
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceViral peptide conjugates for metal-warhead delivery to chromatin†
Lucinda K. Batchelor‡
a,
Louis De Falco‡b,
Paul J. Dyson *a and
Curtis A. Davey*b
*a and
Curtis A. Davey*b
aInstitut des Sciences et Ingénierie Chimiques, École Polytechnique Fédérale de Lausanne (EPFL), 1015 Lausanne, Switzerland. E-mail: paul.dyson@epfl.ch
bSchool of Biological Sciences & NTU Institute of Structural Biology, Nanyang Technological University (NTU), 637551, Singapore. E-mail: cadavey@protonmail.com
First published on 14th March 2024
Abstract
The presence of heavy metal groups can endow compounds with unique structural and chemical attributes beneficial for developing highly potent therapeutic agents and effective molecular labels. However, metallocompound binding site specificity is a major challenge that dictates the level of off-site targeting, which is a limiting factor in finding safer and more effective metal-based drugs. Here we designed and tested a family of metallopeptide conjugates based on two different chromatin-tethering viral proteins and a drug being repurposed for cancer, the Au(I) anti-arthritic auranofin. The viral peptides associate with the acidic patch of the nucleosome while the gold moiety can bind allosterically to the H3 H113 imidazole. To achieve synthesis of the conjugates, we also engineered a sulfur-free, nucleosome-binding Kaposi's sarcoma herpesvirus LANA peptide with a methionine-to-ornithine substitution and coupled the peptide to the metal group in a final step using click chemistry. The four conjugates tested are all selectively cytotoxic towards tumor cell lines, but the choice of viral peptide and mode of linkage to the Au(I) group influences metal binding site preference. Our findings suggest that viral peptide–metalloconjugates have potential for use in chromatin delivery of therapeutic warheads and as nucleosome-specific tags.
Introduction
Given their ability to form covalent bonds with target biomolecules, metal-based compounds have a proclivity for high potency as cancer drugs. Nonetheless, by virtue of promoting toxicity or other side effects, off-site targeting can be a major disadvantage that limits safety and efficacy. Different methods have been employed to alter or enhance molecular site discrimination that include the attachment of a protein or peptide to the metalloagent.1 This strategy can be used to improve cell uptake, bioavailability, and/or site selectivity, depending on the nature of the peptide.2 Here, we focused on conjugates comprising nucleosome-binding viral peptides, which are based on the chromatin-tethering proteins that certain viruses express in order to maintain their genomes in, or deliver their genomes to, the cell nuclei.3As the instructional and regulatory foundation of the cell, chromatin presides over an extraordinary variety of DNA and epigenetic therapeutic targets. Many chromatin sites are targets of metal-based drugs, notably anticancer agents like cisplatin and oxaliplatin.4 Nonetheless, beyond a preference for solvent-accessible N7 sites of guanine, these platinum drugs bind otherwise indiscriminately to the DNA.5 Much recent work has focused on finding more site selective metallocompounds, and we earlier discovered that RAPTA [(arene)Ru(1,3,5-triaza-7-phosphaadamantane)Cl2] anti-metastasis/tumor drugs6,7 and RAPTA-like agents have a preference for binding to defined histone protein sites, as opposed to the DNA, of chromatin.8–10
RAPTA compounds bind to an epitomal regulatory site of chromatin, the “acidic patch” of the nucleosome8–10—a cleft region on the surface of the H2A–H2B dimer having an abundance of aspartate and glutamate residues.11 We subsequently discovered that RAPTA-T (T = toluene) binding at the acidic patch promotes reaction of auranofin [(3,4,5-triacetyloxy-6-acteyloxymethyl,oxane-2-thio-late)Au(triethyl-phosphanium)], through an allosteric mechanism, at two 36+ Å-distant symmetry-related imidazole sites, H3 H113 (AU1) and H3′ H113 (AU1′), on the H3–H4 tetramer.12 Auranofin is an antiarthritic drug that is being repurposed for the treatment of cancer and other diseases,13 and we found that, in combination with RAPTA-T, it also has a synergistic effect in killing tumor cells.12 More recently, we demonstrated that conjugating a RAPTA group to an auranofin-like group, ([Au(4-diphenylphosphanyl-benzoic acid)X], AuDPPBX; where X = Cl), via a long polyethylene glycol (PEG) linker yields a potent heterobimetallic compound that crosslinks the H2A–H2B dimer with the H3–H4 tetramer in chromatin.14 In the current study, we aimed to develop a system for highly specific targeting of nucleosomes with a non-covalent binding module that could be used to deliver a reactive metal group to chromatin.
