
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis and antiproliferative evaluation of tetrahydro-β-carboline histone deacetylase inhibitors bearing an aliphatic chain linker†
Jing Shiab,
Jiayun Wangc,
Xingjie Wangd,
Chao Qu d,
Changchun Yed,
Xiuli Lib,
Xin Chen*c and
Zhengshui Xu*a
d,
Changchun Yed,
Xiuli Lib,
Xin Chen*c and
Zhengshui Xu*a
aDepartment of Thoracic Surgery, The Second Affiliated Hospital of Xi'an Jiaotong University, Xi'an, Shaanxi 710004, China. E-mail: xuzhengshui@xjtu.edu.cn; Fax: +86-029-87679000
bDepartment of Respiratory and Endocrinology, The Second Affiliated Hospital of Xi'an Jiaotong University, Xi'an 710004, Shaanxi, China
cShaanxi Key Labotory of Natural Products & Chemical Biology, College of Chemistry & Pharmacy, Northwest A&F University, Yangling 712100, China. E-mail: chenxin1888@nwsuaf.edu.cn; Fax: +86-029-87092335
dDepartment of General Surgery, The First Affiliated Hospital of Xi'an Jiaotong University, Xi'an, 710061, Shaanxi, China
First published on 19th April 2024
Abstract
The use of histone deacetylase inhibitors (HDACis) is an effective approach for cancer treatment. In this work, a series of hydroxamic acid-based HDACis with a tetrahydro-β-carboline core and aliphatic linker have been designed and synthesized. The optimal compound 13d potently inhibited HDAC1 and showed good antiproliferative activity against different tumor cell lines in vitro. Molecular docking of 13d was conducted to rationalize the high binding affinity for HDAC1. Therefore, this work provides a new structure design for HDAC inhibitors and also offers a promising treatment for solid tumors.
1. Introduction
The regulation of histone modifications, including histone acetylation, methylation, or phosphorylation represent desirable drug targets due to aberrant epigenetic modulation in many cancers.1,2 Histone deacetylases (HDACs), which control the acetylation levels of nuclear proteins and cytoplasmic proteins, have attracted wide attention for their crucial roles in the genesis and development of neoplasms.3–5 The 11 classical zinc-dependent isoforms of mammalian HDAC are divided into four categories: class I (HDAC1, HDAC2, HDAC3, HDAC8), class IIa (HDAC4, 5, 7, 9), class IIb (HDAC6, 10), and class IV (HDAC11).6,7 Class I HDACs, especially HDAC1, are important regulators of cell proliferation and a key oncology target.8 Currently, many HDAC inhibitors are clinical, and five have been approved, namely, vorinostat,9 belinostat,10 panobinostat,11 romidepsin12 and chidamide.13,14 The pharmacophore of HDAC inhibitors is well established: a capping motif occupying the outside of the protein's active pocket, a zinc-binding group (ZBG) chelating the catalytically active zinc ion, and a linker chain connecting the two parts (Fig. 1).15–21 Hydroxamic acid is the most frequently used ZBG. Vorinostat (1), which was approved for the treatment of cutaneous T-cell lymphoma. Belinostat (2) gained U.S. Food and Drug Administration approval for the treatment of peripheral T-cell lymphoma. Panobinostat (3) was approved for the treatment of multiple myeloma. Selective HDAC6i ACY-1215 (4) was clinically evaluated for the treatment of multiple types of tumors.22 It preferentially inhibited HDAC6 with an IC50 of 4.7 nM, but also showed potent HDAC1 inhibition at 58 nM. However, most solid tumors have shown limited response to HDAC inhibitors, in contrast to the success with lymphoma in the clinic.23 Hence, the development of either selective or pan-HDACis with better efficacy against solid tumors is of importance.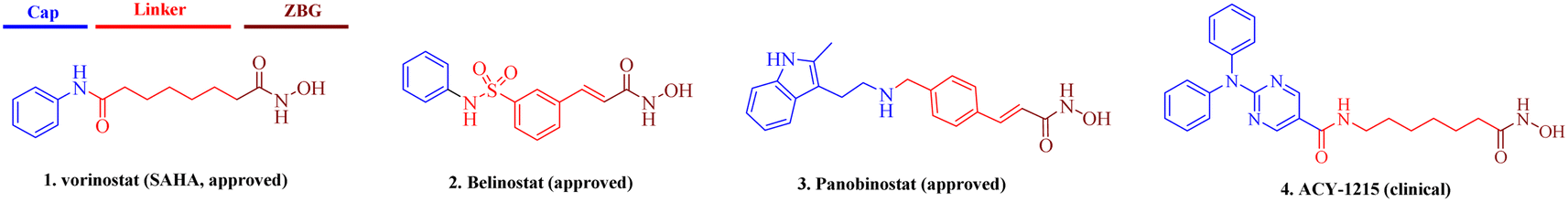 | ||
| Fig. 1 Representative approved and clinical hydroxamic-acid-based HDAC inhibitors. | ||
Tetrahydro-β-carboline (THβC, 5) exists in a large number of natural compounds with different biological properties, such as antiviral, antimalarial, antithrombotic, antithrombotic, analgesic, anticancer and neurodegenerative activities.24 Its semi-rigid structure (active indole ring and flexible piperidine motif) shows preferential affinities against different receptors and can be used as both an H-bond receptor and H-bond donor. This privileged scaffold also matches the characteristics of a capping group, which generally consists of hydrophobic and aromatic structures. Moreover, the THβC scaffold could be obtained through easy steps and is convenient for the construction of compound libraries. Hence, structural optimization on the THβC scaffold is a promising strategy for drug discovery.25,26 In this paper, we attached a typical aliphatic linker and the ZBG hydroxamic acid present in multiple HDACis to this scaffold, and the chirality of the two chiral centers (C1 and C3) of the THβC derivatives were also adjusted through the introduction of various substituents (Fig. 2). Here, we report the synthesis, structure–activity relationship analysis and antiproliferative activity of these new molecules.
