
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiodegradable polyester copolymers: synthesis based on the Biginelli reaction, characterization, and evaluation of their application properties†
Kai Lu‡
a,
Xinyi Shen‡a,
Yunhai Shia,
Zejian He*bc,
Dahong Zhang*d and
Mi Zhou *a
*a
aCollege of Materials Science and Engineering, Zhejiang University of Technology, Hangzhou, Zhejiang 310014, China
bStoddart Institute of Molecular Science, Department of Chemistry, Zhejiang University, Hangzhou, Zhejiang 310027, China. E-mail: hezejian@zju.edu.cn
cZJU-Hangzhou Global Scientific and Technological Innovation Center, Hangzhou, Zhejiang 311215, China
dDepartment of Urology, Zhejiang Provincial People's Hospital, Hangzhou, Zhejiang 310014, China. E-mail: zhangdahong88@126.com
First published on 29th May 2024
Abstract
The Biginelli reaction, a three-component cyclocondensation reaction, is an important member of the multicomponent reaction (MCR) family. In this study, we conducted end-group modifications on a variety of biodegradable polyesters, including poly(1,4-butylene adipate) (PBA), poly(ε-caprolactone) (PCL), polylactic acid (PLA), and poly(p-dioxanone) (PPDO), based on the precursor polyethylene glycol (PEG). By combining two polymers through the Biginelli multi-component reaction, four new biodegradable polyester copolymers, namely DHPM–PBA, DHPM–PCL, DHPM–PLA, and DHPM–PPDO, were formed. These Biginelli reactions demonstrated exceptional completeness, validating the efficiency of the synthesis strategy. Although the introduction of various polyesters lead to different properties, such as crystallinity and cytotoxicity, the newly synthesized 3,4-dihydro-2(H)-pyrimidinone compounds (DHPMs) exhibit enhanced hydrophilicity and can self-assemble in water and N,N-dimethylformamide (DMF) solution to form micelles with a controllable size. Furthermore, DHPM–PPDO promotes cellular growth and has potential applications in wound healing and tissue engineering. In conclusion, this method demonstrates great universality and methodological significance and offers insights into the medical applications of polyethylene glycol.
Introduction
In recent years, research on biodegradable polymer materials has gained popularity, and they have been widely utilized in various biomedical fields.1–3 The applications include in vivo implantable materials,4–6 drug delivery systems,7–9 wound dressings,10–12 and burn dressings. Among the polymer materials, aliphatic polyesters, for example, polyglycolic acid (PGA), polyhydroxyalkanoates (PHA), PBA, PCL, PLA, and PPDO, have become the preferred biomaterials owing to their excellent biocompatibility and biodegradability.13–15However, in practical applications, it is challenging for a single material to satisfy multifaceted requirements. For example, PCL exhibits excellent biocompatibility, hemocompatibility, and drug permeability and has been approved by the Food and Drug Administration (FDA) for use in the human body.16–18 However, its hydrophobic character significantly hampers its application within the pharmaceutical sector. Similarly, other polymers such as PLA, PPDO, and PBA also have restricted applicability owing to their inherent hydrophobicity.19,20 To satisfy the requirements of practical applications, hydrophilic chain segments are customarily integrated into molecular chains. A prototypical example is PEG, a versatile hydrophilic polymer renowned for its biological attributes such as non-toxicity, non-immunogenicity, and non-thrombogenicity, which collectively contribute to its minimal risk of rejection within biological environments.21–25
Amphiphilic block copolymers are synthesized by connecting hydrophobic and hydrophilic chain segments. These copolymers hold considerable promise, finding utility in the fabrication of nanoparticles, micelles, and nanogels.26–28 In 2022, Su et al. fabricated a composite bone scaffold integrating CuS, PEG, and PCL. The PCL moiety functioned as a robust structural backbone, providing the required mechanical strength necessary for load-bearing applications. Concurrently, the hydrophilic PEG segment played a critical role in enhancing the scaffold's interface compatibility with bone tissue, thereby promoting enhanced cellular adhesion and proliferation.29 In 2014, He et al. demonstrated that PEG-PLGA micelles, owing to their distinctively low critical micelle concentration (CMC) attributes and diminutive particle dimensions, exhibited enhanced colloidal stability. This feature not only prolonged the circulation lifetime within living organisms but also facilitated the realization of a controlled, sustained, and gradual drug release profile, thereby optimizing therapeutic outcomes.30
The Biginelli reaction is a type of MCR. The reaction involves a three-component cyclocondensation using aldehydes, 1,3-diketones, and urea as building blocks. It is valuable for synthesizing DHPM compounds in a single reaction vessel. The Biginelli reaction continues to be of interest due to its potential applications in organic synthesis and the pharmaceutical industry. Like other multicomponent reactions, the Biginelli reaction is a powerful method for synthesizing a wide variety of compounds due to its simplicity, efficiency, scalability, and low cost.31–33 Tao Lei et al. synthesized a polymeric antioxidant with a ferrocene moiety by achieving high yields of ferrocene monomers through a stabilized three-component Biginelli reaction.34 Tao Lei et al. have conveniently synthesized a series of monomers containing functional groups with potential anticancer activity through the Biginelli reaction. The monomers were screened using a convenient free radical copolymerization process to produce water-soluble polymers, which could effectively inhibit mitosis in various types of cancer cells.35 The Biginelli reaction can also be used to synthesize various functional polymers, such as condensation polymers, hyperbranched polymers, and dendrimer macromers.36–39
In this study, a series of biodegradable materials with convenient and stable reactions were synthesized using the Biginelli reaction. First, four functionalized segments, namely, PBA, PCL, PLA, and PPDO, were synthesized. Subsequently, PEG was esterified with tert-butylacetoacetate (t-BAA) to obtain PEG-AA. The functionalized chain segments were ester exchanged with 4-formylbenzoic acid to obtain PBA-Bz, PCL-Bz, PLA-Bz, and PPDO-Bz. Leveraging the Biginelli reaction mechanism, these tailored chain segments were further manipulated to fabricate DHPMs, culminating in the creation of a novel class of polyester-based copolymers that exhibit exceptional biocompatibility and biodegradability profiles. The resulting materials were denoted as DHPM–PBA, DHPM–PCL, DHPM–PLA, and DHPM–PPDO. The comprehensive characterization of these copolymers encompassed the assessment of their crystallinity, thermal properties, hydrophilicity, biodegradability, and biocompatibility.
