
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mesoporous Co3O4@CdS nanorods as anode for high-performance lithium ion batteries with improved lithium storage capacity and cycle life†
Hamza Waleedai,
Haroon Ur Rasheedai,
Faisal Faizb,
Amina Zafarc,
Saqib Javedd,
Yanguo Liue,
Shafqat Karima,
Hongyu Sun e,
Yasir Faizf,
Shafqat Hussaina,
Atia Khalidg,
Yanlong Yu*h,
Amjad Nisar*a and
Mashkoor Ahmad*a
e,
Yasir Faizf,
Shafqat Hussaina,
Atia Khalidg,
Yanlong Yu*h,
Amjad Nisar*a and
Mashkoor Ahmad*a
aNanomaterials Research Group, Physics Division, PINSTECH, Islamabad 44000, Pakistan. E-mail: mashkoorahmad2003@yahoo.com; chempk@gmail.com
bCollege of Electronics and Information Engineering, Shenzhen University, Shenzhen, PR China
cCentral Analytical Facility Division, PINSTECH, Islamabad 44000, Pakistan
dTheoretical Physics Division, PINSTECH, Islamabad 44000, Pakistan
eSchool of Resources and Materials, Northeastern University at Qinhuangdao, Qinhuangdao, 066004, PR China
fChemistry Division, PINSTECH, Islamabad 44000, Pakistan
gSchool of Materials Science and Engineering, Tsinghua University, Beijing, China
hCollege of Chemistry and Chemical Engineering, Northeast Petroleum University, Daqing, 163318, PR China. E-mail: ylyu66@163.com
iDepartment of Physics, Faculty of Basic & Applied Sciences, IIU, Islamabad, 44000, Pakistan
First published on 15th April 2024
Abstract
Transition metal oxides based anodes are facing crucial problems of capacity fading at long cycles and high rates due to electrode degradations. In this prospective, an effective strategy is employed to develop advanced electrode materials for lithium-ion batteries (LIBs). In the present work, a mesoporous Co3O4@CdS hybrid sructure is developed and investigated as anode for LiBs. The hybrid structure owning porous nature and large specific surface area, provides an opportunity to boost the lithium storage capabilities of Co3O4 nanorods. The Co3O4@CdS electrode delivers an initial discharge capacity of 1292 mA h g−1 at 0.1C and a very stable reversible capacity of 760 mA h g−1 over 200 cycles with a capacity retention rate of 92.7%. In addition, the electrode exhibits excellent cyclic stability even after 800 cycles and good rate performance as compared to previously reported electrodes. Moreover, density functional theory (DFT) and electrochemical impedance spectroscopy (EIS) confirm the enhanced kinetics of the Co3O4@CdS electrode. The efficient performance of the electrode may be due to the increased surface reactivity, abundant active sites/interfaces for rapid Li+ ion diffusion and the synergy between Co3O4 and CdS NPs. This work demonstrates that Co3O4@CdS hybrid structures have great potential for high performance batteries.
1 Introduction
With the rapid increase in the global energy crisis and environmental pollution, researchers directed their research activities to explore effective ways for store energy. Currently, nations around the world are working hard to produce clean, sustainable, and renewable energy sources including solar, wind, and ocean power. However, these sources need energy storage systems to regulate energy production. So far various energy storage devices such as batteries, supercapacitors and fuel cell etc. have been fabricated to store energy from renewable sources. Among these devices, rechargeable lithium-ion batteries (LIBs) is crucial to meet future needs for industries from personal devices to automobiles.1 However, durability and energy densities of current LIBs are restricted by electrode material.2 Therefore, it remains a great challenge to develop advanced electrode materials for LIBs that exhibits improved cycling stability and larger specific capacities. The current progress in the field of energy storage materials provides more innovative solutions to resolve the energy storage issues.So far, various transition metal oxides (TMOs) such as MnO2,3 V2O5,4 ZnO,5 WO3,6 Co3O4,7 etc. have been exploited as the anode materials for LIBs. Among these, cobalt oxide (Co3O4) is regarded as one of the most promising anode materials due to its high theoretical capacity (∼890 mA h g−1), environmental friendliness, and superior electrochemical properties. However, its poor cyclic stability, irreversible capacity loss and slow kinetics of Li-ion and electron transport hinder its practical application. In order to overcome these drawbacks, many attempts have been made such as the use of conductive polymers,8 doping with transition metals,9 and making composites.10 On the other hand, recent work on metal sulphides has drawn considerable attention for being the most promising electrode materials for LIBs due to their high electrical conductivity, thermal durability and rich redox chemistry than their metal oxides equivalents. To explore novel materials for energy storage devices, metal sulphides provide better option due to its outstanding features. Among various metal sulphides, cadmium sulfide (CdS) has received less attention as an electrode material for LIBs. Therefore, in order to explore the energy storage features of Co3O4 and CdS in a single system, their combination is found to be a good approach for the development of novel electrode material. Such hybrid systems are very appealing for energy storage applications