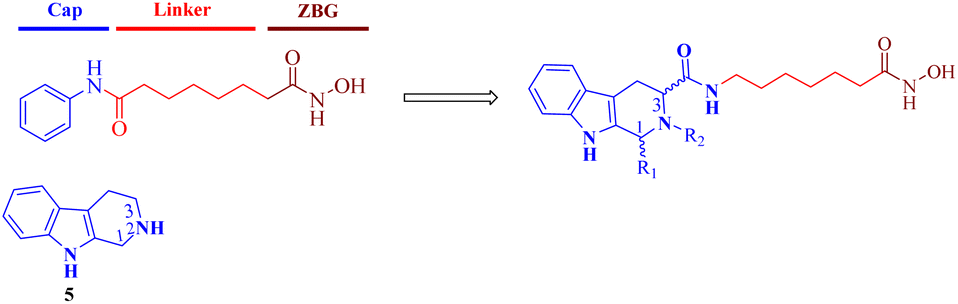 | ||
| Fig. 2 Design of THβC-based HDAC inhibitors. | ||
2. Chemistry
2.1 Chemistry
As outlined in Schemes 1 and 2, the key procedure for the synthesis of THβC derivatives was the application of the Pictet–Spengler cyclization from chiral tryptophan and aldehyde precursors according to our previous reports.26,27 Under acidic conditions, the use of L-tryptophan (6) or D-tryptophan (10) with different aldehydes gave predominantly the desired thermodynamically stable trans-THβCs 7a–g or 11a–g. Alkylation of the N2 position with allyl bromide gave intermediates 8a–b using potassium carbonate as the base (Scheme 1). The building blocks 7a–g and 8a–b were then hydrolyzed and subsequently coupled with commercially available 6-amino-N-hydroxyheptanamide or 7-amino-N-hydroxyhexanamide to provide the corresponding esters, which upon treatment with an aqueous NH2OH/NaOH solution yielded the targeted (1R,3S)-THβCs 9a–i. A similar chemical route was also used to prepare (1S,3R)-THβCs 13a–j in Scheme 2.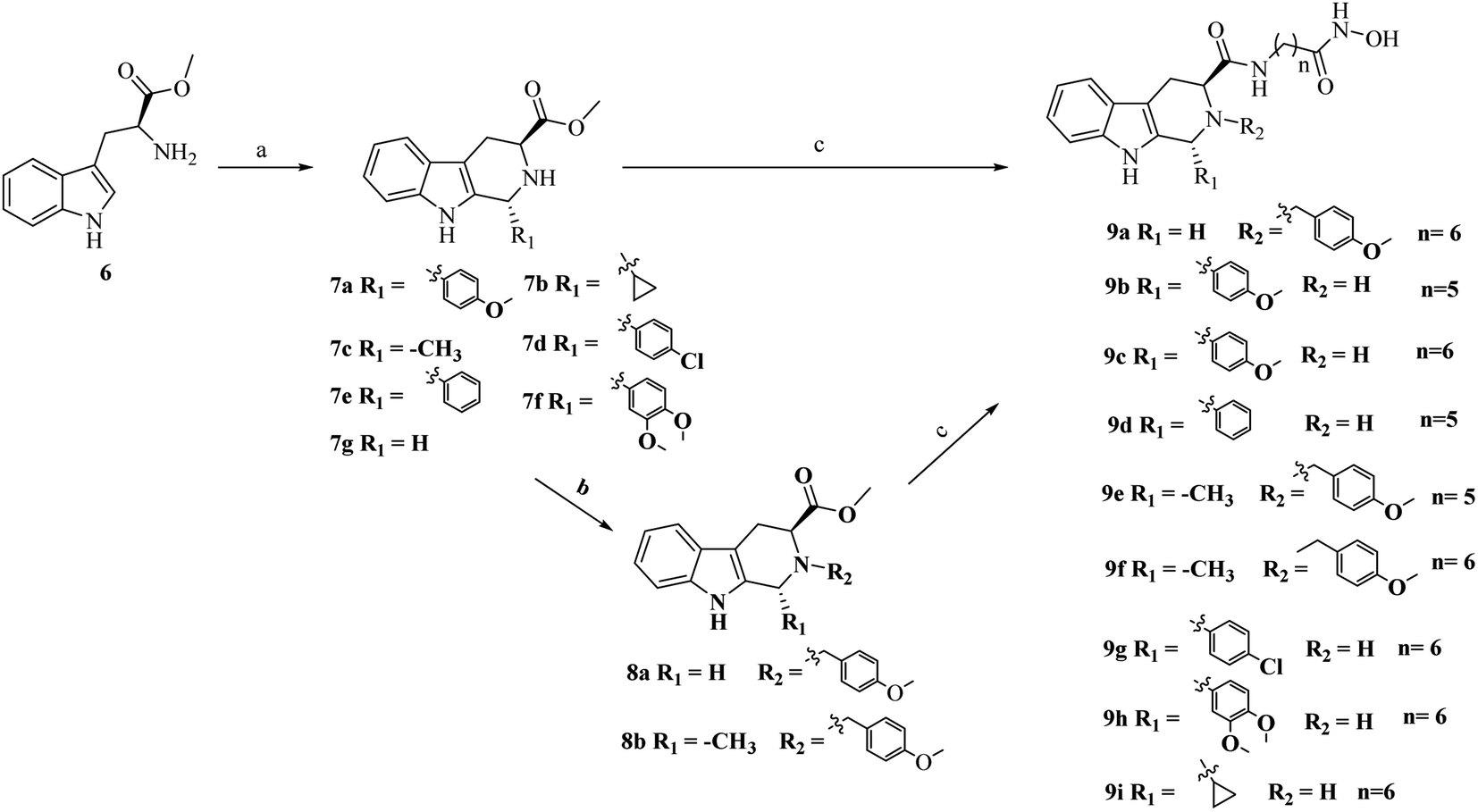 | ||
Scheme 1 Reagents and conditions: (a) isopropanol (i-PrOH), various aldehydes, reflux, overnight; (b) K2CO3, CH3CN, 4-methoxybenzyl bromide, r.t., overnight; (c) (i) LiOH, MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (V:V = 5:1), r.t., overnight; (ii) 2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU), N,N-diisopropylethylamine (DIPEA), N,N-dimethylformamide (DMF), 7-amino-N-hydroxyheptanamide or 6-amino-N-hydroxyhexanamide, 0 °C, 6 h; (iii) NH2OH (aq), KOH, MeOH, 0 °C, 6 h. :H2O (V:V = 5:1), r.t., overnight; (ii) 2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU), N,N-diisopropylethylamine (DIPEA), N,N-dimethylformamide (DMF), 7-amino-N-hydroxyheptanamide or 6-amino-N-hydroxyhexanamide, 0 °C, 6 h; (iii) NH2OH (aq), KOH, MeOH, 0 °C, 6 h. | ||
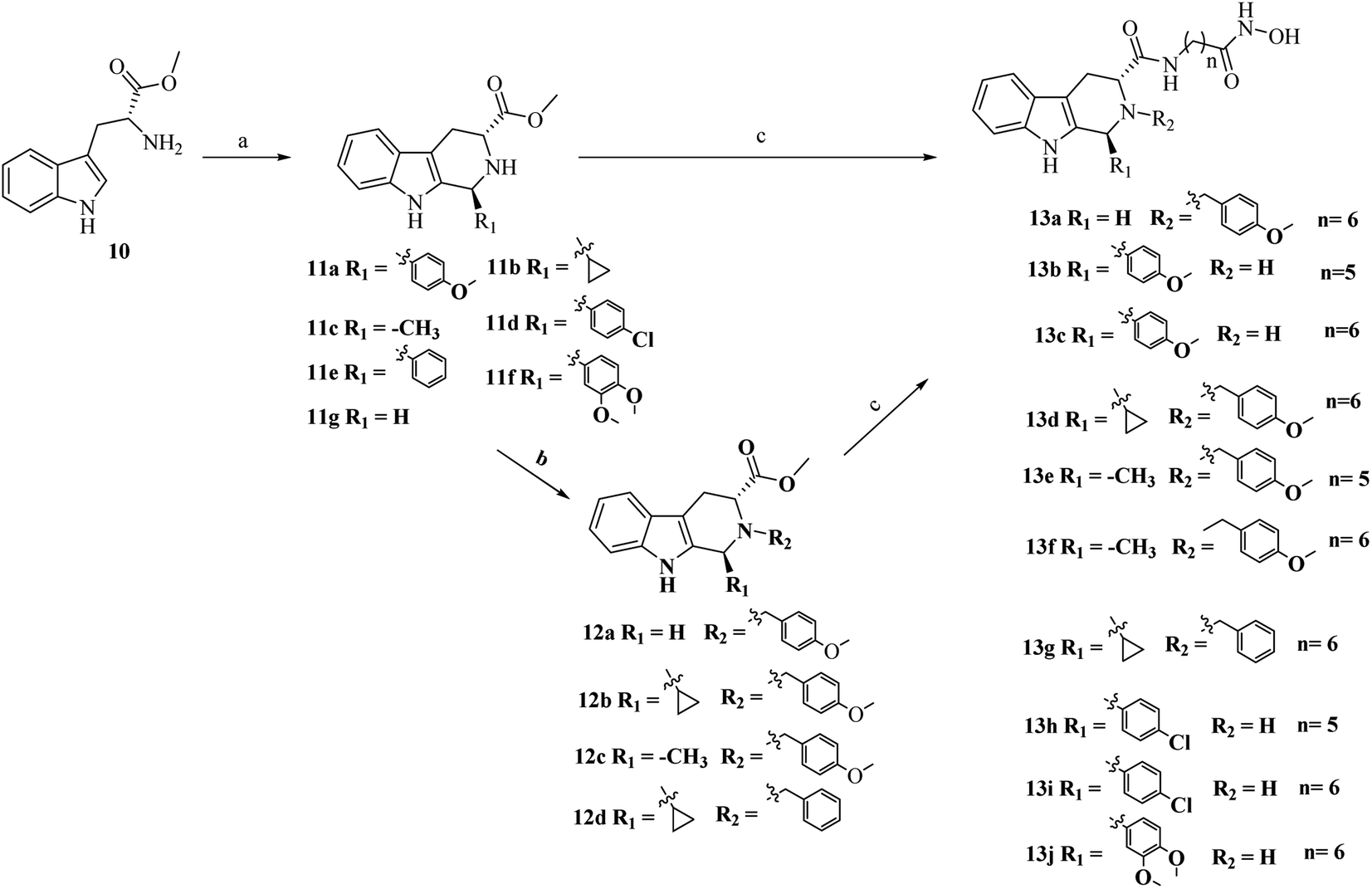 | ||
| Scheme 2 Reagents and conditions: (a) i-PrOH, various aldehydes, reflux, overnight; (b) K2CO3, CH3CN, benzyl bromide or 4-methoxybenzyl bromide, r.t., overnight; (c) (i) LiOH, MeOH:H2O (V:V = 5:1), r.t.; (ii) HATU, DIPEA, DMF, 7-amino-N-hydroxyheptanamide or 6-amino-N-hydroxyhexanamide, 0 °C, 6 h; (iii) NH2OH (aq), KOH, MeOH, 0 °C, 6 h. | ||
3. Results and discussion
3.1 HDAC1 activities and SAR study of the THβC derivatives
All the synthesized compounds were preliminarily evaluated for their inhibition rate (IR) against HDAC1 at 500 nM with pan-HDACi SAHA as reference. For compounds with an IR of over 80%, the IC50 values were further calculated. As shown in Table 1, the (1S,3R)-THβC analogues such as 13a (44.3%, IR) and 13f (91.4%, IR) were superior to the corresponding (1R,3S)-isomers such as 9a (31.8%, IR) and 9f (40.8%, IR). In comparison with 13b (49.2%, IR) and 13e (84.8%, IR; 53.6 nM, IC50), compounds 13c (66%) and 13f (91.4%, 22.4 nM) with an aliphatic linker of six carbon atoms had better HDAC1 inhibition, indicating the importance of the length of the spacers separating the hydroxamic acid and THβC motifs. Structural optimization was also performed to explore the impact of different substituents on the C1 and N2 positions. C1-substituted THβCs 13i (4-chlorophenyl) and 13j (3,4-dimethoxyphenyl) showed IRs of 66% and 72.5% for HDAC1, respectively. Compound 13c with a 4-methoxyphenyl group showed a moderate IR of 81.6%, while the simple N2-substituted 13a, lacking a chiral center, showed a weak IR of 44.3%. Interestingly, 13d–g with simultaneous C1 and N2 functionalization showed distinctly improved potency with IRs between 84.8–99.2%. The IC50 test showed that the best compound 13d inhibited HDAC1 with an IC50 of 8.73 nM. In comparison to the compounds with a hydrogen (13a) or methyl (13f) on the C1 position, the cyclopropyl-substituted THβC (13d) was a better choice, which indicated that the specific substituent on the C1 position maintained the active spatial conformation of total molecule.| Compound | Inhibition% | IC50 | Compound | Inhibition% | IC50 |
|---|---|---|---|---|---|
| a We ran experiments in duplicate, SD < 15%. Assays were performed by Reaction Biology Corporation (Malvern, PA, USA).b / to —: not tested. | |||||
| 9a | 31.8% | —b | 13a | 44.3% | — |
| 9b | 23.2% | — | 13b | 49.2% | — |
| 9c | 41.3% | — | 13c | 81.6% | 47.0 ± 1.25 |
| 9d | 39.6% | — | 13d | 99.2% | 8.73 ± 0.46 |
| 9e | 58.1% | — | 13e | 84.8% | 53.6 ± 2.50 |
| 9f | 40.8% | — | 13f | 91.4% | 22.4 ± 0.84 |
| 9g | 65.4% | — | 13g | 98.2% | 10.4 ± 0.65 |
| 9h | 46.5% | — | 13h | 51.3% | — |
| 9i | 77.3% | — | 13i | 66.0% | — |
| SAHA | — | 4.24 ± 0.23 | 13j | 72.5% | — |
13d was further tested against other classic HDAC isozymes. As shown in Table 2, 13d inhibited HDAC2 and HDAC3 with IC50 values of 23.5 nM and 32.1 nM, respectively. 13d also potently inhibited HDAC6 with an IC50 of 4.5 nM. Additionally, weak inhibition of HDAC11 was observed. For HDAC4/5/7/8/9, 13d showed no activity.
