
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExpanded graphite incorporated with Li4Ti5O12 nanoparticles as a high-rate lithium-ion battery anode†
Junkang Zhao ab,
Xiayu Zhubc,
Wenfeng Zhangbc,
Jingyi Qiubc,
Feiyue Zhaibd,
Huimin Zhangbc,
Gaoping Caobc,
Shengji Gaoab,
Fei Ding*a and
Yu Xiang*bc
ab,
Xiayu Zhubc,
Wenfeng Zhangbc,
Jingyi Qiubc,
Feiyue Zhaibd,
Huimin Zhangbc,
Gaoping Caobc,
Shengji Gaoab,
Fei Ding*a and
Yu Xiang*bc
aHebei University of Technology, Tianjin 100131, China. E-mail: hilldingfei@163.com
bChemical Defense Institute, Academy of Military Sciences, Beijing 100191, China. E-mail: drxiangyu2016@126.com
cBeijing Key Laboratory of Advanced Chemical Energy Storage Technology and Materials, Beijing 100191, China
dBeihang University, Beijing 100191, China
First published on 9th April 2024
Abstract
Due to their small interlayer spacing and a low lithiation potential close to Li+ deposition, current graphite anodes suffer from weak kinetics, and lithium deposition in a fast-charging process, hindering their practical application in high-power lithium-ion batteries (LIBs). In this work, expanded graphite incorporated with Li4Ti5O12 nanoparticles (EG/LTO) was synthesized via moderate oxidization of artificial graphite following a solution coating process. The EG/LTO has sufficient porosity for fast Li+ diffusion and a dense Li4Ti5O12 layer for decreased interface reaction resistance, resulting in excellent fast-charging properties. EG/LTO presented a high reversible capacity of 272.8 mA h g−1 at 3.74 A g−1 (10C), much higher than that of the original commercial graphite (50.1 mA h g−1 at 10C) and even superior to that of hard carbon. In addition, EG/LTO exhibited capacity retention rate of 98.4% after 500 cycles at 10C, demonstrating high structural stability during a long cycling process. This study provides a protocol for a solution chemistry method to prepare fast-charging graphite anode materials with high stability for high-power LIBs.
Introduction
Lithium-ion batteries (LIBs) are widely applied in plenty of consumer electronic devices because of their exceptional performance. However, with the growing demand for fast-charge characteristics and high energy density in electric vehicles and renewable energy storage, LIBs are required with improved power capability.1–5 At present, various artificial graphite materials are still the most widely applied anodes in LIBs due to their distinctive advantages, such as cheap price, long cycle life, and low potential platform.6–8 However, graphite anodes have encountered several limitations in high-rate operating mode. Due to the working potential of graphite (0.1 V vs. Li/Li+) being so close to that of lithium metal deposition (0 V vs. Li/Li+), Li dendrites can easily emerge when charging at high rates or low temperatures, causing severe safety risks.9 Owing to interfacial side reactions, solid electrolyte interface (SEI) films are generated on the graphite surface and gradually become thicker over long cycle operation, leading to increasing interface reaction impedance and a decay in power capability.10To improve the fast-charging performance and long-cycle stability, it is necessary to enhance the dynamic character of the graphite anode at high rates.11 Various strategies have been researched, including chemical oxidation, surface coating treatment, and porous-structure design.12–19 By coating an artificial Li+ conductor on the graphite surface, the interface Li+ diffusion resistance could be obviously decreased, resulting in an enhanced kinetic character.20 Lee et al. applied Li4Ti5O12 (LTO) as a coating layer for MCMB (middle phase carbon microsphere) surface modification, achieving smaller interface resistance than that of the original MCMB after 40 cycles, due to the LTO layer inhibiting the successive decomposition of electrolyte at the electrode/electrolyte interface during long cycles.21,22 Cheng et al.14 prepared porous graphene-like films (GLG), where micropores on the surface can provide additional intercalation or deintercalation sites for Li+, and internal holes can also promote the diffusion of Li+. The results showed that the reversible capacities of GLG at 1, 5 and 10C were about 220, 150 and 100 mA h g−1, respectively, which were much higher than those of the original graphite. Ma et al.13 synthesized graphite/Al2O3 composites via facile high-energy ball milling. The ball milling treatment not only introduced porous structures for Li+ adsorption but also expanded the layer spacing for fast Li+ diffusion in the bulk. Meanwhile, the inert Al2O3 layer further ensured interface stability in a fast charging process. The optimized graphite/Al2O3 displayed a capacity of 200 mA h g−1 after 500 cycles at 3 A g−1. Mahajan et al.19 synthesized a three-dimensional in situ carbon-coated cubic carbide, vanadium carbide (VC@C), that displayed a capacity of 328 mA h g−1 at 2 A g−1 and kept an excellent reversible capacity of 640 mA h g−1 after 100 cycles at 0.1 A g−1. To achieve excellent fast-charging performance, an ideal structural model of graphite material should combine a rapid ion transport channel, expanded interlayer spacing and an optimized SEI property. However, due to the inert surface of graphite, there is a need to search for a distinctive modification method to achieve such functional goals simultaneously.
In this paper, LTO nanoparticles incorporated into expanded graphite were synthesized. First, artificial commercial graphite was pre-treated via moderate oxidization to increase the interlayer spacing and introduce abundant functional sites on the graphite surface. Then, LTO nanoparticles were in situ deposited on the graphite surface under the effect of functional groups in a solution coating process. Finally, the obtained anode composite (EG/LTO) has sufficient porosity for fast Li+ diffusion and a dense Li+ conductor layer for decreased resistance to interface reaction, resulting in excellent fast-charging properties. EG/LTO presented a high reversible capacity of 272.8 mA h g−1 at 3.74 A g−1 (10C), much higher than that of the original commercial graphite (50.1 mA h g−1 at 10C) and even superior to hard carbon. In addition, EG/LTO exhibited a capacity retention rate of 98.4% after 500 cycles at 10C, demonstrating high structural stability during a long cycling process. This study provides a protocol for a solution chemistry method to prepare fast-charging graphite anode materials with high stability for high-power LIBs.