due to their porous nature and ability to absorb guest species such as lithium ion on their surfaces and in the pore spaces. Up till now, according to our knowledge, there have been no reports found on the investigation of Co3O4@CdS nanorods as an electrode material for LIBs. For example, D. S. Patil, et al., have reported the use of the core–shell structure of Co3O4@CdS for high-performance supercapacitors,11 F. Q. Liu, et al., prepared Co3O4/CdS photoelectrode for photoelectrochemical cathodic protection in the dark12 and Z. Qin et al., have developed Co3O4/CdS p–n heterojunction for enhancing photocatalytic hydrogen production.13 C. Jiang et al., prepared Co3O4@CdS Hollow Spheres by the template-removal method with the assistance of the ZIF-67 material and obtained high phenol and dye photodegradation activity.14
In this work, mesoporous Co3O4@CdS nanorods are developed and investigated as an anode material for LIBs. The hybrid structure shows enhanced physical and chemical properties superior to a single counterpart. The porous nature, large specific surface area and excellent kinetics of the synthesized nanorods leads to rapid lithium storage. The developed electrode exhibits enhanced cycling stability and high-rate capability as compared to pristine Co3O4 nanorods. Therefore, Co3O4@CdS structure can be considered suitable candidate as an anode material for LIBs.
2 Experimental
The experimental detail is given in the (ESI).†3 Results and discussion
3.1. Morphological, structural and compositional analysis
XRD analysis was performed in order to investigate the phase and crystallinity of the as-prepared Co3O4 and Co3O4@CdS structures as shown in Fig. 1(a). In pristine Co3O4 pattern, the diffraction peaks at 19°, 31.3°, 37°, 38.5° and 44.7° are assigned to (111), (220), (311), (222) and (400) planes of Co3O4 according to JCPDS # 00-042-1467.15 The diffraction pattern of Co3O4@CdS structure shows the additional peaks located at 25.2°, 26.8°, 27.6°, 43.7°and 52.1° attributed to the (100), (002), (101), (110) and (112) planes of CdS according to (JCPDS # 00-041-1049) and confirms the deposition of CdS.16 No other peaks related to impurities are observed, demonstrating the purity of both samples. | ||
| Fig. 1 (a) XRD patterns of pure Co3O4 and Co3O4@CdS structures. (b) Raman spectra of Co3O4 and Co3O4@CdS NPs structures prepared through deposition by modified SILAR method. (c) FE-SEM image of Co3O4@CdS structures; (d) corresponding EDX spectrum of Co3O4@CdS structures (e) TEM image of Co3O4@CdS structure (f) HRTEM of Co3O4@CdS structure. | ||
In order to understand the vibrational behaviour of the prepared structures, Raman spectra of Co3O4 and Co3O4@CdS structures were recorded in the range of (100–1000) cm−1 as shown in Fig. 1(b). In Co3O4 spectrum, the bands located at 185, 478, 520, and 686 cm−1are associated to F2g, Eg, F2g and A1g modes respectively.17 In Co3O4@CdS spectrum, besides Co3O4 bands, two characteristic bands of CdS at 300 cm−1 and 600 cm−1 were also recorded which are attributed to the fundamental longitudinal optical phonon (1LO) and its overtone longitudinal phonon (2LO) respectively.18 In comparison with Co3O4 spectrum, a small peak located at 413 cm−1 is also observed, which ascribed to the Co–S bond formation on the surface of Co3O4@CdS nanorods.19 With the close observation, a slight shift was also observed in the position of 1LO and 2LO modes which is due to the shape of the nanostructures.20 In addition, the composite spectrum exhibits broadening associated with the slight changes in the crystalline structure of pristine Co3O4 as mentioned by previous reports.21
To examine the morphology of the as-prepared Co3O4@CdS structure, SEM was performed. Fig. 1(c) shows the SEM images of Co3O4@CdS nanorods. As observed from the images, the Co3O4 nanorods are composed of highly dense CdS NPs interconnected with one another in uniformly ordered arrays. For comparison, the SEM images of the pristine Co3O4 nanorods and Co3O4@CdS nanorods are also recorded as shown in Fig. S1 (ESI), S2(a and b)† respectively. The surface of the nanorods appear to be uniform and smooth. Fig. 1(d) represents the corresponding EDX spectrum of Co3O4@CdS which consists of Cd, S, Co, and O peaks, confirm the formation of Co3O4@CdS nanorods. The existence of Cd and S peaks further confirms the successful deposition of CdS NPs. In order to clarify the detail structure and elemental distribution of the prepared composite, EDX elemental mapping are performed. Fig. S3(a–f)† displays the STEM images of Co3O4@CdS structure and a corresponding elemental mapping of Co, O, Cd and S respectively. The presence of Cd and S confirms the deposition of CdS and no other impurities formed other than Co3O4 and CdS. The detailed structural analysis was conducted via TEM and HRTEM. Fig. 1(e) exhibits a low magnification TEM image of Co3O4@CdS structure, demonstrating the deposition of CdS NPs on the whole surface of Co3O4 nanorods. Fig. 1(f) displays the HRTEM analysis of the Co3O4@CdS structure and exhibits a polycrystalline nature. The well resolved lattice fringes of 0.24, 0.47 are observed corresponding to (311) and (111) planes of Co3O4 nanorods. The lattice fringe of 0.33 nm belongs to (002) plane of CdS is also identified.