| Compound | 13d | SAHA | Compound | 13d | SAHA |
|---|---|---|---|---|---|
| a IC50 values for enzymatic inhibition of HDAC enzymes. We ran experiments in duplicate. Assays were performed by Reaction Biology Corporation (Malvern, PA, USA). | |||||
| HDAC1 | 8.73 ± 0.46 | 4.24 ± 0.23 | HDAC6 | 4.50 ± 0.21 | 7.80 ± 0.38 |
| HDAC2 | 23.5 ± 1.03 | 12.1 ± 0.92 | HDAC7 | >50000 |
>50000 |
| HDAC3 | 32.1 ± 1.44 | 3.34 ± 0.17 | HDAC8 | >50000 |
1033 ± 54.5 |
| HDAC4 | >50000 |
>50000 |
HDAC9 | >50000 |
>50000 |
| HDAC5 | >50000 |
>50000 |
HDAC11 | 1800 ± 106 | 895 ± 34.7 |
3.2 Molecular simulation
We docked the most potent compound 13d into the crystal structure of human HDAC1 (PDB code: 5ICN) to probe the potential interaction between the molecule and target. As shown in Fig. 3A, 13d was predicted to bind to Zn2+ with its hydroxamate group chelating in a bidentate manner. Additionally, a hydrogen bond interaction was formed between hydroxamic acid and Tyr303. Moreover, the aliphatic chain linker of 13d occupied the narrow 11 Å hydrophobic tunnel and exhibited π-interactions with both Phe150 and Phe205. The amide at the entrance of this tunnel could generate a hydrogen bond with Gly149. As shown in Fig. 3B, the THβC group was positioned at the exposed hydrophobic surface of the enzyme. The cyclopropyl on the THβC scaffold extended into the upper region of capping part and avoided the cramped space below, indicating S-configuration of chiral C1 position was better adapted to the active pocket of HDAC1 protein. Besides, 4-methoxyphenyl ring on N2 was embedded in a branched pocket and formed an H-bond with Glu98. Such a binding mode matched well with the SAR of THβC derivatives in Table 1.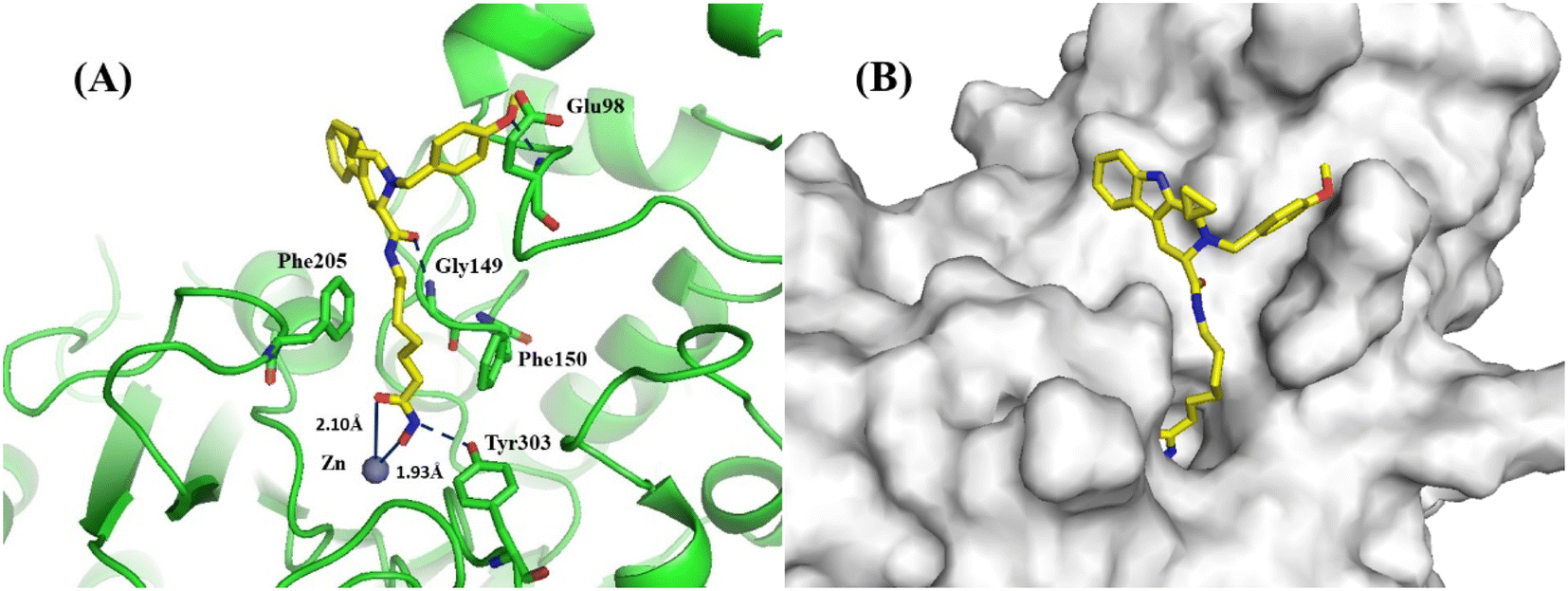 | ||
| Fig. 3 (A) Binding model of 13d (yellow) in the catalytic pocket of human HDAC1 (green cartoon). Key residues are labeled and shown as green sticks. Metal bonds and hydrogen bonds are labeled in dark blue. Zinc ions are shown in brown. (B) Surface map of 13d with HDAC1. The deep active-site pocket is visualized with a transparent light-grey surface. | ||
3.3 Antiproliferative evaluation
In view of the enzymatic potency, the preferred compounds 13d–g and 13i were screened against the colon cancer cell line HCT-116 and melanoma cell line SK-MEL-2. As shown in Table 3, all five THβCs showed potent antiproliferative activity with IC50 values at the single-digit micromolar or submicromolar level. The foremost 13d inhibited HCT116 and SK-MEL-2 with IC50 values of 1.54 and 0.48 μM, outperforming the positive ACY-1215. Importantly, the potency of 13d against SK-MEL-2 cells was obviously better than that of the approved compound SAHA. In addition, all four derivatives showed no influence on normal bone marrow cells HS-5.| Compound | HCT116 | SK-MEL-2 | HS-5 | Compound | HCT116 | SK-MEL-2 | HS-5 |
|---|---|---|---|---|---|---|---|
| a IC50 values are averages of three independent experiments, SD < 10%. | |||||||
| 13d | 1.54 ± 0.03 | 0.48 ± 0.01 | >50 | 13i | 7.55 ± 0.18 | 7.84 ± 0.26 | >50 |
| 13e | 8.34 ± 0.24 | 9.65 ± 0.46 | >50 | SAHA | 1.30 ± 0.07 | 2.45 ± 0.08 | — |
| 13f | 4.22 ± 0.12 | 5.70 ± 0.12 | >50 | ACY-1215 | 3.87 ± 0.11 | 4.50 ± 0.14 | — |
| 13g | 3.20 ± 0.09 | 3.16 ± 0.08 | >50 | ||||
Then, 13d was submitted to NCI-60 for detailed examination of its antiproliferative activity against fifty-nine cancer cell lines. The cancer types in the NCI-60 program include leukemia, non-small-cell lung cancer, colon cancer, CNS cancer, melanoma, ovary cancer, renal cancer, prostate cancer and breast cancer. As shown in Table 4, 13d displayed a broad-spectrum anticancer effect against leukemia and various solid tumors (>80% percent inhibition of 55 cell lines), especially colon cancer and melanoma.