Experimental
Materials
Polyethylene Glycol-4000, p-dioxanone (PDO), lactide (LA), ε-caprolactone (ε-CL), adipic acid, 4-dimethylaminopyridine (DMAP), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide (hydrochloride) (EDC), and 1,4-butanediol (BDO) were procured from Shanghai Aladdin Biochemical Technology Co., Ltd in Shanghai, China. Tert-butylacetoacetate (t-BAA), 4-formylbenzoic acid, and urea were obtained from Sinopharm Chemical Reagent Co., Ltd. All other chemicals were of analytical grade and used without additional purification.Synthesis of PEG-AA
5 g of PEG-4000 and an excess of t-BAA were completely dissolved in DMF at 100 °C. To ensure consistent Biginelli reaction products, the reaction duration was extended to 48 h to facilitate the majority of PEG chain segments undergoing bipartite functionalization. The crude product was subsequently precipitated using chilled anhydrous ether and vacuum-dried.Synthesis of functionalized segments
The synthesis of PPDO with a molecular weight of 2000 was based on the literature.40 5 g of PDO WAS mixed with BDO (0.217 mL, 2.44 mmol) in a flask. Thereafter, 1 mL of 0.019 M stannous octoate solution in toluene was syringe-injected under an argon atmosphere, followed by evacuation with a vacuum pump to eliminate toluene from the system. The reaction proceeded at 80 °C for 72 hours. Upon completion, the product was precipitated in methanol and vacuum-dried. PLA and PCL were synthesized in a manner similar to PPDO.41,42 PBA with a molecular weight of 2000 was synthesized following the procedures outlined in the literature.43PPDO (Mn = 2000), 4-formylbenzoic acid, DMAP, and EDC were completely dissolved in trichloromethane at room temperature. The system was activated by stirring in an ice-water bath for 30 minutes under an argon atmosphere. The reaction was conducted at room temperature for 48 hours. Following this, extraction was performed with an equal volume of saturated brine solution. The transparent lower layer was collected, and the product was precipitated using methanol before being vacuum-dried to yield PPDO-Bz. PBA-Bz, PCL-Bz, and PLA-Bz were synthesized adopting the same methodology.
Synthesis of DHPMs
PEG-AA (1.76 g, 0.44 mmol) and PPDO-Bz (0.8 g, 0.4 mmol) were dissolved in 15 mL of glacial acetic acid at 100 °C together with urea and anhydrous MgCl2. Under an argon atmosphere, the blend was stirred continuously for 48 hours. Upon completion, the product was subjected to dialysis against deionized water (14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da) for three days, followed by freeze-drying, yielding the yellow DHPM–PPDO powder. DHPM–PBA, DHPM–PCL, and DHPM–PLA were synthesized applying the identical procedure.
000 Da) for three days, followed by freeze-drying, yielding the yellow DHPM–PPDO powder. DHPM–PBA, DHPM–PCL, and DHPM–PLA were synthesized applying the identical procedure.
Characterization
The proton nuclear magnetic resonance (NMR) spectra were recorded using a Bruker Avance-400 NMR spectrometer. The sample was dissolved in deuterated chloroform (CDCl3) or deuterium oxide (D2O) as needed, with tetramethylsilane serving as the internal standard for the solvent. The infrared spectra of the samples were measured using a Nicolet 6700 Fourier transform infrared (FT-IR) spectrometer. The measurement range was 4000–400 cm−1, and testing and analysis were conducted. The molecular weight and molecular weight distribution (PDI) of the DHPM copolymer were measured using a Waters 1525 and Agilent PL-GPC220 gel chromatograph. Tetrahydrofuran was used as the eluent at a flow rate of 1 mL min−1 at 35 °C, and polystyrene was used as the standard sample. The changes in the crystallization properties of the products at each stage before and after copolymerization were observed using a Japanese Ultima IV series composite X-ray diffractometer. The samples were dried at 25 °C, and X-ray diffraction patterns were obtained using Cu-Kα radiation generated at 40 kV and 40 mA. The scanning speed was 2° min−1, and the diffraction angle range (2θ) was 3–50°. Elemental analysis data of the final product DHPMs were obtained using an elemental analyzer called UNICUBE. The polymer's hydrodynamic size was determined using dynamic light scattering (DLS). The preparation process was as follows: the DHPMs were dissolved in deionized water to create a solution with a concentration of 1 mg mL−1. The solution was then sonicated for 10 minutes using an ultrasonic bath before being filtered through a 0.22 μm aqueous filter. The prepared solution was diluted as necessary for use in subsequent experiments. The micelle morphology was observed and recorded using a JEM1200 transmission electron microscope operated at 80 kV. The process of sampling with the electron microscope was as follows: the micelle solution, prepared as described above, was deposited onto a copper grid and then dried in a vacuum oven. The thermal stability of the polymers at each stage was determined using the TA Instruments Q5000IR thermal analyzer. Under a nitrogen flow of 20 mL min−1, the samples were heated from room temperature to 800 °C at a rate of 20 °C min−1, and the data were recorded by a computer. An L-700 differential scanning calorimeter (Mettler Toledo, Switzerland) was utilized to examine the non-isothermal crystallization behavior of each polymer. The temperature increased from room temperature to a specific level at a rate of 10 °C min−1 in a nitrogen atmosphere with a gas flow rate of 50 mL min−1 and then