FTIR analysis was carried out to examine and investigate the structural molecular changes and presence of different functional groups in as-prepared Co3O4@CdS structures. Fig. S4 (ESI)† illustrates the FTIR spectra of the Co3O4 and Co3O4@CdS structures. The bands sited at 555 cm−1 and 657 cm−1 reveals the stretching vibrations which show the presence of Co–O bonding. The band located at 1039 cm−1 is illustrating the presence of Cd–S interaction and manifests the formation of the Co3O4@CdS composite. A sharp peak sited at 1458 cm−1 represents the bending vibrations of monodispersed Co3O4 structure. Furthermore, the significant peaks sited at 2851 cm−1 and 2918 cm−1 shows the symmetric and asymmetric stretching modes of –CH2 which comprises from HMT which plays a significant role as a nucleation controlled reagent and thus, favouring the surface modification of Co3O4 nanorods.
The surface of the nanostructures plays an important role in the electrochemical performance of the material. Therefore, XPS of Co3O4@CdS is measured to examine the surface-localized information of the elements. Fig. 2(a) shows the deconvoluted XPS spectrum of Co 2p, which consists of two regions at 796.7 eV and 780.9 eV corresponding to Co 2p1/2 and Co 2p3/2 states respectively. In the Co 2p3/2 region, two coexisting peaks at 781.1 eV and 783.0 eV can be seen which confirms the Co3+ and Co2+ states. Moreover, Co 2p1/2 region also composed of two peaks at 797.3 eV and 798.3 eV related to Co3+ and Co2+ states. Furthermore, two satellites peaks at 786.7 eV and 803.3 eV corresponding to Co 2p3/2 and Co 2p1/2 regions, reconfirms the formation of Co2+ and Co3+ states and agrees well with the previous report.22 For comparison, XPS spectra of Co 2p region of Co3O4 structures is also recorded as shown in Fig. S5 (ESI).† The area ratio of Co3+ to Co2+ of both structures are calculated. As observed the relative area ratio of Co3O4@CdS (1.14) is significantly higher as compare to Co3O4 (1.07) confirming that the Co3+ states are more exposed on the surface of Co3O4@CdS structure. It is already reported that the nanostructure with dominant Co3+ sites exhibit superior electrochemical performance.23–25 Thus Co3O4@CdS structure with dominant Co3+ active sites are considering more suitable for energy storage applications. Fig. 2(b) depicts the high-resolution spectrum of O 1s. The spectrum fitted into three peaks located at 531.4, 532.1 and 533.5 eV corresponding to the O–Co bond, oxygen vacancy defects and O–H species due to the surface absorption of water. The existence of Cd2+ is confirmed by the high-resolution spectrum of Cd 3d peak as shown in Fig. 2(c). The spectrum reveals two binding energy peaks at 406.41 and 411.14 eV correspond to the electronic states of Cd 3d5/2 and Cd 3d3/2, respectively. The energy difference between two peaks is 4.73 eV, which agrees well with the reported study.26 Fig. 2(d) demonstrates the spectrum of S 2p peaks which comprised two major spin–orbit peaks located at 162.3 eV and 163.9 eV corresponding to the S 2p3/2 and S 2p1/2 states respectively. In addition, two peaks at 163.2 eV and 164.7 eV corresponding to the S 2p3/2 and S 2p1/2 states can be seen which ascribed to the formation of Co–S bond on the surface of Co3O4@CdS nanorods.