| Cancer type | Cell line | 13d | Cancer type | Cell line | 13d |
|---|---|---|---|---|---|
| a Assays were performed by National Cancer Institute (NCI) at a single concentration (10 μM). | |||||
| Leukemia | CCRF-CEM | 80.28 | M14 | 82.4 | |
| HL60 | 96.56 | MDA-MB-435 | 100 | ||
| K-562 | 92.5 | SK-MEL-2 | 100 | ||
| MOLT-4 | 87.83 | SK-MEL-28 | 90.16 | ||
| RPMI-8226 | 84.29 | SK-MEL-5 | 100 | ||
| SR | 84.0 | UACC-257 | 90.1 | ||
| Non-small-cell lung cancer | A549/ATCC | 82.73 | UACC-62 | 100 | |
| EKVX | 83.23 | Ovarian cancer | IGROV1 | 100 | |
| HOP-62 | 86 | OVCAR-3 | 96.77 | ||
| HOP-92 | 100 | OVCAR-4 | 59.44 | ||
| NCI-H226 | 71.9 | OVCAR-5 | 100 | ||
| NCI-H23 | 100 | OVCAR-8 | 97.31 | ||
| NCI-H322M | 90.5 | NCI/ADR-RES | 38.0 | ||
| NCI-H460 | 93.6 | SK-OV-3 | 100 | ||
| NCI-H522 | 100 | Renal cancer | 786-0 | 82.65 | |
| Colon cancer | COLO 205 | 100 | A498 | 100 | |
| HCC-2998 | 100 | ACHN | 99.3 | ||
| HCT-116 | 97.9 | CAKI-1 | 96 | ||
| SN12C | 92.15 | ||||
| HCT-15 | 91.15 | TK-10 | 100 | ||
| HT29 | 93.7 | UO-31 | 92.33 | ||
| KM12 | 87.5 | PC-3 | 80.88 | ||
| SW-620 | 100 | Prostate cancer | DU-145 | 90.64 | |
| CNS cancer | SF-268 | 85.46 | MCF7 | 93.61 | |
| SF-295 | 100 | Breast cancer | MDA-MB-231/ATCC | 100 | |
| SF-539 | 97.13 | HS 578T | 100 | ||
| SNB-19 | 93.3 | BT-549 | 74.54 | ||
| SNB-75 | 89.8 | T-47D | 100 | ||
| U251 | 100 | MDA-MB-468 | 95.94 | ||
| Melanoma | LOX IMVI | 100 | |||
| MALME-3M | 98.55 | ||||
4. Conclusion
THβC, a natural privileged skeleton, has unique advantages for drug development. In this paper, nineteen THβC derivatives were obtained, several of which showed potent HDAC1 inhibition. In a cellular assay, five THβCs exhibited micromolar antiproliferative activity against the colon cancer cell line HCT-116 and melanoma cell line SK-MEL-2. Further screening of representative 13d showed promising anti-proliferative activity against solid tumor cell lines such as colon, melanoma, and breast cancer. Currently, these THβCs are being further studied in our lab.5. Experimental section
5.1 Chemistry
Unless otherwise noted, all reagents and solvents were obtained commercially, were of the highest commercial quality, and were used without further purification. All reactions were monitored using thin layer chromatography (TLC) on silica-gel-coated plates (Merck 60 F254), pre-coated silicone plates, and UV visualization. The crude product was purified using silica gel column chromatography using the standard technique (200−300 mesh, Qingdao Marine Chemical Ltd, PR China). All reported volumes are for separated products and were not optimized. The 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded using a Bruker DRX-400 (1H-NMR 400 MHz, 13C-NMR 101 MHz) using TMS as an internal standard. Chemical shifts (δ) are expressed in parts per million (PPM), and coupling constants (J) are given in Hertz (Hz). The melting points of all products were determined using an x-4 instrument (Beijing Tech Instrument Co., Beijing, PR China) without calibration.The chemical information for compounds 7a, 7g and 11a–g can be found in ref. 25. The chemical information for compounds 7b–f can be found in ref. 26.
The chemical information for compounds 8a and 12a–c can be found in ref. 25. The chemical information for compound 8b can be found in ref. 26.
5.1.2.1 Methyl (1S,3R)-2-benzyl-1-cyclopropyl-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (12d). White solid, 56.3% yield. 1H NMR (400 MHz, DMSO-d6) δ: 10.56 (s, 1H), 7.82–7.77 (m, 1H), 7.48 (d, J = 7.7 Hz, 1H), 7.42 (d, J = 7.4 Hz, 2H), 7.35–7.20 (m, 3H), 7.03 (t, J = 7.4 Hz, 1H), 6.99 (t, J = 7.4 Hz, 1H), 4.09 (d, J = 15.0 Hz, 1H), 3.75 (d, J = 15.0 Hz, 1H), 3.92 (t, J = 5.3 Hz, 1H), 3.58 (s, 3H), 3.21 (d, J = 9.6 Hz, 1H), 3.11 (dd, J = 15.6, 4.4 Hz, 1H), 2.85 (dd, J = 15.4, 5.8 Hz, 1H), 0.77 (m, 1H), 0.51 (m, 1H), 0.43 (m, 1H), 0.28 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ: 164.07, 143.18, 138.47, 136.15, 131.41, 131.18, 128.13, 127.01, 126.66, 120.73, 117.74, 111.03, 105.84, 59.52, 57.11, 51.02, 42.02, 18.93, 15.61, 3.72. [α]20D = 20.55 (0.05 mg mL−1, MeOH). ESI-MS m/z: 361.5 [M + H]+.
:V = 5:1) were added various esters (1 equiv.) and LiOH (2 equiv.). The mixture was stirred at r.t. overnight. After completion of the reaction as monitored using TLC, the mixture was concentrated under reduced pressure and diluted with excess water. After being neutralized with acetic acid, the formed precipitate was collected using filtration, washed with water, and dried in vacuo. The acids could be used directly for the next step. (ii) To a stirred mixture of the acid (1 equiv.) obtained from step (i), HATU (1.2 equiv.), and DIPEA (2 equiv.) in DMF was added the corresponding ester (1 equiv.) at 0 °C. After being stirred at r.t. for 6 h, saturated sodium chloride was added. The mixture was then extracted with EtOAc. The combined organic extracts were dried over anhydrous Na2SO4 and concentrated under reduced pressure. The product was obtained using chromatography on a silica gel column. (iii) To a solution of hydroxylamine hydrochloride (1.96 g, 28 mmol) in 20 mL of methanol, KOH (1.58 g, 28 mmol) was added and allowed to react at 40 °C for 10 min. The reaction mixture was cooled to 0 °C and filtered. The various esters obtained from step ii were added to the filtrate, followed by KOH, and allowed to react at r.t. overnight. The reaction mixture was extracted with EtOAc. The organic layer was washed with saturated NH4Cl aqueous solution and brine, dried over Na2SO4, filtered, and concentrated. The target compound was obtained using chromatography on a silica gel column.
5.1.3.1 (S)-N-(7-(Hydroxyamino)-7-oxoheptyl)-2-(4-methoxybenzyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxamide (9a). White solid, yield: 39.2%, m.p.: 102.1–105.3 °C. 1H-NMR (400 MHz, DMSO-d6) δ: 10.53 (s, 1H), 10.26 (s, 1H), 8.61 (d, J = 1.5 Hz, 1H), 7.92 (t, J = 5.8 Hz, 1H), 7.33 (d, J = 7.7 Hz, 1H), 7.25 (d, J = 8.6 Hz, 2H), 7.18 (d, J = 7.7 Hz, 1H), 6.94 (t, J = 7.5 Hz, 1H), 6.88 (t, J = 7.5 Hz, 1H), 6.84 (d, J = 8.6 Hz, 2H), 3.75 (d, J = 16.3 Hz, 1H), 3.68 (s, 3H), 3.63–3.48 (m, 4H), 3.13–3.04 (m, 1H), 3.02–2.89 (m, 2H), 2.77 (dd, J = 15.7, 5.3 Hz, 1H), 1.83 (t, J = 7.4 Hz, 2H), 1.45–1.32 (m, 4H), 1.24–1.07 (m, 4H). 13C-NMR (101 MHz, DMSO-d6) δ: 171.48, 169.14, 158.43, 135.90, 131.47, 130.70, 129.85, 126.89, 120.42, 118.31, 113.69, 110.86, 105.08, 61.04, 55.25, 55.07, 46.01, 38.43, 32.26, 29.18, 28.35, 26.19, 25.12, 20.54. [α]20D = −6.9 (0.9 mg mL−1, MeOH). ESI-MS m/z: 479.6 [M + H]+.