Experimental details
Materials
Commercial artificial graphite powder was purchased from ShanShan Technology Company. Potassium permanganate (KMnO4, chemical purity grade agent), concentrated sulfuric acid (H2SO4, chemical purity grade agent), tetrabutyl titanate (TBT, chemical purity grade agent) and lithium acetate (LiAC, chemical purity grade agent) were bought from Damao (Tianjin) Chemical Reagent Factory. For performance comparison, commercial hard carbon was purchased from KURARY Co., Ltd. All chemicals were used as received without purification.Sample preparation
Commercial artificial graphite powder (denoted as G) was moderately oxidized. Briefly, G was mixed with a certain amount of KMnO4 and H2SO4 in an ice-water bath to obtain a homogeneous solution. The above mixture was put into a water bath at 30 °C for 30 min. Then, an amount of distilled water was added dropwise to form a suspension. The suspension was subsequently filtered and washed with distilled water to obtain a gray powder. This gray powder was further calcined at 750 °C in an argon atmosphere for 1 h to obtain expanded graphite, which was marked as EG.Subsequently, the above-synthesized moderately oxidized graphite powder (without calcination treatment) was dispersed in a certain amount of ethanol. TBT and LiAC were added with a Li4Ti5O12-to-EG weight stoichiometric ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100. Briefly, 4.0 g of EG was dispersed in 60 mL of anhydrous ethanol as suspension A. Then, 0.742 g of TBT was dissolved in 40 mL of anhydrous ethanol as solution B. The whole of solution B was slowly added into solution A drop by drop and continuously stirred for 12 h at 35 °C. Subsequently, the resulting suspension was filtrated to obtain a grey powder. The powder was mixed with AcLi under ball milling for 30 min at 300 rpm. Finally, the solid mixture was calcined in a tubular furnace at 850 °C for 2 h under an Ar atmosphere to obtain expanded graphite coated with Li4Ti5O12, which was marked EG/LTO.
:100. Briefly, 4.0 g of EG was dispersed in 60 mL of anhydrous ethanol as suspension A. Then, 0.742 g of TBT was dissolved in 40 mL of anhydrous ethanol as solution B. The whole of solution B was slowly added into solution A drop by drop and continuously stirred for 12 h at 35 °C. Subsequently, the resulting suspension was filtrated to obtain a grey powder. The powder was mixed with AcLi under ball milling for 30 min at 300 rpm. Finally, the solid mixture was calcined in a tubular furnace at 850 °C for 2 h under an Ar atmosphere to obtain expanded graphite coated with Li4Ti5O12, which was marked EG/LTO.
Material characterization
X-Ray diffraction (XRD, Rigaku SmartLab SE, Japan) analysis was performed to study the crystal structure and lattice parameters of the powders. The measurement was operated using Cu Kα (λ = 1.5406 Å) radiation at a voltage of 40 kV and current of 30 mA. The surface morphology of the powders was observed with a field emission scanning electron microscope (SEM, ZEISS GeminiSEM 300, Germany). A high-resolution transmission electron microscope (TEM, JEOL JEM-F200, Japan) was used to photograph EG/LTO particles, then DigitalMicrograph software was used to program selected area electron diffraction (SAED) images. The valence states of elements for G, EG and EG/LTO were determined by X-ray photoelectron spectroscopy (XPS, Thermo Scientific K-Alpha, USA). The degrees of graphitization of G, EG and EG/LTO were examined with a Raman spectrometer (Raman, HORIBA HR Evolution, Japan). The specific surface area and mesopore distribution of G, EG and EG/LTO were determined with an automatic specific surface and porosity analyzer (BET, Quantachrome Nova 4000e, USA).Electrochemical performance evaluation
First, active materials, Super P, and polyvinylidene fluoride (PVDF) were weighed and mixed at a mass ratio of 8:1:1 with N-methylpyrrolidone (NMP) to make different electrodes. The slurry was evenly coated on the current collector with a doctor blade, followed by drying overnight at 120 °C in a vacuum oven. Then, coin cells were assembled using lithium metal as the counter electrode, polypropylene as the separator, simultaneously with the electrolyte (1 M LiPF6 EC:DEC = 1:1, v/v) in an Ar-filled glove box. Next, the galvanostatic charge–discharge, rate and cycle tests were conducted using a battery testing system (XINWEI, CT-4008Tn, China) at 30 °C in the potential window of 0.001–2 V. Cyclic voltammetry was performed on an electrochemical workstation (BioLogic, VMP3, France) at different scan rates in the same potential range. Finally, electrochemical impedance spectroscopy (EIS) was applied to the electrochemical impedance of the electrode in the frequency range from 0.01 Hz to 100 kHz at a voltage of 10 mV.