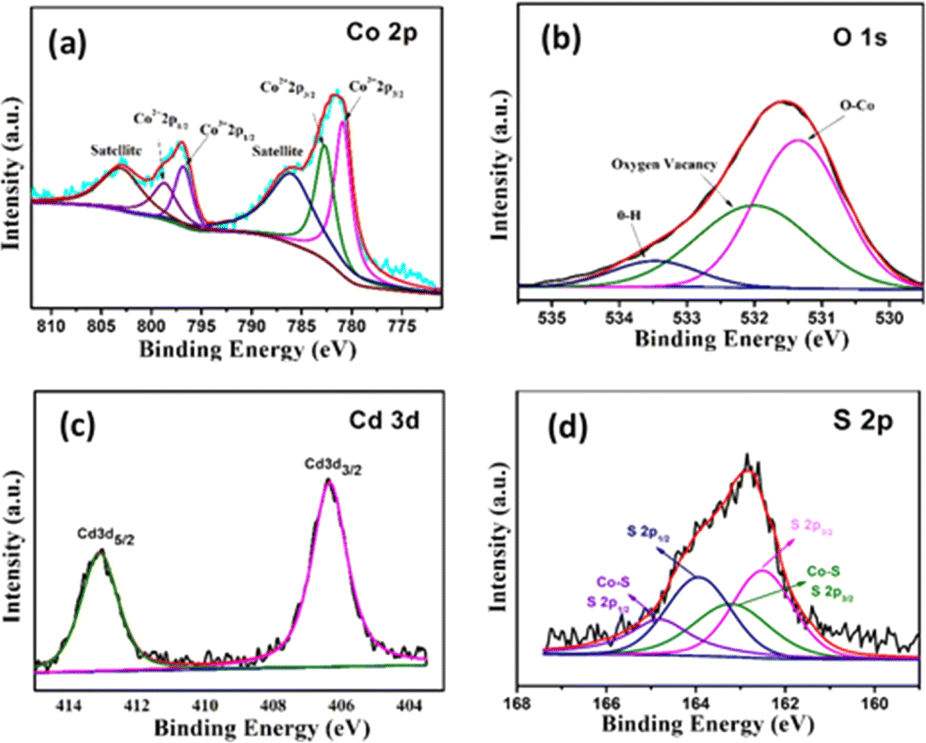 | ||
| Fig. 2 High resolution XPS spectrum of (a) Co 2p (b) O 1s (c) Cd 3d and (d) S 2p peak. | ||
Fig. S6 (ESI)† shows the N2 adsorption–desorption isotherm of Co3O4 and Co3O4@CdS structures determined at 77 K. It can be observed that the isotherm of Co3O4@CdS structures, presents a broad hysteresis loop as compared to pristine Co3O4 structure. The corresponding surface area of Co3O4 and Co3O4@CdS structures was calculated to be 43 m2 g−1 and 81 m2 g−1 respectively. The increased surface area of the hybrid structure is considered due to the CdS NPs and the porous nature of Co3O4@CdS structures. Moreover, the pore size of Co3O4@CdS structures, based on Barrett–Joyner–Halenda (BJH) Model, is also measured by the pore size distribution curve as illustrated in the inset of Fig. S6 (ESI).† The calculated average pore size is ∼11 nm. The interconnected primary nanoparticles and the void spaces within a single nanorod is the primary sources for the formation of mesopores. It is well-known that the porous structure with a high electroactive surface area, plays a vital role in the electrochemical processes.27 Thus, the provision of such good mesoporous surface, accompanying abundant active sites, proves to be beneficial for lithium ion battery.
