
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRestriction of reaction sites on metal-sulfide cores induced by steric repulsion of bis-N-heterocyclic carbene ligands in trinuclear complexes bearing triply bridging sulfide ligands†
Natsuki Yabunea,
Hiroshi Nakajima b and
Takanori Nishioka*b
b and
Takanori Nishioka*b
aDivision of Molecular Materials Science, Graduate School of Science, Osaka City University, Osaka 558-8585, Japan
bDepartment of Chemistry, Graduate School of Science, Osaka Metropolitan University, Osaka 558-8585, Japan. E-mail: nishioka@omu.ac.jp
First published on 16th September 2024
Abstract
Mixed-ligand and mixed-metal trinuclear complexes bearing two {Pt-bisNHC-C1} moieties, [{Pt(bisNHC-C1)}2(ML)(μ3-S)2]n+ (ML = Pt(bisNHC-C2), n = 2; ML = Pt(bisNHC-C3), n = 2; ML = Rh(cod), n = 1; ML = RhCp*, n = 2), where bisNHC-C1, bisNHC-C2 and bisNHC-C3 represent methylene-, ethylene- and propylene-bridged bis-NHC ligands, respectively, were synthesised and structurally characterised. Reactions of these complexes with a half eq. of Ag(I) ions were examined using 1H and 195Pt NMR spectroscopy. The results exhibit that the trinuclear complexes react with Ag(I) ions accompanied by the formation of Ag–Pt bonds with the {Pt-bisNHC-C1} moieties in the first step affording corresponding heptanuclear complexes except for the RhCp* complex. The RhCp* complex gave a tetranuclear complex bearing the trinuclear unit with an Ag(I) ion. Further addition of Ag(I) ions for the other complexes resulted in disassembling of the heptanuclear clusters affording tetranuclear complexes confirmed by the observation of exchange of the Ag(I) ions in 195Pt NMR measurements, which exhibited signals with no coupling to Ag nuclei.
Introduction
In organic reactions catalysed by metal complexes, ancillary ligands of the complexes serve important roles for the selectivity of products in terms of steric effects. For the evaluation of steric bulkiness for phosphines and N-heterocyclic carbenes (NHCs) as ancillary ligands in their metal complexes, which have been used as catalysts for organic reactions, the cone angles1 and the percent buried volume (%Vbur)2 are useful parameters. However, these symmetric evaluations are not suitable to explain the differences in steric effects in some cases because of the planar structures of NHC ligands affording unsymmetric steric repulsion. For example, change of a substituent for i-Pr to H, which derives only the 1% difference in %Vbur, resulted in drastic improvement of the TONs 550 to 2200 for ethenolysis reactions of methyl oleate catalysed by mononuclear Ru complexes with monodentate NHC ligands.3 Furthermore, in the case of bidentate chelating bis-NHC ligands, the dihedral angles between a coordination plane and each of the NHC planes in their square planar complexes change depending on the lengths of the alkylene linkers of the bis-NHC ligands (Fig. 1).4 This feature of the bis-NHCs causes variation in steric hindrance around coordination sites of the metal centres of their complexes. Moreover, the bis-NHC ligands are rather flexible due to the alkylene linkers, and this character in structure affects steric repulsion around coordination sites. Especially for square planar complexes, in which the coordination sites on the coordination plane and the virtual axial positions located vertically over the coordination plane are in different environments, arrangements of bis-NHC ligands form the coordination sphere with unsymmetric steric bulkiness.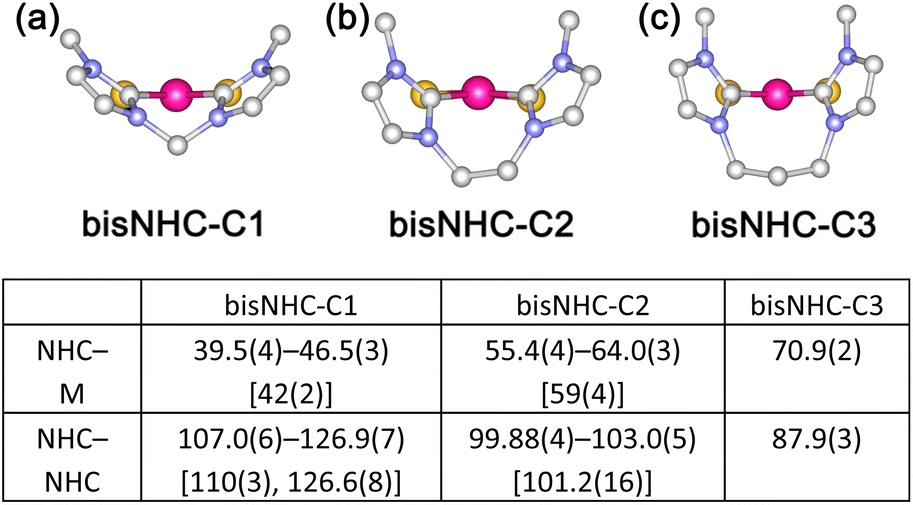 | ||
| Fig. 1 Ligand geometries of {PtC2S2} units in (a) bisNHC-C1, (b) bisNHC-C2 and (c) bisNHC-C3 triplatinum complexes and table of dihedral angles (°, range [average]) between each NHC plane and coordination plane (NHC–M) and between NHC planes (NHC–NHC) obtained from crystallographic analyses of trinuclear complexes. | ||
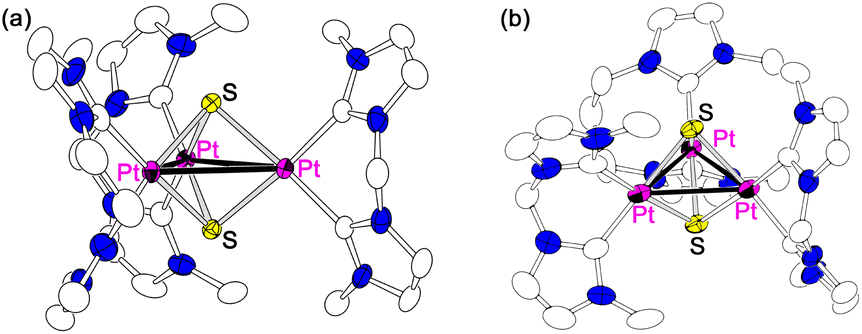
We previously reported triplatinum complexes composed of three bis-NHC-Pt units bridged by two triply bridging sulfide ligands.5 In the triplatinum complexes, each Pt ion has the square planar coordination geometry with two NHC carbon atoms and two sulfides (Fig. 2, red quadrilateral), and the neighbouring Pt ions connected by Pt–Pt bonds exist near the virtual axial positions (Fig. 2, blue). These triplatinum complexes react with Ag(I) ions to afford two kinds of heptanuclear clusters. Both heptanuclear clusters consist of two triplatinum moieties, which involve two sulfides and three bis-NHC ligands, and one Ag(I) ion as a bridging part. One of the heptanuclear clusters involves four Ag–Pt bonds (Fig. 3(a))6 and the other possesses two Ag–S bonds (Fig. 3(b)).7 The difference of the products is attributed to the steric bulkiness of the bis-NHC ligands. The methylene-bridged bis-NHC ligands with methyl substituents (bisNHC-C1), which have large dihedral angle between two NHC planes and then nearly planar structure, prevent the approach of Ag(I) ions to the sulfide ligands by the steric repulsion of the methyl substituents. In this situation, one of the Pt–Pt bonds locates in the less hindered space resulting in the formation of the Ag–Pt bonds. On the other hand, the ethylene-bridged bis-NHC ligands (bisNHC-C2) have the bent structure and cover the Pt–Pt bonds in the triplatinum complex to inhibit the Ag–Pt bond formation. In this case, the sulfide ligands are rather exposed due to the tilted methyl groups in the bis-NHC ligands resulting in the formation of the Ag–S bonds. These clearly appeared steric effects are the results of the enhanced steric repulsion by the crowded environment of the ancillary bis-NHC ligands in the trinuclear system.
 | ||
| Fig. 2 Coordination plane of one of three Pt(II) ions in trinuclear complex bearing sulfide and NHC ligands. | ||
 | ||
| Fig. 3 Heptanuclear complexes (a) with Ag–Pt and (b) with Ag–S bonds. | ||
These results suggest that the bis-NHC trinuclear system is one of the best candidates for investigation of the steric repulsion of bis-NHC ligands in square planar complexes, which are often used as catalysts. In this study, we focus on the reactions of Ag(I) ions and trinuclear complexes bearing the diplatinum unit with the bisNHC-C1 ligands, which could be an essential component for the formation of the Ag–Pt bonds, and another metal–ligand moiety such as platinum with the bisNHC-C2 or propylene-bridged bis-NHC (bisNHC-C3) ligands and rhodium with 1,5-cyclooctadiene (cod) or 1,2,3,4,5-pentamethyl cyclopentadienyl (Cp*) ligands. The bisNHC-C1 ligands in the diplatinum moiety of the trinuclear complexes can move away from each other for the formation of the Ag–Pt bonds. The incorporated other metal–ligand units into the trinuclear complexes would work to tune the steric effects of the bisNHC-C1 ligands on the reactions with Ag(I) ions via steric repulsion of the ancillary ligands in the other metal–ligand units with the bisNHC-C1 ligands. In this study, we describe the unsymmetric steric effects of bis-NHC ligands for square planar complexes, which appeared for the reaction sites of the trinuclear complexes in the reactions with Ag(I) ions.