Results and discussion
Material characterization
The EG/LTO material was synthesized via a solution reaction process, as presented in Fig. 1a. First, raw graphite was treated by a mild oxidization method, and the original graphite bulk (Fig. 1b) was expanded into porous particles (Fig. 1c) covered with an abundance of active oxygen-containing groups. These groups could induce the in situ incorporation of Li4Ti5O12 nanoparticles on the surface of the expanded graphite during the solution process, for which the mechanism of nucleophilic catalysis of nano-Li4Ti5O12 was demonstrated in our previous work.23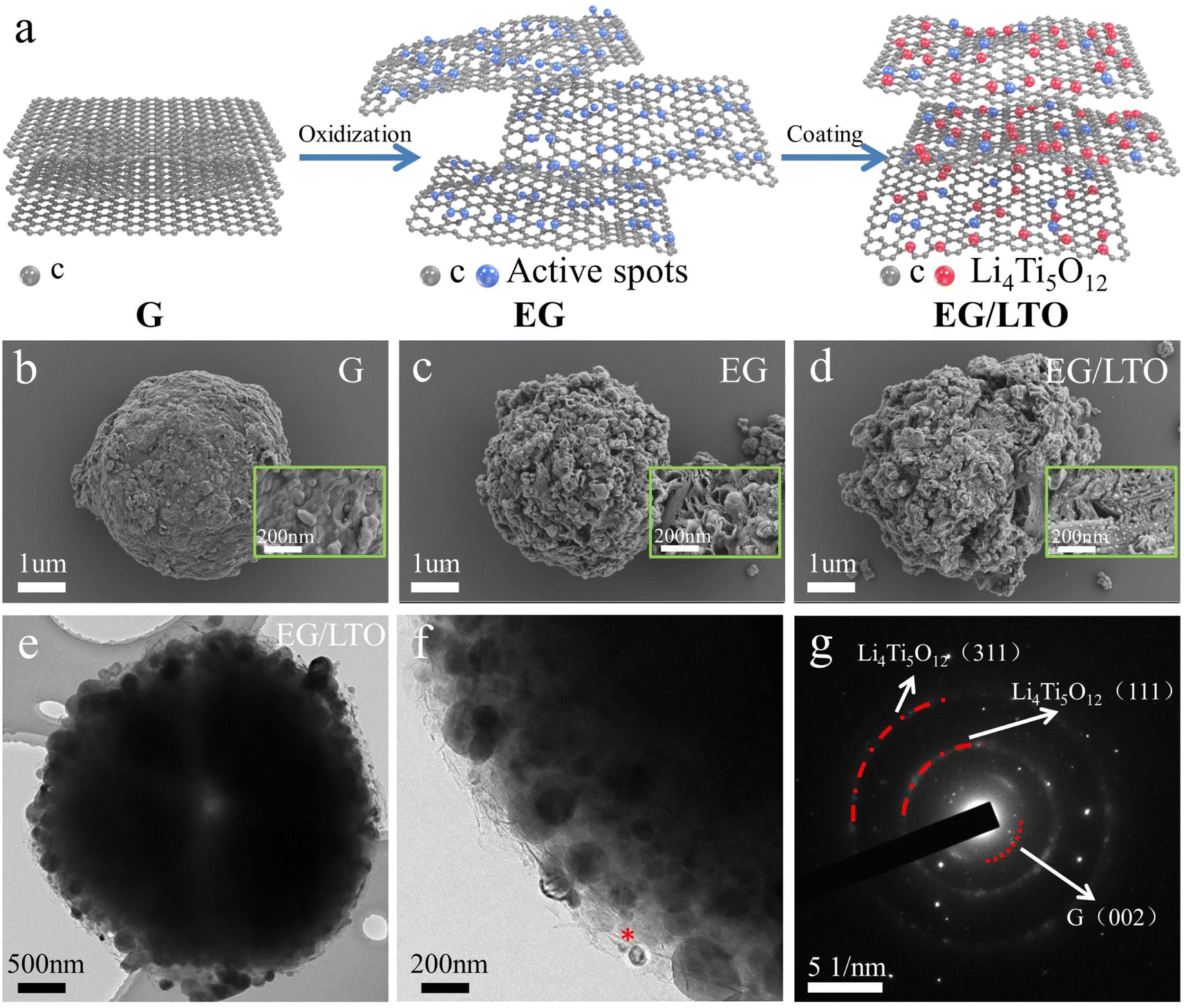 | ||
| Fig. 1 (a) Schematic illustration of EG/LTO prepared by the moderate oxidization method and liquid phase method. (b–d) SEM micrographs of bare G, EG and EG/LTO particles. (e and f) High-resolution TEM images of EG/LTO particles. (g) Selected area electron diffraction (SAED) images of EG/LTO. | ||
Compared with raw graphite (G) and expanded graphite (EG), the EG/LTO composite material displays an observed porous sphere coated with Li4Ti5O12 nanoparticles. The porous structure of EG/LTO is inherited from EG, mainly caused by the intercalation of nitric and sulphuric acids into the graphite bulk during the oxidation process (Fig. 1d). Conversely, a large number of nucleophilic sites induce the generation of nanosized Li4Ti5O12 particles in the sol–gel hydrolysis of tetrabutyl titanate. As confirmed by energy dispersive X-ray analysis (EDX) cross-section mapping of Ti (Fig. S1†), it is demonstrated that an Li4Ti5O12 layer is coated on the surface of EG/LTO. TEM was performed to investigate in depth the microstructure of EG/LTO. After oxidation, the edges of graphite are wrinkled, suggesting an enhanced interlayer spacing and shortened Li+ transport channel (Fig. 1e). The existence of Li4Ti5O12 nanocrystals can be clearly detected from high-resolution TEM images (Fig. 1f). The lattice spacings of 0.483 nm can be indexed to the (111) planes of Li4Ti5O12 (Fig. S2†). The graphite host manifests a long-range disordered feature without a highly ordered graphite layer feature. Moreover, the SAED image (Fig. 1g) exhibits the (111) and (311) planes of Li4Ti5O12 and the (002) plane of graphite. Based on above results, the EG/LTO composite material displays a porous structure coated with a nano-Li4Ti5O12 layer, and the activity of such irregular edges is stronger than that of the flat surface, which provides mountains of active Li+ sites.
In Fig. 2a, the XRD pattern demonstrates that the (002) peak of graphite in EG/LTO shifts from 26.50° to a low angle of 26.28°. According to the Bragg's equation,15 the interplanar spacing of the graphite host in EG/LTO is 0.351 nm, which is higher than that of raw graphite (0.338 nm), demonstrating the expanded interlayer spacing and wide Li+ pathway. Moreover, an obviously broadened peak width is observed, confirming decreased grain interspaces. The conspicuous peaks at 18.36° and 34.24° for EG/LTO samples correspond to the characteristic (111) and (311) peaks of Li4Ti5O12, respectively (PDF#49-0207).
