DOI:
10.1039/D4RA01626B
(Paper)
RSC Adv., 2024,
14, 12634-12638
Effect of defect-healing treatment on layered silicate precursors toward well-defined crosslinked frameworks†
Received
2nd March 2024
, Accepted 12th April 2024
First published on 19th April 2024
Abstract
The synthesis of zeolites from two-dimensional layered precursors through interlayer crosslinking of the layers is a promising avenue for realizing meticulously designed synthesis routes. However, the presence of defective silanol species in the precursors hinders the achievement of desirable synthesis outcomes. This study focuses on PREFER—a layered precursor for FER-type zeolites—which was synthesized and subjected to a liquid-mediated defect-healing treatment that we recently developed. The defect-healing process involves the use of fluoride compounds for reconstruction and organic pore fillers to stabilize the framework. The effects of the treatment on the structure, composition, and iron insertion behavior of PREFER were examined. Characterization results revealed a reduction in the number of intralayer silanol defects, whereas interlayer silanols were unaffected by the defect-healing treatment. Furthermore, the subsequent alterations observed in the crosslinking behavior with iron atoms indicated that the defect-healing treatment may enhance the insertion of iron species between the layers in more homogeneous environments compared with the untreated precursor. These findings provide valuable insights into the prospects of controlled interlayer linkage in two-dimensional zeolite materials.
Introduction
Zeolites—crystalline microporous aluminosilicates—are an industrially important class of materials with three-dimensional framework structures.1,2 They are well known as shape-selective catalysts and adsorbents because of their active sites, such as strong Brønsted acid sites and metal cations dispersed within regular pores of molecular size and limited access to the internal surfaces.3–5 Additionally, thin two-dimensional zeolite nanosheets with a single unit cell or near-single unit cell thickness (2–3 nm) can be synthesized6,7 and have a large external surface area (approximately 50% of the total surface area, compared with 2% for micrometer-sized three-dimensional zeolites) and a large number of active sites exposed on the outer surface. This renders them advantageous for adsorption and reactions involving bulky molecules. There have been many reports on the excellent performance of two-dimensional zeolite materials in catalytic reactions.8–10
In recent years, the synthesis of three-dimensional zeolite frameworks from two-dimensional zeolite nanosheets using bonding layers has been actively pursued. Topotactic conversion,11 assembly–disassembly–organization–reassembly (ADOR),12 and interlayer-expanded zeolite (IEZ)13 are the main methods. Metal species such as iron and tin14 bridging the layers in the ADOR or IEZ methods are of particular interest because they can generate new catalytically active sites that differ from those in three-dimensional zeolite frameworks. However, the sites and distribution of the metal species in the interlayers have not been precisely controlled. Two main problems have been reported: (1) insertion of metal species into the framework of layers and (2) cross-linkage by silicate species, which migrate from the layer frameworks. This causes the metal species in the layers and silicate species in the interlayers to have a random distribution. In fact, three-dimensional zeolites synthesized from two-dimensional zeolites have not been converted back into two-dimensional zeolites by removing the crosslinker between layers.15 Defect sites such as silanol nests due to the lack of silicon atoms are considered to be the main reason for this problem, as metal species can be inserted into the defects, and migration of silicate species is likely to occur from defects in the zeolite framework.
We recently reported a simple liquid-mediated defect-healing method via self-defect-healing process16 that can remove most defects in high-silica zeolites and realize extremely stable zeolites. Fluoride compounds that break siloxane bonds and promote the reconstruction of zeolite frameworks and organic pore fillers, which can stabilize the frameworks and prevent structural collapse, worked cooperatively during treatment. Although this method has only been adopted for zeolitic three-dimensional frameworks, it is also expected to be applicable to two-dimensional zeolite nanosheets, and defect-free zeolite nanosheets are ideal precursors for interlayer linkages.
