
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnticancer potential of novel symmetrical and asymmetrical dihydropyridines against breast cancer via EGFR inhibition: molecular design, synthesis, analysis and screening†
Syed Faizan a,
Sirajunisa Talathb,
Adil Farooq Walib,
Umme Hanic,
Nazima Haiderd,
Subhankar P. Mandala and
B. R. Prashantha Kumar*a
a,
Sirajunisa Talathb,
Adil Farooq Walib,
Umme Hanic,
Nazima Haiderd,
Subhankar P. Mandala and
B. R. Prashantha Kumar*a
aDepartment of Pharmaceutical Chemistry, JSS College of Pharmacy, Constituent College of JSS Academy of Higher Education & Research, Mysuru 570015, India. E-mail: brprashanthkumar@jssuni.edu.in; Fax: +91-821-2548359; Tel: +91-821-2548353
bDepartment of Pharmaceutical Chemistry, RAK Medical & Health Sciences University, Ras Al Khaimah, UAE
cDepartment of Pharmaceutics, College of Pharmacy, King Khalid University, Abha, Saudi Arabia
dDepartment of Pathology, College of Medicine, King Khalid University, Abha, Saudi Arabia
First published on 9th April 2024
Abstract
A series of novel symmetrical and asymmetrical dihydropyridines (HD 1-15) were designed, subjected to in silico ADMET prediction, synthesized, analyzed by IR, NMR, Mass analytical techniques and evaluated against epidermal growth factor receptor (EGFR) as inhibitors against Breast cancer. The results of predicted ADMET studies demonstrated the drug-likeness properties of the reported compounds. The in vitro cytotoxicity assessment of the synthesized compounds revealed that all of them showed good activity (IC50 ranging from 16.75 to 66.54 μM) towards MCF-7 breast cancer cells compared to the standard drug, Lapatinib (IC50 = 2.02 μM). Among these, compounds HD-6, HD-7, and HD-8 displayed the most potent activity with IC50 value of 21.26, 16.75, and 18.33 μM, respectively. Cytotoxicity of all compounds was tested on normal vero cells for comparison at different concentrations using the MTT assay. In addition to the MTT assay, the potent dihydropyridines derivatives were screened for EGFRwt kinase inhibition assay at concentrations ranging from 1 nM to 360 nM. Among the three compounds tested, HD-8 showed reasonably good inhibition with an IC50 value of 15.90 ± 1.20 nM compared to a standard Lapatinib IC50 value of 10.28 ± 1.01 nM. Based on the molecular docking study against EGFR, the most active derivatives HD-7 and HD-8 were docked against the active site of the protein and showed better binding affinity than the standard lapatinib. Additionally, molecular dynamics (MD) simulations were performed to explore the stability of the protein–ligand complex, its dynamic behavior, and the binding affinity.
1. Introduction
Drug discovery research is a multidisciplinary field that leads to the development of novel therapeutic agents. It involves the design, synthesis, and optimization of molecules to target specific biological mechanisms or disease processes.1 Diverse methods are employed to comprehend the molecular basis of disease, to identify drug targets, and to design molecules that interact with these drug targets with therapeutic advantages.2,3 Studies of structure–activity relationships (SARs) advise the modification of lead compounds to increase their therapeutic efficacy and decrease their toxicity.4 Through iterative cycles of synthesis, evaluation, and optimization, medicinal chemists seek to create molecules with optimal drug-like properties, including high bioavailability, metabolic stability, and favorable pharmacokinetic profiles.5 Computational modeling and molecular simulations are valuable tools in drug discovery.6,7 In virtual screening, vast compound libraries are screened using computer algorithms to anticipate potential interactions with the target.8 Molecular docking techniques are employed to investigate how small molecules bind to the target protein, allowing researchers to evaluate the binding affinity and stability of potential drug candidates.9 This computational method facilitates the selection of lead compounds with the highest probability of success in subsequent drug development stages. In recent years, data-driven approaches in designing have emerged due to technological advancements and the availability of large biological data.10,11 The discovery of new drug targets, the prediction of ligand-receptor interactions, and the exploration of chemical space for prospective lead compounds are aided using bioinformatics and cheminformatics approaches to explore and analyze enormous databases of biological and chemical information.12Multicomponent reactions (MCRs) are key for lead discovery and drug design due to their efficiency, variety, and ability to rapidly generate diverse molecular scaffolds.13,14 MCRs leads to a product with varied functional groups and structural motifs by reaction with at least three building blocks.15 This synthetic approach efficiently compiles different chemical libraries, which helps to explore chemical space and identify lead compounds with required pharmacological activity.16 MCRs play key roles in the discovery of potential hits, leads and lead optimization.17,18 Furthermore, MCRs simplify synthetic methods to produce target compounds. This effectiveness helps lead discovery by allowing quick analog synthesis and SAR assessment.19,20 Hantzsch reaction is one of such multi-component reactions, it is a versatile and commonly used method for the synthesis of dihydropyridine derivatives, a class of compounds with a wide range of biological activities.21,22 Dihydropyridine derivatives' structural flexibility and pharmacological potential have made Hantzsch dihydropyridine synthesis a key tool in lead discovery and drug design. The pharmacological activities of these compounds include calcium channel modulation,23 anti-inflammatory properties,24 antihypertensive effects,25 analgesic,26 antidiabetic,27 antimicrobial,28 antiviral,29 AT1 receptor blocker,30 antitumor,31 anticancer.32 The ability to synthesize dihydropyridines via the Hantzsch synthesis allows researchers access to a diverse library of compounds for evaluation in drug discovery.