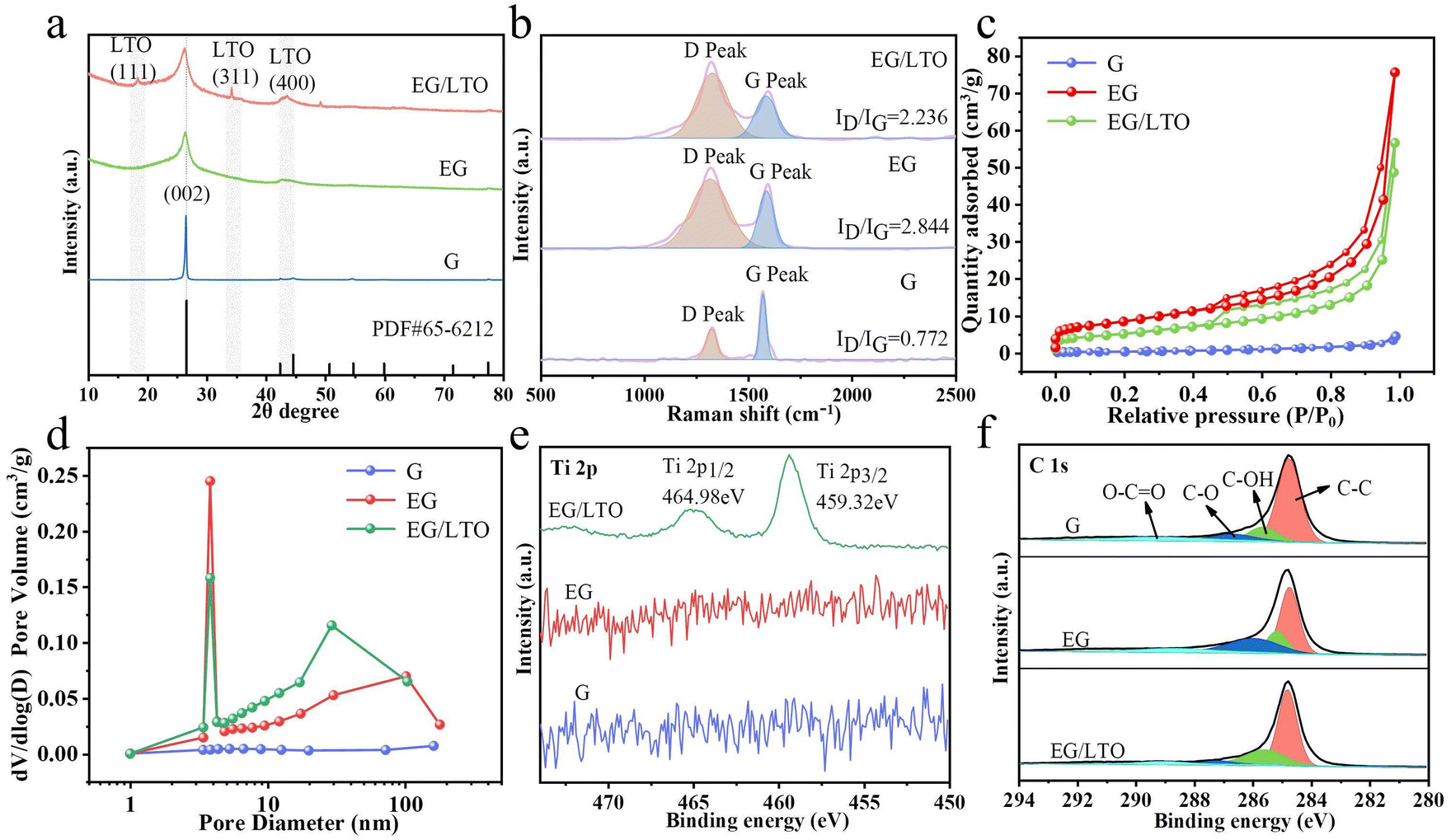 | ||
| Fig. 2 (a) XRD patterns of G, EG and EG/LTO powders. (b) Raman spectra of G, EG and EG/LTO powders. (c) N2 adsorption–desorption isotherm curves of G, EG and EG/LTO powders. (d) Pore size distribution curves of G, EG and EG/LTO powders. (e) Ti 2p and (f) C 1s spectra of G, EG and EG/LTO powders. | ||
To investigate the deficient situation of oxidized samples, Raman spectra were characterized (Fig. 2b). The D band (around 1350 cm−1) of graphite is related to the disordered vibrational mode of the carbon material, whereas the G band at around 1580 cm−1 corresponds to the planar vibrational peak of C–C.24 Compared to the ID/IG value (0.772) of raw graphite, the oxidized EG samples manifest an enhanced ID/IG of 2.844, indicating a high number of defects generated during the chemical oxidization process. However, the ID/IG value of EG/LTO (2.236) is much lower than that of EG, demonstrating the exhaustion of active sites for the in situ deposition of Li4Ti5O12 nanoparticles.
To identify the porosity structure, N2 adsorption/desorption isotherms are shown in Fig. 2c. Obviously, the original graphite (G) reveals I type Langmuir isotherms, indicating a non-porous character. However, EG and EG/LTO are attributed to classical IV type isotherms with H4 hysteresis loops, demonstrating that there are large quantities of mesopores. It is calculated that the BET areas of G, EG and EG/LTO are 0.991 m2 g−1, 29.946 m2 g−1 and 18.579 m2 g−1, respectively; see Table S1.† The DFT pore size distribution of the three samples is listed in Fig. 2d. Obviously, there is nearly no pore volume in G. However, the other two samples exhibit mesopore-dominated features and have a centralized pore size distribution of 3–5 nm. There also exist some macrospores in EG and EG/LTO. The proportion of mesopores is significantly higher than that of macropores. It is well known that a mesopore-dominated structure plus an extent of macropores is very favourable for electrolyte transport and ion diffusion, which are beneficial for improving the high-rate performances of EG and EG/LTO materials.
The chemical compositions of the G, EG and EG/LTO samples were confirmed by XPS, and their full spectra are shown in Fig. S5.† It is evident that only EG/LTO displays the characteristic peaks of Ti element, further proving the existence of an LTO coated layer. The high-resolution spectra of the G, EG, and EG/LTO samples at binding energies ranging from 450 eV to 500 eV are shown in Fig. 2e. For the Ti 2p spectrum of EG/LTO, peaks at 459.32 and 464.98 eV correspond to typical Ti 2p3/2 and Ti 2p1/2 subpeaks, respectively. The C 1 s spectrum (Fig. 2f) can be deconvoluted into four peaks: C–C (284.8 eV), C–OH (285.8 eV), C–O (286.7 eV), and O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (289.6 eV). The C–C bond always takes the dominant role in the C 1s spectrum of the three samples, suggesting that the ordered bulk structure of graphite is still retained.
O (289.6 eV). The C–C bond always takes the dominant role in the C 1s spectrum of the three samples, suggesting that the ordered bulk structure of graphite is still retained.