In this study, PREFER was selected as the two-dimensional zeolite precursor. PREFER is a layered precursor of FER-type zeolites; and calcination of PREFER at 550 °C dehydrates and condenses the silanol groups between the layers, yielding FER-type zeolite.17,18 Iron species were used for the crosslinking of PREFER because of their stability and superior catalytic activity. The states of the iron species in three-dimensional frameworks obtained from the precursors with and without the defect-healing treatment were compared, and significant differences were observed, which may open up the new way to control the position of heteroatoms in interlayer crosslinked materials.
Experimental
Synthesis of PREFER
PREFER was synthesized as described in a previous study.13 4-Amino-2,2,6,6-tetramethylpiperidine (FUJIFILM Wako Pure Chemical Industries) was used as the organic structure-directing agent (OSDA), and the molar composition of the reaction mixture was 1.0 SiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.0 OSDA:1.5 NH4F:1.0 HF:15H2O. NH4F (Wako) was dissolved in H2O to obtain a solution. OSDA and SiO2 (precipitated silica P707, Mizusawa Industrial Chemicals) were added to the solution, followed by stirring until homogenization. The reaction mixture was mixed with an HF solution (Wako), stirred well, and allowed to stand for 1 h. The resulting mixture was heated in a Teflon®-lined stainless-steel autoclave (#4749, Parr Instruments) at 170 °C for 7 days. The solid product was collected via filtration, washed with distilled water, and dried at 80 °C overnight.
:1.0 OSDA:1.5 NH4F:1.0 HF:15H2O. NH4F (Wako) was dissolved in H2O to obtain a solution. OSDA and SiO2 (precipitated silica P707, Mizusawa Industrial Chemicals) were added to the solution, followed by stirring until homogenization. The reaction mixture was mixed with an HF solution (Wako), stirred well, and allowed to stand for 1 h. The resulting mixture was heated in a Teflon®-lined stainless-steel autoclave (#4749, Parr Instruments) at 170 °C for 7 days. The solid product was collected via filtration, washed with distilled water, and dried at 80 °C overnight.
Defect-healing treatment of PREFER
In accordance with our previous report,16 NH4F (Wako) was dissolved in a dilute solution of tetraethylammonium hydroxide (TEAOH, Sigma-Aldrich) to prepare a solution with a molar composition of 0.1 TEAOH:0.1 NH4F:15H2O. The synthesized PREFER powder was slowly added to this solution at a solid-to-liquid weight ratio of 1:2. The resulting mixture was heated in an autoclave at 170 °C for 24 h. The solid product was collected via filtration, washed with pure water, and dried at 80 °C overnight. The product obtained after the defect-healing treatment is denoted as PREFER_healed. The samples before and after the defect-healing treatment were calcined at 550 °C for 6 h and are denoted as PREFER_calcined and PREFER_healed_calcined, respectively.
Crosslinking layers of PREFER with iron atoms
Referring to a previous paper14 on interlayer expansion using metal-linker units, experiments were conducted that involved a hydrothermal reaction under acidic conditions using iron(III) chloride hexahydrate (FeCl·6H2O, Wako) as the metal source. 2.63 g of 1 M HCl aq. (Wako), 6.30 g of H2O, and 0.500 g of PREFER or PREFER_healed were added sequentially to a Teflon® cup in this order and stirred for 10 min. 0.214 g of FeCl3·6H2O was added to the mixture, and the mixture was stirred for another 10 min. The mixture was sealed in an autoclave and heated at 175 °C for 24 h under autogenous pressure. The solid product was collected via filtration, washed with pure water, and dried at 80 °C overnight.