Hantzsch adducts suppress proliferation in breast, ovary, kidney, non-small lung, prostate, colon, and glioma cancer cells.33 ErbB receptor tyrosine kinases are essential for breast cancer development. Breast cancer progression involves HER2/ErbB2 and EGFR/ErbB1.34 Tyrosine kinase inhibitors (TKIs) are used to treat various types of cancer, such as breast, gastric, lung, kidney, and pancreatic cancers. There are three generations of TKIs, including first-generation TKIs like lapatinib, erlotinib, gefitinib, and icotinib; second-generation EGFR inhibitors like afatinib, neratinib, and dacomitinib; and third-generation TKIs like osimertinib, olmutinib, and almonertinib.35 EGFR inhibitors are effective in breast cancer subgroups with EGFR overexpression or mutations but have limited benefit in random patient populations as single agents. They fail due to genomic instability and tumor heterogeneity, causing subsequent mutations in the EGFR gene, activation of alternative signaling pathways resulting to drug resistance.36
In this context, to address the resistance of EGFR towards current TKIs while minimizing their adverse effects, our study focuses on the design, synthesis, cytotoxic assessment, EGFR inhibition, molecular docking, and molecular dynamics simulation of novel symmetrical and asymmetrical Hantzsch dihydropyridines targeting breast cancer. This comprehensive investigation not only presents promising candidates for overcoming EGFR resistance but also offers insights into their potential as safer and more effective therapeutic agents for breast cancer treatment.
2. Results and discussion
2.1. Molecular design
The novel symmetrical and asymmetrical dihydropyridine-based tyrosine kinase inhibitors (TKIs) were designed through scaffold hybridization of gefitinib, afatinib, and axitinib, representing the 1st, 2nd, and 3rd generations of TKIs, respectively (Fig. 1). The dihydropyridine moiety serves as the central scaffold for binding with the tyrosine kinase receptor, with structural optimization achieved by incorporating various substituents onto the benzene ring of the dihydropyridine to ensure an optimal balance of hydrogen bond donors, acceptors, hydrophobic interactions, and functional groups for enhanced target selectivity. Our drug design approach involved the strategic integration of structural elements from first, second, and third generation TKIs to leverage their advantageous pharmacological properties while overcoming limitations such as resistance mechanisms and off-target effects. The synthesized compounds were characterized and evaluated for their activity against MCF-7 breast cancer cells and EGFRwt.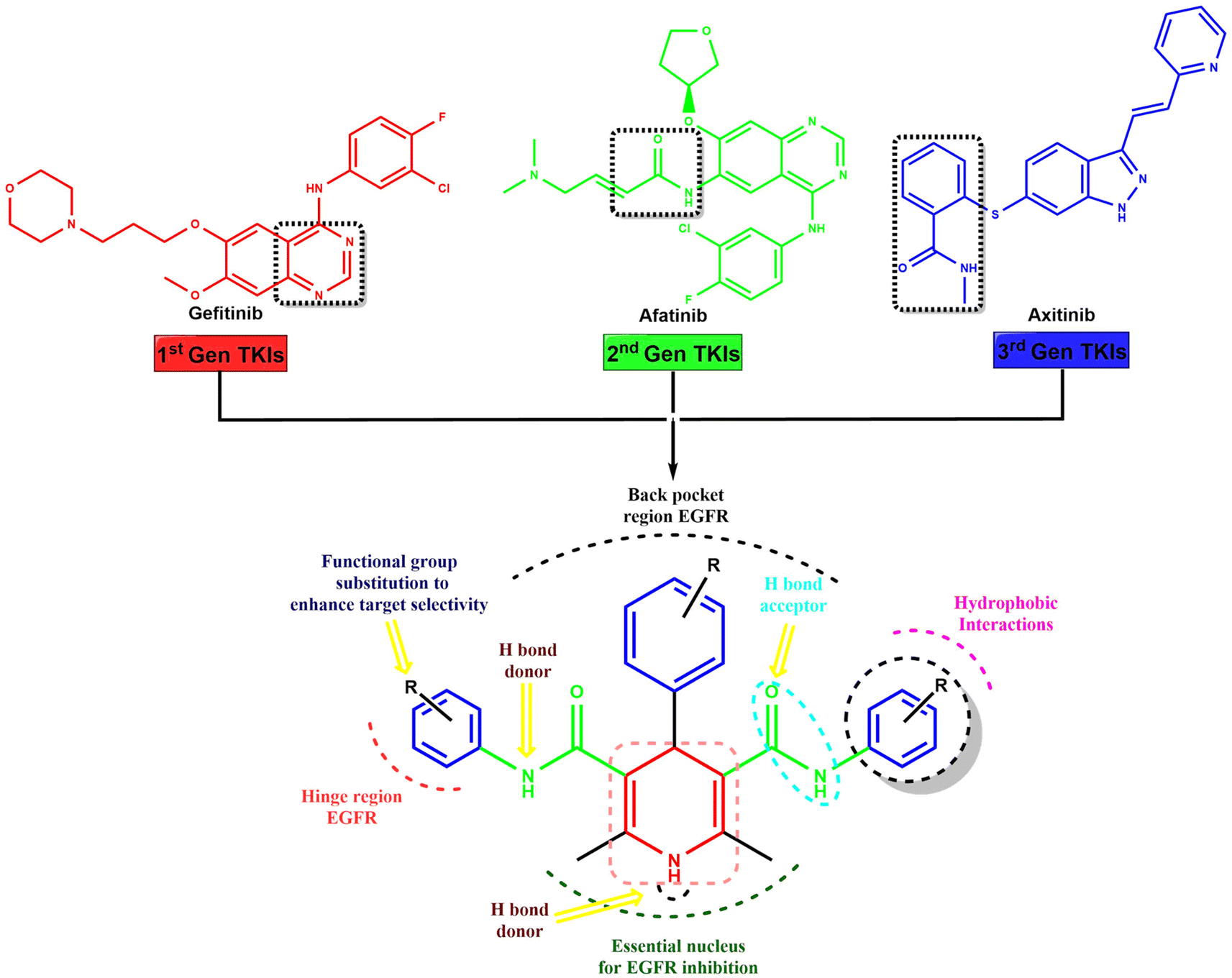 | ||
| Fig. 1 Scaffold hybridization based rational design strategy of the novel dihydropyridines for EGFR inhibition via combining the structural features of 1st, 2nd, and 3rd generation tyrosine kinase inhibitors gefitinib, afatinib and axitinib. | ||
2.2. Chemistry
In the synthesis of acetoacetanilide derivatives, compound A1 (N-(2,4-dimethoxyphenyl)-3-oxobutanamide) was prepared by refluxing 2,4-dimethoxy aniline with ethyl acetoacetate under solvent-free conditions for 2 hours. Similarly, compound A2 (N-(4-chloro-2,5-dimethoxyphenyl)-3-oxobutanamide) was synthesized by refluxing 4-chloro-2,5-dimethoxy aniline with ethyl acetoacetate under identical solvent-free conditions for 2 hours. Subsequently, a parallel synthesis approach was employed for the generation of symmetrical dihydropyridines (HD 1-10). The synthesis involved refluxing acetoacetanilide A1 or A2 with the corresponding aldehyde and ammonia solution in iso-propanol for 24–30 hours, resulting in the formation of dihydropyridines. Compounds HD 1-5 were synthesized by refluxing acetoacetanilide A1 with the appropriate aldehyde and ammonia solution in iso-propanol for 24–30 hours, while compounds HD 6-10 were synthesized by refluxing acetoacetanilide A2 under the same conditions. Additionally, an analogous procedure was employed for the parallel synthesis of asymmetrical dihydropyridines (HD 11-15), achieved by refluxing an equimolar mixture of both acetoacetanilide A1 and A2 with the corresponding aldehyde and ammonia solution in iso-propanol for 24–30 hours. These synthetic protocols demonstrate the successful preparation of diverse dihydropyridine derivatives as shown in Scheme 1, paving the way for subsequent analysis and cytotoxicity assessments against MCF-7 breast cancer cells, as well as elucidating their potential as EGFR inhibitors.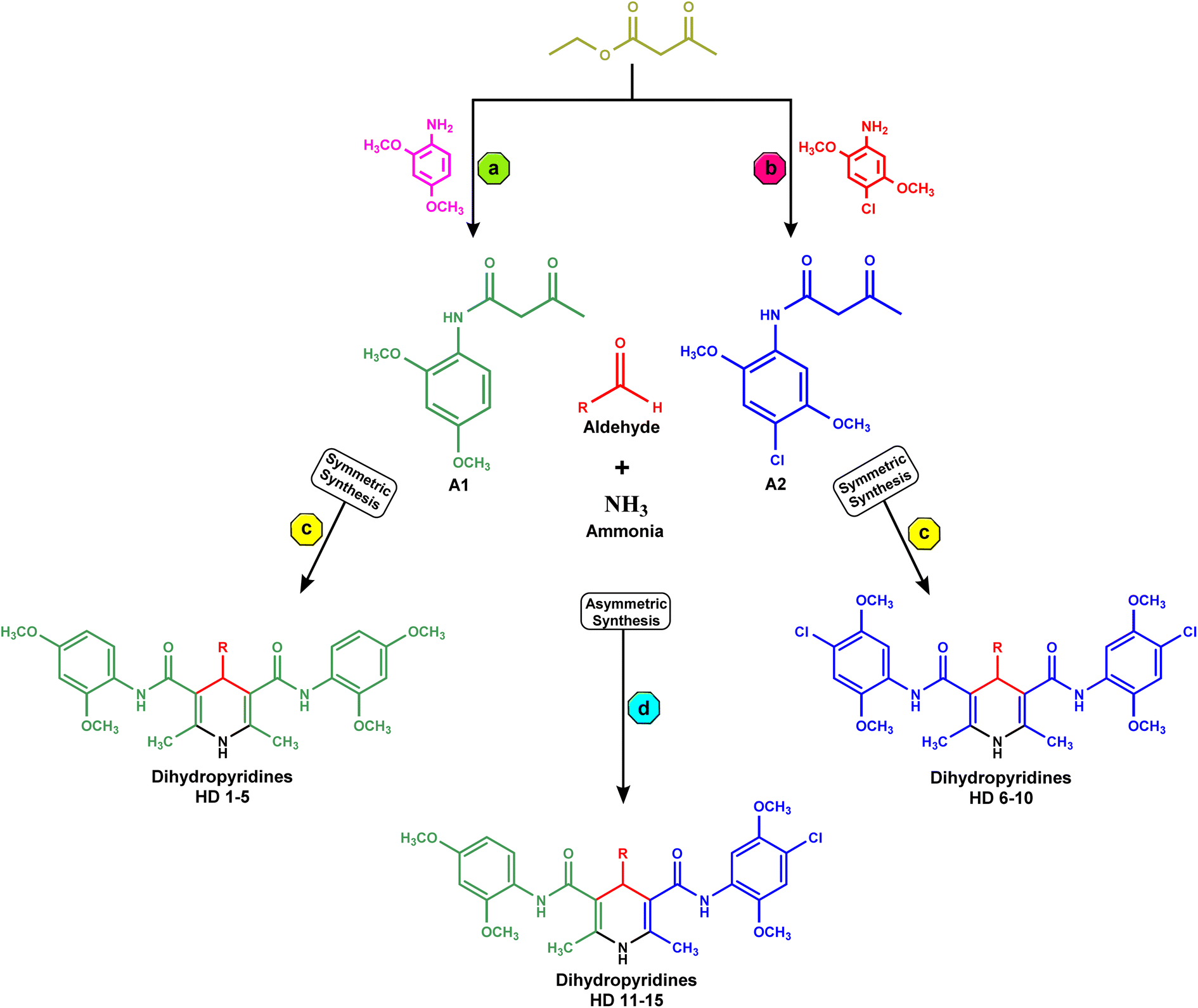 | ||
| Scheme 1 (a) Synthesis of A1, reflux, solvent free condition for 2 h; (b) synthesis of A2, reflux, solvent free condition for 2 h; (c) parallel synthesis of symmetrical dihydropyridines (HD 1-10) by refluxing acetoacetanilide A1 or A2 with appropriate aldehyde and ammonia solution in iso-propanol for 24–30 h; (d) parallel synthesis of asymmetrical dihydropyridines (HD 11-15) by refluxing equimolar mixture of acetoacetanilide A1 and A2 with appropriate aldehyde and ammonia solution in iso-propanol for 24–30 h. | ||
2.3. In vitro anticancer activity
Synthesized molecules were subjected for their anti-cancer activity against MCF-7 breast cancer cells and the cytotoxicity was determined through MTT assay. So far there is much evidence for the screening of dihydropyridines for anti-cancer activity especially for EGFR expression in MCF-7 breast cancer cells.37 The novel compounds are tested for their cytotoxicity and the IC50 values were listed in Table 1 and Fig. 2 below. The results indicate that the selectivity of dihydropyrimidines toward the MCF-7 breast cancer cells compared to the normal vero cells based on their IC50 values. Among the tested compounds, HD-6 (21.26), HD-7 (16.75), and HD-8 (18.33) exhibited significant cytotoxicity. HD-1 (66.54), HD-3 (59.35) and HD-5 (62.12) exhibited relatively weak cytotoxicity. The compounds with electron donating groups such as OH, OCH3 showed less activity whereas electron withdrawing groups such as Cl showed good activity in terms of cytotoxicity against MCF-7 breast cancer cells.| Compound code | Structure | MCF-7 (IC50 μM ml−1) | Vero (IC50 μM ml−1) |
|---|---|---|---|
| A1 | 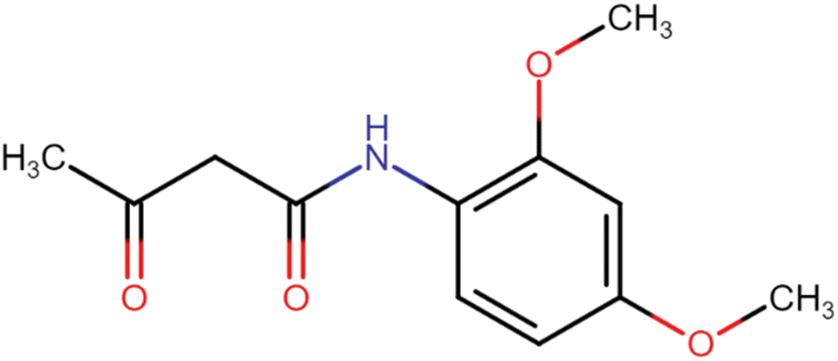 |
