
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel isatin–triazole based thiosemicarbazones as potential anticancer agents: synthesis, DFT and molecular docking studies†
Alia Mushtaqa,
Rabbia Asifa,
Waqar Ahmed Humayunb and
Muhammad Moazzam Naseer *a
*a
aDepartment of Chemistry, Quaid-i-Azam University, Islamabad 45320, Pakistan. E-mail: moazzam@qau.edu.pk
bDepartment of Medical Oncology & Radiotherapy, King Edward Medical University, Lahore 54000, Pakistan
First published on 29th April 2024
Abstract
Thiosemicarbazones of isatin have been found to exhibit versatile bioactivities. In this study, two distinct types of isatin–triazole hybrids 3a and 3b were accessed via copper-catalyzed azide–alkyne cycloaddition reaction (CuAAC), together with their mono and bis-thiosemicarbazone derivatives 4a–h and 5a–h. In addition to the characterization by physical, spectral and analytical data, a DFT study was carried out to obtain the optimized geometries of all thiosemicarbazones. The global reactivity values showed that among the synthesized derivatives, 4c, 4g and 5c having nitro substituents are the most soft compounds, with compound 5c having the highest electronegativity and electrophilicity index values among the synthesized series, thus possessing strong binding ability with biomolecules. Molecular docking studies were performed to explore the inhibitory ability of the selected compounds against the active sites of the anticancer protein of phosphoinositide 3-kinase (PI3K). Among the synthesized derivatives, 4-nitro substituted bisthiosemicarbazone 5c showed the highest binding energy of −10.3 kcal mol−1. These findings demonstrated that compound 5c could be used as a favored anticancer scaffold via the mechanism of inhibition against the PI3K signaling pathways.
1. Introduction
Heterocyclic scaffolds are important synthetic precursors for a wide range of biologically active compounds.1 They play an important role as a core moiety in a diverse range of natural products, including hemoglobin, biomolecules, RNA, DNA, proteins, vitamins, and biologically active compounds.2 Isatin (1H indole-2,3-dione) has attracted the interest of researchers in both synthetic and medicinal chemistry due to its chemical reactivity and broad range of applications, which include antimicrobial,3,4 anti-cancer,5,6 anti-HIV,7 anticonvulsant,8 anti-oxidant,9,10 anti-inflammatory,3,11 antitubercular,12–14 and antidiabetic11,15,16 properties. Sutent (SU11248), also known as Sunitinib, is a drug derived from 5-fluoro isatin that is used for targeted therapy in gastrointestinal stromal tumors, advanced renal cancer, and pancreatic neuroendocrine carcinoma.17,18 Fig. 1 shows some biologically active compounds containing isatin scaffold that have been recently reported for their anticancer potential.19–21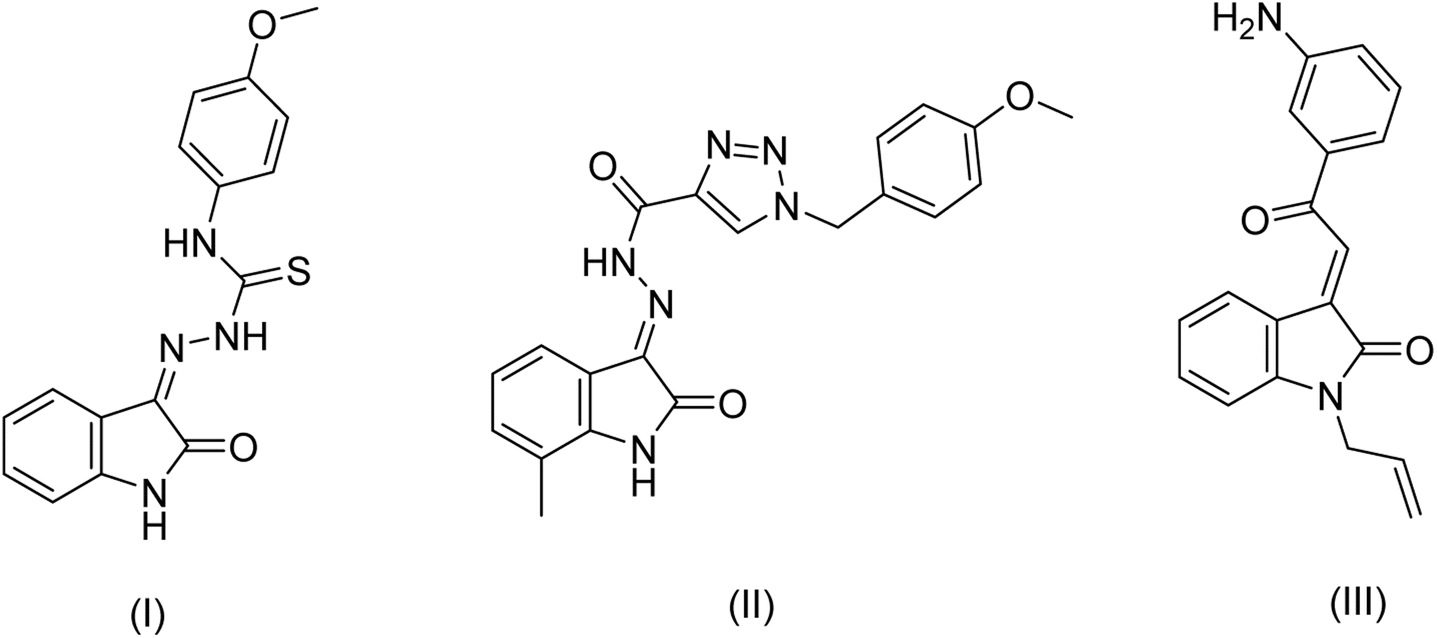 | ||
| Fig. 1 Some biologically active isatin derivatives as anticancer agents.19–21 | ||
Triazoles, specifically 1,2,3-triazole and 1,2,4-triazole, stand out as highly significant nitrogen-containing heterocycles. The incorporation of triazole can enhance solubility and facilitate binding to biomolecular targets through a range of non-covalent interactions.22 Triazoles exhibit a broad spectrum of biological activities, encompassing antimicrobial,23 antiviral,24 analgesic,25 anti-inflammatory,26 antileishmanial,26,27 and anticancer28,29 activities. The development of resistance to drugs used to treat different diseases is a serious problem which needs to be addressed.30 Hence, extensive efforts have been made for the synthesis of safe, more effective, selective and multi-targeted drugs by utilizing pharmacophore hybridization approach.31,32 Several N-1,2,3-triazole–isatin hybrids have been reported for their potential as tumour anti-proliferative,33,34 antitubercular,13,35 and antimicrobial36 agents. Furthermore, thiosemicarbazones (TSCs) being a type of Schiff bases (typically synthesized by condensation of thiosemicarbazides with a suitable aldehyde or ketone)37 have also captured the interest of the chemical and biological communities owing to their potent metal chelating ability and diverse pharmacological effects.38,39 TSCs have displayed a broad clinical antitumor spectrum, with efficacy against leukemia, pancreatic cancer, breast cancer, non-small cell lung cancer, cervical cancer, prostate cancer, and bladder cancer.38,40–48
The strategy of linking two pharmacophores together in order to improve the biological and pharmacological potency of the resulting molecules is now widely used in pharmaceutical and medicinal chemistry.49 When compared to individual moieties, this pharmacophore hybridization approach holds promise for overcoming drug-resistance and increasing potency. Motivated by these considerations and our desire to find more potent anticancer agents to combat drug resistance, we present a facile synthetic protocol for synthesizing novel mono and bis-thiosemicarbazone derivatives of isatin–triazole hybrids through the well known Cu(I)-catalyzed alkyne–azide cycloaddition (CuAAC) reaction. In the present study, the DFT studies of all the synthesized compounds have been explored in order to forecast the structure–reactivity relationship of the synthesized compounds. The most reactive compounds among the series, having the least band gap energy have been selected for the molecular docking studies and explored their probable binding interactions with phosphoinositide 3-kinases (PI3K) which is one of the key therapeutic targets for cancer treatment. The results of this study clearly indicate the potential of the synthesized compounds as anticancer agents, thus providing important guidelines for in vitro and in vivo studies of the synthesized compounds and for the design and development of efficient anticancer agents. It is also worth mentioning here that a considerable number of clinically approved anticancer drugs either in current use or undergoing trials are derived from isatin, 1,2,3-triazoles and thiosemicarbazones.50–54
2. Results and discussion
2.1 Synthesis
The synthesis of two distinct types of desired isatin–triazole hybrid intermediates 3a and 3b was initiated from the terminal alkynes 1b and 2a (Scheme 1), both of which were prepared via N-propargylation of commercially available isatin and 4-hydroxybenzaldehyde respectively with the propargyl bromide in the presence of K2CO3 in DMF.55 The targeted alkynes, 1-(prop-2-ynyl)indoline-2,3-dione 1b and 4-(prop-2-ynyloxy)benzaldehyde 2a were obtained after recrystallization from ethanol as brown and orange color precipitates in appreciable yield of 92% and 84%, respectively (Scheme 1). Similarly, the azide precursors 1a and 2b were synthesized in good yields of 92% and 78% from intermediates 1-(2-chloroethyl)isatin and 4-(2-chloroethoxy)benzaldehyde respectively,56 by their reaction with sodium azide. Finally, the conditions of “Click” chemistry i.e. Cu(I) catalyzed 1,3-dipolar cycloaddition reaction (CuAAC)55 was employed to synthesize isatin–triazole hybrid intermediates 3a and 3b. The reaction between the respective alkynes 1b and 2a and the azide precursors 1a and 2b in the presence of copper sulphate as a catalyst and sodium ascorbate as a reducing agent for copper sulphate provided the desired isatin–triazole hybrid intermediates 3a and 3b in 89% and 85% yield, respectively (Scheme 1).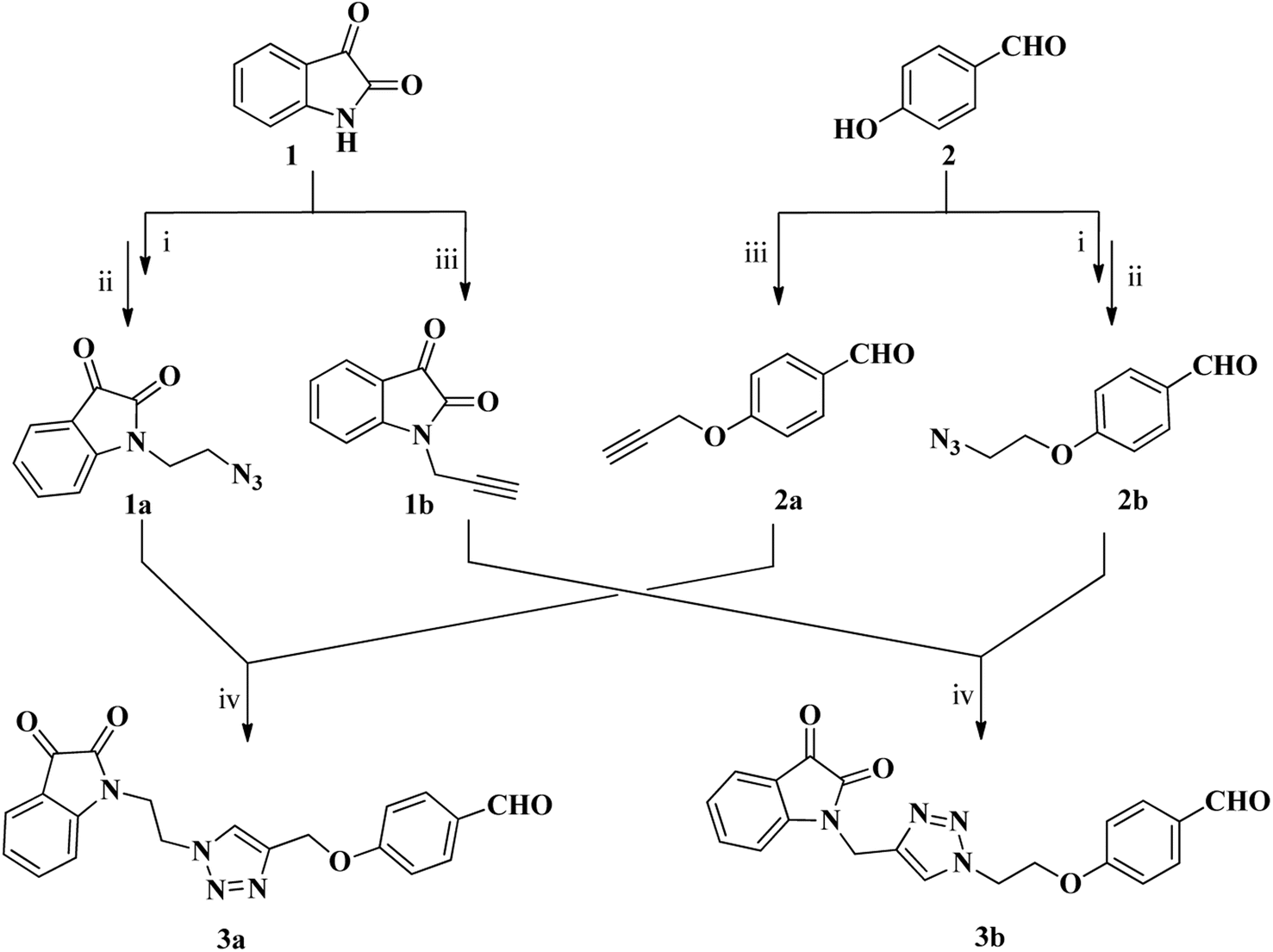 | ||
Scheme 1 Synthesis of isatin–triazole hybrids 3a and 3b; reagents and conditions: (i) dibromoethane (2.1 eq.), DMF, K2CO3 (ii) NaN3 (1.2 eq.), DMF(iii) propargyl bromide, DMF, K2CO3 (iv) CuSO4·5H2O, sodium ascorbate, EtOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (9:1), rt. :H2O (9:1), rt. | ||
In the next step, series of mono and bis-thiosemicarbazones 4a–h and 5a–h (Scheme 2) were synthesized from isatin–triazole hybrid intermediates 3a and 3b by using suitable N4-substituted thiosemicarbazides in different stoichiometric ratios. The synthesis of mono-thiosemicarbazone derivatives 4a–h was achieved by stirring 3a and respective N4-substituted thiosemicarbazide (1.1 eq.) for 4–8 h in ethanol solvent at room temperature. Similarly, bis-thiosemicarbones of isatin–triazole hybrids 5a–h were obtained by reacting 3a and 3b with 2.1 equivalents of suitable N4-substituted thiosemicarbazide under reflux conditions (Scheme 2).