Characterization
Powder X-ray diffraction (XRD) patterns of the samples were recorded using an Ultima IV X-ray diffractometer (Rigaku) with CuKα radiation (λ = 0.15406 nm, 40 kV, 40 mA) in the 2θ range of 3–50° at a scan rate of 10° min−1. The size and morphology of the products were examined using scanning electron microscopy (SEM, JSM-7000F, JEOL) at an accelerating voltage of 5 keV. The elemental compositions of the samples were determined using a Thermo iCAP 6300 inductively coupled plasma atomic emission spectrometer. Ultraviolet-visible (UV-Vis) spectroscopy was performed to measure the absorbance and transmittance of the samples using a JASCO V-670 spectrophotometer in the wavelength range of 190–800 nm. The 29Si dipolar decoupling (DD) magic angle spinning (MAS) nuclear magnetic resonance (NMR) spectra were recorded by collecting 2048 scans at 99.37 MHz with a π/2 pulse length of 5.0 μs, a recycle delay of 60 s, and a spinning frequency of 10 kHz. Infrared (IR) spectra were acquired using a JASCO FT/IR-6600 spectrometer equipped with a mercury cadmium telluride detector. The samples were activated at 500 °C for 1 h under a nitrogen flow. Spectra were collected in the diffuse reflectance mode (DRIFT). The weights of organic materials in the samples were determined via thermogravimetry-differential thermal analysis (TG-DTA) using a Rigaku EV02G ThermoMass Photo/H instrument in the temperature range of 25–800 °C at a heating rate of 10 K min−1 under a flow of 10% O2/90% He mixed gas. The carbon and nitrogen contents of the samples were measured using a CE-440F CHN/O/S elemental analyzer (Systems Engineering). The framework structure of the zeolite was observed using a Scanning Transmission Electron Microscope (JEM-ARM200F NEOARM, JEOL) operating at an accelerating voltage of 200 keV and a convergence semi-angle of 12 mrad. This microscope was equipped with a segmented annular all-field detector (SAAF-Quad, JEOL) for the OBF system, which maximizes the signal-to-noise ratio for light elements and enables the visualization of electron-beam-sensitive materials such as zeolites. The TEM samples were initially picked up as bulk fragments under a stereo microscope, then embedded in a photo-cured resin. Subsequently, approximately 35 nm ultrathin cross-section slices were prepared using an ultramicrotome and mounted on carbon film-coated lacey carbon TEM grids.