— | — |
| A2 | 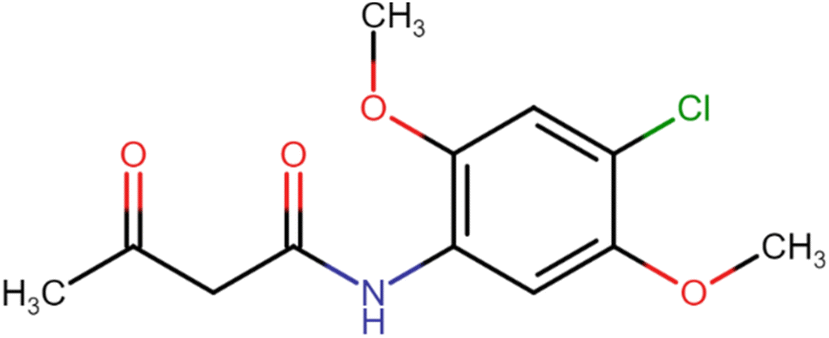 |
— | — |
| HD-1 | 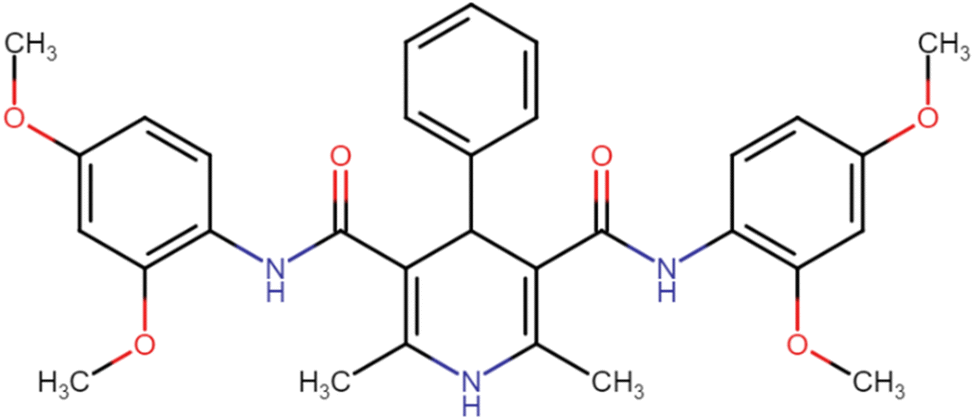 |
66.54 | 86.52 |
| HD-2 | 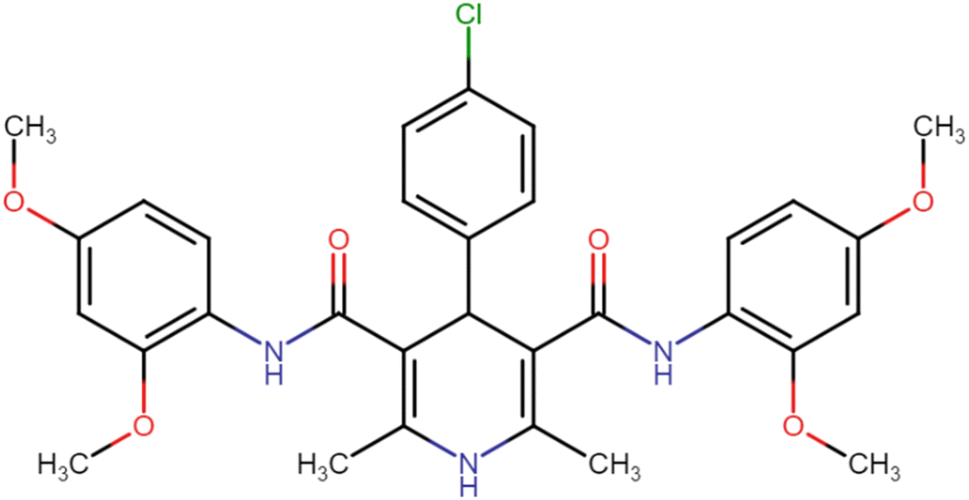 |
51.36 | 76.38 |
| HD-3 | 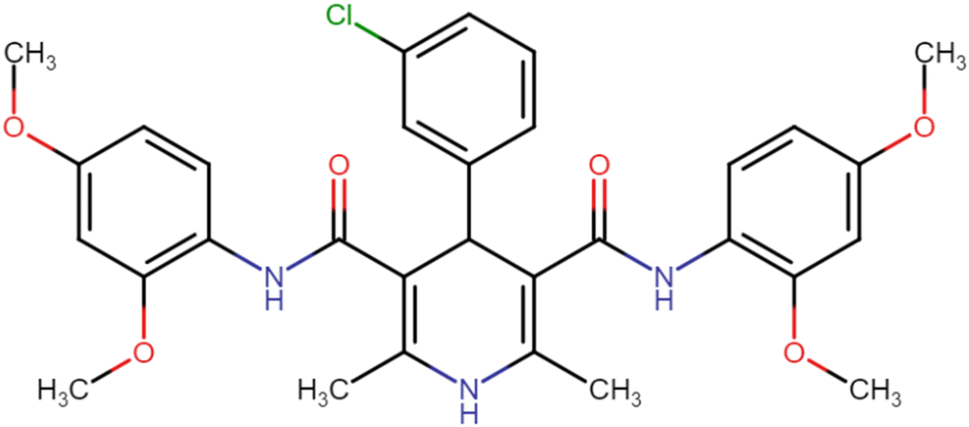 |
59.35 | 67.46 |
| HD-4 | 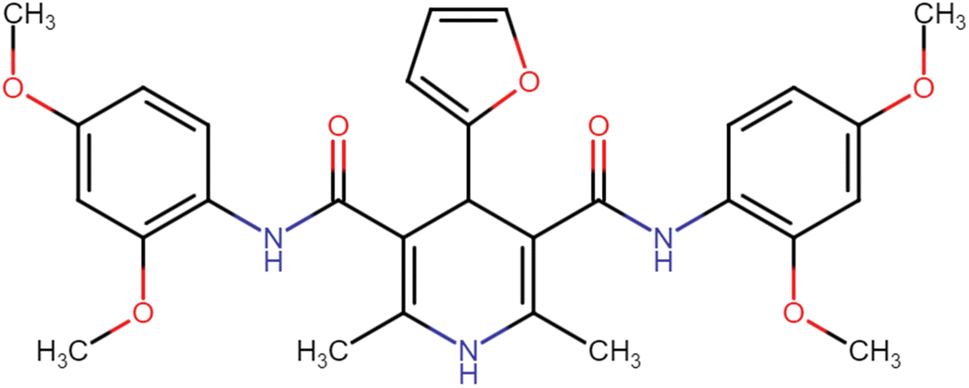 |
36.04 | 51.64 |
| HD-5 | 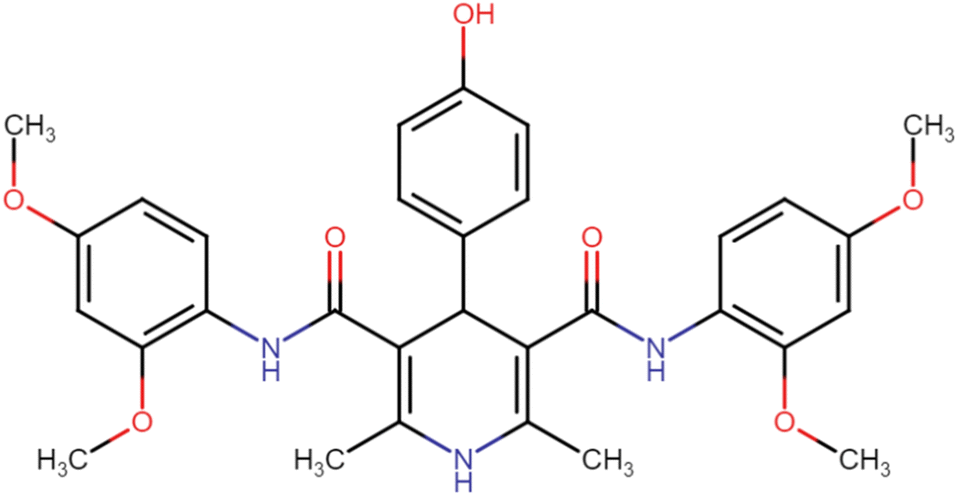 |
62.12 | 70.33 |
| HD-6 | 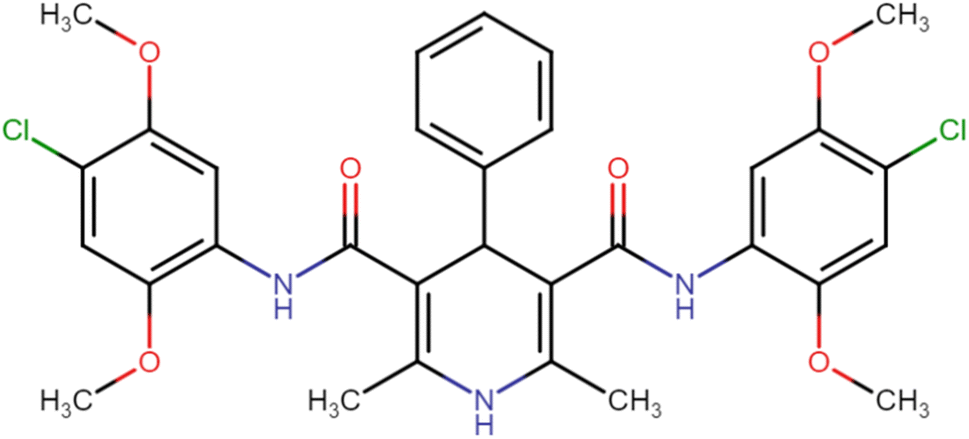 |
21.26 | 42.23 |
| HD-7 | 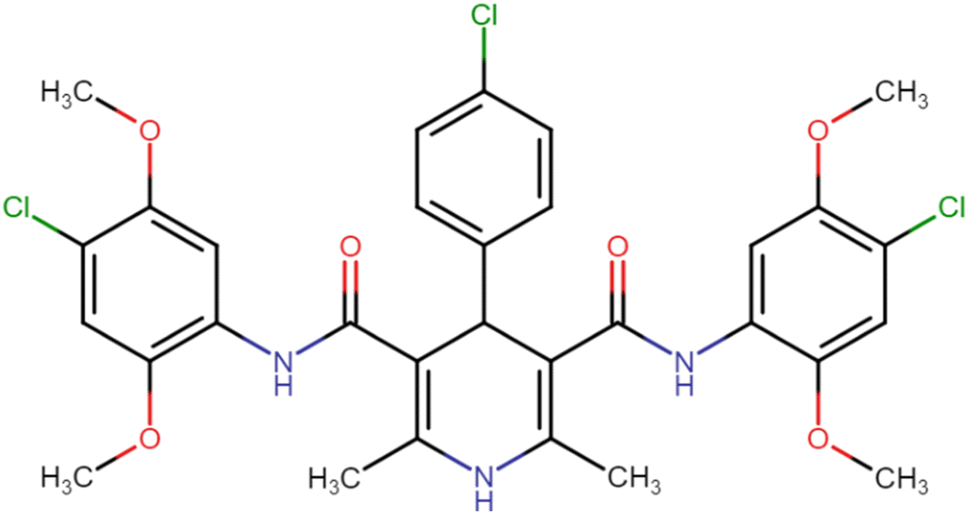 |
16.75 | 27.35 |
| HD-8 | 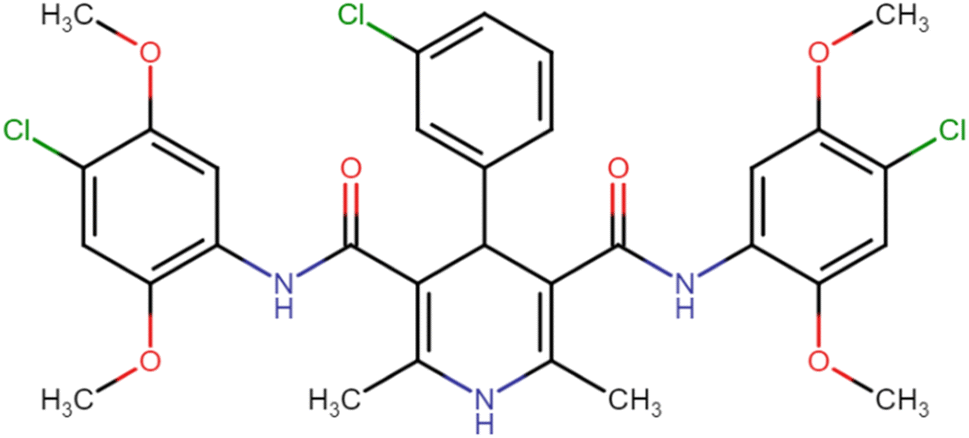 |
18.33 | 28.41 |
| HD-9 |  |
57.66 | 69.84 |
| HD-10 |  |
39.60 | 51.76 |
| HD-11 |  |
42.44 | 59.62 |
| HD-12 |  |
37.46 | 54.01 |
| HD-13 | 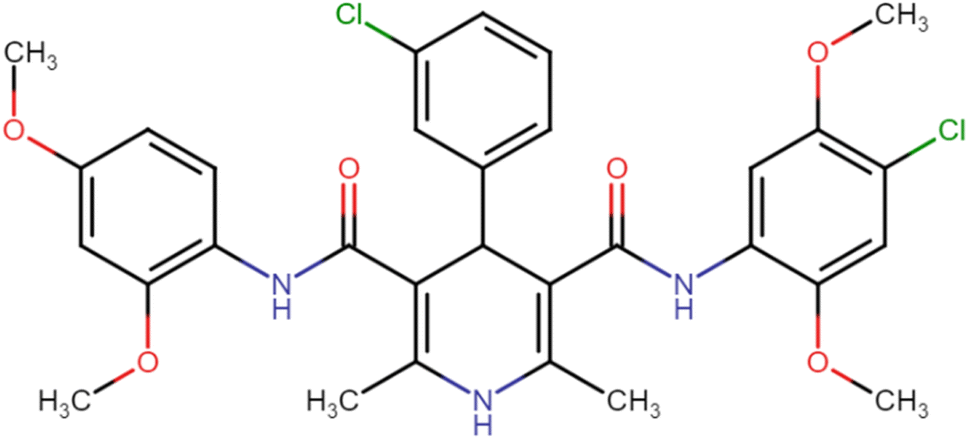 |
44.07 | 49.59 |
| HD-14 | 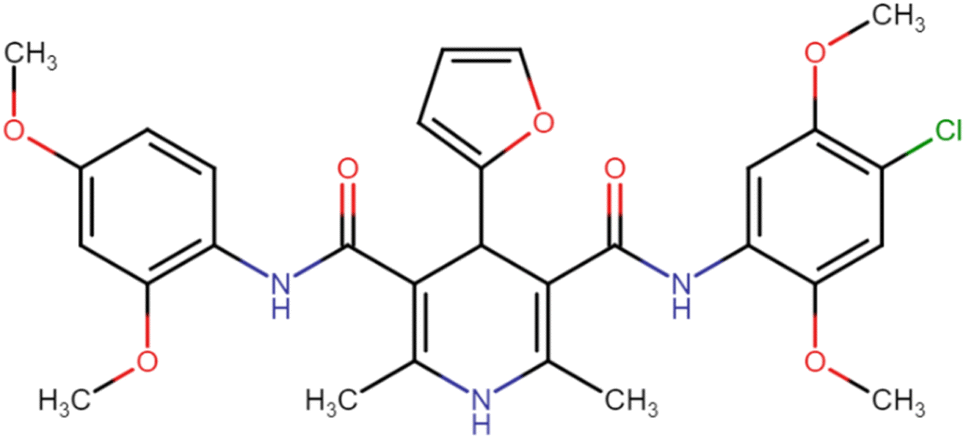 |
52.29 | 68.13 |
| HD-15 | 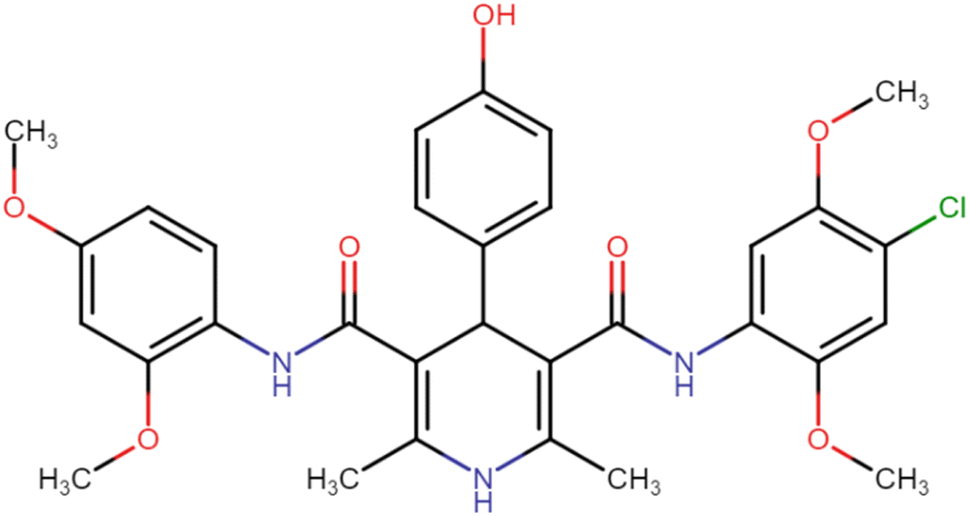 |
42.19 | 60.25 |
| STD | Lapatinib | 2.02 | 17.46 |
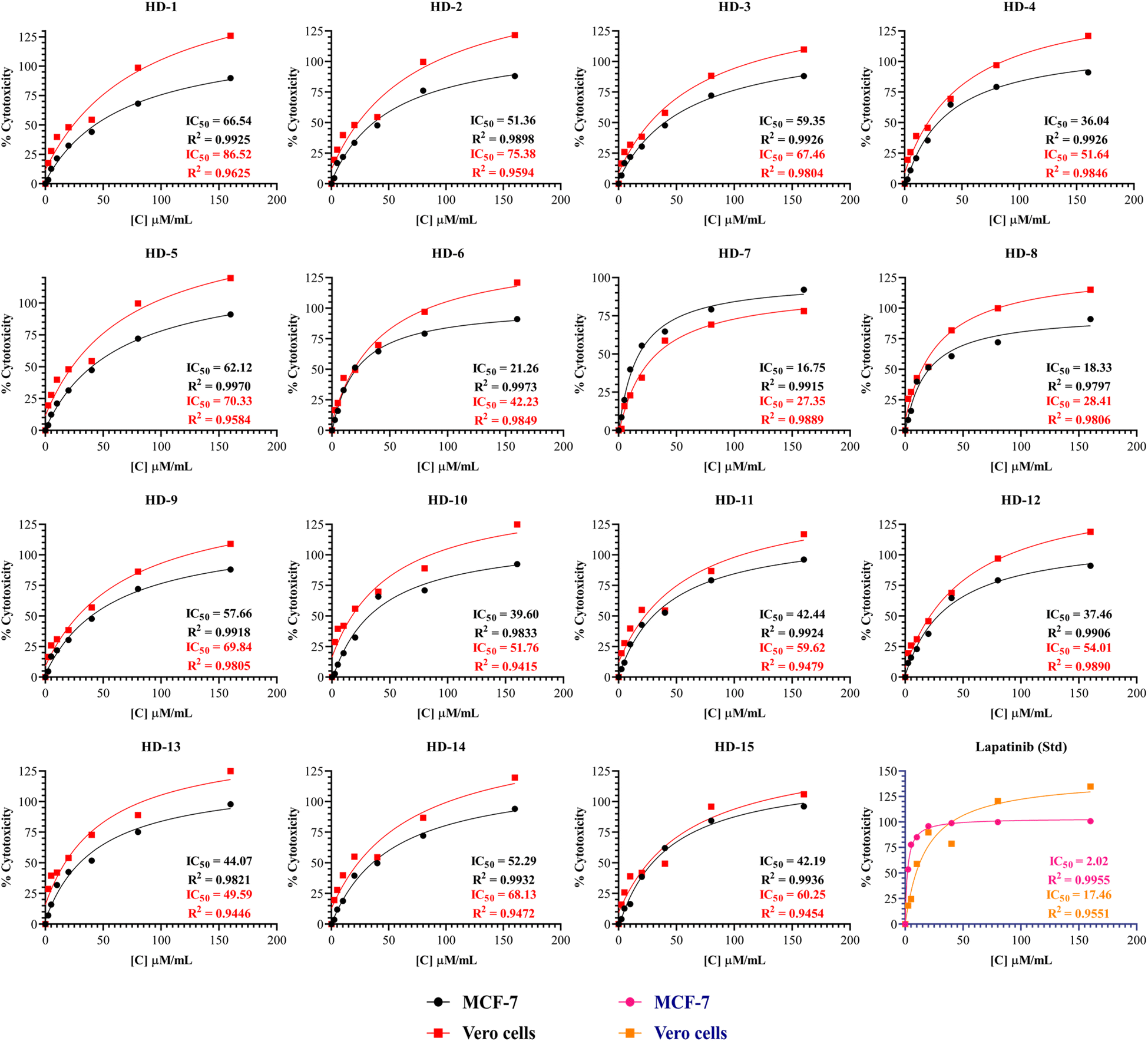 | ||
| Fig. 2 The percentage cytotoxicity at different concentrations of synthesized dihydropyridines and lapatinib (Std) against MCF-7 and Vero cells with its IC50. | ||
A comprehensive structure-activity relationship (SAR) analysis of symmetrical and asymmetrical dihydropyridine derivatives was derived from simple observation to correlate their anticancer activity against the MCF-7 cell line. Symmetrical dihydropyridines (HD 1-10) showed a mean IC50 value of 40.879 μM ml−1, characterized by symmetrically positioned substituents around the dihydropyridine ring. Compounds with electron-withdrawing substituents such as Cl exhibited lower IC50 values, indicating increased potency. In contrast, asymmetrical dihydropyridines (HD 11-15) exhibited a mean IC50 value of 43.09 μM ml−1, showcasing structural diversity with dissimilar substituents on each side of the dihydropyridine ring. While electron-withdrawing Cl groups contributed to enhanced potency in certain asymmetrical derivatives, overall activity varied widely among the compounds. Steric hindrance effects were more prominent in symmetrical derivatives due to identical substituent proximity, while asymmetrical derivatives demonstrated a broader range of steric interactions, influencing their binding affinity and activity.
2.4. Molecular docking studies
In this study, we used Biovia Discovery Studio to run molecular docking simulations while looking at the interactions that occur when a receptor protein and a ligand molecule are bound together. The preparation of the receptor included the removal of non-interacting water molecules, charging, and structural optimization. Ligands were prepared by sketching the 2D structures of synthesized molecules in ChemDraw professional software and later converted into 3D. The CDOCKER method was used for docking, with the grid generation, scoring function, and other parameters adjusted appropriately. The binding energy was used to rank and grade the docking postures. Specific interactions between the receptor and ligand, including hydrogen bonds and hydrophobic interactions, were discovered by analyzing the top-ranked postures. By redocking well-known inhibitor lapatinib and comparing the outcomes with experimental data, the docking process was validated, demonstrating its accuracy and dependability. The binding energy was calculated selecting the top poses of each interacting complex.To narrow down the hits and explore their binding modes, molecular docking was performed. The affinity and the binding mode of all compounds and standard drug with target were evaluated, and the docking results are listed in Table 2. Among all the observed compounds, HD-8 exhibited the better binding energy (−113.607 kcal mol−1) followed by HD-7 and HD-6 with −113.529, −47.2839 kcal mol−1 respectively while the standard compound (lapatinib) depicts −90.8851 binding affinity respectively. Interestingly, the synthesized derivatives HD-7 and HD-8 exhibited higher binding energies with the protein as compared to the lapatinib. The binding mode interactions of receptor-ligand were examined in 2D and 3D, shown in Fig. 3.
| Ligand | Binding energy (kcal mol−1) | -CDOCKER interaction energy |
|---|---|---|
| HD-6 | −47.2839 | 53.1584 |
| HD-7 | −113.529 | 45.3379 |
| HD-8 | −113.607 | 62.1555 |
| Std (lapatinib) | −90.8851 | 70.6095 |
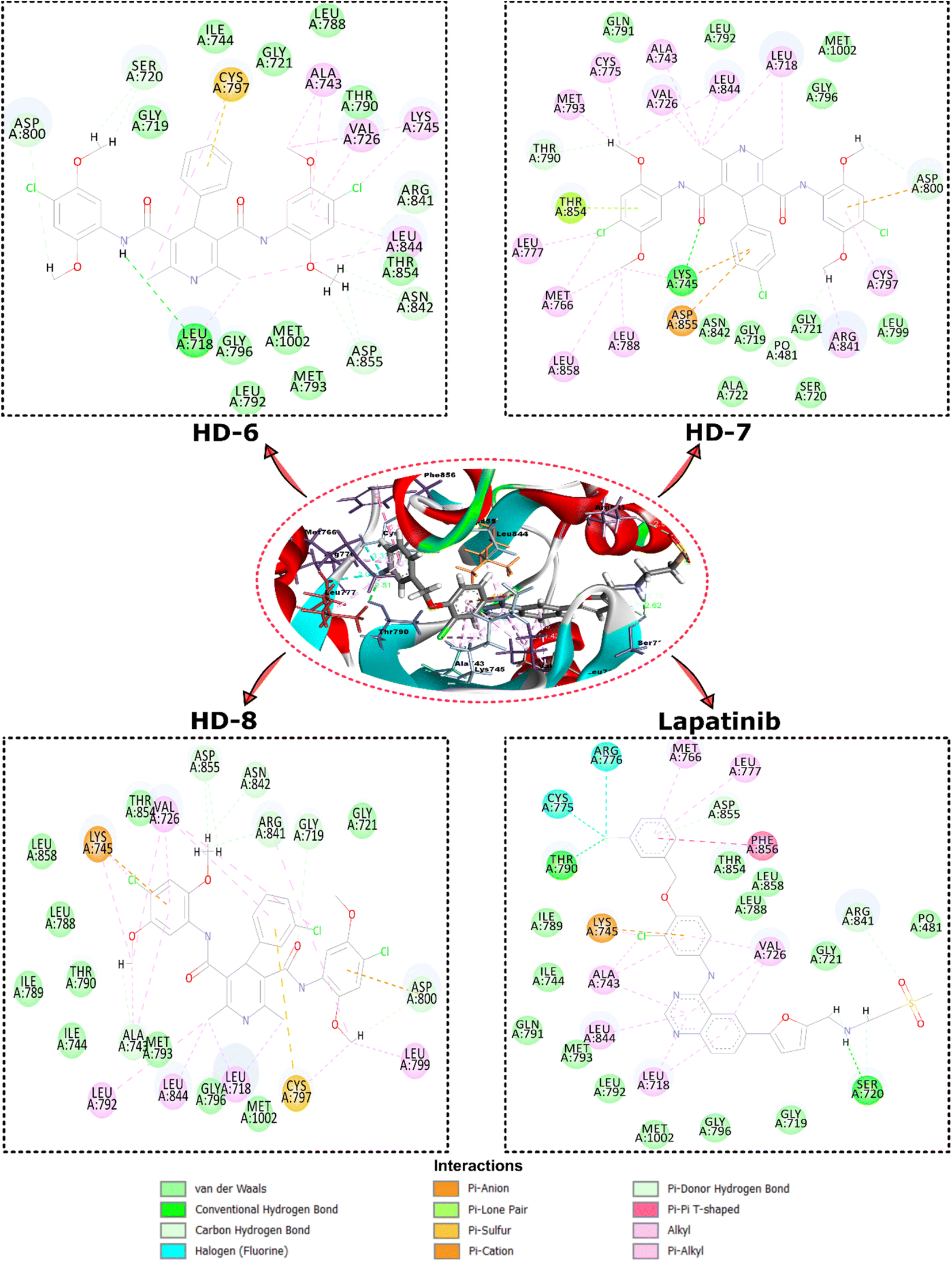 | ||
| Fig. 3 Depicts the top view binding of compounds at the active site of the drug target protein EGFR kinase domain (PDB ID: 1XKK). The figure showcases the docking poses of compounds with the highest dock scores (HD-6, HD-7, and HD-8) and lapatinib. | ||
Further analysis of the molecular interactions shed light on the binding modes of these compounds. Lapatinib exhibited a diverse interaction profile, forming π-cation interactions with LYS A:745, π-donor interactions with ASP A:855, ARG A:841, and conventional hydrogen bonds with SER A:720 and THR A:790. Additionally, it engaged in halogen interactions with ASG A:776 and CYS A:775, as well as π-alkyl interactions with LEU A:718, ALA A:743, LEU A:844, and VAL A:726. These interactions suggested lapatinib's versatility in engaging with various amino acid residues. HD-8 primarily engaged in conventional hydrogen bonding, notably with GLY A:719, ALA A:743, GLY A:796, ARG A:841, ASN A:842, ASP A:855, and MET A:1002 along with π-cation interaction with LYS A:745. It also formed π-alkyl interactions with LEU A:718, VAL A:726, ALA A:743, and LEU A:844, indicating its preference for hydrophobic interactions. HD-7, on the other hand, showed distinctive interactions, including conventional hydrogen bonding with LYS A:745, and van der Waals interactions with GLN A:791, LEU A:792, GLY A:796, MET A:1002 and CH bond interactions with GLY A:719, SER A:720, GLY A:721, ALA A:722, LEU A:799, and π-alkyl interactions with CYS A:797, LEU A:788, ARG A:841, LEU A:858. HD-6 also formed conventional hydrogen bonds, particularly with LEU A:718 and π-alkyl bonds with LYS A:745 and LEU A:844. These findings suggest that HD-8 and HD-7 may have a unique binding mechanism, potentially making it a promising candidate for further study.
