE2 receptor, Molecules, 2021, 26(8), 2383 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2024 |