Results and discussion
Synthesis of PREFER
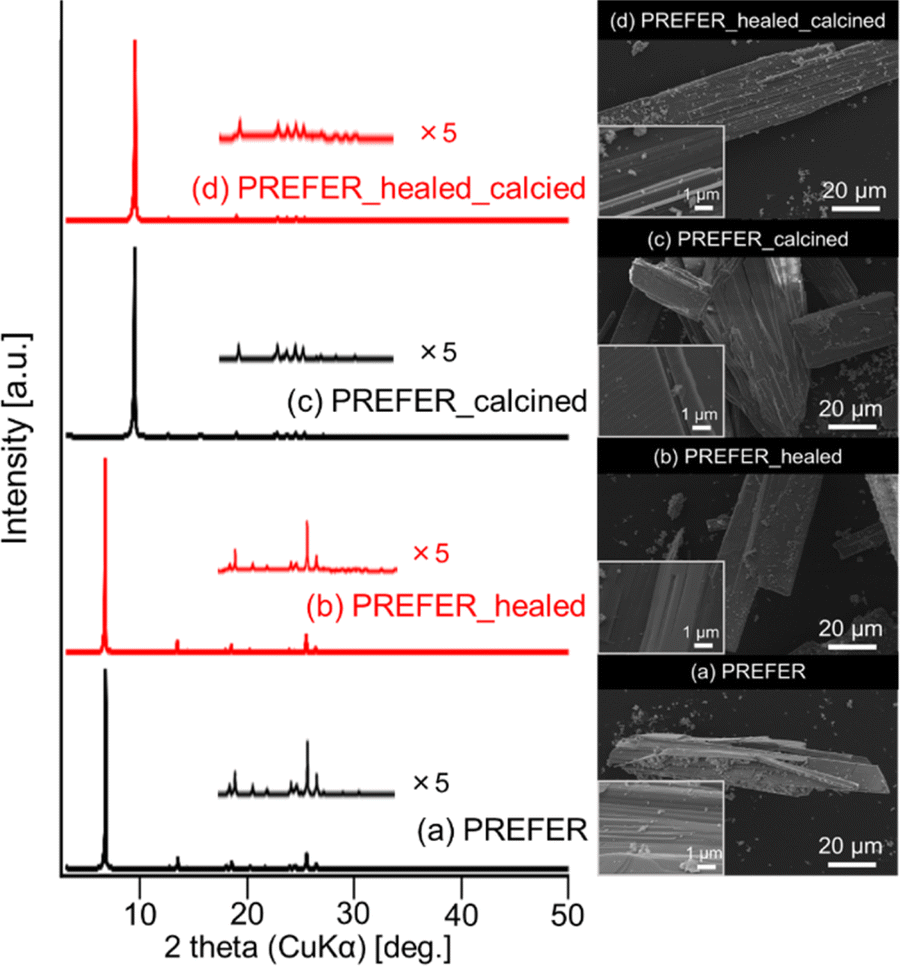
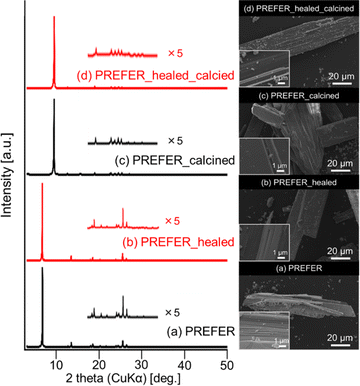
The XRD patterns and SEM images of PREFER and its calcined samples are shown in Fig. 1(a) and (c), respectively. The XRD pattern of the obtained PREFER matched the previously reported diffraction pattern of PREFER.18 The XRD patterns of the calcined sample were consistent with the diffraction pattern of FER-type zeolite, suggesting the topotactic condensation of layers due to calcination. The SEM images also confirmed that the products had layered structures, which are characteristic of the PREFER-layered precursor and FER-type zeolite synthesized from PREFER, suggesting topotactic condensation.
 |
| | Fig. 1 XRD patterns (left) and SEM images (right) of the (a) PREFER, (b) PREFER_healed, (c) PREFER_calcined, and (d) PREFER_healed_calcined samples. | |
Defect-healing treatment of PREFER
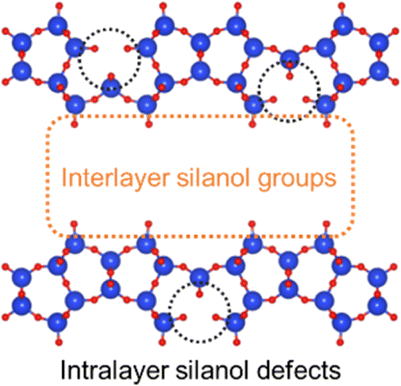
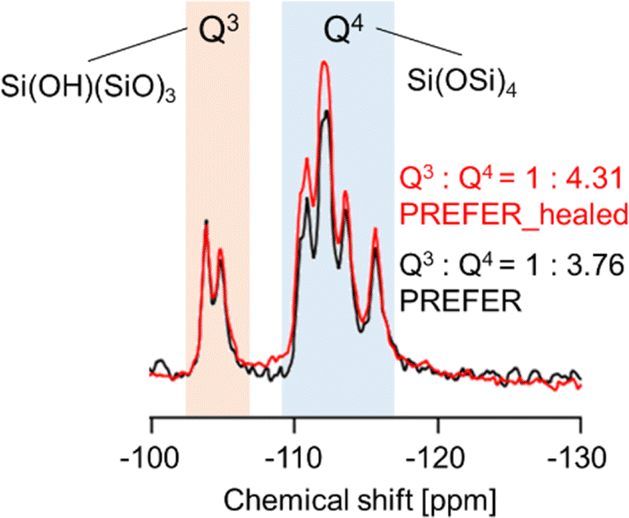
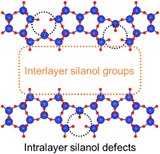
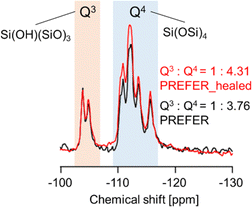
The XRD patterns and SEM images of the defect-healed samples are shown in Fig. 1(b) and (d), respectively. No differences in the XRD patterns due to the defect-healing treatment were observed, indicating that no structural changes occurred during the treatment. Notably, the layers did not condense and maintained the interlayer distance, according to the d value of the first peaks. The SEM images confirmed that the defect-healed samples had layered structures similar to those of the PREFER and PREFER_calcined samples. For the following discussion, the types of silanol species are categorized into intralayer silanol defects and interlayer silanol groups (Fig. 2). The 29Si DD MAS NMR spectra shown in Fig. 3 indicate that the integrated intensity of the Q3 (Si(OH)(OSi)3) signals, interlayer silanol groups and silanol defects, were reduced after the defect-healing treatment relative to that of the Q4 (Si(OSi)4) signals in the frameworks. The presence of a certain amount of the Q3 signal after the defect-healing treatment suggests that the intralayer silanol defects were healed while the interlayer silanol groups in PREFER were maintained. This is because the interlayer silanol groups were separated from each other and stabilized by the OSDA by balancing the charges.