3.2. Electrochemical performance of Co3O4@CdS nanorod/electrode
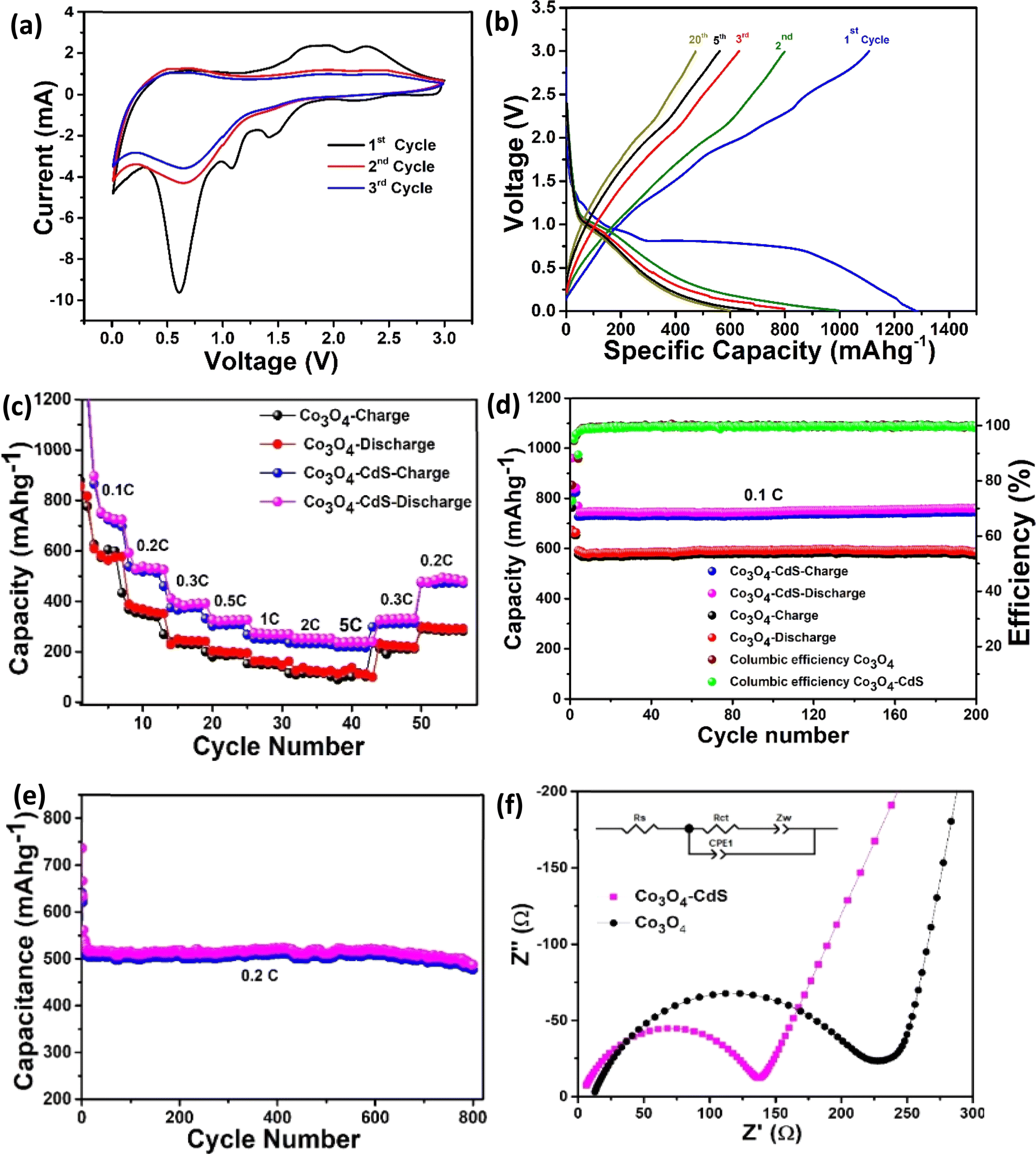 | ||
| Fig. 3 (a) CV curves of the Co3O4@CdS electrode for 1st, 3rd and 5th cycle at a scan rate of 0.5 mV s−1. (b) Galvanostatic charge/discharge profiles of the Co3O4@CdS electrode (c) rate performance (d) performance of electrodes at 0.1C for 200 cycles and (e) cyclic stability of Co3O4@CdS electrode at 0.2C for 800 cycles (f) comparison of the EIS curves of Co3O4@CdS and Co3O4 electrodes; inset is the kinetic parameters of both electrodes. | ||
| Materials | Synthesis methods | Initial discharge capacities (mA h g−1) | Current densities (A g−1/C-rate) | Reversible capacity (mA h g−1) | Shelf life (cycles) | Ref. |
|---|---|---|---|---|---|---|
| Nanoporous TiO2/Co3O4 composite | One-step dealloying method | 998 | 100 mA g−1 | 295 | 500 | 31 |
| Co3O4 NWs | Decomposition of CoC2O4·2H2O NWs | 1027 | 0.11 A g−1 | 611 | 50 | 32 |
| Co3O4/graphene composite | Facile synthesis | 1097 | 50 A g−1 | 541 | 30 | 33 |
| CoTiO3/Co3O4/TiO2 | Mechanical milling | 480 | 100 mA g−1 | 722 | 250 | 34 |
| Co3O4@TiO2 CSNFs | Hydrothermal method | 1034 | 0.2C | 632 | 100 | 35 |
| PNF Co3O4 | Self-combustion | 1108 | 0.5C | 661 | 100 | 36 |
| Co3O4@CNT | Nano-casting method | 1260 | 0.1 A g−1 | 453 | 30 | 37 |
| Co3O4 nanoparticles | Facile synthesis | 1105 | 50 A g−1 | 541 | 30 | 33 |
| Co3O4@C | MOF-derived strategy | 1112 | 250 mA g−1 | 721 | 500 | 38 |
| Co3O4@CdS NRs | Hydrothermal method | 1292 | 0.1C | 760 | 800 (0.2C) | This work |
The rate performance of both cells was also investigated. Fig. 3(c) exhibits the comparison of rate performance of Co3O4 and Co3O4@CdS cells at current rates ranging between 0.1C to 0.5C. As the current rates increase from 0.1C to 5C, it can be observed that the reversible capacity of the Co3O4@CdS electrode steadily decreases from 760 mA h g−1 to 258 mA h g−1. Comparatively, the reversible capacities of the Co3O4 electrode drop sharply from 620 mA h g−1 to 101 mA h g−1 at the similar rates (0.1C to 5C). Interestingly, when the current rate retunes at 0.2C, the Co3O4@CdS electrode restores its original reversible capacity more effectively than the Co3O4 electrode after 50 cycles. The stability of the electrodes is investigated by testing the cyclic performance of the Co3O4 and Co3O4@CdS electrodes over 200 cycles at 0.1C as shown in Fig. 3(d). As observed, both electrodes exhibit good cycling performance. After 200 cycles, the Co3O4@CdS electrode shows the reversible capacity of 760 mA h g−1 with a capacity retention rate of 83.7% and 92.7% respectively. In comparison, the reversible capacity of Co3O4 electrode is 580 mA h g−1 which is much smaller that Co3O4@CdS electrode. In order to further evaluate the long cyclic stability of the Co3O4@CdS electrode, galvanostatic charge/discharge are performed for 800 cycles at 