5.1.3.2 (1R,3S)-N-(6-(Hydroxyamino)-6-oxohexyl)-1-(4-methoxyphenyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxamide (9b). White solid, yield of 48.3%, m.p.: 141.4–143.7 °C. 1H NMR (400 MHz, DMSO-d6) δ: 10.71 (s, 1H), 10.34 (s, 1H), 8.68 (s, 1H), 7.83 (t, J = 5.5 Hz, 1H), 7.44 (d, J = 7.7 Hz, 1H), 7.25 (d, J = 7.8 Hz, 1H), 7.12 (d, J = 8.6 Hz, 2H), 7.03 (t, J = 7.5 Hz, 1H), 6.99–6.93 (m, 2H), 6.88 (d, J = 8.6 Hz, 2H), 5.17 (s, 1H), 3.72 (s, 3H), 3.44–3.40 (m, 1H), 3.04 (m, 2H), 2.94 (dd, J = 15.2, 4.6 Hz, 1H), 2.69 (dd, J = 15.2, 9.5 Hz, 2H), 1.93 (t, J = 7.3 Hz, 2H), 1.52–1.43 (m, 2H), 1.44–1.35 (m, 2H), 1.24–1.18 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ: 172.57, 169.09, 158.39, 136.05, 129.42, 126.71, 120.77, 118.29, 117.62, 113.46, 111.00, 107.77, 55.11, 53.34, 51.69, 38.35, 32.23, 28.86, 26.04, 24.89. [α]20D = −12.14 (0.07 g/100 mL, MeOH). ESI-MS m/z: 451.5 [M + H]+.
5.1.3.3 (1R,3S)-N-(7-(Hydroxyamino)-7-oxoheptyl)-1-(4-methoxyphenyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxamide (9c). Yellow solid, yield: 42.7%, m.p. 125.2–127.1 °C. 1H NMR (400 MHz, DMSO-d6) δ: 10.71 (s, 1H), 10.34 (s, 1H), 8.67 (s, 1H), 7.82 (t, J = 5.6 Hz, 1H), 7.44 (d, J = 7.7 Hz, 1H), 7.25 (d, J = 7.7 Hz, 1H), 7.13 (d, J = 8.6 Hz, 2H), 7.03 (t, J = 7.5 Hz, 1H), 6.96 (t, J = 7.5 Hz, 1H), 6.88 (d, J = 8.6 Hz, 2H), 5.17 (s, 1H), 3.72 (s, 3H), 3.41 (dd, J = 9.5, 4.6 Hz, 1H), 3.07–3.03 (q, J = 6.6 Hz, 2H), 2.94 (dd, J = 15.2, 4.6 Hz, 1H), 2.70 (dd, J = 15.2, 9.5 Hz, 1H), 1.93 (t, J = 7.3 Hz, 2H), 1.52–1.44 (m, 2H), 1.43–1.36 (m, 2H), 1.27–1.21 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ: 172.59, 169.15, 158.40, 136.05, 135.00, 134.77, 129.42, 126.73, 120.78, 118.30, 117.62, 113.46, 111.01, 107.78, 55.11, 54.94, 53.33, 51.71, 38.39, 32.26, 29.01, 28.32, 26.14, 25.16, 25.11. [α]20D = −13.2 (1 mg mL−1, MeOH). ESI-MS m/z: 479.6 [M + H]+.
5.1.3.4 (1R,3S)-N-(6-(Hydroxyamino)-6-oxohexyl)-1-phenyl-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxamide (9d). Yellow solid, yield of 55.3%, m.p. 133.1–134.7 °C. 1H NMR (400 MHz, DMSO-d6) δ: 10.74 (s, 1H), 10.34 (s, 1H), 8.67 (s, 1H), 7.84 (t, J = 5.6 Hz, 1H), 7.45 (d, J = 7.7 Hz, 1H), 7.35–7.28 (m, 2H), 7.28–7.19 (m, 4H), 7.04 (t, J = 7.4 Hz, 1H), 6.97 (t, J = 7.4 Hz, 1H), 5.22 (s, 1H), 3.44–3.40 (m, 1H), 3.04 (q, J = 6.7 Hz, 2H), 2.95 (dd, J = 15.2, 4.6 Hz, 1H), 2.70 (dd, J = 15.2, 8.1 Hz, 2H), 1.95–1.89 (m, 2H), 1.55–1.34 (m, 4H), 1.29–1.17 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ: 172.59, 169.11, 143.02, 136.08, 134.42, 128.33, 128.12, 127.08, 126.71, 120.84, 118.34, 117.67, 111.04, 107.92, 53.90, 51.79, 38.36, 32.23, 28.87, 26.04, 25.25, 24.90. [α]20D = −40.5 (1 mg mL−1, MeOH). ESI-MS m/z: 421.5 [M + H]+.
5.1.3.5 (1R,3S)-N-(6-(Hydroxyamino)-6-oxohexyl)-2-(4-methoxybenzyl)-1-methyl-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxamide (9e). Yellow solid, yield of 50.2%, m.p. 128.2–130.2 °C. 1H NMR (400 MHz, DMSO-d6) δ: 10.63 (s, 1H), 10.30 (s, 1H), 8.64 (s, 1H), 7.86 (t, J = 5.8 Hz, 1H), 7.43 (d, J = 7.9 Hz, 1H), 7.32 (d, J = 8.5 Hz, 2H), 7.25 (d, J = 7.9 Hz, 1H), 7.02 (t, J = 7.1 Hz, 1H), 6.96 (t, J = 7.1 Hz, 1H), 6.88 (d, J = 8.5 Hz, 2H), 3.87 (dd, J = 10.5, 4.8 Hz, 1H), 3.82–3.75 (m, 1H), 3.74 (s, 3H), 3.59 (d, J = 13.9 Hz, 1H), 3.38 (d, J = 14.3 Hz, 1H), 3.31–3.22 (m, 1H), 3.09–2.95 (m, 1H), 2.90–2.83 (m, 1H), 2.80–2.74 (m, 1H), 2.69 (s, 2H), 1.86 (t, J = 7.4 Hz, 2H), 1.48–1.35 (m, 6H), 1.29–1.14 (m, 3H). 13C NMR (101 MHz, DMSO-d6) δ: 171.65, 169.01, 158.25, 136.13, 136.00, 131.15, 129.15, 126.84, 120.49, 118.28, 117.64, 113.69, 110.82, 105.25, 56.25, 55.01, 51.07, 50.28, 38.34, 32.16, 29.09, 26.00, 24.84, 20.74, 18.76. [α]20D = 9.72 (1 mg mL−1, MeOH). ESI-MS m/z: 479.6 [M + H]+.
5.1.3.6 (1R,3S)-N-(7-(Hydroxyamino)-7-oxoheptyl)-2-(4-methoxybenzyl)-1-methyl-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxamide (9f). White solid, yield of 27.5%, m.p.: 158.2–159.3 °C. 1H NMR (400 MHz, DMSO-d6) δ: 10.63 (s, 1H), 10.30 (s, 1H), 8.64 (s, 1H), 7.85 (t, J = 5.9 Hz, 1H), 7.43 (d, J = 7.6 Hz, 1H), 7.32 (d, J = 8.5 Hz, 2H), 7.25 (d, J = 7.9 Hz, 1H), 7.02 (t, J = 7.3 Hz, 1H), 6.96 (t, J = 7.3 Hz, 1H), 6.88 (d, J = 8.5 Hz, 2H), 3.87 (dd, J = 10.1, 4.9 Hz, 1H), 3.79 (d, J = 6.8 Hz, 1H), 3.75 (s, 1H), 3.58 (d, J = 14.0 Hz, 1H), 3.29 (m, 1H), 3.01 (m, 1H), 2.88 (dd, J = 16.0, 10.1 Hz, 1H), 2.76 (dd, J = 16.0, 4.9 Hz, 1H), 1.87 (t, J = 7.4 Hz, 2H), 1.45–1.31 (m, 4H), 1.26–1.09 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ: 171.68, 169.09, 158.27, 136.15, 136.02, 131.15, 129.13, 126.85, 120.52, 118.31, 117.66, 113.70, 110.84, 105.27, 56.28, 55.04, 51.09, 50.33, 38.42, 32.22, 29.27, 28.35, 26.12, 25.05, 20.78, 18.78. [α]20D = 11.3 (1 mg mL−1, MeOH). ESI-MS m/z: 493.6 [M + H]+.
5.1.3.7 (1R,3S)-1-(4-Chlorophenyl)-N-(7-(hydroxyamino)-7-oxoheptyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxamide (9g). White solid, yield of 62.9%, m.p.: 135.2–136.2 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.76 (s, 1H), 10.34 (s, 1H), 8.67 (s, 1H), 7.84 (t, J = 5.6 Hz, 1H), 7.45 (d, J = 7.6 Hz, 1H), 7.38 (d, J = 8.4 Hz, 2H), 7.29–7.18 (m, 3H), 7.05 (t, J = 7.4 Hz, 1H), 6.97 (t, J = 7.4 Hz, 1H), 5.22 (s, 1H), 3.05 (q, J = 6.6 Hz, 2H), 2.92 (dd, J = 15.3,4.7 Hz, 1H), 2.71 (dd, J = 15.3, 9.6 Hz, 1H), 1.93 (t, J = 7.3 Hz, 2H), 1.54–1.42 (m, 2H), 1.42–1.32 (m, 2H), 1.30–1.17 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ: 172.46, 169.13, 142.03, 136.06, 133.97, 131.63, 130.14, 128.03, 126.68, 120.95, 118.41, 117.72, 111.07, 108.03, 53.16, 51.77, 38.40, 32.26, 28.99, 28.32, 26.14, 25.10. [α]20D = −8.00 (1 mg mL−1, MeOH). ESI-MS m/z: 470 [M + H]+.