 |
| | Fig. 2 Types of silanol species. | |
 |
| | Fig. 3 29Si DD MAS NMR spectra of PREFER and PREFER_healed. | |
DRIFT spectra measured at 550 and 250 °C are shown in Fig. 4 and S1,† respectively. The spectra were normalized to the bending vibrations of the Si–O bonds. As shown, the intensities of the absorption peaks corresponding to the stretching vibration of silanol groups observed in the region of 3750–3690 cm−1 were reduced by the defect-healing treatment. According to previous studies,19,20 the peaks at 3691 and 3724 cm−1 can be assigned to interlayer and intralayer silanol groups, respectively, because interlayer silanol groups form strong hydrogen bonds, and their stretching vibrations are considered to shift to lower frequencies.21 The spectra indicated that the number of intralayer silanol groups was significantly reduced by the defect-healing treatment, while the interlayer silanol groups were maintained. These assignments were confirmed by comparing DRIFT spectra obtained at different temperatures. The intensity of the absorption peak assigned to the interlayer silanol groups observed at 3691 cm−1 started to decrease as the temperature increased owing to interlayer condensation, whereas that of the peak assigned to intralayer silanol groups at 3724 cm−1 was maintained even after heating. The TG-DTA curves shown in Fig. S2 and S3† indicate that decomposition of organic matter and condensation of layers started at approximately 300 °C, in agreement with the DRIFT spectra. The C/N ratios obtained via elemental analysis were not affected by the defect-healing treatment, suggesting that the organics between the layers did not change (Table S1†). The results indicated that the TEAOH in the treatment liquid did not enter the layers. This is because the presence of OSDAs between layers is sufficient to stabilize PREFER. Notably, the defect-healing treatment significantly reduced the number of defects in the parent PREFER, which was synthesized in fluoride media and is expected to have few defects.