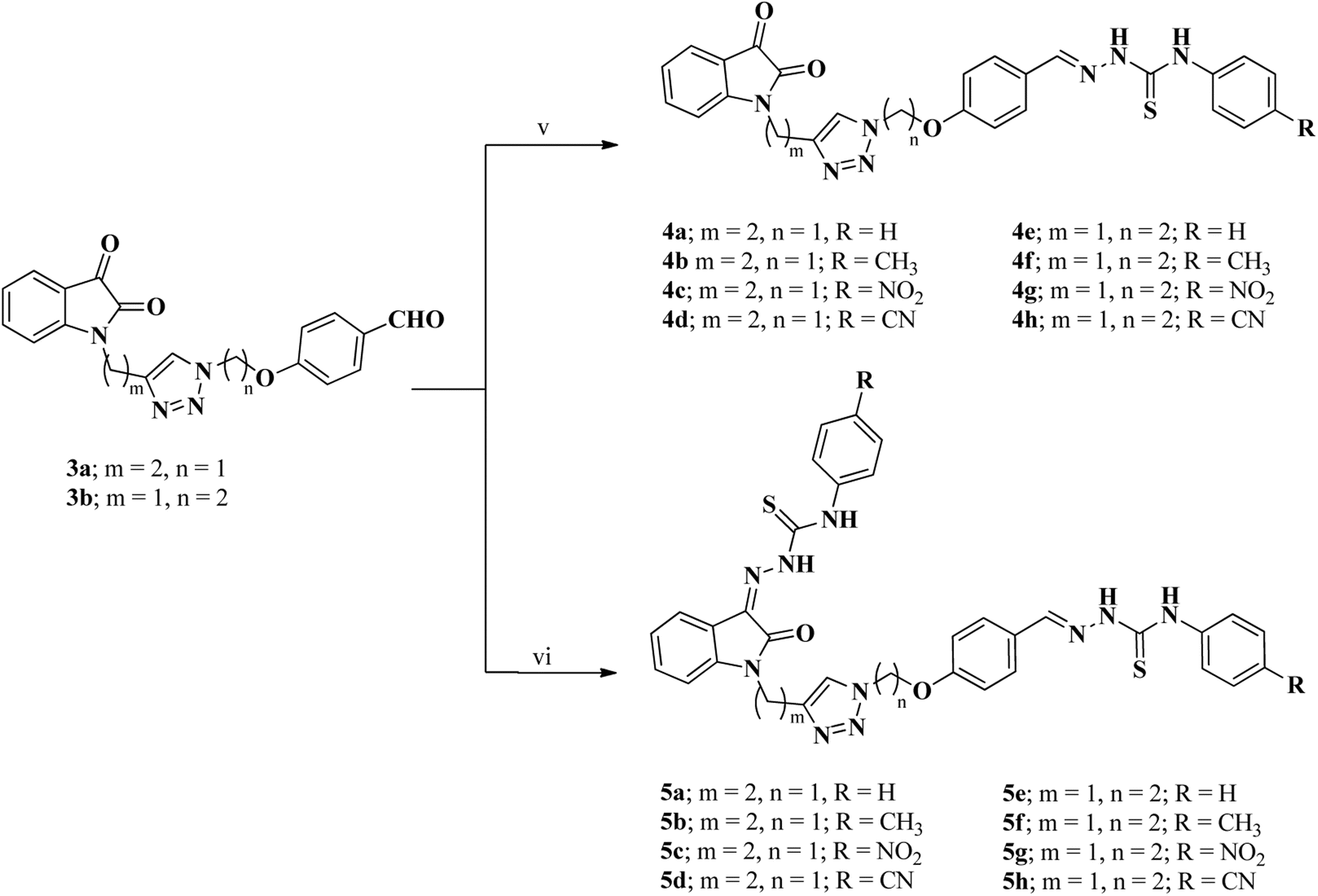 | ||
| Scheme 2 Synthesis of mono and bis-thiosemicarbazones (4a–h and 5a–h) of isatin–triazole hybrids (3a and 3b); reagents and conditions: (v) N4-substituted thiosemicarbazides (1.1 eq.), EtOH, rt; (vi) N4-substituted thiosemicarbazides (2.1 eq.), EtOH, CH3COOH (cat.), reflux. | ||
2.2 Characterization
The formation of isatin–triazole hybrid intermediates 3a & 3b and mono and bis-thiosemicarbazones 4a–h & 5a–h was confirmed by their FT-IR, 1H & 13C NMR and LC-MS data. In the FT-IR spectrum, the absence of the absorption peaks for azide and alkyne functional groups and the presence of three absorption bands at (1732 cm−1, 1692 cm−1 and 1598 cm−1) and (1728 cm−1, 1673 cm−1 and 1600 cm−1) corresponding to the three carbonyls of the lactam, aldehyde and ketone functional groups clearly indicate the successful formation of intermediates 3a & 3b, respectively. Moreover, the presence of absorption bands for ether, methylene and aromatic rings in their respective regions also establishes the desired structures.In the 1H NMR spectrum, the only singlet present in the aromatic region at 8.25 ppm and 7.85 ppm, respectively corresponds to the single proton of triazole ring of 3a & 3b. The presence of two triplets in 1H NMR of 3a at 4.30 ppm (3J = 6 Hz) and 4.84 ppm (3J = 6 Hz) for two methylene protons attached to isatin and triazole, respectively and a singlet present downfield at 5.25 ppm for oxymethylene present next to triazole further support its formation. Similarly, two triplets for two methylene protons attached to triazole and phenoxy ring respectively appeared at 4.45 ppm and 4.80 ppm and a singlet for the methylene that links isatin and triazole appeared downfield at 5.05 ppm supports the formation of 3b. Finally, the presence of singlet at 9.91 ppm and 9.88 ppm, respectively for one proton integration corresponding to aldehyde proton of 3a & 3b, respectively confirms their formation. The 13C NMR spectrum showed two characteristic signals one at 124.4 ppm and 124.0 ppm respectively and a relatively less intense signal at 142.9 ppm and 141.9 ppm respectively due to C-5 and C-4 of triazole ring further confirms the formation of 1,4-disubstituted triazole ring in 3a & 3b. Moreover, three signals at (40.3 ppm, 47.4 ppm, 61.6 ppm) and (35.3 ppm, 49.7 ppm, 66.3 ppm), respectively due to two carbons of ethyl linker and one carbon of –OCH2 group also indicated the formation of desired products. The presence of carbonyl carbons of aldehyde and ketone at 182.9–190.6 ppm provided the final evidence for the formation of intermediates 3a & 3b. Finally, the peak at m/z 412 [M + 36]− for C20H16O4N4 in the LCMS spectrum also support the proposed structures of 3a & 3b.
Similarly, the presence of two signals for NH protons at 11.68 ppm and 9.98 ppm and a singlet at 8.25 ppm for the azomethine proton in 1H NMR spectrum of representative mono-thiosemicarbazone 4f clearly suggests the selective and successful condensation of thiosemicarbazide at aldehydic position. Moreover, a singlet at 2.30 ppm for methyl protons further confirms the formation of 4f. In contrast to the 1H NMR spectrum of 4f, the bis-thiosemicarbazone 5e shows four signals for NH protons at 12.76 ppm, 11.73 ppm, 10.89 ppm and 10.05 ppm and a singlet of azomethine proton at 8.27 ppm. In the 13C NMR spectrum, an upfield signal of 4f appeared at 142.9 ppm and two signals of 5e observed at 143.8 ppm and 142.9 ppm corresponding to imine moieties of thiosemicarbazones strongly support the formation of mono- and bis-thiosemicarbazones 4f and 5e respectively. The absence of signal for aldehydic carbonyl and both aldehyde and ketone carbonyls observed in the respective starting materials and the appearance of signal for thiocarbonyl at 176.2 ppm and two signals at 176.8 and 176.1 ppm for 4f and 5e respectively also support the formation of desired thiosemicarbazones. The final evidence for the formation of mono- and bis-thiosemicarbazones was obtained from their LCMS data. The observation of peaks at 539 m/z and 673 m/z respectively corresponding to [M]− clearly confirms the formation of 4f and 5e. All the other compounds of the series are characterized in a similar manner (see Experimental section for more details).
2.3 Computational study
The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) are two popular quantum chemical parameters.60 The energies of these molecular orbitals, also known as frontier molecular orbitals (FMOs), are important parameters for predicting the reactivity of a chemical species.61 These parameters are critical in molecular reactivity, because HOMO (highest energy orbital with electrons) can act as an electron donor, while LUMO orbital serves as an electron acceptor because it has a lower energy than the other orbitals and can accept electrons.62,63 The energies of the HOMO and LUMO orbital values are shown in Table 1. Higher EHOMO value of compound 3b indicated the greater tendency for electron donation to a suitable acceptor molecule with a low energy and empty molecular orbital.64 Negative EHOMO and ELUMO values, on the other hand, indicated the stability of the compounds under investigation.65,66 Quantum mechanical calculations67 (Table 2) have been used to investigate the properties of donors and acceptors in molecules. The LUMO–HOMO energy differences were measured, demonstrating that the energy gap represents the chemical behavior of a molecule.68 The ELUMO–HOMO energy gap separation was used to assess the kinetic stability of the compounds.69 Frontier molecular orbital energies along with the energy gap between HOMO and LUMO of compounds (3a–b, 4a–h and 5a–h) is given in Table 1.
| S. No. | Compound (R) | ELUMO (eV) | EHOMO (eV) | ΔE (eV) |
|---|---|---|---|---|
| 1 | 3a | 0.73 | −1.57 | 2.31 |
| 2 | 3b | 0.95 | −1.22 | 2.18 |
| 3 | 4a (H) | 0.76 | −4.91 | 5.68 |
| 4 | 4b (CH3) | 0.78 | −4.87 | 5.66 |
| 5 | 4c (NO2) | −0.36 | −0.51 | 0.14 |
| 6 | 4d (CN) | 0.52 | −5.21 | 5.74 |
| 7 | 4e (H) | 0.72 | −3.05 | 3.78 |
| 8 | 4f (CH3) | 0.82 | −3.04 | 3.86 |
| 9 | 4g (NO2) | −0.24 | −0.33 | 0.09 |
| 10 | 4h (CN) | 0.09 | −3.12 | 3.21 |
| 11 | 5a (H) | 0.84 | −5.01 | 5.85 |
| 12 | 5b (CH3) | 0.48 | −4.80 | 5.28 |
| 13 | 5c (NO2) | −1.76 | −3.58 | 1.82 |
| 14 | 5d (CN) | 0.68 | −5.65 | 6.34 |
| 15 | 5e (H) | 0.71 | −3.11 | 3.82 |
| 16 | 5f (CH3) | 0.84 | −3.18 | 4.02 |
| 17 | 5g (NO2) | −0.03 | −3.64 | 3.61 |
| 18 | 5h (CN) | 0.05 | −3.28 | 3.34 |
| S. No. | Compound | EAa | IPb | χc | ηd | Se | μf | ωg |
|---|---|---|---|---|---|---|---|---|
| a Electron affinity.b Ionization Energy.c Electronegativity.d Chemical hardness.e Chemical softness.f Chemical potential.g Electrophilicity. | ||||||||
| 1 | 3a | −0.73 | 1.57 | 0.42 | 1.15 | 0.43 | −0.42 | −0.07 |
| 2 | 3b | −0.95 | 1.22 | 0.13 | 1.09 | 0.45 | −0.13 | 0.07 |
| 3 | 4a | −0.76 | 4.91 | 2.07 | 2.84 | 0.17 | −2.07 | 0.75 |
| 4 | 4b | −0.78 | 4.87 | 2.04 | 2.83 | 0.17 | −2.04 | 0.73 |
| 5 | 4c | 0.36 | 0.51 | 0.43 | 0.08 | 6.25 | −0.43 | 1.15 |
| 6 | 4d | −0.52 | 5.21 | 2.34 | 2.87 | 0.17 | −2.34 | 0.95 |
| 7 | 4e | −0.72 | 3.05 | 1.16 | 1.89 | 0.26 | −1.16 | 0.35 |
| 8 | 4f | −0.82 | 3.04 | 1.11 | 1.93 | 0.25 | −1.11 | 0.31 |
| 9 | 4g | 0.24 | 0.33 | 0.28 | 0.04 | 12.5 | −0.28 | 0.98 |
| 10 | 4h | −0.09 | 3.12 | 1.51 | 1.61 | 0.31 | −1.51 | 0.70 |
| 11 | 5a | −0.84 | 5.01 | 2.08 | 2.92 | 0.17 | −2.08 | 0.74 |
| 12 | 5b | −0.48 | 4.80 | 2.15 | 2.64 | 0.18 | −2.15 | 0.87 |
| 13 | 5c | 1.76 | 3.58 | 2.67 | 0.91 | 0.54 | −2.67 | 3.91 |
| 14 | 5d | −0.68 | 5.65 | 2.48 | 3.17 | 0.15 | −2.48 | 0.97 |
| 15 | 5e | −0.71 | 3.12 | 1.20 | 1.91 | 0.26 | −1.20 | 0.37 |
| 16 | 5f | −0.84 | 3.18 | 1.17 | 2.01 | 0.24 | −1.17 | 0.34 |
| 17 | 5g | 0.03 | 3.64 | 1.83 | 1.80 | 0.27 | −1.83 | 0.93 |
| 18 | 5h | −0.05 | 3.28 | 1.61 | 1.67 | 0.29 | −1.61 | 0.77 |
FMOs are studied in order to investigate the bioactivity and catalytic behavior of the compounds.17,70 According to FMO theory, both HOMO and LUMO are important factors in understanding electronic transitions, molecular reactivities, and intermolecular interactions, and thus provide insight into bioactivity.71–73 Furthermore, the presence of FMOs on the same side of the molecule significantly reduces biological activity.74 The distribution patterns of FMOs (HOMOs and LUMOs) of synthesized triazole hybrids at ground states are shown in Fig. 2. The green color represents a low electron density while the red represents the high electron density.75 The addition of substituents to benzene ring has little effect on the pi-electron cloud of HOMO and LUMO. Interestingly, the energy gap (ELUMO–HOMO) values of the synthesized derivatives of hybrid 3b differ only slightly, with all derivatives falling within the range of 0.09–4.02 eV while those for hybrid 3a derivatives lies in the range of 0.14–6.34 eV. Results showed that incorporating nitro substituents on the phenyl ring lowers the energy separation gap (ELUMO–HOMO). In fact, nitro compounds 4c, 4g and 5c (0.14, 0.09, 1.82 eV, respectively) are found to be the lowest among all the synthesized derivatives (see Table 1). The lower ELUMO–HOMO corresponds to the greater the biological potential,17 by increasing the chemical reactivity and catalytic activity of these synthesized hybrids. Recent studies indicated that molecules with an FMO energy gap of less than 2.5 eV are more likely to bind to specific protein targets.76 Herein, compounds 3b, 4c, 4g and 5c displayed the FMO energy gap of less than 2.2 eV as shown in Fig. 2, hence expected to show the best binding interactions with the biological targets.
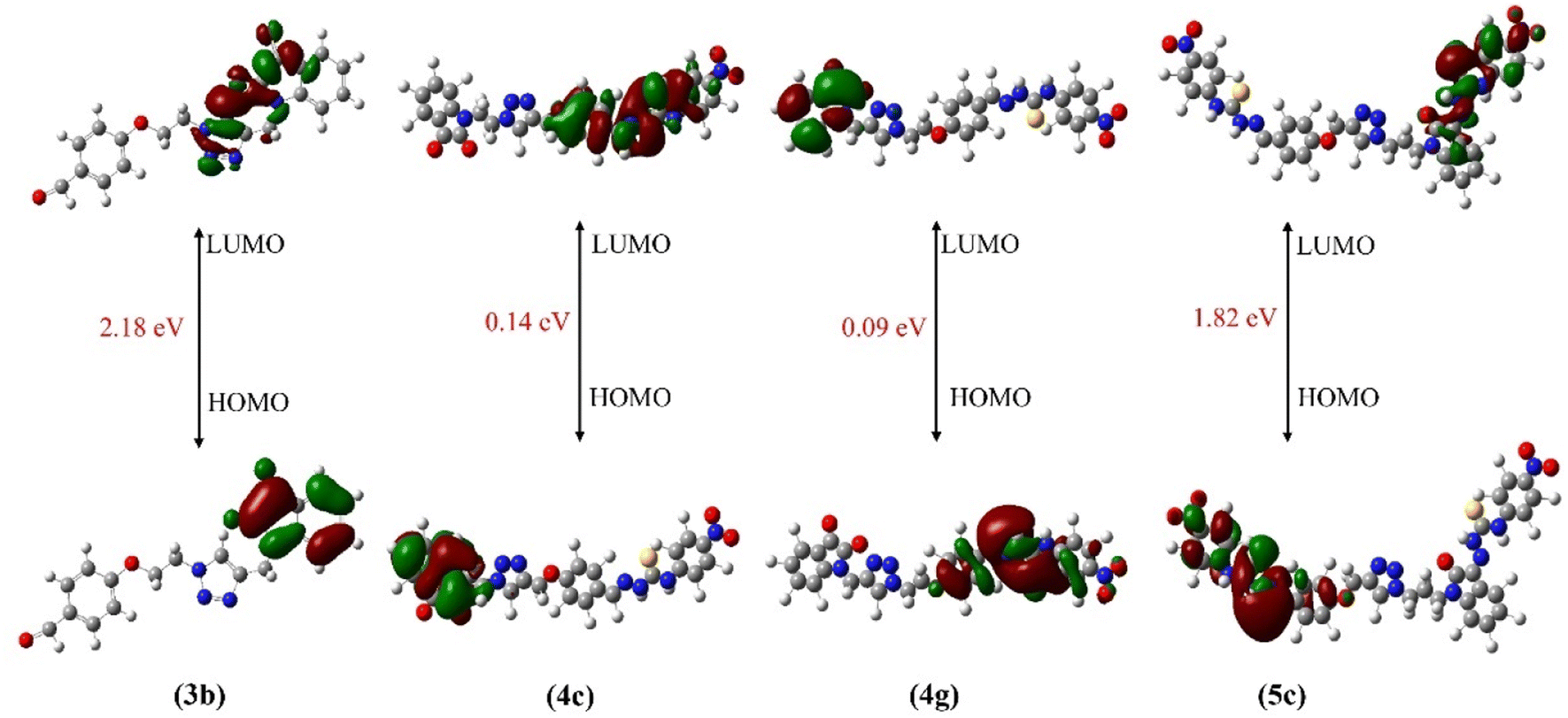 | ||
| Fig. 2 HOMO and LUMO of 3b, 4c, 4g and 5c. | ||
Beside the evaluation of quantum chemical parameters77 i.e. frontier molecular orbitals and separation energies Δ Egap; global reactivity descriptors78 such as ionization potentials (IP = −EHOMO), electron affinity (EA = −ELUMO), electronegativities [χ = (IP + EA)/2], chemical potentials (μ = −χ), chemical hardness [η = (IP − EA)/2], chemical softness (S = 1/2η), global electrophilicity or electrophilicity index (ω = μ2/2η) have also been calculated.79–82 Chemical descriptors are used to analyze drug properties and interactions with biological targets.83–85 Results showed that chemically harder and more stable molecules have a larger LUMO–HOMO energy gap than softer and less stable molecules.62 It was also observed that chemically reactive molecules showed greater chemical potential.86 The lower LUMO value corresponds to the higher electron acceptance ability,87 and the compound 5c showed the highest electron affinity value among the series (Table 2). Similarly, the higher HOMO value corresponds to the higher electron donor ability,88 and the compound 4g displayed the least value of ionization potential, thus indicating the highest electron donor ability of the compound among the synthesized derivatives (Table 2).