5.1.3.8 (1R,3S)-1-(3,4-Dimethoxyphenyl)-N-(7-(hydroxyamino)-7-oxoheptyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxamide (9h). White solid, 45.4% yield, m.p.: 145.1–146.7 °C. 1H NMR (400 MHz, DMSO-d6) δ: 10.72 (s, 1H), 10.35 (s, 1H), 8.68 (s, 1H), 7.87 (t, J = 5.4 Hz, 1H), 7.44 (d, J = 7.6 Hz, 1H), 7.26 (d, J = 7.9 Hz, 1H), 7.07–7.00 (m, 2H), 6.96 (t, J = 7.4 Hz, 1H), 6.85 (d, J = 8.3 Hz, 1H), 6.58 (d, J = 8.2 Hz, 1H), 5.17 (s, 1H), 3.73 (s, 3H), 3.71 (s, 3H), 3.45 (dd, J = 9.4, 4.8 Hz, 2H), 3.10–3.03 (m, 2H), 2.94 (dd, J = 15.1, 4.8 Hz, 1H), 2.72 (dd, J = 15.1, 9.4 Hz, 1H), 1.93 (t, J = 7.3 Hz, 2H), 1.50–1.37 (m, 4H), 1.26–1.23 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ: 172.64, 169.20, 148.69, 148.07, 136.09, 135.37, 134.77, 126.73, 120.83, 120.25, 118.34, 117.68, 112.32, 111.25, 111.05, 107.75, 55.61, 55.55, 53.69, 51.92, 38.42, 32.29, 29.07, 28.37, 26.18, 25.14. [α]20D = −15.2 (1 mg mL−1, MeOH). ESI-MS m/z: 495.6 [M + H]+.
5.1.3.9 (1R,3S)-1-Cyclopropyl-N-(7-(hydroxyamino)-7-oxoheptyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxamide (9i). White solid, yield of 36.2%, m.p.: 135.1–135.8 °C. 1H NMR (400 MHz, DMSO-d6) δ: 10.55 (s, 1H), 10.37 (s, 1H), 8.71 (s, 1H), 7.87 (t, J = 5.4 Hz, 1H), 7.41–7.32 (m, 2H), 7.03 (t, J = 7.5 Hz, 1H), 6.94 (t, J = 7.5 Hz, 1H), 3.38–3.36 (m, 1H), 3.27 (d, J = 8.8 Hz, 1H), 3.12 (q, J = 6.6 Hz, 2H), 2.89 (dd, J = 14.7, 3.2 Hz, 1H), 2.65–2.53 (m, 1H), 1.95 (t, J = 7.3 Hz, 2H), 1.56–1.38 (m, 4H), 1.35–1.18 (m, 4H), 0.98 (m, 1H), 0.75–0.57 (m, 2H), 0.55–0.35 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ: 172.47, 169.22, 136.47, 136.19, 126.75, 120.63, 118.41, 117.48, 111.37, 106.60, 57.72, 57.32, 38.46, 32.30, 29.11, 28.38, 26.20, 25.87, 25.15, 15.48, 3.31, 2.23. [α]20D = −16.9 (0.75 mg mL−1, MeOH). ESI-MS m/z: 399.5 [M + H]+.
5.1.3.10 (R)-N-(7-(Hydroxyamino)-7-oxoheptyl)-2-(4-methoxybenzyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxamide (13a). White solid, yield of 71.2%, m.p.: 139.8–141.8 °C. 1H-NMR (400 MHz, DMSO-d6) δ: 10.60 (s, 1H), 10.32 (d, J = 1.8 Hz, 1H), 8.66 (d, J = 1.8 Hz, 1H), 7.98 (t, J = 5.9 Hz, 1H), 7.39 (d, J = 7.6 Hz, 1H), 7.35–7.27 (m, 2H), 7.28–7.20 (m, 1H), 7.06–6.86 (m, 4H), 3.82 (d, J = 16.1 Hz, 1H), 3.74 (s, 3H), 3.72–3.62 (m, 3H), 3.64–3.54 (m, 3H), 3.17 (dq, J = 13.3, 6.8 Hz, 1H), 3.11–2.95 (m, 2H), 2.84 (dd, J = 15.7, 5.6 Hz, 1H), 1.90 (t, J = 7.4 Hz, 2H), 1.42 (q, J = 7.0 Hz, 5H), 1.22 (d, J = 8.3 Hz, 6H). 13C-NMR (101 MHz, DMSO-d6) δ: 171.93, 169.59, 158.88, 136.35, 131.91, 131.14, 130.30, 127.34, 120.87, 118.76, 117.85, 114.14, 111.30, 105.53, 61.48, 55.69, 55.51, 46.45, 39.63, 38.87, 32.69, 29.62, 28.79, 26.62, 25.56, 20.98. [α]20D = 21.87 (0.065 g/100 mL, MeOH). ESI-MS m/z: 479.6 [M + H]+.
5.1.3.11 (1S,3R)-N-(6-(Hydroxyamino)-6-oxohexyl)-1-(4-methoxyphenyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxamide (13b). White solid, yield of 55.8%, m.p.: 101.3–103.7 °C. 1H NMR (400 MHz, DMSO-d6) δ: 10.71 (s, 1H), 10.34 (s, 1H), 8.67 (s, 1H), 7.82 (t, J = 5.6 Hz, 1H), 7.44 (d, J = 7.7 Hz, 1H), 7.25 (d, J = 7.7 Hz, 1H), 7.13 (d, J = 8.7 Hz, 2H), 7.03 (t, J = 7.1 Hz, 1H), 6.96 (t, J = 7.1 Hz, 1H), 6.88 (d, J = 8.7 Hz, 2H), 5.17 (s, 1H), 3.72 (s, 3H), 3.41 (dd, J = 9.5, 4.7 Hz, 1H), 3.05 (q, J = 6.6 Hz, 2H), 2.94 (dd, J = 15.2, 4.6 Hz, 1H), 2.70 (dd, J = 15.2, 9.5 Hz, 1H), 1.93 (t, J = 7.4 Hz, 2H), 1.48 (p, J = 7.4 Hz, 2H), 1.39 (p, J = 7.4 Hz, 2H), 1.30–1.19 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ: 172.56, 169.07, 158.38, 136.04, 134.99, 134.97, 134.75, 129.41, 126.71, 118.28, 117.61, 113.45, 110.99, 107.78, 55.09, 53.33, 51.68, 38.35, 32.23, 28.87, 26.04, 25.19, 24.89. [α]20D = 42.66 (0.10 mg mL−1, MeOH). ESI-MS m/z: 450.5 [M + H]+.
5.1.3.12 (1S,3R)-N-(7-(Hydroxyamino)-7-oxoheptyl)-1-(4-methoxyphenyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxamide (13c). White solid, yield of 48.3%, m.p.: 104.2–106.4 °C. 1H NMR (400 MHz, DMSO-d6) δ: 10.71 (s, 1H), 10.34 (s, 1H), 8.67 (s, 1H), 7.82 (t, J = 5.6 Hz, 1H), 7.44 (d, J = 7.7 Hz, 1H), 7.25 (d, J = 7.7 Hz, 1H), 7.13 (d, J = 8.6 Hz, 2H), 7.03 (t, J = 7.4 Hz, 1H), 6.96 (t, J = 7.4 Hz, 1H), 6.88 (d, J = 8.6 Hz, 2H), 5.17 (s, 1H), 3.72 (s, 3H), 3.41 (dd, J = 9.5, 4.9 Hz, 1H), 3.04 (dt, J = 10.9, 5.8 Hz, 2H), 2.94 (dd, J = 15.1, 4.7 Hz, 1H), 2.70 (dd, J = 15.1, 9.3 Hz, 1H), 1.93 (t, J = 7.4 Hz, 2H), 1.52–1.43 (m, 2H), 1.43–1.34 (m, 2H), 1.28–1.20 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ: 172.56, 169.11, 158.38, 136.03, 134.99, 134.76, 129.40, 126.71, 120.75, 118.28, 117.60, 113.44, 110.98, 107.76, 55.09, 53.31, 51.70, 38.38, 32.25, 29.00, 28.31, 26.13, 25.16, 25.10. [α]20D = 43.20 (0.09 mg mL−1, MeOH). ESI-MS m/z: 465.6 [M + H]+.
5.1.3.13 (1S,3R)-1-Cyclopropyl-N-(7-(hydroxyamino)-7-oxoheptyl)-2-(4-methoxybenzyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxamide (13d). White solid, yield of 52.1%, m.p.: 113.2–115.6 °C. 1H NMR (400 MHz, DMSO-d6) δ: 10.53 (s, 1H), 10.31 (s, 1H), 8.64 (s, 1H), 7.87–7.66 (m, 1H), 7.45 (d, J = 7.6 Hz, 1H), 7.33–7.23 (m, 3H), 7.07–6.93 (m, 2H), 6.87 (d, J = 7.2 Hz, 2H), 4.17–4.01 (m, 1H), 3.74 (s, 3H), 3.63–3.58 (m, 1H), 3.04–2.96 (m, 1H), 2.91–2.78 (m, 2H), 2.71 (dd, J = 14.1, 3.7 Hz, 1H), 1.93–1.82 (m, 2H), 1.46–1.33 (m, 4H), 1.29–1.11 (m, 4H), 0.54–0.41 (m, 2H), 0.39–0.31 (m, 1H), 0.17–0.07 (m, 1H). 13C NMR (101 MHz, DMSO-d6) δ: 171.65, 169.12, 158.33, 138.83, 136.15, 129.37, 126.48, 120.68, 117.73, 113.65, 111.30, 111.01, 110.11, 88.26, 65.44, 55.04, 38.47, 32.23, 29.31, 28.35, 28.34, 26.15, 25.08, 15.58, 3.47. [α]20D = −17.20 (0.05 mg mL−1, MeOH). ESI-MS m/z: 519.7 [M + H]+.