remained constant for 5 minutes to eliminate any thermal history. The temperature was then lowered to −20 °C at a cooling rate of 10 °C min−1. The temperature was then increased to the specified level at a rate of 10 °C min−1, and the non-isothermal differential scanning calorimetry (DSC) curve of the sample was recorded. The DHPM product was sequentially washed with anhydrous ethanol, dried, gold-sprayed on small sections, and finally subjected to scanning electron microscopy (ZEISS GeminiSEM 300) for morphological assessment. The water contact angle of DHPM products was measured using a Dataphysics OCA30 contact angle analyzer from Germany. The films were prepared using the solution casting method by dissolving 50 mg of DHPM product in 2 mL of tetrahydrofuran (THF). The solution was then dropped onto a horizontal glass plate and left in a fume hood for 1 hour to obtain the DHPM product film. Afterward, 2 μL of water was deposited at three different positions on the film, and the contact angle was measured and averaged. In the biodegradability testing, first, a Tris–HCl solution was prepared by weighing 12.11 g of Tris and placing it in a 100 mL beaker. Approximately 80 mL of deionized water was added and stirred until completely dissolved. Then, approximately 6 mL of concentrated hydrochloric acid (HCl) was added to adjust the pH to 7.6, and the solution was finally diluted to 100 mL. It is important to note that the pH of the Tris–HCl solution varies significantly with temperature. For every 1 °C increase in temperature, the pH of the solution decreases by 0.03 units. Therefore, the solution was prepared in a 37 °C water bath to maintain the necessary temperature sensitivity. Subsequently, 500 mg of DHPM product was placed in a glass vial, and 10 mL of Tris–HCl solution containing proteinase K (1 mg mL−1) was added for degradation at 37 °C. After seven days, the solution was filtered, washed with deionized water, freeze-dried, weighed, and then the aforementioned steps were repeated. (The products are insoluble in water, and any losses are negligible.) The cytotoxicity was evaluated using the CCK-8 method. Human normal bladder epithelial cells (SV-HUC-1) were obtained, and the cell suspension was adjusted to a concentration of approximately 1 × 105 cells per mL. Subsequently, 100 μL of the cell suspension was added to each well of a 96-well culture plate. The plates were incubated in a 5% CO2 environment at 37 °C for approximately 24 hours. The supernatant was then removed from the wells, and the polymer solutions were diluted with serum-free medium to concentrations of 0, 50, 100, and 200 μg mL−1. Six replicate wells were prepared for each concentration, and 100 μL of polymer solution was added to each well. The cell plates were incubated in the CO2 incubator for 24, 48, or 72 hours. Afterward, the liquid above the sediment was removed, and the cells were washed with a PBS solution. To each well, 10 μL of CCK-8 solution and 90 μL of serum-free medium were added and incubated for 2 hours. The absorbance (OD) value at 450 nm was measured for each well using an enzyme assay. The cell viability was determined using the following formula:| Cell viability (%) = (ODsamples − ODvoid)/(ODcompare − ODvoid) × 100. |
Results and discussion
Synthesis and characterization
The PEG segment was synthesized from PEG and tert-butyl acetoacetate through an esterification reaction. PBA-Bz, PCL-Bz, and PLA-Bz functionalized segments were synthesized through the transesterification reaction of PPDO, PBA, PCL, and PLA with 4-formylbenzoic acid. 1H NMR (Fig. S1 in the ESI†) spectra revealed that the peaks at 2.30 ppm and 3.51 ppm in the 1H NMR spectra of the PEG segment were attributed to the methyl and methylene groups in the acetoacetyl group. The peaks at 7.98 ppm and 8.20 ppm in the 1H NMR spectra of functionalized segments were assigned to the aromatic hydrogen (Ar–H) groups. The peaks at 1710 cm−1 and 1740 cm−1 in the FT-IR spectrum (Fig. S3(a and b)†) were observed for PEG-AA, and these were attributed to the acetoacetate groups. The peaks at 750 cm−1 in the FT-IR spectrum were observed in functionalized segments and were attributed to the benzene ring. To assure homogeneity in the Biginelli reaction products, the reaction duration was extended to 48 hours, thereby ensuring majority completion of the bifunctionalization process on the chain ends. The integral transformation analysis in 1H NMR (For example, PCL-Bz:I8.19:I7.98:I4.09 = 1:1.01:8.83) confirmed the successful achievement of over 95% end-group bifunctionalization for the polymer chain segments. These results demonstrate the successful preparation of PEG and functionalized segment polymers.
The synthetic pathway for the Biginelli reaction is illustrated in Fig. 1(a). The reactants for the Biginelli reaction include PEG-AA, functional polymers, ureas, anhydrous magnesium chloride, and acetic acid, while the resulting products are DHPMs. Fig. 1(b) illustrates the reaction mechanism, wherein the acid-catalyzed condensation of urea and an aromatic aldehyde yields an acetal amine, which promptly dehydrates to form an N-imide cation. Simultaneously, an amino acid also dehydrates to form an N-imide intermediate. The ensuing step involves the insertion of a β-keto ester, triggering an enol-mediated attack on the imide cation, thereby forming an open-chain acyl urea. Ultimately, acid-catalyzed intramolecular dehydration and condensation lead to the synthesis of a hexahydropyridine derivative.