Electronegativity refers to a ligand's ability to bind to a protein, which is important for successful binding.89 A direct relationship was observed between electronegativity values (χ) values and tendency to accept electrons i.e. electron affinity (Table 2). Results showed that compound 5c (bisthiosemicarbazone having two nitro substituents) (Table 2) displayed the highest electronegativity value, thus making it more susceptible to accept electrons, through the electron withdrawing effect, hence showing its higher catalytic ability and binding affinity with the target protein.90–92
The electrophilicity index (ω) indicates the compound's ability to interact with electron-rich sites of biological targets and measures the tendency to accept electrons from the environment.93 The most stable compound is the one with a low electrophilicity value i.e. 3a among the studied substrates (Table 2). As we moved from electron donating to electron withdrawing substituents in the substrate, the electrophilicity index value increased. Compound 5c having the p-nitro substituent displayed the highest electrophilicity index value and is found to the most reactive among the series (Table 2), thus showed greater potential to bind with the biological target. Compounds with a higher electrophilicity index value are more reactive and less stable, whereas compounds with a lower electrophilicity index value are more stable and less reactive.94 Furthermore, lower chemical potential (μ) value makes a substituent more electrophilic (electron-seeking), and in this case, the value for the compound 5c having the p-nitro substituent is the lowest i.e. −2.67 eV among the series (Table 2).
Hardness refers to a resistance of compound to electron donation or acceptance and plays an important role in water solubility.89 Overall range of global hardness (η) was found to be 0.04–0.17 eV while the hardest character was shown by the compound 5d within the series (Tabe 2). Softness, the inverse of hardness, refers to a compound's ability to donate or accept electrons, as well as their reactivity with biological targets.95 Considering the soft nature of the synthesized compounds, the compound 4g having nitro substituent showed the highest value of chemical softness (S) (Table 2). Compound 4g being the most reactive compound among the series (Table 2), appeared as a favorable candidate for efficient applications in different synthetic routes and further theoretical calculations, possibly due to the extended conjugation and electron withdrawing nature of nitro group. Moreover, the lowest hardness (η) and the highest softness (S) value of compound 4g (Table 2) indicate that it is less likely to undergo rapid structural changes during binding, which is crucial for stable protein interactions. Concludingly, the DFT studies provided comprehensive analysis of theoretical structure stability and mechanistic explanations of chemical properties of the synthesized compounds for their ability to interact with biological targets.
PASS online (prediction of activity of spectra for substances)100 predicted the profound anticancer potential of synthesized thiosemicarbazones of isatin triazole hybrids. Phosphoinositide 3-kinase PI3K protein was chosen for this study because of its well-known roles in regulating cell growth and survival, particularly in cancer.89,101 The selected active site of the PI3K inhibitor of PDB ID (4TV3)102,103 was downloaded from RCSB104 protein data bank. The structure of desired PI3K protein was prepared for docking using AutoDock Tools.105,106
The primary goal of the molecular docking studies was to evaluate the binding potential of the synthesized compounds as well as molecular interaction with various amino acid residues of the membrane protein PI3K. The Autodock Vina 1.5.7 (ref. 99) was used to obtain docking simulations and the visualization of protein-ligand complex was performed using the Discovery Studio.107 The most stable anchoring conformations having the lowest binding energy values, as well as interacting residues were visualized through three-dimensional and two-dimensional models, as depicted in Fig. 3, which were created using the Discovery Studio Visualizer.108 It has been demonstrated that receptors have active sites that are capable to behave both as hydrogen bond donor and acceptor as well.75
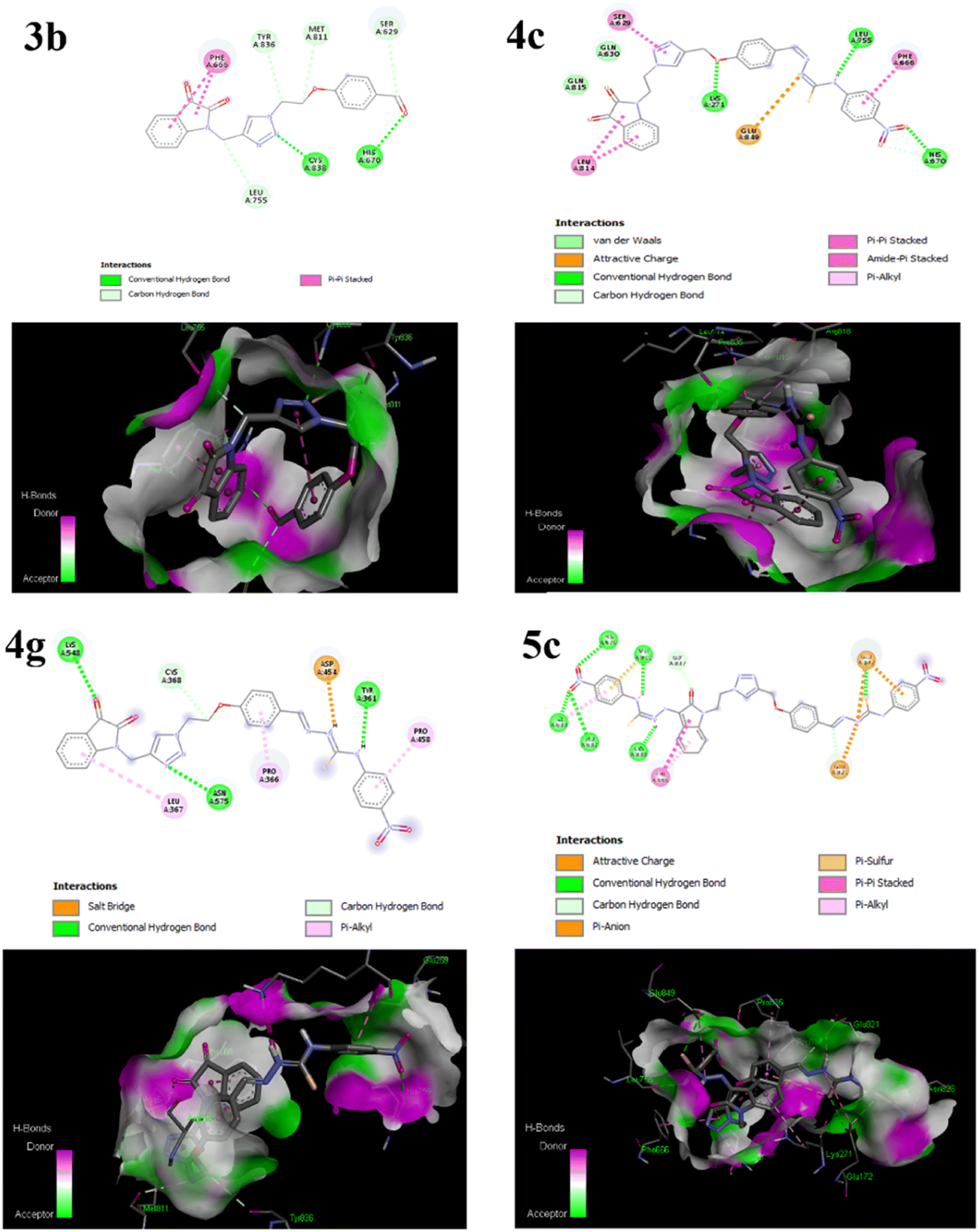 | ||
| Fig. 3 Diagrammatic representation of molecular interactions of the ligand molecules (3b, 4c, 4g and 5c) with various amino acid residues in the binding pocket of the protein PI3K. | ||
The 2D representation in Fig. 3 helps to clearly visualize the types of interactions including hydrophobic interactions, hydrogen bonding, donor atoms, acceptor atoms, and π–π stacking interactions between the studied compounds and the cancer protein's active sites. Table 3 shows the binding energy of enzymes on the ligand, hydrostatic interactions, and hydrogen bonding in the selective host–guest systems. Results displayed the following decreasing order of binding affinity score for the studied compounds 5c > 4g > 4c > 3b (Table 3).
| S. No. | Drug name (ligand) | Binding energy (kcal mol−1) | Hydrogen bond interaction | Hydrostatic interaction with amino acids |
|---|---|---|---|---|
| 1 | 3b | −8.3 | Cys 838(A), His 670(A), Ser 629(A), Met 811(A), Tyr 836(A) | Leu 755(A), Phe 666(A) |
| 2 | 4c | −8.6 | Gln 630 (A), Gln 815 (A), Lys 271 (A), Leu 755 (A), His 670 (A) | Phe 666(A), Leu 814(A), Glu 849 (A), Ser 629 (A) |
| 3 | 4g | −8.9 | Lys 548(A), Asn 575(A), Tyr 361(A) | Leu 367(A), Cys 368(A), Pro 366(A), Asp 454(A), Pro 458(A) |
| 4 | 5c | −10.3 | His 670(A), Met 811(A), Ile 633(A), Leu 632(A), Cys 838(A) | Phe 666(A), Gly 837(A), Glu 172(A), Glu 821(A) |
Among the selected compounds, the compound 5c having two nitro substituents displayed the highest binding energy score of −10.3 kcal mol−1. The amino His 670(A), Met 811(A), Ile 633(A), Leu 632(A), Cys 838(A) formed hydrogen bonding with the nitrogen of –NH of thiosemicarbazones and oxygen of nitro substituents in compound 5c (Fig. 3). Moreover, carbon–hydrogen bond formation was observed with Gly 837(A) and the amino acids Glu 172(A), Glu 821(A) showed π-sulphur and π-anion interactions with the ligand and the amino acid Phe 666(A) formed π-alkyl and π–π stacking interactions with the benzene ring. Similarly, the compound 4g which is 4-nitro substituted mono-thiosemicarbazone derivative of isatin triazole hybrid 3b displayed the second best binding score of −8.9 kcal mol−1, alongwith several binding interactions such as hydrogen bonding of carbonyl oxygen of isatin, nitrogen of 1,2,3-triazole and –NH of thiosemicarbazone moiety with the Lys 548(A), Asn 575(A), Tyr 361(A) residues respectively (Table 3). Furthermore, carbon hydrogen bond, π-alkyl and salt bridge interactions with the active site residues of PI3K protein were also depicted in 2D model (Fig. 3) of compound 4g.
The docking scores suggested that isatin–triazole hybrid 3b posses lesser binding affinity with target protein, as compared to its 4-nitro substituted mono-thiosemicarbone derivative 4g. While the 4-nitro substituted bis-thiosemicarbone derivative 5c displayed even superior binding affinity with the target protein. These results reveal the importance of substitution on benzene ring and presence of thiosemicarzaone scaffold on the isatin–triazole hybrid for enhanced binding interaction of the drug molecules which is also supported by the literature.22,109 Molecular docking results are in good agreement with the findings of DFT analyses and these computational explanations suggested that the synthesized thiosemicarbazones of isatin–triazole hybrids may act against PI3K as anti-cancer agents.
3. Conclusions
A series of mono- and bis-thiosemicarbazones have been synthesized starting from two novel isatin–triazole hybrid intermediates, in turn obtained through the Cu(I)-sodium ascorbate catalysed click reaction, in excellent yield. All the synthesized compounds are characterized by their spectral (FT-IR, 1H & 13C NMR, LCMS) and analytical data. The DFT studies carried out on these compounds showed that the compound 5c (4-nitro-substituted bis-thiosemicarbazone) exhibited the highest electron affinity, electronegativity and electrophilicity index values among the series, hence exhibiting the greater potential to bind with the target protein. The outcome of DFT studies was further validated by molecular docking analysis against the active sites of the phosphoinositide 3-kinases (PI3Ks), indicating the potential of compound 5c (block the active site of targeted enzyme with the lowest binding score of −10.3 kcal mol−1) to be studied as an anticancer agent.4. Experimental
4.1 Chemicals and materials
The used chemicals and materials were supplied as ESI.†4.2 Computational details
The molecular structure of all the synthesized compounds were optimized using Gaussian 09 Package.110 The optimized structures were used to calculate the electronic properties of the synthesized compounds. The frontier molecular orbitals HOMO and LUMO alongwith other descriptors were calculated using Gauss View to understand the effect of substituents on the reactivity of compounds. For molecular docking studies the Autodock Vina 1.5.7 (ref. 99) was used for docking simulations, and the Discovery Studio107 was used for visualization.4.3 Synthesis of 4-((1-(2-(2,3-dioxoindolin-1-yl)ethyl)-1H-1,2,3-triazol-4-yl)-methoxy)benzaldehyde (3a)
For the synthesis of 3a, the reported conditions55 of the click reaction were employed. In a stirred solution of the 1-(2-azidoethyl)isatin 1a (0.10 g, 0.46 mmol) and 4-(prop-2-ynyloxy)benzaldehyde 2a (0.07 g, 0.46 mmol) in 10 mL ethanol:water (9:1), addition of copper sulphate (0.00625 g, 0.025 mmol) was followed by sodium ascorbate (0.013 g, 0.066 mmol) addition at room temperature. Upon the completion of the reaction, as indicated by the TLC (n-hexane: EtOAc, 1:1), ice-cold water was added to the reaction mixture and the precipitates formed were filtered out and dried. Recrystallization from ethanol was performed to obtain the purified product.