Results and discussion
Syntheses of mixed-ligand and mixed-metal trinuclear complexes
Mixed-ligand triplatinum complexes, [{Pt(bisNHC-C1)}2{Pt (bisNHC-Cn)}(μ3-S)2]2+ (n = 2, [PtC12PtC2]2+; n = 3, [PtC12PtC3]2+), were synthesised by the one-pot reactions of cis-[Pt(bisNHC-C2)(SH)2] or cis-[Pt(bisNHC-C3)(SH)2] and 2 eq. of cis-[Pt(bisNHC-C1)Cl2] in the presence of KHCO3 as a proton scavenger (Scheme 1(a)). On the other hand, mixed-metal trinuclear complexes, [{Pt(bisNHC-C1)}2(ML)(μ3-S)2]n+ (ML = Rh(cod), n = 1, [PtC12Rhcod]+; ML = RhCp*, n = 2, [PtC12RhCp*]2+), were synthesised by two-step reactions (Scheme 1(b) and (c)). In the first step for the synthesis of [PtC12Rhcod]+, a bis-sulfide-bridged dinuclear platinum complex with the bisNHC-C1 ligands was generated by the reaction of mononuclear complexes, cis-[Pt(bisNHC-C1)(SH)2] and cis-[Pt(bisNHC-C1)Cl2]. The addition of 0.5 eq. of [{Rh(cod)}2(μ-Cl)2] to the solution in the second step afforded [PtC12Rhcod]+. In the case of [PtC12RhCp*]2+, the bis-(acetonitrile)platinum complex, cis-[Pt(bisNHC-C1)(NCCH3)2]2+, was used instead of the dichloro complex to obtain the bis-sulfide-bridged dinuclear complex in the first step. In the second step, [RhCp*(NCCH3)3]2+ was added to give [PtC12RhCp*]2+. Removal of the chloride ions from the reaction system improved the yield of [PtC12RhCp*]2+ probably because the unfavourable interaction between the Rh(III) centre and chloride ions, which disturbs the reaction with sulfide ligands, could be avoided. Syntheses of mixed metal–ligand trinuclear complexes with two triply bridging sulfide ligands using the reactions of a bis-sulfide-bridged diplatinum complex and an additional metal–ligand unit were reported for diplatinum phosphine and arsine complexes.8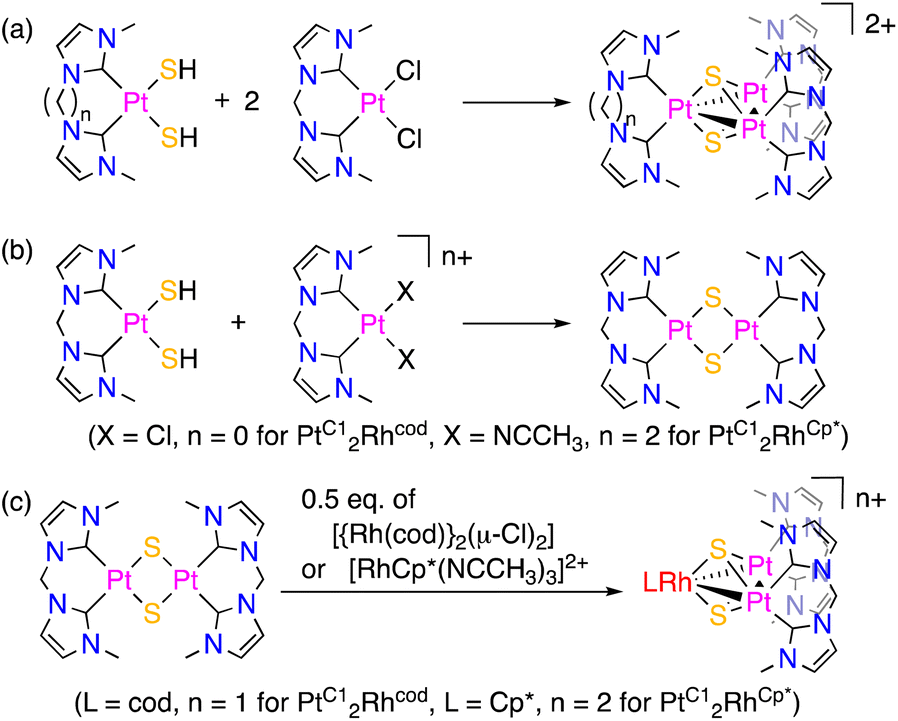 | ||
| Scheme 1 (a) One pot reaction for mixed-ligand triplatinum complexes and (b) the first and (c) second reactions of two-step syntheses for mixed-metal trinuclear complexes. | ||
Structures of trinuclear complexes in the solid state
The structures of [PtC13]Cl(OTf)6 and [PtC23](BPh4)25 were previously analysed by X-ray crystallography. In this study, X-ray crystallographic analyses for [PtC23](PF6)2, [PtC33](PF6)2, [PtC12PtC2](PF6)2, [PtC12PtC3](PF6)2, [PtC12Rhcod](PF6) and [PtC12RhCp*](BPh4)2 were performed showing that crystals containing one of the isomers of each complex were obtained, although there are some possible isomers attributed to the orientation of the bis-NHC ligands (Fig. 4). The crystallographic analysis of the PF6 salt of [PtC23]2+ indicates that the complex cation adopts the Cs-isomeric form (Fig. 5(a)), whereas the previously reported analysis for the BPh4 salt exhibited that the crystal contained only the C3h-isomer.5 On the other hand, [PtC33](PF6)2 crystallised as the C3h-isomer (Fig. 5(b)). These Cs- and C3h-isomers are in equilibrium in solution,5 and the crystal-packing with the counter anions leads the predominant crystallisation of each salt. Mixed-ligand triplatinum complexes, [PtC12PtC2](PF6)2 (Fig. 6(a)) and [PtC12PtC3](PF6)2 (Fig. 6(b)), crystallised as the (MeC1-MeC1)- and (MeC1-AlkC1, MeC1-MeC3)-isomers (Fig. 4(b)), respectively. Both of the crystallised Pt2Rh mixed-metal complexes, [PtC12Rhcod](PF6) (Fig. 7(a)) and [PtC12RhCp*](BPh4)2 (Fig. 7(b)), are the (AlkC1-AlkC1)-isomers (Fig. 4(c)). | ||
| Fig. 4 Possible isomers for (a) triplatinum, (b) mixed-ligand triplatinum and (c) mixed-metal trinuclear complexes bearing bis-NHC ligands. “Me” and “Alk” show relative positions of the methyl substituents and alkylene bridges of bisN-HC ligands, respectively. Red squares represent observed isomers in X-ray crystallography. | ||
 | ||
| Fig. 5 Structures of triplatinum complexes with (a) bisNHC-C2 and (b) bisNHC-C3 ligands. | ||
 | ||
| Fig. 6 Structures of mixed-ligand triplatinum complexes bearing two bisNHC-C1 and (a) bisNHC-C2 or (b) bisNHC-C3 ligands. | ||
 | ||
| Fig. 7 Structures of mixed-metal trinuclear complexes bearing two {Pt-bisNHC-C1} units and (a) {Rh(cod)} or (b) {RhCp*} moieties. One of the positionally disordered cod or Cp* ligands is omitted for clarity (see ESI†). | ||
Structures of trinuclear complexes in solution
On the other hand, [PtC12PtC3]2+ exhibits different behaviour in 1H NMR measurements. For [PtC12PtC3]2+, only the signals for two bisNHC-C1 and one bisNHC-C3 ligands were observed in the 1H NMR spectra at both 293 K (Fig. S3(a) in ESI) and 253 K (Fig. S3(b)†). While [PtC12PtC3]2+ complex can also adopt four isomeric structures similar to [PtC12PtC2]2+, the results indicate that the equilibrium shifts drastically to one of the four isomers in solution. The broadening of the signals at higher temperature suggests that the interconversion between the isomers is slower than the NMR time scale. This result is consistent with the observation of one set of three signals with satellite peaks at −3637 (1JPt–Pt = 735 Hz), −3697 (1JPt–Pt = 723 Hz) and −3791 ppm (1JPt–Pt = 771 Hz) in the 195Pt NMR spectrum (Fig. S15 bottom in ESI†). The observation of only one of the isomers existing in solution probably ascribes to the larger steric repulsion of the bisNHC-C3 ligands along the Pt3 plane in the triplatinum complex than the bisNHC-C2 in the corresponding trinuclear complex. To avoid the large steric repulsion with the methyl groups or the propylene bridge of the bisNHC-3 ligands, the methyl groups in one of the bisNHC-C1 ligands face to the methyl groups of the bisNHC-C3 ligand with the offset orientations. This offset orientation of the methyl groups between the bisNHC-C1 and bisNHC-C3 ligands and the smaller steric repulsion between the methyl groups and the methylene bridge of the bisNHC-C1 ligands result in the stabilisation of the (MeC1-AlkC1, MeC1-MeC3)-isomeric form of the complex, which is observed in the crystallographic analysis.