2.5. ADMET studies
The ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) studies conducted for this research utilized the comprehensive platform of BIOVIA Discovery Studio. Chemical structures of the target compounds were collected and prepared, ensuring compatibility with the software. Assessing the ADMET and toxic characteristics of small molecules is a crucial component of their evaluation. The recently developed dihydropyridine derivatives have been identified as safe for hepatic cells, do not hinder the CYP2D6 metabolizing enzyme, exhibit non-mutagenic properties, and do not induce carcinogenic effects in both male and female rat models, as summarized in Table 3. Additionally, all the compounds have notably low (level = 1) aqueous solubility, indicating their hydrophobic nature.| Drug | Solubility | BBB | CYP2D6 | HIA | NTP rat | Ames mutagen | |
|---|---|---|---|---|---|---|---|
| Male | Female | ||||||
| a Abbreviations: NI: non-inhibitor, NC: non-carcinogen, NM: non-mutagen. • Aqueous solubility: 1,2,3,4,5,6 → level 3 – good • BBB penetrations: 0-very high, 1-high, 2-medium, 3-low, 4-undefined. • CYP2D6 binding (for metabolism present in liver & brain): inhibitor-true, non-inhibitor-false. • Hepatotoxicity: true-toxic, false-non-toxic. • HIA; 0-very good, 1-good. | |||||||
| HD-6 | 1 | 4 | NI | 0 | NC | NC | NM |
| HD-7 | 1 | 4 | NI | 0 | NC | NC | NM |
| HD-8 | 1 | 4 | NI | 0 | NC | NC | NM |
Table 4 illustrates that the observed oral bioavailability of these compounds, consistent with Lipinski's Rule of 5, surpasses 60%. This implies that these compounds are likely to be effectively absorbed and distributed when taken orally. It is important to note that a compound deviating from Lipinski's Rule of 5 in terms of hydrophobicity may encounter challenges in absorption and distribution. In such instances, alternative formulation approaches, like the use of solubilizing agents, can be employed to enhance solubility and bioavailability.
| Drug | Alogp | MW | HBA | HBD | Rat oral LD50 g per kg body weight | Rat Inhalation LC50 mg m−3 h−1 | Carcinogenic potency TD50 rat mg per kg body weight per day |
|---|---|---|---|---|---|---|---|
| a Abbreviations: MW: molecular weight, HBA: hydrogen bond acceptor, HBD: hydrogen bond donor. | |||||||
| HD-6 | 4.993 | 612.50 | 9 | 3 | 1.371 | 600.936 | 8.734 |
| HD-7 | 5.658 | 646.95 | 9 | 3 | 1.096 | 523.487 | 4.483 |
| HD-8 | 5.658 | 646.95 | 9 | 3 | 1.263 | 496.885 | 5.134 |
2.6. EGFRwt kinase inhibition assay
The inhibitory activities of compounds HD-6, HD-7, and HD-8 against EGFRwt kinases were assessed. Lapatinib was used as standard with IC50 = 10.28 ± 1.01 nM. Compounds HD-6, HD-7, and HD-8 excellently inhibited EGFRwt activity with IC50 = 30.53 ± 1.48, 26.53 ± 1.42 and 15.90 ± 1.20 nM respectively. The dose response curve and IC50 values of the compounds tested are depicted in Fig. 4 below.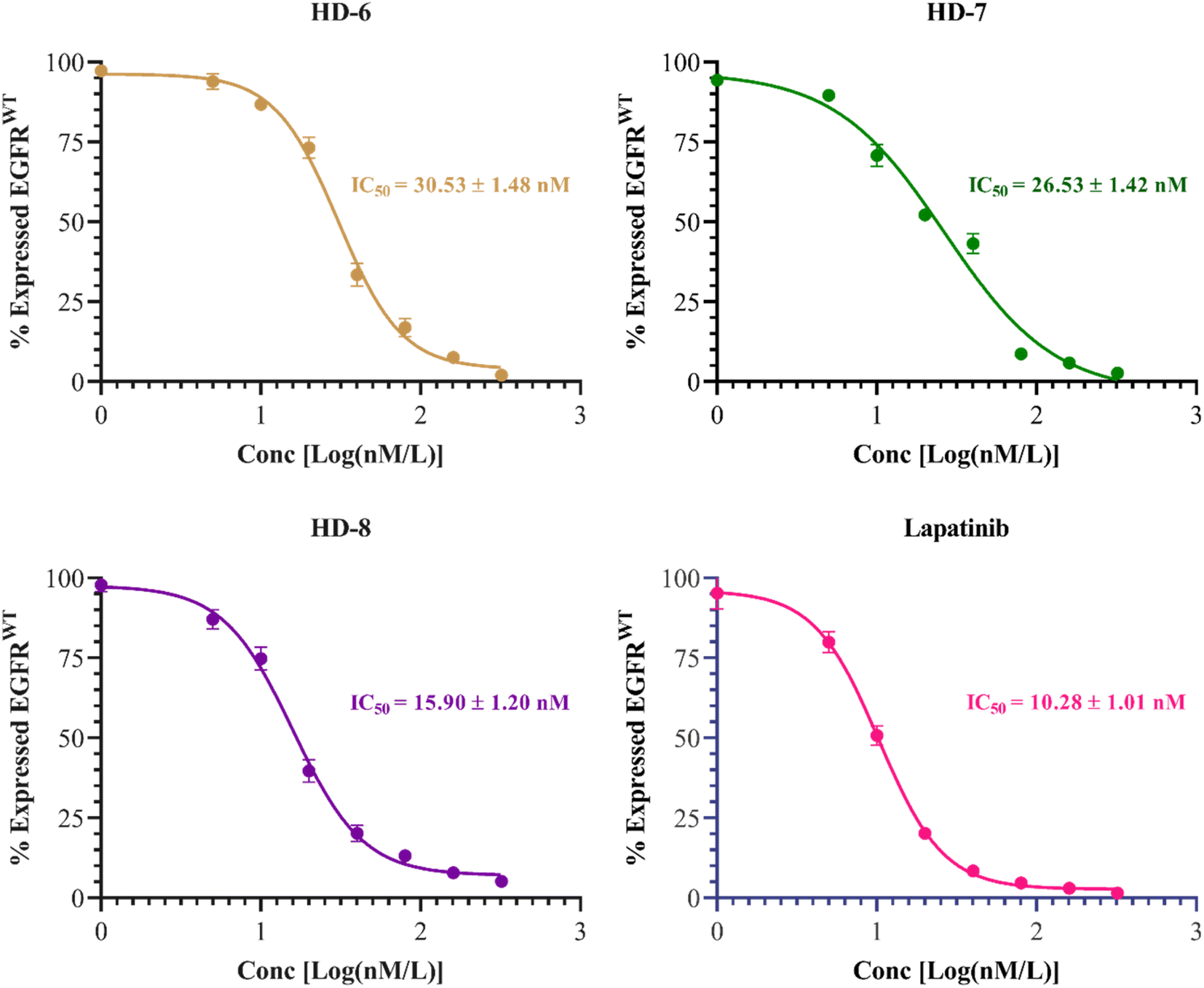 | ||
| Fig. 4 Dose response inhibitory curves and IC50 of synthesized derivatives (HD-6, HD-7, and HD-8) and standard drug lapatinib against EGFRwt. | ||
2.7. Molecular dynamics simulation
A stable bioactive conformer of the synthesized ligands (HD-7 and HD-8) were identified in the binding pocket of the EGFR protein through molecular docking experiments. Nevertheless, the stability of crucial binding interactions could not be determined due to the flexibility of residues and corresponding secondary structure fluctuations in static mode of molecular docking. To overcome these challenges, we conducted molecular dynamics (MD) simulations, which effectively examined the stability of the protein–ligand complex. Thermodynamically optimized solvated ligand-bound protein complexes were studied for dynamic behavior and stability through a 100 ns production run. Additionally, the trajectory of the entire simulation run was analyzed for protein conformation changes by superimposing the structures of different complexes of ligand-free protein and ligand-bound protein at time intervals of 0 ns, 25 ns, 50 ns, 75 ns, and 100 ns as shown in Fig. 5. Initially, the unbound and ligand-bound protein structures aligned perfectly with each other at the beginning of the simulation run. However, a deviation in terms of Root Mean Square Deviation (RMSD) was observed when the simulation was extended while maintaining the same secondary structure pattern and ligand binding. This deviation was due to the flexibility of the protein system in a solvated environment.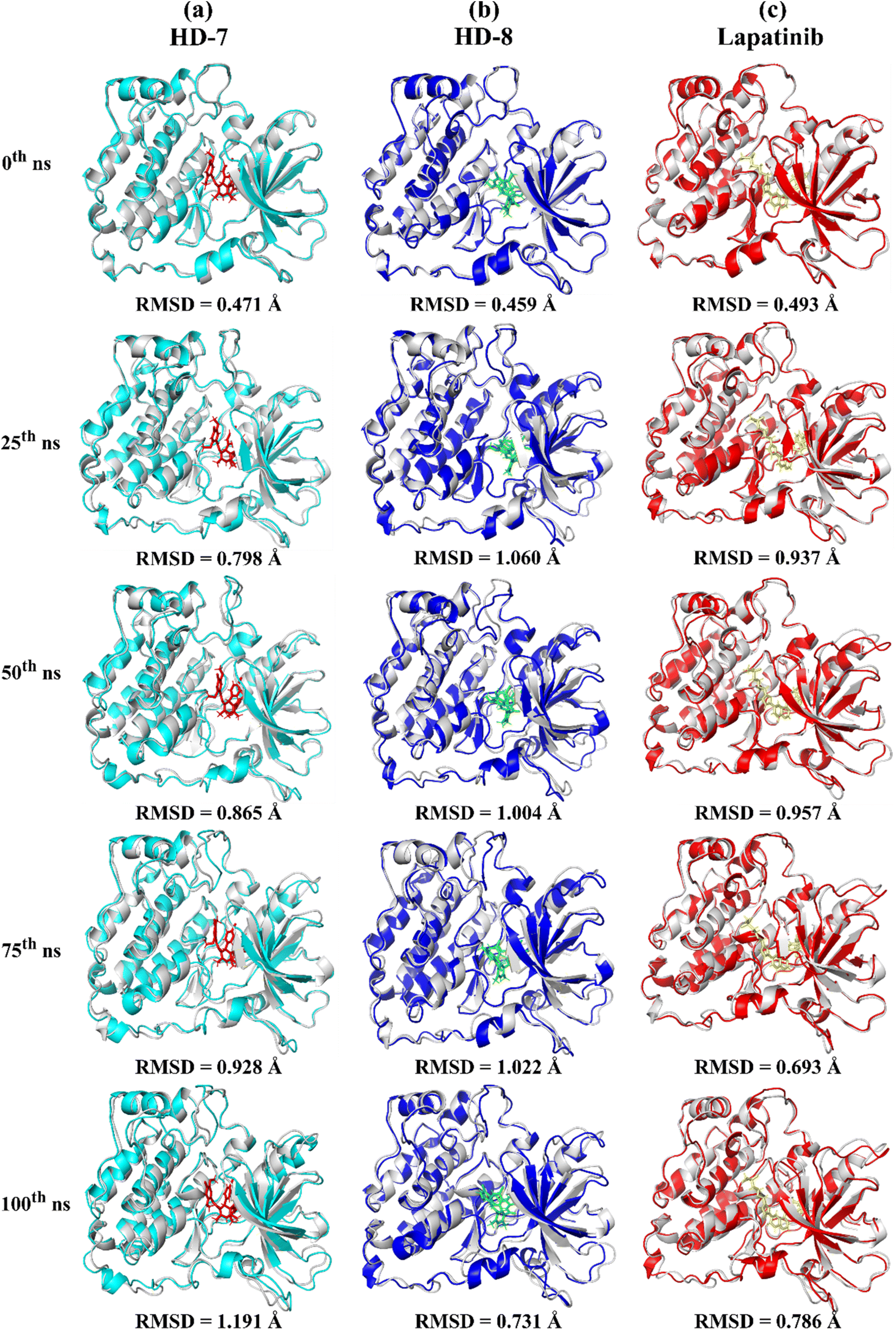 | ||
| Fig. 5 Gradual superimposed conformations of trajectory of (a) ligand-free protein EGFR and ligand (HD-7)-bound EGFR complex, (b) ligand-free protein EGFR and ligand (HD-8)-bound EGFR complex, (c) ligand-free protein EGFR and ligand (lapatinib)-bound EGFR complex, in time-lapse at 0th, 25th, 50th, 75th, and at 100th ns time. | ||
An RMSD plot was created by considering the C-α backbone fluctuations of the protein, which indicated the overall stability and equilibrium of the complex. The RMSD plot (Fig. 6a) showed the comparative stability between the ligand-free protein and ligand-bound protein complexes. Throughout all the simulation runs, a stable trajectory with minimal deviations was only observed after the system had reached equilibrium at around 15 ns. Additionally, it was discovered that the structural rigidity of the ligand-free protein was lower than that of its ligand-bound form. However, stable trajectories were only achieved after initial fluctuations in all simulation runs. To further analyze the motion of the system, the radius of gyration (Rg) was plotted, considering the varying masses of the system, and calculating the root mean square distances around the central axis of rotation. This plot effectively demonstrated the system's ability to maintain its shape and secondary structure folding during motion, as shown in Fig. 6b at the 15 ns time scale. In comparison to ligand-free proteins, ligand-bound protein complexes exhibited a higher gyration scale. Electrostatic interactions, including hydrogen bonds, are crucial for the stability and biological functions of any protein–ligand complex. During the 100 ns simulation run, intermolecular hydrogen bonds were observed (Fig. 6c), which is consistent with the results of molecular docking experiments. The hydrophobic surface area around the ligand-bound protein complexes was mapped using solvent accessible surface area (SASA) plots (Fig. 6d), which were consistent throughout the simulation run. Root mean square fluctuation (RMSF) plots (Fig. 6e and f) were used to map the fluctuation of crucial amino acids during the simulation. The plots revealed that the flexibility of the crucial interacting amino acids was increased in the complex compared to their innate state in the protein.