Appearance: bright orange solid; yield: 89%; melting point: 157–160 °C; Rf: 0.53 (n-hexane: EtOAc, 1:1); FT-IR ![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) (cm−1): 3129, 3049 (Csp2–H stretch), 2928, 2873 (Csp3–H stretch), 2823, 2724 (Csp2–H stretch, aldehyde), 1731 (C
(cm−1): 3129, 3049 (Csp2–H stretch), 2928, 2873 (Csp3–H stretch), 2823, 2724 (Csp2–H stretch, aldehyde), 1731 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch, ketone), 1692 (CO stretch, aldehyde), 1598 (CO stretch, lactam), 1509 (CC stretch, aromatic ring), 1471 (Csp3–H bend, methylene), 1258 (Csp2–O stretch, ether), 1217 (C–N stretch, lactam), 1160 (Csp3–O stretch, ether), 810 (C–H bend, 1,4-disubstitution), 754 (C–H bend, 1,2-disubstitution); 1H NMR (300 MHz, CDCl3) δ (ppm): 9.91 (s, 1H, –CHO), 8.25 (s, 1H, triazole-H), 7.88 (d, 3J = 8.7 Hz, 2H, Ar–H), 7.52 (d, 3J = 7.3 Hz, 1H, Ar–H), 7.43 (t, 3J = 7.5 Hz, 1H, Ar–H), 7.10–7.04 (t, 1H, 3J = 7.3 Hz, Ar–H), 7.06–7.03 (d, 2H, 3J = 8.7 Hz, Ar–H), 6.84 (d, 3J = 7.9 Hz, 1H, Ar–H), 5.25 (s, 2H, –OCH2), 4.84 (t, 3J = 6 Hz, 2H, –CH2-triazole),4.30 (t, 3J = 6 Hz, 2H, –CH2-isatin); 13C NMR (75 MHz, CDCl3) δ (ppm): 190.3 (CO aldehyde), 182.9 (CO ketone), 163.3, 158.1(CO lactum), 150.6, 142.9 (C-5 triazole), 138.1, 131.6, 130.4, 125.0, 124.4 (C-4 triazole), 123.4, 117.6, 115.1, 109.9, 61.6 (–CH2 phenoxy), 47.4 (–CH2 triazole), 40.3 (–CH2 isatin); LC-MS m/z 412 [M + 36]; anal. calcd for C20H16N4O4: C, 63.83; H, 4.29; N, 14.89; O, 17.00; found: C, 63.98; H, 4.15; N, 14.93.
O stretch, ketone), 1692 (CO stretch, aldehyde), 1598 (CO stretch, lactam), 1509 (CC stretch, aromatic ring), 1471 (Csp3–H bend, methylene), 1258 (Csp2–O stretch, ether), 1217 (C–N stretch, lactam), 1160 (Csp3–O stretch, ether), 810 (C–H bend, 1,4-disubstitution), 754 (C–H bend, 1,2-disubstitution); 1H NMR (300 MHz, CDCl3) δ (ppm): 9.91 (s, 1H, –CHO), 8.25 (s, 1H, triazole-H), 7.88 (d, 3J = 8.7 Hz, 2H, Ar–H), 7.52 (d, 3J = 7.3 Hz, 1H, Ar–H), 7.43 (t, 3J = 7.5 Hz, 1H, Ar–H), 7.10–7.04 (t, 1H, 3J = 7.3 Hz, Ar–H), 7.06–7.03 (d, 2H, 3J = 8.7 Hz, Ar–H), 6.84 (d, 3J = 7.9 Hz, 1H, Ar–H), 5.25 (s, 2H, –OCH2), 4.84 (t, 3J = 6 Hz, 2H, –CH2-triazole),4.30 (t, 3J = 6 Hz, 2H, –CH2-isatin); 13C NMR (75 MHz, CDCl3) δ (ppm): 190.3 (CO aldehyde), 182.9 (CO ketone), 163.3, 158.1(CO lactum), 150.6, 142.9 (C-5 triazole), 138.1, 131.6, 130.4, 125.0, 124.4 (C-4 triazole), 123.4, 117.6, 115.1, 109.9, 61.6 (–CH2 phenoxy), 47.4 (–CH2 triazole), 40.3 (–CH2 isatin); LC-MS m/z 412 [M + 36]; anal. calcd for C20H16N4O4: C, 63.83; H, 4.29; N, 14.89; O, 17.00; found: C, 63.98; H, 4.15; N, 14.93.
4.4 Synthesis of 4-(2-(4-((2,3-dioxoindolin-1-yl)methyl)-1H-1,2,3-trizol-1-yl)-ethoxy)-benzaldehyde (3b)
For the synthesis of 3b, reported reaction conditions for click reaction were utilized. Copper sulphate (0.04 g, 0.16 mmol) and sodium ascorbate (0.08 g, 0.23 mmol) were added in succession, to the stirred solution of previously synthesized 1-(prop-2-ynyl)indoline-2,3-dione 1b (0.20 g, 1.08 mmol) and 4-(2-azidoethoxy)benzaldehyde 2b (0.21 g, 1.08 mmol) in 10 mL ethanol: water (9:1) at room temperature. Progress of reaction was monitored by TLC (n-hexane: EtOAc, 1:1).After the reaction completion, ice cold water (15 mL) was added to the reaction mixture and the precipitates formed were filtered off, dried and recrystallized from ethanol to obtain the pure product.
Appearance: orange solid; yield: 85%; melting point: 168–170 °C; Rf: 0.52 (n-hexane: EtOAc, 1:1); FT-IR (cm−1): 3141, 3031 (Csp2–H stretch), 2970, 2942 (Csp3–H stretch), 2847, 2759 (Csp2–H stretch, aldehyde), 1728 (CO stretch, ketone), 1673 (CO stretch, aldehyde), 1600 (CO stretch, lactam), 1508 (CC stretch, aromatic), 1469 (Csp3–H bend, methylene), 1244 (Csp2–O stretch, ether), 1213 (C–N stretch, lactam), 1159 (Csp3–O stretch, ether), 831 (C–H bend, 1,4-disubstitution), 755 (C–H bend, 1,2-disubstitution); 1H NMR (300 MHz, CDCl3) δ (ppm): 9.88 (s, 1H, –CHO) 7.85 (s, 1H, triazole-H) 7.81 (d, 3J = 7.5 Hz, 2H, Ar–H), 7.57 (m, 2H, Ar–H), 7.31 (d, 3J = 8.1 Hz, 2H, Ar–H), 7.11 (t, 3J = 7.5 Hz, 1H, Ar–H), 6.95 (d, 3J = 8.7 Hz, 2H, Ar–H), 5.03 (s, 2H, –CH2-isatin), 4.80(t, 3J = 4.2 Hz, 2H, –CH2-trizaole), 4.45 (t, 3J = 4.2 Hz, 2H, –OCH2); 13C NMR (75 MHz, CDCl3) δ (ppm): 190.6 (CO aldehyde), 183.0 (CO ketone), 162.4, 157.9 (CO lactum), 150.1, 141.9 (C-5 triazole), 138.6, 132.0, 130.7, 125.3, 124.2 (C-4 triazole), 124.0, 117.5, 114.7, 111.4, 66.3 (–CH2 phenoxy), 49.7 (–CH2 triazole), 35.3 (–CH2 isatin); LC-MS m/z 412 [M + 36]; anal. calcd for C20H16N4O4: C, 63.83; H, 4.29; N, 14.89; O, 17.00; found: C, 63.92; H, 4.41; N, 14.74.
4.5 General procedure for the synthesis of isatin–triazole derived mono-thiosemicarbazones (4a–h)
For the synthesis of mono-thiosemicarbazones 4a–h of two different types of isatin–triazole hybrids 3a and 3b, same procedure was followed. To the stirred ethanolic solution of isatin–triazole hybrid (0.1 g, 0.27 mmol), suitable N4-substituted thiosemicarbazide 3c (1.1 eq., 0.30 mmol) was added under vigorous stirring. The reaction was carried out at room temperature for 6 hours and progress of reaction was monitored after regular intervals by TLC (n-hexane: ethyl acetate 7:3). The precipitates formed upon completion, were filtered, washed, dried and recrystallized when required to obtain the pure product.
By following the above procedure, a series of mono-thiosemicarbazone derivatives 4a–h having different thiosemicarbazide moieties were synthesized.
:6); FT-IR (cm−1):3300 (N–H stretch), 3129, 3049 (Csp2–H stretch), 2928, 2873 (Csp3–H stretch), 1737 (CO stretch, ketone), 1600 (CO stretch, lactam), 1525 (CN stretch, imine),1508 (CC stretch, aromatic ring), 1467 (Csp3–H bend, methylene), 1274 (Csp2–O stretch, ether), 1201 (C–N stretch, lactam), 1165 (Csp3–O stretch, ether), 833 (C–H bend, 1,4-disubstitution), 755 (C–H bend, 1,2-disubstitution); 1H NMR (300 MHz, DMSO-d6) δ (ppm): 11.82 (s, 1H, –NH), 9.98 (s, 1H, –NH), 8.38 (s, 1H, –CHN), 7.36 (s, 1H, –CH triazole), 8.37–6.89 (m, 13H, Ar–H), 5.13 (s, 2H, –CH2 isatin), 4.71 (t, 2H, 3J = 6 Hz, –OCH2), 4.14 (t, 2H, 3J = 6 Hz, –CH2 triazole); 13C NMR (75 MHz, DMSO-d6) δ (ppm): 183.4, 161.8 (CS), 160.2, 158.6, 150.6, 149.6, 142.9 (CN azomethine), 139.7, 138.6, 129.6, 129.4, 127.3, 125.9, 124.9, 124.1, 123.7, 117.8, 115.5, 110.7, 61.6, 47.4, 35.4; LC-MS m/z 525 [M−]; anal. calcd for C27H23N7O3S: C, 61.70; H, 4.41; N, 18.66; found: C, 61.63; H, 4.29; N, 18.77.:6); FT-IR (cm−1):3300 (N–H stretch), 3129, 3049 (Csp2–H stretch), 2928, 2873 (Csp3–H stretch), 1731 (CO stretch, ketone), 1612 (CO stretch, lactam), 1541 (CN stretch, imine),1506 (CC stretch, aromatic ring), 1469 (Csp3–H bend, methylene), 1271 (Csp2–O stretch, ether), 1214 (C–N stretch, lactam), 1170 (Csp3–O stretch, ether), 815 (C–H bend, 1,4-disubstitution), 756 (C–H bend, 1,2-disubstitution); 1H NMR (300 MHz, DMSO-d6) δ (ppm): 11.68 (s, 1H, –NH), 9.98 (s, 1H, –NH), 8.25 (s, 1H, –CHN), 8.07 (s, 1H, –CH triazole), 7.81–6.89 (m, 12H, Ar–H), 4.97 (s, 2H, –CH2 isatin), 4.74 (t, 2H, 3J = 6 Hz, –OCH2), 4.42 (t, 2H, 3J = 6 Hz, –CH2 triazole), 2.31 (s, 3H, –CH3); 13C NMR (75 MHz, DMSO-d6) δ (ppm): 183.5, 176.2 (CS), 159.7, 158.2, 150.6, 142.9 (CN azomethine), 141.9, 138.5, 137.0, 134.8, 129.7, 129.3, 128.9, 127.5, 126.3, 124.9, 124.7, 123.8, 118.0, 115.4, 115.1, 111.6, 66.6, 49.5, 35.4, 21.0 (CH3); LC-MS m/z 539 [M−]; anal. calcd for C20H16N4O4: C, 63.83; H, 4.29; N, 14.89; found: C, 63.88; H, 4.17; N, 14.96.:6); FT-IR (cm−1):3300 (N–H stretch), 3129, 3049 (Csp2–H stretch), 2928, 2873 (Csp3–H stretch), 1732 (CO stretch, ketone), 1683 (CO stretch, lactam), 1600 (CN stretch, imine), 1543 (N–O stretch, asymmetric, nitro), 1506 (CC stretch, aromatic ring), 1465 (Csp3–H bend, methylene), 1328 (N–O stretch, symmetric, nitro), 1249 (Csp2–O stretch, ether), 1203 (C–N stretch, lactam), 1166 (Csp3–O stretch, ether), 831 (C–H bend, 1,4-disubstitution), 758 (C–H bend, 1,2-disubstitution); 1H NMR (300 MHz, DMSO-d6) δ (ppm): 12.03 (s, 1H, –NH), 9.98 (s, 1H, –NH), 8.42–6.87 (m, 15H, CHN, –CH triazole, Ar–H), 5.15 (s, 2H, –CH2 isatin), 4.68 (t, 2H, 3J = 6 Hz, –OCH2), 4.14 (t, 2H, 3J = 6 Hz, –CH2 triazole); 13C NMR (75 MHz, DMSO-d6) δ (ppm): 183.4, 161.7 (CS), 160.2, 158.6, 150.6, 149.6, 142.9 (CN azomethine), 139.6, 138.6, 129.6, 129.4, 127.4, 125.9, 124.9, 124.1, 123.7, 117.8, 115.5, 110.6, 61.6, 47.4, 35.4; LC-MS m/z 570 [M−]; anal. calcd for C27H22N8O5S: C, 56.84; H, 3.89; N, 19.64; found: C, 56.78; H, 3.69; N, 19.47.:6); FT-IR (cm−1):3300 (N–H stretch), 3129, 3049 (Csp2–H stretch), 2928, 2873 (Csp3–H stretch), 2250 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretch, cyano) 1735 (CO stretch, ketone), 1606 (CO stretch, lactam), 1537 (CN stretch, imine),1510 (CC stretch, aromatic ring), 1467 (Csp3–H bend, methylene), 1249 (Csp2–O stretch, ether), 1195 (C–N stretch, lactam), 1169 (Csp3–O stretch, ether), 837 (C–H bend, 1,4-disubstitution), 752 (C–H bend, 1,2-disubstitution); 1H NMR (300 MHz, DMSO-d6) δ (ppm): 11.68 (s, 1H, –NH), 9.99 (s, 1H, –NH), 8.29–6.89 (m, 14H, –CHN, –CH triazole Ar–H), 4.97 (s, 2H, –CH2 isatin), 4.74 (t, 2H, 3J = 6 Hz, –OCH2), 4.42 (t, 2H, 3J = 6 Hz, –CH2 triazole); 13C NMR (75 MHz, DMSO-d6) δ (ppm): 183.5, 176.2 (CS), 159.7, 158.2, 150.6, 142.9 (CN azomethine), 141.9, 138.5, 137.0, 134.8, 129,7, 129.3, 128.9, 127.5, 126.3, 124.9, 124.7, 123.8, 118.0 (CN), 115.4, 115.1, 111.6, 66.6, 49.5, 35.4; LC-MS m/z 550 [M−]; anal. calcd for C28H22N8O3S: C, 61.08; H, 4.03; N, 20.35; found: C, 60.97; H, 3.89; N, 20.18.:6); FT-IR (cm−1):3300 (N–H stretch), 3129, 3049 (Csp2–H stretch), 2928, 2873 (Csp3–H stretch), 1737 (CO stretch, ketone), 1600 (CO stretch, lactam), 1525 (CN stretch, imine),1508 (CC stretch, aromatic ring), 1467 (Csp3–H bend, methylene), 1274 (Csp2–O stretch, ether), 1201 (C–N stretch, lactam), 1165 (Csp3–O stretch, ether), 833 (C–H bend, 1,4-disubstitution), 755 (C–H bend, 1,2-disubstitution); 1H NMR (300 MHz, DMSO-d6) δ (ppm): 11.82 (s, 1H, –NH), 9.98 (s, 1H, –NH), 8.38 (s, 1H, –CHN), 7.36 (s, 1H, –CH triazole), 8.37–6.89 (m, 13H, Ar–H), 5.13 (s, 2H, –CH2 isatin), 4.71 (t, 2H, 3J = 6 Hz, –OCH2), 4.14 (t, 2H, 3J = 6 Hz, –CH2 triazole); 13C NMR (75 MHz, DMSO-d6) δ (ppm): 183.4, 161.8 (CS), 160.2, 158.6, 150.6, 149.6, 142.9 (CN azomethine), 139.7, 138.6, 129.6, 129.4, 127.3, 125.9, 124.9, 124.1, 123.7, 117.8, 115.5, 110.7, 61.6, 47.4, 35.4; LC-MS m/z 525 [M−]; anal. calcd for C27H23N7O3S: C, 61.70; H, 4.41; N, 18.66; found: C, 61.56; H, 4.32; N, 18.43.:6); FT-IR (cm−1):3300 (N–H stretch), 3129, 3049 (Csp2–H stretch), 2928, 2873 (Csp3–H stretch), 1731 (CO stretch, ketone), 1612 (CO stretch, lactam), 1541 (CN stretch, imine), 1506 (CC stretch, aromatic ring), 1469 (Csp3–H bend, methylene), 1271 (Csp2–O stretch, ether), 1214 (C–N stretch, lactam), 1170 (Csp3–O stretch, ether), 815 (C–H bend, 1,4-disubstitution), 756 (C–H bend, 1,2-disubstitution); 1H NMR (300 MHz, DMSO-d6) δ (ppm): 11.68 (s, 1H, –NH), 9.98 (s, 1H, –NH), 8.25 (s, 1H, –CHN), 8.07 (s, 1H, –CH triazole), 7.81–6.89 (m, 12H, Ar–H), 4.97 (s, 2H, –CH2 isatin), 4.74 (t, 2H, 3J = 6 Hz, –OCH2), 4.42 (t, 2H, 3J = 6 Hz, –CH2 triazole), 2.31 (s, 3H, –CH3); 13C NMR (75 MHz, DMSO-d6) δ (ppm): 183.5, 176.2 (CS), 159.7, 158.2, 150.6, 142.9 (CN azomethine), 141.9, 138.5, 137.0, 134.8, 129.7, 129.3, 128.9, 127.5, 126.3, 124.9, 124.7, 123.8, 118.0, 115.4, 115.1, 111.6, 66.6, 49.5, 35.4, 21.0 (CH3); LC-MS m/z 539 [M−]; anal. calcd for C28H25N7O3S: C, 62.32; H, 4.67; N, 18.17; found: C, 62.13; H, 4.51; N, 18.01.:6); FT-IR (cm−1):3300 (N–H stretch), 3129, 3049 (Csp2–H stretch), 2928, 2873 (Csp3–H stretch), 1732 (CO stretch, ketone), 1683 (CO stretch, lactam), 1579 (CN stretch, imine), 1556 (N–O stretch, asymmetric, nitro), 1508 (CC stretch, aromatic ring), 1467 (Csp3–H bend, methylene), 1354 (N–O stretch, symmetric, nitro), 1244 (Csp2–O stretch, ether), 1219 (C–N stretch, lactam), 1165 (Csp3–O stretch, ether), 831 (C–H bend, 1,4-disubstitution), 758 (C–H bend, 1,2-disubstitution); 1H NMR (300 MHz, DMSO-d6) δ (ppm): 12.03 (s, 1H, –NH), 9.98 (s, 1H, –NH), 8.42–6.87 (m, 14H, CHN, –CH triazole, Ar–H), 5.15 (s, 2H, –CH2 isatin), 4.68 (t, 2H, 3J = 6 Hz, –OCH2), 4.14 (t, 2H, 3J = 6 Hz, –CH2 triazole); 13C NMR (75 MHz, DMSO-d6) δ (ppm): 183.4, 161.7 (CS), 160.2, 158.6, 150.6, 149.6, 142.9 (CN azomethine), 139.6, 138.6, 129.6, 129.4, 127.4, 125.9, 124.9, 124.1, 123.7, 117.8, 115.5, 110.6, 61.6, 47.4, 35.4; LC-MS m/z 570 [M−]; anal. calcd for C27H22N8O5S: C, 56.84; H, 3.89; N, 19.64; found: C, 56.65; H, 3.71; N, 19.66.:6); FT-IR (cm−1):3300 (N–H stretch), 3129, 3049 (Csp2–H stretch), 2928, 2873 (Csp3–H stretch), 2250 (CN stretch, cyano) 1737 (CO stretch, ketone), 1600 (CO stretch, lactam), 1525 (CN stretch, imine),1508 (CC stretch, aromatic ring), 1467 (Csp3–H bend, methylene), 1274 (Csp2–O stretch, ether), 1201 (C–N stretch, lactam), 1165 (Csp3–O stretch, ether), 833 (C–H bend, 1,4-disubtitution), 755 (C–H bend, 1,2-disubstitution); 1H NMR (300 MHz, DMSO-d6) δ (ppm): 11.68 (s, 1H, –NH), 9.99 (s, 1H, –NH), 8.29–6.89 (m, 14H, –CHN, –CH triazole Ar–H), 4.97 (s, 2H, –CH2 isatin), 4.74 (t, 2H, 3J = 6 Hz, –OCH2), 4.42 (t, 2H, 3J = 6 Hz, –CH2 triazole); 13C NMR (75 MHz, DMSO-d6) δ (ppm): 183.5, 176.2 (CS), 159.7, 158.2, 150.6, 142.9 (CN azomethine), 141.9, 138.5, 137.0, 134.8, 129,7, 129.3, 128.9, 127.5, 126.3, 124.9, 124.7, 123.8, 118.0 (CN), 115.4, 115.1, 111.6, 66.6, 49.5, 35.4; LC-MS m/z 550 [M−]; anal. calcd for C28H22N8O3S: C, 61.08; H, 4.03; N, 20.35; found: C, 60.86; H, 3.93; N, 20.54.