Results and discussion
Synthesis of Au(I)–peptide conjugates
Four Au(I)–peptide conjugates were generated by connecting the nucleosome-binding region of either the Kaposi's sarcoma-associated herpesvirus LANA protein15 or foamy virus GAG protein16 to a AuDPPBX group via a long PEG linker (Fig. 1). Each of two configurations were synthesized, wherein the Au–PEG linker group was attached to the peptide N-terminus (Au–LANA and Au–GAG) versus C-terminus (LANA–Au and GAG–Au). In order to avoid potential interactions with the Au(I) cation, the Met residue in the wild type LANA peptide was replaced by ornithine (Orn; Fig. 1 and below). To produce the conjugates, the first synthetic route tested entailed typical amide coupling approaches to connect the Au-PEG group to the peptide, but this resulted in extensive product degradation (Scheme S1†).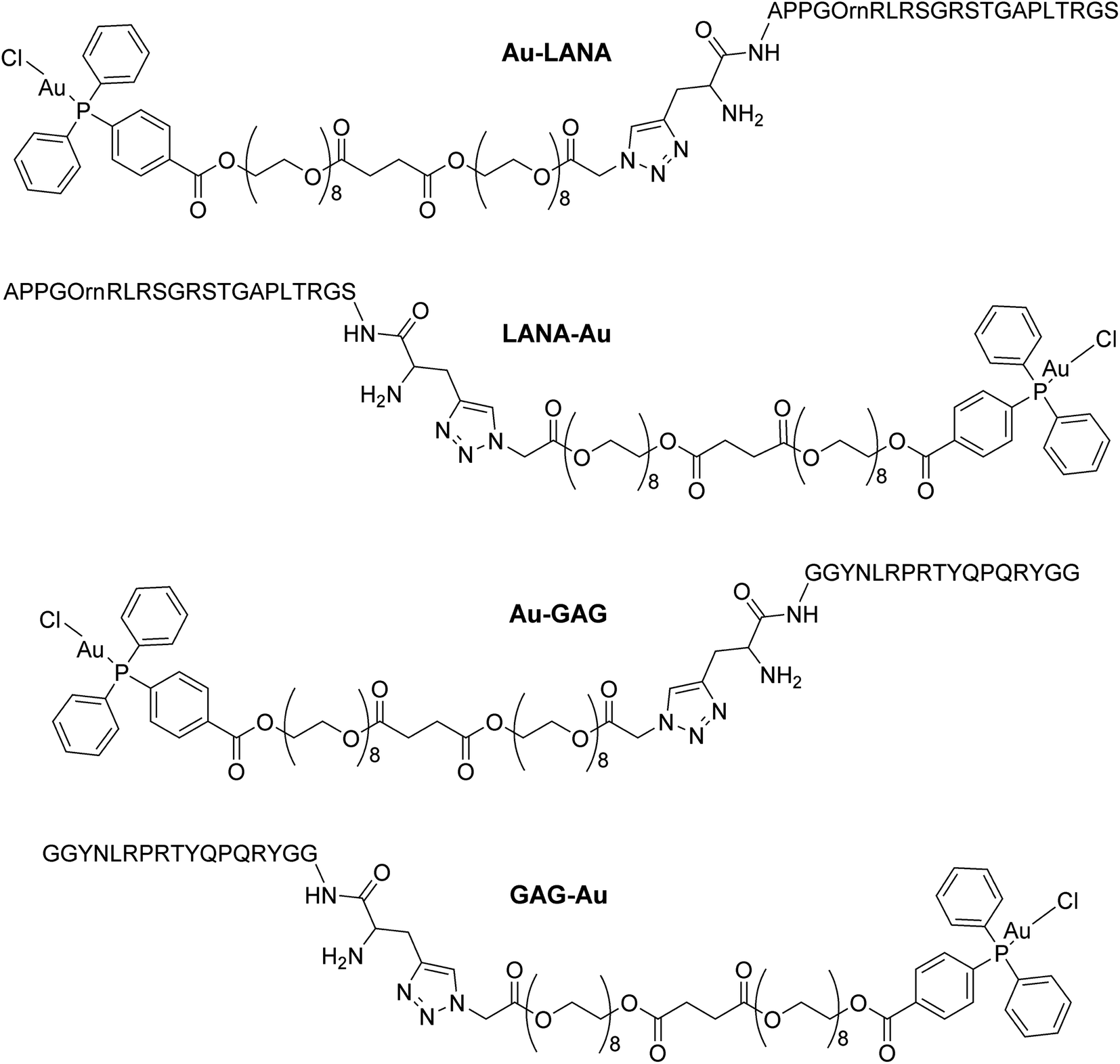 | ||
| Fig. 1 Structures of the four Au(I)–viral peptide conjugates. | ||
In order to circumvent the problematic deprotection step involving the Au(I) center, a synthetic route was devised in which the complex and the deprotected peptide were conjugated using copper click chemistry.17 This route involves the introduction of a propargylglycine residue to the peptide and a terminal azide to the complex. The Au(I) complex, containing a long polyethylene glycol linker and a terminal azide, was prepared in five steps (Scheme 1; ESI Experimental details†). The phosphorus ligand, 4-(diphenylphosphino)benzoic acid, was coupled to octaethylene glycol via an esterification reaction with N-ethyl-N′-(3-dimethlaminopropyl)carbodiimide hydrochloride (EDCI) and 4-(dimethylamino)pyridine (DMAP) acting as coupling reagent and base, respectively. The equivalents of reagents were adapted, 1 equiv. 4-(diphenylphosphino)benzoic acid versus 1.5 equiv. octaethylene glycol, to promote the formation of the mono-phosphine product, 1. A carboxylic acid, necessary for the addition of a second octaethylene glycol to the linker, was introduced to the ligand using succinic anhydride under basic, triethylamine conditions to afford 2. The second octaethylene glycol unit was introduced under the same EDCI/DMAP coupling conditions to yield 3, substantially increasing the length of the linker. The phosphorus ligand was coordinated to the gold centre via a freshly prepared Au(I) intermediate, AuCl(tht) (tht = tetrahydrothiophene). Finally, the terminal alkyne was introduced via an esterification reaction between 4 and 2-azidoacetic acid under EDCI/DMAP conditions in N,N-dimethylformamide (DMF) to yield 5. Each product was purified via flash column chromatography conducted in the normal phase using CH2Cl2/CH3OH as eluent and isolated as viscous oils.