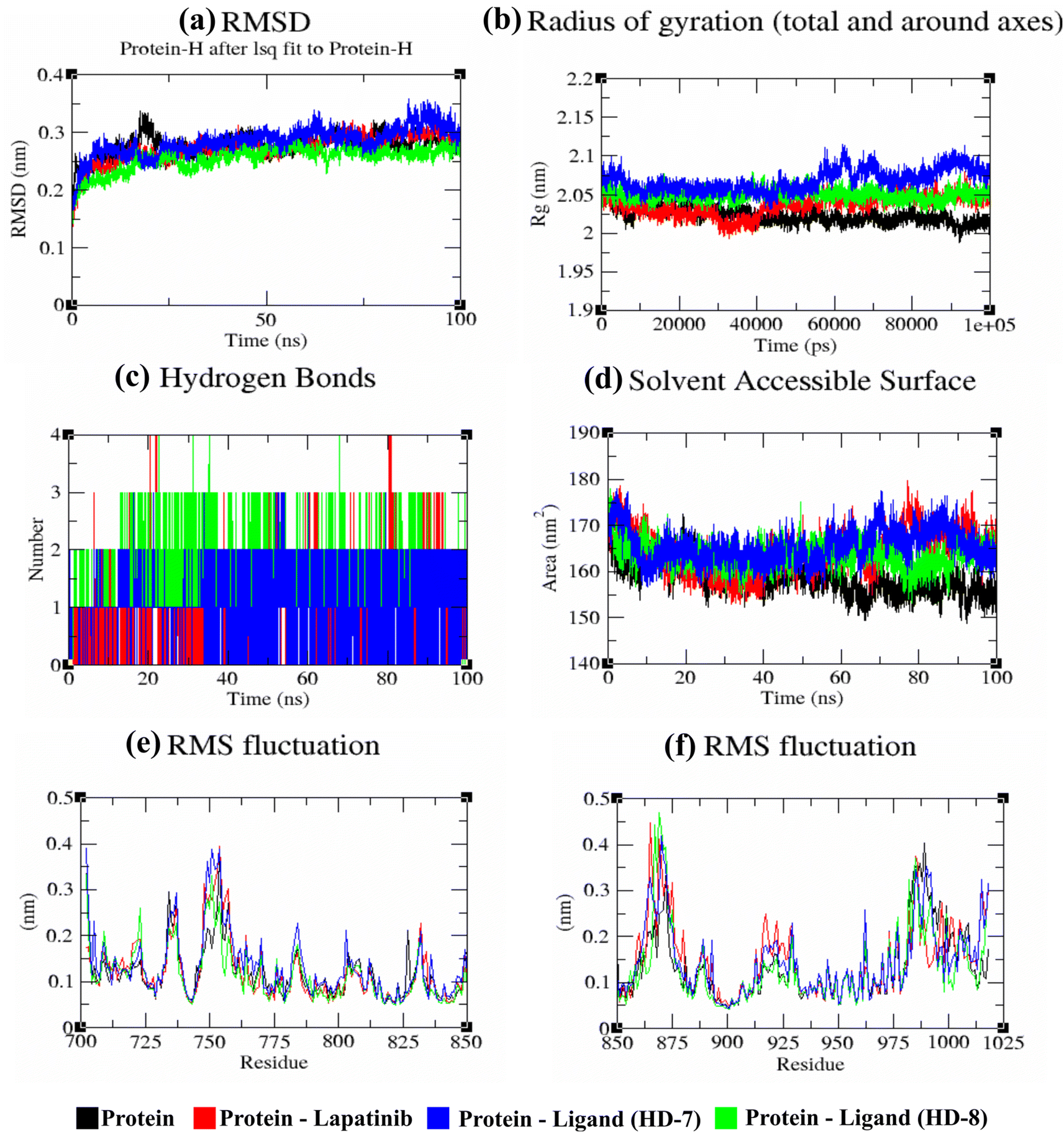 | ||
Fig. 6 Investigation of EGFR (epidermal growth factor receptor) protein complexes with HD-7, HD-8, and pioglitazone ligands at a simulation duration of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ps (100 ns). (a) Plot illustrating the temporal evolution of backbone RMSD (Root Mean Square Deviation) for EGFR protein both in isolation and in complex with the ligands. (b) Graph depicting the change in protein backbone Radius of Gyration (Rg) between its unbound state and when bound to the ligands throughout the simulation. The y-axis represents Rg in nanometers, while the x-axis denotes the simulation time in picoseconds. (c) Analysis of hydrogen bond formation between the protein and ligands across the simulation time in nanoseconds. (d) Representation of Solvent Accessible Surface Area (SASA) over time, where the y-axis indicates SASA in nanometers and the x-axis represents simulation time in nanoseconds. (e) and (f) Plots showcasing the average RMSF (Root Mean Square Fluctuation) of protein residues in their native state and when bound to the ligands. 000 ps (100 ns). (a) Plot illustrating the temporal evolution of backbone RMSD (Root Mean Square Deviation) for EGFR protein both in isolation and in complex with the ligands. (b) Graph depicting the change in protein backbone Radius of Gyration (Rg) between its unbound state and when bound to the ligands throughout the simulation. The y-axis represents Rg in nanometers, while the x-axis denotes the simulation time in picoseconds. (c) Analysis of hydrogen bond formation between the protein and ligands across the simulation time in nanoseconds. (d) Representation of Solvent Accessible Surface Area (SASA) over time, where the y-axis indicates SASA in nanometers and the x-axis represents simulation time in nanoseconds. (e) and (f) Plots showcasing the average RMSF (Root Mean Square Fluctuation) of protein residues in their native state and when bound to the ligands. | ||
3. Summary and conclusions
The primary objective of this study was to develop symmetrical and asymmetrical dihydropyridines targeting the EGFR receptor. A meticulous approach was undertaken in the synthesis and purification processes of these compounds, employing various analytical techniques including TLC, MP, IR, NMR, and Mass for comprehensive characterization. Novel Hantzsch dihydropyridines were designed utilizing scaffold hybridization techniques tailored specifically for targeting the EGFR. Through the incorporation of various aldehyde substitutions, a total of 10 symmetrical and 5 asymmetrical molecules were synthesized and subsequently purified. The cytotoxic effects of these compounds on MCF-7 breast cancer cells were assessed, revealing three noteworthy molecules (HD-6, HD-7, and HD-8) that exhibited significant potential. Molecular docking studies against the EGFR protein, complemented by in silico molecular dynamics (MD) investigations, provided further insights into the ligand–receptor interactions, affirming these compounds as potential candidates for subsequent preclinical assessments targeting the EGFR pathway in breast cancer. Notably, these molecules exhibited interactions consistent with those observed with the ligand co-crystallized with EGFR. In addition, in silico investigations including Lipinski's rule of 5, ADME, and toxicity prediction were performed. HD-7 and HD-8 successfully inhibit ligand binding and stops the EGFR complex from forming dimer, which stops the PI3/AKT signaling pathways (Fig. 7). Based on our comprehensive findings, we have identified two promising dihydropyridines candidates for further preclinical evaluations against breast cancer by targeting EGFR pathways. Our findings also indicate that these compounds exhibit promising anticancer properties by effectively inhibiting the epidermal growth factor receptor at varying concentrations. Notably, HD-7 and HD-8 emerged as a candidate molecule for further investigation using in vivo models. Overall, the results of this study contribute to the growing knowledge regarding the development of dihydropyridine-based EGFR inhibitors with potential implications for cancer therapy.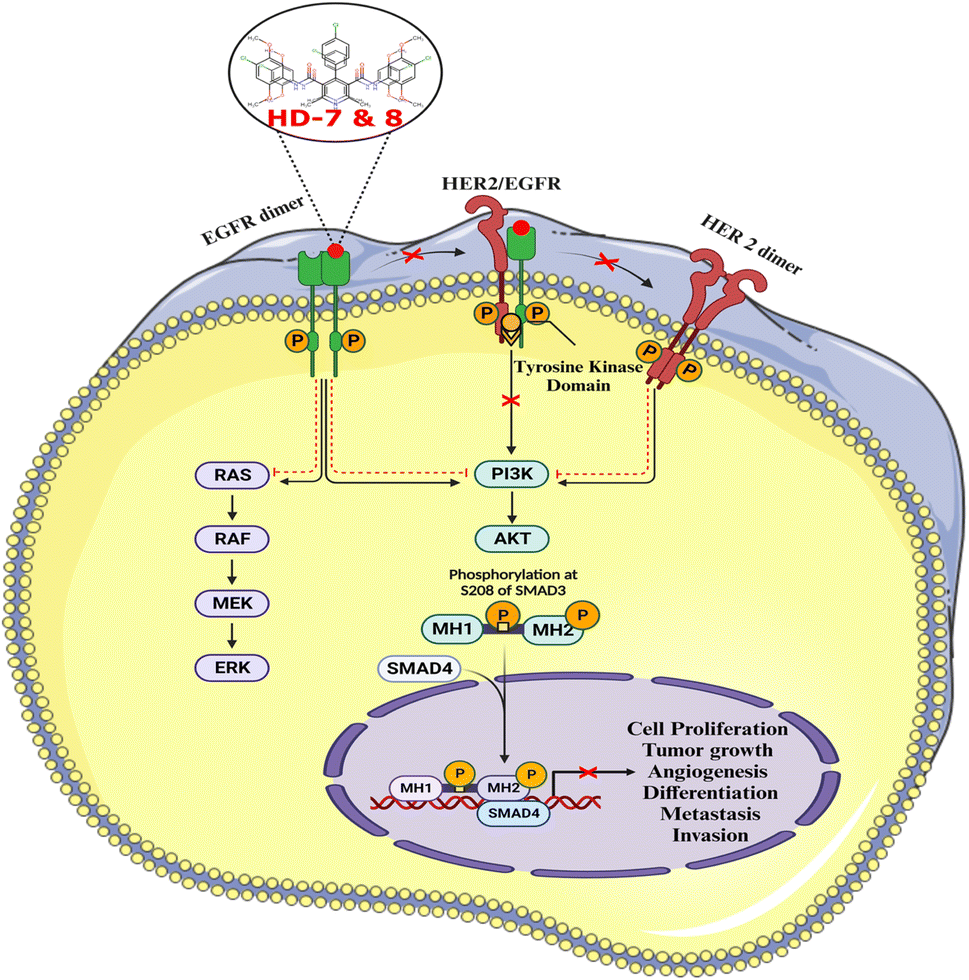 | ||
| Fig. 7 The activation of several components that support cell proliferation, oncogenic activity, and immunological regulation is necessary for the continuation of EGFR receptor signaling. This initiates MCF-7 breast cancer cells and activates signaling pathways such as PI3/AKT. HD-7 & HD-8 inhibit the signaling system which regulates tumor development, cell differentiation, atypical cell proliferation, production of new blood vessels, and cancer cell dissemination. | ||
4. Experimental
4.1. Chemicals and instruments
All reagents and solvents, including 2,4-dimethoxyaniline, 4-chloro-2,5-dimethoxyaniline, iso-propanol, ethanol, ethyl acetoacetate, ethyl acetate, and dichloromethane, were obtained from TCS and Merck. These reagents and solvents were utilized without being purified further. TLC results were obtained using precoated TLC plates with a solvent system of dichloromethane and ethyl acetate in a 9:1 ratio. A digital melting point apparatus was used to record the melting points in Celsius. Using an IR Spirit Shimadzu ATR spectrophotometer, the IR spectra were recorded. Bruker 400 MHz FT-NMR spectrophotometers were used to record 1H-NMR and 13C-NMR in DMSO-d6 using tetramethylsilane (TMS) as an internal standard. The chemical shifts were quoted in parts per million (ppm). Sciex API-4000 LC-MS/MS mass spectrometer with an electron channel multiplier detector and cyano column, was utilized to acquire mass spectra of the synthesized compounds. Elemental analysis of carbon, hydrogen, and nitrogen were analyzed using an Elementar Vario (EL III Carl Erba11080).
:2) mobile phase was then used to characterize the resulting compound. The yield obtained was about 44%.
4.1.1.1. N-(2,4-Dimethoxyphenyl)-3-oxobutanamide (A1). Brown amorphous solid; yield: 38%; m.p. 94–96 °C; Rf = 0.35 (n-Hexane/EtOAc 8
:2); IR (cm−1): 3408.33 (N–H), 3275.24 (ArC–H), 3009.05 (ArC–H), 2949.26 (aliph. C–H), 1712.85 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1035.81 (C–O).
:2) mobile phase was then used to characterize the resulting compound. The yield obtained was about 52%.
O), 1035.81 (C–O).
:2) mobile phase was then used to characterize the resulting compound. The yield obtained was about 52%.
4.1.2.1. N-(4-Chloro-2,5-dimethoxyphenyl)-3-oxobutanamide (A2). White amorphous solid, yield: 46%; m.p. 107–109 °C; Rf = 0.31 (n-hexane/EtOAc 8
:2); IR (cm−1): 3408.33 (N–H), 3267.52 (ArC–H), 3124.79 (ArC–H), 2958.90 (aliph. C–H), 1726.35 (CO), 1031.95 (C–O), 696.33 (C–Cl).
:4). The resulting yields ranged from 46 to 84 percent, indicating favorable outcomes.
4.1.3.1. N3,N5-Bis(2,4-dimethoxyphenyl)-2,6-dimethyl-4-phenyl-1,4-dihydropyridine-3,5-dicarboxamide (HD-1). Cream amorphous solid, yield: 79%, m.p. 192–194 °C; Rf = 0.29 (CH2Cl2/EtOAc 9
:1); IR (cm−1): 3385.18 (N–H), 3306.10 (N–H), 3084.28 (ArC–H), 3001.34 (ArC–H), 2960.83 (aliph. C–H), 1660.77 (CO), 1608.69 (CO), 1525.74 (ArCC), 1211.34 (C–O), 1211.34 (C–O); 1H NMR (δ ppm, 400 mHz, DMSO-d6): 2.19 (s, 6H, CH3), 3.72 (s, 12H, O–CH3), 3.73 (s, 3H, O–CH3), 4.90 (bs, 1H, CH), 6.43 (d, 2H, ArH J = 8.8 Hz), 6.54 (s, 2H, ArH J = 6 Hz), 7.33 (d, t, 4H, ArH), 7.71 (d, 2H, ArH J = 8.8 Hz), 8.17 (s, 2H, NH), 8.26 (s, 1H, NH); 13C NMR (δ ppm, 400 MHz, DMSO-d6): 18.10 (CH3), 55.73 (CH), 56.05 (OCH3), 56.17 (OCH3), 99.11 (ArC), 104.44 (ArC), 105.46 (ArC), 121.53 (ArC), 123.12 (ArC), 127.10 (ArC), 127.74 (ArC), 128.98 (ArC), 140.21 (ArC), 146.82 (ArC), 151.36 (ArC–O), 156.70 (CO), 166.75 (ArC–O); Mass (m/z): [Peak Found (M−1): 542.32 (calculated (M−1): 542.24)]. Anal. Calcd. for C31H33N3O6: C, 68.49; H, 6.12; N, 7.73; Found: C, 68.88; H, 6.19; N, 7.64.
4.1.3.2. 4-(4-Chlorophenyl)-N3,N5-bis(2,4-dimethoxyphenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxamide (HD-2). White crystals; yield: 71%; m.p. 201–203 °C; Rf = 0.30 (CH2Cl2/EtOAc 9
:1); IR (cm−1): 3429.55 (N–H), 3300.31 (N–H), 3265.59 (ArC–H), 3126.71 (ArC–H), 2997.48 (ArC–H), 2937.68 (aliph. C–H), 1664.62 (CO), 1608.69 (CO), 1523.82 (ArCC), 1222.91 (C–O), 785.05 (C–Cl); 1H NMR (δ ppm, 400 mHz, DMSO-d6): 2.19 (s, 6H, CH3), 3.73 (s, 6H, O–CH3), 3.73 (s, 6H, O–CH3), 4.96 (bs, 1H, CH), 6.44 (d, 2H, ArH J = 8.0 Hz), 6.57 (s, 2H, ArH J = 8.8 Hz), 7.34 (d, 4H, ArH), 7.36 (t, 2H, ArH J = 8.4 Hz), 7.67 (d, 2H, ArH J = 8.8 Hz), 8.20 (s, 2H, NH), 8.28 (s, 1H, NH); 13C NMR (δ ppm, 400 MHz, DMSO-d6): 18.08 (CH3), 41.74 (CH), 55.73 (OCH3), 56.06 (OCH3), 99.13 (ArC), 104.45 (ArC), 105.16 (ArC), 121.33 (ArC), 123.54 (ArC), 128.87 (ArC), 129.62 (ArC), 131.65 (ArC), 140.15 (ArC), 145.78 (ArC), 151.60 (ArC–O), 156.89 (CO), 166.69 (ArC–O); mass (m/z): [peak found (M−1): 576.24 (calculated (M−1): 576.20)]. anal. calcd. for C31H32ClN3O6: C, 64.41; H, 5.58; N, 7.27; Found: C, 64.62; H, 5.69; N, 7.24.