N stretch, cyano) 1735 (CO stretch, ketone), 1606 (CO stretch, lactam), 1537 (CN stretch, imine),1510 (CC stretch, aromatic ring), 1467 (Csp3–H bend, methylene), 1249 (Csp2–O stretch, ether), 1195 (C–N stretch, lactam), 1169 (Csp3–O stretch, ether), 837 (C–H bend, 1,4-disubstitution), 752 (C–H bend, 1,2-disubstitution); 1H NMR (300 MHz, DMSO-d6) δ (ppm): 11.68 (s, 1H, –NH), 9.99 (s, 1H, –NH), 8.29–6.89 (m, 14H, –CHN, –CH triazole Ar–H), 4.97 (s, 2H, –CH2 isatin), 4.74 (t, 2H, 3J = 6 Hz, –OCH2), 4.42 (t, 2H, 3J = 6 Hz, –CH2 triazole); 13C NMR (75 MHz, DMSO-d6) δ (ppm): 183.5, 176.2 (CS), 159.7, 158.2, 150.6, 142.9 (CN azomethine), 141.9, 138.5, 137.0, 134.8, 129,7, 129.3, 128.9, 127.5, 126.3, 124.9, 124.7, 123.8, 118.0 (CN), 115.4, 115.1, 111.6, 66.6, 49.5, 35.4; LC-MS m/z 550 [M−]; anal. calcd for C28H22N8O3S: C, 61.08; H, 4.03; N, 20.35; found: C, 60.97; H, 3.89; N, 20.18.:6); FT-IR (cm−1):3300 (N–H stretch), 3129, 3049 (Csp2–H stretch), 2928, 2873 (Csp3–H stretch), 1737 (CO stretch, ketone), 1600 (CO stretch, lactam), 1525 (CN stretch, imine),1508 (CC stretch, aromatic ring), 1467 (Csp3–H bend, methylene), 1274 (Csp2–O stretch, ether), 1201 (C–N stretch, lactam), 1165 (Csp3–O stretch, ether), 833 (C–H bend, 1,4-disubstitution), 755 (C–H bend, 1,2-disubstitution); 1H NMR (300 MHz, DMSO-d6) δ (ppm): 11.82 (s, 1H, –NH), 9.98 (s, 1H, –NH), 8.38 (s, 1H, –CHN), 7.36 (s, 1H, –CH triazole), 8.37–6.89 (m, 13H, Ar–H), 5.13 (s, 2H, –CH2 isatin), 4.71 (t, 2H, 3J = 6 Hz, –OCH2), 4.14 (t, 2H, 3J = 6 Hz, –CH2 triazole); 13C NMR (75 MHz, DMSO-d6) δ (ppm): 183.4, 161.8 (CS), 160.2, 158.6, 150.6, 149.6, 142.9 (CN azomethine), 139.7, 138.6, 129.6, 129.4, 127.3, 125.9, 124.9, 124.1, 123.7, 117.8, 115.5, 110.7, 61.6, 47.4, 35.4; LC-MS m/z 525 [M−]; anal. calcd for C27H23N7O3S: C, 61.70; H, 4.41; N, 18.66; found: C, 61.56; H, 4.32; N, 18.43.:6); FT-IR (cm−1):3300 (N–H stretch), 3129, 3049 (Csp2–H stretch), 2928, 2873 (Csp3–H stretch), 1731 (CO stretch, ketone), 1612 (CO stretch, lactam), 1541 (CN stretch, imine), 1506 (CC stretch, aromatic ring), 1469 (Csp3–H bend, methylene), 1271 (Csp2–O stretch, ether), 1214 (C–N stretch, lactam), 1170 (Csp3–O stretch, ether), 815 (C–H bend, 1,4-disubstitution), 756 (C–H bend, 1,2-disubstitution); 1H NMR (300 MHz, DMSO-d6) δ (ppm): 11.68 (s, 1H, –NH), 9.98 (s, 1H, –NH), 8.25 (s, 1H, –CHN), 8.07 (s, 1H, –CH triazole), 7.81–6.89 (m, 12H, Ar–H), 4.97 (s, 2H, –CH2 isatin), 4.74 (t, 2H, 3J = 6 Hz, –OCH2), 4.42 (t, 2H, 3J = 6 Hz, –CH2 triazole), 2.31 (s, 3H, –CH3); 13C NMR (75 MHz, DMSO-d6) δ (ppm): 183.5, 176.2 (CS), 159.7, 158.2, 150.6, 142.9 (CN azomethine), 141.9, 138.5, 137.0, 134.8, 129.7, 129.3, 128.9, 127.5, 126.3, 124.9, 124.7, 123.8, 118.0, 115.4, 115.1, 111.6, 66.6, 49.5, 35.4, 21.0 (CH3); LC-MS m/z 539 [M−]; anal. calcd for C28H25N7O3S: C, 62.32; H, 4.67; N, 18.17; found: C, 62.13; H, 4.51; N, 18.01.:6); FT-IR (cm−1):3300 (N–H stretch), 3129, 3049 (Csp2–H stretch), 2928, 2873 (Csp3–H stretch), 1732 (CO stretch, ketone), 1683 (CO stretch, lactam), 1579 (CN stretch, imine), 1556 (N–O stretch, asymmetric, nitro), 1508 (CC stretch, aromatic ring), 1467 (Csp3–H bend, methylene), 1354 (N–O stretch, symmetric, nitro), 1244 (Csp2–O stretch, ether), 1219 (C–N stretch, lactam), 1165 (Csp3–O stretch, ether), 831 (C–H bend, 1,4-disubstitution), 758 (C–H bend, 1,2-disubstitution); 1H NMR (300 MHz, DMSO-d6) δ (ppm): 12.03 (s, 1H, –NH), 9.98 (s, 1H, –NH), 8.42–6.87 (m, 14H, CHN, –CH triazole, Ar–H), 5.15 (s, 2H, –CH2 isatin), 4.68 (t, 2H, 3J = 6 Hz, –OCH2), 4.14 (t, 2H, 3J = 6 Hz, –CH2 triazole); 13C NMR (75 MHz, DMSO-d6) δ (ppm): 183.4, 161.7 (CS), 160.2, 158.6, 150.6, 149.6, 142.9 (CN azomethine), 139.6, 138.6, 129.6, 129.4, 127.4, 125.9, 124.9, 124.1, 123.7, 117.8, 115.5, 110.6, 61.6, 47.4, 35.4; LC-MS m/z 570 [M−]; anal. calcd for C27H22N8O5S: C, 56.84; H, 3.89; N, 19.64; found: C, 56.65; H, 3.71; N, 19.66.:6); FT-IR (cm−1):3300 (N–H stretch), 3129, 3049 (Csp2–H stretch), 2928, 2873 (Csp3–H stretch), 2250 (CN stretch, cyano) 1737 (CO stretch, ketone), 1600 (CO stretch, lactam), 1525 (CN stretch, imine),1508 (CC stretch, aromatic ring), 1467 (Csp3–H bend, methylene), 1274 (Csp2–O stretch, ether), 1201 (C–N stretch, lactam), 1165 (Csp3–O stretch, ether), 833 (C–H bend, 1,4-disubtitution), 755 (C–H bend, 1,2-disubstitution); 1H NMR (300 MHz, DMSO-d6) δ (ppm): 11.68 (s, 1H, –NH), 9.99 (s, 1H, –NH), 8.29–6.89 (m, 14H, –CHN, –CH triazole Ar–H), 4.97 (s, 2H, –CH2 isatin), 4.74 (t, 2H, 3J = 6 Hz, –OCH2), 4.42 (t, 2H, 3J = 6 Hz, –CH2 triazole); 13C NMR (75 MHz, DMSO-d6) δ (ppm): 183.5, 176.2 (CS), 159.7, 158.2, 150.6, 142.9 (CN azomethine), 141.9, 138.5, 137.0, 134.8, 129,7, 129.3, 128.9, 127.5, 126.3, 124.9, 124.7, 123.8, 118.0 (CN), 115.4, 115.1, 111.6, 66.6, 49.5, 35.4; LC-MS m/z 550 [M−]; anal. calcd for C28H22N8O3S: C, 61.08; H, 4.03; N, 20.35; found: C, 60.86; H, 3.93; N, 20.54.4.6 General procedure for the synthesis of isatin–triazole derived bis-thiosemicarbazones (5a–h)
To the stirred solution of isatin–triazole hybrids 3a & 3b (0.1 g, 0.27 mmol) respectively in ethanol, suitable N4-substituted thiosemicarbazide 3c (2.1 eq., 0.56 mmol) was added under vigorous stirring. The reaction was carried out for 6 hours heating under reflux and progress of reaction was monitored after regular intervals by TLC (n-hexane: EtOAc, 7:3). The precipitates formed upon completion, were filtered, washed, dried and recrystallized when required to give the pure product. By following this procedure, a series of bis-thiosemicarbazone derivatives 5a–h of isatin–triazole hybrids 3a & 3b having different thiosemicarbazide moieties were synthesized.