Elucidation of steric effects of ligands in trinuclear complexes on their reactions with Ag(I) salt
As previously reported, the reactions of the triplatinum complexes bearing the bisNHC-C1 or bisNHC-C2 ligands with Ag(I) ions yielded the Pt3AgPt3 heptanuclear complexes, in which each Ag(I) ion bridges two triplatinum units via four Ag–Pt6 or two Ag–S bonds,7 respectively. The formation of the different products is attributed to the steric repulsion along the Pt3 plane or around the sulfide ligands depending on the geometry of the bis-NHC ligands, which are determined by the lengths of the alkylene bridges. The bisNHC-C1 ligands coordinating to the platinum centres allow the Ag(I) ions to approach the platinum centres and prevent the coordination of the sulfide ligands, while the bisNHC-C2 ligands act the opposite roles. Here, the steric effects of the bis-NHC ligands were elucidated using reactions of trinuclear complexes bearing two {Pt(bisNHC-C1)} moieties, which could be essential part for the Ag–Pt bond formation, and another metal–ligand unit with Ag(I) ions. In addition, to assess the steric effects of the bisNHC-C3 ligand, reaction with Ag(I) ions was also examined for a triplatinum complex with three bisNHC-C3 ligands. The ligands bring larger steric repulsion along the Pt3 plane and afford more space around the sulfide ligands due to its rather folded structure derived from the nearly vertical dihedral angle between each NHC moiety and the coordination plane. The reactions were monitored by 1H and 195Pt NMR spectroscopy using a mixture of each trinuclear complex with an appropriate amount of silver salt to give Ag-adducts of the trinuclear complexes. The chemical shifts and coupling constants of the 195Pt NMR signals for the trinuclear complexes and their reaction mixtures with Ag(I) ions are listed in Table 1. Here, the reactions of the trinuclear complexes with 0.5 eq. of Ag(I) salt are described first to exhibit the initial step of the reactions. In the second, the rather complicated equilibrium involving the trinuclear complexes with more Ag(I) salt showing the other reaction sites on the metal sulfide cores in the complexes affected by the shapes of their ligands.| Bold: predominant signals, red: Ag-bound Pt centre, blue: Ag-unbound Pt centre in complexes with Ag–Pt bonds. *C3h-isomer. **Unknown products. |
|---|
 |
Reactions of trinuclear complexes with 0.5 eq. of Ag(I) salt
195Pt NMR spectrum of [PtC13]2+ exhibited one singlet signal at −3729 ppm and three singlet signals with satellite peaks by the coupling with the neighbouring 195Pt nuclei at −3891 (1JPt–Pt = 771 Hz, 1JPt–Pt = 1052 Hz), −3644 (1JPt–Pt = 1020 Hz) and −3608 (1JPt–Pt = 779 Hz) ppm (Fig. S11 bottom in ESI†). While the symmetric C3h-isomer, which has three equivalent Pt centres, showed no satellite peaks, the unsymmetric Cs-isomer, which involves three inequivalent Pt centres, exhibited the satellite peaks due to the coupling with the neighbouring 195Pt nuclei. In the 195Pt NMR spectrum of [PtC13]2+ with 0.5 eq. of Ag(I) ions, two doublets at −3155 (1JPt–Ag = 554 Hz) and −3446 ppm (1JPt–Ag = 526 Hz) and one singlet with satellite peaks at −3828 ppm (1JPt–Pt = 2020 Hz, 1JPt–Pt = 1008 Hz) were observed (Fig. S11 middle in ESI†). This observation is consistent with the formation of the Pt3AgPt3 heptanuclear complex with four Pt–Ag bonds, which involves the triplatinum units with the Cs-isomeric form bearing three inequivalent Pt centres (Scheme 2). The doublet signals are assigned to the Pt centres forming the Ag–Pt bonds and the singlet with satellite peaks is assignable to the Ag-unbound Pt centre. Additionally, satellite peaks for the doublet signals could not be detected due to the relatively diluted concentration of the sample to prevent the formation of precipitates of the product. Above mentioned results reveal that the formation of the Ag–Pt bonds causes the downfield shift by hundreds ppm and the splitting by the coupling with Ag nuclei for the 195Pt NMR signals.
 | ||
| Scheme 2 Reactions of triplatinum complex with bisNHC-C1 ligands, [PtC13]2+, with Ag(I) ions. | ||
In the case of [PtC23]2+, its 195Pt NMR spectrum showed one intense singlet at −3612 ppm assignable to the C3h-isomer and three weak singlets at −3738, −3517 and −3471 ppm for the CS-isomers (Fig. S12 bottom in ESI†). Although the 195Pt NMR signals for the Cs-isomer should appear with satellite peaks, no satellite signals could be detected in this concentration because of the reduced intensities of the signals due to the three inequivalent Pt centres of the Cs-isomeric form and the equilibrium shifted to the C3h-isomer. In the presence of 0.5 eq. of Ag(I) ions, [PtC23]2+ showed only one singlet signal at −3587 ppm in the 195Pt NMR spectrum (Fig. S12 middle in ESI†). The downfield shift by dozens ppm is attributed to the formation of the Ag–S bonds.
 | ||
| Scheme 3 Reactions of triplatinum complexes with bisNHC-C2 (n = 2), [PtC23]2+, or bisNHC-C3 ligands (n = 3), [PtC33]2+, with 0.5 eq. of Ag(I) ions. | ||
These results of 1H and 195Pt NMR spectroscopy and crystallography indicate that the bisNHC-C2 and bisNHC-C3 ligands prevent the Ag(I) ions approaching the platinum centres in their trinuclear complexes and some of the Ag-adducts with Ag–S bonds exist in equilibrium in solution.