4.1.3.3. 4-(3-Chlorophenyl)-N3,N5-bis(2,4-dimethoxyphenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxamide (HD-3). Cream amorphous solid; yield: 84%; m.p. 217–219 °C; Rf = 0.32 (CH2Cl2/EtOAc 9
:1); IR (cm−1): 3404.47 (N–H), 3329.25 (N–H), 3003.27 (ArC–H), 2941.54 (ArC–H), 2837.38 (aliph. C–H), 1662.69 (CO), 1610.61 (CO), 1523.82 (ArCC), 1213.27 (C–O), 794.70 (C–Cl); 1H NMR (δ ppm, 400 mHz, DMSO-d6): 2.18 (s, 6H, CH3), 3.72 (s, 6H, O–CH3), 3.73 (s, 6H, O–CH3), 4.97 (bs, 1H, CH), 6.44 (d, 2H, ArH J = 6.8 Hz), 6.56 (s, 2H, ArH), 7.26 (d, 2H, ArH J = 7.6 Hz), 7.34 (t, 2H, ArH J = 8.0 Hz), 7.61 (d, 2H, ArH J = 8.8 Hz), 8.26 (s, 2H, NH), 8.32 (s, 1H, NH); 13C NMR (δ ppm, 400 MHz, DMSO-d6): 18.05 (CH3), 42.05 (CH), 55.73 (OCH3), 56.12 (OCH3), 99.13 (ArC), 99.37 (ArC), 104.48 (ArC), 105.01 (ArC), 121.14 (ArC), 123.85 (ArC), 127.02 (ArC), 130.90 (ArC), 133.49 (ArC), 140.40 (ArC), 149.21 (ArC), 151.83 (ArC–O), 157.01 (CO), 166.72 (ArC–O); mass (m/z): [peak found (M−1): 576.25 (calculated (M−1): 576.20)]. anal. calcd. for C31H32ClN3O6: C, 64.41; H, 5.58; N, 7.27; found: C, 64.56; H, 5.43; N, 7.31.
4.1.3.4. N3,N5-Bis(2,4-dimethoxyphenyl)-4-(furan-2-yl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxamide (HD-4). Gray amorphous solid, yield: 82%, m.p. 196–198 °C; Rf = 0.29 (CH2Cl2/EtOAc 9
:1); IR (cm−1): 3389.04 (N–H), 3001.43 (ArC–H), 2941.54 (ArC–H), 2837.38 (aliph. C–H), 1662.69 (CO), 1610.61 (CO), 1525.74 (ArCC), 1284.63 (C–O), 1211.34 (C–O); 1H NMR (δ ppm, 400 mHz, DMSO-d6): 2.17 (s, 6H, CH3), 3.73 (s, 6H, O–CH3), 3.77 (s, 6H, O–CH3), 4.94 (bs, 1H, CH), 6.19 (t, 1H, ArH), 6.40 (d, 1H, ArH), 6.52 (d, 2H, ArH J = 8.8 Hz), 7.57 (d, 1H, ArH), 7.73 (d, 2H, ArH J = 8.8 Hz), 8.34 (s, 2H, NH), 8.47 (s, 1H, NH); 13C NMR (δ ppm, 400 MHz, DMSO-d6): 18.18 (CH3), 35.62 (CH), 55.74 (OCH3), 56.23 (OCH3), 99.13 (ArC), 102.14 (ArC), 104.49 (ArC), 105.88 (ArC), 110.83 (ArC), 121.62 (ArC), 122.97 (ArC), 141.88 (ArC), 142.65 (ArC), 151.26 (ArC–O), 156.77 (CO), 157.89 (ArC–O), 166.44 (ArC–O); Mass (m/z): [Peak Found (M−1): 532.26 (calculated (M−1): 532.22)]. Anal. Calcd. for C29H31N3O7: C, 65.28; H, 5.86; N, 7.88; Found: C, 65.21; H, 5.99; N, 7.75.
4.1.3.5. N3,N5-Bis(2,4-dimethoxyphenyl)-4-(4-hydroxyphenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxamide (HD-5). Brown amorphous solid; yield: 62%; m.p. 206–208 °C; Rf = 0.28 (CH2Cl2/EtOAc 9
:1); IR (cm−1): 3379.40 (N–H), 3281.02 (N–H), 3007.12 (ArC–H), 2958.90 (ArC–H), 2837.38 (aliph. C–H), 1608.69 (CO), 1519.96 (CO), 1413.87 (ArCC), 1213.27 (C–O); 1H NMR (δ ppm, 400 mHz, DMSO-d6): 2.17 (s, 6H, CH3), 3.71 (s, 12H, O–CH3), 4.73 (bs, 1H, CH), 6.42 (d, 2H, ArH J = 8.4 Hz), 6.55 (s, 2H, ArH J = 6.4 Hz), 6.69 (d, 4H, ArH), 6.69 (d, 2H, ArH J = 8.4 Hz), 7.11 (d, 2H, ArH J = 8.8 Hz), 7.74 (s, 1H, NH), 8.12 (s, 1H, NH), 8.22 (s, 1H, NH), 9.32 (s, 1H, OH); 13C NMR (δ ppm, 400 MHz, DMSO-d6): 18.10 (CH3), 41.34 (CH), 55.75 (OCH3), 55.78 (OCH3), 56.01 (OCH3), 56.15 (OCH3), 99.04 (ArC), 104.52 (ArC), 105.01 (ArC), 115.75 (ArC), 119.64 (ArC), 121.59 (ArC), 122.84 (ArC), 128.80 (ArC), 130.16 (ArC), 131.16 (ArC), 139.88 (ArC), 151.15 (ArC), 153.63 (ArC–O), 156.60 (CO), 156.70 (CO), 166.64 (ArC–O); Mass (m/z): [Peak Found (M−1): 558.31 (calculated (M−1): 558.23)]. Anal. Calcd. for C31H32ClN3O6: C, 66.53; H, 5.94; N, 7.51; Found: C, 66.61; H, 5.82; N, 7.43.
4.1.3.6. N3,N5-Bis(4-chloro-2,5-dimethoxyphenyl)-2,6-dimethyl-4-phenyl-1,4-dihydropyridine-3,5-dicarboxamide (HD-6). White crystals; yield: 58%; m.p. 213–215 °C; Rf = 0.30 (CH2Cl2/EtOAc 9
:1); IR (cm−1): 3419.90 (N–H), 3371.68 (N–H), 3136.36 (ArC–H), 3076.56 (ArC–H), 2964.69 (ArC–H), 2848.96 (aliph. C–H), 1660.77 (CO), 1589.40 (CO), 1516.10 (ArCC), 1222.91 (C–O), 736.83 (C–Cl), 698.25 (C–Cl); 1H NMR (δ ppm, 400 mHz, DMSO-d6): 2.23 (s, 6H, CH3), 3.71 (s, 6H, O–CH3), 3.74 (s, 6H, O–CH3), 4.90 (bs, 1H, CH), 7.06 (d, 2H, ArH), 7.20 (s, 1H, ArH), 7.35 (d, 4H, ArH), 7.95 (d, 2H, ArH), 8.36 (s, 2H, NH), 8.57 (s, 1H, NH); 13C NMR (δ ppm, 400 MHz, DMSO-d6): 18.30 (CH3), 42.04 (CH), 56.78 (OCH3), 56.89 (OCH3), 105.32 (ArC), 106.37 (ArC), 113.22 (ArC), 127.45 (ArC), 129.21 (ArC), 141.66 (ArC), 143.59 (ArC), 146.48 (ArC–O), 148.56 (CO), 166.87 (ArC–O); Mass (m/z): [Peak Found (M−1): 610.21 (calculated (M−1): 610.16)]. Anal. Calcd. for C31H31Cl2N3O6: C, 60.79; H, 5.10; N, 6.86; Found: C, 60.88; H, 5.20; N, 6.83.
4.1.3.7. N3,N5-Bis(4-chloro-2,5-dimethoxyphenyl)-4-(4-chlorophenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxamide (HD-7). White amorphous solid; yield: 78%; m.p. 211–213 °C; Rf = 0.32 (CH2Cl2/EtOAc 9
:1); IR (cm−1): 3460.41 (N–H), 3323.46 (N–H), 2997.48 (ArC–H), 2937.68 (ArC–H), 2837.38 (aliph. C–H), 1660.77 (CO), 1629.90 (CO), 1518.03 (ArCC), 1213.27 (C–O), 821.70 (C–Cl), 773.48 (C–Cl), 734.90 (C–Cl); 1H NMR (δ ppm, 400 mHz, DMSO-d6): 2.20 (s, 6H, CH3), 3.72 (s, 6H, O–CH3), 3.74 (s, 6H, O–CH3), 4.92 (bs, 1H, CH), 7.08 (d, 2H, ArH), 7.31 (d, 2H, ArH), 7.41 (d, 2H, ArH), 7.88 (d, 2H, ArH), 8.37 (s, 2H, NH), 8.58 (s, 1H, NH); 13C NMR (δ ppm, 400 MHz, DMSO-d6): 18.25 (CH3), 41.51 (CH), 56.81 (OCH3), 56.94 (OCH3), 105.03 (ArC), 106.67 (ArC), 113.30 (ArC), 114.20 (ArC), 127.91 (ArC), 129.10 (ArC), 131.96 (ArC), 141.53 (ArC), 143.79 (ArC), 145.41 (ArC–O), 148.54 (CO), 166.71 (ArC–O); Mass (m/z): [Peak Found (M+1): 646.20 (calculated (M+1): 646.12)]. Anal. Calcd. for C31H30Cl3N3O6: C, 57.55; H, 4.67; N, 6.50; Found: C, 57.72; H, 4.55; N, 6.61.
4.1.3.8. N3,N5-Bis(4-chloro-2,5-dimethoxyphenyl)-4-(3-chlorophenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxamide (HD-8). White crystals; yield: 67%; m.p. 197–199 °C; Rf = 0.33 (CH2Cl2/EtOAc 9
:1); IR (cm−1): 3421.83 (N–H), 3379.40 (N–H), 3076.56 (ArC–H), 3001.34 (ArC–H), 2852.81 (aliph. C–H), 1658.84 (CO), 1589.40 (CO), 1392.65 (ArCC), 1159.26 (C–O), 769.62 (C–Cl), 732.97 (C–Cl), 696.33 (C–Cl); 1H NMR (δ ppm, 400 mHz, DMSO-d6): 2.22 (s, 6H, CH3), 3.73 (s, 6H, O–CH3), 3.75 (s, 6H, O–CH3), 4.98 (bs, 1H, CH), 7.08 (d, 1H, ArH), 7.27–7.41 (m, 6H, ArH), 7.85 (d, 1H, ArH), 8.43 (s, 2H, NH), 8.59 (s, 1H, NH); 13C NMR (δ ppm, 400 MHz, DMSO-d6): 18.25 (CH3), 41.87 (CH), 56.81 (OCH3), 56.99 (OCH3), 104.79 (ArC), 106.94 (ArC), 113.33 (ArC), 115.13 (ArC), 126.42 (ArH), 127.31 (ArC), 127.84 (ArC), 131.15 (ArC), 141.75 (ArC), 144.01 (ArC), 145.29 (ArC–O), 148.55 (CO), 148.86 (CO), 166.68 (ArC–O); Mass (m/z): [Peak Found (M+1): 646.17 (calculated (M+1): 646.12)]. Anal. Calcd. for C31H30Cl3N3O6: C, 57.55; H, 4.67; N, 6.50; Found: C, 57.45; H, 4.71; N, 6.49.
4.1.3.9. N3,N5-Bis(4-chloro-2,5-dimethoxyphenyl)-4-(furan-2-yl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxamide (HD-9). Brown crystals; yield: 52%; m.p. 202–204 °C; Rf = 0.31 (CH2Cl2/EtOAc 9
:1); IR (cm−1): 3454.62 (N–H), 3410.26 (N–H), 3134.43 (ArC–H), 2943.47 (ArC–H), 2854.74 (aliph. C–H), 1670.41 (CO), 1523.82 (CO), 1392.82 (ArCC), 1301.99 (C–O), 800.49 (C–Cl), 731.05 (C–Cl); 1H NMR (δ ppm, 400 mHz, DMSO-d6): 2.22 (s, 6H, CH3), 3.76 (s, 6H, O–CH3), 3.78 (s, 6H, O–CH3), 4.94 (bs, 1H, CH), 6.22 (t, 1H, ArH), 6.41 (d, 2H, ArH), 7.60 (s, 2H, ArH), 7.60 (d, 1H, ArH), 8.02 (d, 2H, ArH), 8.54 (s, 2H, NH), 8.77 (s, 1H, NH); 13C NMR (δ ppm, 400 MHz, DMSO-d6): 18.37 (CH3), 35.25 (CH), 56.80 (OCH3), 57.14 (OCH3), 101.97 (ArC), 106.11 (ArC), 106.21 (ArC), 110.96 (ArC), 113.35 (ArH), 114.60 (ArC), 128.30 (ArC), 142.98 (ArC), 143.44 (ArC), 148.63 (ArC–O), 157.46 (CO), 166.37 (ArC–O); Mass (m/z): [Peak Found (M−1): 600.15 (calculated (M−1): 600.14)]. Anal. Calcd. for C29H29Cl2N3O7: C, 57.82; H, 4.85; N, 6.97; Found: C, 57.71; H, 4.79; N, 6.88.
4.1.3.10. N3,N5-Bis(4-chloro-2,5-dimethoxyphenyl)-4-(4-hydroxyphenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxamide (HD-10). Yellow crystals; yield: 56%; m.p. 214–216 °C; Rf = 0.34 (CH2Cl2/EtOAc 9
:1); IR (cm−1): 3572.29 (N–H), 3446.91 (N–H), 3088.14 (ArC–H), 2966.62 (ArC–H), 2841.24 (aliph. C–H), 1688.48 (CO), 1610.61 (CO), 1458.24 (ArCC), 1215.19 (C–O), 767.69 (C–Cl), 731.05 (C–Cl); 1H NMR (δ ppm, 400 mHz, DMSO-d6): 2.20 (s, 6H, CH3), 3.72 (s, 6H, O–CH3), 3.75 (s, 6H, O–CH3), 4.73 (bs, 1H, CH), 6.74 (d, 2H, ArH J = 8.4 Hz), 7.06 (s, 2H, ArH), 7.14 (d, 2H, ArH J = 8.4 Hz), 7.97 (s, 2H, ArH), 8.30 (s, 2H, NH), 8.49 (s, 1H, NH), 9.39 (s, 1H, OH); 13C NMR (δ ppm, 400 MHz, DMSO-d6): 18.31 (CH3), 41.15 (CH), 56.79 (OCH3), 56.86 (OCH3), 105.73 (ArC), 106.21 (ArC), 113.17 (ArC), 114.60 (ArC), 115.94 (ArH), 128.16 (ArC), 128.76 (ArC), 141.28 (ArC), 143.45 (ArC), 148.56 (ArC–O), 156.92 (CO), 166.91 (ArC–O); Mass (m/z): [Peak Found (M−1): 626.15 (calculated (M−1): 626.15)]. Anal. Calcd. for C31H31Cl2N3O7: C, 59.24; H, 4.97; N, 6.69; Found: C, 59.41; H, 4.79; N, 6.60.
4.1.3.11. N3-(4-Chloro-2,5-dimethoxyphenyl)-N5-(2,4-dimethoxyphenyl)-2,6-dimethyl-4-phenyl-1,4-dihydropyridine-3,5-dicarboxamide (HD-11). White amorphous solid; yield: 46%; m.p. 223–225 °C; Rf = 0.32 (CH2Cl2/EtOAc 9
:1); IR (cm−1): 3367.82 (N–H), 3140.22 (N–H), 3007.22 (ArC–H), 2937.68 (ArC–H), 2841.24 (aliph. C–H), 1660.77 (CO), 1604.83 (CO), 1523.82 (ArCC), 1213.27 (C–O), 1159.26 (C–O), 705.97 (C–Cl); 1H NMR (δ ppm, 400 mHz, DMSO-d6): 2.15 (s, 3H, CH3), 2.23 (s, 3H, CH3), 3.68 (s, 6H, O–CH3), 3.73 (s, 6H, O–CH3), 4.91 (bs, 1H, CH), 6.45 (d, 1H, ArH), 6.55 (s, 1H, ArH), 7.33–7.36 (3, 5H, ArH), 7.96 (s, 1H, ArH), 8.05 (s, 1H, ArH), 8.42 (s, 2H, NH), 8.55 (s, 1H, NH); 13C NMR (δ ppm, 400 MHz, DMSO-d6): 18.32 (CH3), 18.59 (CH3), 42.04 (CH), 55.77 (OCH3), 56.04 (OCH3), 56.08 (OCH3), 56.15 (OCH3), 99.16 (ArC), 104.48 (ArC), 106.38 (ArC), 107.14 (ArC), 113.13 (ArC), 114.26 (ArC), 121.22 (ArH), 128.14 (ArC), 128.35 (ArC), 141.63 (ArC), 143.13 (ArC), 148.57 (ArC–O), 166.64 (CO), 166.77 (ArC–O); Mass (m/z): [Peak Found (M−1): 576.24 (calculated (M−1): 576.20)]. Anal. Calcd. for C31H32ClN3O6: C, 64.41; H, 5.58; N, 7.27; Found: C, 64.55; H, 5.63; N, 7.14.