0.2C as shown in Fig. 3(e). It can be observed that after 800 cycles, the electrode shows a stable reversible capacity of 520 mA h g−1 corresponding to the 90% of the initial capacity. The improved and stable performance of the Co3O4@CdS electrode is associated with its mesoporous nature which provides more active sites for Li+ insertion/extraction process. These findings demonstrate that the Co3O4@CdS structure is a suitable choice for the construction of high-rate performance batteries.
| Material | Rs (Ω) | Rct (Ω) |
|---|---|---|
| Co3O4@CdS | 2.615 | 135.1 |
| Co3O4 | 10.88 | 223.6 |
3.3. DFT calculations
To further understand the experimental findings, DFT calculations is performed. The optimized structure for Co3O4 (111)/CdS (002) interface is shown in Fig. 4(a). Evidently, there is a strong interaction at the interface between Co3O4 and CdS (002) atoms, dominated by Co–S and Cd–S bonds. The average Co3O4 (111)–CdS (002) separation is ∼2.30 Å suggesting the presence of chemical bonding besides weak vdW interactions. Besides covalent bonding, interfacial interactions also have ionic character; Bader analysis28 indicates that there is charge transfer of ∼0.65e from Co3O4 to CdS. These findings are consistent with the recent experimental work where both the presence of Co–S interfacial bonds as well as an increase (reduction) in electronic density was observed for CdS (Co3O4) surface within Co3O4@CdS.29 | ||
| Fig. 4 (a) Schematic description of the optimized Co3O4 (111)/CdS (002) interface. Colours; blue (Co), red (O), purple (Cd) and yellow (S), (b) density of states (DOS) contributions of Co3O4 and CdS surfaces within Co3O4@CdS hybrid structure. DOS is normalized to the total number of atoms within each surface to provide a clear comparison. Dashed vertical line presents Fermi level (EF). | ||
The impact of strong interfacial interactions on electronic structure is highlighted in Fig. 4(b) where density of states (DOS) contributions of Co3O4 and CdS within Co3O4@CdS are presented. Interestingly, CdS has states around Fermi level (EF) suggesting a metallic behaviour. This is in contrast to pristine CdS layer which has a large band gap of ∼2.0 eV Fig. S9 (ESI).† Therefore, it is evident that transport character of CdS layer on Co3O4 changes from semiconducting to metallic, owing to strong interfacial interactions. Moreover, conductivity of Co3O4@CdS will be higher as compared to the individual pristine surfaces, especially close to the interface region. Overall, both the increase in conductivity of Co3O4@CdS as well as higher electronic density of CdS due to interfacial charge transfer will improve the reaction kinetics at the electrode.30 This is indeed observed in the impedance spectroscopy (Table 2) where a noticeable reduction in charge transfer resistance (Rct) is observed for Co3O4@CdS in comparison to that of pristine Co3O4.