:6); FT-IR (cm−1):3300 (N–H stretch), 3129, 3049 (Csp2–H stretch), 2928, 2873 (Csp3–H stretch), 1600 (CO stretch, lactam), 1544, 1525 (CN stretch, imine),1487 (CC stretch, aromatic ring), 1467 (Csp3–H bend, methylene), 1249 (Csp2–O stretch, ether), 1192 (C–N stretch, lactam), 1151 (Csp3–O stretch, ether), 827 (C–H bend, 1,4-disubstitution), 746 (C–H bend, 1,2-disubstitution); 1H NMR (300 MHz, DMSO-d6) δ (ppm): 12.56 (s, 1H, –NH), 11.73 (s, 1H, –NH), 10.88 (s, 1H, –NH), 10.05 (s, 1H, –NH), 8.29 (s, 1H, –CHN), 8.11 (s, 1H, –CH triazole), 7.86–6.98 (m, 16H, Ar–H), 5.13 (s, 2H, –OCH2), 4.73 (t, 2H, 3J = 6 Hz, –CH2-triazole), 4.25 (t, 2H, 3J = 6 Hz, –CH2-isatin); 13C NMR ((75 MHz, DMSO-d6) δ (ppm)): 183.3, 176.8 (CS), 176.1 (CS), 161.3, 160.1, 150.6, 143.2 (CN imine), 142.9 (CN azomethine), 139.5, 138.9, 131.7, 129.7, 128.9, 128.5, 127.3, 126.6, 126.3, 124.9, 123.7, 123.4, 121.6, 119.6, 117.8, 115.3, 110.7, 110.1, 61.5, 49.5, 35.1; LC-MS m/z 674 [M−]; anal. calcd for C34H30N10O2S2: C, 60.52; H, 4.48; N, 20.76; found: C, 60.41; H, 4.25; N, 20.81.:6); FT-IR (cm−1):3300 (N–H stretch), 3129, 3049 (Csp2–H stretch), 2928, 2873 (Csp3–H stretch), 1610 (CO stretch, lactam), 1540, 1516 (CN stretch, imine), 1487 (CC stretch, aromatic ring), 1467 (Csp3–H bend, methylene), 1249 (Csp2–O stretch, ether), 1197 (C–N stretch, lactam), 1151 (Csp3–O stretch, ether), 813 (C–H bend, 1,4-disubstitution), 746 (C–H bend, 1,2-disubstitution); 1H NMR (300 MHz, DMSO-d6) δ (ppm): 12.53 (s, 1H, –NH), 11.68 (s, 1H, –NH), 10.88 (s, 1H, –NH), 9.98 (s, 1H, –NH), 8.29 (s, 1H, –CHN), 8.10 (s, 1H, –CH triazole), 7.86–6.98 (m, 16H, Ar–H), 5.13 (s, 2H, –OCH2), 4.73 (t, 2H, 3J = 6 Hz, –CH2-triazole), 4.25 (t, 2H, 3J = 6 Hz, –CH2-isatin), 2.32 (s, 6H, –CH3); 13C NMR (75 MHz, DMSO-d6) δ (ppm): 176.7 (CS), 176.1 (CS), 161.2, 160.0, 143.0 (CN imine), 142.8 (CN azomethine), 137.0, 136.3, 135.9, 134.8, 131.6, 131.2, 129.7, 129.3, 128.9, 127.3, 126.2, 126.1, 125.7, 123.4, 121.6, 119.6, 115.3, 110.1, 61.6, 47.4, 21.6, 21.1 (CH3), 21.1 (CH3); LC-MS m/z 702 [M−]; anal. calcd for C36H34N10O2S2: C, 61.52; H, 4.88; N, 19.93; found: C, 61.37; H, 4.96; N, 20.16.:6); FT-IR (cm−1):3300 (N–H stretch), 3129, 3049 (Csp2–H stretch), 2928, 2873 (Csp3–H stretch), 1600 (CO stretch, lactam), 1544, 1525 (CN stretch, imine), 1556 (N–O stretch, asymmetric, nitro), 1508 (CC stretch, aromatic ring), 1467 (Csp3–H bend, methylene), 1354 (N–O stretch, symmetric, nitro), 1249 (Csp2–O stretch, ether), 1192 (C–N stretch, lactam), 1151 (Csp3–O stretch, ether), 827 (C–H bend, 1,4-disubstitution), 746 (C–H bend, 1,2-disubstitution); 1H NMR (300 MHz, DMSO-d6) δ (ppm): 12.71 (s, 1H, –NH), 11.73 (s, 1H, –NH), 10.90 (s, 1H, –NH), 10.05 (s, 1H, –NH), 8.27 (s, 1H, –CHN), 8.25 (s, 1H, –CH triazole), 8.08–6.91 (m, 16H, Ar–H), 5.07 (s, 2H, –CH2 isatin), 4.75 (t, 2H, 3J = 6 Hz, –OCH2), 4.43 (t, 2H, 3J = 6 Hz, –CH2 triazole); 13C NMR (75 MHz, DMSO-d6) δ (ppm): 176.5 (CS), 175.4 (CS), 172.5, 160.9, 160.0, 144.1, 143.9 (CN imine), 143.2 (CN azomethine), 141.9, 133.1, 132.7, 131.9, 129.9, 127.2, 125.6, 125.4, 124.9, 123.5, 121.8, 119.6, 119.5, 119.2, 115.2, 108.2, 107.1, 66.6, 49.5, 35.1; LC-MS m/z 764 [M−]; anal. calcd for C34H28N12O6S2: C, 53.40; H, 3.69; N, 21.98; found: C, 53.32; H, 3.57; N, 22.11.:6); FT-IR (cm−1):3300 (N–H stretch), 3129, 3049 (Csp2–H stretch), 2928, 2873 (Csp3–H stretch), 2223 (CN stretch, cyano), 1604 (CO stretch, lactam), 1585, 1537 (CN stretch, imine), 1508 (CC stretch, aromatic ring), 1465 (Csp3–H bend, methylene), 1249 (Csp2–O stretch, ether), 1192 (C–N stretch, lactam), 1151 (Csp3–O stretch, ether), 829 (C–H bend, 1,4-disubstitution), 750 (C–H bend, 1,2-disubstitution); 1H NMR (300 MHz, DMSO-d6) δ (ppm): 12.71 (s, 1H, –NH), 12.01 (s, 1H, –NH), 11.06 (s, 1H, –NH), 10.26 (s, 1H, –NH), 8.32 (s, 1H, –CHN), 8.30 (s, 1H, –CH triazole), 8.14–6.99 (m, 16H, Ar–H), 5.14 (s, 2H, –OCH2), 4.73 (t, 2H, 3J = 6 Hz, –CH2-triazole), 4.24 (t, 2H, 3J = 6 Hz, –CH2-isatin); 13C NMR (75 MHz, DMSO-d6) δ (ppm): 176.5 (CS), 175.4 (CS), 172.5, 160.9, 160.0, 144.1, 143.9 (CN imine), 143.2 (CN azomethine), 141.9, 133.1, 132.7, 131.9, 129.9, 127.2, 125.6, 125.4, 124.9, 123.5, 121.8, 119.6 (CN), 119.5 (CN), 119.2, 115.2, 108.2, 107.1, 66.7, 49.5, 35.1; LC-MS m/z 724 [M−]; anal. calcd for C36H28N12O2S2: C, 59.65; H, 3.89; N, 23.19; found: C, 59.49; H, 3.99; N, 23.32.:6); FT-IR (cm−1):3300 (N–H stretch), 3129, 3049 (Csp2–H stretch), 2928, 2873 (Csp3–H stretch), 1608 (CO stretch, lactam), 1544, 1525 (CN stretch, imine),1492 (CC stretch, aromatic ring), 1465 (Csp3–H bend, methylene), 1244 (Csp2–O stretch, ether), 1192 (C–N stretch, lactam), 1141 (Csp3–O stretch, ether), 827 (C–H bend, 1,4-disubstitution), 740 (C–H bend, 1,2-disubstitution); 1H NMR (300 MHz, DMSO-d6) δ (ppm): 12.71 (s, 1H, –NH), 11.73 (s, 1H, –NH), 10.89 (s, 1H, –NH), 10.05 (s, 1H, –NH), 8.27 (s, 1H, –CHN), 8.09 (s, 1H, –CH triazole), 7.83–6.91 (m, 18H, Ar–H), 5.07 (s, 2H, –CH2 isatin), 4.75 (t, 2H, 3J = 6 Hz, –OCH2), 4.43 (t, 2H, 3J = 6 Hz, –CH2 triazole); 13C NMR (75 MHz, DMSO-d6) δ (ppm): 176.8 (CS), 176.1 (CS), 160.9, 159.8, 143.1 (CN imine), 142.9 (CN azomethine), 141.9, 139.5, 138.8, 131.6, 131.5, 129.7, 128.9, 128.4, 127.5, 126.6, 126.3, 126.1, 125.7, 124.8, 123.5, 121.6, 119.8, 115.2, 110.9, 66.6, 49.5, 35.1; LC-MS m/z 674 [M−]; anal. calcd for C34H30N10O2S2: C, 60.52; H, 4.48; N, 20.76; found: C, 60.61; H, 4.29; N, 20.87.:6); FT-IR (cm−1):3300 (N–H stretch), 3129, 3049 (Csp2–H stretch), 2928, 2873 (Csp3–H stretch), 1612 (CO stretch, lactam), 1544, 1539 (CN stretch, imine), 1487 (CC stretch, aromatic ring), 1467 (Csp3–H bend, methylene), 1271 (Csp2–O stretch, ether), 1193 (C–N stretch, lactam), 1151 (Csp3–O stretch, ether), 813 (C–H bend, 1,4-disubstitution), 750 (C–H bend, 1,2-disubstitution); 1H NMR (300 MHz, DMSO-d6) δ (ppm): 12.68 (s, 1H, –NH), 11.68 (s, 1H, –NH), 10.83 (s, 1H, –NH), 9.98 (s, 1H, –NH), 8.27 (s, 1H, –CHN), 8.25 (s, 1H, –CH triazole), 8.07–6.89 (m, 16H, Ar–H), 5.07 (s, 2H, 3J = 6 Hz, –CH2 isatin), 4.75 (t, 2H, 3J = 6 Hz, –OCH2), 4.43 (t, 2H, –CH2 triazole), 2.32 (s, 6H, –CH3); 13C NMR (75 MHz, DMSO-d6) δ (ppm): 176.2 (CS), 176.1 (CS), 160.9, 159.8, 158.2, 150.6, 142.9 (CN imine), 141.9 (CN azomethine), 138.5, 137.0, 136.3, 135.9, 134.8, 131.6, 129.7, 129.3, 128.9, 127.5, 126.3, 126.1, 124.9, 124.7, 123.8, 123.5, 118.0, 115.2, 111.6, 66.7, 49.5, 35.1, 21.6 (CH3), 21.0 (CH3); LC-MS m/z 702 [M−]; anal. calcd for C36H34N10O2S2: C, 61.52; H, 4.88; N, 19.93; found: C, 61.35; H, 4.74; N, 19.89.:6); FT-IR (cm−1):3300 (N–H stretch), 3129, 3049 (Csp2–H stretch), 2928, 2873 (Csp3–H stretch), 1612 (CO stretch, lactam), 1544, 1539 (CN stretch, imine), 1556 (N–O stretch, asymmetric, nitro), 1508 (CC stretch, aromatic ring), 1467 (Csp3–H bend, methylene), 1354 (N–O stretch, symmetric, nitro), 1271 (Csp2–O stretch, ether), 1193 (C–N stretch, lactam), 1151 (Csp3–O stretch, ether), 813 (C–H bend, 1,4-disubstitution), 750 (C–H bend, 1,2-disubstitution); 1H NMR (300 MHz, DMSO-d6) δ (ppm): 12.90 (s, 1H, –NH), 12.09 (s, 1H, –NH), 11.18 (s, 1H, –NH), 10.37 (s, 1H, –NH), 8.31–6.89 (m, 18H, –CHN, –CH triazole, Ar–H), 5.07 (s, 2H, –CH2 isatin), 4.97 (t, 2H, 3J = 6 Hz, –OCH2), 4.76 (t, 2H, 3J = 6 Hz, –CH2 triazole); 13C NMR (75 MHz, DMSO-d6) δ (ppm): 176.5 (CS), 175.4 (CS), 172.5, 160.9, 160.0, 144.1, 143.9 (CN imine), 143.2 (CN azomethine), 141.9, 133.1, 132.7, 131.9, 129.9, 127.2, 125.6, 125.4, 124.9, 123.5, 121.8, 119.6, 119.5, 119.2, 115.2, 108.2, 107.1, 66.7, 49.5, 35.1; LC-MS m/z 764 [M−]; anal. calcd for C34H28N12O6S2: C, 53.40; H, 3.69; N, 21.98; found: C, 53.53; H, 3.78; N, 21.69.:6); FT-IR (cm−1):3300 (N–H stretch), 3129, 3049 (Csp2–H stretch), 2928, 2873 (Csp3–H stretch), 2223 (CN stretch, cyano), 1604 (CO stretch, lactam), 1585, 1537 (CN stretch, imine),1508 (CC stretch, aromatic ring), 1465 (Csp3–H bend, methylene), 1249 (Csp2–O stretch, ether), 1192 (C–N stretch, lactam), 1151 (Csp3–O stretch, ether), 829 (C–H bend, 1,4-disubstitution), 750 (C–H bend, 1,2-disubstitution); 1H NMR (300 MHz, DMSO-d6) δ (ppm): 12.86 (s, 1H, –NH), 12.01 (s, 1H, –NH), 11.08 (s, 1H, –NH), 10.26 (s, 1H, –NH), 8.27 (s, 1H, –CHN), 8.25 (s, 1H, –CH triazole), 8.12–6.93 (m, 16H, Ar–H), 5.07 (s, 2H, –CH2 isatin), 4.77 (t, 2H, 3J = 6 Hz, –OCH2), 4.44 (t, 2H, 3J = 6 Hz, –CH2 triazole); 13C NMR (75 MHz, DMSO-d6) δ (ppm): 176.5 (CS), 175.4 (CS), 172.5, 160.9, 160.0, 144.1, 143.9 (CN imine), 143.2 (CN azomethine), 141.9, 133.1, 132.7, 131.9, 129.9, 127.2, 125.6, 125.4, 124.9, 123.5, 121.8, 119.6 (CN), 119.5 (CN), 119.2, 115.2, 108.2, 107.0, 66.6, 49.5, 35.1; LC-MS m/z 724 [M−]; anal. calcd for C36H28N12O2S2: C, 59.65; H, 3.89; N, 23.19; found: C, 59.48; H, 3.75; N, 23.33.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are highly grateful to the Department of Chemistry, Quaid-i-Azam University for financial support under URF; we also thank “The World Academy of Sciences (TWAS)” for financial support (Project No. 13–419 RG/PHA/AS_CUNESCO FR: 3240279216).References
- A. Mushtaq, P. Wu and M. M. Naseer, Recent drug design strategies and identification of key heterocyclic scaffolds for promising anticancer targets, Pharmacol. Ther., 2024, 254, 108579 CrossRef CAS PubMed.
- B. Eftekhari-Sis, M. Zirak and A. Akbari, Arylglyoxals in synthesis of heterocyclic compounds, Chem. Rev., 2013, 113(5), 2958–3043 CrossRef CAS PubMed.
- T. Al-Warhi, D. M. Elimam, Z. M. Elsayed, M. M. Abdel-Aziz, R. M. Maklad, A. A. Al-Karmalawy, K. Afarinkia, M. A. Abourehab, H. A. Abdel-Aziz and W. M. Eldehna, Development of novel isatin thiazolyl-pyrazoline hybrids as promising antimicrobials in MDR pathogens, RSC Adv., 2022, 12(48), 31466–31477 RSC.
- Y. Wang, Z. Liang, Y. Zheng, A. S.-L. Leung, S.-C. Yan, P.-K. So, Y.-C. Leung, W.-L. Wong and K.-Y. Wong, Rational structural modification of the isatin scaffold to develop new and potent antimicrobial agents targeting bacterial peptidoglycan glycosyltransferase, RSC Adv., 2021, 11(29), 18122–18130 RSC.
- A. Katiyar, M. Hegde, S. Kumar, V. Gopalakrishnan, K. D. Bhatelia, K. Ananthaswamy, S. A. Ramareddy, E. De Clercq, B. Choudhary and D. Schols, Synthesis and evaluation of the biological activity of N′-[2-oxo-1, 2 dihydro-3 H-indol-3-ylidene] benzohydrazides as potential anticancer agents, RSC Adv., 2015, 5(56), 45492–45501 RSC.
- L. Zhang, F. Chen, J. Wang, Y. Chen, Z. Zhang, Y. Lin and X. Zhu, Novel isatin derivatives of podophyllotoxin: synthesis and cytotoxic evaluation against human leukaemia cancer cells as potent anti-MDR agents, RSC Adv., 2015, 5(118), 97816–97823 RSC.
- A. M. Hamdy, N. A. Al-Masoudi, C. Pannecouque, Q. Rahman, A. Villinger and P. Langer, Regioselective Suzuki–Miyaura reactions of 4, 7-dichloro-N-methylisatin. Synthesis, anti-HIV activity and modeling study, RSC Adv., 2015, 5(130), 107360–107369 RSC.
- L. Emami, L. Moezi, L. Amiri-Zirtol, F. Pirsalami, M. Divar, A. Solhjoo and S. Khabnadideh, Anticonvulsant activity, molecular modeling and synthesis of spirooxindole-4 H-pyran derivatives using a novel reusable organocatalyst, Mol. Diversity, 2022, 26(6), 3129–3141 CrossRef CAS PubMed.
- R. Nath, S. Pathania, G. Grover and M. J. Akhtar, Isatin containing heterocycles for different biological activities: Analysis of structure activity relationship, J. Mol. Struct., 2020, 1222, 128900 CrossRef CAS.
- J. Haribabu, G. Subhashree, S. Saranya, K. Gomathi, R. Karvembu and D. Gayathri, Isatin based thiosemicarbazone derivatives as potential bioactive agents: Anti-oxidant and molecular docking studies, J. Mol. Struct., 2016, 1110, 185–195 CrossRef CAS.
- A. S. Hassan, N. M. Morsy, W. M. Aboulthana and A. Ragab, Exploring novel derivatives of isatin-based Schiff bases as multi-target agents: design, synthesis, in vitro biological evaluation, and in silico ADMET analysis with molecular modeling simulations, RSC Adv., 2023, 13(14), 9281–9303 RSC.