5.1.3.14 (1S,3R)-N-(6-(Hydroxyamino)-6-oxohexyl)-2-(4-methoxybenzyl)-1-methyl-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxamide (13e). Yellow solid, yield of 47.5%, m.p.: 92.4–94.1 °C. 1H NMR (400 MHz, DMSO-d6) δ: 10.63 (s, 1H), 10.30 (s, 1H), 8.65 (s, 1H), 7.86 (t, J = 5.9 Hz, 1H), 7.43 (d, J = 7.9 Hz, 1H), 7.32 (d, J = 8.5 Hz, 2H), 7.25 (d, J = 7.9 Hz, 1H), 7.02 (t, J = 7.4 Hz, 1H), 6.96 (t, J = 7.4 Hz, 1H), 6.88 (d, J = 8.5 Hz, 2H), 3.87 (dd, J = 10.4, 4.9 Hz, 1H), 3.81–3.76 (m, 1H), 3.74 (s, 3H), 3.58 (d, J = 14.0 Hz, 1H), 3.01 (dd, J = 12.7, 6.9 Hz, 1H), 2.93–2.83 (m, 1H), 2.76 (dd, J = 16.7, 5.2 Hz, 1H), 1.85 (t, J = 7.4 Hz, 2H), 1.46–1.37 (m, 6H), 1.24–1.14 (m, 3H). 13C NMR (101 MHz, DMSO-d6) δ: 171.65, 169.01, 158.25, 136.13, 136.00, 131.15, 129.15, 126.84, 120.49, 118.28, 117.64, 113.69, 110.82, 105.25, 56.25, 55.01, 51.07, 50.28, 38.34, 32.16, 29.09, 26.00, 24.84, 20.74, 18.76. [α]20D = −88.39 (0.05 mg mL−1, MeOH). ESI-MS m/z: 478.6 [M + H]+.
5.1.3.15 (1S,3R)-N-(7-(Hydroxyamino)-7-oxoheptyl)-2-(4-methoxybenzyl)-1-methyl-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxamide (13f). White solid, yield of 47.5%, m.p.: 97.6–99.5 °C. 1H NMR (400 MHz, DMSO-d6) δ: 10.63 (s, 1H), 10.30 (s, 1H), 8.63 (s, 1H), 7.85 (t, J = 5.8 Hz, 1H), 7.43 (d, J = 7.9 Hz, 1H), 7.32 (d, J = 8.4 Hz, 2H), 7.25 (d, J = 7.9 Hz, 1H), 7.02 (t, J = 7.4 Hz, 1H), 6.96 (t, J = 7.3 Hz, 1H), 6.88 (d, J = 8.6 Hz, 2H), 3.87 (dd, J = 10.5, 4.8 Hz, 1H), 3.83–3.75 (m, 2H), 3.74 (s, 3H), 3.59 (d, J = 14.0 Hz, 1H), 3.44–3.37 (m, 2H), 3.01 (m, 1H), 2.92–2.84 (m, 1H), 2.80–2.72 (m, 1H), 1.87 (t, J = 7.3 Hz, 2H), 1.39 (d, J = 6.7 Hz, 4H), 1.23 (m, 2H), 1.21–1.17 (m, 3H). 13C NMR (101 MHz, DMSO-d6) δ: 171.69, 169.10, 158.28, 136.16, 136.02, 131.15, 129.14, 126.86, 120.53, 118.31, 117.67, 113.70, 110.85, 105.27, 56.27, 55.03, 51.09, 50.33, 38.40, 32.22, 29.26, 28.34, 26.12, 25.05, 20.78, 18.78. [α]20D = −48.30 (0.05 mg mL−1, MeOH). ESI-MS m/z: 493.6 [M + H]+.
5.1.3.16 (1S,3R)-2-Benzyl-1-cyclopropyl-N-(7-(hydroxyamino)-7-oxoheptyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxamide (13g). White solid, yield of 48.3%, m.p.: 103.6–105.3 °C. 1H NMR (400 MHz, DMSO-d6) δ: 10.54 (s, 1H), 10.32 (s, 1H), 8.66 (s, 1H), 7.87–7.75 (m, 1H), 7.46 (d, J = 7.7 Hz, 1H), 7.40 (d, J = 7.4 Hz, 2H), 7.34–7.20 (m, 4H), 7.04 (t, J = 7.4 Hz, 1H), 6.97 (t, J = 7.4 Hz, 1H), 4.12 (dd, J = 10.6, 5.3 Hz, 1H), 3.70 (d, J = 14.2 Hz, 1H), 3.35–3.28 (m, 2H), 3.10–3.03 (m, 1H), 3.01 (d, J = 7.7 Hz, 1H), 2.97–2.79 (m, 2H), 1.86 (t, J = 7.4 Hz, 2H), 1.46–1.31 (m, 4H), 1.27–1.09 (m, 4H), 0.54–0.43 (m, 2H), 0.43–0.29 (m, 1H), 0.24–0.10 (m, 1H). 13C NMR (101 MHz, DMSO-d6) δ: 171.60, 169.13, 139.32, 136.16, 133.72, 128.26, 128.17, 126.99, 126.69, 120.71, 118.37, 117.76, 111.03, 106.03, 59.31, 57.14, 51.47, 38.49, 32.25, 29.31, 28.37, 26.16, 25.08, 18.72, 15.61, 3.53, 3.45. [α]20D = 19.67 (0.06 mg mL−1, MeOH). ESI-MS m/z: 489.6 [M + H]+.
5.1.3.17 (1S,3R)-1-(4-Chlorophenyl)-N-(6-(hydroxyamino)-6-oxohexyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxamide (13h). White solid, yield of 55.4%, m.p.: 117.6–119.8 °C. 1H NMR (400 MHz, DMSO-d6) δ: 10.75 (s, 1H), 10.33 (s, 1H), 8.66 (s, 1H), 7.83 (t, J = 5.7 Hz, 1H), 7.45 (d, J = 7.7 Hz, 1H), 7.38 (d, J = 8.5 Hz, 2H), 7.29–7.21 (m, 3H), 7.04 (t, J = 7.5 Hz, 1H), 6.97 (t, J = 7.4 Hz, 1H), 5.21 (s, 1H), 3.04 (q, J = 6.7 Hz, 2H), 2.92 (dd, J = 15.3,4.7 Hz, 1H), 2.70 (dd, J = 15.3, 9.6 Hz, 1H), 1.92 (t, J = 7.4 Hz, 2H), 1.47 (p, J = 7.5 Hz, 2H), 1.39 (p, J = 7.2 Hz, 2H), 1.29–1.18 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ: 172.45, 169.07, 142.03, 136.06, 133.96, 131.61, 128.02, 126.67, 120.94, 118.39, 117.71, 111.05, 108.04, 53.16, 51.75, 38.35, 32.22, 28.84, 26.03, 25.16, 24.88. [α]20D = 52.78 (0.11 mg mL−1, MeOH). ESI-MS m/z: 454.9 [M + H]+.
5.1.3.18 (1S,3R)-1-(4-Chlorophenyl)-N-(7-(hydroxyamino)-7-oxoheptyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxamide (13i). White solid, yield of 39.6%, m.p.: 99.8–101.4 °C. 1H NMR (400 MHz, DMSO-d6) δ: 10.76 (s, 1H), 10.34 (s, 1H), 8.67 (s, 1H), 7.83 (t, J = 5.7 Hz, 1H), 7.45 (d, J = 7.7 Hz, 1H), 7.38 (d, J = 8.4 Hz, 2H), 7.30–7.22 (m, 3H), 7.05 (t, J = 7.1 Hz, 1H), 6.97 (t, J = 7.1 Hz, 1H), 5.22 (s, 1H), 3.41–3.36 (m, 1H), 3.05 (q, J = 6.7 Hz, 2H), 2.93 (dd, J = 15.2, 4.7 Hz, 1H), 2.71 (dd, J = 15.2, 9.5 Hz, 1H), 1.93 (t, J = 7.2 Hz, 2H), 1.48 (q, J = 7.2 Hz, 2H), 1.38 (q, J = 6.9 Hz, 2H), 1.29–1.20 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ: 172.47, 169.12, 142.06, 136.08, 134.00, 131.63, 130.14, 128.03, 126.69, 120.95, 118.40, 117.72, 111.07, 108.05, 53.17, 51.79, 38.41, 32.26, 29.00, 28.33, 26.14, 25.17, 25.11. [α]20D = 41.60 (0.10 mg mL−1, MeOH). ESI-MS m/z: 468.9 [M + H]+.