Electrochemical performance evaluation
To assist in the discussion of fast-charging performance, the cyclic voltammetry (CV) curves of G and EG/LTO are shown in Fig. 3a and b. For cathode scans, the curves started from 2 to 0.001 V (lithiation process). Then, for anode scans, the curves proceed from 0.001 to 2 V (de-lithiation process). The irreversible cathode peak at 0.71 V corresponds to the formation of a solid–electrolyte interface phase (SEI reaction).25 The reversible cathode peak at 0.16 V and anode peak at 0.26 V can be attributed to Li+ intercalation into and deintercalation from the graphite layer. While for EG/LTO, the cathode peak at 1.51 V and anode peak at 1.59 V can be related to the lithiation and de-lithiation of Li4Ti5O12.26 The cathodic peak related to SEI drifts to 0.64 V, suggesting that the SEI reaction has been restrained to an extent. However, for EG/LTO, the typical (de)-interaction peak of graphite is relatively smooth, confirming the different reaction mechanism after oxidization and coating. The quasi-rectangular curve displays possible pseudo-capacitance behaviour in the EG/LTO electrode, suggesting the accelerated Li+ transportation process.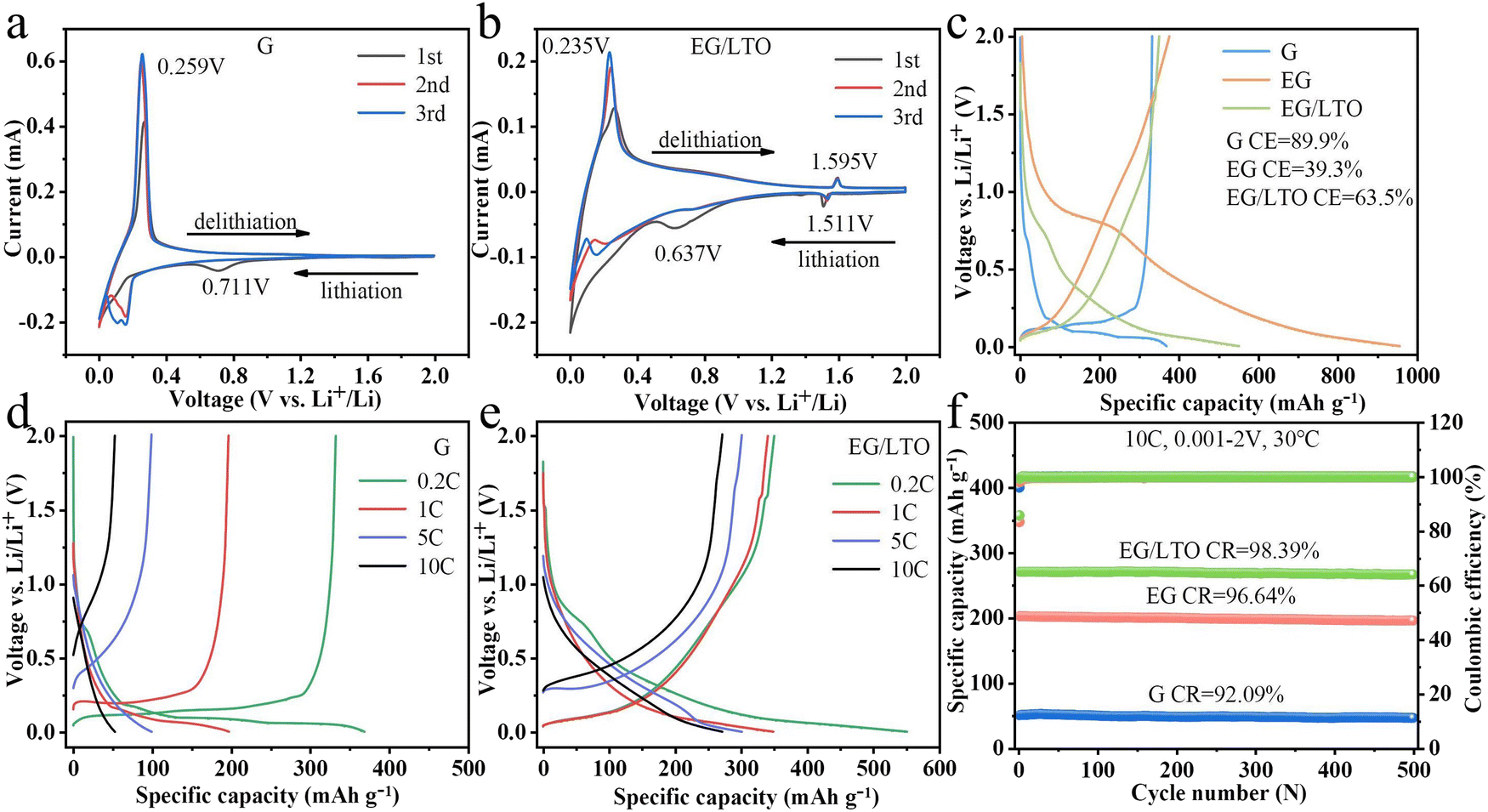 | ||
| Fig. 3 Cycle voltammogram curves of (a) G and (b) EG/LTO at 0.1 mV s−1 scan rates for the initial three cycles. (c) Initial charge and discharge curves of G, EG and EG/LTO at 0.2C in the voltage range from 0.001 to 2.0 V. Rate capability of G (d) and EG/LTO (e). (f) Cycle test of G, EG and EG/LTO at 10C after activation. | ||
The first charge–discharge profiles of all the samples are shown in Fig. 3c. The initial charge capacity of commercial graphite is 332.6 mA h g−1 with first coulombic efficiency of 89.9%. With its porous structure, EG displays a higher initial reversible specific capacity of about 375 mA h g−1. However, due to the existence of defects and residual oxygen-containing functional groups, the coulombic efficiency of EG in the first cycle is only 39.3%. Combing the unique porous structure and nano-Li4Ti5O12 coated layer, the initial reversible capacity of EG/LTO is still as high as 350.3 mA h g−1, and the first coulombic efficiency meanwhile increases to 63.5% under the protective layer. In contrast to raw G, the charge and discharge platform of EG/LTO is sloped, indicating that there is likely to be a pseudo-capacitance effect in EG/LTO. In other words, EG/LTO could endure higher polarization and is very suitable for operating at high current density.