Addition of 0.5 eq. of Ag(I) ions to a solution of mixed-ligand triplatinum complex [PtC12PtC2]2+, which shows broad signals in the 1H NMR spectrum (Fig. S7(a) in ESI†), afforded the observation of sharp signals (Fig. S7(b)†). This fact implies that the reaction of the complex with 0.5 eq. of Ag(I) ions affords the corresponding Pt3AgPt3 heptanuclear complex with the Ag–Pt bonds (Scheme 4). The 195Pt NMR spectrum of the reaction mixture exhibited two doublets at −3168 (1JPt–Ag = 530 Hz) and −3412 ppm (1JPt–Ag = 538 Hz) and one singlet at −3739 ppm (Fig. S14 middle in ESI†). The observation of the downfield shift of the signals and the coupling with the Ag nucleus clearly shows the Ag–Pt bond formation with two of the three Pt centres.
 | ||
| Scheme 4 Reactions of mixed-ligand triplatinum complexes, [PtC12PtC2]2+ (ML = Pt(bisNHC-C2), n = 0) and [PtC12PtC3]2+ (ML = Pt(bisNHC-C3), n = 0), and mixed-metal complex with the {Rh(cod)} moiety, [PtC12Rhcod]+ (ML = Rh(cod), n = 1), with 0.5 eq. of Ag(I) ions. | ||
The mixed-ligand complex with bisNHC-C3, [PtC12PtC3]2+, exhibited similar spectral changes upon the addition of 0.5 eq. of Ag(I) ions (Fig. S8(b) in ESI†) to the bisNHC-C2 complex, [PtC12PtC2]2+, indicating that the Ag-adduct with the Ag–Pt bonds forms at the first stage (Scheme 4). The 195Pt NMR spectrum of the mixture of [PtC12PtC3]2+ with 0.5 eq. of Ag(I) ions exhibited two doublets at −3263 (1JPt–Ag = 454 Hz) and −3342 ppm (1JPt–Ag = 518 Hz) and one singlet with satellite peaks at −3827 ppm (1JPt–Pt = 1743 Hz, 1JPt–Pt = 1444 Hz) besides a weak broad signal at −3620 ppm (Fig. S15 middle in ESI†). The trinuclear core of [PtC12PtC3]2+ contains three inequivalent Pt centres. The two doublet signals showing the coupling with the Ag nucleus are assigned to the Pt centres forming the Ag–Pt bonds in the heptanuclear complex and the singlet with the satellite peaks is attributed to the Ag-unbound Pt centre. Although the other broad signal should be accompanied with two other signals, they would probably be too weak to be detected due to the low concentration, equilibrium and/or the coupling with Ag and Pt nuclei.
On the other hand, the Pt2Rh mixed-metal complex with a {RhCp*} moiety, [PtC12RhCp*]2+, shows different behaviour in the reaction with Ag(I) ions (Fig. S10 in ESI†). With the addition of 0.5 eq. of Ag(I) ions, at least two species exist in equilibrium (Fig. S10(b)†), whereas the other complexes with the {[Pt(bisNHC-C1)]2} moiety form the corresponding heptanuclear complexes with Ag–Pt bonds. The 195Pt NMR spectrum for [PtC12RhCp*]2+ with 0.5 eq. of Ag(I) ions clearly showed a double doublet signal at −3554 ppm (1JPt–Ag = 422 Hz, 1JPt–Rh = 68 Hz) exhibiting the Ag–Pt bond formation (Fig. S17 middle in ESI†). The observation of the Pt–Rh coupling after the addition of Ag(I) ions also indicates the enhancement of the intermetallic interaction between the Pt and Rh centres while [PtC12RhCp*]2+ exhibits no Pt–Rh coupling in the 195Pt NMR spectrum (Fig. S17† bottom) due to the weaker intermetallic interaction. Although the 1H NMR spectrum exhibited the signals for more than two species, the 195Pt NMR measurement could not detect the other species probably due to equilibrium of them and the difference of the time scale for 1H and 195Pt NMR spectroscopy. Furthermore, in the case of [PtC12RhCp*]2+, the extraction of Ag(I) ions by the addition of chloride ions proceeded and reproduced the 1H NMR signals for the trinuclear complex (Fig. S10(f),† Scheme 5), otherwise the cod complex, [PtC12Rhcod]+, affords the heptanuclear complex in the same condition. This difference is probably ascribed to the steric bulkiness of the incorporated metal–ligand units. The less hindered cod ligand in the trinuclear complex gives more room for the bisNHC-C1 ligands moving to provide the sufficient space in between the methylene bridges for the access of an Ag(I) ion (Fig. 8(a) and (b)). On the other hand, the Cp* complex gives smaller space and the rotation of the Cp* ligand causes further steric repulsion with the methyl substituents (Fig. 8(c) and (d)). As a result, the bisNHC-C1 ligands cannot provide sufficient space to form a stable heptanuclear complex with four Ag–Pt bonds.
 | ||
| Scheme 5 Reactions of Pt2Rh complex with a {RhCp*} moiety, [PtC12RhCp*]2+ with 0.5 eq. of Ag(I) ions. | ||
 | ||
| Fig. 8 CPK models of trinuclear complexes bearing {Rh(cod)} ([PtC12Rhcod]+ (a) top view, (b) side view or {RhCp*} ([PtC12RhCp*]2+ (c) top view, (d) side view. Structures were optimised by DFT calculations. | ||
Reactions of trinuclear complexes with excess Ag(I) salt
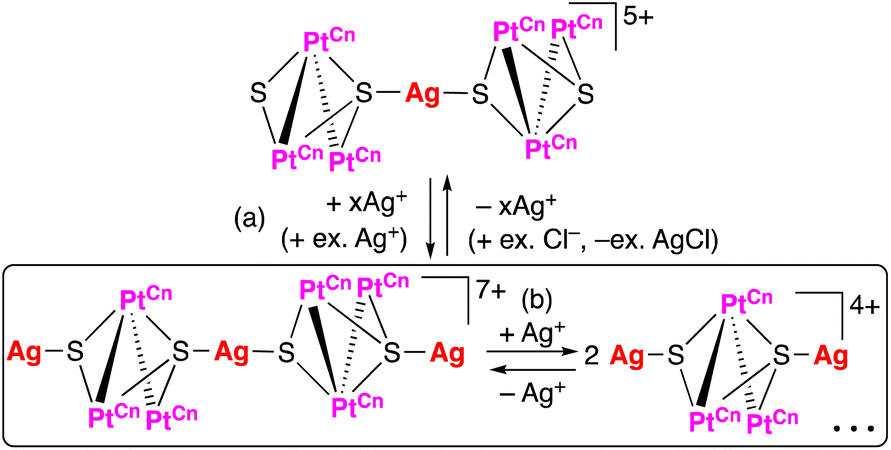 | ||
| Scheme 6 (a) Coordination of Ag(I) ions to uncoordinated sulfide ligands of heptanuclear complexes with bisNHC-C2 (n = 2), [PtC23]2+, or bisNHC-C3 ligands (n = 3), [PtC33]2+, and (b) dissociation of trinuclear units via further coordination of Ag(I) ions to sulfide ligands by the addition of excess Ag(I) ions. | ||
From the reaction mixture of [PtC33]2+ with excess Ag(I) ions, single crystals were obtained and were analysed by crystallography. Although the quality of the crystals was not suitable for full structural characterisation, the analysis clearly revealed a pentanuclear Ag–Pt3–Ag framework, in which both sulfide ligands of [PtC33]2+ coordinate to the Ag(I) ions (Fig. S18 in ESI†). In addition, a nonanuclear Ag-adduct containing two triplatinum moieties with the bisNHC-C2 ligands also crystallised from the reaction mixture with excess Ag(I) ions. In the nonanuclear Ag-adduct, one of the three Ag(I) ions bridges one of the two sulfide ligands of each triplatinum unit and the other sulfide ligands coordinate to the other Ag(I) ions bearing acetonitrile ligands (Fig. 9). Although the bulk preparations of these complexes could not be achieved to collect the data for their characterisation and the results show no proof whether these complexes actually exist in solution, the existence of these Ag-adducts strongly suggests that the species in solution could have the Ag–S bonds and both the sulfide ligands in each triplatinum complex, [PtC23]2+ or [PtC33]2+, can coordinate to Ag(I) ions.
