
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual-functional nano-photosensitizers: Eosin-Y decorated gold nanorods for plasmon-enhanced fluorescence and singlet oxygen generation†
Sravani Kaja,
Ashin Varghese Mathews and
Amit Nag *
*
Department of Chemistry, Birla Institute of Technology and Science (BITS) Pilani, Hyderabad Campus, Hyderabad 500078, India. E-mail: amitnag@hyderabad.bits-pilani.ac.in
First published on 17th April 2024
Abstract
Photosensitizer (PS) with enhanced fluorescence is attractive for image-guided photodynamic therapy (PDT) due to its dual functional role in Singlet Oxygen Generation (SOG) and producing high fluorescence signals. Here, Eosin-Y (Ey) decorated polymer coated gold nanorods (GNRs) of different aspect ratios are synthesized and introduced as novel plasmon-enhanced nano-photosensitizers for this purpose. We show, upon excitation at 519 nm, simultaneous enhancement in fluorescence and SOG was achieved for the hybrid nanostructure. The best enhancement factors of 110 and 18 for metal-enhanced fluorescence and metal-enhanced SOG, respectively, are obtained with GNRs of length 133 nm and width 45 nm, where Ey is positioned at 12.6 nm from the metal core using layer-by-layer assembly of oppositely charged polymers. The observed plasmonic effect is critically analysed by comparing the near field damping rate along with decay length, far field scattering and nonradiative energy transfer of the nanohybrids.
1. Introduction
Cancer is the most lethal disease, and is one of the major causes of increasing mortality rates in humans. While surgery, chemotherapy, and radiotherapy treat well the early stages of the cancer, the efficacy of these methods for different cancer types are doubtful when the cancer reaches metastasis stage due to their unwanted and serious side effects. In search for a sustainable method, photodynamic therapy (PDT) is a non-invasive and selective method that has shown promising performances against different cancer types, with the help of nanoscience and nanotechnology.1,2 PDT needs three main components, a photosensitizer (PS), suitable light source, and cellular oxygen, for achieving successful cytotoxic effects to kill the cancer cells.3,4 Moreover, there is an urgent need to develop theranostics materials that provide a diagnosis and therapy together. Therefore, photosensitizer (PS) with enhanced fluorescence can be a game-changer due to its superior efficacy compared to traditional PS, as it results into singlet oxygen generation (SOG), as well as it produces high fluorescence signal.5,6 Thus, image-guided PDT is possible with PS with bright fluorescence as it provides a therapeutic outcome with simultaneous ability to trace the disease remission/progression. But traditional PSs exhibit few drawbacks such as very weak fluorescence. Moreover, when the loading of such PS is high to compensate for the low fluorescence yield, production of SOG and fluorescence becomes minimal due to aggregation-caused quenching effects. In addition, as most of the PS molecule absorbs in the visible region, except two-photon PDT, the technique may encounter nontrivial visible light scattering by tissues.7 It leads to poor imaging contrast and spatial resolution during the imaging, as well as photodamage to biological samples. Thus, it is essential to simultaneously enhance the fluorescence and SOG of the PS molecules, for successfully utilising them in theranostics applications but with minimal incident light intensity.On the other hand, upon irradiation by an incident electromagnetic field, plasmonic metallic nanoparticles made of gold, silver, copper etc. produce localized surface plasmon resonances from the collective oscillation of the conduction band electrons. The plasmon decays in ultrafast time scale, however it produces strong near- and far-field effects around the nanoparticle. By virtue of the plasmonic nanoantenna effect, the incident light can be localized in the near field, producing a strong local enhanced electric field that extends from the nanoparticle's surface to few nm distance from the surface.8 On the other hand, the far-field properties are explained by the extinction of the light interacting with the localized surface plasmon of the nanoparticles, which results from contributions of two processes, namely, absorption and scattering. The radiative decay of the plasmon leads to scattering, i.e., re-radiation of light into the far-field (hundreds of nanometers) that can increase the level of light trapping in the whole sample and act as a secondary light source.9,10 Plasmonic metallic nanoparticles (MNPs) can interact very strongly with the fluorophores next to them causing enhancement in their fluorescence and the phenomenon is known as metal-enhanced fluorescence (MEF).11,12 Similarly, metal-enhanced singlet oxygen (1O2) generation (ME-SOG) is also another emerging application of plasmonics. It has been shown from different plasmon based optical experiments individually that the production of SOG by a photosensitizer or fluorescence of an emitter can be dramatically enhanced, by varying several parameters, including the nanostructure morphology,13 size,14 composition,15 and most importantly photosensitizer-metal or emitter-metal distance.16 The conjugation of organic molecules via a spacer with various plasmonic nanostructures such as sphere, rod, star, bipyramid, sheet, cluster etc. made up of mainly Ag and Au, was achieved using various strategies, namely, core–shell technique, polyelectrolyte coating, polymer coating etc.17
Among the various types of plasmonic nanostructures explored in theranostics, gold nanorods (GNRs) were widely studied due to their several advantages including good biocompatibility, tunability in the localized surface plasmon resonance maxima and excellent photothermal property.18,19 The dipolar coupling of the electric field and surface plasmons along the short and long axis of the nanorods results into two bands, namely short-wavelength transverse surface plasmon resonance (TSPR) and long wavelength longitudinal surface plasmon resonance (LSPR).20,21 Due to the asymmetric shape of the GNRs, the near- and far-field plasmonic effects of them are much superior compared to symmetric nanoparticles, that had been successfully used in various plasmon enhanced optical processes such as plasmonic catalysis,22,23 plasmonic energy conversion,24 photothermal therapy,25–27 two photon absorption and emission,28,29 and photodynamic therapy.30–34 However, there are only a handful of reports available on simultaneous enhancement of fluorescence and SOG by hybrid plasmon-enhanced photosensitizer with GNR. Huang et al. noticed modest 3-fold and 1.4-fold enhancement in fluorescence and SOG, respectively, for chlorin e6 adsorbed on bare GNRs.35 Xuebin et al. observed co-enhancement in fluorescence and SOG by silica coated GNRs and also identified that the rates of MEF and SOG of AlC4Pc varied with increase in silica thickness from 2.1 nm to 28.6 nm. They recorded a maximum of 7-fold and 2.1-fold enhancement in MEF and SOG, respectively, when the silica thickness is ∼10.6 nm.36 Novikova et al. synthesized GNRs with various aspect ratios, coated them with silica shell and reported 6.7- and 13-fold enhancement in luminescence and SOG respectively using MO6 clusters.37 Therefore, the current challenge in MEF and ME-SOG is to increase the hitherto observed small enhancement factors for realizable applications. Also, to achieve the desired results, an ideal PS should be water soluble, less cytotoxic and its fluorescence spectrum shouldn't demonstrate a significant overlap with the extinction spectra of the nanocores.38,39 Because, more spectral overlap will lead to higher non-radiative energy transfer from PS to metal and the desirable effect might not be achieved. Hence, there is a quest for a rational design of a hybrid theranostics GNR in which GNRs are conjugated with a suitable PS at an appropriate distance, with an aim to significantly enhance the fluorescence and SOG of the PS molecule.
