
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical oxidative decarboxylative of α-oxocarboxylic acids towards the synthesis of quinazolines and quinazolinones†
Jiwei Wua,
Mengru Zhanga,
Jun Hea,
Kaixuan Lia,
Longqiang Ye*a,
Jie Zhoub,
Xiaolan Xu*c,
Zirong Li*a and
Huajian Xu *b
*b
aCollege of Chemistry and Materials Engineering, Anhui Science and Technology University, Fengyang, 233100, China. E-mail: lizir@ahstu.edu.cn; yelq@ahstu.edu.cn
bSchool of Food and Biological Engineering, Hefei University of Technology, Hefei, 230009, China. E-mail: hjxu@hfut.edu.cn
cSchool of Medical Science, Anhui Medical University, Hefei, 230009, China. E-mail: xlxu@ustc.edu.cn
First published on 4th March 2024
Abstract
A mild and environmentally electrochemical method for the synthesis of quinazolines and quinazolinones has been developed through anodic oxidation decarboxylative of α-oxocarboxylic acids. The present reaction was efficiently conducted by using simple and cheap NH4I as the N-source and electrolyte in an undivided cell. The desired products, quinazolines and quinazolinones, were isolated in high yield under chemical oxidant free conditions.
Nitrogen-containing heterocycles,1 like quinazolines2 and quinazolinones,3 represent important classes of compounds that are widely found in natural products and biologically active molecules. Furthermore, some compounds such as prazosin, iressa, methaqualone, rutaecarpine, bouchardatine etc., containing quinazolines and quinazolinones scaffolds, exhibit various pharmaceutical activities and have been used in daily life (Fig. 1).4 Due to their importance, various methods for the synthesis of quinazolines5 and quinazolinones6 have been developed. Although several good results have been achieved, most of these methods require either metal catalysts or chemical oxidants. Recently, Wang's7 group reported a prominent research study on the electrosynthesis of quinazolines and quinazolinones using tetramethylethane-1,2-diamine as the methyl source (Scheme 1, I). However, the long reaction times and finite substrate scope have limited the application of the reaction.
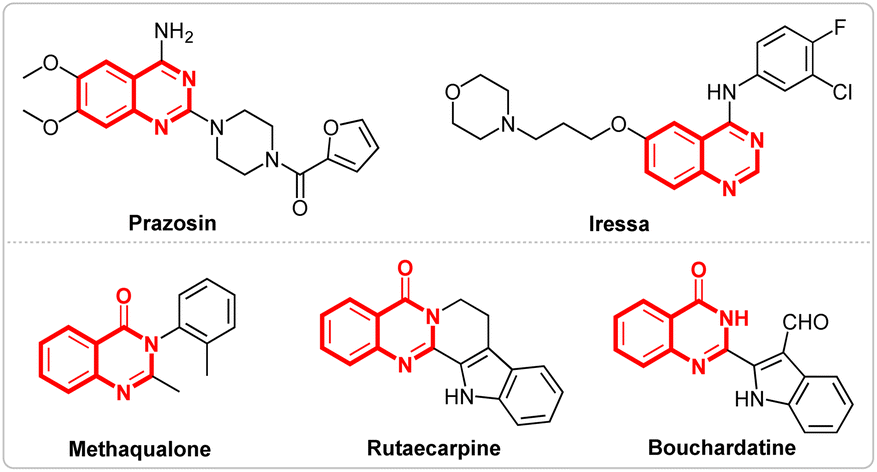 | ||
| Fig. 1 Structures of selected bioactive quinazolines and quinazolinones. | ||
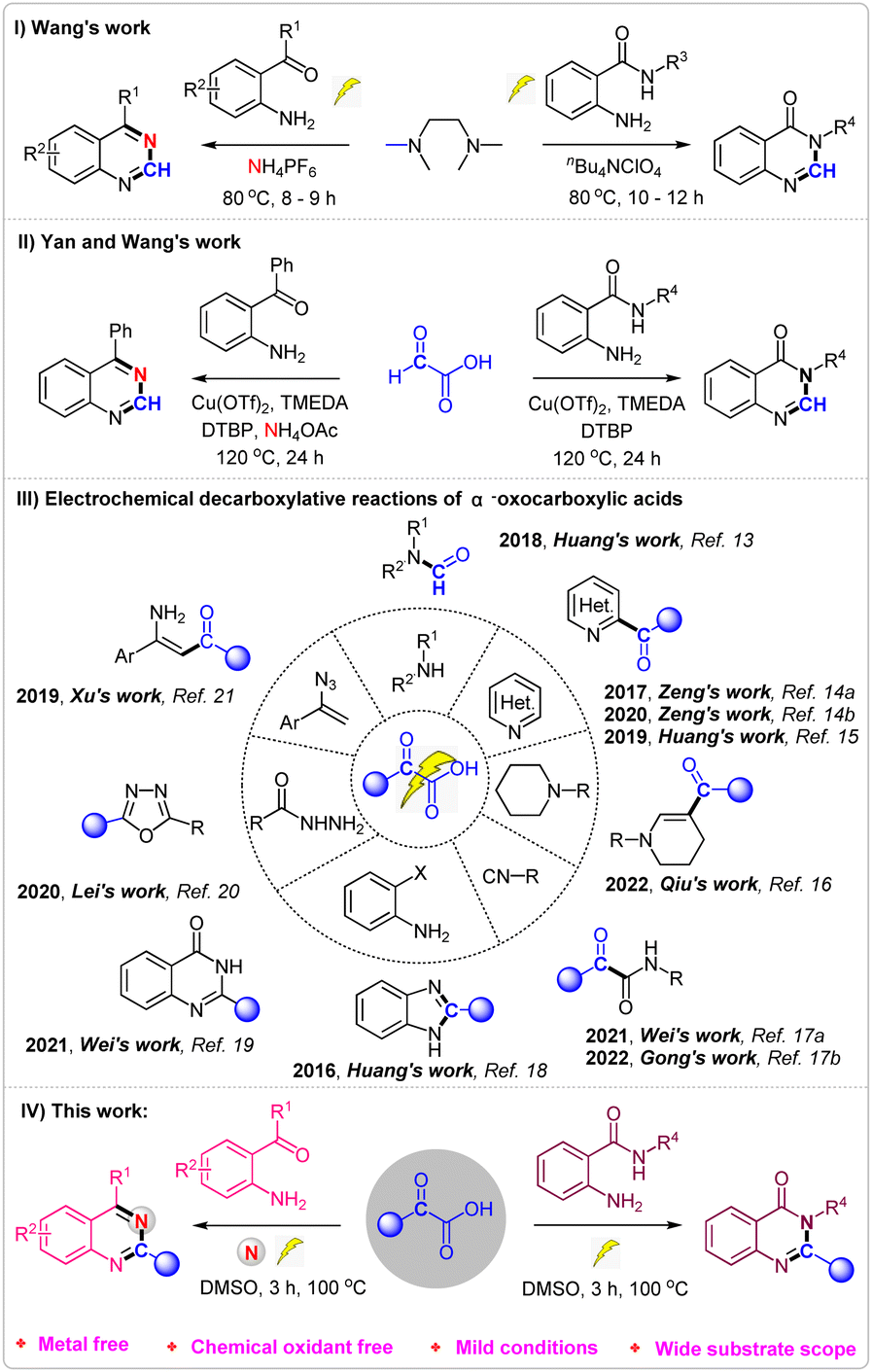 | ||
| Scheme 1 Various methods for the synthesis of quinazolines and quinazolinones and electrochemical decarboxylative reactions of α-oxocarboxylic acids. | ||
α-Oxocarboxylic acids, known for their low toxicity, high stability, and ease of storage, have been widely used as acyl reagents or C1 synthon through decarboxylation in the field of organic synthesis.8 In 2019, Yan and Wang reported a copper-catalyzed oxidative decarboxylative amination of glyoxylic acid for synthesis of quinazolines and quinazolinones9 (Scheme 1, II). Nevertheless, this method involves metal catalysts, chemical oxidants and long reaction time.