 | ||
| Fig. 9 Structure of nonanuclear Ag-adduct of triplatinum complex bearing three bisNHC-C2 ligands. | ||
 | ||
| Scheme 7 (a) Dissociation of trimetallic moieties of heptanuclear complexes, (b) exchange reaction of Ag(I) ions for tetranuclear complexes and (c) coordination of Ag(I) ions to sulfide ligands of mixed-ligand triplatinum complexes, [PtC12PtC2]2+ (ML = Pt(bisNHC-C2), n = 0) and [PtC12PtC3]2+ (ML = Pt(bisNHC-C3), n = 0), and mixed-metal complex with the {Rh(cod)} moiety, [PtC12Rhcod]+ (ML = Rh(cod), n = 1), with excess Ag(I) ions. | ||
The addition of more than 0.5 eq. Ag(I) ions to a solution of [PtC12PtC3]2+ afforded broad 1H NMR signals showing some Ag-adducts in equilibrium in solution (Fig. S8(c)–(e) in ESI†) as similar to the case for [PtC12PtC2]2+, even though the shapes of the 1H NMR signals for the NHC protons are slightly different. The addition of chloride ions extracts the Ag(I) ions from the adducts leading the regeneration of the heptanuclear complex with Ag–Pt bonds (Fig. S8(f)†). Moreover, in the 195Pt NMR spectrum of the complex with excess Ag(I) ions (Fig. S15 top in ESI†), three singlet signals with satellite peaks appeared at −3359 (1JPt–Pt = 1179, 1JPt–Pt = 478 Hz), −3427 (1JPt–Pt = 1165, 1JPt–Pt = 426 Hz) and −3848 ppm (1JPt–Pt = 1205 Hz) besides one weak singlet at −3602 ppm and two small signals at −3943 and −4019 ppm. The signals at −3359 and −3427 ppm appeared in more downfield if they are compared with those for the trinuclear complex indicating the existence of an Ag-adduct with the Ag–Pt bonds, although no Ag–Pt coupling was observed. The missing of the coupling with Ag nuclei reflects the exchange of the Ag(I) ion in the Ag-adduct (Scheme 7(b)) and suggests the disassembling of the heptanuclear framework affording an Ag-adduct involving one triplatinum unit with two Ag–Pt bonds (Scheme 7(a)). The disassembling of the heptanuclear complex is probably triggered by the coordination of the sulfide ligands to Ag(I) ions and mediated via formation of an intermediate bearing Ag–S and Ag–Pt bonds such as a pentanuclear complex described below (Fig. 10). Additionally, Ag-adducts with the Ag–S bonds could exist in the presence of excess Ag(I) ions due to the accessible space around the sulfide ligands (Scheme 7(c)).
 | ||
Fig. 10 Structure (left) and framework of metal-sulfide core (right) of pentanuclear Ag-adduct of triplatinum complex bearing two bisNHC-C1 and one bisNHC-C3 ligands. One of the terminal ligands for the Ag(I) ions is disordered CH3O− and CH3CN with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. Selected bond distances (Å): Ag–Ag 2.9696(10); Ag–Pt 2.8895(8), 2.8943(8), 2.8907(8), 2.9003(8); Pt–Pt 3.2786(6), 3.1257(5), 3.1048(5); Ag–S 2.757(3), 2.650(3). :1 ratio. Selected bond distances (Å): Ag–Ag 2.9696(10); Ag–Pt 2.8895(8), 2.8943(8), 2.8907(8), 2.9003(8); Pt–Pt 3.2786(6), 3.1257(5), 3.1048(5); Ag–S 2.757(3), 2.650(3). | ||
Single crystals were obtained from the reaction mixture of [PtC12PtC3]2+ with excess Ag(I) ions and analysed by crystallography to reveal a pentanuclear structure that consists of one trinuclear moiety and two Ag(I) ions binding to both of the platinum centres and the sulfide ligands (Fig. 10). The Ag(I) ions are coordinated by bridging and terminal difluorophosphates (PO2F2−), which were presumably generated by partial hydrolysis of hexafluorophosphate counter anions during the crystallisation. Some examples for the partial hydrolysis of hexafluorophosphate anion to afford difluorophosphate were reported.11 Another terminal ligand for the Ag(I) ion was disordered methoxy and acetonitrile ligands with 1:1 ratio. The crystallographic result indicates that the cluster cation possesses +1.5 averaged positive charge because the P atom of one of the hexafluorophosphate anion locates at the crystallographic inversion centre. The averaged positive charge of the adduct supports the disordered terminal ligands with equal amounts of the methoxy monoanion and the acetonitrile molecule. The Ag–Ag distance (2.9696(10) Å) is slightly longer than the interatomic distance in metallic silver (2.89 Å) and shorter than the sum of the van der Waals radii of two Ag atoms (3.44 Å)12 showing the argentophilic interaction.13 Such interaction for Ag–Ag units bridged by substituted phosphates with an Ag–O–P–O–Ag fashion was reported for Ag–Ag distances of 2.9865(5)–3.1596(3) Å in some Ag clusters.14 The Ag–Ag unit locates at the least hindered space in between two methylene bridges of the bisNHC-C1 ligands similar to the case for the heptanuclear cluster with the bisNHC-C1 triplatinum moieties. The Pt–Pt distance near the Ag(I) ions in the Pt3Ag2 pentanuclear complex (3.2786(6) Å) is close to that for the Pt3AgPt3 heptanuclear cluster with the bisNHC-C1 ligands (3.2958(5) Å). The Ag–S distances (2.650(3) and 2.757(3) Å) are significantly longer than the typical Ag–S bond lengths (2.34(1)–2.38(1) Å) observed in a silver alkylthiolate complex,15 for example. However, the Ag–S distances are below the sum of the van der Waals radii of Ag and S atoms (3.52 Å)12 suggesting a weak interaction between Ag and S atoms. Although the bulk synthesis of the pentanuclear complex could not be achieved, the structure of this adduct clearly indicates that two Ag(I) ions are accessible to the Pt–Pt bond in the {[Pt(bisNHC-C1)]2} dimeric unit of [PtC12PtC3]2+.
Summarising the above results of the NMR spectroscopic and crystallographic analyses, the replacement of one of the three bisNHC-C1 ligands in [PtC13]2+ with the bisNHC-C2 or bisNHC-C3 ligand provides the combined steric characters of accessibility to the Pt centres and the sulfide ligands derived from the bisNHC-C1 and bisNHC-C2 or bisNHC-C3 ligands, respectively, for the mixed-ligand triplatinum complexes.
On the other hand, 195Pt NMR spectrum of [PtC12RhCp*]2+ in the presence of excess Ag(I) ions showed one doublet signal at −3589 ppm (1JPt–Rh = 120 Hz) besides two weak signals at −3676 and 3603 ppm (Fig. S17 top in ESI†). Because the Pt–Ag coupling is missing due to the exchange of the Ag(I) ion and the formation of the Ag–Pt bonds leads downfield shift of 195Pt NMR signals by hundreds ppm, the doublet signal is assignable to the Ag-bound Pt centres (Scheme 8(b)). These results imply the formation of a AgPt2Rh tetranuclear complex. The two weak signals are probably attributed to the unsymmetric isomer of the tetranuclear complex bearing the trinuclear unit with the (MeC1-AlkC1)-isomeric form, which involves two inequivalent Pt centres. Additionally, in the presence of excess Ag(I) ions, the Ag-adducts with the Ag–S bonds possibly form because of the accessible spaces around the sulfide ligands (Scheme 8(c)).
 | ||
| Scheme 8 (a) Addition of excess Ag(I) ions affording (b) exchange reaction of Ag(I) ions and (c) coordination of Ag(I) ions to sulfide ligands of Pt2Rh complex with a {RhCp*} moiety, [PtC12RhCp*]2+, with excess Ag(I) ions. | ||
Conclusions
In this study, we demonstrated that NHC ligands with strong σ-donation ability enhance the reactivity of trinuclear complexes bearing bisNHC-platinum units with Ag(I) ions. We also evaluated the steric effects of bisNHC ligands with a variety of alkylene bridges, which affect the dihedral angles between each of two NHC planes and the coordination plane, using reactions of Ag(I) ions with trinuclear complexes exhibiting striking steric repulsions. The bisNHC-C1 ligands in the trinuclear complexes make large steric bulkiness around the sulfide ligands but provide sufficient space for an Ag(I) ion to form Ag–Pt bonds. On the other hand, bisNHC-C2 and bisNHC-C3 ligands, which have more-folded structures, provide sufficient space for Ag(I) ions approaching to the sulfide ligands but prevent the Ag–Pt bond formation. In the mixed-ligand system, the complexes have the characters of both bisNHC-C1 and the other bis-NHC complexes. For the mixed-metal complexes with the {[Pt(bisNHC-C1)]2} unit, the larger Cp* ligand in the complex prevents the formation of the corresponding heptanuclear complex, whereas the complex with the relatively smaller cod ligand exhibits similar reactivity to the mixed-ligand complexes to form the heptanuclear complex.The steric bulkiness of the bis-NHC ligands around the Pt–Pt bond and the sulfide ligands corresponds to that around the unoccupied virtual axial positions and the coordination sites of the square planar Pt(II) centre, respectively. In this point of view, the bisNHC-C1 ligand causes more steric repulsion than the bisNHC-C2 and bisNHC-C3 ligands around the coordination sites of square planar complexes such as palladium complexes, which are used for catalysts of coupling reactions. On the other hand, bisNHC-C2 and bisNHC-C3 ligands greatly affect the steric bulkiness around the unoccupied virtual axial positions of square planar complexes. The results obtained in this study will provide useful information for designing novel NHC complex catalysts for selective reactions.
Experimental section
General procedures
All chemicals were purchased from Sigma-Aldrich, Nacalai Tesque and Wako Pure Chemical Industries. All reagents and solvents were used as received. cis-[Pt(bisNHC-C1)Cl2], cis-[Pt(bisNHC-C1)(NCCH3)2](OTf)2, cis-[Pt(bisNHC-C3)(NCCH3)2] (PF6)2 and cis-[Pt(bisNHC-Cn)(SH)2] (bisNHC-Cn = 1,1′-dimethyl-3,3′-X-4-diimidazolilydene (n = 1, X = methylene; n = 2, X = ethylene; n = 3, propylene)), were prepared according to the reported procedures.16 [(RhCp*Cl)2(μ-Cl)2] was synthesised using a previously reported procedure.17 1H, 13C and 195Pt NMR spectra were recorded on Bruker AVANCE 400 or 600 FT-NMR spectrometers. Chemical shifts (δ in ppm, coupling constants J in Hz) for 1H and 13C NMR signals are expressed from SiMe4 and referenced to residual solvent resonances (Fig. S19–S28 in ESI†). Chemical shifts for 195Pt signals were externally referenced to the signal of 1.2 mol L−1 Na2[PtCl6] in D2O (Fig. S11–S17 in ESI†). Elemental analyses were performed on a J-Science Lab JM-10 elemental analyser by the Analytical Research Centre at Osaka Metropolitan University. Electrospray ionisation mass spectrometry was performed on JEOL AccuTOF LC-plus JMS-T100LP spectrometer.Synthesis of [{Pt(bisNHC-C3)}3(μ3-S)2](PF6)2 ([PtC33](PF6)2)
A solution of cis-[Pt(bisNHC-C3)}(SH)2] (0.10 g, 0.22 mmol) and cis-[{Pt(bisNHC-C3)}(NCCH3)2](PF6)2 (0.35 g, 0.45 mmol) in DMSO (35 mL) was stirred for 25 min and heated at 60 °C for 2 h to give a yellow solution. KHCO3 (0.58 g, 5.8 mmol) was added to the solution and the mixture was stirred for 20 min. The solvent was removed under reduced pressure to give a white-yellow solid. The solid was re-dissolved in water (10 mL). To the resulted solution was added a solution of NH4PF6 (0.84 g 5.1 mmol) in water (5 mL) to give a white-yellow solid. The solid was collected by suction filtration, washed with water (14 mL) and air-dried. Yield: 0.32 g, 96%. Single crystals suitable for X-ray crystallography were obtained by slow evaporation of the solvents from a solution in a mixture of acetonitrile and toluene. Anal. calcd for [{Pt(bisNHC-C3)}3(μ3-S)2](PF6)2 (C33H48F12N12P2Pt3 S2): C, 25.54; H, 3.12; N, 10.83, found: C, 26.03 H, 3.59; N, 10.72. MS (ESI+, CH3CN): m/z = 631 ([PtC33]2+), 1406 ([[PtC33](PF6)]+).Synthesis of [{Pt(bisNHC-C1)}2{Pt(bisNHC-C2)}(μ3-S)2](PF6)2 ([PtC12PtC2](PF6)2)
A mixture of cis-[Pt(bisNHC-C2)(SH)2] (0.023 g, 0.05 mmol), cis-[Pt(bisNHC-C1)Cl2] (0.046 g, 0.10 mmol) and KHCO3 (0.26 g, 2.6 mmol) in DMSO (19 mL) was stirred for 10 min to give a yellow mixture, which was heated at 60 °C for 4 h to afford a pale-yellow mixture. The solvent was removed under reduced pressure to give a pale-yellow solid. The solid was re-dissolved in water (5 mL) and added a solution of NH4PF6 (0.21 g 1.3 mmol) in water (10 mL) to give a white solid, which was collected by suction filtration, washed with water (15 mL) and air-dried. Yield: 0.057 g, 74%. The sample for elemental analysis was purified by the addition of diethyl ether to a solution of the complex in acetonitrile. Single crystals suitable for X-ray crystallography were obtained from a solution in a mixture of acetonitrile and toluene by slow evaporation of the solvents. Anal. calcd for [{Pt(bisNHC-C1)}2{Pt(bisNHC-C2)}(μ3-S)2](PF6)2·0.5Et2O (C30H43F12N12O0.5P2Pt3S2): C, 23.72; H, 2.85; N, 11.07, found: C, 23.76; H, 2.83; N, 11.02. MS (ESI+, CH3CN): m/z = 596 ([PtC12PtC2]2+), 1336 ([[PtC12PtC2](PF6)]+).Synthesis of [{Pt(bisNHC-C1)}2{Pt(bisNHC-C3)}(μ3-S)2](PF6)2 ([PtC12PtC3](PF6)2)
A mixture of cis-[Pt(bisNHC-C3)(SH)2] (0.024 g, 0.05 mmol), cis-[Pt(bisNHC-C1)Cl2] (0.046 g, 0.10 mmol) and KHCO3 (0.11 g, 1.1 mmol) in DMSO (19 mL) was stirred for 10 min to give a yellow mixture, which was heated at 60 °C for 4 h to afford a pale-yellow mixture. The solvent was removed under reduced pressure to give a pale-yellow solid. The solid was re-dissolved in water (5 mL) and added a solution of NH4PF6 (0.071 g 0.43 mmol) in water (10 mL) to give a white solid, which was collected by suction filtration, washed with water (15 mL) and air-dried. Yield: 0.040 g, 53%. Single crystals suitable for X-ray crystallography were obtained by slow evaporation of the solvents from a solution in a mixture of acetonitrile, methanol and toluene. Anal. calcd for [{Pt(bisNHC-C1)}2{Pt(bisNHC-C3)}(μ3-S)2](PF6)2·2H2O (C29H44F12N12O2P2Pt3S2): C, 22.74; H, 2.89; N, 10.97, found: C, 22.82; H, 2.82; N, 11.05. MS (ESI+, CH3CN): m/z = 603 ([PtC12PtC3]2+), 1350 ([[PtC12PtC3](PF6)]+).Synthesis of [{Pt(bisNHC-C1)}2{Rh(cod)}(μ3-S)2](PF6) ([PtC12Rhcod](PF6))
A solution of cis-[Pt(bisNHC-C1)Cl2] (0.023 g, 0.05 mmol) in DMSO (2 mL) was added to a solution of cis-[Pt(bisNHC-C1)(SH)2] (0.021 g, 0.05 mmol) in DMSO (5 mL) to give a yellow solution. The solution was stirred for 10 min. A solution of [{Rh(cod)}2{μ-Cl}2}] (0.013 g, 0.026 mmol) in DMSO (2 mL) was added to the yellow solution, and then KHCO3 (0.15 g, 1.5 mmol) was added to the solution to give a blown mixture. After the mixture was stirred for 30 min, the solvent was removed under reduced pressure to give a yellow solid. The solid was re-dissolved in water (5 mL) and added a solution of NH4PF6 (0.11 g, 0.69 mmol) in water (5 mL) to give a dark-brown solid, which was collected by suction filtration, washed with water (6 mL) and air-dried. Yield: 0.047 g, 84%. Single crystals suitable for X-ray crystallography were obtained by slow diffusion of diethyl ether into a solution in acetonitrile. Anal. calcd for [{Pt(bisNHC-C1)}2{Rh(cod)}(μ3-S)2](PF6)·0.75(Et2O)·H2O (C29H45.5F6N8O1.75P Pt2RhS2): C, 28.17; H, 3.71; N, 9.06. Found: C, 28.32; H, 3.81; N, 9.29. MS (ESI+, CH3CN): m/z = 1017 ([PtC12Rhcod]+).Synthesis of [{Pt(bisNHC-C1)}2(RhCp*)(μ3-S)2](PF6)2 ([PtC12RhCp*](PF6)2)
A solution of AgOTf (0.026 g, 0.10 mmol) in CH3CN (5 mL) was added to a solution of [(RhCp*Cl)2(μ-Cl)2] (0.016 g, 0.026 mmol) and the mixture was stirred for 2 h to give an AgCl solid. The solid was removed by centrifugation to afford an orange solution. Diethyl ether was added to the solution to give red and grey precipitates. The solid was re-dissolved in CH3CN (5 mL) and the insoluble solid was removed using a membrane filter. The obtained orange filtrate and KHCO3 (0.027 g, 0.27 mmol) were added to a reaction mixture, which was obtained by the reaction of cis-[Pt(bisNHC-C1)(SH)2] (0.023 g, 0.053 mmol) and cis-[Pt(bisNHC-C1)(NCCH3)2](OTf)2 (0.037 g, 0.049 mmol) in DMSO (10 mL) with stirring for 10 min. After the mixture was stirred for 2 h, the solvent was removed under reduced pressure to give a dark-green solid. Water (15 mL) was added to the solid and an insoluble solid was collected by suction filtration, washed with diethylether and air-dried. Yield: 0.034 g, 57%.A solution of NaBPh4 (0.36 g, 1.0 mmol) in CH3OH (2 mL) was added to a solution of [PtC12RhCp*](OTf)2 (0.067 g, 0.05 mmol) in methanol (2 mL) to give [PtC12RhCp*](BPh4)2 as a white-green solid. The solid was collected by suction filtration. Yield: 0.072 g, 85%.
A solution of PPh4Cl (0.031 g, 0.083 mmol) in dichloromethane (2 mL) was added to a mixture of the [PtC12RhCp*](BPh4)2 (0.072 g, 0.043 mmol) in dichloromethane (10 mL) and acetonitrile (2 mL) and the mixture was stirred for 30 min. The solvent was removed under reduced pressure to give a white-green solid. The solid was dissolved in water (10 mL) and an insoluble white-solid was removed by suction filtration. The solvent was removed under reduced pressure to give [PtC12RhCp*]Cl2 as a green filmed solid. The solid was collected by suction filtration using acetone and diethyl ether. Yield: 0.021 g, 44%.
A solution of NH4PF6 (0.033 g, 0.20 mmol) in water (5 mL) was added to a solution of [PtC12RhCp*]Cl2 (0.021 g, 0.019 mmol) in water (5 mL) to give [PtC12RhCp*](PF6)2 as a green solid. The solid was collected by suction filtration and washed with water. Yield: 0.0065 g, 23%.
Single crystals of the BPh4 salt suitable for X-ray crystallography were obtained by slow diffusion of cyclohexane into a solution of the BPh4 salt in a mixture of acetonitrile, dichloromethane and a small amount of water. Anal. calcd for [{Pt(bisNHC-C1)}2(RhCp*)(μ3-S)2](PF6)2·CH3CN·H2O (C30H44F12 N9OP2Pt2RhS2): C, 25.85; H, 3.18; N, 9.04. Found: C, 25.65; H, 3.24; N, 8.75. MS (ESI+, CH3CN): m/z = 522 ([PtC12RhCp*]2+), 1189 ([[PtC12RhCp*](PF6)]+).
Reaction of trinuclear complexes with Ag(I) ions
For the investigation of the reactions of the trinuclear complexes and Ag+, samples for 1H NMR measurements were prepared using solutions of trinuclear complexes in CD3CN (10 mmol L−1, 2 mL) with 0.5, 1, 5 or 20 eq. of AgPF6. For the extraction of Ag(I) ions from the reaction system, 40 eq. of NH4Cl was added to the solutions with excess Ag+.Single crystals of the Ag-adducts with triplatinum complexes with three bisNHC-C2 or bisNHC-C3 ligands were obtained from respective solutions of the complexes in acetonitrile with excess amount of AgPF6 by diffusion of diethyl ether after the addition of a small amount of methanol.
In the case of the Ag-adduct containing a triplatinum moiety with two bisNHC-C1 and bisNHC-C3 ligands and two Ag(I) ions coordinated by difluorophosphate ligands, single crystals for the X-ray crystallographic measurement were obtained as follows. To a solution of the mixed-ligand triplatinum complex (9.7 mmol L−1, 1 mL) with (9.2 mg, 3.8 μmol), methanol (ca. 0.5 mL) was added followed by the addition of a small amount of toluene to afford a tiny amount of white solids. After the solids were removed using a membrane filter, the filtrate was allowed to evaporate the solvents resulting in crystallisation of the Ag-adduct.
X-ray crystallography
Single crystals of [PtC23](PF6)2, [PtC33](PF6)2, [PtC12PtC2](PF6)2, [PtC12PtC3](PF6)2, [PtC12Rhcod](PF6), [PtC12RhCp*](BPh4)2 and Ag-adducts of [PtC23]2+, [PtC12PtC3]2+ and [PtC33]2+ were mounted on a loop using Paratone. Diffraction data were collected on a Rigaku Varimax Saturn724 diffractometer using a rotation method with 0.5° frame widths. The data were integrated, scaled, sorted, and averaged using the CrystalClear18 software. Absorption corrections were applied using the multi-scan method. The structures were solved using SHELXS97 (ref. 19) and refined with SHELXL Version 2018/3 (ref. 20) using the CrystalStructure software21 or version of olex2.refine 1.5-dev.22 All hydrogen atoms were located at the calculated positions and refined as riding models. Crystallographic data are summarised in Tables S1–S9 in ESI.†DFT calculations
DFT calculations were carried out on the isomers of trinuclear complex cations, which were observed in crystallographic analyses for [PtC12Rhcod]+ and [PtC12RhCp*]2+ using Gaussian 09.23 Atomic coordinates were optimised at the level of B3LYP/LanL2DZ. Structural optimisations were started from the structures obtained from crystallographic analyses. Vibrational frequencies were calculated for all converged structures, and no imaginary frequencies appeared showing that these structures lie on minima. The atomic coordinates of the optimised structures of the complexes are listed in Tables S10 and S11 in ESI.†Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for [PtC23](PF6)2, [PtC33](PF6)2, [PtC12PtC2](PF6)2, [PtC12PtC3](PF6)2, [PtC12RhCp*](BPh4)2, [PtC12Rhcod](PF6), [Ag{[Pt(bisNHC-C2)]3[Ag(NCCH3)3](m4-S)2}2](PF6)7 and [{Pt(bisNHC-C1)}2{Pt(bisNHC-C3)}(m4-S)2{Ag (O2PF2)}{Ag(NCCH3)0.5(OCH3)0.5}(m-O2PF2)](PF6)1.5 has been deposited at the CCDC under 2238719–2238726 and can be obtained from https://www.ccdc.cam.ac.uk.Author contributions
Natsuki Yabune: conceptualisation, funding acquisition, investigation, visualisation and writing – original draft. Hiroshi Nakajima: writing – review & editing. Takanori Nishioka: funding acquisition, project administration, supervision, visualisation and writing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers N22K05146 and K22KJ2607.Notes and references
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS.
- (a) A. Poster, B. Cosenza, A. Correa, S. Giudice, F. Ragone, V. Scarano and L. Cavallo, Eur. J. Inorg. Chem., 2009, 1759–1766 Search PubMed; (b) L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Olive, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef CAS; (c) A. Collado, J. Balogh, S. Meiries, A. M. Z. Slawin, L. Falivene, L. Cavallo and S. P. Nolan, Organometallics, 2013, 32, 3249–3252 CrossRef CAS; (d) M. S. Viciu, O. Navarro, R. F. Germaneau, R. A. Kelly III, W. Sommer, N. Marion, E. D. Stevens, L. Cavallo and S. P. Nolan, Organometallics, 2004, 23, 1629–1635 CrossRef CAS; (e) S. Gaillard, X. Bantreil, A. M. Z. Slawin and S. P. Nolan, Dalton Trans., 2009, 6967–6971 RSC; (f) H. Clavier, A. Correa, L. Cavallo, E. C. Escudero-Adán, J. Benet-Buchholz, A. M. Z. Slawin and S. P. Nolan, Eur. J. Inorg. Chem., 2009, 13, 1767–1773 CrossRef; (g) C. A. Urbina-Blanco, X. Bantreil, H. Clavier, A. M. Z. Slawin and S. P. Nolan, Beilstein J. Org. Chem., 2010, 6, 1120–1126 CrossRef CAS; (h) X. Bantreil, R. A. M. Randall, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2010, 29, 3007–3011 CrossRef CAS; (i) H. Clavier and S. P. Nolan, Chem. Commun., 2010, 46, 841–861 RSC; (j) S. Gaillard, A. M. Z. Slawin, A. T. Bonura, E. D. Stevens and S. P. Nolan, Organometallics, 2010, 29, 394–402 CrossRef CAS; (k) S. R. Patrick, A. Collado, S. Meiries, A. M. Z. Slawin and S. P. Nolan, J. Organomet. Chem., 2015, 775, 152–154 CrossRef CAS; (l) A. Gómez-Suárez, D. J. Nelson and S. P. Nolan, Chem. Commun., 2017, 53, 2650–2660 RSC; (m) D. Gasperini, M. D. Greenhalgh, R. Imad, S. Siddiqui, A. Malik, F. Arshad, M. I. Choudhary, A. M. Al-Majid, D. B. Cordes, A. M. Z. Slawin, S. P. Nolan and A. D. Smith, Chem.–Eur. J., 2019, 25, 1064–1075 CrossRef CAS PubMed.