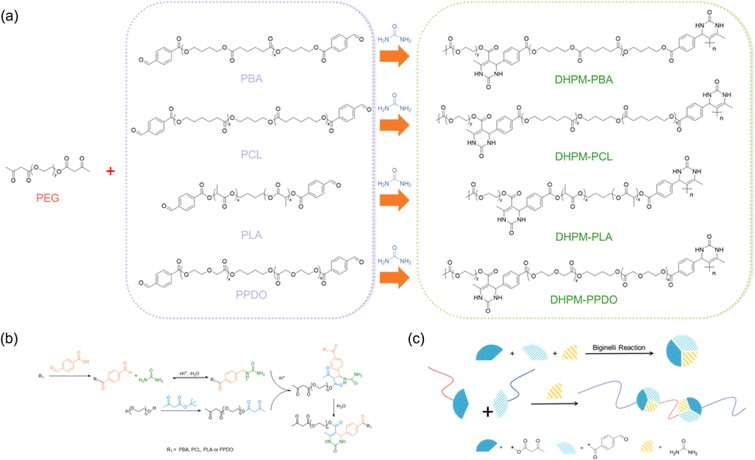 | ||
| Fig. 1 (a) PEG-based Biginelli reaction. (b) Mechanism of the Biginelli Reaction. (c) Schematic of synthesis of DHPMs products. | ||
The 1H NMR spectra in Fig. S2† indicate that the peak at 2.30 ppm, corresponding to the original methyl group in the acetoacetic acid, disappears once the reaction is completed. In the newly formed DHPMs, the peaks at 10.10 and 8.18 ppm corresponding to the –N–H– group in the DHPM ring, and a peak at 2.40 ppm representing the methyl group, were observed. The FT-IR spectrum of the final product is depicted in Fig. S3(c).† The presence of the peak at 3240 cm−1 in the product is attributed to the –N–H– group on the DHPM ring. These changes indicate the disappearance of the acetoacetic acid group and the success of the Biginelli reaction. The integral transformation analysis in 1H NMR, elemental analysis (Table S1†), and GPC trace (Fig. S4†) substantiate the successful synthesis of the four DHPM materials with molecular weights of nearly 25 kDa.
Compatibility studies of chain segments
From the XRD curves depicted in Fig. 2(a), it is evident that all the products exhibit distinct diffraction peaks. But PLA shows no discernible diffraction peaks due to its low crystallization ability. The DHPMs exhibit diffraction peaks corresponding to their PEG and functionalized segment. The degree of crystallinity is calculated as 72.60% for DHPM–PBA, 55.31% for DHPM–PCL, 34.82% for DHPM–PLA, and 58.19% for DHPM–PPDO. These results partially demonstrate the good compatibility between PEG and the functionalized segment.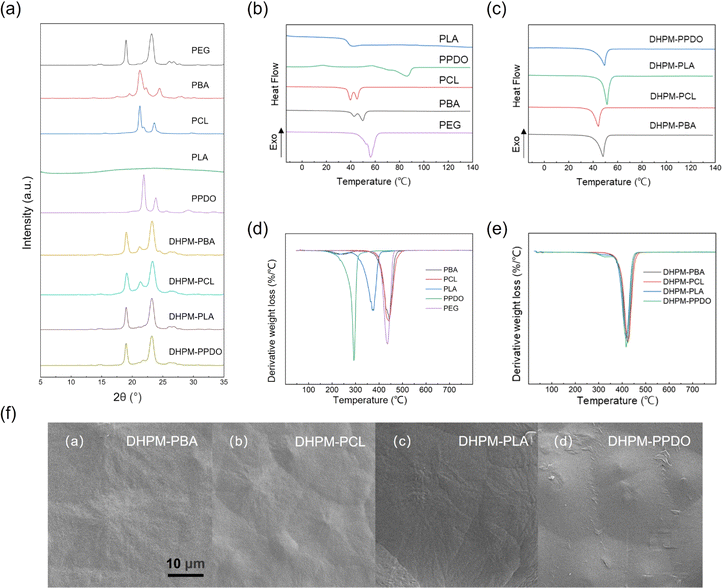 | ||
| Fig. 2 (a) XRD curves, (b and c) DSC heating curves, and (d and e) DTG curves of PEG, PBA, PCL, PPDO, PLA, DHPM–PBA, DHPM–PCL, DHPM–PLA, and DHPM–PPDO. (f) SEM images of DHPM–PBA, DHPM–PCL, DHPM–PLA, and DHPM–PPDO. | ||
The DSC curves for all the samples are presented in Fig. 2. Non-isothermal crystallization parameters are shown in Table S2.† Fig. 2(b and c) display the heating curves of all samples at a rate of 10 °C min−1. It can be observed that PPDO exhibits an exothermic peak of crystallization at 17.5 °C. PBA and PCL exhibit two melting peaks, which were not evident during the initial heating process. The gradual warming process may cause previously formed crystals to partially melt, and the unmelted portion can serve as a nucleation point for the recrystallization of the melt. This situation results in the formation of two melting peaks, with the recrystallized melt melting at higher temperatures. PLA exhibits a glass transition at 37.84 °C, but no glass transition was observed in the subsequent DHPM–PLA. This phenomenon arises from the block copolymer nature of DHPMs, with the Tg of such a material being governed by the segment with the lowest Tg among its constituent polymer chains. Considering that PEG-4000, a key component in DHPMs, exhibits an extremely low Tg (−60 °C), falling outside the scope of standard testing methodologies, no definitive Tg could be discerned for DHPMs within the constraints of conventional assays. The Tg of PCL, PPDO, and PBA is −60 °C, −30 °C, and −42 °C, respectively.44–48
Moreover, during heating, DHPMs exhibit convergent melting peaks at 48 °C, indicative of the substantial influence of PEG on the functionalized segments. The melting temperatures (Tm) show varying degrees of decline, a characteristic of block copolymers, thus corroborating the favorable compatibility between PEG and the functional polymer chains.
The TGA curves for all the samples are presented in Fig. 2. The thermal performance data are shown in Table S3.† In Fig. 2(d), it can be observed that PPDO exhibits relatively poor thermal stability, followed by PLA, while PCL, PBA, and PEG demonstrate similar performances. Besides, the TGA curves of PBA reveal two distinct degradation phases, wherein the initial decomposition stage terminates at 262.6 °C, accompanied by a mass loss of 9.83%. This early degradation is attributed to the reduced thermal stability of the butanediol units, leading to their premature disintegration at relatively low temperatures. Owing to the higher content of butanediol units compared to other polymers, PBA manifests a dual-stage degradation pattern. The molecular weight of PLA may not be controlled sufficiently, resulting in the presence of the small molecular weight PLA that decomposes first.