Herein, to achieve this goal, hybrid theranostics nano-photosensitizers were designed, consisting a photosensitizer Eosin Y (Ey) conjugated to layer-by-layer (LBL) assembled GNRs of different aspect ratios. We report a comprehensive experimental and theoretical investigation of the extent of the near- and far-field plasmonic effect of GNRs on MEF and ME-SOG of the photosensitizer molecule Ey. Ey is a water-soluble PS molecule, which is known for photodynamic inactivation of bacterial cells.40,41 Moreover, the transverse surface plasmon resonance bands of GNRs overlap with the absorbance and fluorescence spectra of Ey, which may significantly modify the optical properties of the molecule in the GNR-Ey hybrid. The LBL strategy was inspired by our previously reported metal-enhanced nanoplatforms.17 The negative end of the polarizable molecule Ey binds electrostatically to the positively charged polyelectrolyte coating on GNRs, to extract the best plasmonic effect. Moreover, using LBL method the lowest and highest spacer thicknesses were tightly controlled such that the PS remains within the domain of near field decay length of the GNRs at all times. Among all, the best theranostics property, i.e., highest fluorescence and 1O2 yield, was demonstrated by the PS-GNR assembly which showed the highest theoretical near-field effect at the absorption maximum of Ey i.e., 519 nm, as well as highest far-field effect in form of maximum scattering of light, measured experimentally.
2. Materials and methods
2.1. Materials
Gold(III) chloride trihydrate (HAuCl4·3H2O), cetyltrimethylammonium bromide (CTAB), silver nitrate (AgNO3), Hydroquinone, Eosin Y, Poly (sodium 4-styrene sulfonate) (PSS, MW = 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), 1,3-diphenylisobenzofuran (DPBF), and 3-aminopropyl triethoxysilane (APTES) were purchased from Sigma Aldrich. Poly (allylaminehydrochloride) (PAH) was procured from Alfa Aesar. Sodium borohydride (NaBH4), 5,5 Dimethyl-1-pyrroline-N-oxide (DMPO), Tetra ethyl Orthosilicate (TEOS) was brought from TCI chemicals India. Sodium Azide (NaN3), Sodium chloride (NaCl), and Ammonium hydroxide (NH4OH) were obtained from SRL Chemicals, India. All the chemicals were used as obtained without any further purification.
000), 1,3-diphenylisobenzofuran (DPBF), and 3-aminopropyl triethoxysilane (APTES) were purchased from Sigma Aldrich. Poly (allylaminehydrochloride) (PAH) was procured from Alfa Aesar. Sodium borohydride (NaBH4), 5,5 Dimethyl-1-pyrroline-N-oxide (DMPO), Tetra ethyl Orthosilicate (TEOS) was brought from TCI chemicals India. Sodium Azide (NaN3), Sodium chloride (NaCl), and Ammonium hydroxide (NH4OH) were obtained from SRL Chemicals, India. All the chemicals were used as obtained without any further purification.
2.2. Experimental methods
000 rpm for 10 min and the supernatant was discarded, the pellet was again dispersed in 100 mL of Millipore water and was used for further studies.000 rpm and redispersed in pure water to remove excess CTAB. Later, 60 mL purified GNRs were taken and added to 60 mL of negatively charged 2 mg mL−1 PSS in 1 mM NaCl solution. The pH of the solution was found to be 6. The solution was stirred for 3 hours and then centrifuged at 15000 rpm for 20 minutes. The pellet containing GNR-PSS was dispersed in 60 mL Millipore water and the supernatant containing unreacted PSS was discarded. The centrifugation step was repeated three times to remove the excess unbound PSS. The solution was labelled as LBL a. In the next step, we have coated the obtained 50 mL LBL a with 50 mL positively charged 2 mg mL−1 PAH polymer in 1 mM NaCl solution and the whole process is repeated and the formed NRs were labelled as LBL b. Like this we have coated 6 polymer layers on GNRs as shown in the scheme below and labelled as LBL a to LBL f.
 | ||
| Scheme 1 A schematic representation of layer-by-layer (LBL) assembly on GNRs. | ||
000 rpm for 30 min and the pellet was dispersed in water and the supernatant containing unreacted Ey was collected and stored. The UV-Visible spectra of supernatant were recorded to quantify the unbound Ey. The purification process was repeated several times until the supernatant was devoid of Ey absorption peak and finally the obtained GNR-LBL-Ey conjugate was dispersed in Millipore water and used for further studies. Other experimental details such as synthesis of amine functionalized silica nanoparticle (SiNPs), synthesis of SiNP-LBL NPs, Eosin Y conjugation on SiNP-LBL NPs as well as the details of COMSOL simulation are provided in the ESI (Experimental section, ESI†).2.3. Characterization
The size and structure of all the synthesized GNRs, GNR-LBL was determined by using a Field emission scanning electron microscope (FE-SEM) (FEI Apreo Lo Vac equipped with Retractable STEM 3+ Detector, DBS Detector). The samples were diluted and about 10 μL of sample was drop casted on a clean silicon wafer and samples were dried properly before taking the images. The images of GNRs in STEM mode were collected using Copper grids with 200 mesh size. Around 5 μL of solution was dropped on the Cu grid, allowed to dry and then images were collected in STEM mode. The average particle size of NRs was calculated using ImageJ software using 50 GNRs. The extinction spectra of the NPs, absorbance of Ey, and DPBF were recorded by using Shimadzu UV 3600 Plus spectrophotometer. Zeta potential measurements were carried out using a Zetasizer Nano-ZS 90 from Malvern Instruments Ltd. Metal enhanced fluorescence studies were performed using Hitachi F-7000 Spectrofluorometer by keeping excitation slit, emission slit and PMT voltage constant for all the measurements. The experimental scattering studies were carried out on Horiba fluorolog spectrofluorometer equipped with an integrating sphere in synchronous mode within the range of 300–800 nm with an offset of 20 nm. Excited state fluorescence life time measurements were carried out using Time Correlated Single Photon Counting (TCSPC) technique (Horiba, Deltaflex model 1), 510 nm LED and the peak preset was set to 10000. Further details such as calibration curve for Ey quantification, sample preparation for MEF, Lifetime and MESOG studies, experimental setup for determination of scattering component of the nanorods are provided in ESI (Sample preparation, ESI†).