- Q. U. Ain, A. Singh, I. Singh, R. Carmieli and R. Sharma, Synthesis, characterization and anti-tubercular activities of copper (II) complexes of substituted 2, 3-isatin bisthiosemicarbazones: An experimental and theoretical approach, Results Chem., 2023, 6, 101171 CrossRef CAS.
- S. K. Kancharla, S. Birudaraju, A. Pal, L. K. Reddy, E. R. Reddy, S. K. Vagolu, D. Sriram, K. B. Bonige and R. B. Korupolu, Synthesis and biological evaluation of isatin oxime ether-tethered aryl 1 H-1, 2, 3-triazoles as inhibitors of Mycobacterium tuberculosis, New J. Chem., 2022, 46(6), 2863–2874 RSC.
- M. Ahmad, H. Pervez, S. Zaib, M. Yaqub, M. M. Naseer, S. U. Khan and J. Iqbal, Synthesis, biological evaluation and docking studies of some novel isatin-3-hydrazonothiazolines, RSC Adv., 2016, 6(65), 60826–60844 RSC.
- S. Mehreen, M. Zia, A. Khan, J. Hussain, S. Ullah, M. U. Anwar, A. Al-Harrasi and M. M. Naseer, Phenoxy pendant isatins as potent α-glucosidase inhibitors: reciprocal carbonyl⋯ carbonyl interactions, antiparallel π⋯ π stacking driven solid state self-assembly and biological evaluation, RSC Adv., 2022, 12(32), 20919–20928 RSC.
- A. Mushtaq, U. Azam, S. Mehreen and M. M. Naseer, Synthetic α-glucosidase inhibitors as promising anti-diabetic agents: Recent developments and future challenges, Eur. J. Med. Chem., 2023, 249, 115119 CrossRef CAS PubMed.
- S. Deswal, R. K. Tittal, D. G. Vikas, K. Lal and A. Kumar, 5-Fluoro-1H-indole-2, 3-dione-triazoles-synthesis, biological activity, molecular docking, and DFT study, J. Mol. Struct., 2020, 1209, 127982 CrossRef CAS.
- B. Posocco, M. Buzzo, L. Giodini, S. Crotti, S. D'Aronco, P. Traldi, M. Agostini, E. Marangon and G. Toffoli, Analytical aspects of sunitinib and its geometric isomerism towards therapeutic drug monitoring in clinical routine, J. Pharm. Biomed. Anal., 2018, 160, 360–367 CrossRef CAS PubMed.
- M. D. Hall, K. R. Brimacombe, M. S. Varonka, K. M. Pluchino, J. K. Monda, J. Li, M. J. Walsh, M. B. Boxer, T. H. Warren, H. M. Fales and M. M. Gottesman, Synthesis and Structure–Activity Evaluation of Isatin-β-thiosemicarbazones with Improved Selective Activity toward Multidrug-Resistant Cells Expressing P-Glycoprotein, J. Med. Chem., 2011, 54(16), 5878–5889 CrossRef CAS PubMed.
- B. Aneja, N. S. Khan, P. Khan, A. Queen, A. Hussain, M. T. Rehman, M. F. Alajmi, H. R. El-Seedi, S. Ali, M. I. Hassan and M. Abid, Design and development of Isatin-triazole hydrazones as potential inhibitors of microtubule affinity-regulating kinase 4 for the therapeutic management of cell proliferation and metastasis, Eur. J. Med. Chem., 2019, 163, 840–852 CrossRef CAS PubMed.
- S. N. A. Bukhari, T. G. Alsahli, H. Ejaz, N. Ahmed, W. Ahmad, M. A. Elsherif, N. H. Alotaibi, K. Junaid and N. Janković, Dual activity of indolin-2-ones containing an arylidene motif: DNA and BSA interaction, RSC Adv., 2023, 13(40), 28139–28147 RSC.
- K. K. Saini, R. K. Upadhyay, R. Kant, A. Vajpayee, K. Jain, A. Kumar, L. S. Kumar and R. Kumar, Design, synthesis, molecular docking and DFT studies on novel melatonin and isatin based azole derivatives, RSC Adv., 2023, 13(39), 27525–27534 RSC.
- H. A. Radwan, I. Ahmad, I. M. Othman, M. A. Gad-Elkareem, H. Patel, K. Aouadi, M. Snoussi and A. Kadri, Design, synthesis, in vitro anticancer and antimicrobial evaluation, SAR analysis, molecular docking and dynamic simulation of new pyrazoles, triazoles and pyridazines based isoxazole, J. Mol. Struct., 2022, 1264, 133312 CrossRef CAS.
- P. Chopade and A. Shaikh, Advancements and Future Perspectives of 1, 2, 3 Triazole Scaffold as Promising Antiviral Agent in Drug Discovery, Int. J. Pharm. Sci. Res., 2022, 4805–4818 CAS.
- N. Jangir, S. K. Bagaria and D. K. Jangid, Nanocatalysts: applications for the synthesis of N-containing five-membered heterocycles, RSC Adv., 2022, 12(30), 19640–19666 RSC.
- R. Dharavath, N. Nagaraju, M. R. Reddy, D. Ashok, M. Sarasija, M. Vijjulatha, T. Vani, K. Jyothi and G. Prashanthi, Microwave-assisted synthesis, biological evaluation and molecular docking studies of new coumarin-based 1, 2, 3-triazoles, RSC Adv., 2020, 10(20), 11615–11623 RSC.
- A. Nandikolla, S. Srinivasarao, B. K. Kumar, S. Murugesan, H. Aggarwal, L. L. Major, T. K. Smith and K. V. G. C. Sekhar, Synthesis, study of antileishmanial and antitrypanosomal activity of imidazo pyridine fused triazole analogues, RSC Adv., 2020, 10(63), 38328–38343 RSC.
- A. Rani, G. Singh, A. Singh, U. Maqbool, G. Kaur and J. Singh, CuAAC-ensembled 1, 2, 3-triazole-linked isosteres as pharmacophores in drug discovery, RSC Adv., 2020, 10(10), 5610–5635 RSC.
- H. A. Abuelizz, H. M. Awad, M. Marzouk, F. A. Nasr, A. S. Alqahtani, A. H. Bakheit, A. M. Naglah and R. Al-Salahi, Synthesis and biological evaluation of 4-(1 H-1, 2, 4-triazol-1-yl) benzoic acid hybrids as anticancer agents, RSC Adv., 2019, 9(33), 19065–19074 RSC.
- K. Bhagat, J. V. Singh, A. Sharma, A. Kaur, N. Kumar, H. K. Gulati, A. Singh, H. Singh and P. M. S. Bedi, Novel series of triazole containing coumarin and isatin based hybrid molecules as acetylcholinesterase inhibitors, J. Mol. Struct., 2021, 1245, 131085 CrossRef CAS.
- A. Kumar, Y. Rohila, V. Kumar and K. Lal, A Mini Review on Pharmacological Significance of Isatin-1, 2, 3-Triazole Hybrids, Curr. Top. Med. Chem., 2023, 23(10), 833–847 CrossRef CAS PubMed.
- B. Aneja, N. S. Khan, P. Khan, A. Queen, A. Hussain, M. T. Rehman, M. F. Alajmi, H. R. El-Seedi, S. Ali and M. I. Hassan, Design and development of Isatin-triazole hydrazones as potential inhibitors of microtubule affinity-regulating kinase 4 for the therapeutic management of cell proliferation and metastasis, Eur. J. Med. Chem., 2019, 163, 840–852 CrossRef CAS PubMed.
- N. Busto, J. Leitão-Castro, A. T. García-Sosa, F. Cadete, C. S. Marques, R. Freitas and A. J. Burke, N-1, 2, 3-Triazole–isatin derivatives: anti-proliferation effects and target identification in solid tumour cell lines, RSC Med. Chem., 2022, 13(8), 970–977 RSC.
- N. Rezki, M. A. Almehmadi, S. Ihmaid, A. M. Shehata, A. M. Omar, H. E. Ahmed and M. R. Aouad, Novel scaffold hopping of potent benzothiazole and isatin analogues linked to 1, 2, 3-triazole fragment that mimic quinazoline epidermal growth factor receptor inhibitors: Synthesis, antitumor and mechanistic analyses, Bioorg. Chem., 2020, 103, 104133 CrossRef CAS PubMed.
- R. Kumar and P. Takkar, Repositioning of Isatin hybrids as novel anti-tubercular agents overcoming pre-existing antibiotics resistance, Med. Chem. Res., 2021, 30, 847–876 CrossRef CAS.
- A. Kumar, K. Lal, M. Yadav, S. Kumar, M. B. Singh and K. Kumari, Isatin-semicarbazone linked acetamide 1, 2, 3-triazole hybrids: Synthesis, antimicrobial evaluation and docking simulations, J. Mol. Struct., 2023, 1287, 135660 CrossRef CAS.
- B. Shakya and P. N. Yadav, Thiosemicarbazones as potent anticancer agents and their modes of action, Mini-Rev. Med. Chem., 2020, 20(8), 638–661 CrossRef CAS PubMed.
- N. Balakrishnan, J. Haribabu, M. Dharmasivam, J. P. Jayadharini, D. Anandakrishnan, S. Swaminathan, N. Bhuvanesh, C. Echeverria and R. Karvembu, Influence of Indole-N substitution of thiosemicarbazones in cationic Ru (II)(η6-Benzene) complexes on their anticancer activity, Organometallics, 2023, 42(3), 259–275 CrossRef CAS.
- D. L. Sun, S. Poddar, R. D. Pan, E. W. Rosser, E. R. Abt, J. Van Valkenburgh, T. M. Le, V. Lok, S. P. Hernandez and J. Song, Isoquinoline thiosemicarbazone displays potent anticancer activity with in vivo efficacy against aggressive leukemias, RSC Med. Chem., 2020, 11(3), 392–410 RSC.
- B. Z. Sibuh, P. K. Gupta, P. Taneja, S. Khanna, P. Sarkar, S. Pachisia, A. A. Khan, N. K. Jha, K. Dua and S. K. Singh, Synthesis, in silico study, and anti-cancer activity of thiosemicarbazone derivatives, Biomedicines, 2021, 9(10), 1375 CrossRef CAS PubMed.
- F. Pape Veronika, A. Enyedy Éva, K. Keppler Bernhard and R. Kowol Christian, Anticancer thiosemicarbazones: chemical properties, interaction with iron metabolism, and resistance development, Antioxid. Redox Signaling, 2019, 30, 1062–1082 CrossRef PubMed.
- N. K. Singh, A. A. Kumbhar, Y. R. Pokharel and P. N. Yadav, Anticancer potency of copper (II) complexes of thiosemicarbazones, J. Inorg. Biochem., 2020, 210, 111134 CrossRef CAS PubMed.
- B. Sever, G. Akalın Çiftçi, A. Özdemir and M. Altıntop, Design, synthesis and in vitro evaluation of new thiosemicarbazone derivatives as potential anticancer agents, J. Res. Pharm., 2019, 23(1), 16–24 CAS.
- A. Mirzaahmadi, S. A. Hosseini-Yazdi, E. Safarzadeh, B. Baradaran, E. Samolova and M. Dusek, New series of water-soluble thiosemicarbazones and their copper (II) complexes as potentially promising anticancer compounds, J. Mol. Liq., 2019, 293, 111412 CrossRef CAS.
- S. J. Sardroud, S. A. Hosseini-Yazdi, M. Mahdavi, M. Poupon and E. Skorepova, Synthesis, characterization and in vitro evaluation of anticancer activity of a new water-soluble thiosemicarbazone ligand and its complexes, Polyhedron, 2020, 175, 114218 CrossRef.
- V. Manakkadan, J. Haribabu, V. N. V. Palakkeezhillam, P. Rasin, M. Mandal, V. S. Kumar, N. Bhuvanesh, R. Udayabhaskar and A. Sreekanth, Synthesis and characterization of N4-substituted thiosemicarbazones: DNA/BSA binding, molecular docking, anticancer activity, ADME study and computational investigations, J. Mol. Struct., 2023, 1285, 135494 CrossRef CAS.
- J. Haribabu, N. Balakrishnan, S. Swaminathan, D. P. Dorairaj, M. Azam, M. K. M. Subarkhan, Y.-L. Chang, S. C. Hsu, P. Štarha and R. Karvembu, Michael addition-driven synthesis of cytotoxic palladium (ii) complexes from chromone thiosemicarbazones: investigation of anticancer activity through in vitro and in vivo studies, New J. Chem., 2023, 47(33), 15748–15759 RSC.
- K. Preetha, E. Seena, P. K. Maniyampara, E. Manoj and M. P. Kurup, Synthesis, crystal structure, Hirshfeld surface analysis, DFT, molecular docking and in vitro antitumor studies of (2E)-2-[4-(diethylamino) benzylidene]-N-ethylhydrazinecarbothioamide, J. Mol. Struct., 2024, 1295, 136700 CrossRef CAS.
- Y. Zou, Q. Zhao, J. Liao, H. Hu, S. Yu, X. Chai, M. Xu and Q. Wu, New triazole derivatives as antifungal agents: Synthesis via click reaction, in vitro evaluation and molecular docking studies, Bioorg. Med. Chem. Lett., 2012, 22(8), 2959–2962 CrossRef CAS PubMed.
- K. Vine, L. Matesic, J. Locke, M. Ranson and D. Skropeta, Cytotoxic and anticancer activities of isatin and its derivatives: a comprehensive review from 2000-2008, Anti-Cancer Agents Med. Chem., 2009, 9(4), 397–414 CrossRef CAS PubMed.
- A. K. Gupta, S. Tulsyan, M. Bharadwaj and R. Mehrotra, Systematic review on cytotoxic and anticancer potential of n-substituted isatins as novel class of compounds useful in multidrug-resistant cancer therapy: In silico and in vitro analysis, Top. Curr. Chem., 2019, 377, 1–21 CrossRef CAS PubMed.
- M. Serafini, T. Pirali and G. C. Tron, Click 1, 2, 3-triazoles in drug discovery and development: From the flask to the clinic?, Adv. Heterocycl. Chem., 2021, 134, 101–148 CrossRef.
- E. M. Othman, E. A. Fayed, E. M. Husseiny and H. S. Abulkhair, Rationale design, synthesis, cytotoxicity evaluation, and in silico mechanistic studies of novel 1, 2, 3-triazoles with potential anticancer activity, New J. Chem., 2022, 46(25), 12206–12216 RSC.
- Y. Yu, D. S. Kalinowski, Z. Kovacevic, A. R. Siafakas, P. J. Jansson, C. Stefani, D. B. Lovejoy, P. C. Sharpe, P. V. Bernhardt and D. R. Richardson, Thiosemicarbazones from the old to new: iron chelators that are more than just ribonucleotide reductase inhibitors, J. Med. Chem., 2009, 52(17), 5271–5294 CrossRef CAS PubMed.
- P. Singh, P. Sharma, A. Anand, P. Bedi, T. Kaur, A. Saxena and V. Kumar, Azide-alkyne cycloaddition en route to novel 1H-1, 2, 3-triazole tethered isatin conjugates with in vitro cytotoxic evaluation, Eur. J. Med. Chem., 2012, 55, 455–461 CrossRef CAS PubMed.
- A. Shukla, Synthesis and biological screening of benzimidazole derivatives, Int. J. Pharm. Sci. Res., 2012, 3(3), 922 CAS.
- S. J. Pitman, A. K. Evans, R. T. Ireland, F. Lempriere and L. K. McKemmish, Benchmarking basis sets for density functional theory thermochemistry calculations: Why unpolarized basis sets and the polarized 6-311G family should be avoided, J. Phys. Chem. A, 2023, 127(48), 10295–10306 CrossRef CAS PubMed.
- N. Johnee Britto, M. Panneerselvam, M. Deepan Kumar, A. Kathiravan and M. Jaccob, Substituent Effect on the Photophysics and ESIPT Mechanism of N, N′-Bis (salicylidene)-p-phenylenediamine: A DFT/TD-DFT Analysis, J. Chem. Inf. Model., 2021, 61(4), 1825–1839 CrossRef CAS PubMed.