In our experiment, a half-cell is applied to test the intrinsic rate performance of EG/LTO. It is worth noting that the resistance of a half-cell is much higher than those of real batteries (such as soft-packs or 18650 types). Hence, the data for rate capability at a much higher mass loading is severely distorted if tested in a half-cell. On the contrary, controlling the mass loading is an important way to design high-power LIBs. Finally, we examined the variation trend of rate performance of EG/LTO at mass loading of 0.5–2 mg cm−2, as shown in Fig. S10.† The rate capabilities of G and EG/LTO are shown in Fig. 3d and e, respectively. The EG/LTO material manifests high reversible capacities of 305.6 mA h g−1 at 5C and 272.8 mA h g−1 at 10C. However, the original graphite displays only 100.3 mA h g−1 at 5C and 50.1 mA h g−1 at 10C. The charging and discharging platforms in raw G nearly disappear after 5C, but the galvanostatic curve of EG/LTO at 10C is similar to that at 0.2C. It is demonstrated that the polarization effect of EG/LTO is much lower than that of G.
In addition, Table S2† reveals a comparison of high-rate performance between this work and some relative studies. It can be seen that rate performance and coulombic efficiency are comparable to those reported in other literature. It is demonstrated that the reversible capacity of this work is much superior to that in the majority of other literature at high rate. Though the specific capacity of Al2O3@graphite and TiO2 coated graphite seem to be larger than in this work, yet the original graphite of the two studies displayed excellent rate performances, both exhibiting above 330 mA h g−1 at 3 A g−1 to 4 A g−1. However, the original graphite in this work only shows below 100 mA h g−1 at 1.8 A g−1. In this work, the specific capacity of the modified graphite has increased by nearly 3 times, but the specific capacities of Al2O3@graphite and TiO2 coated graphite are only improved by 10%. Hence, it is demonstrated that the proposed modification method has an obvious positive influence on enhancing the rate capability of the graphite anode.
LTO has some capacity as an anode material for LIBs in terms of CV curves (Fig. 3b), so it is essential to calculate the authentic reversible capacity of EG in EG/LTO. The mass ratio of Li4Ti5O12 in EG/LTO was calculated to be 9.06 wt% according to the thermogravimetric analysis; see Fig. S6.† Obviously, Li4Ti5O12 certainly provides an amount of reversible capacity in EG/LTO. Based on test data, a pure Li4Ti5O12 anode revealed 225.7, 211.3 and 203.0 mA h g−1 in the potential range of 0.001–2 V at 0.2, 5 and 10C, respectively, whose galvanostatic curves are shown in Fig. S7.† In our work, EG/LTO manifested high reversible capacities of 350.3, 305.6 and 272.8 mA h g−1 in the potential range of 0.001–2 V at 0.2, 5 and 10C. In terms of eqn (1), the capacity contributions (%) of Li4Ti5O12 and EG were calculated, as shown in Table S3,† where Q(EG/LTO), Q(EG) and Q(LTO) are used to represent the capacities of EG/LTO, EG and LTO, and m represents the mass of EG/LTO. From eqn (1), the capacity contributions of LTO in EG/LTO are 5.84, 6.26 and 6.69% in the potential range of 0.001–2 V at 0.2, 5 and 10C, respectively.
| Q(EG/LTO) × m = Q(EG) × 90.94%m + Q(LTO) × 9.06%m | (1) |
To investigate the cyclic stability of EG/LTO under high current density, the cycling performances at 10C of G and EG/LTO were further characterized (Fig. 3f). Although there is a difference in reversible capacity among the three samples, they all exhibit extremely strong cycling stability. This not only exhibited the structural stability of commercial graphite G but also proved that the porous structure and nano-Li4Ti5O12 layer of EG/LTO can be retained in the high-rate cycle. An SEM characterization experiment was used to check inactive lithium and lithium dendrites; see Fig. S3.† For original G, there existed some lithium dendrites and a large amount of inactive lithium. For EG/LTO, no lithium dendrites or inactive lithium were observed. It is known that hard carbon is a type of high-rate anode material, and it displayed excellent power capability in previous research. To further evaluate the application of synthesized EG/LTO as a high-rate anode material, we also compared its rate performance with commercial hard carbon (Fig. S11†). The commercial hard carbon displays a reversible capacity of 150.9 mA h g−1 at 10C and retention of 94.5% after 500 cycles at 10C (Fig. S12†). The initial reversible capacity and coulombic efficiency of the hard carbon are 283.2 mA h g−1 and 75.4%, respectively (Fig. S9†). Except for the first coulombic efficiency of EG/LTO being a little lower, other high-rate performances are superior to those of the commercial hard carbon. In this work, the proposed method for modifying commercial graphite has potential for large-scale production. But, only if the manufacturing cost is lower than that of hard carbon will it be a potential modification protocol to produce high-rate anode materials to power LIBs.
To better understand the kinetic variations of the three samples, EIS measurements were performed, as shown in Fig. 4a. All Nyquist plots of the three samples consist of two semicircles and one sloping line. The two semicircles suggest that there exist two distinct interfaces. One corresponds to the counter metal Li electrode/electrolyte interface in the half-cell. The other is ascribed to the graphite anode/electrolyte interface. In the applied equivalent circuit, R1 is the series resistance, R2 and R3 are the charge transfer resistance for the metal Li counter electrode and graphite anode, respectively. W1 is used to represent the solid diffusion process of Li+ inside the particles of the active material. The calculated values of all paramenters are listed in Table 1.
