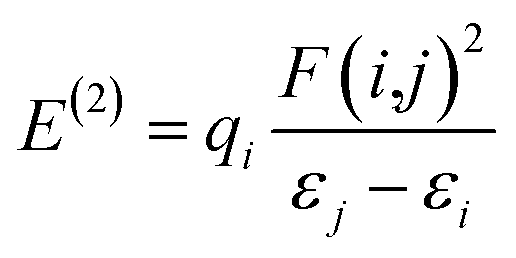DOI:
10.1039/D4RA01846J
(Paper)
RSC Adv., 2024,
14, 14992-15007
Synthesis, characterization, crystal structure, and fabrication of photosensitive Schottky device of a binuclear Cu(II)-Salen complex: a DFT investigations†
Received
10th March 2024
, Accepted 26th April 2024
First published on 8th May 2024
Abstract
This work explores one centrosymmetric binuclear Cu(II)-Salen complex synthesis, characterization, photosensitive Schottky barrier diode (PSBD) function, and DFT spectrum. The crystal growth involves H2LSAL and Cu(NO3)2·3H2O in CH3OH + ACN (acetonitrile) solvent medium. Herein, structural characterization employs elemental, IR/Raman, NMR, UV-VIS, DRS, SEM-EDX, PXRD, SCXRD, and XPS analyses. The complex crystal size is 0.2 × 0.2 × 0.2, showing monoclinic space group C2/c. The dimeric unit contains two Cu(II) centres with distorted square pyramidal (SQP) geometries. The crystal packing consists of weak C–H⋯O interactions. DFT and Hirshfeld surface (HS) further substantiated the packing interactions, providing valuable insights into the underlying mechanisms. The 2-D fingerprint plots showed the presence of N⋯H (3%) and O⋯H (8.2%) contacts in the molecular arrangement. NBO, QTAIM, ELF-LOL, and energy frameworks are utilized to investigate the bonding features of the complex. We extensively studied electrical conductivity and PSBD for H2LSAL and the complex based on band gap (3.09 and 3.07 eV). Like an SBD, the complex has better electrical conductivity, evidencing potentiality in optoelectronic device applications. Optical response enhances conductivity, according to I–V characteristics. Complex Schottky diode has lower barrier height, resistance, and higher conductivity under light. The complex transports charge carriers through space and is rationalized by the ‘hopping process’ and ‘structure–activity-relationship’ (SAR). The charge transport mechanism was analysed by estimating complex mobility (μeff), lifetime (τ), and diffusion length (LD). The experimental and theoretical DOS/PDOS plots provide evidence for the Schottky diode function of the complex.
Introduction
Salen ligands (H2LSAL)/(H2LRSAL), (SAL = salicylaldehyde, R = substituted group), which are well-received Schiff bases (SBs) containing nitrogen (N) and oxygen (O) donor atoms, have been extensively researched over the last few years due to their ubiquitous properties1 and wide range of physicochemical characteristics.2–7 The ligands' N/O-donor atom versatility makes stabilizing components via complex formations across multiple oxidation states.4,5,8 Furthermore, a recent eye-catching trend is investigating (H2LSAL)/(H2LRSAL)-metal complexes or coordination polymers (CPs) electrical conductivity and charge transport properties9–14 (Scheme S1†). Several reasons exist for selecting Salen-metal complexes or their CPs for photo-switching Schottky barrier diode function (SBDF). In the current research, scientists are interested in using metal–organic hybrid materials (MOHM) to discover novel electronic devices for everyday use.15–23 One of the most exciting features of complex molecular solid-state materials is how their unique properties can be modified to meet daily needs like SBD utility. Noteworthy, the conducting properties of a complex material depend on the judicious choice of Mn+ centres, used ligand natures and linkers (SCN−, N3−, DCA (dicyanamide ion), OCN−, SeCN−, etc.) and the extent of resonance within its structural architecture.24–26 Concerning the SBDF using linkers choice, one must be careful with energy bandgap engineering to create effective SBD devices. Fortunately, the bandgap of Salen metal complexes/CPs can be adjusted by controlling linker size. It generally expands the size or conjugation of organic linkers, effectively reducing the band gaps.27 Salen ligands containing aromatic ring π-conjugated systems have also enabled metal complexes to exhibit fascinating electrical conducting properties.28–31 Salen ligand's simplistic synthesis route makes the development of metal complexes time-efficient and less expensive. Incorporating different R-groups within ligand molecules modulates significant characteristics of resulting metal complexes. Therefore, Salen complexes or CPs are ideal for photosensitive switching devices due to their electrical conductivity.32–34 Furthermore, M(II)-Salen (M = versatile metal ions) complexes are quickly produced, yields are satisfactory, and they are in high demand in contemporary photo-switching devices.14 However, due to the numerous limitations, preparing successful semiconducting Salen complexes can be challenging. Notably, binuclear complexes containing two Mn+ ions have undoubtedly piqued the interest of researchers due to their significant research areas, like electrical conductivity, catalytic applications, and magnetism.35–37 It is well-established through recent research that Cu(II) complexes possess fascinating electrical and optoelectronic properties.38–40 An extensive literature survey revealed that loosely bound d9 electrons of Cu(II) centres with square-pyramidal (SQP) shapes enhance conductivity.41,42 Cu(II) complexes are popular due to their structural versatility and ion flexibility.43,44 Due to this incredible attraction of Cu(II) ions, scientists successfully applied it to a metal-based Schottky diode (MBSD) (Table S1†). Henceforth, the intention of novel copper complex synthesis could create electronic materials for the semiconductor industry. In addition, SBDF research is not limited to copper complexes. It has been observed that only a few Cu(II)-complexes exhibit SBDF (Table S1†).27,45 Dincă et al. reported 2-D nature Cu(II)-CP46 with high electrical conductivity. The SBD features under dark and illuminated current–voltage conditions were analysed to provide evidence. Recently, Cd/Zn/Cu complexes have received attention for their excellent electrical and optoelectronic properties (Table S3†).47,48 Although the crystal structure of the Cu(II)-Salen complex has been previously studied in the literature, its properties remain undisclosed.49 We aim to explore the copper complex unveiled PSBD function and analyse electronic properties using DFT.50–53
This research study presents a sound report on the synthesis, crystal structure, and characterizations, including SEM-EDX, DRS, and XPS analysis of a Cu(II)-Salen complex. DFT analysis explores novel bonding features in the complex. We will be discussing the SBD function of H2LSAL compared to complex. The complex better displays electrical conductivity comparable to an SBD, indicating its potential as a semiconductor. The conductivity increased from the dark (2.41 × 10−7 S m−1) to the light phase (4.27 × 10−7 S m−1). The SD device increases the rectification ratio (Ion/Ioff) from 32.59 in the dark phase to 55.17 in the light phase.
Experimental section
Materials and measurements
All the research chemicals utilized are reagent-grade and purchased from Sigma Aldrich, TCI, and Hi Media. PerkinElmer measures the elemental CHN, and the PerkinElmer Spectrum RX 1 instrument analyses IR spectra. The Bruker RFS 27 model is used to analyse the Raman spectra. NMR spectra were collected using a Bruker FT-NMR spectrometer (400 MHz–75.45 MHz). The Oxford XMX N model was used to manage the EDX data, while the JEOL JSM-6390LV and BRUKER AXS were used to present the SEM figures and PXRD. U-3501 spectrophotometer model is used for UV-visible spectra. The PerkinElmer Lambda 365 model (200–1000 nm) was utilized for the Diffuse reflectance spectroscopic study (DRS). XPS spectra were recorded on a model thermo-scientific NEXA Surface analyser.
DFT methodology
The stability of the copper complex was assessed using the DFT/B3LYP-D3/lanL2DZ level of theory, which was implemented in the Gaussian09 software.54–58 Electronic plots, including Density of States (DOS), partial density of plots (PDOS), and Frontier Molecular Orbitals (FMO), were calculated using TD-DFT theory.59,60 Multiwfn package61 generated LOL and ELF. QTAIM topological analyses revealed that a series of electrostatic interactions stabilize the complex. Additionally, the natural bond orbital (NBO) analysis highlighted hyperconjugation interactions that contribute to the stability of the complex.62
X-Ray crystallography
We collected SCXRD data using a Bruker Smart CCD diffractometer (RT). The diffractometer used Mo Kα radiation with a wavelength (λ) of 0.71073 Å. We utilized multiple crystallographic programs to decipher the intricate crystal structure. SMART was used to perform information frames, index reflections, and collect scale and lattice parameters.63 We used the SAINT64 software to reduce the crystal data. The multi-scan technique SADABS65 was utilized to apply scaling and absorption corrections. We solved and refined the structure using the direct F2 method with SHELXS and SHELXL-2014 programs.66 We used the latest Mercury software to represent the crystal structures visually. We refined the first non-H atoms iso-tropically. For the 2nd step, anisotropic refinement was performed using SHELXL's full-matrix least-square method67 based on F2. The crystal structure in the checkcif report shows no A-level alert (R = 0.03), indicating high-quality crystal data. The CCDC number 2329455 is linked to the complex. The crystallographic information and structure refinement parameters are presented in Table 1.
Table 1 Crystal data and full structure refinement
| Empirical formula |
C32H28Cu2N4O4 |
| Formula weight |
659.68 |
| Temperature/K |
293(2) |
| Crystal system |
Monoclinic |
| Space group |
C2/c |
| a/Å |
26.628(3) |
| b/Å |
6.9771(7) |
| c/Å |
14.7128(15) |
| α/° |
90 |
| β/° |
97.479(2) |
| γ/° |
90 |
| Volume/Å3 |
2710.2(5) |
| Z |
4 |
| ρcalc g cm−3 |
1.617 |
| μ/mm−1 |
1.617 |
| F(000) |
1352.0 |
| Crystal size/mm3 |
0.2 × 0.2 × 0.2 |
| Radiation |
MoKα (λ = 0.71073) |
| 2Θ range for data collection/° |
5.586 to 49.362 |
| Index ranges |
−31 ≤ h ≤ 30, −8 ≤ k ≤ 8, −17 ≤ l ≤ 17 |
| Reflections collected |
39![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 865 865 |
| Independent reflections |
2295 [Rint = 0.0549, Rsigma = 0.0196] |
| Data/restraints/parameters |
2295/0/190 |
| Goodness-of-fit on F2 |
1.181 |
| Final R indexes [I ≥ 2σ (I)] |
R1 = 0.0307, wR2 = 0.0799 |
| Final R indexes [all data] |
R1 = 0.0346, wR2 = 0.0822 |
| Largest diff. peak/hole/e Å−3 |
0.34/−0.54 |
Optical characterization
The UV-vis absorption spectrum of the synthesized ligand (H2LSAL) and the complex characterized the optical properties. The absorption spectra of H2LSAL and the copper complex were collected in the 270–700 nm (Fig. S1A,† inset). Applying Tauc's equation on the obtained absorbance spectra, the optical direct band gap (Eg) was computed as 3.09 eV and 3.07 eV for H2LSAL and the complex, respectively (Fig. S1A†) (eqn (S1)†)68 (ESI, Section 1.1†). The values mentioned are like those of copper complexes characterized by X-ray and published in the literature. Also, ligand (H2LSAL) optical bandgap values are comparable to the bandgap of the published Schiff base (Table S1†). The copper complex shows a better semiconductor-like band gap. Therefore, we intend to investigate its electrical conductivity accordingly.
Device fabrication
The synthesized ligand (H2LSAL) and copper complex will be semiconducting, as the calculated optical band gap (Eg) indicates. A thin film metal–semiconductor junction device, abbreviated as LTFD and CTFD, is developed following a previously reported procedure (ESI, Section 1.2†).
Electrical characterization
The Eg that was just calculated makes sense of specific semiconducting properties of the synthesized ligand (H2LSAL) and the copper complex. Therefore, we needed to perform some electrical tests to analyse their characteristics. Two thin-film metal–semiconductor junction devices were created using the previously reported procedure69 (ESI, Section 1.3†). In this regard, CTFD is the thin film device made from the complex, and LTFD is the thin film device made from the H2LSAL. The active devices were fabricated on an ITO-coated glass substrate of size 2 cm × 2 cm, with a circular Al electrode of diameter 1.5 cm. The I–V properties of the thin film devices were analysed under dark and illumination conditions while applying ±2 V bias voltage. The illuminator had an intensity of 100 mW cm−2, and the temperature was 300 K. The charge transport phenomenon was explored by collecting current–voltage (I–V) data using Keithley Model No: 2400 source.
Synthesis of H2LSAL
The ligand was prepared using the commonly published method.48 Yield: (92%), Anal. Calc. for C16H16N2O2: C, 71.62; H, 6.01; N, 10.44. Found: C, 71.95; H, 6.50; N, 10.36%. IR (KBr cm−1) selected bands: ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 1642, ν(C–Ophenolic) 1280, ν(CC), 1587, ν(C–H), 2900, ν(O–H), 3660, 1H NMR (DMSO-d6, 400 MHz): δ (ppm): 13.25 (1H, OH), 8.34 (1H, NCH), 6.84–7.30 (8H, Ar-H), 3.92 (2H, N–CH2), 13C NMR (DMSO-d6, 75.45 MHz): δ (ppm): 116.97–132.43 (Arom-C), 161.01 (C–OH), 166.53 (CHN), UV-Vis λmax (CH3OH): 278, and 368 nm.
N), 1642, ν(C–Ophenolic) 1280, ν(CC), 1587, ν(C–H), 2900, ν(O–H), 3660, 1H NMR (DMSO-d6, 400 MHz): δ (ppm): 13.25 (1H, OH), 8.34 (1H, NCH), 6.84–7.30 (8H, Ar-H), 3.92 (2H, N–CH2), 13C NMR (DMSO-d6, 75.45 MHz): δ (ppm): 116.97–132.43 (Arom-C), 161.01 (C–OH), 166.53 (CHN), UV-Vis λmax (CH3OH): 278, and 368 nm.
Synthesis and crystal growth of [Cu2(LSAL)2]
The complex was synthesized in situ using a mixed solvent mixture of methanol (CH3OH) and acetonitrile (ACN). After being gently boiled, ligand H2LSAL (0.269 g, 1 mmol) was dissolved in a methanol solvent (15 mL). After that, Cu(NO3)2·3H2O in 10 mL ACN (0.242 g, 1 mmol) was charged into the ligand solution. Immediately, a green solution was the result. The solution mixture was refluxed for about one hour. Then, the solution is cooled and stirred with a magnetic stirrer for 30 minutes. The solution is kept at room temperature for 25 minutes and then filtered. Under this condition, the green solution crystallization began under freezing conditions. The plate-shaped green single-crystals suitable for SCXRD were obtained after 25 days. The crystals were delicately stored and dried in the air using a desiccator. Yield: 70%, Anal. Calc. for C32H28Cu2N4O4: C, 58.26; H, 4.28; N, 8.49, Cu, 19.27. Found: C, 57.99; H, 4.09; N, 8.52, Cu, 20.03%. FT-IR (KBr cm−1) selected bands: ν(CN), 1638, ν(Cu–N), 425, ν(Cu–O), 452–510, ν(CC), 1590, ν(Ar-O), 1275, Raman spectra (cm−1) selected bands: ν(CN), 1642, ν(Cu–N), 512, ν(CC), 1605, ν(Ar-O), 1298, 1H NMR (DMSO-d6, 400 MHz): δ (ppm): 6.57–7.03 (Arom-H), 8.33 (–HCN), UV-Vis λmax (DMF): 291, 349, and 705 nm. DRS: 279, 387, and 774 nm.
Results and discussion
Research overview
The H2LSAL was expertly synthesized using the well-established method described in the previous literature.48 The H2LSAL and Cu(NO3)2·3H2O in CH3OH + ACN (1:1 M) self-assembled at room temperature to yield a water-insoluble green binuclear copper complex in crystal form with a satisfactory yield (Scheme 1). The compound has been structurally characterized using various analytical techniques like IR/Raman/NMR spectroscopy, CHN, SCXRD, PXRD, SEM/EDX, and XPS analysis. SCXRD ensures that the asymmetric unit of the copper complex comprises two Cu(II) metal ions and deprotonated [LSAL]2−. The PXRD analysis has confirmed the phase's purity and crystallinity. The XPS studies supported complex formation between H2LSAL and Cu(II) metal ions. The literature contains limited information on the function of photosensitive Schottky barrier diode (PSBD) in copper complexes (Tables S1 & S3†).27,45 In contrast, the copper complex's crystal structure has already been explored.49 Herein, we divulge the PSBD function of Cu(II)-Salen complex entirely in a new dimension. We implemented DFT to characterize the complex bonding features, including NBO, QTAIM, ELF-LOL, and interaction energy (IE) and energy frameworks (EF).
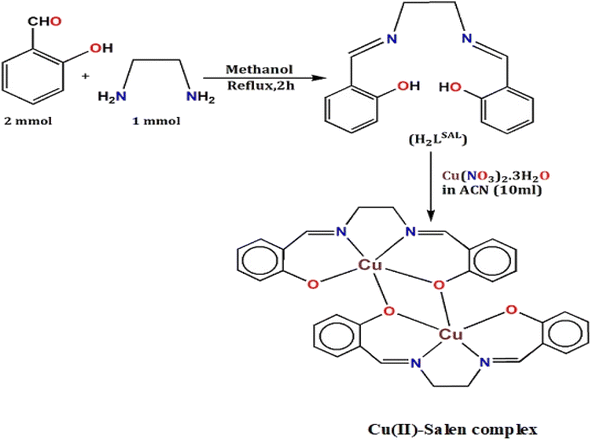 |
| | Scheme 1 Synthetic routes for H2LSAL and the copper complex. | |
Structural characterization
FT-IR/Raman spectroscopy. IR and Raman spectroscopic studies accomplished the structural characterizations of the ligand (H2LSAL) and the copper complex (Fig. S1–S3†). Accordingly, the significant IR/Raman bands in the complex are identified ν(CHN), ν(C–O), ν(M1–O)/ν(M1–N) (M1Cu), and ν(CC). The complex spectra showed a significant shift in connection of ν(CHN) compared to the free H2LSAL. The peak H2LSAL was observed at 1642 cm−1, while changes were observed at 1638 cm−1 in the complex. The modification above explores that the azomethine N-atom participates in the complexation.70,71 This IR shift value may be caused by the ν(CHN) bond weakening following complexation.72 The above behaviour is explained by donating imine N-atom e → Cu(II) ion partially vacant d orbital.73,74 The complex's ν(C–O) stretching bands shifted significantly to 1275 cm−1 compared to H2LSAL. In the complex, the vibrational bands of the aromatic ring's ν(CC) appeared at 1590 cm−1. The bands nearer 452–510 and 425 cm−1 correspond to the ν(M1–O)/ν(M1–N). The coordination between the deprotonated form of H2LSAL and Cu(II) ions is facilitated by the N-atom of (CHN), as confirmed by the SCXRD structure. Additionally, Raman spectra of the complex were characterized, and the following Raman stretching bands in cm−1 were identified: ν(CHN), ν(C–O), ν(M1–N), and ν(CC), at 1642, 1298, 512, 1605, cm−1, respectively. Therefore, IR and Raman spectra are an excellent choice for characterizing H2LSAL and copper complex structural frameworks.
UV-visible and DRS spectrum. The H2LSAL and copper complexes were investigated using UV-visible absorption and diffuse reflectance spectra (DRS) to identify possible optical transitions (Fig. S4–S6†). Generally, the UV region shows transition due to the ligand (H2LSAL), while the visible domain shows d–d transition.75,76 The UV spectra of the H2LSAL and complex were analysed in methanol and DMF solvents. The H2LSAL absorption bands indicate π → π* and n → π* transitions near 278 and 368 nm. The UV spectrum of the complex in DMF exhibits bands at 291, 349, and 705 nm, respectively. The first UV bands arise from the π → π* intra-ligand charge transfer (IL → CT) transition.77–79 The band at 349 nm is due to the CT from the ligand H2LSAL → M, known as L → M charge transfer (L → MCT). Notably, ligand (H2LSAL) chelation by N and O-donor atoms with Cu(II) ions causes a shift towards longer wavelengths than the free ligand.80 A broad weak peak is identified at 705 nm for the complex. It was assigned due to d–d transitions (2Eg → 2T2g) for the Cu(II) complex.43,81 The absorption bands of these copper complexes are like those characterized by X-ray.82,83 The copper complex diffuse reflectance spectroscopy (DRS) further checks for any shoulder peaks. Two slightly shifted bands are observed here, along with a shoulder. The absorption bands observed at 279 nm and 387 nm result from intra-ligand transitions of the π → π* or n → π* type. A complete broad shoulder peak is also identified at 774 nm due to d–d transitions (Cu(II) 3d9 system). Henceforth, the nature of the DRS spectrum ensures the coordination mode of the ligand using N/O-donor centres with Cu(II) metal ions.
1H/13C ∼ NMR spectra. An NMR spectroscopic study was conducted separately to characterize the H2LSAL structural frameworks and copper complex (Fig. S7–S9†). In the case of the H2LSAL, we utilized a combined approach using 1H/13C-NMR spectroscopy, whereas, for the complex, we only conducted a proton NMR study. We began by examining the 1H NMR spectra of both compounds to establish their characteristics. The absence of the –NH2 group in the H2LSAL formation is confirmed by the lack of peak values between δ 5.0 to 8.1 ppm. The δ 13.25 ppm value is evidence of unbound O–H in the ligand (H2LSAL). Again, δ 6.84–7.30 ppm indicates the presence of aromatic hydrogens in the ligand. The azomethine proton peak is observed in the H2LSAL at a value of 8.34 ppm. We further characterized the H2LSAL using 13C NMR spectral data. The 13C NMR spectra of the H2LSAL showed peaks at (CHN)–C 166.53 ppm, Aro-C 116.97–132.43 ppm, and C–OH 161.01 ppm. Concerning the complex, 1H NMR peaks for aromatic and azomethine protons are δ 6.57–7.03 ppm and 8.33 ppm, respectively. Furthermore, the proton peak at O–H vanished in the 1H NMR spectra of the complex, indicating coordination between the O-atom and Cu(II) metal ions after deprotonation of the ligand (H2LSAL).84,85
SEM-EDX. In principle, the elemental composition in the complex is characterized using an EDX profile. According to the EDX analysis (Fig. S10†), the compound comprises carbon (C), oxygen (O), nitrogen (N), and Cu metal ions. The EDX spectrum's highest peak is C, followed by O and Cu. The weight percentage contribution of carbon (C), oxygen (O), and copper (Cu) metal reflect the energy-dispersive X-ray (EDX) profiles. The elements C, O and N come from the ligand H2LSAL. Therefore, the EDX profile confirms the interaction between Cu(II) ions with H2LSAL. The SEM micrograph of the ligand (H2LSAL) and the copper complex (Fig. S11 and S12†) has tested the surface morphology and the sample's homogeneity.86 The SEM image of H2LSAL shows irregular ice-like morphology (Fig. S12†) with a distributed structure. In contrast, the surface of the complex is smooth and continuous, with a well-organized ice morphology.
PXRD. The powder X-ray diffraction (PXRD) study verified the phase purity and crystallinity of the copper complex (Fig. S13†). In our complex, the experimental XRD pattern of our bulk product, both before and after deposition, matches the simulated XRD pattern very closely. It indicates that they are in excellent agreement with each other. The CCDC Mercury software generated the simulated pattern from single-crystal X-ray diffraction (SCXRD) data (CIF). Therefore, proceeding according to the PXRD, it has been found that the bulk material is a single crystal.
X-Ray photoelectron spectroscopy. X-Ray photoelectron spectroscopy (XPS) studies strongly support the formation of the Cu(II) complex between H2LSAL and Cu(II) metal ions (Table S4/Fig. S14†). This information is informative and thoughtfully presented. The structure H2LSAL demonstrates the formation of a primary coordination core with N2O2 to Cu(II) ions. The XPS scan is conducted for the synthesized complex, which includes C 1s, O 1s, N 1s, and Cu 2p.87 We have compared the free ligand's XPS binding energy data (eV) with that of the Cu(II)-complex. The ligand is a standard Salen ligand, and its usual XPS binding energy (eV) will be considered. Our study reveals Cu(II) peak shifts in the complex compared to the free ligand for Cu, N, and O XPS peaks.87 The peak position in the C 1s spectrum of ligand shifted to 285.74 eV upon complexation with Cu(II) ions.87,88 The N 1s spectra peak shifts from 399.64–402.01 when the ligand coordinates with copper metal ions. The XPS N1s peak in the complex has been separated into two distinct peak components through deconvolution. The imine and amine correspond to peak components at 399.2 and 400.2 eV, respectively.89 The binding energy of the amine remained almost unchanged. The imine binding energy shifted slightly, representing only the ligand N-atom of the imine group coordinated with copper metal ions.90 The outcome was in line with the UV and IR spectral information. Similarly to the O 1s spectra, the peak of the ligand was shifted to 532.16 eV in its complex.90 Copper was detected via a Cu 2p XPS image, with a binding energy range of 935.31 and 954.80 eV.88,90 In the complex, the divalent state of copper metal ions ensures this BE value.90 No Cu(I) is present in the complex as the Cu 2p XPS image didn't find the applicable BE value 931.8 eV.90 Therefore, during XPS analysis, the complex does not undergo a reduction of Cu(II) to Cu(I) caused by X-ray irradiation.88 The Cu 2p satellite peak has broadened, possibly due to a distortion in planar geometry.88 Thus, XPS reveals the binding of H2LSAL through N/O-donor centers by Cu(II) ions.
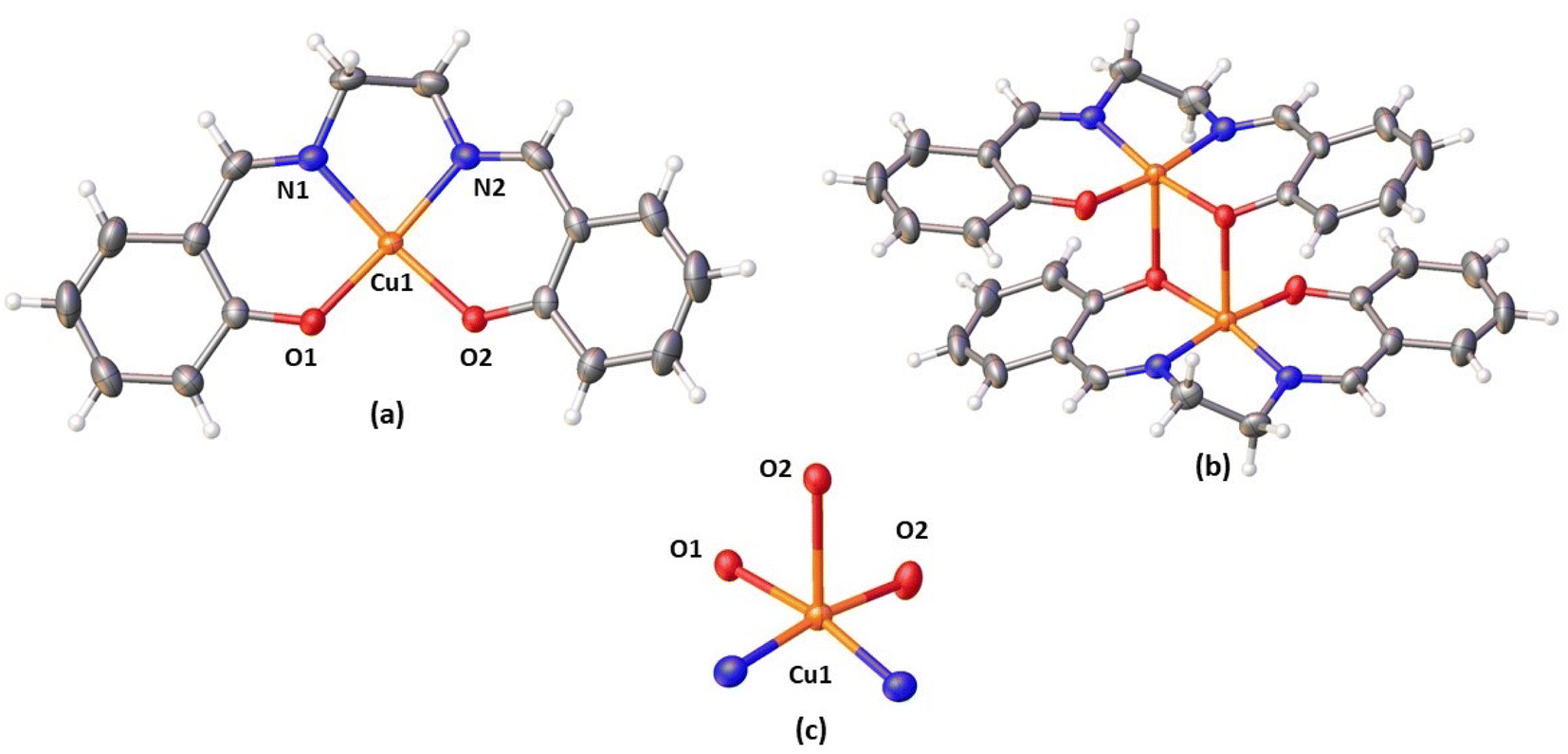
X-ray crystal structure. The copper complex exhibits monoclinic space group C2/c. According to the SCXRD, the complex has a centrosymmetric binuclear structure [Cu2L2SAL] with a double phenoxide bridge. The copper complex cell parameters were determined through crystallography as: a = 26.628 (3) Å, b = 6.9771 (7) Å, c = 14.7128 (15) Å, and α = γ = 90° and β = 97.479 (2), Z = 4, and volume = 2710.2 (5). Table S2† contains important crystallographic parameters that have been collected for the complex. Fig. 1 explores the complex perspective view with thermal ellipsoid probability 50%. ORTEP view of the complex is shown in Fig. S15† (atoms are shown as 50% thermal ellipsoids). Both copper(II) centres have distorted square pyramidal (SQP) geometries in the dimeric unit. The central metal atom, Cu(II), is surrounded by two imines (CHN) N-atoms [N(1), N(2)] and two phenoxide (Ar-O−) O-atoms [O(1) and O(2)]. All come after the [LSAL]2−, constituting the basal plane. For the complex, the coordinating atoms in their respective basal plane deviate only slightly from the mean plane: just 0.030(2), 0.024(2), 0.0274(19), and 0.0318(17) Å for N(1), N(2), O(1) and O(2). The Cu(1) deviated value is −0.1138(3) Å away from its average position. Each copper(II) centre is connected to a phenoxide (Ar-O−) O-atoms [O(2)] of the H2LSAL ligand from another unit, which is symmetry-related (1.5 − x, 1/2 + y, 1.5 − z). Doing so fulfils the penta-coordinate geometry of each copper(II) centre, and the H2LSAL ligand bridges the two copper(II) centres. The Addison parameter (τ) = 0.128 (τ = (β − α)/60) indicates a distorted-square-pyramidal geometry.91–93 In the basal plane, M–O and M–N distances are 1.908(2) and 1.941(2) Å, and 1.948(2) and 1.953(2) Å, whereas the axial bond length M(1)–O(2)* [* = 1.5 − x, 1/2 + y, 1.5 − z] is 2.4122) Å (M = Cu). Two copper(II) atoms with a penta-coordinated geometry are connected by two phenoxide O-atoms, which bridge the copper atoms together to form a Cu2O2 core. Two copper(II) centres in the Cu2O2 core are separated by 3.1950(6) Å. The bridging angle is 93.83(6), with the formula [M(1)–O(1)–M(1)*, * = 1.5 − x, 1/2 + y, 1.5 − z]. Cu2O2 core is symmetric, a standard feature for similar dimer complexes, as bridging angles and bond lengths are equal.93–95 The bond distances and angles of the synthesized copper complex are comparable to those of the published analogous complexes (Tables S1 & S3†).
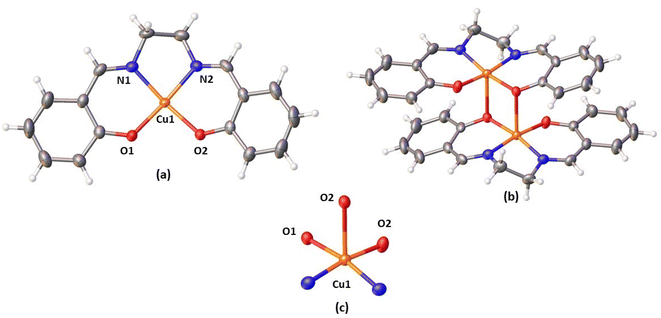 |
| | Fig. 1 Perspective view of (a) asymmetric unit with selective atom numbering scheme, (b) complex, (c) square pyramidal geometry around Cu(II) (thermal ellipsoid probability 50%). | |
Supramolecular interactions. In the copper complex, the crystal packing consists of weak C–H⋯O interactions. The hydrogen atom, H(8A), attached to the carbon atom, C(8), forms C–H⋯O interaction with an oxygen atom, O(2). These interactions create a 1-D structure (Fig. 2). The details of the geometric features are explored in Table 2.
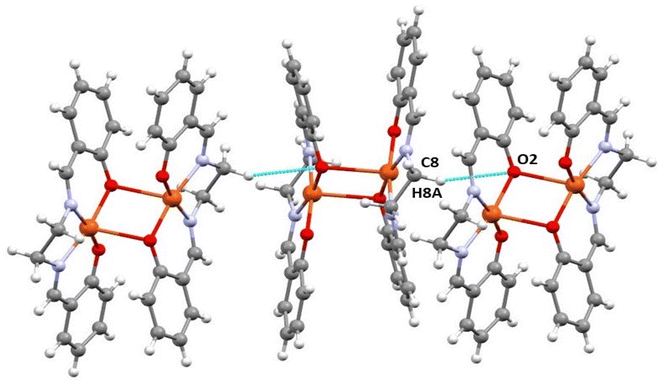 |
| | Fig. 2 Perspective view of C–H⋯O interactions in the complex (selective atom numbering scheme). | |
Table 2 Hydrogen bond distances (Å) and angles (°) of the complexa
| D–H⋯A |
D–H |
H⋯A |
D⋯A |
∠D–H⋯A |
| D = donor; H = hydrogen; A = acceptor. a = 1.5 − x, 1/2 + y, 1.5 − z. |
| C(8)–H(8)–O(2)a |
0.97(3) |
2.71(2) |
3.61(3) |
155.6(2) |
DFT investigations.
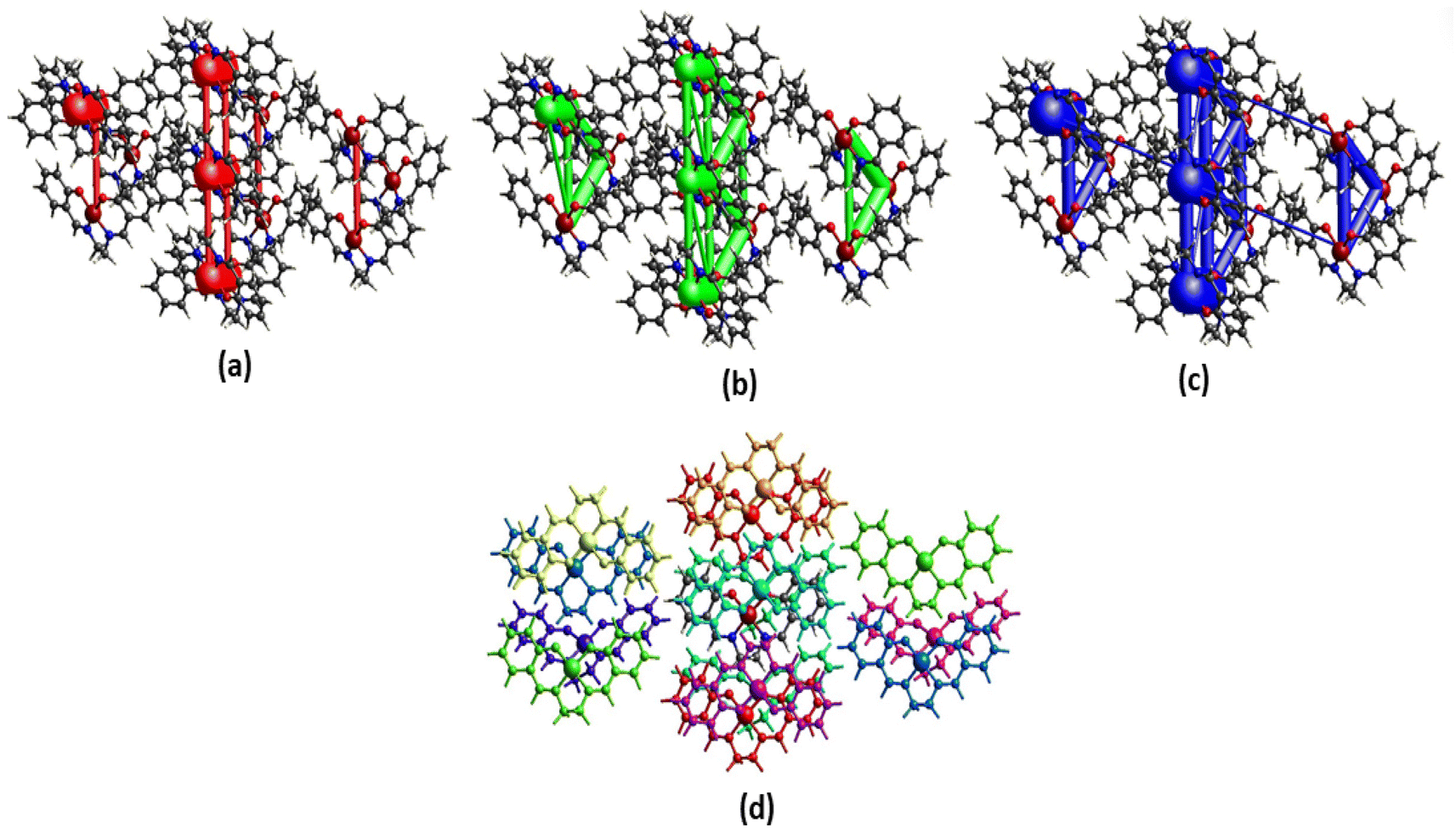
Hirshfeld surface. Hirshfeld surface analysis is theoretically utilized to study different forms of supramolecular contacts. HS has been expertly mapped using dnorm (−0.5 → 1.5 Å), shape index (SI) (−1.0 → 1.0 Å), curvedness (C) (−4.0 → 0.4 Å), and fragment patch (FP) (0 → 15 Å) (Fig. 3). HS are colour-coded system representing the distances between atoms in the crystal lattice. Specifically, the blue circular zones indicate shorter interatomic distances, while the red circular zones represent longer interatomic distances. The white zone that appears on surfaces is a representation of the van der Waals radii contacts. The Hirshfeld surface plots show significant red regions due to N⋯H (3%) and O⋯H (8.2%) contacts. The 2-D fingerprint plots were generated to effectively represent the intermolecular contacts/interactions based on de and di.
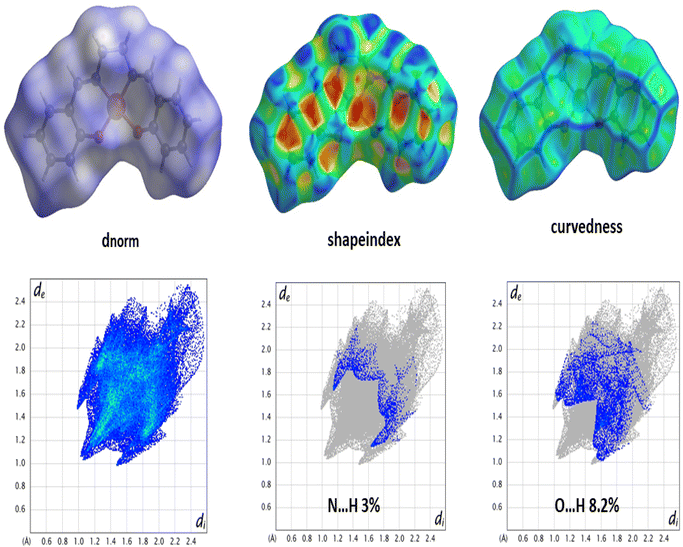 |
| | Fig. 3 HS mapped with dnorm, shape index, curvedness (top) and 2-D fingerprint plot for the corresponding interactions (bottom). | |
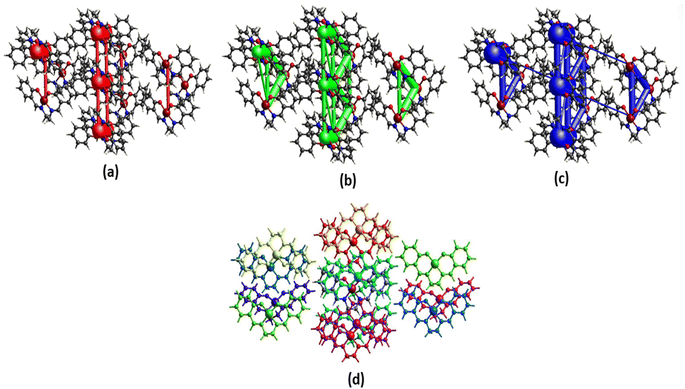 |
| | Fig. 4 Perspective views of electrostatic energy (a), dispersion energy (b), total energy diagrams (c), and different interaction energies of the molecular pairs (d) energy framework for a cluster of molecules in the complex. The cylindrical radius is proportional to the relative strength of the corresponding energies. The scale factor used is 50 with cut-off values of 10 kJ mol−1. | |
LOL-ELF. The LOL and ELF are highly effective methods for comprehending chemical bonding. They enable the identification of key locations in molecular space where electrons are concentrated, including bonding, nonbonding, and lone pair regions.100 LOL uses η(r), and ELF uses τ(r) to quantify excess kinetic energy density due to Pauli repulsion.101 ELF is based on electron pair density, while LOL recognizes that localized orbital gradients are maximized when they overlap.100 Contour maps and shaded surfaces illustrating the ELF and LOL for the studied complex are carried out using the Multiwfn_3.7 program61 and presented in Fig. 5(a)–(c). Our ELF values, τ(r), range from 0.0 to 1.0, with higher values (0.5 to 1.0) indicating the presence of bonding and nonbonding localized electrons. Lower values (<0.5) suggest that electrons may be delocalized. Meanwhile, LOL, η(r), achieves significant values (>0.5) in areas dominated by electron localization.102 Excitingly, the ELF maps reveal the high regions around H15, H33, and H50, indicating the existence of both bonding and nonbonding electrons. A faint red spot surrounding these selected H atoms suggests the presence of an excess of delocalized electrons, which are blocked in their regions by O–Cu groups. This finding indicates that a high charge transfer occurs on the surface and between ligands (H2LSAL), as confirmed by the localization of molecular orbitals. Furthermore, blue areas surrounding specific C atoms suggest the existence of a delocalized electron cloud. It indicates that O–Cu groups may have a tunnelling effect that causes the electrons to localize in these active regions, thereby enhancing the accumulation of electrons in all the rings. Upon examination of the LOL maps, it's clear that the central region of the H atom (H15) is incredibly dense with electrons, as shown by the pure white colour. This density exceeds the maximum value on the colour scale (0.80). C atoms exhibit covalent regions readily identifiable through their high LOL value, highlighted in red. Excitingly, we can observe electron depletion regions, represented by blue circles around C and O nuclei, which show the distance between the valence shell and the inner shell.
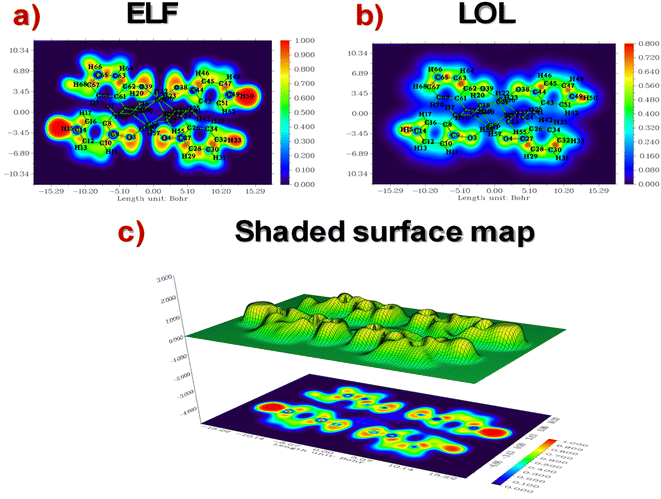 |
| | Fig. 5 ELF-LOL contour maps and shade surfaces of copper complex. | |
QTAIM. The QTAIM theory, created by Bader103,104 is a dependable approach for examining interactions within and between molecules, especially hydrogen bonding. Recent research suggests that within each molecule lies a crucial point called the bond critical point (BCP). This point is vital in determining the unique nature of chemical interactions within the molecular structure.105,106 At this point, various topological parameters can be computed, including the electronic density (ρ(r)) and its Laplacian (∇2ρ(r)), the kinetic energy density (KED) (G(r)), the potential energy density (PED) (V(r)), the total energy density (TED) (H(r)) where H = G(r) + V(r), ellipticity (ε), Hessian eigenvalues (λ1, λ2, and λ3), and the interaction energy (Eint = V(r)/2). The AIM graph and the calculated topological parameters are shown in Fig. S16† and summarized in Table 3. Based on the symmetrical inversion among identical molecules, we chose the seven BCPs, as shown in the graph. Atomic interactions are classified as shared (covalent and polar bonds) with charge density contracting between nuclei (∇2ρ(r) < 0) and closed shell (H-bonds, ionic bonds, and van der Waals forces) with charge density contracting towards each nucleus (∇2ρ(r) > 0).107 Various types of interactions were observed within the copper complex, including Cu⋯Cu, N⋯O, O⋯H, and C⋯H interactions. The values for ρ(r) and ∇2ρ(r) range from 0.005 to 0.014 (a.u.) and 0.015 to 0.65 (a.u.), respectively. Importantly, all ∇2ρ(r) values are positive, indicating the presence of closed-shell interactions. Furthermore, all H(r) values, as per the criteria proposed by Rozas et al.,108 are positive, indicating weak and electrostatic interactions, except for BCP1, which shows medium and partially covalent characteristics. The energy of interactions is also employed to assess bond strength at the BCPs, with Eint values below 50 kJ mol−1 (12 kcal mol−1), indicating weak interactions within the studied complex. The most substantial interaction occurs when bonding the two metal atoms (Cu). Ellipticity (ε) is a key metric reflecting the stability of a bond, where a decrease in its value correlates with an increase in bond stability.109 This parameter is characterized by low values ranging from 0.076 a.u. to 0.826 a.u. for 2/2′, 4/4′, and 6/6′ BCPs, indicating the excellent stability of these bindings. In conclusion, the two symmetrical ligands in the complex are stabilized by a total of fourteen van der Waals interactions, which significantly contribute to the overall stability of the complex. This level of stabilization is not frequently reported in the literature for complexes stabilized by such a specific number of electrostatic interactions. The high stability of our newly synthesized compound makes it promising for a wide range of applications, including electronic and optical devices. Furthermore, the numerous interactions between the ligands facilitate a substantial charge transfer, as evidenced by both Frontier Molecular Orbital (FMO) and Electron Localization Function (ELF) analyses.
Table 3 Selected QTAIM topological parameters of the copper complex
| BCPs |
ρ(r) |
∇2ρ(r) |
G(r) |
V(r) |
H(r) |
λ1 |
λ2 |
λ3 |
ε(r) |
Eint (kJ mol−1) |
| 1 |
0.008 |
0.015 |
0.005 |
−0.006 |
−0.001 |
−0.0018 |
0.022 |
−0.004 |
1.409 |
−7.876 |
| 2/2′ |
0.014 |
0.65 |
0.013 |
−0.010 |
0.0027 |
−0.016 |
−0.015 |
0.096 |
0.076 |
−13.127 |
| 3/3′ |
0.006 |
0.024 |
0.004 |
−0.003 |
0.0012 |
−0.0009 |
0.028 |
−0.003 |
2.262 |
−3.938 |
| 4/4′ |
0.005 |
0.019 |
0.003 |
−0.003 |
0.0008 |
−0.0016 |
0.023 |
−0.0023 |
0.826 |
−3.938 |
| 5/5′ |
0.005 |
0.019 |
0.003 |
−0.002 |
0.001 |
−0.0008 |
−0.0025 |
0.023 |
2.034 |
−2.625 |
| 6/6′ |
0.006 |
0.021 |
0.004 |
−0.003 |
0.001 |
−0.002 |
0.02 |
−0.003 |
0.461 |
−3.938 |
| 7/7′ |
0.005 |
0.019 |
0.003 |
−0.002 |
0.001 |
−0.0004 |
0.02 |
−0.003 |
6.005 |
−2.625 |
Natural bond orbital (NBO). Implementing natural bond orbital (NBO) analysis110 can explore the hyperconjugation interactions within a molecule. These interactions stabilize the system by transferring electron density from a donor to an acceptor orbital.111 The strength of these interactions is directly proportional to the level of stabilization energy: stronger molecular interactions correspond to higher associated energy. To explore intermolecular delocalization, a comprehensive analysis utilizing NBO and second order Fock matrix perturbation theory was conducted for the studied complex.112 The crucial hyper conjugative interactions in our complex are outlined in Table S6.† We estimate the SE (stabilization energy) E(2), an essential parameter linked to delocalization from D (donor) (i) →A (acceptor) (j), using the method.| |
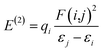 | (1) |
According to eqn (1), each term has its usual significance. The E(2) value indicates the strength and extent of conjugation within the system; however, only interactions with E(2) values exceeding 10 kcal mol−1 are considered in our discussion. The NBO results indicate the presence of six types of interactions: LP → RY*, LP → LP*, π → π, π → π*, π → π, and LP → π*, which collectively contribute to the stabilization of the analyzed complex. The notable hyper conjugative interactions identified include π*(C8–C9) → π*(C26–C27)/(C10–C12)/(C14–C16), π*(C43–C44) → π*(C45–C47)/(C49–C51), and π*(C61–C62) → π*(C67–C69). These interactions exhibit high perturbation energies, specifically 287.72, 260.58, and 262.99 kcal mol−1, respectively, highlighting the robust conjugative nature of the CC bonds involved.
Schottky device and photo-sensing function
Ligand (H2LSAL) vs. complex. Fig. 6(A) shows the I–V curves of a ligand-based (H2LSAL) thin film device (LTFD) and a copper complex-based thin film device (CTFD) in both light and dark conditions. The I–V characteristic of the thin film complex-based device is shown in Fig. 6(B) with a logarithmic scale. The electrical conductivity values were computed as 2.79 × 10−6 S m−1 and 2.37 × 10−7 S m−1 for CTFD and LTFD under without illumination conditions, respectively. At illumination conditions, the electrical conductivity seems to be improved for CTFD as 4.27 × 10−5 S m−1, but for LTFD, it remains the same as 2.41 × 10−7 S m−1. This thin film device shows improved electrical conductivity under illumination, indicating photo-induced charge generation. It is noteworthy that the I–V graphs obtained from the bare ligand-based (H2LSAL) device (LTFD) exhibits no Schottky behaviour and lacks photo-sensing characteristics under both dark and illuminated conditions (Fig. 6(A). Therefore, concerning to the H2LSAL, our copper-complex-made devices (CTFD) exhibit both Schottky and photo-sensing characteristics. As shown in Fig. 6(B), the device exhibits nonlinear rectifying characteristics, indicating that it is a Schottky Barrier Diode (SBD). We have measured specific device parameters to compare the performance of CTFD and LTFD. Without light irradiation at the bias voltage ±2 V, the on/off ratio (Ion/Ioff) has been calculated as 32.39 and 1.02 for CTFD and LTFD. Meanwhile, under irradiation conditions, it has been computed as 55.17 and 1.03 for CTFD and LTFD, respectively. The CTFD devices have a photo-responsivity of 3.59, while LTFD devices have a photo-responsivity of 1.07. Table 4 compares the conductivity and photosensitivity of the bare ligand systems CTFD and LTFD.
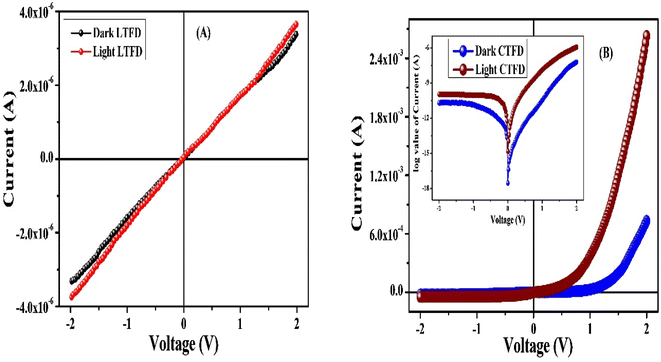 |
| | Fig. 6 (A) I–V graph (under light and dark conditions) of our fabricated thin film devices made from the ligand (H2LSAL), and (B) the complex, and the corresponding characteristic in log value (inset of (B)). | |
Table 4 Calculated device parameters for CTFD and LTFD
| Device |
Condition |
Rectification ratios (Ion/Ioff) |
Conductivity (S cm−1) |
Photosensitivity |
Ideality factor (η) |
Barrier height (∅B) (eV) |
Series resistance (RS) |
| From dV/dlnI (KΩ) |
From H (KΩ) |
| LTFD |
Dark |
1.02 |
2.37 × 10−7 |
1.07 |
— |
— |
— |
— |
| Light |
1.03 |
2.79 × 10−6 |
— |
— |
— |
— |
| CTFD |
Dark |
32.59 |
2.41 × 10−7 |
3.59 |
2.05 |
0.68 |
64.26 |
64.78 |
| Light |
55.17 |
4.27 × 10−5 |
1.43 |
0.45 |
9.44 |
9.55 |
Schottky barrier diode activities. All the I–V characteristics of CTFD were analysed using the thermionic emission theory (TET)-Cheung's method utilized113–116 (ESI, Section 1.3†) to calculate some major diode parameters. The ideality factor (η) for the CTFD is 2.05. Table S7† lists the intercept of the dV/dlnI vs. I plot (Fig. S17†), calculated under low light (dark) conditions. The η value has been accurately calculated under irradiation conditions using the same reliable and proven method as 1.43. The series resistances (RS) were calculated by determining the precise slope of the graph depicted in Fig. S17(A).† Table 4 shows a deviation in the calculated ideality factor. Due to the inhomogeneity of Schottky barrier height (SBH), interface states, and series resistance near the junction, the device's performance may be affected.117,118 When there is enough illumination, the ideality factor approaches the ideal value of 1, which is a remarkable discovery. Due to less recombination of interfacial charge carriers, better homogeneity is observed at the junctions of the Schottky device.113 The CTFD barrier height (∅B) can be computed from the intercept of the H(I) vs. I plot (Fig. S17(B)†) [eqn (S6)†]. The reduction of the height of the barrier at the junction in the presence of light conditions is a significant observation. This phenomenon directly results from generating and assembling PICC (photo-induced charge carriers) near the conduction band. The series resistance (RS) of the device is again computed from the slope of the H(I) vs. I plot (Fig. S17(B)†). All the calculated values of barrier height (∅B), ideality factor (η), and series resistance (RS) for the CTFD are presented in Table S7† for all conditions. The results from both processes demonstrate outstanding consistency. Light's presence decreased RS's calculated values, indicating its significance in optoelectronics device fabrication (Table S7†). A complete study of CTFD I–V graphs understanding the charge transport at MS junction. The I–V graphs under combined environments show two distinct slopes in the logarithmic scale (Fig. 7). The 1st region (region-I) (Fig. 7(A)) represents the ohmic regime, and it possesses a slope value of ∼1. In region-I, the produced current obeys the ohmic law of I ∝ V. The slope value of the 2nd region, also known as region-II, is approximately 2. In this regime, the current generated is proportional to the voltage square (I vs. V2) (Fig. 7(A). In simple terms, it refers to a condition where the flow of electric current is limited by the amount of charge present in a space without any hindrance or obstruction. It is commonly known as the space charge limited current (SCLC) regime.113,119 Here, the inserted carriers circulated in the whole device as the number of inserted carriers is more significant than the background carriers. It produces a SCF (space charge field). The newly established field effectively monitors the currents in this context and is commonly called SCLC.113,119 We used SCLC theory to investigate device performance. Adequate carrier mobility was calculated using the Mott–Gurney equation from the higher voltage region of the I vs. V2 plot (Fig. 7(B)).113,119| |
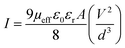 | (2) |
where, I = the current, ε0 = permittivity of free space, εr = relative dielectric constant of the synthesized material, and μeff stand for the effective mobility.
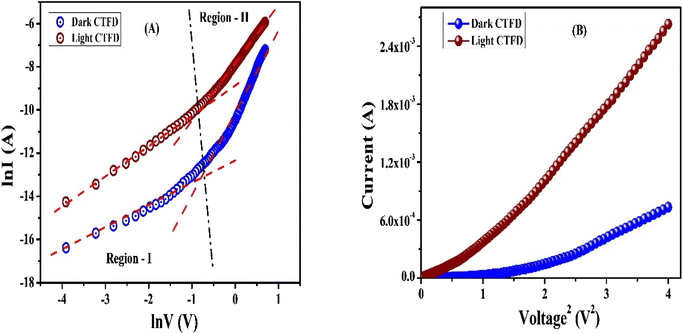 |
| | Fig. 7 (A) LnI vs. lnV, and (B) I vs. V2 curves for CTFD under dark and irradiation conditions. | |
The capacitance versus frequency graph at constant bias potential was used to evaluate the relative dielectric constant of CTFD, which was found to be 5.35 × 10−1 (Fig. S1C†) (ESI, Section 1.3†). CTFD's transit time (τ) and diffusion coefficient (D) were calculated to estimate charge transport near the metal–semiconductor junction. The value of τ is calculated from eqn (3) using the slope of the SCLC region (region II) in the logarithmic representation of the forward I–V curve shown in Fig. 7(A).113
| |
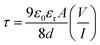 | (3) |
| |
 | (4) |
where,
D is the diffusion coefficient, and it is determined by Einstein–Smoluchowski equation (
eqn (4)).
113 All parameters calculated in the SCLC region show an enhancement in charge transport properties of CTFD in the presence of light, as presented in Table S7.
†
Charge-carrier transport: a fruitful mechanism. The copper complex's higher mobility indicated a greater transport rate during photo-irradiation, which suggests an increase in charge carrier generation under similar conditions. The complex device exhibits a photosensitivity of 3.59, which is appreciable. Light exposure improved the ideality factor ∼1 of complex-based devices (CTFD), resulting in an ideal device with better Schottky junction homogeneity and reduced charge recombination.120 Therefore, discussing charge-carrier transport concerning our Salen metal complex is vital. The charge-carrier transport in hybrid organic–inorganic complex (OIC) structures has been successfully operated through the following mechanisms:121 (a) through-bond conduction, (b) guest molecules, and (c) space conduction. Notably, the transportation of charge carriers induced by light occurs through a through-bond conduction mechanism. Through-bond conduction involves charges moving through covalent and coordination bonds in a complex material (CTFD), allowing the transmission of electrical signals. The process usually involves a ‘hopping transport'.122 Light absorption excites electrons, creating a photocurrent as they move from H → L (HOMO → LUMO). This photocurrent response reveals the semiconductor's conductance and free carrier count during light exposure.122 Besides, the complex materials' (CTFD) secondary metal–ligand interaction and structural features are crucial elements that significantly determine electrical conductivity. In some cases, exposure to light can cause slight changes in molecular structure and bond angles, affecting the material's ability to conduct charge.14 In a true sense, extensive research is needed to understand the complete phase of the charge transportation mechanism. The improved photo-irradiation performance of CTFD over LTFD suggests a more efficient charge transfer, making it an attractive option for semiconductor devices.
Structure–activity-relationship. The copper complex exhibits higher conductivity in the presence of light compared to that in the dark, indicating light sensitivity. The conductivity is compared concerning the ligand (H2LSAL) and can be explained based on structure–activity-relationship (SAR).123 The copper complex is a hybrid composite material that combines organic and inorganic components. It is often referred to as an Organic–Inorganic Composite Material (OICM). The complex's potential dual donor–acceptor properties (D–A) can explain photosensitivity behaviour. Upon photoexcitation, the organic ligand (H2LSAL) generously donates electrons (ED) while the inorganic Cu–OH–Cu moiety eagerly accepts (EA) them. Moreover, supramolecular frameworks of copper complexes aid in facilitating the photosensitivity process. It can also function as an optoelectronic material, specifically a Schottky barrier diode (SBD). Fig. S18† provides a brief explanation of the photosensitivity mechanism of the complex.
Theoretical rationalization: DOS and PDOS approach. Analysing the density of states (DOS) and partial density of states (PDOS) is a powerful tool for gaining insights into the intricate interactions and charge transfer processes between molecules.124 By revealing the diverse states that electrons can occupy at various energy levels, DOS plays a pivotal role in understanding the electronic structure of materials. This understanding extends to the HOMO and LUMO regions, where significant alterations occur, providing crucial information about the sensing abilities of conducting materials.125 The DOS and PDOS spectra were generated using the Gauss Sum program126 and are depicted in Fig. 8(a) and 8(b). An analysis of the DOS/PDOS spectra of the compound under study reveals the HOMO and LUMO energy levels at −4.4 eV and −3.7 eV, respectively, indicating a gap energy of 0.7 eV. A material's high conductivity arises due to its small band gap energy, which enables easy flow of electrons from the valence band to the conduction band of complex materials. The characteristic is extremely advantageous for the performance of Schottky diodes. Additionally, this result suggests a significant intra-charge/energy transfer (ICT) occurring between the ligands. The presence of oxygen acceptor atoms around the copper metal ensures the availability of free electrons, which are crucial for facilitating the movement of electrons back and forth. This phenomenon indicates the high charge density coexisting on the surface of the ligand. Such specificity is advantageous for the development of new Schottky diode devices. Furthermore, this analysis illuminates the intricate interplay between the individual molecular orbitals of each molecule, culminating in the formation of new electronic states within the combined system.
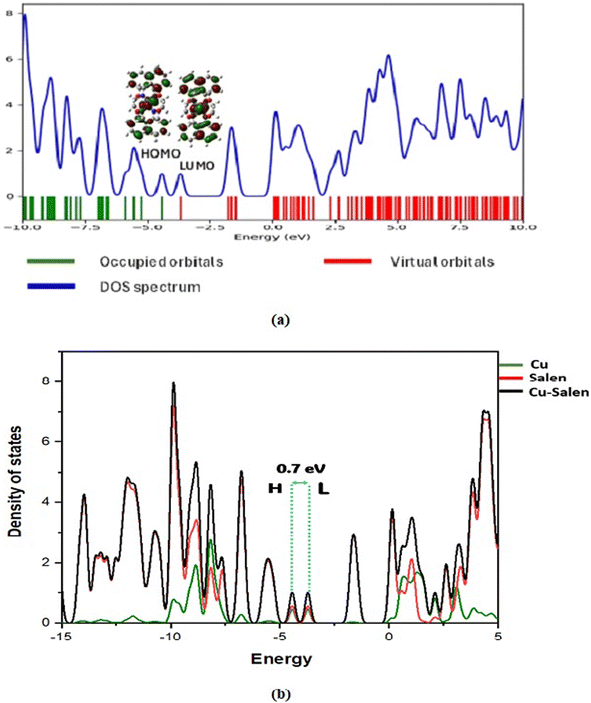 |
| | Fig. 8 DOS and PDOS spectra of the studied complex. | |
Conclusion
In summary, a centrosymmetric binuclear Cu(II)-Salen complex was synthesized by self-assembly method with ligand (H2LSAL) and Cu(NO3)2·3H2O in the CH3OH + ACN solvent mixture. The complex is structurally characterized using various analytical techniques, including DRS, SEM-EDX, PXRD, and XPS analyses. The dimeric unit in SCXRD has two Cu(II) centres with distorted square pyramidal geometries (SQP) supported by the tau value (τ = 0.128). The XPS analysis supports the Salen ligand N/O-donor centres' binding with Cu(II) metal ions. HS and 2-D fingerprint plots authenticate the crystal structure supramolecular contacts. The interaction energy and energy frameworks are utilized to investigate the complex crystal packing. NBO, QTAIM, and ELF-LOL are employed to analyse complex bonding features. The PSBD complex was tested for its potential use in optoelectronic devices. The results showed that it had a significantly higher level of electrical conductivity (4.27 × 10−5 S m−1) in light conditions compared to dark conditions (2.41 × 10−7 S m−1). The findings are comparable to the literature published by Schottky behaved X-characterized complexes. Additionally, it was found to be approximately two times more photosensitive during the light phase than the dark phase. Theoretical DOS/PDOS investigations substantiate the experimental Schottky diode behaviour. Therefore, the copper complex exhibits promising potential in optoelectronic device applications.
Author contributions
Dr Dhrubajyoti Majumdar was the project's author and conceived the whole research idea, performedchemdata curation, conceptualization, methodology, research investigation, formal analysis, contributed reagents, materials, software visualization, writing review, initial draft preparation, and editing. Dr Bouzid Gassoumi performed DFT experiments. Dr Arka Dey performed conductivity-based experiments. Dr Sourav Roy was involved in the X-ray crystal structure and graphics preparation. Dr Sahbi Ayachi performed DFT experiments. Suman Hazra formal analysis, research investigation, graphics preparation, and software visualization. Prof. Dr Sudipta Dalai supervise, conceptualization, methodology, research investigation, and formal analysis. All authors in the manuscript carefully read and approved the final version before submission.
Conflicts of interest
No conflicts of interest or personal relationships influenced this work.
Acknowledgements
This research study has not received funding from any public, commercial, or non-profit funding agency. All authors thank the Central Laboratory of Tamralipta Mahavidyalaya, Tamluk, West Bengal, India. We acknowledge the Raman spectra, UV-visible, Diffuse reflectance spectroscopy (DRS), SEM-EDX, and XPS analysis conducted by SAIF, IIT Madras in Chennai-600036, SAIC Tezpur university-784028, Tezpur, STIC, Cochin in Kerala-682022, and IIT Jammu-181121, India.
References
- G. Venkatesh, P. Vennila, S. Kaya, S. B. Ahmed, P. Sumathi, V. Siva, P. Rajendran and C. Kamal, ACS Omega, 2024, 9(7), 8123–8138 CAS.
- D. Aggoun, Z. Messasma, B. Bouzerafa, R. Berenguer, E. Morallon, Y. Ouennoughi and A. Ourari, J. Mol. Struct., 2021, 1231, 129923 CrossRef CAS.
- L. Chen, L. Wang, W. Ann, R. Wang and L. Tian, Inorg. Nano-Met. Chem., 2020, 50(9), 872–879 CrossRef CAS.
- B. Naureen, G. A. Miana, K. Shahid, M. Asghar, S. Tanveer and A. Sarwar, J. Mol. Struct., 2021, 1231, 129946 CrossRef CAS.
- M. Muthukkumar, C. Kamal, G. Venkatesh, C. Kaya, S. Kaya, I. V. M. V. Enoch, P. Vennila and R. Rajavel, J. Mol. Struct., 2017, 1147, 502–514 CrossRef CAS.
- D. J. Majumdar, A. Dubey, A. Tufail, D. Sutradhar and S. Roy, Heliyon, 2023, 9(3), e16057 CrossRef CAS PubMed.
- D. J. Majumdar, J. E. Philip, S. Roy and B. Tuzun, Results Chem., 2022, 4, 100574 CrossRef CAS.
- H. Kargar, M. Ashfaq, M. Fallah-Mehrjardi, R. Behjatmanesh-Ardakani, K. S. Munawar and M. N. Tahir, Inorg. Chim. Acta, 2022, 536, 120878 CrossRef CAS.
- M. Mladenović and N. Vukmirovic, Adv. Funct. Mater., 2015, 25, 1915–1932 CrossRef.
- J. Terao, A. Wadahama, A. Matono, T. Tada, S. Watanabe, S. Seki, T. Fujihara and Y. Tsuji, Nat. Commun., 2013, 4, 1691–1699 CrossRef PubMed.
- S. Stafstrom, Chem. Soc. Rev., 2010, 39, 2484–2499 RSC.
- B. Dutta and S. Halder, ChemistrySelect, 2023, 8, e202301586 CrossRef CAS.
- H. Wang, Q.-L. Zhu, R. Zou and Q. Xu, Chem, 2017, 2, 52–80 CAS.
- S. Halder, Mater. Adv., 2023, 4, 5033–5049 RSC.
- P. J. Low, Dalton Trans., 2005, 2821–2824 RSC.
- A. A. Talin, A. Centrone, A. C. Ford, M. E. Foster, V. Staliva, P. Haney, R. A. Kenney, V. Szalai, F. E. Gabaly, H. P. Yoon, F. Leonard and M. D. Allendorf, Science, 2013, 343, 66–69 CrossRef PubMed.
- A. Nath, K. S. Asha and S. Mandal, Chem.–Eur. J., 2021, 27, 11482–11538 CrossRef CAS PubMed.
- I. Stassen, N. Burtch, A. Talin, P. Falcaro, M. Allendorf and R. Ameloot, Chem. Soc. Rev., 2017, 46, 3185–3241 RSC.
- J. Wu, J. Chen, C. Wang, Y. Zhou, K. Ba, H. Xu, W. Bao, X. Xu, A. Carlsson, S. Lazar, A. Meingast, Z. Sun and H. Deng, Adv. Sci., 2020, 7, 1903003 CrossRef CAS PubMed.
- L.-T. Zhang, Y. Zhou and S.-T. Han, Angew. Chem., Int. Ed., 2021, 133, 15320–15340 CrossRef.
- J. Nicks, K. Sasitharan, R. R. R. Prasad, D. J. Ashworth and J. A. Foster, Adv. Funct. Mater., 2021, 31, 2103723 CrossRef CAS.
- W. Zhao, J. Peng, W. Wang, S. Liu, Q. Zhao and W. Huang, Coord. Chem. Rev., 2018, 377, 44–63 CrossRef CAS.
- W.-J. Li, J. Liu, Z.-H. Sun, T.-F. Liu, J. Lu, S.-Y. Gao, C. He, R. Cao and J.-H. Luo, Nat. Commun., 2016, 7, 11830 CrossRef CAS PubMed.
- X. Wang, Y. Wang, M. A. Silver, D. Gui, Z. Bai, Y. Wang, W. Liu, L. Chen, J. Diwu, Z. Chai and S. Wang, Chem. Commun., 2018, 54, 4429–4432 RSC.
- M. Sadakiyo, H. Kasai, K. Kato, M. Takata and M. Yamauchi, J. Am. Chem. Soc., 2014, 136, 1702–1705 CrossRef CAS PubMed.
- L. Zhai, J.-W. Yu, J. Zhang, W.-W. Zhang, L. Wang and X.-M. Ren, Dalton Trans., 2019, 48, 12088–12095 RSC.
- M. Ghosh, S. Saha, A. Banerjee, D. Schollmeyer, A. Sarkar and S. Banerjee, New J. Chem., 2019, 43, 16255–16263 RSC.
- Y.-B. Dong, L. Wang, J.-P. Ma, X.-X. Zhao, D.-Z. Shen and R.-Q. Huang, Cryst. Growth Des., 2006, 6, 2475–2485 CrossRef CAS.
- J. Zhang, L. Xu and W.-Y. Wong, Coord. Chem. Rev., 2018, 355, 180–198 CrossRef CAS.
- S. Djebbar-Sid, O. Benali-Baitich and J. P. Deloume, Polyhedron, 1997, 16, 2175–2182 CrossRef CAS.
- X. Li, Y. Jiao and S. Li, Eur. Polym. J., 1991, 27, 1345–1351 CrossRef CAS.
- S. Mahato, A. Mondal, M. Das, M. Joshi, P. P. Ray, A. Roy Choudhury, C. M. Reddy and B. Biswas, Dalton Trans., 2022, 51, 1561–1570 RSC.
- S. Karadeniz, D. E. Yilid, H. H. Gullu, D. A. Kose, A. A. Hussaini and M. Yildirim, J. Mater. Sci.: Mater. Electron., 2022, 33, 18039–18053 CrossRef CAS.
- Q. Li, J. Meng and Z. Li, J. Mater. Chem. A, 2022, 10, 8107–8128 RSC.
- S. İlhan, J. Coord. Chem., 2008, 61, 2884–2895 CrossRef.
- G. Venkatachalam, N. Raja, D. Pandirajan and R. Ramesh, Spectrochim. Acta, Part A, 2008, 71, 884–891 CrossRef CAS.
- K. Geetha, S. K. Tiwary, A. R. Chakravarty and G. Ananthakrishna, J. Chem. Soc., Dalton Trans., 1999, 4463–4467 RSC.
- B. Dutta, A. Dey, K. Naskar, F. Ahmed, R. Purkait, S. Islam, S. Ghosh, C. R. Sinha, P. P. Ray and M. H. Mir, New J. Chem., 2018, 42, 8629–8637 RSC.
- S. Khan, S. Halder, P. P. Ray, S. Herrero, R. González-Prieto, M. G. B. Drew and S. Chattopadhyay, Cryst. Growth Des., 2018, 18, 651–659 CrossRef CAS.
- S. Roy, S. Halder, M. G. B. Drew, P. P. Ray and S. Chattopadhyay, New J. Chem., 2018, 42, 15295–15305 RSC.
- M. G. Campbell, D. Sheberla, S. F. Liu, T. M. Swager and M. Dinca, Angew. Chem., Int. Ed., 2015, 54, 4349–4352 CrossRef CAS PubMed.
- D. Y. Lee, D. V. Shinde, S. J. Yoon, K. N. Cho, W. Lee, N. K. Shrestha and S.-H. Han, J. Phys. Chem. C, 2014, 118, 16328–16334 CrossRef CAS.
- E. Q. Gao, S. Q. Bai, C. F. Wang, Y. F. Yue and C. H. Yan, Inorg. Chem., 2003, 42, 8456–8464 CrossRef CAS PubMed.
- M. Mondal, S. Jana, M. G. B. Drew and A. Ghosh, Polymer, 2020, 204, 122815 CrossRef CAS.
- A. Hossain, A. Dey, S. K. Seth, P. P. Roy, P. Ballester, R. G. Pritchard, J. Ortega-Castro, A. Frontera and S. Mukhopadhyay, ACS Omega, 2018, 3, 9160–9171 CrossRef CAS PubMed.
- M. G. Campbell, D. Sheberla, S. F. Liu, T. M. Swager and M. Dincă, Angew. Chem., Int. Ed., 2015, 54, 4349–4352 CrossRef CAS PubMed.
- D. J. Majumdar, A. Dey, S. Roy, D. Sutradhar and S. Hazra, Inorg. Chem. Commun., 2024, 162, 112155 CrossRef CAS.
- D. J. Majumdar, S. Roy, A. Dey and D. Sutradhar, J. Mol. Struct., 2023, 1294, 136438 CrossRef CAS.
- L. C. Nathan, J. E. Koehne, J. M. Gilmore, K. A. Hannibal, W. E. Dewhirst and T. D. Mai, Polyhedron, 2003, 22, 887–894 CrossRef CAS.
- H. Kargar, R. Behjatmanesh-Ardakani, V. Torabi, M. Kashani, Z. Chavoshpour-Natanzi, Z. Kazemi, V. Mirkhani, A. Sahraei, M. N. Tahir, M. Ashfaq and K. S. Munawar, Polyhedron, 2021, 195, 114988 CrossRef CAS.
- H. Kargar, R. Behjatmanesh-Ardakani, V. Torabi, A. Sarvian, Z. Kazemi, Z. Chavoshpour-Natanzi, V. Mirkhani, A. Sahraei, M. Nawaz Tahir and M. Ashfaq, Inorg. Chim. Acta, 2021, 514, 120004 CrossRef CAS.
- L. Rigamonti, F. Demartin, A. Forni, S. Righetto and A. Pasini, Inorg. Chem., 2006, 45, 10976–10989 CrossRef CAS PubMed.
- S. Celika, S. Yurdakul and B. Erdem, J. Mol. Struct., 2023, 1273, 134279 CrossRef.
- S. Grimme, J. Chem. Phys., 2006, 124(3), 034108 CrossRef PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- G09|Gaussian.com, http://gaussian.com/glossary/g09/.
- B. P. Borah, S. Majumder, P. P. Nath, A. K. Choudhury and J. Bhuyan, J. Mol. Struct., 2023, 1275, 134557 CrossRef CAS.
- B. Abdelaziz, I. Chérif, B. Gassoumi, S. Patanè and S. Ayachi, J. Phys. Chem. A, 2023, 127, 9895–9910 CrossRef CAS PubMed.
- B. Abdelaziz, Z. Mazouz, B. Gassoumi, N. E. I. Boukortt, S. Patanè and S. Ayachi, J. Mol. Liq., 2024, 395, 123934 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- K. M. Al-Ahmary, M. M. Habeeb and S. H. Aljahdali, J. Mol. Liq., 2019, 277, 453–470 CrossRef CAS.
- SMART & SAINT Software Reference Manuals Version 6.45, Bruker Analytical X-ray Systems, Inc., Madison, WI, 2003 Search PubMed.
- Bruker, SAINT, SAINT Bruker AXS InC, Madison, Wisconsin, USA, 2009.
- SHELXTL Reference Manual Ver. 6.1, Bruker Analytical X-ray Systems, Inc., Madison, WI, 2000 Search PubMed.
- G. M. Sheldrick, SHELXTL, a Software for Empirical Absorption Correction Ver.6.12, Bruker AXS Inc., WI. Madison, 2001 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. Dey, S. Middya, R. Jana, M. Das, J. Datta, A. Layek and P. P. Ray, J. Mater. Sci.: Mater. Electron., 2016, 27, 6325–6335 CrossRef CAS.
- G. Bairy, A. Dey, B. Dutta, P. P. Ray and C. Sinha, Cryst. Growth Des., 2022, 22, 3138–3147 CrossRef CAS.
- T. L. Yusuf, S. D. Oladipo, S. Zamisa, H. M. Kumalo, I. A. Lawal, M. M. Lawal and N. Mabuba, ACS Omega, 2021, 6, 13704–13718 CrossRef CAS PubMed.
- M. Hazra, T. Dolai, A. Pandey, S. K. Dey and A. Patra, Bioinorg. Chem. Appl., 2014, 2014, 104046 Search PubMed.
- C. Şenol, Z. Hayvali, H. Dal and T. Hökelek, J. Mol. Struct., 2011, 997, 53–59 CrossRef.
- O. Signorini, E. R. Dockal, G. Castellano and G. Oliva, Polyhedron, 1996, 15, 245–255 CrossRef CAS.
- Z. Rezvani, A. R. Abbasi, K. Nejati and M. Seyedahmadian, Polyhedron, 2005, 24, 1461–1470 CrossRef CAS.
- S. D. Oladipo, B. Omondi and C. Mocktar, Appl. Organomet. Chem., 2020, 34, e5610 CrossRef CAS.
- T. L. Yusuf, S. D. Oladipo, S. Zamisa, H. M. Kumalo, I. A. Lawal, M. M. Lawal and N. Mabuba, ACS Omega, 2021, 6, 13704–13718 CrossRef CAS PubMed.
- M. Salehi, F. Faghani, M. Kubicki and M. Bayat, J. Iran. Chem. Soc., 2018, 15, 2229–2240 CrossRef CAS.
- S. Zolezzi, A. Decinti and E. Spodine, Polyhedron, 1999, 18, 897–904 CrossRef CAS.
- C. E. Satheesh, P. K. Raghavendra, N. Shivakumar, K. Lingaraju, P. Murali Krishna, H. Rajanaika and A. Hosamani, Inorg. Chim. Acta, 2019, 495, 118929 CrossRef CAS.
- N. E. Eltayeb, S. G. Teoh, R. Adnan, J. Bee-Jan The and H.-K. Fun, J. Fluoresc., 2011, 21, 1393–1400 CrossRef CAS PubMed.
- M. Hazra, T. Dolai, A. Pandey, S. K. Dey and A. Patra, Bioinorg. Chem. Appl., 2014, 2014, 104046 Search PubMed.
- H. Kabeer, S. Hanif, A. Arsalan, S. Asmat, H. Younus and M. Shakir, ACS Omega, 2020, 5, 1229–1245 CrossRef CAS PubMed.
- S. Khan, S. Halder, A. Dey, B. Dutta, P. P. Ray and S. Chattopadhay, New J. Chem., 2020, 44, 11622–11630 RSC.
- D. Sadhukhan, A. Roy, G. Rosair, L. Charbonniere and S. Mitra, BCSJ, 2011, 84, 211–217 CrossRef CAS.
- M. Amirnasr, K. J. Schenk, M. Salavati, S. Dehghanpour, A. Taeb and A. Tadjarodi, J. Coord. Chem., 2003, 56, 231–243 CrossRef CAS.
- B. Dutta, S. Paul and S. Halder, Heliyon, 2023, 9, e13504 CrossRef CAS PubMed.
- R. Nag, S. Polepalli, M. A. Hussain and C. P. Rao, ACS Omega, 2019, 4, 13231–13240 CrossRef CAS PubMed.
- G. R. Reddy, S. Balasubramanian and K. Chennakesavulu, J. Mater. Chem. A, 2014, 2, 15598–15610 RSC.
- E. T. Kang, K. G. Neoh and K. L. Tan, Surf. Interface Anal., 1992, 19, 33–37 CrossRef CAS.
- B. Wang, F. Gao and H. Ma, J. Hazard. Mater., 2007, 144, 363–368 CrossRef CAS PubMed.
- S. Roy, I. Mondal, K. Harms and S. Chattopadhyay, Polyhedron, 2019, 159, 265–274 CrossRef CAS.
- A. W. Addison, N. T. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 7, 1349–1356 RSC.
- S. Roy, T. Dutta, M. G. B. Drew and S. Chattopadhyay, Polyhedron, 2020, 178, 114311 CrossRef CAS.
- P. Mahapatra, S. Ghosh, S. Giri, V. Rane, R. Kadam, M. G. B. Drew and A. Ghosh, Inorg. Chem., 2017, 56, 5105–5121 CrossRef CAS PubMed.
- M. Fondo, J. Doejo, A. M. Garcia-Deibe, N. Ocampo and J. Sanmartin, Polyhedron, 2015, 101, 78–85 CrossRef CAS.
- M. J. Turner, S. Grabowsky, D. Jayatilaka and M. A. Spackman, J. Phys. Chem. Lett., 2014, 5, 4249–4255 CrossRef CAS PubMed.
- P. Venkatesan, S. Thamotharan, A. Ilangovan, H. Liang and T. Sundius, Spectrochim. Acta, Part A, 2016, 153, 625–636 CrossRef CAS PubMed.
- M. J. Turner, S. P. Thomas, M. W. Shi, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2015, 51, 3735–3738 RSC.
- G. Yuan, K. Z. Shao, D. Y. Du, X. L. Wang, Z. M. Su and J. F. Ma, CrystEngComm, 2012, 14, 1865–1873 RSC.
- B. Silvi and A. Savin, Nature, 1994, 371, 683–686 CrossRef CAS.
- E. Matito and M. Sola, Coord. Chem. Rev., 2009, 253, 647–665 CrossRef CAS.
- H. Jacobsen, Can. J. Chem., 2008, 86, 695–702 CrossRef CAS.
- R. F. W. Bader, Atoms, in Molecules, A Quantum Theory, Oxford University Press, 1990 Search PubMed.
- R. F. W. Bader and M. A. Austen, J. Chem. Phys., 1997, 107(11), 4271–4285 CrossRef CAS.
- B. Gassoumi, M. Echabaane, F. E. Ben Mohamed, L. Nouar, F. Madi, A. Karayel, H. Ghalla, M. E. Castro, F. J. Melendez, S. Özkınalı, A. Rouis and R. B. Chaabane, Spectrochim. Acta, Part A, 2022, 264, 120242 CrossRef CAS PubMed.
- H. Hadi, G. Bouzid, S. Nasr, H. Ghalla, R. Ben Chaabane and S. Ayachi, Heliyon, 2023, 9, e20206 CrossRef CAS PubMed.
- R. F. W. Bader and H. Essen, J. Chem. Phys., 1984, 80, 1943–1960 CrossRef CAS.
- I. Rozas, I. Alkorta and J. Elguero, J. Am. Chem. Soc., 2000, 122, 11154–11161 CrossRef CAS.
- L. F. Matta and R. J. Boyd, J. Chem. Inf. Model., 2005, 45, 354–359 CrossRef PubMed.
- H. S. Mohamed, A. A. Dahy, G. S. Hassan, S.-S. M. Eid and R. M. Mahfouz, Struct. Chem., 2017, 28, 1093–1109 CrossRef CAS.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- F. J. Weinhold, and J. E. Carpenter, The structure of small molecules and Ions, ed. R. Naaman and Z. Vager, Plenum, 1988, pp. 227–236 Search PubMed.
- A. Dey, S. Middya, R. Jana, M. Das, J. Datta, A. Layek and P. P. Ray, J. Mater. Sci.: Mater. Electron., 2016, 27, 6325–6335 CrossRef CAS.
- E. H. Rhoderick, Metal-Semiconductors Contacts, Oxford University Press, Oxford, 1978 Search PubMed.
- S. K. Cheung and N. W. Cheung, Appl. Phys. Lett., 1986, 4985–4987 Search PubMed.
- A. Dey, A. Layek, A. Roychowdhury, M. Das, J. Datta, S. Middya, D. Das and P. P. Ray, RSC Adv., 2015, 5, 36560–36567 RSC.
- R. K. Gupta and F. Yakuphanoglu, Sol. Energy, 2012, 86, 1539–1545 CrossRef CAS.
- G. X. Miao, S. Tongay, M. K. Petterson, K. Berke, A. G. Rinzler, B. R. Appleton and A. F. Hebard, Nano Lett., 2012, 12, 2745–2750 CrossRef PubMed.
- H. P. W. M. Blom, M. J. M. de Jong and M. G. van Munster, Phys. Rev. B, 1997, 55, R656–R659 CrossRef.
- X. Miao, S. Tongay, M. K. Petterson, K. Berke, A. G. Rinzler, B. R. Appleton and A. F. Hebard, Nano Lett., 2012, 12, 2745–2750 CrossRef CAS PubMed.
- A. Hossain, A. Dey, S. K. Seth, P. P. Roy, P. Ballester, R. G. Pritchard, J. Ortega-Castro, A. Frontera and S. Mukhopadhyay, ACS Omega, 2018, 3, 9160–9171 CrossRef CAS PubMed.
- L. P. Wu, Y. L. Zhang, L. Z. Long, C. P. Cen and X. J. Li, RSC Adv., 2014, 4, 20716–20721 RSC.
- S. Roy, A. Dey, M. G. B. Drew, P. P. Roy and S. Chattopadhyay, New J. Chem., 2019, 43, 5020–5031 RSC.
- D. Recatalá, R. Lusar, A. Barlow, G. Wang, M. Samoc, M. G. Humphrey and A. L. Guschin, Dalton Trans., 2015, 44, 13163–13172 RSC.
- T. Jadoon, T. Mahmood and K. Ayub, J. Mol. Liq., 2020, 306, 112878 CrossRef CAS.
- F. Maddalena, C. Falco, M. Caironi and D. Natali, Org. Electron., 2015, 17, 304–318 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
Sourav Royd,
Sahbi Ayachie,
Suman Hazraaf and
Sudipta Dalai
c,
Sourav Royd,
Sahbi Ayachie,
Suman Hazraaf and
Sudipta Dalai