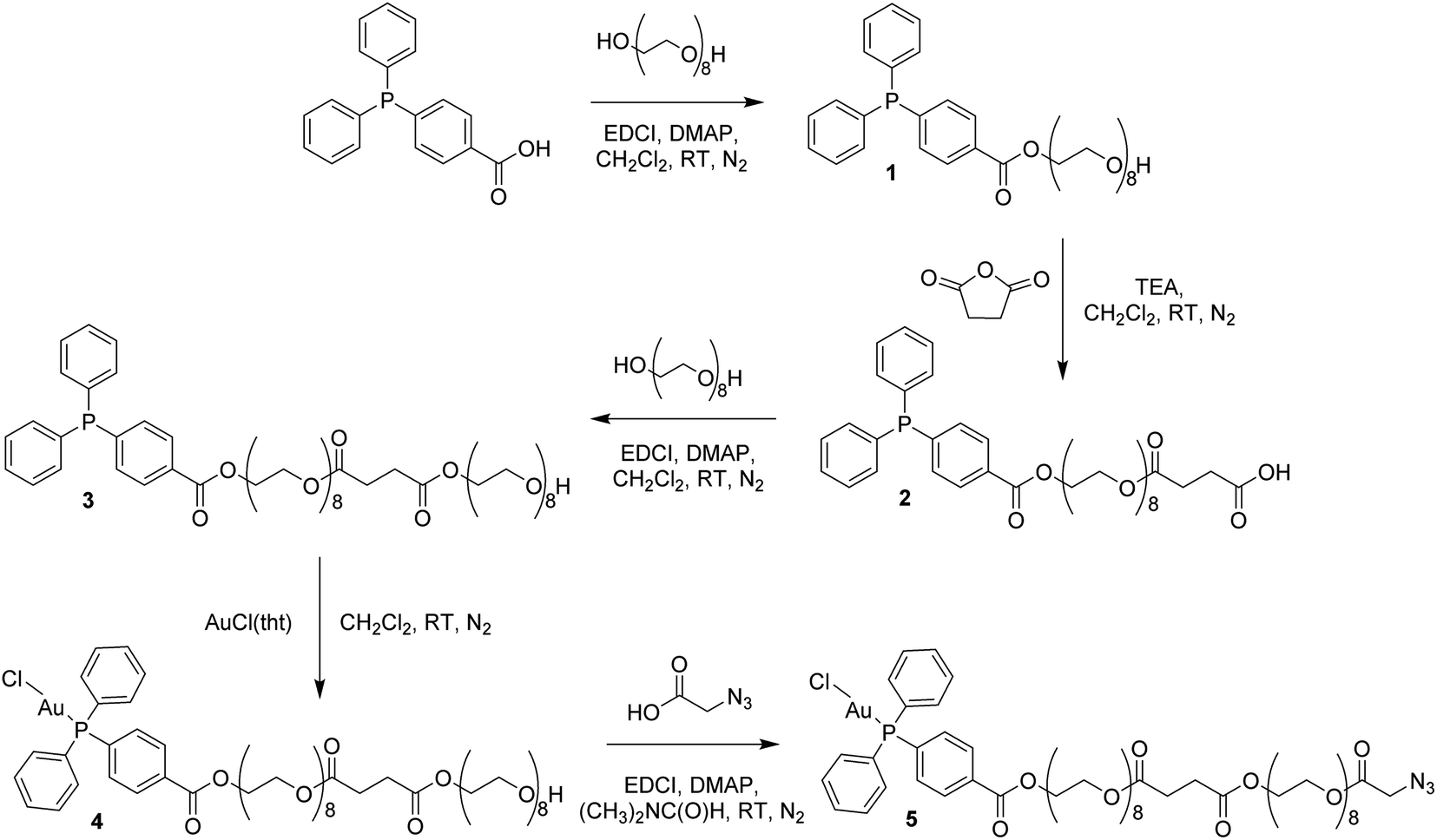 | ||
| Scheme 1 Synthesis of the Au(I) complex containing a terminal azide, 5. | ||
Compounds 1–5 were fully characterized using 1H, 31P and 13C NMR spectroscopy (Fig. S1–S5†), mass spectrometry and elemental analysis. The complexation of the phosphorus ligand to the Au(I) centre was confirmed via a downfield shift in the 31P NMR spectra from −5.07 ppm for 3 to 32.97 ppm for 4. The successful synthesis of 5 was confirmed by NMR spectroscopy, where a characteristic multiplet representing the CH2–O–(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–CH2–N3 protons is observed at 4.31–4.33 ppm in the 1H NMR spectrum. The phosphorus peak is almost unchanged at 33.00 ppm in the 31P NMR spectrum and the [M + Na]+ ion was observed at m/z 1448.4567 in the electrospray ionization (ESI) mass spectrum.
O)–CH2–N3 protons is observed at 4.31–4.33 ppm in the 1H NMR spectrum. The phosphorus peak is almost unchanged at 33.00 ppm in the 31P NMR spectrum and the [M + Na]+ ion was observed at m/z 1448.4567 in the electrospray ionization (ESI) mass spectrum.
The LANA (L1 and L2) and GAG (L3 and L4) peptides, modified at either the N-terminus (L1, L3) or the C-terminus (L2, L4) with a propargylglycine (Fig. S6†), were synthesized with an automatic peptide synthesizer using standard Fmoc solid phase chemistry on Rink Amide MBHA resin (25 μmol scale, 0.3 mmol per g loading). The amide coupling reactions were performed twice for each amino acid using 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b] pyridinium-3-oxide hexafluorophosphate (HATU) and N,N-diisopropylethylamine (DIPEA). The Fmoc protecting groups were removed with pipiridine (20% (v/v)) in DMF and the peptides were capped using acetic anhydride (5% (v/v)) and lutidine (6% (v/v)) in DMF. The peptides were deprotected and cleaved from the resin via incubation with 90% trifluoroacetic acid (TFA), 2.5% H2O, 2.5% thioanisole, 2.5% phenol and 2.5% ethanedithiol (EDT), and the peptides were purified via RP-HPLC. The successful synthesis of the peptides, L1–L4, was confirmed via high-resolution ESI(+) spectrometry, with the [M + 2H]2+ ion observed at m/z 1101.6240 (L1), 1101.6228 (L2), 1039.0308 (L3) and 1039.0299 (L4).
The click reactions between 5 and each of the peptides (L1–L4) were conducted in tert-butyl alcohol and milliQ water in the presence of 1 mol% Cu(II) sulphate and 10 mol% sodium ascorbate in millQ water (Scheme 2).18 The resulting conjugates were purified via RP-HPLC and target fractions were lyophilised. The successful synthesis of the conjugates was confirmed via high resolution mass spectrometry, where the [M–Cl + 3H]4+ ion was observed for each conjugate at m/z 898.9393 (Au–LANA, derived from L1), 898.9382 (LANA–Au, derived from L2), 867.6437 (Au–GAG, derived from L3) and 867.6442 (GAG–Au, derived from L4). A shift in the 31P NMR signals was observed from 33.00 ppm for the free complex to 42.31–42.67 ppm for the conjugates, indicating that the environment of the phosphorus is influenced by the introduction of the peptide. The purity of the conjugates was determined by analytical HPLC (Fig. S7†). The stability of the conjugates was assessed in 100 mM NaCl in D2O for 72 h, monitored via 31P NMR spectroscopy and ESI mass spectrometry. The phosphorus peak in the 31P NMR spectra remained essentially unchanged (Fig. S8–S11†), and the [M–Cl + 3H]4+ peak was observed with no discernible degradation throughout the 72 h incubation. Thus, the Au(I) peptides show good stability with no degradation observed.