Following the Biginelli reaction, the thermal decomposition properties of the DHPMs were analyzed. From Fig. 2(e), the temperatures at which the weight loss was 5% (T5%), the temperature of maximum thermal decomposition (Tmax), and the temperature at which the weight loss was 95% (T95%) of the DHPMs tend to increase overall. Additionally, the DTG curves of the four DHPMs tend to converge, indicating that the inclusion of the PEG segment improves the thermal decomposition properties of the DHPMs.
SEM images serve as a powerful tool for investigating and characterizing the self-assembly behaviors and microstructural attributes of block copolymers. Owing to their unique molecular architectures, these materials typically self-assemble into a range of periodic nanostructures or microphase-separated morphologies in either solution or the melt phase. Such structural nuances are indeed discernible in the SEM images. Powder DHPM products were gold-sputtered and their morphology was observed by scanning electron microscopy. As illustrated in Fig. 2(f), the SEM image of DHPMs manifests periodic, island-like structures, indicative of a uniform microstructure. This visualization is instrumental in unraveling the intricate self-assembly processes and deciphering the material's intrinsic properties.
The Biginelli reaction combines PEG with PBA, PCL, PLA, and PPDO to form PEG-based block copolymers. By characterizing the crystalline and thermal properties of the DHPMs, we discovered that they exhibit combined crystalline diffraction peaks from the respective PEG and functionalized segments. Notably, five initially distinct materials with differing melting points were converted into DHPMs, displaying similar melting temperatures and unified thermal profiles, suggesting the effective merging of the disparate thermal stability. It can be concluded that PEG and the functionalized segments of the DHPMs exhibit better compatibility.
Micelle size regulation and morphology characterization
To elucidate the morphology of DHPMs, they were solubilized in a 1:1 water/DMF cosolvent system. Upon solvent evaporation, they were observed using TEM. Observations showed a dark central core, attributed to the compact, hydrophobic nature of the functionalized polymer, enveloped by a less dense, brighter shell composed of hydrophilic PEG chains. This biphasic pattern indicates efficient surface coverage by PEG segments, stabilizing an aqueous interface, while the functional polymers concentrate in the micellar core.
This finding led to the study of adjusting the micelle sizes. 5 mg, 10 mg, 15 mg, and 20 mg of the DHPMs were dissolved in 2 mL of DMF. During vigorously stirring, 5 mL of deionized water was injected into the solution at a rate of 5 mL h−1 by a microsyringe pump. Next, deionized water was used for dialysis to obtain micelle samples with different concentrations. As shown in Fig. 3(I), analyzing the TEM images from left to right distinctly illustrates the dose-dependent increase in micelle dimensions. The observed size variation corresponds well with the DLS outcomes in Fig. 3(II), reinforcing the precision and reliability of our micelle size control strategy. Consequently, we have effectively achieved tunable sizing in the design of functional polymer assemblies.
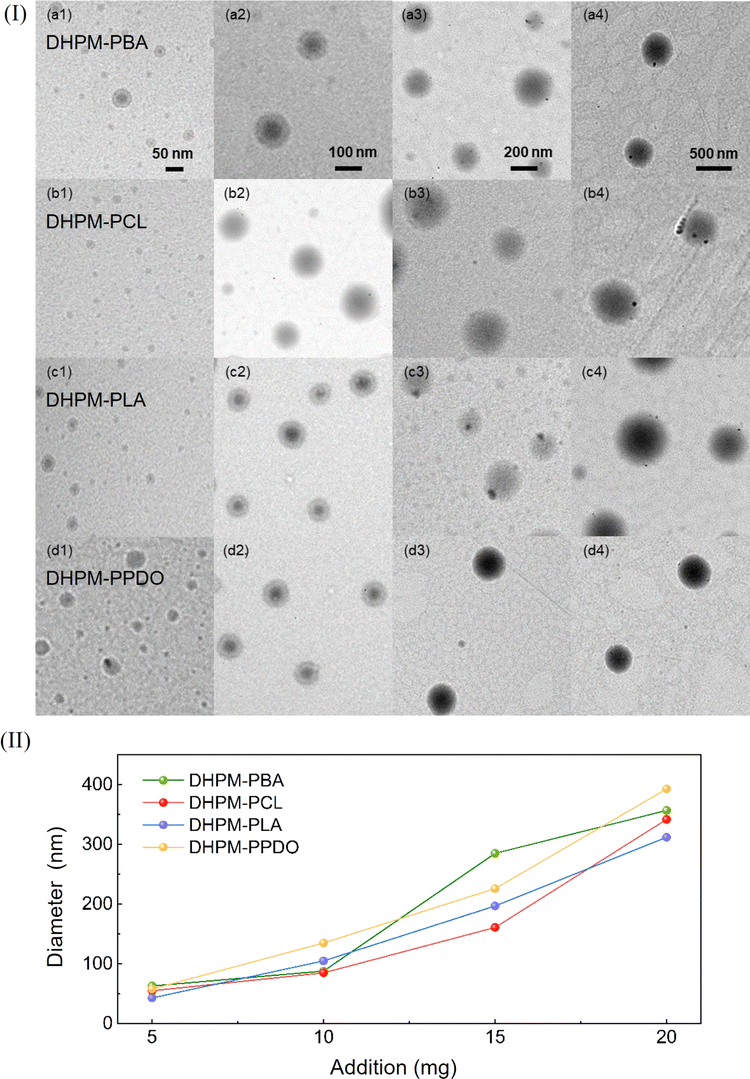 | ||
| Fig. 3 (I) TEM of micelles (a) DHPM–PBA, (b) DHPM–PCL, (c) DHPM–PLA, and (d) DHPM–PPDO. (II) Size regulation of micelles. | ||
Biocompatibility testing
Water contact angle is a critical parameter for medical applications and reflects the material's interaction with water; excessive hydrophobicity can severely restrict its usability. As shown in Fig. 4(a), PEG's main chain hosts abundant ether linkages, oxygen atoms readily engage in strong hydrogen bonding with the hydrogen atoms of water molecules, as evidenced by a low contact angle of 32.3°, allowing the rapid dispersion of water droplets upon contact. In contrast, the four functionalized chain segments displayed poor hydrophilicity. After undergoing the Biginelli reaction, the water contact angles of DHPMs were modulated to converge at a similar level, suggesting an optimization of the hydrophilic–hydrophobic balance. Importantly, the incorporation of PEG significantly enhanced the overall hydrophilicity of the DHPM constructs. This outcome demonstrates that the DHPM products have undergone successful surface property tuning without compromising the intrinsic characteristics of the functionalized polymers.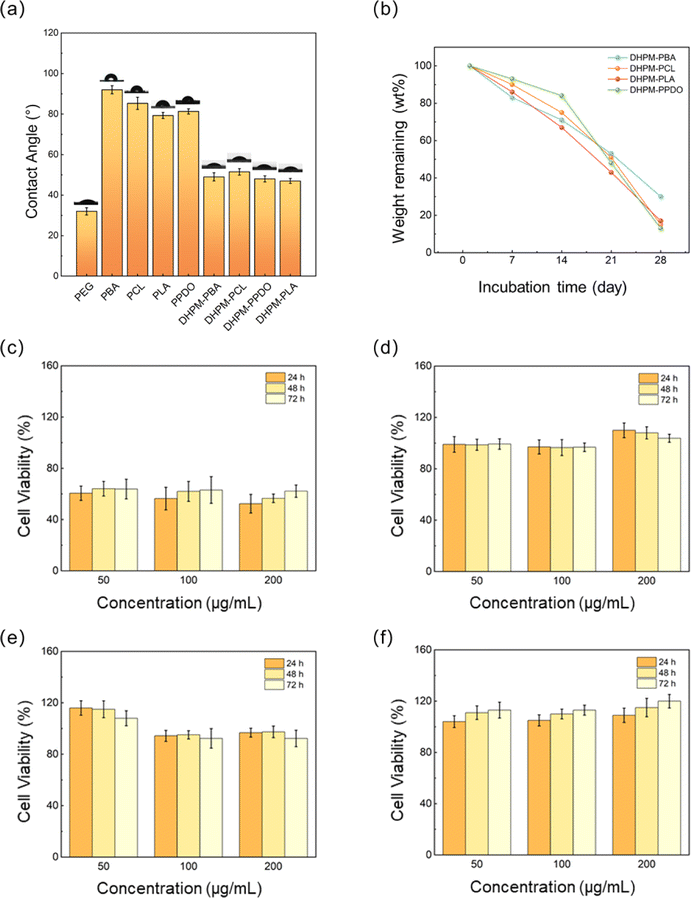 | ||