Since the discovery of Kolbe electrolysis,10 electrochemical oxidative11 decarboxylation has emerged as a practical tool for constructing C–C or C–heteroatom bonds, as it circumvents the need for chemical oxidants.12 Recently, Huang, Zeng, Xu, Lei, Wei and others reported a series of electrochemical decarboxylation reactions of α-oxocarboxylic acids for synthesis of formamides,13 ketone,14 3-formylindoles,15 β-substituted desaturated cyclic amines,16 α-ketoamides,17 2-substituted benzimidazoles,18 quinazolinones,19 2,5-disubstituted 1,3,4-oxadiazoles,20 enaminones21 etc.22 (Scheme 1, III). To further expand the application of electrochemical oxidative decarboxylation of α-oxocarboxylic acids, we hereby report an environmentally friendly electrochemical oxidative decarboxylation protocol for the synthesis of quinazolines and quinazolinones (Scheme 1, IV).
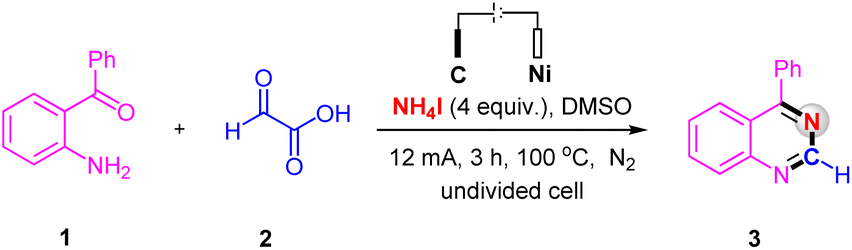
We commenced our studies by using (2-aminophenyl)(phenyl)methanone 1 and glyoxylic acid 2 as the model substrates to optimize the reaction conditions (Table 1). The desired product 3 was isolated in 94% yield by conducted the electrolysis in an undivided cell equipped with a carbon rod anode and a nickel plate cathode with a solvent of DMSO containing NH4I as the N-source and electrolyte at 100 °C under a constant current of 12 mA for 3 h (Table 1, entry 1). Control experiment has confirmed the essential role of electricity, no desired product without current (Table 1, entry 2). Compared with NH4I as the N-source and electrolyte, the yield dropped significantly when employing NH4Cl and CH3COONH4 as the N-source and electrolyte (Table 1, entries 3 and 4). Furthermore, increasing the current to 15 mA did not improve the reaction yield, whereas when the current was decreased to 8 mA, a slight decrease in yield was observed (Table 1, entries 5 and 6). As for the reaction temperature, higher temperature has no effect on the reaction (Table 1, entry 7), but the yield decreased below 100 °C (Table 1, entry 8). Neither prolonging nor shortening the reaction time could improve yield of 3 (Table 1, entries 9 and 10). Different electrode couples such as C(+)/C(−), C(+)/Pt(−) and C(+)/Fe(−) were examined, from which the couple electrode of C(+)/Ni(−) was the best choice for this reaction (Table 1, entries 1 and 11–13). When switching DMSO to other solvents, such as CH3CN and DMF, the yield of 3 was obtained in only 11% and 35%, respectively (Table 1, entries 14 and 15). Only 41% yield of product 3 was obtained, when the reaction was performed under air (Table 1, entry 16).