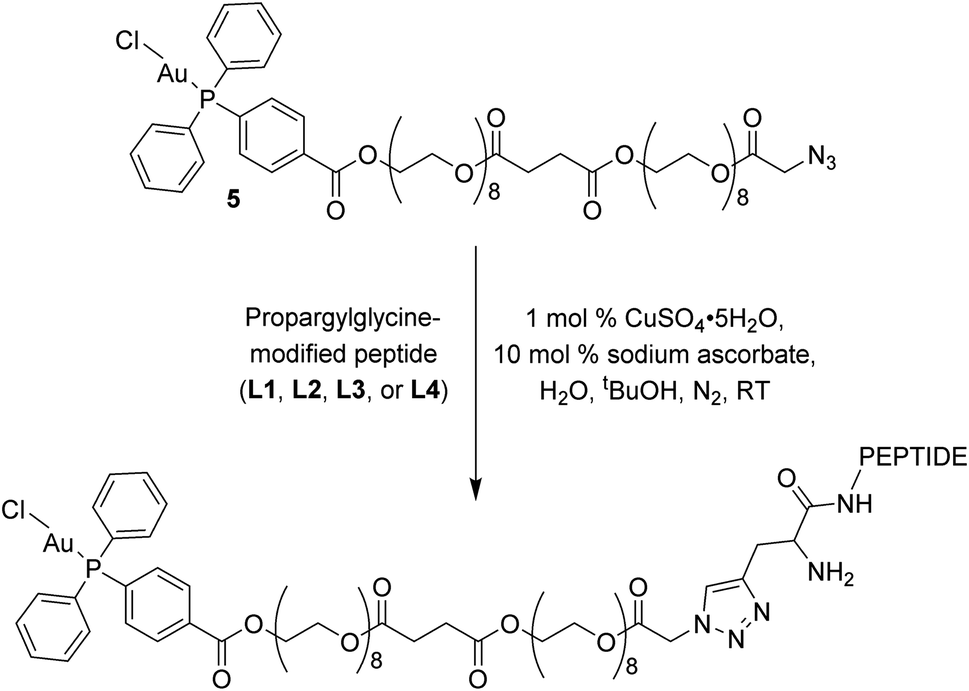 | ||
| Scheme 2 Copper click reaction used to form the final Au–peptide conjugates: Au–LANA (derived from L1), LANA–Au (derived from L2), Au–GAG (derived from L3), and GAG–Au (derived from L4). | ||
Cytotoxicity of Au(I)–peptide conjugates
The cytotoxicity of the Au–peptide conjugates and the free peptide ligands was assessed against cisplatin-sensitive and cisplatin-resistant human ovarian carcinoma (A2780 and A2780cisR) and non-tumoral human embryonic kidney (HEK293) cell lines (Table 1). The Au(I) conjugate group in isolation, AuDPPBX (where X = [3,4,5-triacetyloxy-6-acetyloxymethyl,oxane-2-thiolate]), was also evaluated as a reference alongside auranofin, cisplatin, and RAPTA-C (C = p-cymene) as controls. The free ligands L1–L4 were inactive against all cell lines, with IC50 values >200 μM. Although several-fold less cytotoxic than AuDPPBX, conjugating the Au(I) complex with the peptides yields moderate cytotoxicity in the 35–53 μM range against both cancer cell lines, A2780 and A2780cisR. Notably, the potency of the Au–peptide conjugates against the sensitive versus resistant cell lines is overall nearly equivalent. This is in contrast to the other Au(I) compounds tested, AuDPPBX and auranofin, which both show significant cross-resistance with cisplatin. On the other hand, while AuDPPBX, auranofin, and cisplatin are (equally or) more cytotoxic to the model healthy (HEK293) cell line compared to the A2780cisR tumor cell line, the Au–peptide conjugates are all significantly less cytotoxic in this regard. Any differences observed between the Au–peptides conjugates are modest, but include a slightly higher potency for Au–GAG relative to GAG–Au against the tumoral cell lines. Also, the two GAG-based conjugates are slightly more cytotoxic towards the A2780cisR cell line compared to A2780 cells, whereas the opposite relationship is seen for the LANA-based conjugates.| Compound | A2780 | A2780cisR | HEK293 |
|---|---|---|---|
| a Values are given as the mean ± SD (μM). | |||
| Au–LANA | 35 ± 3 | 42 ± 3 | >100 |
| LANA–Au | 36 ± 3 | 46 ± 3 | >100 |
| Au–GAG | 40 ± 4 | 36 ± 2 | >100 |
| GAG–Au | 53 ± 6 | 50 ± 5 | >100 |
| L1 | >200 | >200 | >200 |
| L2 | >200 | >200 | >200 |
| L3 | >200 | >200 | >200 |
| L4 | >200 | >200 | >200 |
| AuDPPBX | 6.9 ± 0.8 | 12.0 ± 2 | 11.7 ± 0.4 |
| Auranofin | 1.1 ± 0.1 | 2.6 ± 0.3 | 1.7 ± 0.8 |
| Cisplatin | 1.6 ± 0.9 | 17 ± 1.8 | 8 ± 1 |
| RAPTA-C | >200 | >200 | >200 |
Nucleosome binding of Au(I)–peptide conjugates