| Fig. 4 (a) Water contact angles of PEG, PBA, PCL, PPDO, PLA, DHPM–PBA, DHPM–PCL, DHPM–PLA, and DHPM–PPDO. (b) Degradation rates of DHPM–PBA, DHPM–PCL, DHPM–PLA, and DHPM–PPDO. Cell viability of DHPM–PBA (c), DHPM–PCL (d), DHPM–PLA (e), and DHPM–PPDO (f). | ||
The degradation properties of polymeric materials are also important indicators for medical purposes. In this study, we investigated the enzymatic degradation of DHPMs using proteinase K, an enzyme known to accelerate the degradation of both PEG and PLA.49,50 As depicted in Fig. 4(b), DHPM–PCL, DHPM–PLA, and DHPM–PPDO demonstrate similar degradation capabilities. The degradation rate of DHPM–PPDO initially seemed slow, followed by a rapid increase. This behavior may be attributed to the hydrolysis of the ester bond in the PPDO. However, the molecular weight of the fractured products was not small enough to be soluble in the degradation solution, leading to an initially inconspicuous loss of mass. The degradation rate notably accelerated in the third week, attributed to an enhanced autocatalytic effect, which expedited the hydrolytic cleavage of the ester bond. Among the four DHPMs, DHPM–PBA exhibited a notably slower mass loss rate, potentially stemming from its relatively low hydrophilicity, which hampers hydrolytic degradation. Moreover, the elevated crystallinity and the presence of a crystalline phase in DHPM–PBA further impede the hydrolysis process.
The cytotoxicity test is a crucial method for evaluating the biocompatibility of a material. As depicted in Fig. 4(c–f), the cell survival rate of DHPM–PBA is significantly lower than that of the other DHPMs, at approximately 55%. This suggests that DHPM–PBA has good biodegradability but lacks biocompatibility; we postulate that this toxicity may arise from the persistence of low molecular weight PBA oligomers within the system, as evidenced by the shoulder peak discernible on the right, low molecular weight flank of the GPC plot featured in Fig. S6.† Their enhanced water solubility appears to facilitate intracellular circulation, potentially culminating in deleterious effects on the cells. The cell survival rates of DHPM–PCL and DHPM–PLA are close to 95%, indicating that both materials exhibit excellent biocompatibility. The results of DHPM–PPDO are quite surprising. In a serum-free medium, the cell viability surpassed 100% across all tested concentrations, reaching up to 120% at the highest dose and consistently increased with both rising solution concentrations and extended incubation periods. This implies that DHPM–PPDO solutions actively stimulate the proliferation of human bladder epithelial cells, thereby supporting tissue regeneration. Li Yuling et al.,51 Shi Feng et al.,52 and Wang Guannan et al.,53 independently reported that both PEG and PPDO exhibit low cytotoxic effects; however, neither of these materials was observed to actively stimulate cell proliferation. The distinct properties observed in DHPMs may arise from the unique structural configuration introduced by the Biginelli reaction. Given this pro-growth property, DHPM–PPDO holds promise for applications in medical dressing development, tissue engineering constructs, and human implant materials.
The collective evidence from the triad of methods establishes that DHPM–PCL, DHPM–PLA, and DHPM–PPDO exhibit outstanding biodegradation profiles and biocompatibility traits, pointing towards their eligibility for potential deployment in medical tissue engineering and as constituents of human implant materials.