4.1.3.12. N3-(4-Chloro-2,5-dimethoxyphenyl)-4-(4-chlorophenyl)-N5-(2,4-dimethoxyphenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxamide (HD-12). Yellow crystals; yield: 58%; m.p. 217–219 °C; Rf = 0.29 (CH2Cl2/EtOAc 9
:1); IR (cm−1): 3427.62 (N–H), 3381.33 (N–H), 3088.14 (ArC–H), 2999.41 (ArC–H), 2937.68 (aliph. C–H), 1658.84 (CO), 1608.69 (CO), 1394.58 (ArCC), 1211.34 (C–O), 769.62 (C–Cl), 729.12 (C–Cl); 1H NMR (δ ppm, 400 mHz, DMSO-d6): 2.18 (s, 3H, CH3), 2.21 (s, 3H, CH3), 3.69 (s, 6H, O–CH3), 3.74 (s, 6H, O–CH3), 4.93 (bs, 1H, CH), 6.44 (d, 1H, ArH), 6.57 (s, 1H, ArH), 7.05 (s, 1H, ArH), 7.32 (d, 2H, ArH J = 8.4 Hz), 7.41 (d, 2H, ArH J = 8.8 Hz), 7.89 (s, 1H, ArH), 8.16 (d, 1H, ArH), 8.39 (s, 1H, NH), 8.44 (s, 1H, NH), 8.57 (s, 1H, NH); 13C NMR (δ ppm, 400 MHz, DMSO-d6): 18.25 (CH3), 18.53 (CH3), 41.94 (CH), 56.08 (OCH3), 56.79 (OCH3), 56.82 (OCH3), 56.88 (OCH3), 99.14 (ArC), 104.51 (ArC), 106.68 (ArC), 106.79 (ArC), 113.15 (ArC), 114.96 (ArC), 120.95 (ArH), 128.99 (ArC), 129.09 (ArC), 131.83 (ArC), 141.52 (ArC), 143.79 (ArC), 148.56 (ArC–O), 166.58 (CO), 166.72 (ArC–O); Mass (m/z): [Peak Found (M+1): 612.22 (calculated (M+1): 612.16)]. Anal. Calcd. for C31H31Cl2N3O6: C, 60.79; H, 5.10; N, 6.86; Found: C, 60.80; H, 5.21; N, 6.91.
4.1.3.13. N3-(4-Chloro-2,5-dimethoxyphenyl)-4-(3-chlorophenyl)-N5-(2,4-dimethoxyphenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxamide (HD-13). Yellow amorphous solid; yield: 62%; m.p. 227–229 °C; Rf = 0.32 (CH2Cl2/EtOAc 9
:1); IR (cm−1): 3458.48 (N–H), 3398.69 (N–H), 3084.28 (ArC–H), 2943.47 (ArC–H), 2837.38 (aliph. C–H), 1688.48 (CO), 1641.48 (CO), 1523.82 (ArCC), 1209.41 (C–O), 792.77 (C–Cl), 727.19 (C–Cl); 1H NMR (δ ppm, 400 mHz, DMSO-d6): 2.19 (s, 3H, CH3), 2.22 (s, 3H, CH3), 3.73 (s, 6H, O–CH3), 3.75 (s, 6H, O–CH3), 4.97 (bs, 1H, CH), 6.45 (s, 1H, ArH), 6.56 (d, 1H, ArH), 7.06 (d, 1H, ArH), 7.27–7.39 (m, 4H, ArH), 7.85 (s, 1H, ArH), 8.20 (d, 1H, ArH), 8.43 (s, 1H, NH), 8.46 (s, 1H, NH), 8.58 (s, 1H, NH); 13C NMR (δ ppm, 400 MHz, DMSO-d6): 18.24 (CH3), 18.52 (CH3), 42.14 (CH), 56.13 (OCH3), 56.81 (OCH3), 56.97 (OCH3), 57.00 (OCH3), 99.15 (ArC), 104.51 (ArC), 106.94 (ArC), 113.19 (ArC), 113.34 (ArC), 115.12 (ArC), 126.43 (ArH), 127.30 (ArC), 128.06 (ArC), 133.59 (ArC), 141.78 (ArC), 143.47 (ArC), 148.56 (ArC–O), 152.43 (ArC–O), 166.52 (CO), 166.68 (ArC–O); Mass (m/z): [Peak Found (M−1): 610.16 (calculated (M−1): 610.16)]. Anal. Calcd. for C31H31Cl2N3O6: C, 60.79; H, 5.10; N, 6.86; Found: C, 60.65; H, 5.11; N, 6.72.
4.1.3.14. N3-(4-Chloro-2,5-dimethoxyphenyl)-N5-(2,4-dimethoxyphenyl)-4-(furan-2-yl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxamide (HD-14). Cream amorphous solid; yield: 48%; m.p. 214–216 °C; Rf = 0.28 (CH2Cl2/EtOAc 9
:1); IR (cm−1): 3373.61 (N–H), 3308.03 (N–H), 3138.29 (ArC–H), 2999.41 (ArC–H), 2837.38 (aliph. C–H), 1688.48 (CO), 1606.76 (CO), 1525.74 (ArCC), 1211.34 (C–O), 734.90 (C–Cl); 1H NMR (δ ppm, 400 mHz, DMSO-d6): 2.19 (s, 3H, CH3), 2.24 (s, 3H, CH3), 3.74 (s, 3H, O–CH3), 3.75 (s, 3H, O–CH3), 3.79 (s, 3H, O–CH3), 3.82 (s, 3H, O–CH3), 4.96 (bs, 1H, CH), 6.22 (d, 1H, ArH), 6.41 (t, 1H, ArH), 6.60 (d, 1H, ArH), 7.14 (s, 1H, ArH), 8.05 (d, 1H, ArH), 8.35 (s, 2H, NH), 8.61 (s, 1H, NH); 13C NMR (δ ppm, 400 MHz, DMSO-d6): 18.40 (CH3), 18.58 (CH3), 36.69 (CH), 55.72 (OCH3), 56.22 (OCH3), 56.78 (OCH3), 57.13 (OCH3), 99.15 (ArC), 104.49 (ArC), 105.80 (ArC), 106.08 (ArC), 113.29 (ArC), 113.37 (ArC), 121.52 (ArC), 128.37 (ArH), 128.50 (ArC), 142.83 (ArC), 143.21 (ArC), 143.42 (ArC), 148.65 (ArC–O), 151.52 (ArC–O), 166.34 (CO), 166.39 (ArC–O); Mass (m/z): [Peak Found (M+1): 568.25 (calculated (M+1): 568.18)]. Anal. Calcd. for C29H30ClN3O7: C, 61.32; H, 5.32; N, 7.40; Found: C, 61.44; H, 5.27; N, 6.57.
4.1.3.15. N3-(4-Chloro-2,5-dimethoxyphenyl)-N5-(2,4-dimethoxyphenyl)-4-(4-hydroxyphenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxamide (HD-15). Yellow crystals; yield: 51%; m.p. 225–227 °C; Rf = 0.27 (CH2Cl2/EtOAc 9
:1); IR (cm−1): 3570.36 (N–H), 3446.91 (N–H), 3010.98 (ArC–H), 2964.69 (ArC–H), 2837.38 (aliph. C–H), 1688.48 (CO), 1610.61 (CO), 1392.65 (ArCC), 1211.34 (C–O), 731.05 (C–Cl); 1H NMR (δ ppm, 400 mHz, DMSO-d6): 2.20 (s, 3H, CH3), 2.25 (s, 3H, CH3), 3.70 (s, 3H, O–CH3), 3.73 (s, 3H, O–CH3), 3.75 (s, 3H, O–CH3), 3.77 (s, 3H, O–CH3), 4.73 (bs, 1H, CH), 6.57 (d, 1H, ArH), 6.58 (s, 1H, ArH), 6.73 (d, 2H, ArH J = 8.4 Hz), 7.05 (s, 1H, ArH), 7.13 (d, 2H, ArH J = 8.4 Hz), 7.99 (s, 1H, ArH), 8.07 (s, 1H, ArH), 8.13 (s, 2H, NH), 8.46 (s, 1H, NH), 9.28 (s, 1H, OH); 13C NMR (δ ppm, 400 MHz, DMSO-d6): 18.61 (CH3), 41.13 (CH), 55.73 (OCH3), 56.06 (OCH3), 56.80 (OCH3), 56.89 (OCH3), 99.13 (ArC), 104.45 (ArC), 105.54 (ArC), 113.06 (ArC), 113.21 (ArC), 115.84 (ArC), 121.34 (ArH), 128.22 (ArC), 128.44 (ArC), 137.12 (ArC), 141.24 (ArC), 143.45 (ArC), 148.56 (ArC–O), 151.74 (ArC–O), 156.83 (ArCO), 166.72 (CO), 166.87 (ArC–O); Mass (m/z): [Peak Found (M+1): 594.27 (calculated (M+1): 594.19)]. Anal. Calcd. for C31H32ClN3O7: C, 62.68; H, 5.43; N, 7.07; Found: C, 62.52; H, 5.59; N, 7.11.
4.2. In vitro anticancer screening
The National Centre for Cell Sciences (NCCS), Pune, India provided the vero and MCF-7 cell cultures. Each compound is weighed individually and dissolved in dimethyl sulfoxide (DMSO) for cytotoxicity studies. To prepare a stock solution with a known concentration, it is first diluted with Dulbecco's Modified Eagle Medium (DMEM), which has a pH of 7.4 and 2% inactivated foetal bovine serum (FBS), to get the maintenance medium. The stock solution is then sterilized by filtering and kept at −20 °C until it is required. Two-fold serial dilutions of a previous stock solution are created for a lower concentration. Stock cells were cultured at 37 °C in a humid environment containing 5% CO2 in DMEM media supplemented with 10% inactivated FBS, amphotericin B (5 μg ml−1), streptomycin (100 μg ml−1), and penicillin (100 IU ml−1). In TPVG solution (0.05% glucose, 0.2% trypsin, and 0.02% EDTA in PBS), the cells are separated. Each experiment is carried out on 96-well microtiter plates, and all stock cell cultures are produced in 25 cm2 flat flasks.39Cell viability was evaluated using the MTT assay as a measure of the sensitivity of the cells to the synthesized compounds. In 96-well flat-bottom microplates (100 ml per well), exponentially growing cells were added at a density of approximately 1.5 × 105 MCF-7 cells per well and allowed to adhere for 24 hours prior to the inclusion of the compounds. The compounds were dissolved in DMSO and added to the cells at concentrations ranging from 2.5 to 160 μM. They were then incubated for 24 or 72 hours at 37 °C. After this, 10 μL of MTT (5 mg ml−1) was added to each well and incubated for another 2 hours at 37 °C. The culture medium was then eliminated and 100 μL of DMSO was added per well to dissolve the formazan crystals. At 550 nm, optical density was measured with a 96-well multiscanner autoreader (ELISA). Finally, the following formula was used to calculate the percentage of growth inhibition. The data obtained were processed using the GraphPad Prism Version 8. The dose-response curve was used to calculate the IC50 values for each cell (Table 1).
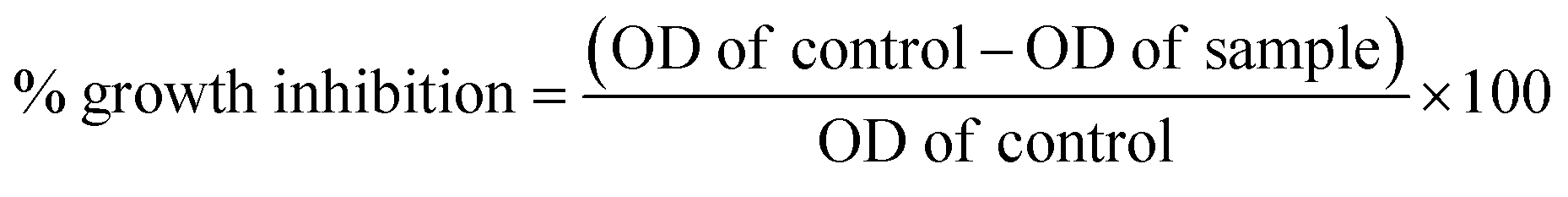
4.3. Molecular docking studies
Epidermal growth factor's crystal structure was retrieved from the Protein Data Bank (PDB: 1XKK, https://www.rcsb.org/structure/1xkk).37 After selecting the protein structure, the discovery studio protein preparation protocol was used to prepare the structure. All water molecules except interacting ones were eliminated from the structure of the protein. The energy of protein structure was minimized, and the active site of the protein was determined. The ligands active site was defined as the binding site and co-crystallized ligand was deleted. The prepared protein was then saved in the .pdb format.We utilized ChemDraw to draw the 2D chemical structure and later converted them to 3D. All ligands were prepared for molecular interaction experiments using discovery studio's small molecules protocol. The default pH 7.4 was utilized by Ligprep for ligand preparation. The structure energy of the ligand was minimized, and partial atomic charges were calculated. The docking was conducted using the CDOCKER protocol of DS 2020 with the default parameters. The CDOCKER protocol was utilized in conjunction with the CDOCKER score scoring function for computational docking. The favorable pose with the most excellent CDOCKER score was then taken for binding energy calculation. The 2D and 3D interactions of the best ligand-receptor complexes were then derived.