3. Results and discussion
3.1. Preparation and detail characterization of Eosin Y decorated GNRs
GNRs of diverse aspect ratios with larger lengths and widths, were synthesized by a seed mediated growth process using CTAB as the stabilizer. We expected higher plasmon enhancement efficiency from them compared to traditional GNRs of 40–50 nm length,44 but it is also important to note that the final nanostructure should be in the optimal range of 100–200 nm for better blood circulation and biodistribution. By changing the concentration of CTAB during the growth process, GNRs of six different aspect ratios were synthesized and were labelled as GNR1 to GNR6. As shown in the FE-SEM images (Fig. 1a–f) and the average size plots (Fig. S1, ESI†) of GNR1 to GNR6, the average length increased gradually from 115 ± 3.2 to 157 ± 4.6 nm and width decreased gradually from 57.5 ± 2.1 to 40.5 ± 1.8 nm, resulting into a variation in the aspect ratio (AR) from 2 ± 0.3 to 3.8 ± 0.9 (Table S1, ESI†). The extinction spectra of the synthesized GNRs exhibited the coexistent TSPR peaks in the visible range, while the LSPR peaks shifted from 699 to 828 nm with an increase in AR (Fig. 1g and Table S1, ESI†). The synthesized GNRs formed stable colloidal suspensions as revealed by their large positive zeta (ζ) potential values (Fig. S2, ESI†). | ||
| Fig. 1 FE-SEM images of GNRs (a) 1, (b) 2, (c) 3, (d) 4, (e) 5, and (f) 6. The scale bars are 200 nm for (a) to (c) and 400 nm for (d) to (f); (g) extinction spectra of GNRs 1–6; (h) schematic illustration of adsorption of polyelectrolytes and Ey on GNR. | ||
Subsequently, using LBL assembly method, the GNRs were coated with polymer layers (Fig. 1h), which relies on the adsorption of oppositely charged polymers and can be used to fine tune the distance between metal core and photosensitizer. Herein, we have used a negatively charged polymer poly styrene sulfonate (PSS) and a positively charged polymer polyallylamine hydrochloride (PAH) to prepare the LBL assembly around the metal core. The positive charge due to the presence of CTAB stabilizer on bare GNR, enable the formation of first LBL layer by the electrostatic attraction between CTAB and PSS. It is to be noted here that the polymer was always taken in NaCl solution as it minimized the electrostatic repulsions, leading to the layer formation with higher thickness.45 In the next step, the second LBL layer was formed by adsorbing positively charged PAH to negatively charged PSS layer. This process was continued until we obtained LBL assembled GNRs of six different thicknesses, which will be labelled as GNR1a to GNR1f for GNR1 and in the same way for other GNRs, while ‘a’ being the smallest and ‘f’ being the highest thickness. The measurement of ζ-potential values indicated a charge reversal at every subsequent layer, confirming the successful formation of LBL coated GNRs (Fig. 2a and S3–S8, ESI†). The extinction spectra of the LBL assembled GNRs are shown in Fig. 2b and S9, ESI.† We noticed a small red shift and peak broadening in the LSPR spectra of GNR after the LBL assembly, which might be due to decrease in inter nanorod distance.45 From the FE-SEM images of LBL coated GNRs, as expected the average size of GNRs increased after each LBL coating. For all the GNRs, the average polymer layer thickness (spacer) was found to be 2.8 ± 0.8 nm, 6.1 ± 1.3 nm, 9.4 ± 1.2 nm, 12.6 ± 1.8 nm, 15.8 ± 1.3 nm, and 19.7 ± 2.1 nm for LBL a–f, respectively (Fig. S10–S21, ESI†). A plot of average spacer length against number of polymer layers for GNR 4, showed a linear increase in average thickness with each new layer (Fig. S22, ESI†).
 | ||
| Fig. 2 (a) A representative histogram showing the charge reversal at every stage of LBL self-assembly for GNR4; (b) extinction spectra of GNR 4a–4f; (c) absorption spectra of Ey, extinction spectra of GNR, GNR-4d, GNR-4d-Ey; (d) STEM image of GNR 4d taken in dark field mode and the inset shows the same along with layer thickness. | ||
The final step in this multilayer architecture is the adsorption of PS molecules to LBL coated GNRs. For this, the GNR-LBL in water were diluted to optical density (OD) value of 0.1 and the appropriate quantity of Ey in ethanol was added to it and the conjugation process was carried out by stirring the reaction mixture for three hours at room temperature in dark. After three hours, the conjugated GNRs were purified by centrifugation. From the pellet, we recorded the absorbance spectra of hybrid nanorods (Fig. 2c and S23, ESI†), whereas the supernatant produced the absorption spectra of unconjugated Ey (Fig. S24, ESI†). The study revealed that the maximum conjugation of Ey was observed for GNRs with LBL b, d, and f, where the outer layer was positively charged due to presence of PAH. Moreover, a comparison of the OD values of Ey from the absorption spectra of pellet and supernatant (Fig. S24, ESI†), indicated about negligible or no conjugation of Ey to the GNRs with LBL a, c and e (PSS coating). Also, bright green emission of Ey was obtained from the supernatant of GNR 4c (Fig. S25, ESI†) under UV excitation, indicating the presence of Ey in negligible amount in the pellet. Under same condition, the supernatant of GNR 4d produced no emission intensity, indicating the strong conjugation of Ey in case of GNR 4d. Further, the measurement of the ζ potential values of GNR 4c and GNR 4d (Fig. S26, ESI†) substantiated the above-mentioned fact. The ζ potential value of GNR 4d was altered substantially after Ey conjugation to become close to neutral (+5 mV) from positive value of +27.2 mV, indicating the unavailability of the free amine groups of PAH as they were conjugated with Ey molecules. Whereas, in case of GNR 4c, the ζ potential value changed from −35 mV to only −20 mV after Ey conjugation, indicating the presence of still substantial free sulfonate groups of PSS on the surface. Subsequently, the amount of conjugated Ey was quantified by using a calibration plot prepared with the absorbance of the known concentrations of Ey (Fig. S27, ESI†), and by comparing it with the absorbance of Ey in supernatant, in each case. For the GNRs with LBL b, d, and f, the concentration of conjugated Ey was found to be in the range ∼1–1.25 μM. However, for the subsequent MEF and ME-SOG experiments, the Ey conjugated GNR solutions were appropriately diluted to match the OD values of Ey to a concentration of 1 μM. Since Ey was preferentially adsorbed on GNR with outer polymer layers as PAH, we compared the MEF and ME-SOG results of GNR-Ey hybrids for b, d, and f layers only. As the absorption maxima of Ey was found to be overlapping with the TSPR band of the hybrid GNR, the coupling of the plasmon resonance of GNR with the absorption spectra of Ey significantly modified the optical processes of the Ey molecules (Fig. 2c). This ‘plasmonic-molecular resonance coupling’ may significantly modify the absorption and fluorescence property of the molecules, present in hybrid GNR.