To confirm compound binding to and reactivity with chromatin, many trials were conducted by incubating nucleosome core particle (NCP) crystals with each of the conjugates at different concentrations and for different durations, followed by analysis with X-ray crystallography (Fig. 2, S12–S14 and Tables S1–S3†). The LANA and GAG viral peptide elements of the four conjugates associate with the nucleosome acidic patch as previously characterized (PDB accession code, 5MLU).15 Since a Met-to-Orn substitution was introduced into Au–LANA and LANA–Au, we also confirmed the ability of the modified LANA peptide in isolation to bind NCP. The Met side chain of the wide type LANA peptide binds in a cavity formed by hydrophobic elements as well as two glutamate side chains.15 In its place, the Orn side chain (having nearly the same length as the Met side chain) of bound Au–LANA and LANA–Au is sandwiched between two carboxylate groups, resulting in ion pairing with the ammonium group (Fig. 2c). Alongside association of the peptide, a substantial degree of coordination of the AuDPPB group at the AU1 site is apparent for Au–LANA, which also binds to a lesser extent at the symmetry-related AU1′ (Fig. 2a and Table S3†). In addition, there is evidence of non-covalently associated AuDPPBX in the form of residual (apparently depot) binding. For the LANA–Au X-ray data, there is no evidence of AuDPPB group coordination, but this may be at least partly attributable to a more pronounced disordering effect on the crystals for this particular conjugate.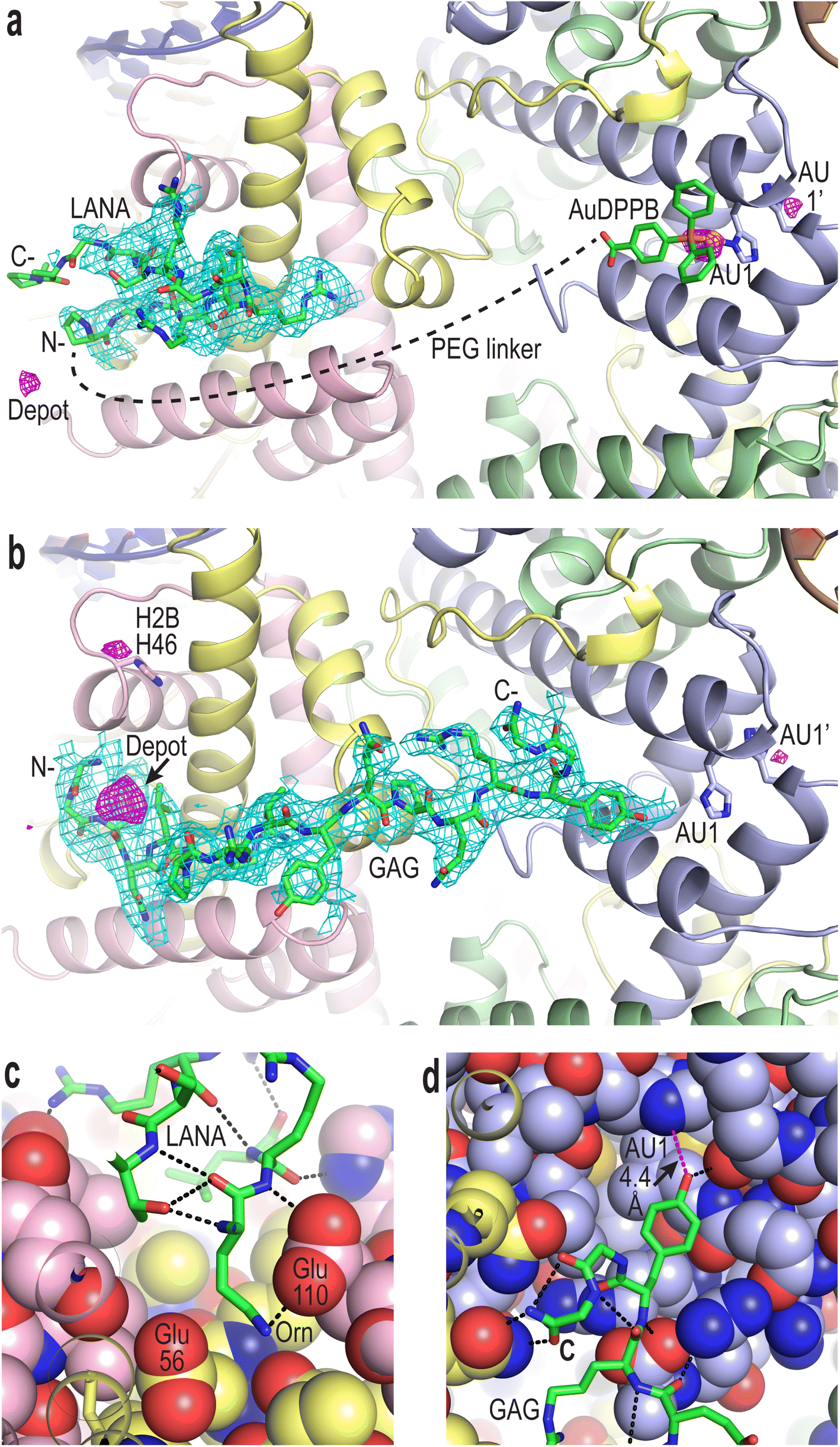 | ||
| Fig. 2 Viral peptide binding and AuDPPBX coordination/localization in the nucleosome. (a and b) X-ray crystal structure of NCP treated with Au–LANA ((a); 2 N- and 5 C-terminal peptide residues not shown/visible in electron density maps) and Au–GAG (b). A FO − FC omit electron density map (cyan; contoured at 2σ; prior to inclusion of viral peptide) and an anomalous difference electron density map (magenta; contoured at 3.5σ; indicates locations of gold atoms) are shown superimposed on the model (N- and C-, peptide N- and C-termini). (c and d) Close view of the LANA (c) and GAG (d) conjugate peptides binding in the NCP crystal structures (H-bonds, dashed black lines; close contact, dashed magenta line; histone protein atoms in space filling representation). (a–d) Histone proteins are shown in purple (H3), pale green (H4), yellow (H2A) and pink (H2B), and DNA strands (a and b) are orange and dark blue. | ||
In the case of the two GAG conjugates, pronounced localization of the AuDPPBX group is mostly evident as depot binding in the vicinity of the N- or C-terminal points of peptide attachment for Au–GAG and GAG–Au, respectively (Fig. 2b and Table S3†). This depot association is likely fostered by hydrophobic interactions with GAG residues. Evidence for gold(I)–histone binding is otherwise limited to low levels of adduction at the AU1′ site on the opposing nucleosome face and an imidazole site (H2B H46) proximal to the peptide N-terminus, for Au–GAG, as well as a weaker gold atom signal for GAG–Au binding to AU1. The substantial depot binding and lack of pronounced adduct formation at AU1 for the GAG conjugates, contrasting what is seen for Au–LANA, is likely attributable to a steric obstacle posed by the C-terminal Tyr residue of GAG, which binds in close proximity to AU1 (Fig. 2d).