The improved lithium storage performance of the Co3O4@CdS hybrid structure may have following possible reasons (i) the functionalization of CdS NPs increase the surface reactivity and porosity which creates abundant active sites/interfaces for the rapid diffusion and transportation of Li+ ions during the electrochemical reaction (ii) the addition of CdS NPs increase the contact area between electrolyte and electrode and protect the Co3O4 nanosheets from degradation during the charge and discharges process. (iii) The synergy between Co3O4 and CdS NPs greatly improved the kinetics of Co3O4@CdS nanosheets for fast lithium storage.
4 Conclusion
In summary, a novel mesoporous Co3O4@CdS hybrid structure were successfully synthesized by employing hydrothermal along with SILAR method. The hybrid structure demonstrates several structural features including the large specific surface area, abundant active sites and excellent kinetics. The developed electrode showed improved lithium storage performance as compare with pristine and previously reported electrodes. The electrode delivers a stable reversible capacity of 760 mA h g−1 at 0.1C over 200 cycles with a capacity retention rate of 92.7%. The electrode can achieve reversible capacity of 520 mA h g−1 at 0.2C even after 800 cycles. The improved lithium storage performance may be due to the increase in kinetics, synergy and abundant active sites that facilitate the storing of more Li+ ions and fast transportation during the lithiation/delithiation. It is suggested that the electrochemical performance of Co3O4@CdS hybrid structure can be further enhanced by structural engineering and optimizing the amount of CdS NPs. This work provides a novel platform to construct a LIBs with long cyclic stability and high-rate capability.Author contributions
M. A. conceived the idea and designed the experiment. H. W. and H. R. synthesized materials. S. K., F. F and A. N. analyzed the XRD and FESEM data. Y. Y., A. K. and H. S. carried out the HRTEM characterization and analysis. S. J. carried out the DFT calculations. H. W., M. A., and A. K. performed coin cell measurements, analyzed the XPS data. S. H. performed the Raman measurements. A. Z. conducted the electrochemical impedance spectroscopic measurements. H. W. and M. A. performed the CV and GCD measurements. H. W., M. A., and A. N. co-wrote the paper. All authors discussed the results and commented on the manuscript. M. A. and A. N. supervised the whole research work.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We are thankful to Pakistan Atomic Energy Commission for supporting this work. The authors acknowledge the contributions of Mr Muhammad Hussain, Materials Division, PINSTECH for XRD data collection.References
- M. Z. Iqbal, M. Shaheen, A. A. Ifseisi, S. Aftab, Z. Ahmad, S. H. Siyal and M. J. Iqbal, RSC Adv., 2023, 13, 18038–18044 RSC.
- J. Xu, X. Cai, S. Cai, Y. Shao, C. Hu, S. Lu and S. Ding, Energy Environ. Mater., 2023, 6, e12450 CrossRef CAS.
- K. M. Racik, A. Manikandan, M. Mahendiran, P. Prabakaran, J. Madhavan and M. V. A. Raj, Phys. E, 2020, 119, 114033 CrossRef.
- B. Yan, X. Li, X. Fu, L. Zhang, Z. Bai and X. Yang, Nano Energy, 2020, 78, 105233 CrossRef CAS.
- E. Quartarone, V. Dall'Asta, A. Resmini, C. Tealdi, I. G. Tredici, U. A. Tamburini and P. Mustarelli, J. Power Sources, 2016, 320, 314–321 CrossRef CAS.
- X. Wu and S. Yao, Nano Energy, 2017, 42, 143–150 CrossRef CAS.
- B. A. Al Jahdaly, A. Abu-Rayyan, M. M. Taher and K. Shoueir, ACS Omega, 2022, 7, 23673–23684 CrossRef CAS PubMed.
- M. N. ur Rehman, T. Munawar, M. S. Nadeem, F. Mukhtar, A. Maqbool, M. Riaz, S. Manzoor, M. N. Ashiq and F. Iqbal, Ceram. Int., 2021, 47, 18497–18509 CrossRef.
- M. Aadil, G. Nazik, S. Zulfiqar, I. Shakir, M. F. A. Aboud, P. O. Agboola, S. Haider and M. F. Warsi, Ceram. Int., 2021, 47, 9225–9233 CrossRef CAS.
- X. Wei, J. Kang, L. Gan, W. Wang, L. Yang, D. Wang, R. Zhong and J. Qi, Nanomaterials, 2023, 13, 1917 CrossRef CAS PubMed.
- D. S. Patil, S. A. Pawar and J. C. Shin, Chem. Eng. J., 2018, 335, 693–702 CrossRef CAS.
- Q.-P. Li, F.-Q. Liu, X.-L. Mu, H.-W. Wang, B. Wu and W. Li, J. Alloys Compd., 2021, 870, 159340 CrossRef CAS.
- L. Chen, X. Xie, T. Su, H. Ji and Z. Qin, Appl. Surf. Sci., 2021, 567, 150849 CrossRef CAS.