3.2. MEF studies on GNR-LBL-Ey nanohybrids
Subsequently, the MEF studies of hybrid GNR-LBL-Ey nano-photosensitizer were performed as a function of different aspect ratios of GNR (Fig. 3a and S28, ESI†) by varying the spacer thickness in each GNR from LBL b–f. The fluorescence of Ey was completely quenched when it was directly adsorbed on bare GNRs and also very less fluorescence of Ey was recorded (not shown) when it was adsorbed on PSS layer (LBL a, c and e). Whereas, with the outer layer as PAH (LBL b, d and f), the fluorescence of Ey was found to be significantly higher compared to PSS. With PAH layers, the maximum fluorescence of GNR-Ey was observed for ‘LBL d’ starting from ‘LBL b’ and it decreased subsequently for ‘LBL f’, emphasizing the distance dependent nature of MEF. The enhancement factor (EF) of MEF is a critical parameter to choose the best plasmonic material. To calculate the EF, an identical MEF study was carried out by adsorbing the same concentration of Ey molecules (10−6 M) on LBL assembled SiNPs (control), which were devoid of any plasmonic metal. LBL assembled SiNPs were synthesized by modified Stober's method and the size of these nanoparticles was found to be 120 nm. The conjugation of Ey to SiNPs was similarly carried out as mentioned earlier and thoroughly characterized (Fig. S29, ESI†). The fluorescence of Ey was found to be negligibly small for SiNP-LBL-Ey hybrids, although the number density of SiNPs (5 × 1015 particles per mL) was calculated to be much higher (calculation S1, ESI†) than the number density of GNRs (9 × 1012 particles per mL, GNR 4). Subsequently, EF of MEF was calculated as the ratio of fluorescence intensity of Hybrid GNR-Ey of various LBL thicknesses to the fluorescence intensity of appropriate control sample SiNP-LBL-Ey. Among all GNR-LBL-Ey hybrids, the highest fluorescence as well as staggering EF of ∼110 for MEF was observed with GNR 4d (Fig. 3a and S30, ESI†), where Ey was positioned at a distance of ∼12.6 nm from the GNR surface (Fig. 2d). The fluorescence lifetime values of free Ey, Ey adsorbed on SiNPs and GNR-LBL-Ey were recorded to understand the mechanism of MEF. The average lifetime of Ey in water was reported earlier as 1.44 ns46,47 which decreased to ∼0.84 ns on direct adsorption on bare GNR (Fig. 3b), while it increased to 2.64 ns on direct adsorption on polymer coated SiNPs, similar to previous reports.17,48 As depicted in Fig. 3b and Tables S2–S7, ESI,† the average life time values for each GNR-Ey hybrid slightly decreased with increase in polymer thickness up to LBL-d and then increased with further increase in polymer coating. Hence, as there is a moderate decrease in the average lifetime of the fluorophore associated with an increase in fluorescence, therefore ‘enhancement in the radiative decay rate’ is one of the mechanisms contributing to MEF, but it may not be the sole effect. Because, the transition rate of the excitation process of Ey is also dependent on the near field generated close to the nanostructure, as the transition rate is proportional to the square of the local electric field at the transition frequency. Therefore, higher excitation rate is another factor, which may lead to generation of higher fluorescence intensity. Finally, GNR-Ey interactions may also open up new nonradiative decay pathways of the excited electrons in the emitter, by which fluorescence intensity can be quenched as energy can be transferred to the GNR. Therefore, MEF of a hybrid nanostructure is a combined effect of the aforementioned three mechanisms (vide infra).
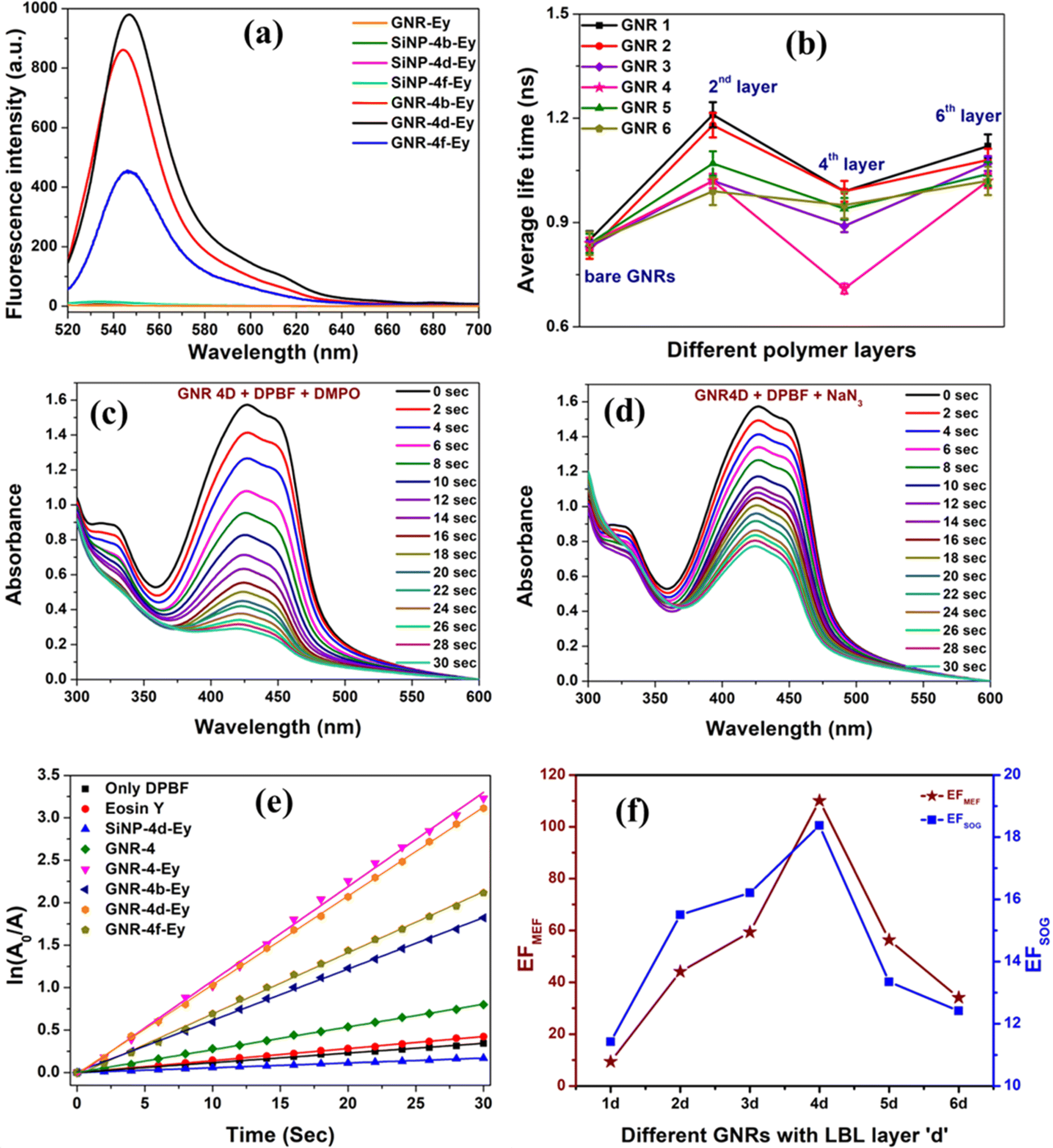 | ||
| Fig. 3 (a) MEF studies of Ey adsorbed on GNR 4b–4f, excitation: 519 nm; (b) average life time plots of Ey adsorbed on bare GNRs and GNRs with different polymer layers; degradation of 50 μM DPBF by GNR-4d-Ey in the presence of (c) DMPO, (d) NaN3; (e) ln(A0/A) plots depicting the degradation rate of 50 μM DPBF by GNR 4b–4f and different controls, under 35 mW cm−2 white light; (f) EFs of MEF and MESOG for GNRs with different aspect ratio with LBL layer ‘d’ only. | ||