4.4. ADMET prediction
ADMET studies play an important role in the pharmacokinetic and toxicological properties of potential drug candidates and serve a crucial role in drug discovery. To facilitate such research, BIOVIA Discovery Studio provides an interface that includes several essential stages. Initially, the chemical structures of the target compounds are compiled and prepared in the appropriate file formats. ADMET property prediction modules then estimate properties such as solubility, permeability, plasma protein binding, and blood–brain barrier penetration using BIOVIA Discovery Studio. The toxicology prediction modules assess potential toxicity, while the metabolism prediction modules simulate biotransformation and identify metabolic pathways. The gathered information is then analyzed and compared to aid in the selection and optimization of lead compounds. Bayesian and regression models were utilized to investigate the recognized dosage range, mutagenic properties, and carcinogenic potential of the compound.40,414.5. EGFRwt kinase inhibition assay
The inhibitory activity of the potent compounds (HD-6, HD-7, HD-8 & lapatinib) against EGFRwt kinase was assessed using the EGFRwt Kinase Assay Kit from BPS biosciences as per the manufacturer's instructions.42,43 To initiate the enzymatic reaction, a master mixture containing EGFRwt enzyme, substrates, ATP, and kinase assay enzymatic buffer was incubated with the tested compounds for 40 minutes at 30 °C. The reaction was halted by adding a detecting reagent (Kinase-Glo Max reagent) and further incubating at room temperature for 15 minutes. Luminescence was then measured using the GloMax® discover microplate Reader. The results were expressed as mean ± SEM (n = 2) percentage enzyme inhibition compared to lapatinib, chosen as the reference drug due to its potent inhibitory activity against EGFRwt. The data obtained were processed using the GraphPad Prism Version 8.4.6. Molecular dynamics simulations
The stability of a protein–ligand complex in a static pose was assessed through molecular dynamics (MD) simulations over a period. MD simulations were conducted on the native protein structure in both the undocked and docked states with ligands, and stability was compared using energy graphs. The GROMACS 5.1.5 software was used on the Ubuntu 16.04.7 platform and other web-based servers. The protein and ligand topology files were generated either by the native GROMACS platform or externally using the SwissParam online server (https://www.swissparam.ch/) with the CHARMm27 force field. The protein systems in their innate and complex forms were virtually placed in a triclinic box, with 1.0 nm distance maintained between the outer surface of the protein and sides of the box. This was done to preserve the van der Waals interactions, with a distance cut-off of 1.0 nm. Coulombic interactions were calculated using the Partial Mesh Ewald (PME) summation method for long-range electrostatics, with a cut-off of 1.0 nm. To explicitly solvate the system, periodic boundary conditions were used in conjunction with the TIP3P water model, and the electroneutrality of the system was maintained by adding the required counter ions (Na+/Cl−).44The entire system was energy-minimized using the steepest descent algorithm for 5000 steps, with a tolerance of 1000 kJ mol−1 nm−1. To equilibrate the system, position restraints were applied to the complex, and two simulations were performed at a constant temperature of 300 K and pressure of 1 bar using the canonical NVT and NPT ensembles. The temperature was coupled using velocity rescaling with a coupling constant of 0.1 ps, and the initial velocities were generated according to the Maxwell distribution. The temperature–pressure coupling process was accomplished using an extended Parrinello–Rahman ensemble algorithm with a coupling constant of 2 ps. The system was subsequently subjected to a 100 ns molecular dynamics production run and optimized for faster processing by integrating the time steps every 20 fs. The trajectories were analyzed using the default GROMACS analysis tools and plotted using the XMGRACE-5.1.22 platform (available at https://plasmagate.weizmann.ac.il/Grace).45,46
Abbreviations
| EGFR | Epidermal growth factor receptor |
| MCF-7 | Michigan cancer foundation-7 |
| TKIs | Tyrosine kinase inhibitors |
| PTSA | p-Toluene sulphonic acid |
| TLC | Thin layer chromatography |
| IC50 | Inhibitory concentration 50 |
| DHPs | Dihydropyridines |
| BBB | Blood–brain barrier |
| NI | Non-inhibitor |
| NC | Non-carcinogen |
| NM | Non-mutagen |
| MD | Molecular dynamics |
| DMSO | Dimethyl sulfoxide |
| DMEM | Dulbecco's modified eagle medium |
| FBS | Fetal bovine serum |
| PBS | Phosphate buffered saline |
| RO5 | Lipinski's rule of 5 |
Author contributions
Conceptualization, B. R. P. K. and S. F.; methodology, S. F.; software, S. F. and S. P. M; validation, B. R. P. K. and S. F.; formal analysis, B. R. P. K.; investigation, S. F.; resources, S. F.; data curation, S. F.; writing-original draft preparation, S. F.; writing-review and editing, S. F., S. T., A. F. W., S. P. M. and B. R. P. K.; visualization, S. F.; supervision, B. R. P. K.; project administration, B. R. P. K.; funding acquisition, S. T., A. F. W., U. H., and N. H. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no financial conflicts of interest or personal affiliations that could have influenced the research conducted in this study.Acknowledgements
This research was funded by King Khalid University through a Large group Research Project under grant number RGP 2/150/45. The authors extend their appreciation to the Deanship of scientific Research at King Khalid University for funding this research work through a large group Research Project under grant number RGP 2/150/45. All the authors are grateful to JSS College of Pharmacy, Mysore, a constituent college of JSS Academy of Higher Education & Research, Mysore for providing the instrumental and infrastructural facilities to carry out the research work.References
- S. Faizan, B. P. Kumar, N. L. Naishima, T. Ashok, A. Justin, M. V. Kumar, R. B. Chandrashekarappa, N. M. Raghavendra, P. Kabadi and L. Adhikary, Design, parallel synthesis of Biginelli 1, 4-dihydropyrimidines using PTSA as a catalyst, evaluation of anticancer activity and structure activity relationships via 3D QSAR studies, Bioorg. Chem., 2021, 117, 105462, DOI:10.1016/j.bioorg.2021.105462.
- A. A. Sadybekov, A. V. Sadybekov, Y. Liu, C. Iliopoulos-Tsoutsouvas, X. P. Huang, J. Pickett, B. Houser, N. Patel, N. K. Tran, F. Tong and N. Zvonok, Synthon-based ligand discovery in virtual libraries of over 11 billion compounds, Nature, 2022, 601(7893), 452–459, DOI:10.1038/s41586-021-04220-9.
- C. A. Lipinski, Lead-and drug-like compounds: the rule-of-five revolution, Drug Discovery Today: Technol., 2004, 1(4), 337–341, DOI:10.1016/j.ddtec.2004.11.007.
- G. M. Whitesides and V. M. Krishnamurthy, Designing ligands to bind proteins, Q. Rev. Biophys., 2005, 38(4), 385–395, DOI:10.1017/S0033583506004240.
- I. D. Kuntz, K. Chen, K. A. Sharp and P. A. Kollman, The maximal affinity of ligands, Proc. Natl. Acad. Sci. U. S. A., 1999, 96(18), 9997–10002, DOI:10.1073/pnas.96.18.9997.
- G. Sliwoski, S. Kothiwale, J. Meiler and E. W. Lowe, Computational methods in drug discovery, Pharmacol. Rev., 2014, 66(1), 334–395, DOI:10.1124/pr.112.007336.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Delivery Rev., 1997, 23(1–3), 3–25, DOI:10.1016/S0169-409X(96)00423-1.
- J. J. Irwin and B. K. Shoichet, ZINC− a free database of commercially available compounds for virtual screening, J. Chem. Inf. Model., 2005, 45(1), 177–182, DOI:10.1021/ci049714+.
- T. Cheng, X. Li, Y. Li, Z. Liu and R. Wang, Comparative assessment of scoring functions on a diverse test set, J. Chem. Inf. Model., 2009, 49(4), 1079–1093, DOI:10.1021/ci9000053.
- A. Tropsha, Best practices for QSAR model development, validation, and exploitation, Mol. Inf., 2010, 29(6-7), 476–488, DOI:10.1002/minf.201000061.
- G. Schneider, Automating drug discovery, Nat. Rev. Drug Discovery, 2018, 17(2), 97–113, DOI:10.1038/nrd.2017.232.
- W. Wang, O. Donini, C. M. Reyes and P. A. Kollman, Biomolecular simulations: recent developments in force fields, simulations of enzyme catalysis, protein-ligand, protein-protein, and protein-nucleic acid noncovalent interactions, Annu. Rev. Biophys. Biomol. Struct., 2001, 30(1), 211–243, DOI:10.1146/annurev.biophys.30.1.211.
- S. E. John, S. Gulati and N. Shankaraiah, Recent advances in multi-component reactions and their mechanistic insights: a triennium review, Org. Chem. Front., 2021, 8(15), 4237–4287, 10.1039/D0QO01480J.
- S. Faizan, T. F. Roohi, R. M. Raju, S. Yuvaraj and B. P. Kumar, A Century-Old One-Pot Multicomponent Biginelli Reaction Products Still Finds a Niche in Drug Discoveries: Synthesis, Mechanistic studies and Diverse Biological Activities of Dihydropyrimidines, J. Mol. Struct., 2023, 16, 136020, DOI:10.1016/j.molstruc.2023.136020.
- J. E. Biggs-Houck, A. Younai and J. T. Shaw, Recent advances in multicomponent reactions for diversity-oriented synthesis, Curr. Opin. Chem. Biol., 2010, 14(3), 371–382, DOI:10.1016/j.cbpa.2010.03.003.
- B. B. Toure and D. G. Hall, Natural product synthesis using multicomponent reaction strategies, Chem. Rev., 2009, 109(9), 4439–4486, DOI:10.1021/cr800296p.
- D. Urselmann, D. Antovic and T. J. Müller, Pseudo five-component synthesis of 2, 5-di (hetero) arylthiophenes via a one-pot Sonogashira–Glaser cyclization sequence, Beilstein J. Org. Chem., 2011, 7(1), 1499–1503, DOI:10.3762/bjoc.7.174.
- A. Domling, W. Wang and K. Wang, Chemistry and biology of multicomponent reactions, Chem. Rev., 2012, 112(6), 3083–3135, DOI:10.1021/cr100233r.
- D. K. Agrafiotis, M. Shemanarev, P. J. Connolly, M. Farnum and V. S. Lobanov, SAR maps: a new SAR visualization technique for medicinal chemists, J. Med. Chem., 2007, 50(24), 5926–5937, DOI:10.1021/jm070845m.
- H. A. Younus, M. Al-Rashida, A. Hameed, M. Uroos, U. Salar, S. Rana and K. M. Khan, Multicomponent reactions (MCR) in medicinal chemistry: a patent review (2010-2020), Expert Opin. Ther. Pat., 2021, 31(3), 267–289, DOI:10.1080/13543776.2021.1858797.
- L. Oehberg and J. Westman, An efficient and fast procedure for the Hantzsch dihydropyridine synthesis under microwave conditions, Synlett, 2001,(08), 1296–1298, DOI:10.1055/s-2001-16043.
- D. Enders, S. Müller and A. S. Demir, Enantioselective hantzsch dihydropyridine synthesis via metalated chiral alkyl acetoacetate hydrazones1, Tetrahedron Lett., 1988, 29(49), 6437–6440, DOI:10.1016/S0040-4039(00)82366-7.
- B. J. Epstein, K. Vogel and B. F. Palmer, Dihydropyridine calcium channel antagonists in the management of hypertension, Drugs, 2007, 67, 1309–1327, DOI:10.2165/00003495-200767090-00005.
- P. Thirumurugan, S. Mahalaxmi and P. T. Perumal, Synthesis and anti-inflammatory activity of 3-indolyl pyridine derivatives through one-pot multi component reaction, J. Chem. Sci., 2010, 122, 819–832, DOI:10.1007/s12039-010-0070-3.
- M. De Luca, G. Ioele and G. Ragno, 1, 4-Dihydropyridine antihypertensive drugs: Recent advances in photostabilization strategies, Pharmaceutics, 2019, 11(2), 85, DOI:10.3390/pharmaceutics11020085.
- S. Ulloora, S. Kumar, R. Shabaraya and A. V. Adhikari, New dihydropyridine derivatives: anti-inflammatory, analgesic and docking studies, Med. Chem. Res., 2013, 22, 1549–1562, DOI:10.1007/s00044-012-0156-1.
- E. Praveenkumar, N. Gurrapu, P. K. Kolluri, V. Yerragunta, B. R. Kunduru and N. J. Subhashini, Synthesis, anti-diabetic evaluation and molecular docking studies of 4-(1-aryl-1H-1, 2, 3-triazol-4-yl)-1, 4-dihydropyridine derivatives as novel 11-β hydroxysteroid dehydrogenase-1 (11β-HSD1) inhibitors, Bioorg. Chem., 2019, 90, 103056, DOI:10.1016/j.bioorg.2019.103056.
- H. M. Refat and A. A. Fadda, Synthesis and antimicrobial activity of some novel hydrazide, benzochromenone, dihydropyridine, pyrrole, thiazole and thiophene derivatives, Eur. J. Med. Chem., 2013, 70, 419–426, DOI:10.1016/j.ejmech.2013.09.003.
- T. S. Ibrahim, H. A. El-Sayed, M. T. Khayat, A. M. Al-Mahmoudy, A. H. Moustafa, A. K. El-Deen, S. A. Rostom and S. S. Panda, Synthesis of Nucleosides and Non-nucleosides Based 4, 6-disubstituted-2-oxo-dihydropyridine-3-carbonitriles as Antiviral Agents, Med. Chem., 2018, 14(8), 791–808, DOI:10.2174/1573406414666180522123524.
- A. Fournier, R. Oprisiu-Fournier, J. M. Serot, O. Godefroy, J. M. Achard, S. Faure, H. Mazouz, M. Temmar, A. Albu, R. Bordet and O. Hanon, Prevention of dementia by antihypertensive drugs: how AT1-receptor-blockers and dihydropyridines better prevent dementia in hypertensive patients than thiazides and ACE-inhibitors, Expert Rev. Neurother., 2009, 9(9), 1413–1431, DOI:10.1586/ern.09.89.
- M. F. Mohamed, N. S. Ibrahim, A. H. Elwahy and I. A. Abdelhamid, Molecular studies on novel antitumor bis 1, 4-dihydropyridine derivatives against lung carcinoma and their limited side effects on normal melanocytes, Anti-Cancer Agents Med. Chem., 2018, 18(15), 2156–2168, DOI:10.2174/1871520618666181019095007.
- P. A. Sidhom, E. El-Bastawissy, A. A. Salama and T. F. El-Moselhy, Revisiting ageless antiques; synthesis, biological evaluation, docking simulation and mechanistic insights of 1, 4-Dihydropyridines as anticancer agents, Bioorg. Chem., 2021, 114, 105054, DOI:10.1016/j.bioorg.2021.105054.
- J. Tenhunen, T. Kučera, M. Huovinen, J. Küblbeck, E. Bisenieks, B. Vigante, Z. Ogle, G. Duburs, M. Doležal, R. Moaddel and M. Lahtela-Kakkonen, Screening of SIRT6 inhibitors and activators: A novel activator has an impact on breast cancer cells, Biomed. Pharmacother., 2021, 138, 111452, DOI:10.1016/j.biopha.2021.111452.
- D. F. Stern, Tyrosine kinase signalling in breast cancer: ErbB family receptor tyrosine kinases, Breast Cancer Res., 2000, 2(3), 1–8, DOI:10.1186/bcr51.
- H. Zhang, Three generations of epidermal growth factor receptor tyrosine kinase inhibitors developed to revolutionize the therapy of lung cancer, Drug Des., Dev. Ther., 2016, 24, 3867–3872, DOI:10.2147/DDDT.S119162.
- X. Du, B. Yang, Q. An, Y. G. Assaraf, X. Cao and J. Xia, Acquired resistance to third-generation EGFR-TKIs and emerging next-generation EGFR inhibitors, The Innovation, 2021, 2(2), 1–17, DOI:10.1016/j.xinn.2021.100103.
- E. R. Wood, A. T. Truesdale, O. B. McDonald, D. Yuan, A. Hassell, S. H. Dickerson, B. Ellis, C. Pennisi, E. Horne, K. Lackey and K. J. Alligood, A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib) relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells, Cancer Res., 2004, 64(18), 6652–6659, DOI:10.1158/0008-5472.CAN-04-1168.
- G. V. Jadhav, Synthesis of acetoacetanilides from aromatic amines, J. Ind. chem. Soc., 1930, 7, 69–684 Search PubMed.
- F. Denizot and R. Lang, Rapid colorimetric assay for cell growth and survival: modifications to the tetrazolium dye procedure giving improved sensitivity and reliability, J. Immunol. Methods, 1986, 89(2), 271–277, DOI:10.1016/0022-1759(86)90368-6.
- A. Daina, O. Michielin and V. Zoete, SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules, Sci. Rep., 2017, 7, 42717, DOI:10.1038/srep42717.
- A. Cheng and K. Merz, Prediction of aqueous solubility of a diverse set of compounds using quantitative structure-property relationships, J. Med. Chem., 2003, 46, 3572–3580, DOI:10.1021/jm020266b.
- J. L. Nakamura, The epidermal growth factor receptor in malignant gliomas: pathogenesis and therapeutic implications, Expert Opin. Ther. Targets, 2007, 11(4), 463–472, DOI:10.1517/14728222.11.4.463.
- N. Reda, A. Elshewy, H. I. El-Askary, K. O. Mohamed and A. A. Helwa, Design, synthesis, and biological evaluation of new pyrimidine-5-carbonitrile derivatives as novel anti-cancer, dual EGFR WT/COX-2 inhibitors with docking studies, RSC Adv., 2023, 13(46), 32296–32320, 10.1039/D3RA06088H.
- A. P. Kumar, S. Mandal, P. Prabitha, S. Faizan, B. P. Kumar, S. P. Dhanabal and A. Justin, Rational design, molecular docking, dynamic simulation, synthesis, PPAR-γ competitive binding and transcription analysis of novel glitazones, J. Mol. Struct., 2022, 1265, 133354, DOI:10.1016/j.molstruc.2022.133354.
- S. Mandal, B. R. P. Kumar, M. T. Alam, P. P. Tripathi and B. Channappa, Novel imidazole phenoxyacetic acids as inhibitors of USP30 for neuroprotection implication via the ubiquitin-Rho-110 fluorometric assay: Design, synthesis, and in silico and biochemical assays, ACS Chem. Neurosci., 2022, 13(9), 1433–1445, DOI:10.1021/acschemneuro.2c00076.
- S. Mandal, S. Faizan, N. M. Raghavendra and B. P. Kumar, Molecular dynamics articulated multilevel virtual screening protocol to discover novel dual PPAR α/γ agonists for anti-diabetic and metabolic applications, Mol. Diversity, 2023, 27(6), 2605–2631, DOI:10.1007/s11030-022-10571-w.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra01424c |
| This journal is © The Royal Society of Chemistry 2024 |