5.1.3.19 (1S,3R)-1-(3,4-Dimethoxyphenyl)-N-(7-(hydroxyamino)-7-oxoheptyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxamide (13j). White solid, yield of 42.8%, m.p.: 113.1–116.7 °C. 1H NMR (400 MHz, DMSO-d6) δ: 10.72 (s, 1H), 10.34 (s, 1H), 8.67 (s, 1H), 7.86 (t, J = 5.5 Hz, 1H), 7.44 (d, J = 7.7 Hz, 1H), 7.25 (d, J = 8.0 Hz, 1H), 7.06–6.93 (m, 3H), 6.85 (d, J = 8.3 Hz, 1H), 6.57 (d, J = 8.3 Hz, 1H), 5.16 (s, 1H), 3.72 (s, 3H), 3.71 (s, 3H), 3.45 (dd, J = 9.3,4.7 Hz, 1H), 3.06 (q, J = 6.6 Hz, 2H), 2.94 (dd, J = 15.2, 4.7 Hz, 1H), 2.77–2.65 (m, 1H), 1.93 (t, J = 7.3 Hz, 2H), 1.50–1.37 (m, 4H), 1.25–1.20 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ: 172.60, 169.15, 148.67, 148.05, 136.06, 126.71, 120.79, 120.23, 118.30, 117.65, 112.31, 111.24, 111.02, 107.73, 55.59, 55.53, 53.66, 51.90, 38.39, 32.27, 29.05, 28.34, 26.16, 25.12. [α]20D = 42.0 (0.07 mg mL−1, MeOH). ESI-MS m/z: 494.6 [M + H]+.
5.2 In vitro HDAC enzyme assay
IC50 testing of compounds was performed by Reaction Biology Corporation. The HDACs were isolated from a baculovirus expression system in Sf9 cells using an acetylated fluorogenic peptide, RHKKAc, as the substrate. The reaction buffer was made up of 50 mM Tris–HCl pH 8.0, 127 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 1 mg per mL BSA, and a final concentration of 1% DMSO. Compounds were delivered in DMSO and delivered to an enzyme mixture with a preincubation of 5–10 min followed by substrate addition and incubation for 2 h at 30 °C. Trichostatin A and a developer were added to quench the reaction and generate fluorescence, respectively. Dose–response curves were generated starting at 30 μM of compound with 3-fold serial dilutions to generate a 10-dose plot. IC50 values were then generated from the resulting plots. The detailed protocol can be found in ref. 25.5.3 Cell culture and cell viability assay
The cells were cultured in IMDM medium with 20% FBS, 100 U per mL penicillin and 100 μg per mL streptomycin. Briefly, a 100 μL cell suspension or completed medium were plated into a 96-well plate. Compounds were added and incubated for 72 h. Subsequently, 22 μL Alamar blue solution (1 mM) was pipetted into each well of the 96-well plate; and the plates were incubated for an additional 5–6 h. The absorbance (OD) was read at 530/590 nm. Data were normalized to vehicle groups and represented as the means of three independent measurements with standard errors of <20%. The IC50 values were calculated using Prism 5.0.For NCI-60 screening, all details of the test method are described on the NCI Web site (https://dtp.cancer.gov/discovery_development/nci-60/methodology.htm).
5.4 Computational methods
Sybyl-X 2.0 software (Saint Louis, MO, USA) was used for simulation. Docking was conducted based on the cocrystal of HDAC1 (PDB: 5ICN). HDAC1 was used as the receptor. The cavity occupied by a polypeptide was selected as binding site. Water molecules outside the binding pocket were excluded. The other docking parameters were kept as default.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This research was funded by National Natural Science Foundation of China (82303811), the Natural Science Basic Research Program of Shaanxi Province (2023-JC-QN-0840), Youth Project of the Second Affiliated Hospital of Xi'an Jiaotong University (YJ-QN-202320) and the Key Research and Development Plan of Shaanxi Province (2022SF-228).References
- M. Shirvaliloo, Epigenomics, 2022, 14, 1465–1477 CrossRef CAS.
- Y. Zhang, Z. Sun, J. Jia, T. Du, N. Zhang, Y. Tang, Y. Fang and D. Fang, Adv. Exp. Med. Biol., 2021, 1283, 1–16 CrossRef CAS PubMed.
- R. Sarkar, S. Banerjee, S. A. Amin, N. Adhikari and T. Jha, Eur. J. Med. Chem., 2020, 192, 112171 CrossRef CAS.
- A. Badie, C. Gaiddon and G. Mellitzer, Cancers, 2022, 14, 5472 CrossRef CAS PubMed.
- E. Shirbhate, R. Veerasamy, S. H. S. Boddu, A. K. Tiwari and H. Rajak, Drug Discovery Today, 2022, 27, 1689–1697 CrossRef CAS PubMed.
- A. J. de Ruijter, A. H. van Gennip, H. N. Caron, S. Kemp and A. B. van Kuilenburg, Biochem. J., 2003, 370, 737–749 CrossRef CAS PubMed.
- I. V. Gregoretti, Y. M. Lee and H. V. Goodson, J. Mol. Biol., 2004, 338, 17–31 CrossRef CAS PubMed.
- G. P. Delcuve, D. H. Khan and J. R. Davie, Expert Opin. Ther. Targets, 2013, 17, 29–41 CrossRef CAS PubMed.
- P. A. Marks and R. Breslow, Nat. Biotechnol., 2007, 25, 84–90 CrossRef CAS PubMed.
- A. Sawas, D. Radeski and O. A. O'Connor, Ther. Adv. Hematol., 2015, 6, 202–208 CrossRef CAS.
- J. P. Laubach, P. Moreau, J. F. San-Miguel and P. G. Richardson, Clin. Cancer Res., 2015, 21, 4767–4773 CrossRef CAS PubMed.
- S. A. Reddy, Crit. Rev. Oncol./Hematol., 2016, 106, 99–107 CrossRef PubMed.
- M. Dong, Z. Q. Ning, P. Y. Xing, J. L. Xu, H. X. Cao, G. F. Dou, Z. Y. Meng, Y. K. Shi, X. P. Lu and F. Y. Feng, Cancer Chemother. Pharmacol., 2012, 69, 1413–1422 CrossRef CAS PubMed.
- Z. Q. Ning, Z. B. Li, M. J. Newman, S. Shan, X. H. Wang, D. S. Pan, J. Zhang, M. Dong, X. Du and X. P. Lu, Cancer Chemother. Pharmacol., 2012, 69, 901–909 CrossRef CAS.
- T. Liang, F. Wang, R. M. Elhassan, Y. Cheng, X. Tang, W. Cheng, H. Fang and X. Hou, Acta Pharm. Sin. B, 2023, 13, 2425–2463 CrossRef CAS PubMed.
- T. Liang, Z. Xie, B. Dang, J. Wang, T. Zhang, X. Luan, T. Lu, C. Cao and X. Chen, Bioorg. Med. Chem. Lett., 2023, 81, 129148 CrossRef CAS PubMed.
- T. C. S. Ho, A. H. Y. Chan and A. Ganesan, J. Med. Chem., 2020, 63, 12460–12484 CrossRef CAS PubMed.
- X. Chen, S. Zhao, H. Li, X. Wang, A. Geng, H. Cui, T. Lu, Y. Chen and Y. Zhu, Eur. J. Med. Chem., 2019, 168, 110–122 CrossRef CAS PubMed.
- X. Chen, X. Chen, R. R. Steimbach, T. Wu, H. Li, W. Dan, P. Shi, C. Cao, D. Li, A. K. Miller, Z. Qiu, J. Gao and Y. Zhu, Eur. J. Med. Chem., 2020, 187, 111950 CrossRef CAS PubMed.
- X. Chen, G. Gong, X. Chen, R. Song, M. Duan, R. Qiao, Y. Jiao, J. Qi, Y. Chen and Y. Zhu, Chem. Pharm. Bull., 2019, 67, 1116–1122 CrossRef CAS PubMed.
- G. Gong, J. Qi, Y. Lv, S. Dong, C. Cao, D. Li, R. Zhao, Z. Li and X. Chen, Chem. Pharm. Bull., 2020, 68, 466–472 CrossRef CAS.
- L. Santo, T. Hideshima, A. L. Kung, J. C. Tseng, D. Tamang, M. Yang, M. Jarpe, J. H. van Duzer, R. Mazitschek, W. C. Ogier, D. Cirstea, S. Rodig, H. Eda, T. Scullen, M. Canavese, J. Bradner, K. C. Anderson, S. S. Jones and N. Raje, Blood, 2012, 119, 2579–2589 CrossRef CAS PubMed.
- N. Arrighetti, C. Corno and L. Gatti, Crit. Rev. Oncog., 2015, 20, 83–117 CrossRef PubMed.
- J. Wang, F. Gong, T. Liang, Z. Xie, Y. Yang, C. Cao, J. Gao, T. Lu and X. Chen, Eur. J. Med. Chem., 2021, 225, 113815 CrossRef CAS.
- X. Chen, J. Wang, P. Zhao, B. Dang, T. Liang, R. R. Steimbach, A. K. Miller, J. Liu, X. Wang, T. Zhang, X. Luan, J. Hu and J. Gao, Eur. J. Med. Chem., 2023, 260, 115776 CrossRef CAS PubMed.
- W. Wen, J. Hu, C. Wang, R. Yang, Y. Zhang, B. Huang, T. Qiao, J. Wang and X. Chen, Bioorg. Med. Chem. Lett., 2024, 129670, DOI:10.1016/j.bmcl.2024.129670.
- S. Khochbin, A. Verdel, C. Lemercier and D. Seigneurin-Berny, Curr. Opin. Genet. Dev., 2001, 11, 162–166 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra01672f |
| This journal is © The Royal Society of Chemistry 2024 |