Conclusions
Here we presented a design principle and synthetic scheme for producing biologically active viral peptide metalloconjugates. The four Au(I)–peptide compounds explored are selectively cytotoxic to cancer cells, including cells with acquired resistance to cisplatin, in contrast to auranofin or the isolated AuDPPBX complex. Although the conjugates display relatively modest cytotoxicity against the tested cancer cell types, some distinctions are evident, and these may be linked to the differences in site selectivity/reactivity and impact on the NCP crystals observed between the four agents. In particular, the overall cancer cell cytotoxicity and extent of adduct formation at AU1/AU1′ is highest for Au–LANA, but the latter is less than that observed for AuDPPBX or auranofin that is allosterically elicited via RAPTA binding in the acidic patch.12,14 For the GAG agents, this can be rationalized by limited steric access due to the bound peptide, but it also suggests that binding of the viral peptides does not bring about the same extent of allosteric activation. Nonetheless, we also observe gold adducts at H2B H46, and considering that the chromatin fiber in vivo would present a variety of nucleosomal structural contexts,19 alternative imidazole sites beyond H3 H113 (AU1/1′) may also be relevant given the cellular activities observed here.This metallopeptide conjugate system has potential for further development towards therapeutic and research tool applications. For instance, alternative metal groups with distinct site selectivity attributes could be explored, in addition to viral peptide modifications that modulate cellular uptake, localization, or stability/lability characteristics. Indeed, the distinctions in adduct formation potential and site discrimination on the nucleosome observed here that result from changes to the viral peptide sequence or disposition of coupling to the metal group may assist further conjugate design efforts; in particular, by providing insight into target site accessibility, proximity, and allosteric impact relationships. Moreover, although we did not observe substantial differences in the impact of the conjugates on cancer cells, further studies could illuminate distinctions with respect to other attributes beyond cytotoxicity.
Experimental section
Materials for synthesis
Chemical reagents were purchased from commercial sources (Sigma Aldrich, ABCR, Acros, and TCI Chemicals) and used without further purification. The reactions were performed under an inert atmosphere (N2) using Schlenk techniques. Dry solvents, dried using a PureSolv solvent purification system (Innovative Technology Inc.), were collected and used under an inert atmosphere (N2). Thin Layer Chromatography was conducted on Merck TLC silica gel coated aluminium sheets 60 F254 and verified by UV lamb at 254 nm. Purifications were achieved by column flash chromatography using a CombiFlash Rf+ automated column machine operated with prepacked Luknova columns.Instrumentation for synthetic procedures
1H (400 MHz), 31P (162 MHz) and 13C (101 MHz) NMR spectra were recorded on a Bruker Avance II 400 spectrometer at 298 K. Chemical shifts are reported in parts per million (ppm) and referenced to deuterated solvent residual peaks (CDCl3: 1H δ 7.26, 13C{1H} δ 77.16 ppm) and coupling constants (J) are reported in Hertz (Hz). High resolution electrospray ionization mass spectra (HR ESI-MS) were obtained on a Xevo G2-S QTOF mass spectrometer coupled to the Acquity UPLC Class Binary Solvent manager and BTN sample manager (Waters, Corporation, Milford, MA). Elemental analyses were determined on a Thermo Scientific Flash 2000 Organic Elemental Analyzer.Compound synthesis
Details for the synthesis and characterization of the Au(I)–peptide conjugates can be found in the ESI.†Cell culture
Human ovarian carcinoma (A2780 and A2780cisR) cell lines were obtained from the European Collection of Cell Cultures. The human embryonic kidney (HEK293) cell line was obtained from ATCC (Sigma, Buchs, Switzerland). Penicillin streptomycin, RPMI 1640 GlutaMAX (where RPMI = Roswell Park Memorial Institute), and DMEM GlutaMAX media (where DMEM = Dulbecco's modified Eagle medium) were obtained from Life Technologies, and fetal bovine serum (FBS) was obtained from Sigma. The cells were cultured in RPMI 1640 GlutaMAX (A2780 and A2780cisR) and DMEM GlutaMAX (HEK-293) media containing 10% heat-inactivated FBS and 1% penicillin streptomycin at 37 °C and CO2 (5%). The A2780cisR cell line was routinely treated with cisplatin (2 μM) in the media to maintain cisplatin resistance.Cell antiproliferation activity assays
Cytotoxicity was determined using the 3-(4,5-dimethyl 2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay.20 Cells were seeded in flat-bottomed 96-well plates as a suspension in a prepared medium (100 μL aliquots and approximately 4300 cells per well) and preincubated for 24 h. Stock solutions of compounds were prepared in MilliQ water and sequentially diluted in the appropriate medium to give a final compound concentration range (0–200 μM). Cisplatin and RAPTA-C were tested as positive (0–100 μM) and negative (>200 μM) controls, respectively. The compounds were added to the preincubated 96-well plates in 100 μL aliquots, and the plates were incubated for a further 72 h. MTT (20 μL, 5 mg mL−1 in Dulbecco's phosphate buffered saline) was added to the cells, and the plates were incubated for a further 4 h. The culture medium was aspirated, and the purple formazan crystals, formed by the mitochondrial dehydrogenase activity of vital cells, were dissolved in DMSO (100 μL per well). The absorbance of the resulting solutions, directly proportional to the number of surviving cells, was quantified at 590 nm using a SpectroMax M5e multimode microplate reader (using SoftMax Pro software, version 6.2.2). The percentage of surviving cells was calculated from the absorbance of wells corresponding to the untreated control cells. The reported IC50 values are based on the means from two independent experiments, each comprising four tests per concentration level.Crystallographic analysis of treated nucleosome
X-ray crystallographic analysis was conducted using NCP assembled with recombinant Homo sapiens histones and a 145 bp DNA fragment.10,21 The hanging droplet method was used to grow NCP crystals from buffers containing MnCl2, KCl and K-cacodylate [pH 6.0].22 Crystals were harvested and transferred into a stabilization buffer (37 mM MnCl2, 40 mM KCl, 20 mM K-cacodylate [pH 6.0], 24% 2-methyl-2,4-pentanediol and 2% trehalose). MgSO4 was substituted in place of MnCl2 by thorough rinsing of crystals with a magnesium buffer (10 mM MgSO4, 20 mM K-cacodylate [pH 6.0], 24% 2-methyl-2,4-pentanediol and 2% trehalose).8To obtain structural data on adduct formation of the Au–peptide conjugates in the nucleosome core, native NCP crystals were subjected to incubation with magnesium buffer containing 0.5–2 mM of the conjugates. Treated crystals were mounted directly into a cryocooling N2 gas stream set at −175 °C.9 X-ray diffraction data were recorded at beam line X06DA of the Swiss Light Source (Paul Scherrer Institute, Villigen, Switzerland) with a Pilatus 2M-F detector at a wavelength corresponding to the X-ray absorption edge of gold (1.04 Å). Diffraction data were indexed, integrated, merged, scaled and evaluated with a combination of iMosflm,23,24 XDS,25 autoPROC,26 SCALA,27 and AIMLESS28 from the CCP4 package29,30 and in-house data processing pipelines, go.com and go.py, developed by the Swiss Light Source macromolecular crystallography beamlines (Paul Scherrer Institute, Villigen, Switzerland).