3.3. ME-SOG studies on GNR-LBL-Ey nanohybrids
GNR-Ey hybrids were evaluated for their ME-SOG efficiency, by an indirect method via analysing the rate of degradation of a SOG selective molecular probe Diphenyl isobenzofuran (DPBF). While, complete quenching of the fluorescence was observed when Ey was adsorbed directly on bare GNR, but the same hybrid yielded maximum rate of SOG (Fig. 3e) because of efficient energy transfer from bare GNRs to adsorbed oxygen molecules via PS.17,49 Despite of its high SOG capacity, Ey directly adsorbed on bare GNR cannot be used for image guided therapies due to its poor or almost zero fluorescence making it practically unimportant. GNR- Ey hybrids with LBL layers b, d, and f provided much higher SOG capacity compared to the control SiNP-LBL-Ey. We also performed other control studies by studying the degradation of DPBF with and without light and in presence of only Ey separately (Fig. S32, ESI†). The self-degradation of DPBF was subtracted from all the samples before calculating its SOG rate (Fig. 3e and S33–S39, ESI†). To clearly understand the SOG efficiency, we have plotted ln(A0/A) Vs time and the slopes obtained from the linearly fitted curves were used to calculate the EF of the ME-SOG of the nanohybrids (Fig. S40 and S41, ESI†). The EF of ME-SOG was calculated as the ratio of slopes of DPBF degradation in presence of GNR-LBL-Ey to SiNP-LBL-Ey. Similar to the results obtained in MEF, GNR-4d demonstrated the maximum SOG and the highest EF value ∼18, when compared to other GNRs (Fig. 3f). In order to confirm that the decrease in DPBF degradation was due to 1O2 only, rather than any other reactive oxygen species, we performed two different experiments using DMPO and NaN3 (Fig. 3c and d). DMPO specifically binds to hydroxyl and superoxide radicals and we did not notice any change in the rate of degradation of DPBF with hybrid GNR 4d-Ey particles with and without the addition of DMPO, ruling out the presence of hydroxyl and superoxide radicals (Fig. 3c).17,50 To confirm the presence of 1O2, we added NaN3 to the GNR 4d-Ey solution and the degradation of DPBF was studied. In this case, the rate of degradation of DPBF was found to be much smaller which confirmed the trapping of in situ generated 1O2 by NaN3, proving the presence of 1O2 as the ROS (Fig. 3d).17,503.4. Theoretical investigation using COMSOL simulations
In order to understand the role of the near field effect on MEF and ME-SOG of Ey conjugated GNR, COMSOL simulations were carried out to visualize the electromagnetic field maps for bare GNRs (Fig. S42, ESI†), GNRs of different AR but same SiO2 thickness (Fig. 4a), as well as with SiO2 spacer of different thicknesses (Fig. S43, ESI†). It is to be noted here that we used dielectric material SiO2 as the spacer (relative permittivity 3.9) instead of PSS and PAH, due to readily available parameters of SiO2 such as refractive index, electrical conductivity, susceptibility etc. Moreover, it has been reported earlier that the relative permittivity values of different polymers, including polystyrene, are found in the range of approximately between 1 to 6,51 which coincides with relative permittivity value of SiO2. Fig. 4b shows, among all the hybrid GNRs with same thickness of the SiO2 coating (12.6 nm, i.e., analogous to LBL d), the near field damping rate was lowest for GNR 4d. Also, when we calculated the decay lengths,52 we noticed that decay length was maximum i.e., ∼108 nm in case of GNR 4d when compared to other GNR with same SiO2 thickness (Table 1 and Fig. S44, ESI†). Therefore, it may be understood that Ey experienced maximum electric field with GNR 4d compared to other hybrid GNRs justifying the earlier obtained experimental results where GNR 4d produced maximum MEF and ME-SOG outcome. Further, by varying the SiO2 thicknesses for only GNR 4, the damping rate was found to decrease with increase in silica spacing (Fig. 4c), indicating stronger near field prevalent for ‘GNR 4 d–f’. However, for the bare GNRs the highest and lowest near field damping rates were recorded for GNR 6 (AR: 3.8) and GNR 1 (AR: 2), respectively (Fig. S45, ESI†). The result is similar to the already reported data in literature, which estimates higher damping rate with nanorod with higher aspect ratio.15,20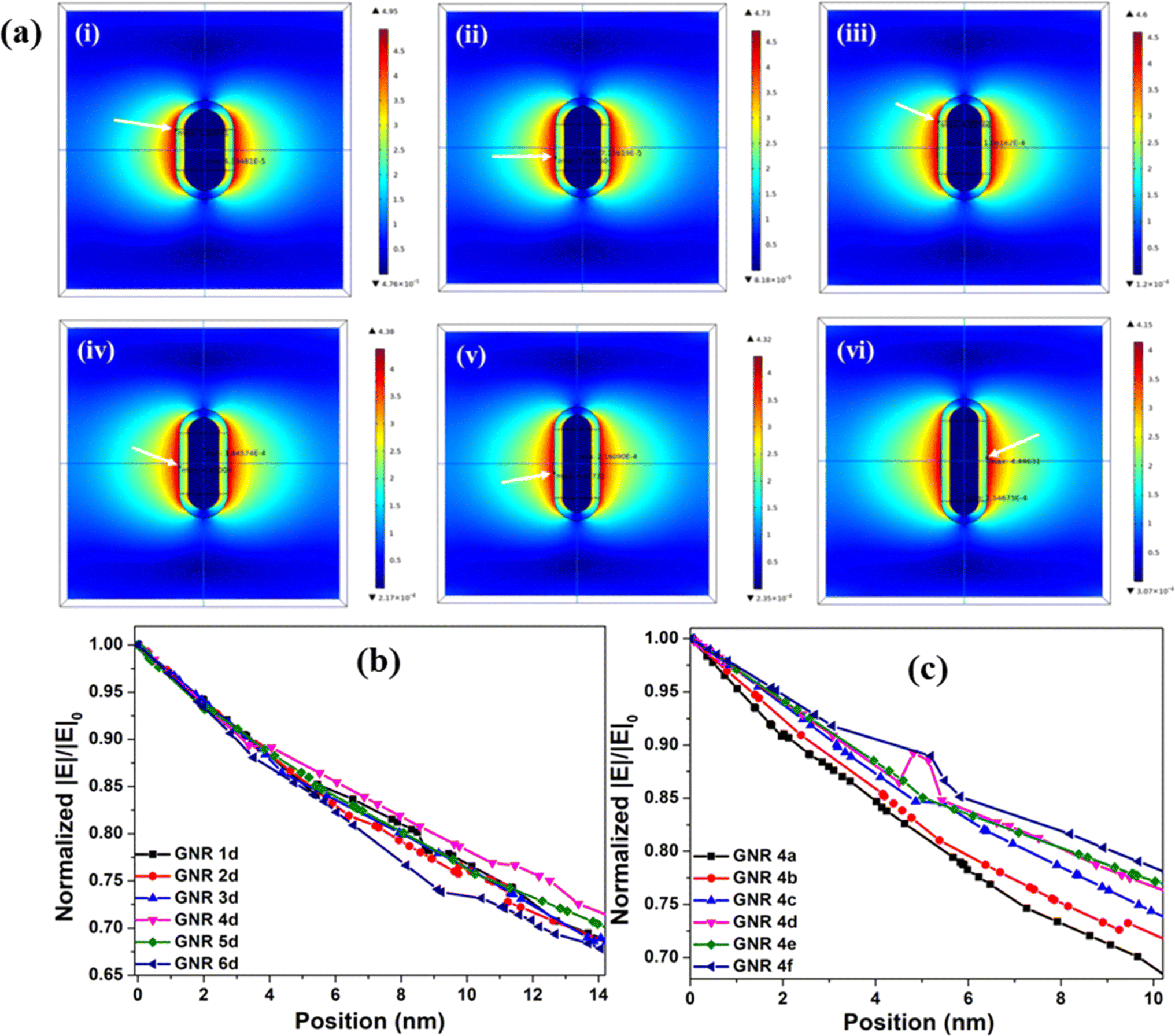 | ||