- S. Byun, H. Seo, J.-H. Choi, J. Y. Ryu, J. Lee, W.-J. Chung and S. Hong, Organometallics, 2019, 38, 4121–4132 CrossRef CAS.
- (a) S. K. U. Riederer, P. Gigler, M. P. Högerl, E. Herdtweck, B. Bechlars, W. A. Herrmann and F. E. Kühn, Organometallics, 2010, 29, 5681–5692 CrossRef CAS; (b) S. Ahrens, A. Zeller, M. Taige and T. Strassner, Organometallics, 2006, 25, 5409–5415 CrossRef CAS; (c) J. A. Mata, M. Poyatos and E. Peris, Coord. Chem. Rev., 2007, 251, 841–859 CrossRef CAS; (d) M. G. Gardiner and C. C. Ho, Coord. Chem. Rev., 2018, 375, 373–388 CrossRef CAS; (e) Y. Maeda, H. Hashimoto, I. Kinoshita and T. Nishioka, Inorg. Chem., 2014, 53, 661–663 CrossRef CAS.
- Y. Maeda, H. Hashimoto and T. Nishioka, Dalton Trans., 2012, 41, 12038–12047 RSC.
- N. Yabune, H. Nakajima and T. Nishioka, Dalton Trans., 2020, 49, 7680–7683 RSC.
- N. Yabune, H. Nakajima and T. Nishioka, Dalton Trans., 2021, 50, 12079–12082 RSC.
- (a) J. Ruiz, V. Rodríguez, A. Pérez, G. López and D. Bautista, J. Organomet. Chem., 2004, 689, 2080–2086 CrossRef CAS; (b) S.-W. A. Fong, T. S. A. Hor, S. M. Devoy, B. A. Waugh, B. K. Nicholson and W. Henderson, Inorg. Chim. Acta, 2004, 357, 2081–2090 CrossRef CAS; (c) B. C. White, D. Harrison, W. Henderson, B. K. Nicholson and T. S. A. Hor, Inorg. Chim. Acta, 2010, 363, 2387–2393 CrossRef CAS; (d) R. B. Sutton and W. Henderson, Inorg. Chim. Acta, 2020, 506, 119557 CrossRef CAS; (e) Z. Li, H. Liu, K. F. Mok, A. S. Batsanov, J. A. K. Howard and T. S. A. Hor, J. Organomet. Chem., 1999, 575, 223–231 CrossRef CAS; (f) R. Mas-Ballesté, P. A. Champkin, W. Clegg, P. González-Duarte, A. Lledós and G. Ujaque, Organometallics, 2004, 23, 2522–2532 CrossRef; (g) S. Jeram, W. Henderson, B. K. Nicholson and T. S. A. Hor, J. Organomet. Chem., 2006, 691, 2827–2838 CrossRef CAS.
- Y. Maeda, H. Hashimoto, I. Kinoshita and T. Nishioka, Inorg. Chem., 2015, 54, 448–459 CrossRef CAS PubMed.
- O. Hiltner, F. J. Boch, L. Brewitz, P. Härter, M. Drees, E. Herdtweck, W. A. Herrmann and F. E. Kühn, Eur. J. Inorg. Chem., 2010, 5284–5293 CrossRef CAS.
- (a) M. Ferrer, A. Gallen, A. Gutiérrez, M. Martínez, E. Ruiz, Y. Lorenz and M. Engeser, Chem.–Eur. J., 2020, 26, 7847–7860 CrossRef CAS; (b) F. Santoro, M. Althaus, C. Bonaccorsi, S. Gischig and A. Mezzetti, Organometallics, 2008, 27, 3866–3878 CrossRef CAS; (c) A. K. Jain, M. R. Gau, P. J. Carroll and K. I. Goldberg, Organometallics, 2022, 41, 3341–3348 CrossRef CAS; (d) A. B. McQuarters, J. W. Kampf, E. E. Alp, M. Hu, J. Zhao and N. Lehnert, Inorg. Chem., 2017, 56, 10513–10528 CrossRef CAS; (e) D. L. Gerlach, S. Siek, D. B. Burks, J. M. Tesh, C. R. Thompson, R. M. Vasquez, N. J. White, M. Zeller, D. B. Grotjahn and E. T. Papish, Inorg. Chim. Acta, 2017, 466, 442–450 CrossRef PubMed; (f) I. T. Chizhevsky, N. V. Rastova, N. E. Kolobova, P. V. Petrovskii, E. I. Fedin, L. E. Vinogradova, L. A. Leites, A. I. Yanovsky and Yu. T. Struchkov, J. Organomet. Chem., 1988, 339, 165–180 CrossRef.
- A. Bondi, J. Phys. Chem., 1965, 68, 441–451 CrossRef.
- (a) M. Jansen, Angew. Chem., Int. Ed., 1987, 26, 1098–1110 CrossRef; (b) P. Majumdar, K. K. Kamar, S. Goswami and A. Castineiras, Chem. Commun., 2001, 1292–1293 RSC; (c) X.-D. Chen, M. Du and T. C. W. Mak, Chem. Commun., 2005, 4417–4419 RSC.
- (a) J. A. Fry, C. R. Samanamu, J.-L. Montchamp and A. F. Richards, Eur. J. Inorg. Chem., 2008, 463–470 CrossRef CAS; (b) C. R. Samanamu, E. N. Zamora, J.-L. Montchamp and A. F. Richards, J. Solid State Chem., 2008, 181, 1462–1471 CrossRef CAS; (c) V. Moodley, L. Mthethwa, M. N. Pillay, B. Omondi and W. E. van Zyl, Polyhedron, 2015, 99, 87–95 CrossRef CAS.
- I. G. Dance, L. J. Fitzpatrick, A. D. Rae and M. L. Scudder, Inorg. Chem., 1983, 22, 3785–3788 Search PubMed.
- (a) M. Muehlhofer, T. Strassner, E. Herdtweck and W. A. Herrmann, J. Organomet. Chem., 2002, 660, 121–126 CrossRef CAS; (b) Y. Unger, A. Zeller, M. A. Taige and T. Strassner, Dalton Trans., 2009, 4786–4794 Search PubMed.
- C. White, A. Yates, P. M. Maitlis and D. M. Heinekey, Inorg. Synth., 1992, 29, 228–234 CrossRef CAS.
- CrystalClear, Rigaku Corporation, CrystalClear Software User’s Guide, Molecular Structure Corporation, 1999 CrossRef CAS; J. W. Pflugrath, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1999, 55, 1718–1725 CrossRef CAS.
- SHELXL97 and G. M. Sheldrick, Program for Crystal Structure Refinement, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS.
- CrystalStructure 4.3, Crystal Structure Analysis Package, Rigaku Corporation, 2000– 2019, Tokyo 196-8666, Japan Search PubMed.
- (a) L. J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard and H. Puschmann, The Anatomy of a Comprehensive Constrained, Restrained, Refinement Program for the Modern Computing Environment – Olex2 Dissected, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 59–71 CrossRef CAS; (b) O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, Olex2: a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- Gaussian 09, Revision A.02, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of 195Pt NMR spectroscopy, characterisation of new complexes (1H and 13C NMR spectra and ESI-mass spectra), DFT calculations and X-ray crystallography. CCDC 2238719–2238726. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra01671h |
| This journal is © The Royal Society of Chemistry 2024 |