Conclusions
In summary, a simple method for synthesizing DHPM derivatives using polyethylene glycol acetoacetate is proposed. Firstly, PEG-AA was synthesized as a precursor through the modification of polyethylene glycol. PBA, PCL, PLA, and PPDO, which are also biodegradable polyesters, were chosen, altered, and then synthesized through the Biginelli reaction. Four different DHPMs were synthesized and confirmed using 1H NMR and FT-IR spectroscopy. The introduction of various functionalized segments in the polymers changed the properties of the DHPMs. On the one hand, the DHPMs exhibit some common changes, such as improved hydrophilicity and thermal properties. Besides, they also exhibit the crystallographic diffraction peaks of the respective polymers and possess great crystalline properties. Furthermore, the DHPMs can self-assemble in water and DMF solution to form micelles, and the sizes of the micelles can be effectively adjusted by controlling the input amount. On the other hand, the various functionalized segments in polymers confer distinct properties to the DHPMs. Some of the DHPMs exhibit excellent biocompatibility and can promote cell growth, making them suitable for use in wound healing and tissue engineering. The outcomes show that by upholding the initial structural integrity, the drawbacks detrimental to medical use have been refined, allowing the material to conform to more sophisticated application settings. This universally applicable synthesis technique signifies a pivotal methodological advance, presenting a fresh avenue for broadening the biomedical scope of biodegradable polyesters. DHPMs typically exhibit anti-cancer and anti-UV characteristics, warranting further investigation into their effects on cancerous cells and antioxidant capacities.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51973194), the National Natural Science Foundation of China (52273051, 51973194 and 51703198), the Zhejiang Provincial Natural Science Foundation of China (LY20E030007), and China Postdoctoral Science Foundation (2022M712815).Notes and references
- I. Ekladious, Y. L. Colson and M. W. Grinstaff, Nat. Rev. Drug Discovery, 2019, 18, 273–294 CrossRef CAS PubMed.
- A. G. Niculescu and A. M. Grumezescu, Polymers, 2022, 14, 421 CrossRef CAS PubMed.
- J. Seppala, B. van Bochove and A. Lendlein, Biomacromolecules, 2020, 21, 273–275 CrossRef CAS PubMed.
- M. S. B. Reddy, D. Ponnamma, R. Choudhary and K. K. Sadasivuni, Polymers, 2021, 13, 1105 CrossRef CAS PubMed.
- M. Santoro, S. R. Shah, J. L. Walker and A. G. Mikos, Adv. Drug Delivery Rev., 2016, 107, 206–212 CrossRef CAS PubMed.
- D. da Silva, M. Kaduri, M. Poley, O. Adir, N. Krinsky, J. Shainsky-Roitman and A. Schroeder, Chem. Eng. J., 2018, 340, 9–14 CrossRef CAS PubMed.
- A. Kumari, S. K. Yadav and S. C. Yadav, Colloids Surf., B, 2010, 75, 1–18 CrossRef CAS PubMed.
- Y. B. Patil, U. S. Toti, A. Khdair, L. Ma and J. Panyam, Biomaterials, 2009, 30, 859–866 CrossRef CAS PubMed.
- A. Kolate, D. Baradia, S. Patil, I. Vhora, G. Kore and A. Misra, J. Controlled Release, 2014, 192, 67–81 CrossRef CAS PubMed.
- G. D. Mogosanu and A. M. Grumezescu, Int. J. Pharm., 2014, 463, 127–136 CrossRef CAS PubMed.
- H. Chen, R. Cheng, X. Zhao, Y. Zhang, A. Tam, Y. Yan, H. Shen, Y. S. Zhang, J. Qi, Y. Feng, L. Liu, G. Pan, W. Cui and L. Deng, NPG Asia Mater., 2019, 11, 3 CrossRef CAS.
- Z. Muwaffak, A. Goyanes, V. Clark, A. W. Basit, S. T. Hilton and S. Gaisford, Int. J. Pharm., 2017, 527, 161–170 CrossRef CAS PubMed.
- I. Manavitehrani, A. Fathi, H. Badr, S. Daly, A. Negahi Shirazi and F. Dehghani, Polymers, 2016, 8, 20 CrossRef PubMed.
- Q. Zhang, M. Song, Y. Xu, W. Wang, Z. Wang and L. Zhang, Prog. Polym. Sci., 2021, 120, 101430 CrossRef CAS.
- C. Vilela, A. F. Sousa, A. C. Fonseca, A. C. Serra, J. F. J. Coelho, C. S. R. Freire and A. J. D. Silvestre, Polym. Chem., 2014, 5, 3119–3141 RSC.
- E. Malikmammadov, T. E. Tanir, A. Kiziltay, V. Hasirci and N. Hasirci, J. Biomater. Sci., Polym. Ed., 2018, 29, 863–893 CrossRef CAS PubMed.
- M. Bartnikowski, T. R. Dargaville, S. Ivanovski and D. W. Hutmacher, Prog. Polym. Sci., 2019, 96, 1–20 CrossRef CAS.
- R. A. Ilyas, M. Y. M. Zuhri, M. N. F. Norrrahim, M. S. M. Misenan, M. A. Jenol, S. A. Samsudin, N. M. Nurazzi, M. R. M. Asyraf, A. B. M. Supian, S. P. Bangar, R. Nadlene, S. Sharma and A. A. B. Omran, Polymers, 2022, 14, 182 CrossRef CAS PubMed.
- K. Hamad, M. Kaseem, H. W. Yang, F. Deri and Y. G. Ko, eXPRESS Polym. Lett., 2015, 9, 435–455 CrossRef CAS.
- K.-K. Yang, X.-L. Wang and Y.-Z. Wang, J. Macromol. Sci., Polym. Rev., 2002, 42, 373–398 CrossRef.
- P. Mishra, B. Nayak and R. K. Dey, Asian J. Pharm. Sci., 2016, 11, 337–348 CrossRef.