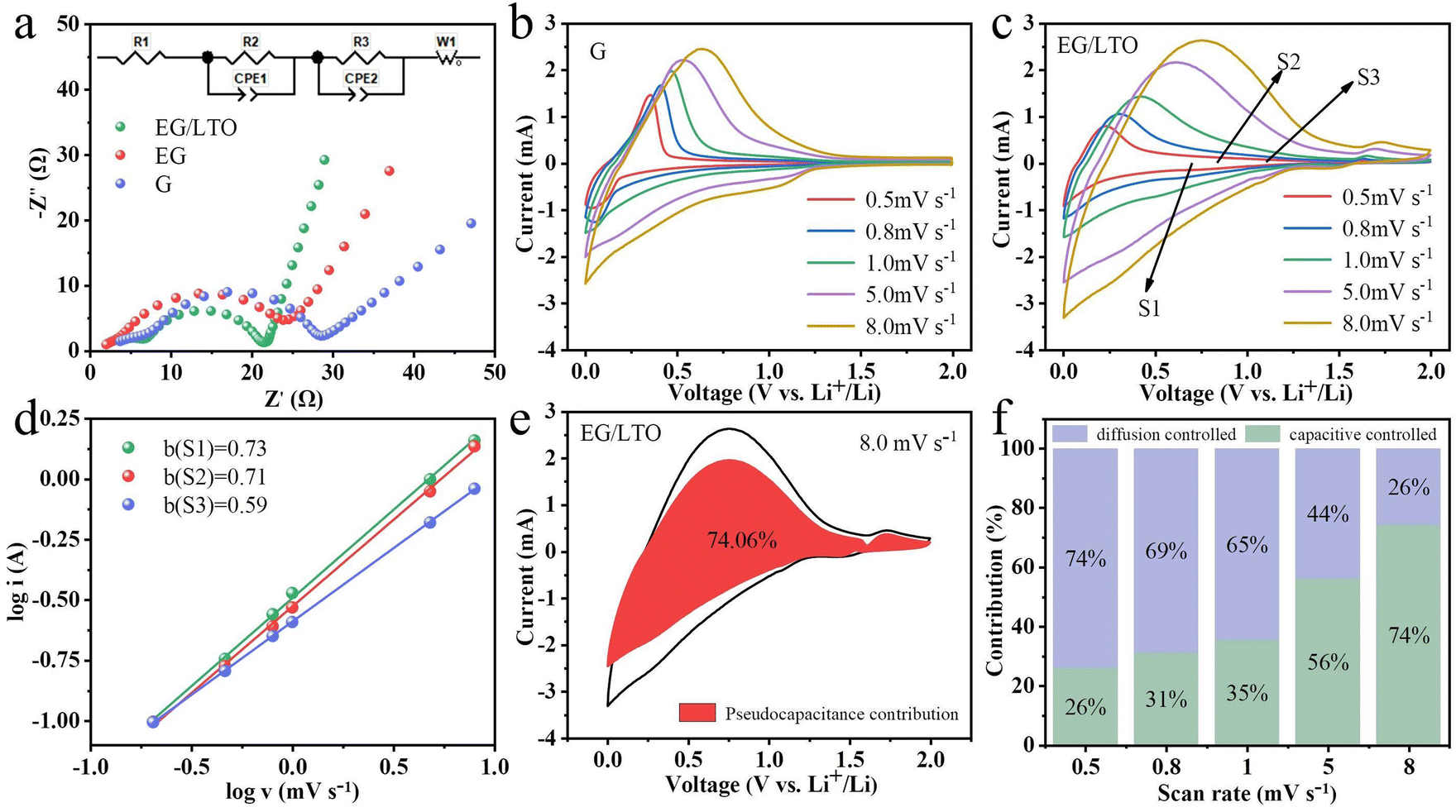 | ||
| Fig. 4 (a) Typical Nyquist plots of the three materials after 50 cycles at 25 °C. CV curves of (b) G and (c) EG/LTO at different scan rates. (d) Fitted curve of logi against logv. (e) Pseudocapacitive contribution of EG/LTO at a scan rate of 8 mV s1. (f) Capacitive contribution ratio of EG/LTO at various scan rates. | ||
| Samples | R1 (Ω) | R2 (Ω) | R3 (Ω) |
|---|---|---|---|
| G | 1.84 | 7.28 | 18.22 |
| EG | 2.30 | 4.78 | 13.51 |
| EG/LTO | 2.41 | 3.08 | 12.03 |
Notably, the R3 of EG/LTO is 12.03 Ω and is the lowest among the three samples, demonstrating that the polarization effect of the electrochemical reaction at the interface is also the weakest. It is proved that an Li4Ti5O12 coating layer could improve the Li+ reaction process at the electrode surface. Additionally, the gradient of the sloping line is attributed to the Li+ diffusion process in the bulk. When the gradient is higher, the Li+ diffusion coefficient is higher. Hence, it could be assessed that EG/LTO shows the fastest Li+ diffusion rate, suggesting the best rate capability among the three samples. The porosity and surface area of EG are higher than those of EG/LTO, but the Li+ diffusion ability of EG is still lower than that of EG/LTO. This further proves that the Li4Ti5O12 coating layer could decrease Li+ diffusion resistance across the electrode interface to bulk and enhance the apparent Li+ diffusion ability.
In addition, the EIS spectra of fresh electrodes of the three samples were examined, and the results are shown in Fig. S13.† Based on the same equivalent circuit, it is calculated that the R2 of G, EG and EG/LTO were 403.4, 355.5 and 182.8 Ω, respectively. Even though the internal resistances of the three fresh electrodes are very large without activation, EG/LTO still has a much lower R2 than the other two samples. It is reconfirmed that the Li4Ti5O12 coating layer could reduce Li+ diffusion resistance at the electrode interface. Meanwhile, the plotted Nyquist curve of EG/LTO in the low-frequency region retains the highest sloping line, revealing the best Li+ diffusion kinetic character.
The cyclic voltammetry curves at different scan rates were tested, as shown in Fig. 4b and c. The relationship between peak current (i) and scan rate (v) is generally fitted to the Randles–Ševčík equation described by eqn (2).27,28
| i = avb | (2) |
It is common knowledge that a and b are constants. When b = 0.5, the electrochemical process is controlled by a diffusion process. When b = 1.0, the electrochemical process is controlled by surface behaviour, representing a capacitive contribution.29 If the b value is between 0.5 and 1.0, the two energy storage mechanisms exist simultaneously.30 The diffusion-contributed and capacitance-contributed capacities could be distinguished according to the equation. The b values of S1, S2 and S3 were 0.73, 0.71 and 0.59, respectively, meaning that EG/LTO displayed perfect lithium storage performance with respect to capacitive contribution (Fig. 4d).