|
||
|---|---|---|
| Entry | Variation from the standard conditions | Yield ofa 3 (%) |
| a Reaction conditions: C anode, Ni cathode, constant current = 12 mA, 1 (0.2 mmol), 2 (0.4 mmol, 50% in water), NH4I (0.8 mmol), in 10.0 mL DMSO at 100 °C under N2 for 3 h. Yields shown are of isolated products. | ||
| 1 | None | 94 |
| 2 | No electric current | Trace |
| 3 | NH4Cl instead of NH4I | 45 |
| 4 | CH3COONH4 instead of NH4I | 72 |
| 5 | 15 mA instead of 12 mA | 81 |
| 6 | 8 mA instead of 12 mA | 77 |
| 7 | 120 °C instead of 100 °C | 93 |
| 8 | 80 °C instead of 100 °C | 61 |
| 9 | 3.5 h instead of 3 h | 88 |
| 10 | 2.5 h instead of 3 h | 76 |
| 11 | Graphite rod instead of nickel plate | 80 |
| 12 | Platinum plate instead of nickel plate | 86 |
| 13 | Iron plate instead of nickel plate | 67 |
| 14 | CH3CN instead of DMSO | 11 |
| 15 | DMF instead of DMSO | 35 |
| 16 | Air instead of N2 | 41 |
With the optimized conditions in hand, we next explored the scope of various o-carbonyl anilines and α-oxocarboxylic acids to illustrate the robustness of the electrochemical reactions for the synthesis of quinazolines. First, we examined the effect of o-carbonyl anilines with different substituents. It was observed that substituent R1 was an aryl group with electron-withdrawing such as F, Cl and Br attached to the phenyl ring were well tolerated, giving the corresponding quinazolines products 4–6 in good yields. When R1 was cyclohexyl, a 73% yield was obtained in this electrochemical strategy (Scheme 2, 7). Unfortunately, only 37% yield was obtained by employing 2-aminobenzaldehyde as the reactant (Scheme 2, 8). For the substrates of Cl and Br substituents on the 5-position of 2-aminobenzophenone, reactions also proceeded smoothly with glyoxylic acid and NH4I to deliver the desired products 9 and 10 in good yields. Then, the α-oxocarboxylic acids were investigated under the standard conditions. For phenylglyoxylic acid substrate, the product 11 was obtained in 84% isolated yield. Ortho-, meta-, and para-methyl substituted phenylglyoxylic acids were effectively converted into corresponding quinazolines product with good to excellent yields (Scheme 2, 12–14). Other electron-donating substituents, such as tert-butyl and methoxy groups, were also tolerated (Scheme 2, 15–16). The halogen group such as F, Cl and Br had little influence on the reaction, providing the products 17, 18 and 19 in 56–76% yields. For multisubstituted substrate 2-(3,5-dimethylphenyl)-2-oxoacetic acid, the product 20 was obtained in 57% isolated yield. Finally, the 2-(naphthalen-1-yl)-2-oxoacetic acid and 2-(naphthalen-2-yl)-2-oxoacetic acid were successfully used as the substrates, and the products 21 and 22 were produced in 73% and 80% yields, respectively.
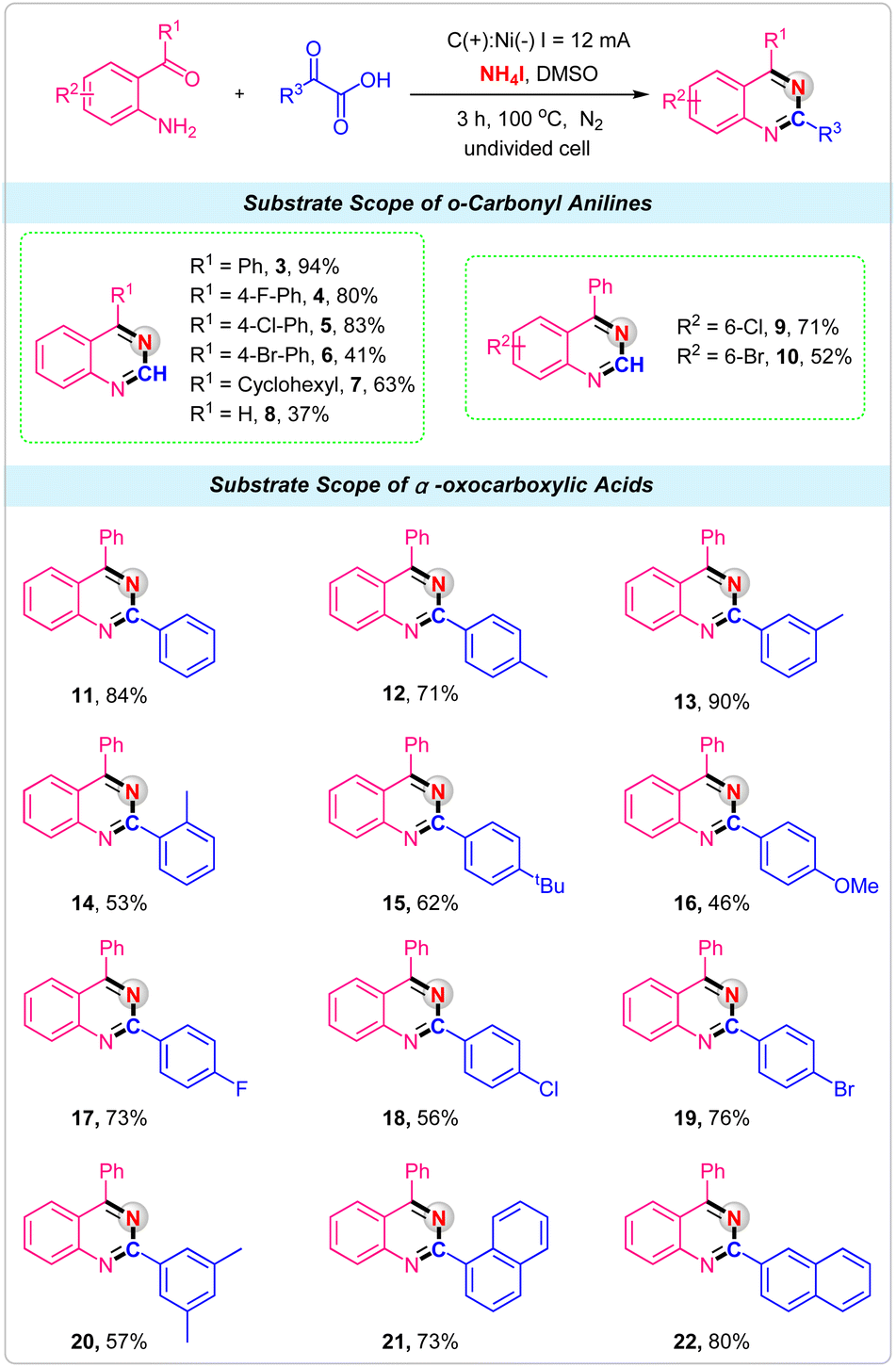 | ||
| Scheme 2 Substrate scope of electrochemical decarboxylative reactions of α-oxocarboxylic acids for synthesis of quinazolines. Reaction conditions: C anode, Ni cathode, constant current = 12 mA, o-carbonyl anilines (0.2 mmol), α-oxocarboxylic acids (0.4 mmol), NH4I (0.8 mmol), in 10.0 mL DMSO at 100 °C under N2 for 3 h. Yields shown are of isolated products. | ||
It is noteworthy to emphasize that the current tactics could also be used to synthesize quinazolinones under the same reaction conditions. Gratifyingly, the reaction of 2-aminobenzamide with glyoxylic acid proceeded smoothly, delivering the desired products 23 in a 74% yield. Then, 2-amino-N-alkylbenzamides were attempted. As shown, when the alkyl was methyl, n-butyl, allyl group, cyclopropyl, cyclohexyl or benzyl showed good to excellent reactivity in the synthesis of quinazolinones (Scheme 3, 24–29). Notably, the heterocyclic substituents such as 2-methylfuran, 2-methylthiophene and tryptamine were well tolerated in this electrochemical reaction giving the corresponding quinazolinones in excellent yields (Scheme 3, 30–32). Similarly, for 2-amino-N-phenylbenzamides, reactions also proceeded well to deliver the corresponding quinazolinones products in moderate to excellent yields, in which electron-donating and electron-withdrawing groups on the benzene ring of 2-amino-N-phenylbenzamides could be well tolerated (Scheme 3, 33–37). 2-Amino-N-(naphthalen-1-yl)benzamide was also suitable substrate to give product 38 in 67% yield. Next, various α-oxocarboxylic acids were tested to reacted with 2-aminobenzamide for synthesis of 2-aryl quinazolinones. From the reaction between 2-aminobenzamide and 2-oxo-2-phenylacetic acid, the product 2-phenylquinazolin-4(3H)-one 39 was achieved in 97% yield. The substituent position did not strongly impact the efficiency of this protocol (Scheme 3, 40–42). When methoxy, F and Cl groups were introduced into the para-position of 2-oxo-2-phenylacetic acid the products 43, 44 and 45 were obtained in good to excellent yields. Notably, 2-(naphthalen-2-yl)-2-oxoacetic acid could also react well with 2-aminobenzamide to form the target product 46 in 63% yield.
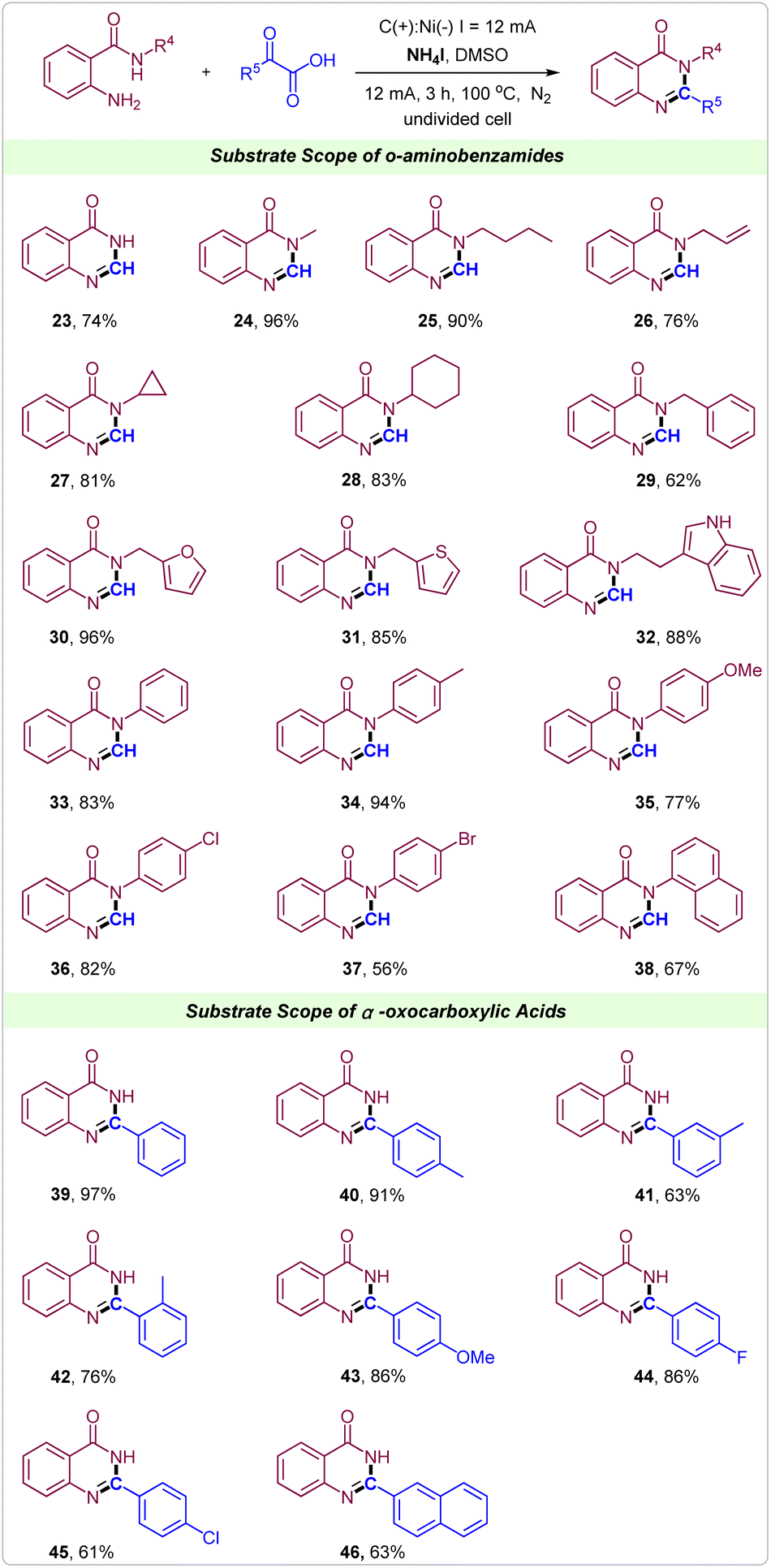 | ||
| Scheme 3 Substrate scope of electrochemical decarboxylative reactions of α-oxocarboxylic acids for synthesis of quinazolinones. Reaction conditions: C anode, Ni cathode, constant current = 12 mA, o-carbonyl anilines (0.2 mmol), α-oxocarboxylic acids (0.4 mmol), NH4I (0.8 mmol), in 10.0 mL DMSO at 100 °C under N2 for 3 h. Yields shown are of isolated products. | ||
The scalability of the electrochemical oxidative decarboxylative of α-oxocarboxylic acids for synthesizing nitrogen-containing heterocycles was evaluated on a 10.0 mmol scale (Scheme 4). The reaction afforded the corresponding product in an 84% yield, substantiating the great applied potential of this electrochemical reaction.
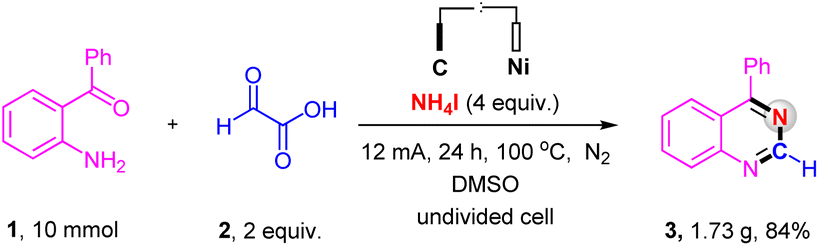 | ||
| Scheme 4 Gram scale synthesis. | ||
To gain more insight into the electrochemical oxidative reaction, a set of mechanistic studies was performed. First, the cyclic voltammetry (CV) experiments on NH4I, 1 and 47 have been carried out and showed that the oxidation peaks of NH4I, 1 and 47 in acetonitrile were 0.60 V, 1.62 V and 1.58 V (Fig. S5†), respectively. Then, the reaction between 1 and 47 was performed with chemical oxidants I2 in the absence of an electrical input and the desired product 11 was generated in 8% yield (Scheme 5A). These results implicated that this reaction probably proceeds by an iodine mediated process. Second, a 3 equiv. radical scavenger TEMPO (2,2,6,6-tetramethylpiperidin-1-oxyl) or BHT (2,6-di-tert-butyl-4-methylphenol) was added in this reaction under standard conditions, the yield of desired product 11 was obtained in yields of 17% and 13%, respectively. And the compounds 49, 50 and 51 were detected by GC-MS (Scheme 5B). These results implicated that the reaction may proceed by a radical pathway with acyl radical and nitrogen radical generating. Additionally, a constant amount of compound 48 was observed under the standard conditions. When, the 48 was test in the absence of 1 and 47, the product 11 was obtained in 41% yield (Scheme 5C), which showed that compound 48 may the inter-mediate in this reaction. Furthermore, compound 52 was detected by GC-MS (Scheme 5D) and benzaldehyde 53 was formed from 2-oxo-2-phenylacetic acid 47 (Scheme 5E) under standard conditions. Last but not least, replacing 1 with 53 under the standard conditions, 11 was obtained in 38% yield (Scheme 5F). However, when the reaction was reacted under the standard conditions without electric current for 3 h and then reacted under the standard conditions the yield of 11 was improved to 61% (Scheme 5G). These results indicating that the aldehyde intermediate may be generated in this electrochemical reaction.
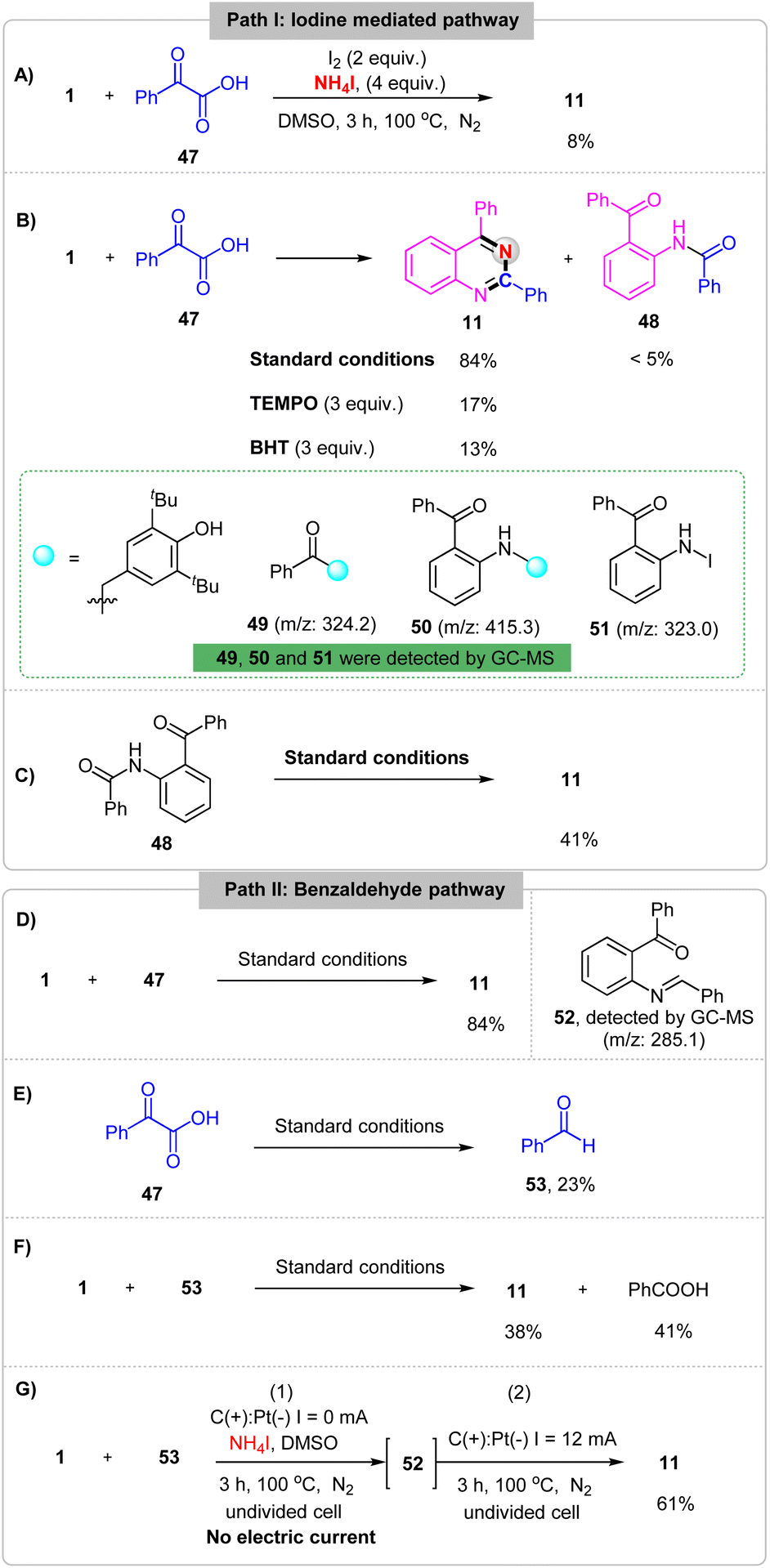 | ||
| Scheme 5 Mechanistic investigations. (A) I2 oxidation experiment. (B) Radical-trapping experiment. (C) Intermediate 48 validation experiment. (D) Intermediate 52 detection experiment. (E) Benzaldehyde 53 generation experiment. (F) Intermediate 53 validation experiment. (G) Intermediate 52 validation experiment. | ||
Taken the above results and the previous literature precedent, we propose two plausible mechanistic pathways for this electrochemical transformation. In path I, iodide ion generates I2 by anodic oxidation.14a,21 Then I2 react with α-keto carboxylate anion 55 to form acyl hypoiodite 56 along with iodide ion.14a,21 At the same time, the substrate 1 reacted with I2 to generated 51. Followed by the 56 and 51 undergoes hemolytic dissociation to give iodine radical, aroyloxy radical 57![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 14a,21 and nitrogen radical 58. Subsequently, intermediate 59 is generated through radical/radical coupling between 57 and 58. Finally, intermediate 59 react with NH4I to generate the target product 63.5b In path II, α-oxocarboxylic acid products aldehyde 60 with decarboxylation.18 Then, imine 61 is formed from 60 and 1 by dehydration condensation.7 On the other hand, +NH4 was reduced on the cathode to generate NH3.7,23 Subsequently, nucleophilic addition of NH3 to imine 61 provides an intermediate 62, which via a tandem condensation-oxidation process to generate the target product 63.5a,7 Noteworthily, acyl radical (57) and primary amine-based radical (58) were both transient radicals, making it difficult to generate 59 from the cross coupling of 57 and 58. Therefore, we propose that path II is the more favorable process for our reaction (Scheme 6).
14a,21 and nitrogen radical 58. Subsequently, intermediate 59 is generated through radical/radical coupling between 57 and 58. Finally, intermediate 59 react with NH4I to generate the target product 63.5b In path II, α-oxocarboxylic acid products aldehyde 60 with decarboxylation.18 Then, imine 61 is formed from 60 and 1 by dehydration condensation.7 On the other hand, +NH4 was reduced on the cathode to generate NH3.7,23 Subsequently, nucleophilic addition of NH3 to imine 61 provides an intermediate 62, which via a tandem condensation-oxidation process to generate the target product 63.5a,7 Noteworthily, acyl radical (57) and primary amine-based radical (58) were both transient radicals, making it difficult to generate 59 from the cross coupling of 57 and 58. Therefore, we propose that path II is the more favorable process for our reaction (Scheme 6).
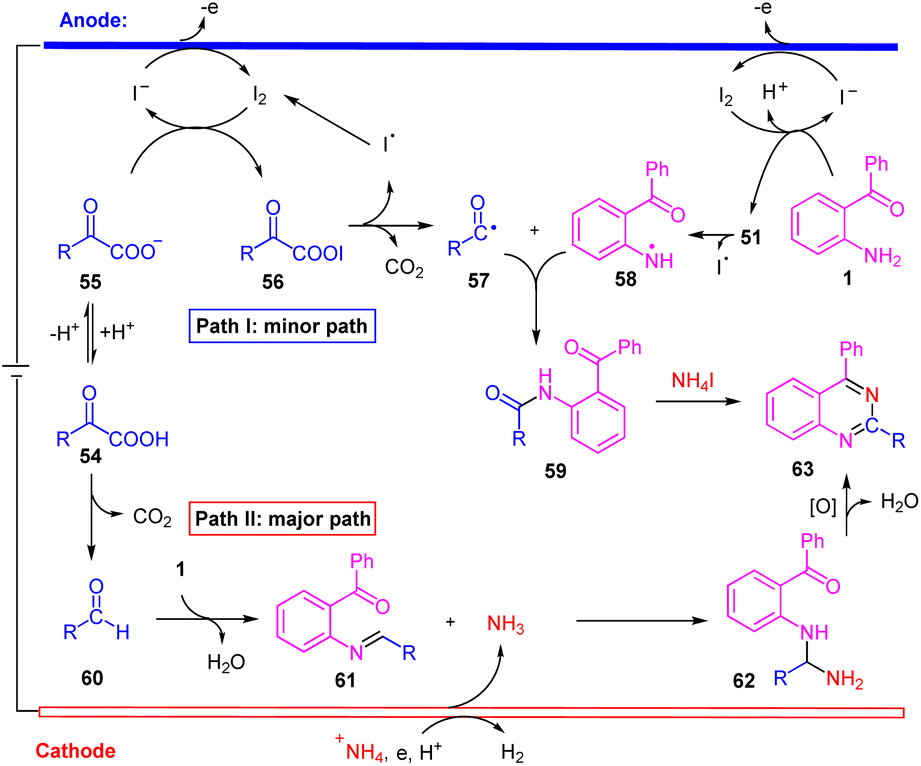 | ||
| Scheme 6 Proposed mechanism. | ||
Conclusions
In conclusion, we have developed a mild and environmentally friendly electrochemical method for the synthesis of quinazolines and quinazolinones via electrochemical oxidative decarboxylation of α-oxocarboxylic acids. This protocol utilizes NH4I as the N-source and electrolyte, which is simple and inexpensive, eliminating the need for transition metals and chemical oxidants. A wide range of substituted quinazolines and quinazolinones were synthesized with good to excellent yields.Author contributions
J. Wu, M. Zhang, J. He and K. Li, conducted the experiments. J. Wu, L. Ye and X. Xu conceived the project, wrote the manuscript and prepared the ESI. J. Zhou prepared part of the starting materials. Z. Li and H. Xu supervised the work and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from the National Nature Science Foundation of China (62205004), the Natural Science Foundation of Anhui Province (2008085QB66), the Talent Foundation of Anhui Science and Technology University (HCYJ201903), the Fundamental Research Funds for the Central Universities (PA2020GDKC0021), College Students' Innovation and Entrepreneurship Training Program (S202310879099).References
- (a) L. F. Tietze and A. Modi, Med. Res. Rev., 2000, 20, 304 CrossRef CAS; (b) V. Polshettiwar and R. S. Varma, Curr. Opin. Drug Discovery Dev., 2007, 10, 723 CAS.
- (a) J. P. Michael, Nat. Prod. Rep., 2008, 25, 166 RSC; (b) B. A. Foster, H. A. Coffrey, M. J. Morin and F. Rastinejad, Science, 1999, 286, 2507 CrossRef CAS; (c) J. F. Mendes da Silva, M. Walters, S. AlDamluji and C. R. Ganellin, Bioorg. Med. Chem., 2008, 16, 7254 CrossRef CAS.
- (a) K. Chen, A. F. Al Aowad, S. J. Adelstein and A. I. Kassis, J. Med. Chem., 2007, 50, 663 CrossRef CAS; (b) J. Ma, P. Li, X. Li, Q. Shi, Z. Wan, D. Hu, L. Jin and B. Song, J. Agric. Food Chem., 2014, 62, 8928 CrossRef CAS; (c) F. Rörsch, E. Buscato, K. Deckmann, G. Schneider, M. Schubert-Zsilavecz, G. Geisslinger, E. Proschak and S. Grösch, J. Med. Chem., 2012, 55, 3792 CrossRef.
- (a) R. Gundla, R. Kazemi, R. Sanam, R. Muttineni, J. A. P. R. Sarma, R. Dayam and N. Neamati, J. Med. Chem., 2008, 51, 3367 CrossRef CAS; (b) D. Fry, A. Kraker, A. McMichael, L. Ambroso, J. Nelson, W. R. Leopold, R. Connors and A. Bridges, Science, 1994, 265, 1093 CrossRef CAS; (c) A. Luth and W. Lowe, Eur. J. Med. Chem., 2008, 43, 1478 CrossRef; (d) J. F. M. Silva, M. Walters, S. Al-Damluji and C. R. Ganellin, Bioorg. Med. Chem., 2008, 16, 7254 CrossRef PubMed.
- For selected examples, see: (a) Y. Yan, Y. Zhang, C. Feng, Z. Zha and Z. Wang, Angew. Chem., Int. Ed., 2012, 51, 8077 CrossRef CAS PubMed; (b) D. Zhao, Q. Shen and J.-X. Lia, Adv. Synth. Catal., 2015, 357, 339 CrossRef CAS; (c) G. Satish, P. Ashok, L. Kota and A. Ilangovan, Org. Biomol. Chem., 2019, 17, 4774 RSC; (d) H.-B. Xu, Y.-Y. Zhu, J.-H. Yang, X.-Y. Chai and L. Dong, Org. Chem. Front., 2020, 7, 1230 RSC; (e) J. K. Laha, S. Panday, M. Tomar and K. V. Patel, Org. Biomol. Chem., 2021, 19, 845 RSC; (f) B. Han, C. Wang, R.-F. Han, W. Yu, X.-Y. Duan, R. Fang and X.-L. Yang, Chem. Commun., 2011, 47, 7818 RSC; (g) S. Yao, K. Zhou, J. Wang, H. Cao, L. Yu, J. Wu, P. Qiu and Q. Xu, Green Chem., 2017, 19, 2945 RSC; (h) J. Zhang, D. Zhu, C. Yu, C. Wan and Z. Wang, Org. Lett., 2010, 12, 2841 CrossRef CAS PubMed; (i) L. Zhang, Z. Hu, J. Dong, X. M. Zhang and X. Xu, Adv. Synth. Catal., 2018, 360, 1938 CrossRef CAS.
- For selected examples, see: (a) W. Yu, X. Zhang, B. Qin, Q. Wang, X. Ren and X. He, Green Chem., 2018, 20, 2449 RSC; (b) L. Cao, H. Huo, H. Zeng, Y. Yu, D. Lu and Y. Gong, Adv. Synth. Catal., 2018, 360, 4764 CrossRef CAS; (c) Y. Yan, Y. Xu, B. Niu, H. Xie and Y. Liu, J. Org. Chem., 2015, 80, 5581 CrossRef CAS PubMed; (d) D. Zhao, T. Wang and J.-X. Li, Chem. Commun., 2014, 50, 6471 RSC; (e) Y. Zhang, Z. Zhou, Z. Li, K. Hu, Z. Zha and Z. Wang, Chem. Commun., 2022, 58, 411 RSC; (f) Y. Zhu, P. Z. Fei, M. C. Liu, F. C. Jia and A. X. Wu, Org. Lett., 2013, 15, 378 CrossRef CAS; (g) K. Govindan, T. Duraisamy, A. Jayaram, G. C. Senadi and W.-Y. Lin, Synthesis, 2022, 54, 1115 CrossRef CAS.