NCP-conjugate structures were solved by molecular replacement (PHASER31 and MOLREP32 from the CCP4 package29,30) using the 1.99 Å resolution crystal structure of native NCP, assembled with the same histones and 145 bp DNA fragment (pdb code 6IPU),33 as the reference model. COOT34 and REFMAC35 from the CCP4 suite29,30 were used to conduct model building and structural refinement. Small molecule crystal structures of [Au(2-isopropylimidazole)(Pcyclohexyl3)]36 and [Au(4-(diphenylphosphanyl)-benzoic acid)Cl]37 were used to compose stereochemical restraint parameters for the AuDPPB–histidine adduct.
Data collection and structural refinement statistics are given in Tables S1 and S2,† and gold cation binding evaluation based on anomalous difference electron density maps is shown in Table S3.† Molecular graphics images were produced with PyMOL (DeLano Scientific LLC, San Carlos, CA, USA). Atomic coordinates and structure factors for the LANA, Au–Lana, GAG, and Au–GAG models have been deposited in the Protein Data Bank under accession codes 8Q3M, 8Q3X, 8Q3E, and 8Q36.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Swiss National Science Foundation, Singapore Ministry of Education Academic Research Fund Tier 1 Program (2021-T1-001-014, 2017-T1-002-020, and 2020-T1-001-128), and Singapore Ministry of Education Academic Research Fund Tier 2 Program (MOE-T2EP30121-0005 and MOE2015-T2-2-089). We thank M. Wang, V. Olieric and staff at the Swiss Light Source (Paul Scherrer Institute, Villigen, Switzerland).References
- B. Albada and N. Metzler-Nolte, Organometallic–Peptide Bioconjugates: Synthetic Strategies and Medicinal Applications, Chem. Rev., 2016, 116, 11797–11839 CrossRef CAS PubMed.
- M. Soler, L. Feliu, M. Planas, X. Ribas and M. Costas, Peptide-mediated vectorization of metal complexes: conjugation strategies and biomedical applications, Dalton Trans., 2016, 45, 12970–12982 RSC.
- I. Aydin and M. Schelhaas, Viral Genome Tethering to Host Cell Chromatin: Cause and Consequences, Traffic, 2016, 17, 327–340 CrossRef CAS.
- G. Palermo, A. Magistrato, T. Riedel, T. von Erlach, C. A. Davey, P. J. Dyson and U. Rothlisberger, Fighting Cancer with Transition Metal Complexes: From Naked DNA to Protein and Chromatin Targeting Strategies, ChemMedChem, 2016, 11, 1199–1210 CrossRef CAS PubMed.
- B. Wu, G. E. Davey, A. Nazarov, P. J. Dyson and C. A. Davey, Specific DNA structural attributes modulate platinum anticancer drug site selection and cross-link generation, Nucleic Acids Res., 2011, 39, 8200–8212 CrossRef CAS.
- A. Bergamo, A. Masi, P. J. Dyson and G. Sava, Modulation of the metastatic progression of breast cancer with an organometallic ruthenium compound, Int. J. Oncol., 2008, 33, 1281–1289 CAS.
- C. Scolaro, A. Bergamo, L. Brescacin, R. Delfino, M. Cocchietto, G. Laurenczy, T. J. Geldbach, G. Sava and P. J. Dyson, In vitro and in vivo evaluation of ruthenium(II)-arene PTA complexes, J. Med. Chem., 2005, 48, 4161–4171 CrossRef CAS PubMed.
- B. Wu, M. S. Ong, M. Groessl, Z. Adhireksan, C. G. Hartinger, P. J. Dyson and C. A. Davey, A ruthenium antimetastasis agent forms specific histone protein adducts in the nucleosome core, Chem.–Eur. J., 2011, 17, 3562–3566 CrossRef CAS PubMed.
- Z. Adhireksan, G. E. Davey, P. Campomanes, M. Groessl, C. M. Clavel, H. Yu, A. A. Nazarov, C. H. F. Yeo, W. H. Ang, P. Dröge, U. Rothlisberger, P. J. Dyson and C. A. Davey, Ligand substitutions between ruthenium-cymene compounds can control protein versus DNA targeting and anticancer activity, Nat. Commun., 2014, 5, 3462 CrossRef PubMed.