- P. L. Turecek, M. J. Bossard, F. Schoetens and I. A. Ivens, J. Pharm. Sci., 2016, 105, 460–475 CrossRef CAS PubMed.
- W. Chen, P. Zhong, F. Meng, R. Cheng, C. Deng, J. Feijen and Z. Zhong, J. Controlled Release, 2013, 169, 171–179 CrossRef CAS PubMed.
- C. Chen, Y. C. Cheng, C. H. Yu, S. W. Chan, M. K. Cheung and P. H. Yu, J. Biomed. Mater. Res., Part A, 2008, 87, 290–298 CrossRef PubMed.
- A. D'Souza A and R. Shegokar, Expert Opin. Drug Delivery, 2016, 13, 1257–1275 CrossRef PubMed.
- Y. Xu, W. T. Chen, T. F. Fan, X. M. Wu and H. X. Gao, Adv. Mater. Res., 2015, 1088, 449–454 Search PubMed.
- Y. Niu, P. Zhang, J. Zhang, L. Xiao, K. Yang and Y. Wang, Polym. Chem., 2012, 3, 2508 RSC.
- A. Salimi, S. Ahmadi, M. Faramarzi and J. Faghihi, Macromol. Res., 2023, 31, 873–881 CrossRef CAS.
- X. Xue, H. Zhang, H. Liu, S. Wang, J. Li, Q. Zhou, X. Chen, X. Ren, Y. Jing, Y. Deng, Z. Geng, X. Wang and J. Su, Adv. Funct. Mater., 2022, 32, 2202470 CrossRef CAS.
- K. Zhang, X. Tang, J. Zhang, W. Lu, X. Lin, Y. Zhang, B. Tian, H. Yang and H. He, J. Controlled Release, 2014, 183, 77–86 CrossRef CAS PubMed.
- Z. Ma, B. Wang and L. Tao, Molecules, 2022, 27, 7886 CrossRef CAS PubMed.
- C. Zhu, B. Yang, Y. Zhao, C. Fu, L. Tao and Y. Wei, Polym. Chem., 2013, 4, 5395–5400 RSC.
- R. Kaur, S. Chaudhary, K. Kumar, M. K. Gupta and R. K. Rawal, Eur. J. Med. Chem., 2017, 132, 108–134 CrossRef CAS PubMed.
- T. Mao, L. Yang, G. Liu, Y. Wei, Y. Gou, J. Wang and L. Tao, ACS Macro Lett., 2019, 8, 639–645 CrossRef CAS PubMed.
- Y. Li, T. Tan, Y. Zhao, Y. Wei, D. Wang, R. Chen and L. Tao, ACS Macro Lett., 2020, 9, 1249–1254 CrossRef CAS PubMed.
- L. Tao, C. Zhu, Y. Wei and Y. Zhao, in Multi-Component and Sequential Reactions in Polymer Synthesis, 2014, DOI: DOI:10.1007/12_2014_301, ch. 301, pp. 43–59.
- T. Mao, X. He, G. Liu, Y. Wei, Y. Gou, X. Zhou and L. Tao, Polym. Chem., 2021, 12, 852–857 RSC.
- A. C. Boukis, A. Llevot and M. A. Meier, Macromol. Rapid Commun., 2016, 37, 643–649 CrossRef CAS PubMed.
- H. Shen, H. Jin, H. Li, H. Wang, J. Duan, Y. Jiao and S. Z. Qiao, Nat. Commun., 2023, 14, 2843 CrossRef CAS PubMed.
- Q. Zeng, K.-K. Yang, S.-C. Chen, X.-L. Wang, J.-B. Zeng and Y.-Z. Wang, Eur. Polym. J., 2008, 44, 465–474 CrossRef CAS.
- G. Li, M. Zhao, F. Xu, B. Yang, X. Li, X. Meng, L. Teng, F. Sun and Y. Li, Molecules, 2020, 25, 5023 CrossRef CAS PubMed.
- T. Xiong and Y.-F. Zhang, e-Polymers, 2022, 22, 147–155 CrossRef CAS.
- X. Liu, T. Wang, J. Li, J. Cheng and J. Zhang, J. Polym. Res., 2015, 22, 149 CrossRef.
- Q. Feng, J. Yang, Y. Liu, H. Xiao and S. Fu, J. Mater. Sci. Technol., 2014, 30, 90–96 CrossRef CAS.
- J. S. Jayan, B. D. S. Deeraj, A. Saritha and K. Joseph, Plastics, Rubber and Copolymers, 2020, vol. 49, 237–244 Search PubMed.
- L. C. Cesteros, C. A. Ramírez, A. Peciña and I. Katime, Macromol. Chem. Phys., 2007, 208, 1764–1772 CrossRef CAS.
- N. Hashim, K. Retenam, S. Somderam and K. Yusoh, Adv. Mater. Res., 2015, 1134, 185–190 Search PubMed.
- Q. Lai, Y.-Z. Wang, K.-K. Yang, X.-L. Wang and Q. Zeng, React. Funct. Polym., 2005, 65, 309–315 CrossRef CAS.
- L. Tong, M. Zhou, Y. Chen, K. Lu, Z. Zhang, Y. Mu and Z. He, Molecules, 2023, 28, 4021 CrossRef CAS PubMed.
- A. Kundys, A. Plichta, Z. Florjańczyk, A. Frydrych and K. Żurawski, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1444–1456 CrossRef CAS.
- P. Zhang, J. Hu, L. Bu, H. Zhang, B. Du, C. Zhu and Y. Li, Polymers, 2019, 11, 262 CrossRef PubMed.
- S. Liu, H. Cao, R. Guo, H. Li, C. Lu, G. Yang, J. Nie, F. Wang, N. Dong, J. Shi and F. Shi, Polym. Degrad. Stab., 2020, 172, 109067 CrossRef CAS.
- T. Fan, J. Qin, F. Dong, X. Meng, Y. Li, Y. Wang, Q. Liu and G. Wang, RSC Adv., 2022, 12, 10711–10724 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra02002b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |