
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProbing capping mechanisms and polymer matrix loading of biogenic vaterite CaCO3–Ag hybrid through X-ray photoelectron spectroscopy (XPS)†
Mohammad Hossein Azarian a,
Supinya Nijpanichb,
Narong Chanlekb and
Wimonlak Sutapun*ac
a,
Supinya Nijpanichb,
Narong Chanlekb and
Wimonlak Sutapun*ac
aResearch Centre for Biocomposite Materials for Medical, Agricultural and Food Industry, Suranaree University of Technology, Nakhon Ratchasima, 30000, Thailand. E-mail: wimonlak@sut.ac.th
bSynchrotron Light Research Institute (Public Organization), 111 University Avenue, Muang District, Nakhon Ratchasima 30000, Thailand
cSchool of Polymer Engineering, Suranaree University of Technology, Nakhon Ratchasima, 30000, Thailand
First published on 3rd May 2024
Abstract
Despite extensive research in the literature, the synthesis of silver nanoparticles (AgNPs) via capping mechanisms remains incompletely understood. This study employs a mechanistic approach to unravel the underlying molecular interactions driving the capping process of biogenic vaterite CaCO3–Ag and explores their interactions with different polymer matrices. X-ray photoelectron spectroscopy (XPS) was used to reveal the capping mechanisms, surface composition alterations, and vaterite polymorph transitions. The oxidation states of AgNPs exhibited distinct changes under different capping agents. The Ag3d spin–orbit splitting profiles revealed the coexistence of Ag+ and Ag0 within CaCO3–Ag, with a significant presence of Ag0 when poly(sodium 4-styrene sulfonate) was employed as the capping agent. Conversely, the use of carboxy methyl cellulose as the capping agent resulted in Ag+ dominance. XPS analysis illuminated the transformation of CaCO3 polymorphs from calcite to vaterite structure, which remained stable following embedding within polymer matrices. Integrating CaCO3–Ag microspheres into polymer matrices and investigating their surface characteristics represents a strategic step toward tailoring material properties for potential applications in active packaging and biomedicine.
Introduction
X-ray photoelectron spectroscopy (XPS) is a widely employed core level spectroscopy technique that provides valuable information about the elemental composition, chemical state, and electronic structure of a broad range of materials. It is particularly effective in investigating the very top surface properties of materials, allowing to gain insights into their surface chemistry, oxidation states, and interactions with surrounding environments.1 XPS offers a powerful tool to study the chemical environment of nanoparticles and elucidate the changes that occur upon embedding them in a polymer matrix. For instance, each element possesses a unique binding energy (BE), and a shift towards higher BE indicates an increase in the electronegativity of neighbouring atoms or a higher oxidation state, and vice versa.2 However, there are three elements that contradict this rule due to relativistic effects.3,4 These elements are often referred to as the ‘coinage metals' and include silver (Ag), copper (Cu) and gold (Au).5,6Among the diverse array of nanomaterials available, silver nanoparticles (AgNPs) have emerged as highly promising candidates due to their exceptional antimicrobial properties. Nevertheless, the inherent instability of AgNPs and their potential adverse effects on living organisms have prompted the exploration for materials capable of safeguarding them and providing improved control over their release.7 Introducing vectors or carriers, such as vaterite calcium carbonate (CaCO3), present a significant solution to these challenges.8–11 By utilizing vaterite CaCO3, the stability issues of AgNPs can be effectively mitigated, enhancing their overall potential. However, to fully harness the benefits of this approach, it is essential to comprehend the interactions between AgNPs and CaCO3, as well as their behaviour after loading into diverse polymer matrices. A comprehensive understanding of these dynamics can be achieved through advanced characterization techniques like XPS, which allows for the analysis of their chemical composition and surface properties.12–15
Capping agents are often used in the synthesis of AgNPs to stabilize the particles and prevent them from agglomerating or oxidizing. These capping agents can be organic molecules such as carboxylic acid of plants derivative/extracts or, synthetic polymer macromolecules containing sulfonate groups which can bind to the surface of the AgNPs through chemical bonds.16,17 For instance, AgNPS capped with polystyrene sulfonate (PSS) macromolecules, the sulphur atom in the sulfonate group can form a chemical bond (electrostatic interactions or coordination bonds) with the Ag surface through a complexation reaction.18 This binding can be quite strong and can provide additional stability to the nanoparticle structure. Similarly, capping agents containing amines, such as chitosan can also bind to the surface of AgNPs and result in the formation of coordination complexes.19 However, using different capping agents can significantly influence the properties of AgNPs in terms of size, release, stability and consequently cytotoxicity and biocompatibility.20,21
This manuscript presents a comprehensive XPS study of vaterite CaCO3–Ag formation using different capping agents and embedding it in two biodegradable polymer matrices, namely sodium carboxymethyl cellulose (CMC) and polyvinyl alcohol (PVA). Vaterite CaCO3, obtained from discarded eggshells, serves as a unique and sustainable precursor for the synthesis of the AgNPs-loaded material.22,23 The utilization of waste materials not only contributes to environmental sustainability but also showcases a novel approach to producing advanced materials.
The choice of CMC and PVA as polymer matrices is based on their desirable properties, such as biocompatibility, mechanical strength, and ease of processing. CMC, a water-soluble cellulose derivative, possesses excellent film-forming properties and has been widely used in various applications, including packaging, drug delivery systems and wound dressings.24 Polyvinyl alcohol, on the other hand, is a synthetic polymer known for its excellent film-forming ability, biocompatibility, and biodegradability.25,26 The combination of these polymer matrices with the vaterite CaCO3–Ag enables the development of functional materials with tailored properties for potential biomedical applications.
In this study, we utilize a mechanistic approach to examine the capping mechanism of silver with calcium carbonate in the presence of various polyelectrolytes. Using X-ray photoelectron spectroscopy (XPS), we aim to reveal the molecular interactions involved. Our work seeks to enhance the current understanding of this mechanism, providing valuable insights into the underlying interactions. By understanding the alterations in the electronic structure and chemical interactions between the CaCO3–Ag and the polymer matrices, we aim to gain valuable insights into the behaviour and potential applications of these materials. Overall, this research highlights the importance of XPS as a valuable technique for characterizing materials and elucidating the changes in oxidation states and chemical environments. Additionally, the use of discarded eggshells as a precursor for vaterite CaCO3, along with the incorporation of biodegradable polymer matrices, adds novelty and significance to this work. By combining waste materials and advanced characterization techniques, this study presents a unique approach to producing advanced materials with potential applications in the active packaging and biomedical field.
Experimental
Materials
Discarded eggshells from a local bakery near Suranree University of Technology (SUT) in Nakhon Ratchasima, Thailand, were collected as the starting material. The eggshells were thoroughly washed with water and then boiled for six hours at 100 °C to remove the eggshell membranes and organic residues. Following this, the eggshells were dried at 60 °C for 24 hours and subsequently ground into a fine powder. The chemicals utilized in the study were procured from the following sources: Silver nitrate (99.0%, ACS reagent), sodium carbonate (powder, 99.5%, ACS reagent) and polyethylene glycol (average Mw 600 g mol−1) were obtained from Sigma-Aldrich. Sodium carboxymethyl cellulose (CMC) was used in powder form, with an average molecular weight of 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1. Poly(sodium 4-styrene sulfonate) (PSS) was also in powder form, with an average molecular weight of 70000 g mol−1. These were likewise obtained from Sigma-Aldrich. Polyvinyl alcohol (PVA), with a high molecular weight and 98–99% hydrolysis level, was purchased from Alfa Aesar. Nitric acid 65% (AR Grade), with a molecular weight of 63.01 g mol−1, was sourced from ANaPURE.
000 g mol−1. Poly(sodium 4-styrene sulfonate) (PSS) was also in powder form, with an average molecular weight of 70000 g mol−1. These were likewise obtained from Sigma-Aldrich. Polyvinyl alcohol (PVA), with a high molecular weight and 98–99% hydrolysis level, was purchased from Alfa Aesar. Nitric acid 65% (AR Grade), with a molecular weight of 63.01 g mol−1, was sourced from ANaPURE.
Synthesis of vaterite CaCO3–Ag in a single pot
To initiate the synthesis, a polyelectrolyte solution was prepared at ambient temperature. This involved dissolving 0.5 g of either CMC or PSS in 20 mL of deionized water (DI water). The polyelectrolyte was completely dissolved before proceeding. Separately, a 0.01 M silver nitrate solution was prepared by dissolving it in 5 mL of DI water. The stable silver colloid was formed by adding the 0.01 M silver nitrate solution to the polyelectrolyte solution while stirring at room temperature. For the calcium nitrate solution, eggshell powders were employed according to the methodology described in our recent publication.22 The molar concentration of the calcium nitrate was adjusted, and the pH was brought close to neutral by adding a 0.1 M of sodium hydroxide solution. A sodium carbonate solution of the same molar concentration was also prepared accordingly. Next, an equal volume of the calcium nitrate and sodium carbonate solutions was rapidly added and thoroughly mixed with the freshly prepared silver colloid/polyelectrolyte suspension under stirring for 30 seconds at ambient temperature. The resulting suspension was then subjected to centrifugation at 5000 rpm for 5 minutes to remove the polyelectrolyte solution and any unreacted silver nanoparticles. The obtained vaterite CaCO3–Ag microspheres were washed through three cycles of centrifugation and subsequently dried in a vacuum oven at 40 °C for 24 hours. The obtained vaterite CaCO3–Ag designated as CaCO3–Ag–CMC and CaCO3–Ag–PSS when using CMC and PSS as AgNPs capping agents, respectively. The physical characteristics of the obtained microspheres are presented in Table 1. Additionally, SEM images and histograms of size distribution are available in Fig. S1 in the ESI file.† It is important to note that, the level the size of particles and their porosity is relevant to the XPS analysis, as it can influence the detection and estimation of atomic percentages and Ag0/Ag+ ratios. The presence of pores may affect the surface accessibility of Ag0 and Ag+ potentially impacting their detectability by XPS.| Sample | Ag PMD (nm) | CaCO3 PMD (μm) | Ag (mg g−1) ICP-OES | BET surface area (m2 g−1) | Pore volume (cm3 g−1) |
|---|---|---|---|---|---|
| a PMD; particles mean diameter. | |||||
| CaCO3–Ag–CMC | 80.23 ± 30.19 | 13.77 ± 3.21 | 13.77 ± 0.32 | 19.69 | 0.040 |
| CaCO3–Ag–PSS | 118.6 ± 36.08 | 3.37 ± 0.29 | 19.02 ± 0.91 | 19.38 | 0.045 |
Preparation of internally cross-linked CMC film embedded with vaterite CaCO3–Ag microspheres
For internal cross-linking, calcium chloride (20% wt/wt, with respect to CMC) was dissolved in 9 mL of DI water. Once complete dissolution occurred, CMC powders were added (5% wt/v), and the mixture was stirred at room temperature until a gel-like solution was obtained. Additionally, 20% w/w PEG 600 (with respect to CMC) was added to the solution as a plasticizer and allowed to stir for another hour at room temperature. The solution was then degassed using an ultrasonic bath for 5 minutes. In a separate step, 7.5% w/w CaCO3–Ag powders (with respect to CMC weight) were dispersed in 1 mL of distilled water using a magnetic stirrer for 30 minutes. Subsequently, the 1 mL of CaCO3–Ag suspension was added to the CMC solution under mild stirring until a homogeneous suspension was achieved before casting the solution. The resulting suspension was poured into a glass Petri dish and dried in a hot air oven at 40 °C for 48 hours. The obtained films designated as CMC/CaCO3–Ag–CMC and CMC/CaCO3–Ag–PSS when CaCO3–Ag–CMC and CaCO3–Ag–PSS microspheres were used, respectively. The complete synthesis process and thorough characterization of the material, including structural, chemical, thermal, mechanical properties, antimicrobial, and cell cytotoxicity analyses, have been previously documented in ref. 27.Preparation of PVA film embedded with vaterite CaCO3–Ag microspheres
Antimicrobial absorbent PVA films were prepared using the solvent casting method. Initially, PVA powders (5% wt/v) were dissolved in DI water at 75 °C under vigorous stirring for 6 hours. The resulting solution was degassed using an ultrasonic bath for 5 minutes. Similarly, 7.5% w/w CaCO3–Ag–CMC or CaCO3–Ag–PSS microspheres (with respect to PVA weight percent) were dispersed in 1 mL of DI water using a magnetic stirrer for 30 minutes. This suspension was then added to the PVA solution under gently stirring for an additional hour. The resulting suspension was poured into a glass Petri dish, and the film was dried in a hot air oven at 40 °C for 48 hours. The material's comprehensive synthesis process and detailed characterization, covering structural, chemical, thermal, mechanical, antimicrobial properties, and cell cytotoxicity analyses, along with a thorough investigation and report on the silver release profile under pH conditions simulating wound environments, have been previously documented in ref. 28.XPS characterization for capping mechanism and surface composition
The surface composition and oxidation state of the samples were analysed using X-ray photoelectron spectroscopy (XPS) at the SUT-NANOTEC-SLRI Joint Research Facility, located at the Synchrotron Light Research Institute (SLRI) in Nakhon Ratchasima, Thailand. The instrument used for the analysis was the PHI5000 VersaProbe II by ULVAC-PHI, Japan. To excite the samples, a monochromatized Al-Kα X-ray source with a photon energy (hγ) of 1486.6 eV was employed. The resulting XPS spectra were analysed using PHI MultiPak XPS software (version 9.6.0.15), utilizing a combination of Gaussian–Lorentzian lines for peak fitting. Prior to peak fitting, a Shirley-type background correction was applied to remove the background from the spectrum. The survey spectra were recorded with an energy step of 1.000 eV and a pass energy of 117.4 eV, while the high-resolution spectra were recorded with an energy step of 0.05 eV and a pass energy of 46.95 eV. For peak referencing, the C1s spectrum was utilized, with the C–C/C–H peak marked at 284.8 eV.The relative atomic percentage of element x was calculated using the following eqn (1):
 | (1) |
Results and discussion
Vaterite CaCO3–Ag–CMC and CaCO3–Ag–PSS capping mechanism and chemical compositions
This section of the study examines the precipitation of eggshell particles (ESP) in a silver colloid, in which the Ag was reduced and stabilized using two different polyelectrolytes (capping agents), namely PSS and CMC. The primary objective of employing these capping agents is to reduce the Ag ions present in the colloid and subsequently protect them, stabilize the CaCO3–Ag after formation, prevent the calcite conversion and thereby enhance the vaterite CaCO3–Ag stability. Furthermore, the study aims to investigate the mechanism of capping by these different polyelectrolytes on AgNPs. XPS survey scans were first conducted on the surface of neat ESP as well as CaCO3–Ag–CMC and CaCO3–Ag–PSS microspheres. The scan detected C, O, Ca and N elements in ESP, and in addition, Ag was detected in CaCO3–Ag–CMC and CaCO3–Ag–PSS, as shown by the survey spectra in Fig. 1. The elemental compositions of the sample surface calculated by XPS high resolution spectra are listed in Table 2. It is interesting to note that despite the careful removal of the protein membrane layers out of the eggshells (ESP), a small amount of nitrogen (1.79%) was detected in ESP powders, likely attributed to protein residues bonded to the surfaces of the eggshell CaCO3 particles from matrix proteins.29 In contrast, the presence of nitrogen was not detected after the precipitation of ESP in CaCO3–Ag–CMC and CaCO3–Ag–PSS microspheres. This observation strongly suggests that the precipitation process not only allows for the control and attainment of desired polymorphs and sizes but also enables the production of purer CaCO3 with minimal impurities. | ||
| Fig. 1 XPS survey scans of (a) ESP (eggshell particles), (b) CaCO3–Ag–CMC and (c) CaCO3–Ag–PSS. | ||
| Atomic concentration (%) | |||||||
|---|---|---|---|---|---|---|---|
| C | O | Ca | N | Ag | Cl | Na | |
| Corrected RSF | 36.823 | 85.994 | 250.487 | 58.53 | 833.710 | 126.513 | 129.358 |
| ESP | 42.99 | 43.55 | 11.68 | 1.79 | n/a | n/a | n/a |
| CaCO3–Ag–CMC | 47.92 | 42.34 | 9.22 | n/a | 0.52 | n/a | n/a |
| CaCO3–Ag–PSS | 53.38 | 38.82 | 7.23 | n/a | 0.58 | n/a | n/a |
| Neat CMC | 61.78 | 36.00 | n/a | n/a | n/a | n/a | 2.22 |
| CMC/CaCO3–Ag–CMC | 67.99 | 27.25 | 1.24 | n/a | 0.25 | 1.99 | 1.27 |
| CMC/CaCO3–Ag–PSS | 76.98 | 20.97 | 1.06 | n/a | 0.18 | 0.79 | 0.02 |
| Neat PVA | 65.64 | 34.36 | n/a | n/a | n/a | n/a | n/a |
| PVA/CaCO3–Ag–CMC | 87.01 | 10.59 | 1.74 | n/a | 0.65 | n/a | n/a |
| PVA/CaCO3–Ag–PSS | 85.54 | 11.92 | 2.05 | n/a | 0.49 | n/a | n/a |
On the other hand, the C atomic percentage slightly increased in CaCO3–Ag–CMC and CaCO3–Ag–PSS, indicating the interaction between the capping polymer and the CaCO3–Ag particles. It is noteworthy that when using PSS as the capping polyelectrolyte, both the C and Ag atomic percentages were higher compared to using CMC polyelectrolyte. This suggests that the interaction mechanisms between Ag and the capping polymer slightly favour PSS over CMC. Moreover, the results obtained from ICP-OES in our previous manuscript,28 where the samples were digested in nitric acid, showed silver content of 13.77 mg g−1 for CaCO3–Ag–CMC and 19.02 mg g−1 for CaCO3–Ag–PSS, which aligns well with the data presented here. Furthermore, the atomic percentage of Ca is slightly lower in both CaCO3–Ag–CMC and CaCO3–Ag–PSS in comparison to EPS, indicating a potential displacement or alteration of the Ca signal due to the presence of the AgNPs and the capping polymer coatings. Therefore, the slight decrease in the atomic percentage of Ca indicates that the capping polymer may coat the CaCO3–Ag particles and affect the XPS signal associated with Ca.
Scheme 1 illustrated the proposed capping mechanism of CMC and PSS with AgNPs. The capping mechanism interaction between CMC or PSS and Ag ions involves several processes. Both CMC and PSS capping agents are polyelectrolytes with charged functional groups. These chains containing charged groups can attract and bind to the Ag ions through electrostatic interactions. The negatively charged carboxylate groups in CMC or the sulfonate groups in PSS can form coordination bonds with the positively charged Ag ions. Furthermore, both CMC and PSS can act as reducing agents in the presence of Ag+. The O loan electron pairs in carboxylate groups of CMC or the sulfonate groups of PSS can donate electrons to the Ag+ ions, resulting in the reduction of Ag+ to Ag0. Once Ag0 nanoparticles are formed, CMC and PSS can further act as stabilizers or capping agents. They can be adsorbed onto the surface of the AgNPs, forming a protective layer. The long chain structure of CMC or PSS provides steric hindrance, preventing the aggregation or growth of the vaterite CaCO3–Ag microspheres. Furthermore, they can also protect the AgNPs from oxidation. Overall, the capping mechanism interactions between CMC or PSS and CaCO3–Ag involve reduction of Ag+ ions, stabilization of microspheres, and protection against oxidation. Scheme 2 illustrates the reaction of calcium nitrate with sodium carbonate in silver colloid and resulted CaCO3–Ag–CMC or CaCO3–Ag–PSS microspheres formation. These interactions contribute to the formation, stability, and functionality of the vaterite CaCO3–Ag microspheres.
 | ||
| Scheme 1 Proposed capping mechanism of CMC (I) and PSS (II) with AgNPs. | ||
 | ||
| Scheme 2 Proposed reactions of calcium nitrate with sodium carbonate in silver colloid and structure of vaterite CaCO3–Ag–CMC or vaterite CaCO3–Ag–PSS. | ||
Results for the narrow scan spectra of C1s, O1s, Ca2p and Ag3d are presented in Fig. 2A–D, respectively. The narrow scans of C1s and O1s showed that those elements are bound in a different chemical environment. Both the C1s and O1s spectra were shifted toward higher binding energy (BE) with precipitation of CaCO3 in silver colloid due to interactions and chemical bonding after the formation of CaCO3–Ag microspheres. The BE here represents the energy required to remove an electron from the S orbital shell. All BEs in our manuscript were initially compared and verified using the NIST X-ray Photoelectron Spectroscopy Database.30 The BE is influenced by the electron density and the local chemical environment. An increase in the BE of orbits suggests a decrease in electron density around the atom. When the BE increases, it indicates that the electron is more tightly bound to the atom, implying a decrease in electron density. This could be due to factors such as an electron donation to neighbouring atoms and changes in oxidation state.31 The increase in the O1s BE from 530.9 eV to 531.5 eV in CaCO3–Ag–CMC and 531.7 eV in CaCO3–Ag–PSS confirms the interaction between O of CaCO3 and AgNPs. This interaction is a result of the donation of electrons from the O atoms to the Ag ions, indicating the formation of chemical bonds between O and Ag which is likely forming a coordination or complex bond. This type of bonding is known as coordination or ligand exchange, where the Ag ions interact with the lone pair of electrons on the O atoms (as illustrated in Scheme 1).32 Furthermore, as shown in Fig. 2C, the Ca2p3/2 BE of ESP increased from 346.3 eV to 347.1 and 347.3 eV in CaCO3–Ag–CMC and CaCO3–Ag–PSS, respectively. This indicates the transformation polymorph of CaCO3 from calcite to vaterite structure. The same observation has been reported by Ni et al.33 As shown in Fig. 2D, the Ag3d5/2 BE of CaCO3–Ag–PSS is 368.7 eV which is slightly higher than CaCO3–Ag–CMC BE i.e., 368.2 eV. This would be due to the presence of different capping mechanisms which can affect the charge transfer processes between the Ag ions and CaCO3. The charge transfer interactions can influence the BE by modifying the electron density around the Ag ions.34 Furthermore, the sulfonate functional groups present in PSS and carboxymethyl functional groups in CMC can interact differently with the silver ions, leading to variations in the coordination chemistry and bond strength. This can result in changes in the electronic environment around the silver ions and, consequently, different BEs were observed.
 | ||
| Fig. 2 C1s (A), O1s (B), Ca2p (C) and Ag3d (D) narrow scan spectra of (a) ESP, (b) CaCO3–Ag–CMC and (c) CaCO3–Ag–PSS. | ||
The high depth deconvolution XPS spectra for the C1s and O1s regions are depicted in Fig. 3. In Fig. 3a, which pertains to ESP, the C1s spectrum was analysed and fitted to five distinct BEs. The peak observed at 281.2 eV is attributed to metal-carbide bond resulting from surface contamination on the ESP. At 283.4 eV, a peak is observed corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds, which can be linked to protoporphyrin. The protoporphyrin is recognized as the primary pigment responsible for the brown coloration in eggshells.35 Another peak at 284.8 eV corresponds to sp3 hybridized carbon, denoting a carbon atom with four substituents bonded to it. The presence of a peak at 287.5 eV can be attributed to the C–O bond. Lastly, the peak with the highest BE, at 289.1 eV, is assigned to the CO bond.36 In Fig. 3b, the XPS high depth deconvolution analysis of the CaCO3–Ag–CMC reveals five distinct BEs. Notably, the protoporphyrin and metal-carbide peaks, observed in Fig. 3a, are no longer present. Instead, two new peaks at BEs of 282.3 eV and 287.6 eV emerge, indicating the influence of the CaCO3–Ag capping with CMC polymer chains. These polymer chains introduce changes in the bonding environment, leading to the appearance of these additional peaks. Moreover, the intensity of the CO peak decreases in CaCO3–Ag–CMC compared to Fig. 3a. This reduction in intensity can be attributed to the coating layer of the capping polyelectrolyte, which modifies the interaction between the Ag+ and the CO groups. Notably, the intensity of the CO peak is even lower in CaCO3–Ag–PSS (Fig. 3c). Additionally, the two BEs corresponding to C–O, arising from the capping polymer and C–O bonds of CaCO3, seem to overlap, indicating a combined contribution. As depicted in Fig. 3d, the untreated ESP displayed three detectable energy levels corresponding to the O1s peaks: 529.2 eV, which suggests O2− specious from inorganic compounds; 531.0 eV, indicating OC bonds; and 532.5 eV, representing the presence of protein oxygen-containing species.37,38 Following the precipitation of CaCO3 and AgNPS loading, the peak associated with protein oxygen-containing species at 532.5 eV was no longer observed. Instead, a new peak emerged at 533.1 and 532.6 eV. This new peak was attributed to the O–C contributions arising from the CMC and PSS capping agents.
C bonds, which can be linked to protoporphyrin. The protoporphyrin is recognized as the primary pigment responsible for the brown coloration in eggshells.35 Another peak at 284.8 eV corresponds to sp3 hybridized carbon, denoting a carbon atom with four substituents bonded to it. The presence of a peak at 287.5 eV can be attributed to the C–O bond. Lastly, the peak with the highest BE, at 289.1 eV, is assigned to the CO bond.36 In Fig. 3b, the XPS high depth deconvolution analysis of the CaCO3–Ag–CMC reveals five distinct BEs. Notably, the protoporphyrin and metal-carbide peaks, observed in Fig. 3a, are no longer present. Instead, two new peaks at BEs of 282.3 eV and 287.6 eV emerge, indicating the influence of the CaCO3–Ag capping with CMC polymer chains. These polymer chains introduce changes in the bonding environment, leading to the appearance of these additional peaks. Moreover, the intensity of the CO peak decreases in CaCO3–Ag–CMC compared to Fig. 3a. This reduction in intensity can be attributed to the coating layer of the capping polyelectrolyte, which modifies the interaction between the Ag+ and the CO groups. Notably, the intensity of the CO peak is even lower in CaCO3–Ag–PSS (Fig. 3c). Additionally, the two BEs corresponding to C–O, arising from the capping polymer and C–O bonds of CaCO3, seem to overlap, indicating a combined contribution. As depicted in Fig. 3d, the untreated ESP displayed three detectable energy levels corresponding to the O1s peaks: 529.2 eV, which suggests O2− specious from inorganic compounds; 531.0 eV, indicating OC bonds; and 532.5 eV, representing the presence of protein oxygen-containing species.37,38 Following the precipitation of CaCO3 and AgNPS loading, the peak associated with protein oxygen-containing species at 532.5 eV was no longer observed. Instead, a new peak emerged at 533.1 and 532.6 eV. This new peak was attributed to the O–C contributions arising from the CMC and PSS capping agents.
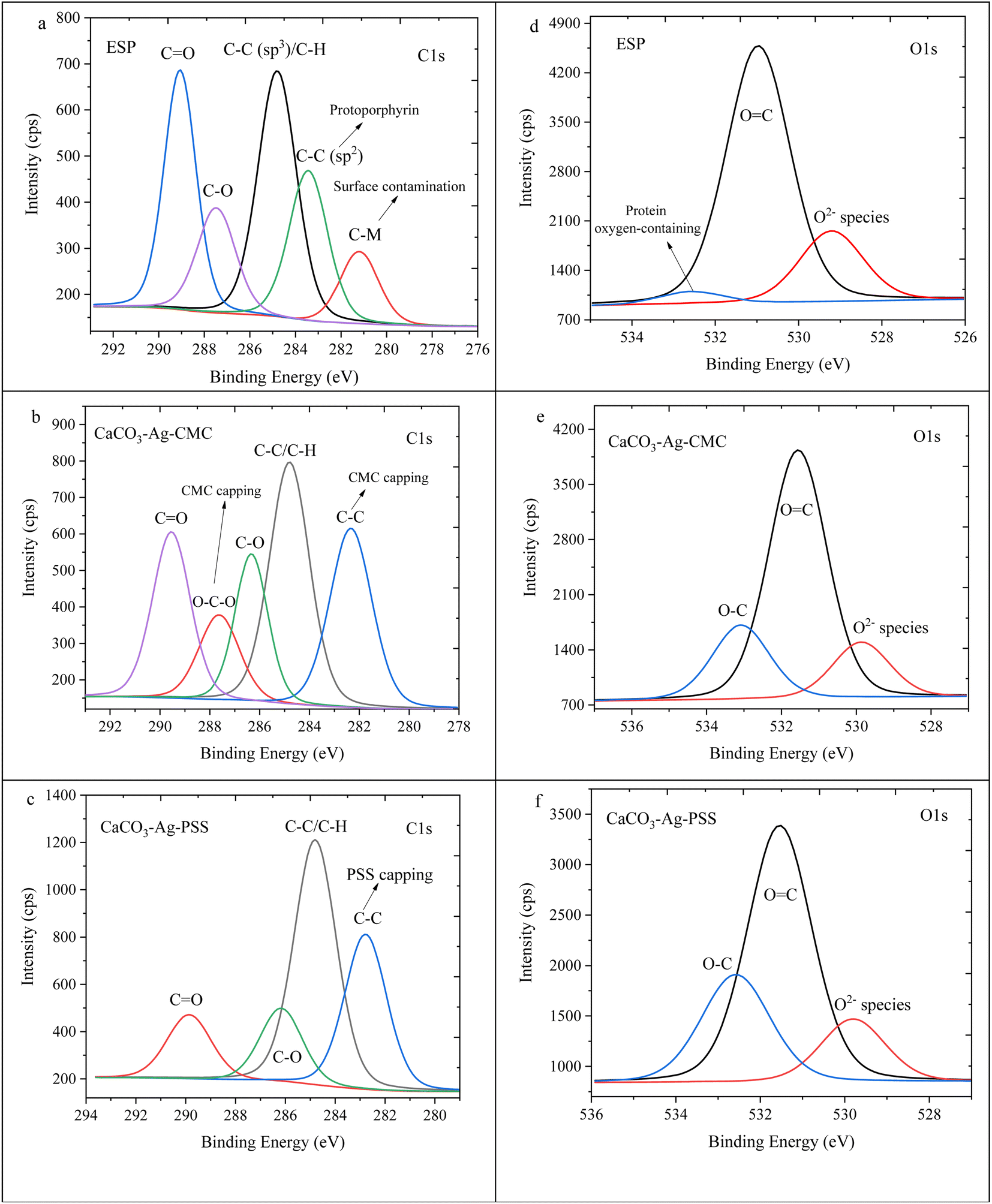 | ||
| Fig. 3 C1s high depth deconvolution spectra of (a) ESP, (b) CaCO3–Ag–CMC and (c) CaCO3–Ag–PSS. O1s high depth deconvolution spectra of (d) ESP, (e) CaCO3–Ag–CMC and (f) CaCO3–Ag–PSS. | ||
The high depth XPS deconvolution presented in Fig. 4 provides detailed information about the Ag3d and Ca2p regions. Fig. 4a and b depict the deconvolution XPS scans of Ag3d, revealing the presence of two spin–orbit split peaks, Ag3d5/2, and Ag3d3/2. The observed BEs assignment are supported by literature.39,40 For the CaCO3–Ag–CMC sample, the BEs for Ag3d3/2 are measured at 374.8 eV and 373.8 eV, while for Ag3d5/2, they are recorded at 368.8 eV and 367.8 eV. In the case of CaCO3–Ag–PSS, the BE values for Ag3d3/2 are observed at 374.9 eV and 373.8 eV, and for Ag3d5/2, they are found at 368.9 eV and 367.8 eV. Those BE values indicate the presence of both Ag+ and Ag0 states in the samples.41 The appearance of Ag3d spectra in higher BE, indicating that there were electrons loss in electron shells of Ag atoms as reported by Zhou et al.42 This suggests that in addition to Ag+, there is also the presence of elemental silver. The observed BE values provide evidence for the coexistence of different valence states of Ag, namely Ag+ and Ag0. A notable observation is that in the CaCO3–Ag–PSS sample, the intensity of the Ag0 spin–orbit splitting is higher compared to Ag+. This result was also mentioned in ref. 28. Contrarily, in the CaCO3–Ag–CMC sample, the intensity of Ag+ is higher compared to Ag0. This difference in intensity can be attributed to the unique capping properties exhibited by both CMC and PSS polyelectrolytes. The CMC polyelectrolyte has a greater affinity for Ag+, facilitating their stabilization and reducing their conversion to Ag0. The PSS polymer demonstrates a more effective capability to convert Ag+ to Ag0 or provide better protection for Ag0, preventing its oxidation to Ag+. The PSS polyelectrolyte potentially contributes to the higher presence and stability of Ag0 in the CaCO3–Ag–PSS microspheres. On the other hand, the CMC polymer might exhibit a lesser ability to convert Ag+ to Ag0 or provide less protection against oxidation, resulting in a lower intensity of Ag0 spin–orbit splitting in the CaCO3–Ag–CMC sample. In the Ca2p deconvolution analysis presented in Fig. 4c–e, only one spin–orbit splitting (Ca2p3/2 and Ca2p1/2) was observed for all the samples, indicating the presence of a single vaterite polymorphs of Ca2p species. This observation is consistent with findings in the literature.36
 | ||
| Fig. 4 Ag3d high depth deconvolution spectra of (a) CaCO3–Ag–CMC and (b) CaCO3–Ag–PSS.28 Ca2p high depth deconvolution spectra of (c) ESP, (d) CaCO3–Ag–CMC and (e) CaCO3–Ag–PSS. | ||
CaCO3–Ag–CMC and CaCO3–Ag–PSS embedded in CMC matrix and internally physical cross-linking
In this section, the study focuses on the incorporation of CaCO3–Ag–CMC and CaCO3–Ag–PSS microspheres into a CMC polymer matrix, which undergoes physical cross-linking with calcium chloride. The purpose of physical cross-linking of CMC is to enhance its stability in the water and improve its water absorption and retention properties. The resulting films of physically cross-linked CMC, containing CaCO3–Ag–CMC or CaCO3–Ag–PSS, have potential applications as superabsorbent pads in active packaging. To analyse the surface composition of the films, XPS survey scans were performed on neat CMC polymer film, as well as CMC/CaCO3–Ag–CMC and CMC/CaCO3–Ag–PSS films. The XPS scan revealed the presence of C, O, and Na elements in the neat CMC film. Additionally, in the CMC/CaCO3–Ag–CMC and CMC/CaCO3–Ag–PSS films, the spectrum displayed the detection of Ag, Ca, and Cl elements, in addition to C, O, and Na, as shown in Fig. 5. Moreover, the elemental compositions of the sample surface calculated by XPS high resolution spectra are listed in Table 2. The schematic for physically crosslinked CMC polymer matrix with incorporated CaCO3–Ag–PSS along with SEM micrographs of magnification 4000× and 1000× was presented in ref. 27.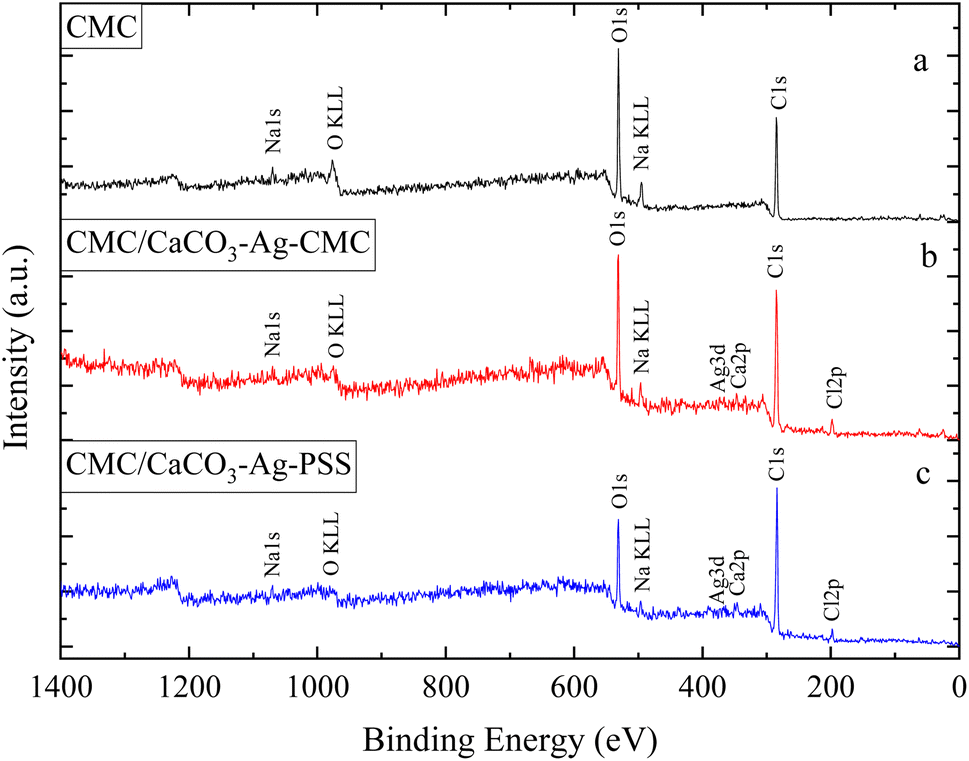 | ||
| Fig. 5 XPS survey scans of (a) neat CMC, (b) CMC/CaCO3–Ag–CMC and (c) CMC/CaCO3–Ag–PSS. | ||
Results for the narrow scan spectra of C1, O1s, Ca2p and Ag3d are presented in Fig. 6. Fig. 6A displays the narrow scan of C1s, revealing a dominant peak in the neat CMC film. However, after embedding CaCO3–Ag–CMC and CaCO3–Ag–PSS and cross-linking, the peak intensity decreased, and it further exhibited splitting with an additional shoulder peak at a lower BE. This phenomenon is likely attributed to the physically cross-linking bonding interaction between polymer chains. On the other hand, the O1s feature peak broadened, and its intensity decreased, indicating the occurrence of physical interactions involving the O from carboxylate functional groups (–COO–) present in the CMC polymer chains as illustrated in Fig. 6B. This suggests that carboxylate groups of CMC can form coordination bonds with Ca ions from CaCl2, leading to the physical cross-linking of the polymer chains. This cross-linking enhances the stability and water absorption properties of the resulting CMC/CaCO3–Ag–CMC or CMC/CaCO3–Ag–PSS films. Furthermore, as shown in Fig. 6C, the BEs of Ca2p3/2 and Ca2p1/2 peaks remain the same as they were before embedding in the CMC matrix, indicating that the CaCO3 polymorphs retain their vaterite structure. As shown in Fig. 6D, the Ag3d3/2 BEs of CMC/CaCO3–Ag–CMC is shifted to the lower BE in compared to the CaCO3–Ag–CMC and CaCO3–Ag–PSS before embedding (Fig. 2D). This indicated the oxidation of AgNPs after embedding and cross-linking of CMC chains.
 | ||
| Fig. 6 C1s (A), O1s (B), Ca2p (C) and Ag3d (D) narrow scan spectra of (a) neat CMC, (b) CMC/CaCO3–Ag–CMC and (c) CMC/CaCO3–Ag–PSS. | ||
Fig. 7 displays high depth deconvolution XPS measurements for the C1s and O1s regions. Fig. 7a, represents the neat CMC polymer with distinct energy levels for the C1s peaks. These levels correspond to specific C components within the CMC polymer film: 284.8 eV indicates C–C or C–H bonds, 286.3 eV signifies C–O bonds, and 287.7 eV suggests CO/C–O–C in the polymer chains.43 Notably, as shown in Fig. 7b and c these distinct peaks remain after introducing CaCO3–Ag particles into the CMC mixture and performing the cross-linking using CaCl2. However, a clear trend emerges as the intensity of the C–C peak increases while the CO peak intensity decreases. This pattern suggests interaction involving the CMC chains and Ca2+ resulted from the cross-linking mechanism. Moreover, in the CMC/CaCO3–Ag–CMC and CMC/CaCO3–Ag–PSS systems, the CO/C–O–C peak, initially at 287.7 eV, gradually shifts to 287.9 eV and 288.2 eV, respectively. This shift likely arises from interactions between CaCO3–Ag microspheres and the carboxylate groups of CMC resulting in adjustments in the behaviour of carbon atoms in the polymer matrix along with changes in electron distribution due to the cross-linking process. On the other hand, as illustrated in Fig. 7d, the pristine CMC polymer exhibited two distinct energy levels for the O1s peaks: 530.9 eV indicating OC bonds and 532.7 eV signifying O–C bonds. Remarkably, as shown in Fig. 7f, the O1s peak at 530.9 eV showed a slight shift towards higher BE due to internal crosslinking reactions.
 | ||
| Fig. 7 C1s high depth deconvolution spectra of (a) neat CMC, (b) CMC/CaCO3–Ag–CMC and (c) CMC/CaCO3–Ag–PSS. O1s high depth deconvolution spectra of (d) neat CMC, (e) CMC/CaCO3–Ag–CMC and (f) CMC/CaCO3–Ag–PSS. | ||
Fig. 8a and b illustrate the Ag3d spin–orbit splitting for CMC/CaCO3–Ag–CMC and CMC/CaCO3–Ag–PSS, respectively. The image of Fig. 8b was presented earlier in ref. 27. Deconvolution of the XPS scan results revealed a distinct peak for Ag3d5/2 and Ag3d3/2, positioned at a lower BE than that of the unembedded CaCO3–Ag. The initial BE values, prior to embedding, indicated the presence of both Ag0 and Ag+. However, following embedding, the lower BE values exclusively indicated the presence of Ag+ valence state, implying surface oxidation of the AgNPs during both the embedding process in the CMC polymer matrix and the subsequent cross-linking reactions. The lower BEs associated with ionic silver align well with those reported in the literature.44,45 Fig. 8c and d depict the Ca2p spin–orbit splitting for CMC/CaCO3–Ag–CMC and CMC/CaCO3–Ag–PSS, respectively. The deconvolution analysis showed the presence of two distinct splitting peaks at 346.8 eV and 347.7 eV for Ca2p3/2, as well as 350.4 eV and 351.3 eV for Ca2p1/2 in the CMC/CaCO3–Ag–CMC film. The Ca2p spin–orbit splitting at the lower BE corresponds to Ca2+ originating from CaCO3. Conversely, the spin–orbit splitting at a higher BE energy corresponds to the Ca2+ involved in cross-linking within the CMC film. Interestingly, the intensity of the crosslinking related Ca2+ signal is higher in CMC/CaCO3–Ag–PSS.
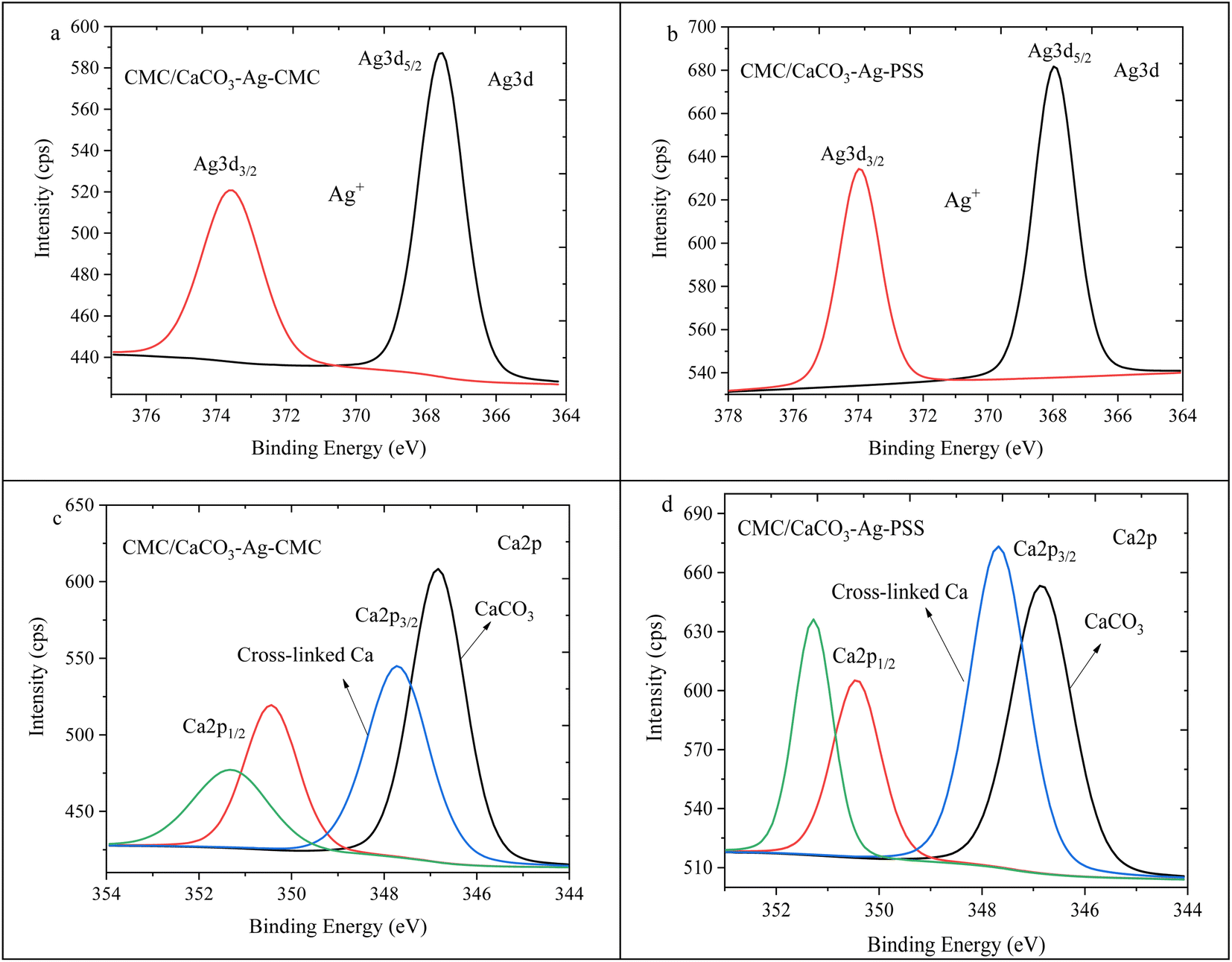 | ||
| Fig. 8 Ag3d high depth deconvolution spectra of (a) CMC/CaCO3–Ag–CMC and (b) CMC/CaCO3–Ag–PSS.27 Ca2p high depth deconvolution spectra of (c) CMC/CaCO3–Ag–CMC and (d) CMC/CaCO3–Ag–PSS. | ||
CaCO3–Ag–CMC and CaCO3–Ag–PSS embedded in PVA film, bonding, and chemical compositions
In this section, the study focuses on the incorporation of CaCO3–Ag–CMC and CaCO3–Ag–PSS microspheres into a fully hydrolysed poly vinyl alcohol (PVA) polymer matrix by an aqueous solution casting technique. The obtained PVA/CaCO3–Ag–CMC or PVA/CaCO3–Ag–PSS films with the water absorbent ability have the potential to be applied for wound dressing application. An XPS survey scans were conducted on the surface of neat PVA, PVA/CaCO3–Ag–CMC and PVA/CaCO3–Ag–PSS films. The scan detected C, and O in neat PVA, and in addition, Ag and Ca were detected in PVA/CaCO3–Ag–CMC and PVA/CaCO3–Ag–PSS, as shown in XPS spectra in Fig. 9. It is noteworthy that the incorporation of either CaCO3–Ag–CMC or CaCO3–Ag–PSS leads to a significant alteration in the atomic composition of the PVA matrix (Table 2). Specifically, the O atomic percentage in the PVA structure experiences a marked reduction from 35% to 10%, accompanied by a corresponding increase in the C atomic percentage from 65% to 86% as numbered in Table 2. These changes are attributed to the presence of CaCO3–Ag microparticles in the PVA film surface. The reduction in the O atomic percentage could potentially result from the interactions between the microspheres and the PVA matrix which reduces the exposure of O atoms in the surface, contributing to the observed decrease. The increase in the C atomic percentage can also be attributed to the incorporation of the microspheres which might lead to a relatively higher carbon-rich regions at the surface being analysed. Furthermore, In the PVA/CaCO3–Ag–CMC film, although the Ca2+ percentage is lower than that of the PVA/CaCO3–Ag–PSS film, the atomic percentage of Ag is slightly higher. This observation may correlate with the size of CaCO3–Ag–CMC microspheres which is 3 times larger than CaCO3–Ag–PSS. | ||
| Fig. 9 XPS survey scans of (a) neat PVA, (b) PVA/CaCO3–Ag–CMC and (c) PVA/CaCO3–Ag–PSS. | ||
Results for the narrow scan spectra of C1s, O1s, Ca2p and Ag3d are presented in Fig. 10A–D, respectively. The C1s narrow peak in neat PVA film becomes sharper and its intensity increased after incorporation of CaCO3–Ag–CMC or CaCO3–Ag–PSS. This would be due to inducing changes in the crystallinity of the PVA film due to reduced disorder structure and enhanced signal-to-noise ratio which result in a sharper C1s peak.46 However, as the CaCO3–Ag–CMC and CaCO3–Ag–PSS microspheres are concentrated at the surface of the PVA film, this could also influence the surface chemistry and molecular arrangement and lead to sharper and more intense peaks due to the altered electronic environment near the surface. Moreover, the O1s narrow peak undergoes broadening, accompanied by a decrease in intensity in PVA after loading the microspheres. This could be due to the new peak arises from O bonded to the Ca2+ in CaCO3–Ag, potentially leading to the displacement of the O1s narrow scan towards the lower BE region. Furthermore, as shown in Fig. 10C, the BE of Ca2p3/2 and Ca2p1/2 remains the same as they were before embedding in the PVA (Fig. 2C), indicating that the CaCO3–Ag polymorphs retain their vaterite structure. As shown in Fig. 10D, the Ag3d BE of both PVA/CaCO3–Ag–CMC and PVA/CaCO3–Ag–PSS are shifted to the lower BE in comparison to the CaCO3–Ag–CMC and CaCO3–Ag–PSS before embedding (Fig. 2D), indicating the change in oxidation state of AgNPs.
 | ||
| Fig. 10 C1s (A), O1s (B), Ca2p (C) and Ag3d (D) narrow scan spectra of (a) neat PVA, (b) PVA/CaCO3–Ag–CMC and (c) PVA/CaCO3–Ag–PSS. | ||
The deconvoluted XPS spectra representing the C1s and O1s regions for pristine PVA, PVA/CaCO3–Ag–CMC, and PVA/CaCO3–Ag–PSS are displayed in Fig. 11. In Fig. 11a, dedicated to the neat PVA film, the C1s peaks revealed two distinctive BE values. The peak detected at 284.8 eV is ascribed to the presence of a C–C bond from the PVA backbone chains. Additionally, the peak at 286.4 eV is attributed to the C–OH group within the PVA structure.47 Interestingly, the subsequent deconvolution scans in Fig. 11b and c showcase a complete absence of the C–OH bond due to the less amount of PVA after embedding the microspheres at the surface of film. This is subsequently accompanied by the appearance of a new peak at 288.1 eV, arising from the CO bonds in the CaCO3–Ag microspheres. As depicted in Fig. 11d, the pristine PVA polymer displayed a single energy level corresponding to the O1s peaks, measured at 532.5 eV, signifying the presence of OH bonds. Contrarily, as indicated in Fig. 11e and f for both the CMC/CaCO3–Ag–CMC and CMC/CaCO3–Ag–PSS films, a new peak emerged at 531.3 eV and 531.2 eV, respectively. This observed peak can be ascribed to the presence of OC bonds within the CaCO3–Ag microspheres. Furthermore, the reduction in the intensity of the O–C band is likely due to the predominance of CaCO3–Ag microspheres concentrated at the surface of the PVA film. The phase morphologies showing vertically uneven distribution of the active particles within the films of PVA/CaCO3–Ag–PSS and PVA/CaCO3–Ag–CMC were revealed elsewhere in ref. 28.
 | ||
| Fig. 11 C1s high depth deconvolution spectra of (a) neat PVA, (b) PVA/CaCO3–Ag–CMC and (c) PVA/CaCO3–Ag–PSS. O1s high depth deconvolution spectra of (d) neat PVA, (e) PVA/CaCO3–Ag–CMC and (f) PVA/CaCO3–Ag–PSS. | ||
Fig. 12a and b showcase the Ag3d5/2 and Ag3d3/2 spin–orbit splitting patterns for PVA/CaCO3–Ag–CMC and PVA/CaCO3–Ag–PSS respectively. These results have been previously reported in ref. 28. These peaks were situated at lower BE compared to the unembedded CaCO3–Ag microspheres. The initial BE values, prior to the embedding process, indicated the coexistence of both metallic and ionic silver states (refer to Fig. 4a & b). However, after embedding into the PVA matrix, the decreased BE values exclusively pointed towards the prevalence of the Ag+ valence state. This observation implies that the AgNPs underwent surface oxidation during their integration into the PVA polymer matrix.
 | ||
| Fig. 12 Ag3d high depth deconvolution spectra of (a) PVA/CaCO3–Ag–CMC and (b) PVA/CaCO3–Ag–PSS.28 Ca2p high depth deconvolution spectra of (c) PVA/CaCO3–Ag–CMC and (d) PVA/CaCO3–Ag–PSS. | ||
Moving to Fig. 12c and d, the Ca2p spin–orbit splitting for PVA/CaCO3–Ag–CMC and PVA/CaCO3–Ag–PSS revealed the presence of a spin–orbit splitting peak similar to that of the vaterite CaCO3 microspheres, as observed in Fig. 4c which indicates no changes in the CaCO3–Ag polymorphic structures.
Conclusions
X-ray photoelectron spectroscopy (XPS) provides valuable insights into the complicated processes involved in capping mechanism, surface composition alterations, polymorph transitions, reduction of Ag ions and stabilization. To summarize, this study has been primarily dedicated to the utilization of XPS for analysing the formation of biogenic vaterite CaCO3–Ag hybrid microspheres in conjunction with various polyelectrolytes serving as capping agents, as well as examining their interactions following incorporation into polymer matrices. The multifunctional role of capping agents, serving as both stabilizers, and reducing agents in the synthesis of vaterite CaCO3–Ag microspheres, was examined. Oxidation states and electronic environments of loaded AgNPs were found to differ before and after embedding in polymer matrices. When employing PSS as the capping agent, Ag0 was the predominant state of silver, while using CMC as the capping agent resulted in Ag+ being the dominant state of the AgNPs loaded onto vaterite CaCO3 microspheres. Additionally, the CaCO3–Ag polymorph structure was investigated by analysing the Ca2p spin–orbit splitting, revealing the stability of the vaterite polymorph. This study offers significant insights into the behaviour of AgNPs when loaded onto vaterite CaCO3–Ag using various capping agents, as well as their integration within different polymer matrices.Data availability
Data will be made available upon request.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by Suranaree University of Technology, Research and Development Fund (IRD7-710-67-12-19). The authors would like to acknowledge Synchrotron Light Research Institute (SLRI) Thailand for conducting XPS analysis.References
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-Ray Photoelectron Spectroscopy: A Reference Book of Standard Spectra for Identification and Interpretation of XPS Data, Physical Electronics Division, Perkin-Elmer Corporation, 1992, https://books.google.co.th/books?id=A_XGQgAACAAJ Search PubMed.
- F. Loi, M. Pozzo, L. Sbuelz, L. Bignardi, P. Lacovig and E. Tosi, et al., Breakdown of the correlation between oxidation states and core electron binding energies at the sub-nanoscale, Appl. Surf. Sci., 2023, 619, 156755, DOI:10.1016/j.apsusc.2023.156755.
- D. Golze, L. Keller and P. Rinke, Accurate Absolute and Relative Core-Level Binding Energies from GW, J. Phys. Chem. Lett., 2020, 11, 1840–1847, DOI:10.1021/acs.jpclett.9b03423.
- R. Meyer, C. Lemire, S. K. Shaikhutdinov and H. J. Freund, Surface chemistry of catalysis by gold, Gold Bull., 2004, 37, 72–124, DOI:10.1007/BF03215519.
- K. A. Johnston, L. M. Stabryla, L. M. Gilbertson and J. E. Millstone, Emerging investigator series: connecting concepts of coinage metal stability across length scales, Environ. Sci.: Nano, 2019, 6, 2674–2696, 10.1039/C9EN00407F.
- A. Mänz, A. A. Hauke and G. Witte, Copper Phthalocyanine as Contact Layers for Pentacene Films Grown on Coinage Metals, J. Phys. Chem. C, 2018, 122, 2165–2172, DOI:10.1021/acs.jpcc.7b10324.
- J. Butler, R. D. Handy, M. Upton and A. Besinis, Review of Antimicrobial Nanocoatings in Medicine and Dentistry: Mechanisms of Action, Biocompatibility Performance, Safety, and Benefits Compared to Antibiotics, ACS Nano, 2023, 17, 7064–7092, DOI:10.1021/acsnano.2c12488.
- D. B. Trushina, T. N. Borodina, S. Belyakov and M. N. Antipina, Calcium carbonate vaterite particles for drug delivery: advances and challenges, Mater. Today Adv., 2022, 14, 100214, DOI:10.1016/j.mtadv.2022.100214.
- Z. Feng, T. Yang, S. Dong, T. Wu, W. Jin and Z. Wu, et al., Industrially synthesized biosafe vaterite hollow CaCO3 for controllable delivery of anticancer drugs, Mater. Today Chem., 2022, 24, 100917, DOI:10.1016/j.mtchem.2022.100917.
- A. M. Ferreira, A. Vikulina, G. W. V. Cave, M. Loughlin, V. Puddu and D. Volodkin, Vaterite vectors for the protection, storage and release of silver nanoparticles, J. Colloid Interface Sci., 2023, 631, 165–180, DOI:10.1016/j.jcis.2022.10.094.
- A. M. Ferreira, A. S. Vikulina and D. Volodkin, CaCO3 crystals as versatile carriers for controlled delivery of antimicrobials, J. Controlled Release, 2020, 328, 470–489, DOI:10.1016/j.jconrel.2020.08.061.
- J. Qu and H. M. Meyer, X-Ray Photoelectron Spectroscopy (XPS), in Encyclopedia of Tribology, ed. Q. J. Wang and Y.-W. Chung, Springer US, Boston, MA, 2013, pp. 4133–4138, DOI:10.1007/978-0-387-92897-5_1222.
- D. Briggs and J. T. Grant, Surface Analysis by Auger and X-Ray Photoelectron Spectroscopy, 2003, pp. 978-1-901019-04-9 Search PubMed.
- F. A. Stevie and C. L. Donley, Introduction to x-ray photoelectron spectroscopy, J. Vac. Sci. Technol., A, 2020, 38, 063204, DOI:10.1116/6.0000412.
- S. Oswald, X-Ray Photoelectron Spectroscopy in Analysis of Surfaces, in Encyclopedia of Analytical Chemistry, John Wiley & Sons, Ltd, Chichester, UK, 2013, DOI:10.1002/9780470027318.a2517.pub2.
- M. Majeed, K. R. Hakeem and R. U. Rehman, Synergistic effect of plant extract coupled silver nanoparticles in various therapeutic applications- present insights and bottlenecks, Chemosphere, 2022, 288, 132527, DOI:10.1016/j.chemosphere.2021.132527.