- G. E. Davey, Z. Adhireksan, Z. Ma, T. Riedel, D. Sharma, S. Padavattan, D. Rhodes, A. Ludwig, S. Sandin, B. S. Murray, P. J. Dyson and C. A. Davey, Nucleosome acidic patch-targeting binuclear ruthenium compounds induce aberrant chromatin condensation, Nat. Commun., 2017, 8, 1575 CrossRef PubMed.
- R. K. McGinty and S. Tan, Principles of nucleosome recognition by chromatin factors and enzymes, Curr. Opin. Struct. Biol., 2021, 71, 16–26 CrossRef CAS PubMed.
- Z. Adhireksan, G. Palermo, T. Riedel, Z. Ma, R. Muhammad, U. Rothlisberger, P. J. Dyson and C. A. Davey, Allosteric cross-talk in chromatin can mediate drug-drug synergy, Nat. Commun., 2017, 8, 14860 CrossRef CAS.
- C. Order and M. J. Thomson, Auranofin: repurposing an old drug for a golden new age, Drugs R&D, 2015, 15, 13–20 CrossRef PubMed.
- L. K. Batchelor, L. De Falco, T. von Erlach, D. Sharma, Z. Adhireksan, U. Roethlisberger, C. A. Davey and P. J. Dyson, Crosslinking allosteric sites on the nucleosome, Angew Chem. Int. Ed. Engl., 2019, 58, 15660–15664 CrossRef CAS PubMed.
- A. J. Barbera, J. V. Chodaparambil, B. Kelley-Clarke, V. Joukov, J. C. Walter, K. Luger and K. M. Kaye, The nucleosomal surface as a docking station for Kaposi's sarcoma herpesvirus LANA, Science, 2006, 311, 856–861 CrossRef CAS PubMed.
- J. Tobaly-Tapiero, P. Bittoun, J. Lehmann-Che, O. Delelis, M. L. Giron, H. de Thé and A. Saïb, Chromatin tethering of incoming foamy virus by the structural Gag protein, Traffic, 2008, 9, 1717–1727 CrossRef CAS PubMed.
- L. Liang and D. Astruc, The copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC) “click” reaction and its applications. An overview, Coord. Chem. Rev., 2011, 255, 2933–2945 CrossRef CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkyne, Angew. Chem., 2002, 114, 2708–2711 CrossRef.
- Z. Adhireksan, D. Sharma, P. L. Lee and C. A. Davey, Near-atomic resolution structures of interdigitated nucleosome fibres, Nat. Commun., 2020, 11, 4747 CrossRef CAS PubMed.
- T. Mosmann, Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- M. S. Ong, T. J. Richmond and C. A. Davey, DNA Stretching and Extreme Kinking in the Nucleosome Core, J. Mol. Biol., 2007, 368, 1067–1074 CrossRef CAS PubMed.
- C. A. Davey, D. F. Sargent, K. Luger, A. W. Maeder and T. J. Richmond, Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 Å resolution, J. Mol. Biol., 2002, 319, 1097–1113 CrossRef CAS PubMed.
- A. G. W. Leslie, The integration of macromolecular diffraction data, Acta Crystallogr., Sect. D, 2006, 62, 48–57 CrossRef.
- H. R. Powell, T. G. G. Battye, L. Kontogiannis, O. Johnson and A. G. W. Leslie, Integrating macromolecular X-ray diffraction data with the graphical user interface iMosflm, Nat. Protoc., 2017, 12, 1310–1325 CrossRef CAS.
- W. Kabsch, XDS, Acta Crystallogr., Sect. D, 2010, 66, 125–132 CrossRef CAS PubMed.
- C. Vonrhein, C. Flensburg, P. Keller, A. Sharff, O. Smart, W. Paciorek, T. Womack and G. Bricogne, Data processing and analysis with the autoPROC toolbox, Acta Crystallogr., Sect. D, 2011, 67, 293–302 CrossRef CAS PubMed.
- P. Evans, Scaling and assessment of data quality, Acta Crystallogr., Sect. D, 2006, 62, 72–82 CrossRef.
- P. R. Evans and G. N. Murshudov, How good are my data and what is the resolution?, Acta Crystallogr., Sect. D, 2013, 69, 1204–1214 CrossRef CAS.
- S. Bailey, The CCP4 suite: programs for protein crystallography, Acta Crystallogr., Sect. D, 1994, 50, 760–763 CrossRef PubMed.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin and K. S. Wilson, Overview of the CCP4 suite and current developments, Acta Crystallogr., Sect. D, 2011, 67, 235–242 CrossRef CAS PubMed.
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, Phaser crystallographic software, J. Appl. Crystallogr., 2007, 40, 658–674 CrossRef CAS PubMed.
- A. Vagin and A. Teplyakov, Molecular replacement with MOLREP, Acta Crystallogr., Sect. D, 2010, 66, 22–25 CrossRef CAS.
- D. Sharma, L. De Falco, S. Padavattan, C. Rao, S. Geifman-Shochat, C. F. Liu and C. A. Davey, PARP1 exhibits enhanced association and catalytic efficiency with γH2A.X-nucleosome, Nat. Commun., 2019, 10, 5751 CrossRef CAS PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Features and development of Coot, Acta Crystallogr., Sect. D, 2010, 66, 486–501 CrossRef CAS PubMed.
- G. N. Murshudov, A. A. Vagin and E. J. Dodson, Refinement of macromolecular structures by the maximum-likelihood method, Acta Crystallogr., Sect. D, 1997, 53, 240–255 CrossRef CAS.
- B. Bovio, F. Bonati, A. Burini and B. R. Pietroni, Linear GoId(I) Derivatives with Bulky Phosphine and Imidazolato Ligands. X-Ray Crystal Structure of 1-(C6H11)3PAu-2-i-Pr-Imidazole·C6H6, Z. Naturforsch. B., 1984, 39, 1747–1754 CrossRef.
- L. K. Batchelor, E. Păunescu, M. Soudani, R. Scopelliti and P. J. Dyson, Influence of the Linker Length on the Cytotoxicity of Homobinuclear Ruthenium(II) and Gold(I) Complexes, Inorg. Chem., 2017, 56, 9617–9633 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra01617c |
| ‡ Equal contributions. |
| This journal is © The Royal Society of Chemistry 2024 |