| Fig. 4 (a) Theoretical electromagnetic field maps for (i) GNR 1d, (ii) GNR 2d, (iii) GNR 3d, (iv) GNR 4d, (v) GNR 5d, (vi) GNR 6d; excited at 519 nm (the white arrow in the COMSOL figure indicates the location of the electric field's maximum value (Emax) and the corresponding enhancement factor is calculated as the square of (Emax − Emin)/Eapplied); 1D line plot showing the normalized electric field damping as a function of distance, starting from the SiO2 surface of (b) GNR 1d–6d and (c) GNR 4a–4f. | ||
| S. No. | Sample | Experimental | COMSOL simulation | Estimated J integral (M−1 cm−1 nm5) | |||
|---|---|---|---|---|---|---|---|
| EF MEF | Average life time (ns) | EF SOG | Electric field EF | Decay length (nm) | |||
| 1 | GNR 1d | 9.41 | 0.99 ± 0.02 | 11.4 | 27.08 | 83.7 | 5.68 × 1010 |
| 2 | GNR 2d | 44.1 | 0.99 ± 0.02 | 15.5 | 26.34 | 87.1 | 2.83× 1010 |
| 3 | GNR 3d | 59.3 | 0.89 ± 0.01 | 16.2 | 24.28 | 92.0 | 2.42 × 1010 |
| 4 | GNR 4d | 110.1 | 0.71 ± 0.01 | 18.3 | 21.43 | 107.7 | 3.02 × 1010 |
| 5 | GNR 5d | 56.3 | 0.94 ± 0.03 | 13.3 | 20.13 | 103.6 | 3.08 × 1010 |
| 6 | GNR 6d | 34.1 | 0.95 ± 0.03 | 12.4 | 19.76 | 100.6 | 3.98 × 1010 |
3.5. Far-field scattering experiment and measurement of overlap integral for the nanohybrids
Finally, to understand the role of the far-field effect on MEF and ME-SOG, we performed an experiment to collect only the scattering component of the GNRs with and without the polymer spacer, using an emission spectrometer coupled with an integrating sphere.14,15 It is known that the extinction of light in the far field, is composed by two optical processes, namely, absorption and scattering of the nanostructures. Scattering in the far field creates a secondary light source which can in-turn enhance the light absorption and subsequently the excitation rate of the emitter. As depicted in Fig. 5a, scattering component of GNR 4 was experimentally found to be maximum compared to any other GNRs, indicating the superiority of GNR 4 over other nanorods in MEF and ME-SOG of Ey. Among different GNRs with same polymer thickness (LBL d), GNR 4d produced maximum scattering intensity (Fig. 5b), agreeing to our earlier observation. However, as expected, when we collected the scattering components of GNR4a – GNR4f, it decreases gradually with increase in polymer layer, indicating an increase in absorbance (Fig. S46, ESI†). | ||
| Fig. 5 (a) and (b) are the experimental scattering spectra obtained from GNR 1–6, and GNR 1d–6d, respectively, blank is the same solution but without GNRs; (c) shows the overlap between emission spectra of Ey and extinction spectra of GNR 1d–6d-Ey hybrids. | ||
At this point, it is important to consider the role of competitive nonradiative energy transfer process enforced by the individual GNR on the emitter, by which fluorescence of the emitter is quenched.53 The extent of nonradiative energy transfer is estimated as the overlap of the emission spectra of Ey and the extinction spectra of different polymer coated GNRs with same spacer thickness (Fig. 5c). Table 1 shows the J integral (overlap) parameter of different GNR with same spacer thickness along with other important values. From the combination of J values and the simulated electric field enhancement factors for each GNR (Table 1), it could be predicted that the plasmonic efficiency of GNR 3d and GNR 4d would be superior compared to the rest. However, as shown earlier, the more scattering component of GNR 4d keeps it ahead of GNR 3d to produce higher plasmonic efficiency (vide supra).