- Z. Zhou, K. Hu, J. Wang, Z. Li, Y. Zhang, Z. Zha and Z. Wang, ACS Omega, 2020, 5, 31963 CrossRef CAS.
- For selected examples, see: (a) F. Penteado, E. F. Lopes, D. Alves, G. Perin, R. G. Jacob and E. J. Lenardão, Chem. Rev., 2019, 119, 7113 CrossRef CAS PubMed; (b) Y. Xiang, G. Zeng, X. Sang, X. Li, Q. Ding and Y. Peng, Tetrahedron, 2021, 91, 132193 CrossRef CAS; (c) L.-N. Guo, H. Wang and X.-H. Duan, Org. Biomol. Chem., 2016, 14, 7380 RSC.
- B. Niu, S. Li, C. Cui, Y. Yan, L. Tang and J. Wang, Eur. J. Org Chem., 2019, 2019, 7800 CrossRef CAS.
- (a) H. Kolbe, Liebigs Ann. Chem., 1848, 64, 339 CrossRef; (b) H. Kolbe, Liebigs Ann. Chem., 1849, 69, 257 CrossRef.
- (a) Y. Yuan, J. Yang and A. Lei, Chem. Soc. Rev., 2021, 50, 10058 RSC; (b) P. Xiong and H.-C. Xu, Acc. Chem. Res., 2019, 52, 3339 CrossRef CAS; (c) K.-J. Jiao, Y.-K. Xing, Q. L. Yang, H. Qiu and T.-S. Mei, Acc. Chem. Res., 2020, 53, 300 CrossRef CAS; (d) S.-H. Shi, Y. Liang and N. Jiao, Chem. Rev., 2021, 121, 485 CrossRef CAS; (e) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230 CrossRef CAS; (f) K. D. Moeller, Chem. Rev., 2018, 118, 4817 CrossRef CAS; (g) S. D. Minteer and P. S. Baran, Acc. Chem. Res., 2020, 53, 545 CrossRef CAS; (h) P. Qian, Z. Zha and Z. Wang, ChemElectroChem, 2020, 7, 2527 CrossRef CAS; (i) J. Yount and D. G. Piercey, Chem. Rev., 2022, 122, 8809 CrossRef CAS; (j) X. Cheng, A. Lei, T.-S. Mei, H.-C. Xu, K. Xu and C. Zeng, CCS Chem., 2022, 4, 1120 CrossRef CAS; (k) B. Kaboudin, M. Behroozi and S. Sadighi, RSC Adv., 2022, 12, 30466 RSC; (l) R. Francke and R. D. Little, Curr. Opin. Electrochem., 2023, 40, 101315 CrossRef CAS; (m) Z. Tan, H. Zhang, K. Xu and C. Zeng, Sci. China: Chem., 2024, 67, 450 CrossRef CAS; (n) S. Imeni, A. Makarem and R. Javahershenas, Asian J. Org. Chem., 2023, 12, e202300303 CrossRef CAS.
- (a) J. Lia, S. Zhang and K. Xu, Chin. Chem. Lett., 2021, 32, 2729 CrossRef; (b) H. Zhang, S. Liang, D. Wei, K. Xu and C. Zeng, Eur. J. Org Chem., 2022, e202200794 CrossRef CAS; (c) V. Ramadoss, Y. Zheng, X. Shao, L. Tian and Y. Wang, Chem.–Eur. J., 2021, 27, 3213 CrossRef CAS.
- D.-Z. Lin and J.-M. Huang, Org. Lett., 2018, 20, 2112 CrossRef CAS.
- (a) Q.-Q. Wang, K. Xu, Y.-Y. Jiang, Y.-G. Liu, B.-G. Sun and C.-C. Zeng, Org. Lett., 2017, 19, 5517 CrossRef CAS; (b) H. Ding, K. Xu and C. C. Zeng, J. Catal., 2020, 381, 38 CrossRef CAS.
- D.-Z. Lin and J.-M. Huang, Org. Lett., 2019, 21, 5862 CrossRef CAS.
- T. Feng, S. Wang, Y. Liu, S. Liu and Y. Qiu, Angew. Chem., Int. Ed., 2022, 61, e20211517 Search PubMed.
- (a) F. Zhao, N. Meng, T. Sun, J. Wen, X. Zhao and W. Wei, Org. Chem. Front., 2021, 8, 6508 RSC; (b) Y. Zhao, X. Meng, C. Cai, L. Wang and H. Gong, Asian J. Org. Chem., 2022, 11, e202100748 CrossRef CAS.
- H.-B. Wang and J.-M. Huang, Adv. Synth. Catal., 2016, 358, 1975 CrossRef CAS.
- Q. Tian, J. Zhang, L. Xu and Y. Wei, Mol. Catal., 2021, 500, 111345 CrossRef CAS.
- F. Lu, F. Gong, L. Li, K. Zhang, Z. Li, X. Zhang, Y. Yin, Y. Wang, Z. Gao, H. Zhang and A. Lei, Eur. J. Org Chem., 2020, 2020, 3257 CrossRef CAS.
- X. Kong, Y. Liu, L. Lin, Q. Chen and B. Xu, Green Chem., 2019, 21, 3796 RSC.
- (a) X. Kong, Y. Chen, X. Chen, Z.-X. Lu, W. Wang, S.-F. Ni and Z.-Y. Cao, Org. Lett., 2022, 24, 2137 CrossRef CAS; (b) X. Wang, S. Wu, Y. Zhong, Y. Wang, Y. Pan and H. Tang, Chin. Chem. Lett., 2023, 34, 107537 CrossRef CAS.
- L.-S. Kang, M.-H. Luo, C. M. Lam, L.-M. Hu, R. D. Little and C.-C. Zeng, Green Chem., 2016, 18, 3767 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra01318b |
| This journal is © The Royal Society of Chemistry 2024 |