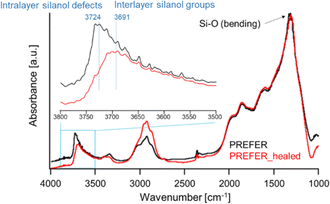 |
| | Fig. 4 DRIFT spectra of PREFER and PREFER_healed at 550 °C. | |
Crosslinking layers of PREFER with iron atoms
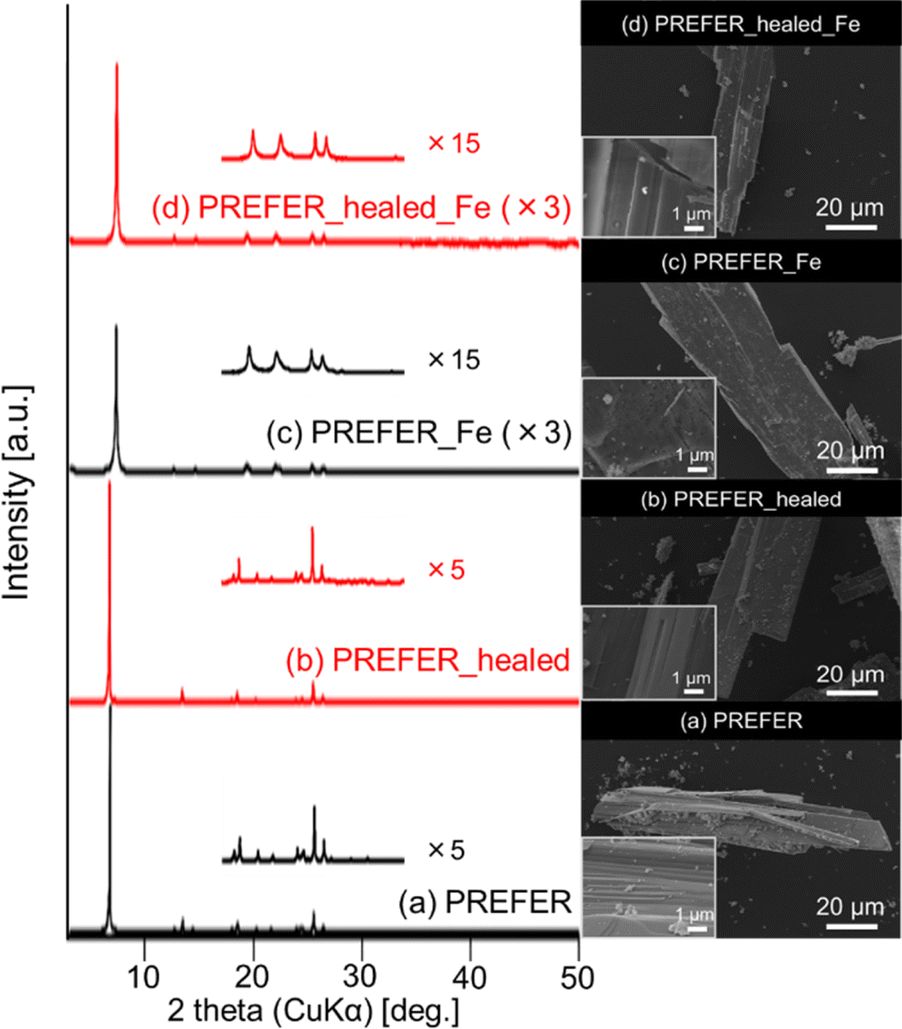
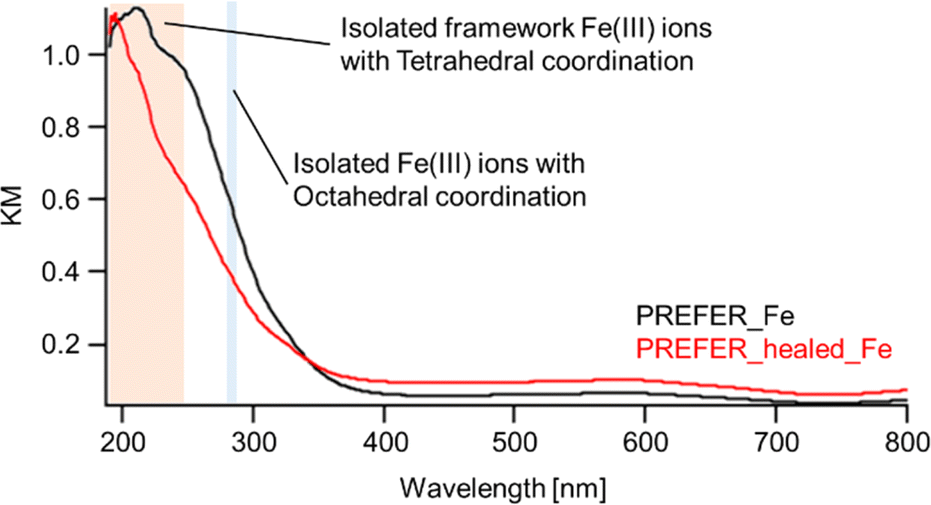
The XRD patterns, SEM images and UV-Vis spectra of the samples crosslinked with iron atoms are shown in Fig. 5(c), (d) and 6 respectively. The first diffraction peaks of the crosslinked samples were shifted to a high angle (low d value) relative to those of PREFER, indicating that the interlayer distance narrowed. The interlayer distance of the crosslinked sample calculated from the d value was shorter than that of PREFER but longer than that of the FER-type zeolite, indicating that the condensed interlayer distances were increased by the insertion of iron atoms. Additionally, the SEM images confirmed that the samples kept layered structures before and after crosslinking with iron atoms. The TEM images are shown in Fig. S4.† The TEM images indicated that the interlayer distance has increased relative to FER structure in both PREFER and PREFER_healed due to interlayer cross-linking, which is consistent with the d values calculated from the XRD patterns. The UV-Vis spectra showed differences in the 200–300 nm peak depending on whether the starting material—PREFER—was defect-healed. This indicates that the numbers of isolated framework Fe(III) ions with tetrahedral coordination and isolated Fe(III) ions with octahedral coordination were reduced22 in the defect-healed samples, which is thought to suppress the reaction of iron atoms outside the interlayer. Thus, the defect-healing treatment on the precursor suppressed the migration of silicate species, and more iron atoms were inserted between the layers of PREFER in homogeneous environment after the defect-healing treatment.
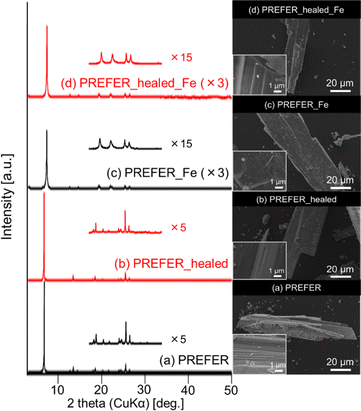 |
| | Fig. 5 XRD patterns (left) and SEM images (right) of samples subjected to defect-healing and crosslinking. | |
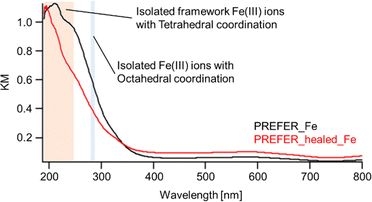 |
| | Fig. 6 UV-Vis spectra of PREFER_Fe and PREFER_healed_Fe. | |
Conclusions
Defect-healing treatment was applied to PREFER—a layered precursor of FER-type zeolite—which was subjected to interlayer crosslinking using iron atoms. The number of intralayer silanol defects was significantly reduced by the treatment, whereas the interlayer silanol groups were maintained. The UV-Vis spectra of the crosslinked samples indicated that the iron atoms were in different states depending on the treatment, and the sample obtained using the defect-healed precursor exhibited enhanced iron atom insertion in more homogeneous environments, in contrast to the untreated precursor. The results indicate the possibility of controlling the atomic environment in layered materials. This study provides a new way to synthesize well-defined crosslinked three-dimensional frameworks.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the Japan Science and Technology Agency (JST) through Precursory Research for Embryonic Science and Technology (PRESTO, grant No. JPMJPR21N3) and the Strategic International Collaborative Research Program (SICORP) EIG CONCERT-Japan (grant No. JPMJSC22C5).
Notes and references
- K. Iyoki, K. Itabashi and T. Okubo, Microporous Mesoporous Mater., 2014, 189, 22–30 CrossRef CAS.
- M. E. Davis, Nature, 2002, 417, 813–821 CrossRef CAS PubMed.
- B. Yilmaz and U. Müller, Top. Catal., 2009, 52, 888–895 CrossRef CAS.
- A. Corma, J. Catal., 2003, 216, 298–312 CrossRef CAS.
- T. Yoshioka, K. Iyoki, Y. Hotta, Y. Kamimura, H. Yamada, Q. Han, T. Kato, C. A. J. Fisher, Z. Liu, R. Ohnishi, Y. Yanaba, K. Ohara, Y. Sasaki, A. Endo, T. Takewaki, T. Sano, T. Okubo and T. Wakihara, Sci. Adv., 2022, 8, 1–14 Search PubMed.
- W. J. Roth and J. Ejka, Catal. Sci. Technol., 2011, 1, 43–53 RSC.
- E. Schulman, W. Wu and D. Liu, Materials, 2020, 13, 1822 CrossRef CAS PubMed.
- Y. Wu, L. Emdadi, Z. Wang, W. Fan and D. Liu, Appl. Catal., A, 2014, 470, 344–354 CrossRef CAS.
- L. Ren, Q. Guo, M. Orazov, D. Xu, D. Politi, P. Kumar, S. M. Alhassan, K. A. Mkhoyan, D. Sidiras, M. E. Davis and M. Tsapatsis, ChemCatChem, 2016, 8, 1274–1278 CrossRef CAS.
- H. W. Lee, S. H. Park, J. K. Jeon, R. Ryoo, W. Kim, D. J. Suh and Y. K. Park, Catal. Today, 2014, 232, 119–126 CrossRef CAS.
- T. Ikeda, Y. Akiyama, Y. Oumi, A. Kawai and F. Mizukami, Angew. Chem., Int. Ed., 2004, 43, 4892–4896 CrossRef CAS PubMed.
- P. Eliášová, M. Opanasenko, P. S. Wheatley, M. Shamzhy, M. Mazur, P. Nachtigall, W. J. Roth, R. E. Morris and J. Čejka, Chem. Soc. Rev., 2015, 44, 7177–7206 RSC.
- J. Ruan, P. Wu, B. Slater, Z. Zhao, L. Wu and O. Terasaki, Chem. Mater., 2009, 21, 2904–2911 CrossRef CAS.
- H. Gies, M. Feyen, T. De Baerdemaeker, D. E. De Vos, B. Yilmaz, U. Müller, X. Meng, F. S. Xiao, W. Zhang, T. Yokoi, T. Tatsumi and X. Bao, Microporous Mesoporous Mater., 2016, 222, 235–240 CrossRef CAS.
- O. Veselý, P. Eliášová, R. E. Morris and J. Čejka, Mater. Adv., 2021, 2, 3862–3870 RSC.
- K. Iyoki, K. Kikumasa, T. Onishi, Y. Yonezawa, A. Chokkalingam, Y. Yanaba, T. Matsumoto, R. Osuga, S. P. Elangovan, J. N. Kondo, A. Endo, T. Okubo and T. Wakihara, J. Am. Chem. Soc., 2020, 142, 3931–3938 CrossRef CAS PubMed.
- L. Schreyeck, P. Caullet, J. C. Mougenel, J. L. Guth and B. Marler, Microporous Mater., 1996, 6, 259–271 CrossRef CAS.
- B. Marler and M. Müller, Microporous Mesoporous Mater., 2019, 288, 109597 CrossRef CAS.
- Y. Asakura, Y. Sakamoto and K. Kuroda, Chem. Mater., 2014, 26, 3796–3803 CrossRef CAS.
- I. C. Medeiros-Costa, E. Dib, N. Nesterenko, J. P. Dath, J. P. Gilson and S. Mintova, Chem. Soc. Rev., 2021, 50, 11156–11179 RSC.
- A. Erigoni, S. H. Newland, G. Paul, L. Marchese, R. Raja and E. Gianotti, ChemCatChem, 2016, 8, 3161–3169 CrossRef CAS.
- J. Zhang, X. Tang, H. Yi, Q. Yu, Y. Zhang, J. Wei and Y. Yuan, Appl. Catal., A, 2022, 630, 118467 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ad,
Tatsuya Okubo
ad,
Tatsuya Okubo