4. Conclusions
In conclusion, we demonstrated that simultaneous enhancement of fluorescence and 1O2 production of a model photosensitizer Ey coupled to GNRs via polymer spacer, crucially depends on the near- and far-field properties of the plasmonic nanostructure. The enhanced fluorescence and SOG production depend significantly on the distance of the PS from the gold core. To the best of our knowledge this is the first time, a library of GNRs with length >100 nm, small variations in their size and aspect ratio were explored to utilize them for image-guided PDT applications. Among all the nano-photosensitizers in our synthesized library, GNR 4d-Ey (length 133 ± 4.7 nm, width 45 ± 1.2 nm, aspect ratio ∼2.95) offered unprecedented ∼110- and 18-fold simultaneous increase in fluorescence and SOG production, respectively, due to its highest experimentally recorded scattering yield, highest theoretically estimated near-field enhancement and highest experimentally determined enhancement of the radiative decay rate. However, the combination of the plasmon-enhanced optical processes with non-radiative energy transfer between the nanorod and the emitter, is also equally important to obtain the best result. Overall, this study would pave away for more rational design and synthesis of nano-photosensitizers for theranostics applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. K. thanks BITS Pilani for the support in form of research fellowship. Authors gratefully acknowledge the support from Department of Science and Technology, India for the PURSE grant SR/PURSE /2020/20 and FIST grant SR/FST/CSI-240/2012. Central Analytical Facility of BITS Pilani Hyderabad Campus is gratefully acknowledged for SEM and STEM analyses. A. N. thanks DST-SERB (CRG/2023/002769) for financial support.References
- D. W. Felsher, Nat. Rev. Cancer, 2003, 3, 375–380 CrossRef CAS PubMed.
- I. J. McDonald and T. J. Dougherty, J. Porphyrins Phthalocyanines, 2001, 5, 105–129 CrossRef.
- H. Zheng, Technol. Cancer Res. Treat., 2005, 4, 283–293 CrossRef PubMed.
- S. Li, F. Yang, Y. Wang, T. Du and X. Hou, Chem. Eng. J., 2023, 451, 138621 CrossRef CAS.
- W. Bäumler, J. Regensburger, A. Knak, A. Felgenträger and T. Maisch, Photochem. Photobiol. Sci., 2012, 11, 107–117 CrossRef PubMed.
- J. M. Fernandez, M. D. Bilgin and L. I. Grossweiner, J. Photochem. Photobiol., B, 1997, 37, 131–140 CAS.
- M. A. Liebert, D. Ph, N. D. Kumar, D. Ph, C. Zhao, D. Ph, P. N. Prasad and D. Ph, J. Clin. Laser Med. Surg., 1997, 15, 201–204 CrossRef PubMed.
- V. Giannini, A. I. Fernández-Domínguez, S. C. Heck and S. A. Maier, Chem. Rev., 2011, 111, 3888–3912 CrossRef CAS PubMed.
- N. Kongsuwan, A. Demetriadou, M. Horton, R. Chikkaraddy, J. J. Baumberg and O. Hess, ACS Photonics, 2020, 7, 463–471 CrossRef CAS.
- B. Gallinet and O. J. F. Martin, Opt. Express, 2011, 19, 22167 CrossRef PubMed.
- C. D. Geddes and J. R. Lakowicz, J. Fluoresc., 2002, 12, 121–129 CrossRef.
- K. Aslan, I. Gryczynski, J. Malicka, E. Matveeva, J. R. Lakowicz and C. D. Geddes, Curr. Opin. Biotechnol., 2005, 16, 55–62 CrossRef CAS PubMed.
- N. MacIa, R. Bresoli-Obach, S. Nonell and B. Heyne, J. Am. Chem. Soc., 2019, 141, 684–692 CrossRef CAS PubMed.
- M. Bregnhøj, S. Rodal-Cedeira, I. Pastoriza-Santos and P. R. Ogilby, J. Phys. Chem. C, 2018, 122, 15625–15634 CrossRef.
- N. Macia, V. Kabanov, M. Côté-Cyr and B. Heyne, J. Phys. Chem. Lett., 2019, 10, 3654–3660 CrossRef CAS PubMed.
- O. Planas, N. Macia, M. Agut, S. Nonell and B. Heyne, J. Am. Chem. Soc., 2016, 138, 2762–2768 CrossRef CAS PubMed.
- S. Kaja, A. Mukherjee, M. Chakravarty and A. Nag, Colloids Surf., A, 2022, 649, 129448 CrossRef CAS.
- N. S. Abadeer, M. R. Brennan, W. L. Wilson and C. J. Murphy, ACS Nano, 2014, 8, 8392–8406 CrossRef CAS PubMed.
- R. Bardhan, N. K. Grady, J. R. Cole, A. Joshi and N. J. Halas, ACS Nano, 2009, 3, 744–752 CrossRef CAS PubMed.
- H. Chen, L. Shao, Q. Li and J. Wang, Chem. Soc. Rev., 2013, 42, 2679–2724 RSC.
- J. Zheng, X. Cheng, H. Zhang, X. Bai, R. Ai, L. Shao and J. Wang, Chem. Rev., 2021, 121, 13342–13453 CrossRef CAS PubMed.