- P. Bianchi and J.-C. M. Monbaliu, Three decades of unveiling the complex chemistry of C-nitroso species with computational chemistry, Org. Chem. Front., 2022, 9(1), 223–264 RSC.
- S. H. Sumrra, F. Mushtaq, F. Ahmad, R. Hussain, W. Zafar, M. Imran and M. N. Zafar, Coordination behavior, structural, statistical and theoretical investigation of biologically active metal-based isatin compounds, Chem. Pap., 2022, 76(6), 3705–3727 CrossRef CAS.
- K. Kaavin, D. Naresh, M. Yogeshkumar, M. K. Prakash, S. Janarthanan, M. M. Krishnan and M. Malathi, In-silico DFT studies and molecular docking evaluation of benzimidazo methoxy quinoline-2-one ligand and its Co, Ni, Cu and Zn complexes as potential inhibitors of Bcl-2, Caspase-3, EGFR, mTOR, and PI3K, cancer-causing proteins, Chem. Phys. Impact., 2024, 8, 100418 CrossRef.
- M. Miar, A. Shiroudi, K. Pourshamsian, A. R. Oliaey and F. Hatamjafari, Theoretical investigations on the HOMO–LUMO gap and global reactivity descriptor studies, natural bond orbital, and nucleus-independent chemical shifts analyses of 3-phenylbenzo [d] thiazole-2 (3 H)-imine and its para-substituted derivatives: Solvent and substituent effects, J. Chem. Res., 2021, 45(1–2), 147–158 CrossRef CAS.
- Y. Wu and W. Zhu, Organic sensitizers from D–π–A to D–A–π–A: effect of the internal electron-withdrawing units on molecular absorption, energy levels and photovoltaic performances, Chem. Soc. Rev., 2013, 42(5), 2039–2058 RSC.
- P. Udhayakalaa, T. V. Rajendiranb and S. Gunasekaran, Density functional theory investigations for the adsorption of some oxadiazole derivatives on mild steel, J. Adv. Sci. Res., 2012, 3(03), 67–74 Search PubMed.
- A. H. Radhi, E. A. Du, F. A. Khazaal, Z. M. Abbas, O. H. Aljelawi, S. D. Hamadan, H. A. Almashhadani and M. M. Kadhim, HOMO-LUMO energies and geometrical structures effecton corrosion inhibition for organic compounds predict by DFT and PM3 methods, NeuroQuantology, 2020, 18(1), 37 Search PubMed.
- M. Akram, M. Adeel, M. Khalid, M. N. Tahir, M. U. Khan, M. A. Asghar, M. A. Ullah and M. Iqbal, A combined experimental and computational study of 3-bromo-5-(2, 5-difluorophenyl) pyridine and 3, 5-bis (naphthalen-1-yl) pyridine: Insight into the synthesis, spectroscopic, single crystal XRD, electronic, nonlinear optical and biological properties, J. Mol. Struct., 2018, 1160, 129–141 CrossRef.
- A. Miličević, G. Miletić and I. N. Jovanović, Electrochemical oxidation of flavonoids: PM6 and DFT for elucidating electronic changes and modelling oxidation potential (part II), J. Mol. Liq., 2019, 295, 111730 CrossRef.
- V. Choudhary, A. Bhatt, D. Dash and N. Sharma, DFT calculations on molecular structures, HOMO–LUMO study, reactivity descriptors and spectral analyses of newly synthesized diorganotin (IV) 2-chloridophenylacetohydroxamate complexes, J. Comput. Chem., 2019, 40(27), 2354–2363 CrossRef CAS PubMed.
- T. Kar, S. Scheiner and A. Sannigrahi, Hardness and Chemical Potential Profiles for Some Open-Shell HAB→ HBA Type Reactions. Ab Initio and Density Functional Study, J. Phys. Chem. A, 1998, 102(29), 5967–5973 CrossRef CAS.
- L. F. Dechouk, A. Bouchoucha, Y. Abdi, K. S. Larbi, A. Bouzaheur and S. Terrachet-Bouaziz, Coordination of new palladium (II) complexes with derived furopyran-3, 4-dione ligands: Synthesis, characterization, redox behaviour, DFT, antimicrobial activity, molecular docking and ADMET studies, J. Mol. Struct., 2022, 1257, 132611 CrossRef CAS.
- N.-B. Sun, J.-Q. Fu, J.-Q. Weng, J.-Z. Jin, C.-X. Tan and X.-H. Liu, Microwave assisted synthesis, antifungal activity and DFT theoretical study of some novel 1, 2, 4-triazole derivatives containing the 1, 2, 3-thiadiazole moiety, Molecules, 2013, 18(10), 12725–12739 CrossRef CAS PubMed.
- M. Asiri, Synthesis, crystal structure and spectroscopic properties of 1, 2-benzothiazine derivatives: An experimental and DFT study, Struct. Chem., 2015, 34(1), 15–25 Search PubMed.
- M. N. Arshad, A. M. Asiri, K. A. Alamry, T. Mahmood, M. A. Gilani, K. Ayub and A. S. Birinji, Synthesis, crystal structure, spectroscopic and density functional theory (DFT) study of N-[3-anthracen-9-yl-1-(4-bromo-phenyl)-allylidene]-N-benzenesulfonohydrazine, Spectrochim. Acta, Part A, 2015, 142, 364–374 CrossRef CAS PubMed.
- R. Galeazzi, C. Marucchini, M. Orena and C. Zadra, Stereoelectronic properties and activity of some imidazolinone herbicides: a computational approach, J. Mol. Struct., 2003, 640(1–3), 191–200 CrossRef CAS.
- A. Mahal, M. Al-Janabi, V. Eyüpoğlu, A. Alkhouri, S. Chtita, M. M. Kadhim, A. J. Obaidullah, J. M. Alotaibi, X. Wei and M. R. F. Pratama, Molecular docking, drug-likeness and DFT study of some modified tetrahydrocurcumins as potential anticancer agents, Saudi Pharm. J., 2024, 32(1), 101889 CrossRef CAS PubMed.
- S. Kumar, V. Saini, I. K. Maurya, J. Sindhu, M. Kumari, R. Kataria and V. Kumar, Design, synthesis, DFT, docking studies and ADME prediction of some new coumarinyl linked pyrazolylthiazoles: Potential standalone or adjuvant antimicrobial agents, PLoS One, 2018, 13(4), 0196016 CrossRef PubMed.
- F. Pereira, K. Xiao, D. A. Latino, C. Wu, Q. Zhang and J. Aires-de-Sousa, Machine learning methods to predict density functional theory B3LYP energies of HOMO and LUMO orbitals, J. Chem. Inf. Model., 2017, 57(1), 11–21 CrossRef CAS PubMed.
- R. Vijayaraj, V. Subramanian and P. Chattaraj, Comparison of global reactivity descriptors calculated using various density functionals: a QSAR perspective, J. Chem. Theory Comput., 2009, 5(10), 2744–2753 CrossRef CAS PubMed.
- R. G. Parr and R. G. Pearson, Absolute hardness: companion parameter to absolute electronegativity, J. Am. Chem. Soc., 1983, 105(26), 7512–7516 CrossRef CAS.
- P. Geerlings, F. De Proft and W. Langenaeker, Conceptual density functional theory, Chem. Rev., 2003, 103(5), 1793–1874 CrossRef CAS PubMed.
- R. G. Parr, L. v. Szentpály and S. Liu, Electrophilicity Index, J. Am. Chem. Soc., 1999, 121(9), 1922–1924 CrossRef CAS.
- P. K. Chattaraj and S. Giri, Stability, Reactivity, and Aromaticity of Compounds of a Multivalent Superatom, J. Phys. Chem. A, 2007, 111(43), 11116–11121 CrossRef CAS PubMed.
- R. Srivastava, Theoretical studies on the molecular properties, toxicity, and biological efficacy of 21 new chemical entities, ACS Omega, 2021, 6(38), 24891–24901 CrossRef CAS PubMed.
- Z. He, J. Zhang, X. H. Shi, L. L. Hu, X. Kong, Y. D. Cai and K. C. Chou, Predicting drug-target interaction networks based on functional groups and biological features, PLoS One, 2010, 5(3), e9603 CrossRef PubMed.
- N. Flores-Holguín, J. Frau and D. Glossman-Mitnik, Computational prediction of bioactivity scores and chemical reactivity properties of the Parasin I therapeutic peptide of marine origin through the calculation of global and local conceptual DFT descriptors, Theor. Chem. Acc., 2019, 138, 1–9 Search PubMed.
- C. Cárdenas, N. Rabi, P. W. Ayers, C. Morell, P. Jaramillo and P. Fuentealba, Chemical reactivity descriptors for ambiphilic reagents: dual descriptor, local hypersoftness, and electrostatic potential, J. Phys. Chem. A, 2009, 113(30), 8660–8667 CrossRef PubMed.
- S. K. Saha, M. Murmu, N. C. Murmu and P. Banerjee, Evaluating electronic structure of quinazolinone and pyrimidinone molecules for its corrosion inhibition effectiveness on target specific mild steel in the acidic medium: a combined DFT and MD simulation study, J. Mol. Liq., 2016, 224, 629–638 CrossRef CAS.
- J. Hossen, M. A. Ali and S. Reza, Theoretical investigations on the antioxidant potential of a non-phenolic compound thymoquinone: a DFT approach, J. Mol. Model., 2021, 27(6), 173 CrossRef CAS PubMed.
- K. Kaavin, D. Naresh, M. R. Yogeshkumar, M. Krishna Prakash, S. Janarthanan, M. Murali Krishnan and M. Malathi, In-silico DFT studies and molecular docking evaluation of benzimidazo methoxy quinoline-2-one ligand and its Co, Ni, Cu and Zn complexes as potential inhibitors of Bcl-2, Caspase-3, EGFR, mTOR, and PI3K, cancer-causing proteins, Chem. Phys. Impact., 2024, 8, 100418 CrossRef.
- T. Nishiyama, N. Hatae, T. Yoshimura, S. Takaki, T. Abe, M. Ishikura, S. Hibino and T. Choshi, Concise synthesis of carbazole-1,4-quinones and evaluation of their antiproliferative activity against HCT-116 and HL-60 cells, Eur. J. Med. Chem., 2016, 121, 561–577 CrossRef CAS PubMed.
- M. Tercel, H. H. Lee, S. Y. Mehta, J. J. Youte Tendoung, S. Y. Bai, H. S. Liyanage and F. B. Pruijn, Influence of a basic side chain on the properties of hypoxia-selective nitro analogues of the duocarmycins: Demonstration of substantial anticancer activity in combination with irradiation or chemotherapy, J. Med. Chem., 2017, 60(13), 5834–5856 CrossRef CAS PubMed.
- S. J. Choi, J. E. Lee, S. Y. Jeong, I. Im, S. D. Lee, E. J. Lee, S. K. Lee, S. M. Kwon, S. G. Ahn and J. H. Yoon, 5, 5′-substituted indirubin-3′-oxime derivatives as potent cyclin-dependent kinase inhibitors with anticancer activity, J. Med. Chem., 2010, 53(9), 3696–3706 CrossRef CAS PubMed.
- O. Noureddine, N. Issaoui and O. Al-Dossary, DFT and molecular docking study of chloroquine derivatives as antiviral to coronavirus COVID-19, J. King Saud Univ. Sci., 2021, 33(1), 101248 CrossRef PubMed.
- A. Mubarik, S. Mahmood, N. Rasool, M. A. Hashmi, M. Ammar, S. Mutahir, K. G. Ali, M. Bilal, M. N. Akhtar and G. A. Ashraf, Computational Study of Benzothiazole Derivatives for Conformational, Thermodynamic and Spectroscopic Features and Their Potential to Act as Antibacterials, Crystals, 2022, 12(7), 912 CrossRef CAS.
- S. Sakthivel, T. Alagesan, S. Muthu, C. S. Abraham and E. Geetha, Quantum mechanical, spectroscopic study (FT-IR and FT - Raman), NBO analysis, HOMO-LUMO, first order hyperpolarizability and docking studies of a non-steroidal anti-inflammatory compound, J. Mol. Struct., 2018, 1156, 645–656 CrossRef CAS.
- M. Sankarganesh, J. D. Raja, N. Revathi, R. V. Solomon and R. S. Kumar, Gold (III) complex from pyrimidine and morpholine analogue Schiff base ligand: synthesis, characterization, DFT, TDDFT, catalytic, anticancer, molecular modeling with DNA and BSA and DNA binding studies, J. Mol. Liq., 2019, 294, 111655 CrossRef CAS.
- B. K. Paul and N. Guchhait, A spectral deciphering of the binding interaction of an intramolecular charge transfer fluorescence probe with a cationic protein: thermodynamic analysis of the binding phenomenon combined with blind docking study, Photochem. Photobiol. Sci., 2011, 10(6), 980–991 CrossRef CAS PubMed.
- V. T. Sabe, T. Ntombela, L. A. Jhamba, G. E. Maguire, T. Govender, T. Naicker and H. G. Kruger, Current trends in computer aided drug design and a highlight of drugs discovered via computational techniques: A review, Eur. J. Med. Chem., 2021, 224, 113705 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, J. Comput. Chem., 2010, 31(2), 455–461 CrossRef CAS PubMed.
- S. C. Palapetta, H. Gurusamy, S. Krishnan and S. Ponnusamy, Facile Multicomponent Synthesis, Computational, and Docking Studies of Spiroindoloquinazoline Compounds, ACS Omega, 2022, 7(9), 7874–7884 CrossRef CAS PubMed.
- V. Asati, D. K. Mahapatra and S. K. Bharti, PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: Structural and pharmacological perspectives, Eur. J. Med. Chem., 2016, 109, 314–341 CrossRef CAS PubMed.
- M. E. Salem, M. Samir, A. H. Elwahy, A. M. Farag, A. M. Selim, A. A. Alsaegh, M. Sharaky, N. Bagato and I. T. Radwan, Design, synthesis, docking study, cytotoxicity evaluation, and PI3K inhibitory activity of Novel di-thiazoles, and bis (di-thiazoles), J. Mol. Struct., 2024, 1301, 137379 CrossRef CAS.
- M. Yu, Q. Gu and J. Xu, Discovering new PI3Kα inhibitors with a strategy of combining ligand-based and structure-based virtual screening, J. Comput.-Aided Mol. Des., 2018, 32(2), 347–361 CrossRef CAS PubMed.
- P. Chen, Y. L. Deng, S. Bergqvist, M. D. Falk, W. Liu, S. Timofeevski and A. Brooun, Engineering of an isolated p110α subunit of PI3Kα permits crystallization and provides a platform for structure-based drug design, Protein Sci., 2014, 23(10), 1332–1340 CrossRef CAS PubMed.
- G. M. Sastry, M. Adzhigirey, T. Day and R. Annabhimoju, Ligand preparation: parameters, protocols, and influence on virtual screening enrichments, J. Comput.-Aided Mol. Des., 2013, 27, 221–234 CrossRef PubMed.
- J. Wang, P. A. Kollman and I. D. Kuntz, Flexible ligand docking: a multistep strategy approach, Proteins: Struct., Funct., Bioinf., 1999, 36(1), 1–19 CrossRef CAS.
- Systèmes, D., Biovia, Discovery Studio Modeling Environment, Dassault Systèmes Biovia, San Diego, CA, USA, 2016 Search PubMed.
- Biovia, D. S., Discovery Studio, Dassault Systèmes BIOVIA, 2016 Search PubMed.
- A. Singh, K. Kaur, H. Kaur, P. Mohana, S. Arora, N. Bedi, R. Chadha and P. M. S. Bedi, Design, synthesis and biological evaluation of isatin-benzotriazole hybrids as new class of anti-Candida agents, J. Mol. Struct., 2023, 1274, 134456 CrossRef CAS.
- M. Caricato, M. J. Frisch, J. Hiscocks and M. J. Frisch, Gaussian 09: IOps Reference, Gaussian Wallingford, CT, USA, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra01937g |
| This journal is © The Royal Society of Chemistry 2024 |