- S. Ahmed, M. Ahmad, B. L. Swami and S. Ikram, A review on plants extract mediated synthesis of silver nanoparticles for antimicrobial applications: a green expertise, J. Adv. Res., 2016, 7, 17–28, DOI:10.1016/j.jare.2015.02.007.
- J. Girard, P. S. Brunetto, O. Braissant, Z. Rajacic, N. Khanna and R. Landmann, et al., Development of a polystyrene sulfonate/silver nanocomposite with self-healing properties for biomaterial applications, C. R. Chim., 2013, 16, 550–556, DOI:10.1016/j.crci.2013.04.010.
- G. J. C. Canama, M. C. L. Delco, R. A. Talandron and N. P. Tan, Synthesis of Chitosan-Silver Nanocomposite and Its Evaluation as an Antibacterial Coating for Mobile Phone Glass Protectors, ACS Omega, 2023, 8, 17699–17711, DOI:10.1021/acsomega.3c00191.
- A. M. Ferreira, A. Vikulina, M. Loughlin and D. Volodkin, How similar is the antibacterial activity of silver nanoparticles coated with different capping agents?, RSC Adv., 2023, 13, 10542–10555, 10.1039/D3RA00917C.
- C. V. Restrepo and C. C. Villa, Synthesis of silver nanoparticles, influence of capping agents, and dependence on size and shape: a review, Environ. Nanotechnol., Monit. Manage., 2021, 15, 100428, DOI:10.1016/j.enmm.2021.100428.
- M. H. Azarian and W. Sutapun, Tuning polymorphs of precipitated calcium carbonate from discarded eggshells: effects of polyelectrolyte and salt concentration, RSC Adv., 2022, 12, 14729–14739, 10.1039/D2RA01673G.
- M. E. E. Zin, P. Moolkaew, T. Junyusen and W. Sutapun, Preparation of hybrid particles of Ag nanoparticles and eggshell calcium carbonate and their antimicrobial efficiency against beef-extracted bacteria, R. Soc. Open Sci., 2023, 10, 221197, DOI:10.1098/rsos.221197.
- M. S. Rahman, M. S. Hasan, A. S. Nitai, S. Nam, A. K. Karmakar and M. S. Ahsan, et al., Recent Developments of Carboxymethyl Cellulose, Polymers, 2021, 13, 1345, DOI:10.3390/polym13081345.
- M. H. Azarian and P. Boochathum, Nanofiber films of chloroacetated natural rubber/poly (vinyl alcohol) by electrospinning technique : Silica effects on biodegradation, J. Appl. Polym. Sci., 2018, 46432, 1–10, DOI:10.1002/app.46432.
- M. H. Azarian, W. A. Kamil Mahmood, E. Kwok, W. F. Bt Wan Fathilah and N. F. Binti Ibrahim, Nanoencapsulation of intercalated montmorillonite-urea within PVA nanofibers: hydrogel fertilizer nanocomposite, J. Appl. Polym. Sci., 2018, 135, 45957, DOI:10.1002/app.45957.
- M. H. Azarian, T. Junyusen and W. Sutapun, Tailoring microstructure of superabsorbent film for active food packaging using carboxy methyl cellulose and biogenic vaterite CaCO3–Ag hybrid microspheres, J. Food Eng., 2024, 369, 111938, DOI:10.1016/j.jfoodeng.2024.111938.
- M. H. Azarian, T. Junyusen and W. Sutapun, Biogenic Vaterite Calcium Carbonate-Silver/Poly(Vinyl Alcohol) Film for Wound Dressing, ACS Omega, 2024, 9, 955–969, DOI:10.1021/acsomega.3c07135.
- R. Huopalahti, R. López-Fandiño, M. Anton, R. Schade, I. Recio and M. Ramos, Bioactive Egg Compounds, Springer Berlin Heidelberg, Berlin, Heidelberg, 2007, DOI:10.1007/978-3-540-37885-3.
- A. V. Naumkin, A. Kraut-Vass, S. W. Gaarenstroom and C. J. Powell, NIST X-ray Photoelectron Spectroscopy Database, SRD 20, V.5, 2012, DOI:10.18434/T4T88K.
- G. Greczynski and L. Hultman, X-ray photoelectron spectroscopy: towards reliable binding energy referencing, Prog. Mater. Sci., 2020, 107, 100591, DOI:10.1016/j.pmatsci.2019.100591.
- L. H. Nguyen, D. P. Tran and T. N. Truong, Computational Study on the Nature of Bonding between Silver Ions and Nitrogen Ligands, ACS Omega, 2022, 7, 45231–45238, DOI:10.1021/acsomega.2c05707.
- M. Ni and B. D. Ratner, Differentiating calcium carbonate polymorphs by surface analysis techniques-an XPS and TOF-SIMS study, Surf. Interface Anal., 2008, 40, 1356–1361, DOI:10.1002/sia.2904.
- R. Otero, A. L. Vázquez de Parga and J. M. Gallego, Electronic, structural and chemical effects of charge-transfer at organic/inorganic interfaces, Surf. Sci. Rep., 2017, 72, 105–145, DOI:10.1016/j.surfrep.2017.03.001.
- S. Samiullah and J. R. Roberts, The location of protoporphyrin in the eggshell of brown-shelled eggs, Poult. Sci., 2013, 92, 2783–2788, DOI:10.3382/ps.2013-03051.
- C. S. Gopinath, S. G. Hegde, A. V. Ramaswamy and S. Mahapatra, Photoemission studies of polymorphic CaCO3 materials, Mater. Res. Bull., 2002, 37, 1323–1332, DOI:10.1016/S0025-5408(02)00763-8.
- L. Q. Guo, S. X. Qin, B. J. Yang, D. Liang and L. J. Qiao, Effect of hydrogen on semiconductive properties of passive film on ferrite and austenite phases in a duplex stainless steel, Sci. Rep., 2017, 7, 3317, DOI:10.1038/s41598-017-03480-8.
- M. Minakshi, S. Higley, C. Baur, D. R. G. Mitchell, R. T. Jones and M. Fichtner, Calcined chicken eggshell electrode for battery and supercapacitor applications, RSC Adv., 2019, 9, 26981–26995, 10.1039/c9ra04289j.
- A. K. Mohamedkhair, Q. A. Drmosh and Z. H. Yamani, Silver Nanoparticle-Decorated Tin Oxide Thin Films: Synthesis, Characterization, and Hydrogen Gas Sensing, Front. Mater., 2019, 6, 1–10, DOI:10.3389/fmats.2019.00188.
- P. Uznanski, J. Zakrzewska, F. Favier, S. Kazmierski and E. Bryszewska, Synthesis and characterization of silver nanoparticles from (bis)alkylamine silver carboxylate precursors, J. Nanopart. Res., 2017, 19, 121, DOI:10.1007/s11051-017-3827-5.
- J. Wang, C. An, M. Zhang, C. Qin, X. Ming and Q. Zhang, Photochemical conversion of AgCl nanocubes to hybrid AgCl–Ag nanoparticles with high activity and long-term stability towards photocatalytic degradation of organic dyes, Can. J. Chem., 2012, 90, 858–864, DOI:10.1139/v2012-079.
- Z. Zhou, Z. Shen, C. Song, M. Li, H. Li and S. Zhan, Boosting the activation of molecular oxygen and the degradation of tetracycline over high loading Ag single atomic catalyst, Water Res., 2021, 201, 117314, DOI:10.1016/j.watres.2021.117314.
- M. Zhao, S. Zhang, G. Fang, C. Huang and T. Wu, Directionally-Grown Carboxymethyl Cellulose/Reduced Graphene Oxide Aerogel with Excellent Structure Stability and Adsorption Capacity, Polymers, 2020, 12, 2219, DOI:10.3390/polym12102219.
- J. A. Jiménez, H. Liu and E. Fachini, X-ray photoelectron spectroscopy of silver nanoparticles in phosphate glass, Mater. Lett., 2010, 64, 2046–2048, DOI:10.1016/j.matlet.2010.07.004.
- P. Kumari and P. Majewski, Adsorption of Albumin on Silica Surfaces Modified by Silver and Copper Nanoparticles, J. Nanomater., 2013, 2013, 1–7, DOI:10.1155/2013/839016.
- G. Greczynski and L. Hultman, A step-by-step guide to perform x-ray photoelectron spectroscopy, J. Appl. Phys., 2022, 132, 011101, DOI:10.1063/5.0086359.
- J. M. Ino, P. Chevallier, D. Letourneur, D. Mantovani and C. Le Visage, Plasma functionalization of poly(vinyl alcohol) hydrogel for cell adhesion enhancement, Biomatter, 2013, 3(4), e25414, DOI:10.4161/biom.25414.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra01710b |
| This journal is © The Royal Society of Chemistry 2024 |