There existed two storage mechanism contributions: pseudocapacitive storage (k1v) and diffusion process (k2v1/2). The relationship of peak current at constant rate (v) followed these equations:31
| i(v) = k1v + k2v1/2 | (3) |
| i(v)/v1/2 = k1v1/2 + k2 | (4) |
Conclusions
In summary, the EG/LTO composite anode has been prepared via moderate oxidation treatment of commercial graphite following a solution coating process, during which Li4Ti5O12 nanoparticles are in situ incorporated onto the surface of expanded graphite. The expanded interlayer spacing and porous structure enable fast Li+ diffusion. Meanwhile, the Li4Ti5O12 layer can further decrease the interface reaction resistance and guarantee the stability of fast charging. Both factors result in obviously improved rate capability. The EG/LTO composite anode presents 272.8 mA h g−1 at 10C, superior to commercial graphite (50.1 mA h g−1 at 10C). In addition, EG/LTO exhibits a capacity retention rate of 98.4% after 500 cycles at 10C, which is much higher than that of commercial graphite (92.1%). This work provides a new perspective for designing high-performance fast-charging graphite anodes, which are significant for the large-scale production of fast-charging electrode materials for lithium-ion batteries.Author contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by Junkang Zhao, Yu Xiang, Fei Ding. The first draft of the manuscript was written by Junkang Zhao and Yu Xiang. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Hebei University of Technology and Chemical Defense Institute for the financial support during this project. We are also grateful for the equipment and academic facility by Chemical Defense Institute and Beijing Key Laboratory of Advanced Chemical Energy Storage Technology and Materials.Notes and references
- T. Kim, W. Song, D.-Y. Son, L. K. Ono and Y. Qi, J. Mater. Chem. A, 2019, 7, 2942–2964 RSC.
- M. Wakihara, Mater. Sci. Eng., R, 2001, 33, 109–134 CrossRef.
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef PubMed.
- M. Weiss, R. Ruess, J. Kasnatscheew, Y. Levartovsky, N. R. Levy, P. Minnmann, L. Stolz, T. Waldmann, M. Wohlfahrt-Mehrens and D. Aurbach, Adv. Energy Mater., 2021, 11, 2101126 CrossRef CAS.
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 1–16 Search PubMed.
- H. Zhang, Y. Yang, D. Ren, L. Wang and X. He, Energy Storage Mater., 2021, 36, 147–170 CrossRef.
- J. Asenbauer, T. Eisenmann, M. Kuenzel, A. Kazzazi, Z. Chen and D. Bresser, Sustainable Energy Fuels, 2020, 4, 5387–5416 RSC.
- Y. Liu, H. Shi and Z.-S. Wu, Energy Environ. Sci., 2023, 16, 4834–4871 RSC.
- E. Logan and J. Dahn, Trends Chem., 2020, 2, 354–366 CrossRef CAS.
- R. Wang, X. Li, Z. Wang and H. Zhang, Nano Energy, 2017, 34, 131–140 CrossRef CAS.
- R. Wang, L. Wang, R. Liu, X. Li, Y. Wu and F. Ran, ACS Nano, 2024, 18, 6500–6512 CrossRef PubMed.
- P. Luo, C. Zheng, J. He, X. Tu, W. Sun, H. Pan, Y. Zhou, X. Rui, B. Zhang and K. Huang, Adv. Funct. Mater., 2022, 32, 2107277 CrossRef CAS.
- H. Ma, Z. Yu, J. Chen, D. Wang, C. Dong and Z. Mao, ACS Appl. Energy Mater., 2023, 6, 1389–1395 CrossRef CAS.
- Q. Cheng, Y. Okamoto, N. Tamura, M. Tsuji, S. Maruyama and Y. Matsuo, Sci. Rep., 2017, 7, 14782 CrossRef PubMed.
- U. Holzwarth and N. Gibson, Nat. Nanotechnol., 2011, 6, 534 CrossRef CAS.
- J. Chen, B. Yao, C. Li and G. Shi, Carbon, 2013, 64, 225–229 CrossRef CAS.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- S. Parmar, S. Pal, A. Biswas, S. Gosavi, S. Chakraborty, M. C. Reddy and S. Ogale, Chem. Commun., 2019, 55, 7562–7565 RSC.
- M. Mahajan, K. Roy, S. Parmar, G. Singla, O. Pandey, K. Singh, R. Vaidhyanathan and S. Ogale, Carbon, 2020, 161, 108–116 CrossRef CAS.
- C. Zhong, S. Weng, Z. Wang, C. Zhan and X. Wang, Nano Energy, 2023, 108894 CrossRef CAS.
- M.-L. Lee, Y.-H. Li, S.-C. Liao, J.-M. Chen, J.-W. Yeh and H. C. Shih, Appl. Surf. Sci., 2012, 258, 5938–5942 CrossRef CAS.
- M.-L. Lee, Y. H. Li, S.-C. Liao, J.-M. Chen, J.-W. Yeh and H. C. Shih, Electrochim. Acta, 2013, 112, 529–534 CrossRef CAS.
- Y. Xiang, W. Zhang, B. Chen, Z. Jin, H. Zhang, P. Zhao, G. Cao and Q. Meng, J. Power Sources, 2020, 447, 227372 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095 CrossRef CAS.
- E. Peled and S. Menkin, J. Electrochem. Soc., 2017, 164, A1703 CrossRef CAS.
- M. M. Thackeray and K. Amine, Nat. Energy, 2021, 6, 683 CrossRef CAS.
- Y.-H. Du, X.-Y. Liu, X.-Y. Wang, J.-C. Sun, Q.-Q. Lu, J.-Z. Wang, A. Omar and D. Mikhailova, Rare Met., 2022, 41, 415–424 CrossRef CAS.
- M. Zukalová, M. Kalbác, L. Kavan, I. Exnar and M. Graetzel, Chem. Mater., 2005, 17, 1248–1255 CrossRef.
- F. Yu, T. Huang, P. Zhang, Y. Tao, F.-Z. Cui, Q. Xie, S. Yao and F. Wang, Energy Storage Mater., 2019, 22, 235–255 CrossRef.
- M. Yin, X. Feng, D. Zhao, Y. Zhao, H. Li, W. Zhou, H. Liu, X. Bai, H. Wang and C. Feng, ACS Sustain. Chem. Eng., 2019, 7, 6122–6130 CrossRef CAS.
- J. Wang, J. Polleux, J. Lim and B. Dunn, J. Phys. Chem. C, 2007, 111, 14925–14931 CrossRef CAS.
- G. Fang, Z. Wu, J. Zhou, C. Zhu, X. Cao, T. Lin, Y. Chen, C. Wang, A. Pan and S. Liang, Adv. Energy Mater., 2018, 8, 1703155 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra00832d |
| This journal is © The Royal Society of Chemistry 2024 |