- W. Xiong, D. Sikdar, L. W. Yap, P. Guo, M. Premaratne, X. Li and W. Cheng, Nano Res., 2016, 9, 415–423 CrossRef CAS.
- P. Priecel, H. A. Salami, R. H. Padilla, Z. Zhong and J. A. Lopez-Sanchez, Chin. J. Catal., 2016, 37, 1619–1650 CrossRef CAS.
- S. Chang, Q. Li, X. Xiao, K. Y. Wong and T. Chen, Energy Environ. Sci., 2012, 5, 9444–9448 RSC.
- X. Huang, I. H. El-Sayed, W. Qian and M. A. El-Sayed, J. Am. Chem. Soc., 2006, 128, 2115–2120 CrossRef CAS PubMed.
- W. Il Choi, J. Y. Kim, C. Kang, C. C. Byeon, Y. H. Kim and G. Tae, ACS Nano, 2011, 5, 1995–2003 CrossRef PubMed.
- R. Mooney, L. Roma, D. Zhao, D. Van Haute, E. Garcia, S. U. Kim, A. J. Annala, K. S. Aboody and J. M. Berlin, ACS Nano, 2014, 8, 12450–12460 CrossRef CAS PubMed.
- T. Zhao, L. Li, S. Li, X. F. Jiang, C. Jiang, N. Zhou, N. Gao and Q. H. Xu, J. Mater. Chem. C, 2019, 7, 14693–14700 RSC.
- K. Imura, T. Nagahara and H. Okamoto, J. Phys. Chem. B, 2005, 109, 13214–13220 CrossRef CAS PubMed.
- W. S. Kuo, C. N. Chang, Y. T. Chang, M. H. Yang, Y. H. Chien, S. J. Chen and C. S. Yeh, Angew. Chem., Int. Ed., 2010, 49, 2711–2715 CrossRef CAS PubMed.
- R. Vankayala, Y. K. Huang, P. Kalluru, C. S. Chiang and K. C. Hwang, Small, 2014, 10, 1612–1622 CrossRef CAS PubMed.
- E. L. L. Yeo, J. U. J. Cheah, D. J. H. Neo, W. I. Goh, P. Kanchanawong, K. C. Soo, P. S. P. Thong and J. C. Y. Kah, J. Mater. Chem. B, 2017, 5, 254–268 RSC.
- L. Luo, W. Sun, Y. Feng, R. Qin, J. Zhang, D. Ding, T. Shi, X. Liu, X. Chen and H. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 12591–12599 CrossRef CAS PubMed.
- B. Huang, J. Tian, D. Jiang, Y. Gao and W. Zhang, Biomacromolecules, 2019, 20, 3873–3883 CrossRef CAS PubMed.
- X. Huang, X. J. Tian, W. L. Yang, B. Ehrenberg and J. Y. Chen, Phys. Chem. Chem. Phys., 2013, 15, 15727–15733 RSC.
- X. Ke, D. Wang, C. Chen, A. Yang, Y. Han, L. Ren, D. Li and H. Wang, Nanoscale Res. Lett., 2014, 9, 1–8 CrossRef CAS PubMed.
- E. D. Novikova, Y. A. Vorotnikov, N. A. Nikolaev, A. R. Tsygankova, M. A. Shestopalov and O. A. Efremova, Chem. Commun., 2021, 57, 7770–7773 RSC.
- T. Mthethwa and T. Nyokong, J. Lumin., 2015, 157, 207–214 CrossRef CAS.
- X. Zhou, X. He, S. Wei, K. Jia and X. Liu, J. Colloid Interface Sci., 2016, 482, 252–259 CrossRef CAS PubMed.
- B. Fan, W. Peng, Y. Zhang, P. Liu and J. Shen, Biomater. Sci., 2023, 11, 4930–4937 RSC.
- F. Freire, A. C. B. P. Costa, C. A. Pereira, M. Beltrame, J. C. Junqueira and A. O. C. Jorge, Lasers Med. Sci., 2014, 29, 949–955 CrossRef PubMed.
- S. Picciolini, D. Mehn, I. Ojea-Jiménez, F. Gramatica and C. Morasso, J. Visualized Exp., 2016, 2016, 1–7 Search PubMed.
- B. Wang, J. H. Wang, Q. Liu, H. Huang, M. Chen, K. Li, C. Li, X. F. Yu and P. K. Chu, Biomaterials, 2014, 35, 1954–1966 CrossRef CAS PubMed.
- C. Liang, J. Luan, Z. Wang, Q. Jiang, R. Gupta, S. Cao, K. K. Liu, J. J. Morrissey, E. D. Kharasch, R. R. Naik and S. Singamaneni, ACS Appl. Mater. Interfaces, 2021, 13, 11414–11423 CrossRef CAS PubMed.
- J. Liang, K. Li, G. G. Gurzadyan, X. Lu and B. Liu, Langmuir, 2012, 28, 11302–11309 CrossRef CAS PubMed.
- Z. Wei, D. Chen, Z. Guo, P. Jia and H. Xing, Inorg. Chem., 2020, 59, 5386–5393 CrossRef CAS PubMed.
- G. R. Fleming, A. W. E. Knight, J. M. Morris, R. J. S. Morrison and G. W. Robinson, J. Am. Chem. Soc., 1977, 99, 4306–4311 CrossRef CAS.
- M. Tavakkoli Yaraki, F. Hu, S. Daqiqeh Rezaei, B. Liu and Y. N. Tan, Nanoscale Adv., 2020, 2, 2859–2869 RSC.
- Y. Zhang, K. Aslan, M. J. R. Previte and C. D. Geddes, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 1798–1802 CrossRef CAS PubMed.
- D. P. Damera, V. Krishna, V. V. K. Venuganti and A. Nag, J. Photochem. Photobiol., B, 2021, 225, 112335 CrossRef CAS PubMed.
- Z. Ahmad, Recent Advances in Dielectric Materials, Intechopen, 2012 Search PubMed.
- N. Ganesh, P. C. Mathias, W. Zhang and B. T. Cunningham, J. Appl. Phys., 2008, 103, 1–6 Search PubMed.
- G. Rezanejade Bardajee, Z. Hooshyar and M. Khanjari, J. Photochem. Photobiol., A, 2014, 276, 113–121 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: The content of ESI includes details of experimental methods and COMSOL simulations, FE-SEM images, average size plots, zeta potential graphs of bare GNRs and LBL coated GNRs. It also includes fluorescence plots, lifetime studies, average life time tables and SOG studies. See DOI: https://doi.org/10.1039/d4ra01551g |
| This journal is © The Royal Society of Chemistry 2024 |