- H. Yang, J. Fan, C. Zhou, R. Luo, H. Liu, Y. Wan, J. Zhang, J. Chen, G. Wang, R. Wang and C. Jiang, ACS Omega, 2020, 5, 17160–17169 CrossRef CAS PubMed.
- S. R. Bhalerao, S. S. Arbuj, S. B. Rane, J. D. Ambekar and U. P. Mulik, Nanosci. Nanotechnol. Lett., 2014, 6, 204–209 CrossRef CAS.
- K. S. Kim and Y. J. Park, Nanoscale Res. Lett., 2012, 7, 1–6 CrossRef PubMed.
- S. Rengaraj, S. H. Jee, S. Venkataraj, Y. Kim, S. Vijayalakshmi, E. Repo, A. Koistinen and M. Sillanpää, J. Nanosci. Nanotechnol., 2011, 11, 2090–2099 CrossRef CAS PubMed.
- S. Deng, N. Chen, D. Deng, Y. Li, X. Xing and Y. Wang, Ceram. Int., 2015, 41, 11004–11012 CrossRef CAS.
- H. Sheng, E. D. Hermes, X. Yang, D. Ying, A. N. Janes, W. Li, J. Schmidt and S. Jin, ACS Catal., 2019, 9, 8433–8442 CrossRef CAS.
- S. Naranthatta, P. Janardhanan, R. Pilankatta and S. S. Nair, ACS Omega, 2021, 6, 8646–8655 CrossRef CAS PubMed.
- S. Deng, N. Chen, D. Deng, Y. Li, X. Xing and Y. Wang, Ceram. Int., 2015, 41, 11004–11012 CrossRef CAS.
- M. Hussain, A. Nisar, L. Qian, S. Karim, M. Khan, Y. Liu, H. Sun and M. Ahmad, Nanotechnology, 2021, 32, 205501 CrossRef CAS PubMed.
- X. Han, G. He, Y. He, J. Zhang, X. Zheng, L. Li, C. Zhong, W. Hu, Y. Deng and T. Y. Ma, Adv. Energy Mater., 2018, 8, 1702222 CrossRef.
- D. Su, S. Dou and G. Wang, Sci. Rep., 2014, 4, 5767 CrossRef PubMed.
- K. Song, E. Cho and Y.-M. Kang, ACS Catal., 2015, 5, 5116–5122 CrossRef CAS.
- B. Poornaprakash, R. Mangiri, A. A. Al-Kheraif, D. D. Divakar, Y. Kim, M. Kumar and M. S. P. Reddy, Appl. Phys. A: Mater. Sci. Process., 2021, 127, 1–7 CrossRef.
- L. Zu, W. Zhang, L. Qu, L. Liu, W. Li, A. Yu and D. Zhao, Adv. Energy Mater., 2020, 10, 2002152 CrossRef CAS.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- L. Chen, X. Xie, T. Su, H. Ji and Z. Qin, Appl. Surf. Sci., 2021, 567, 150849 CrossRef CAS.
- Q. He, B. Yu, Z. Li and Y. Zhao, Energy Environ. Mater., 2019, 2, 264–279 CrossRef CAS.
- D. Zhao, Q. Hao and C. Xu, Electrochim. Acta, 2016, 211, 83–91 CrossRef CAS.
- X. Yao, X. Xin, Y. Zhang, J. Wang and Z. Liu, J. Alloys Compd., 2012, 521, 95–100 CrossRef CAS.
- Z.-S. Wu, W. Ren, L. Wen, L. Gao, J. Zhao, Z. Chen, G. Zhou, F. Li and H.-M. Cheng, ACS Nano, 2010, 4, 3187–3194 CrossRef CAS PubMed.
- H. Liu, X. Wu, E. Guo and Q. Lu, Energy Technol., 2020, 8, 1900774 CrossRef CAS.
- X. Tong, M. Zeng, J. Li and Z. Liu, J. Alloys Compd., 2017, 723, 129–138 CrossRef CAS.
- J. Wen, L. Xu, J. Wang, Y. Xiong, J. Ma, C. Jiang, L. Cao, J. Li and M. Zeng, J. Power Sources, 2020, 474, 228491 CrossRef CAS.
- J. Wang, C. Wang and M. Zhen, Chem. Eng. J., 2019, 356, 1–10 CrossRef CAS.
- J. Zhang, R. Chu, Y. Chen, H. Jiang, Y. Zeng, X. Chen, Y. Zhang, N. M. Huang and H. Guo, J. Alloys Compd., 2019, 797, 83–91 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra01028k |
| This journal is © The Royal Society of Chemistry 2024 |