
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn silico identification of multi-target inhibitors from medicinal fungal metabolites against the base excision repair pathway proteins of African swine fever virus†
Mark Andrian B. Macalalad a and
Fredmoore L. Orosco*abc
a and
Fredmoore L. Orosco*abc
aVirology and Vaccine Research and Development Program, Department of Science and Technology - Industrial Technology Development Institute, Taguig, Metro Manila 1632, Philippines. E-mail: orosco.fredmoore@gmail.com; florosco@up.edu.ph
bS&T Fellows Program, Department of Science and Technology, Taguig, Metro Manila 1632, Philippines
cDepartment of Biology, College of Arts and Sciences, University of the Philippines Manila, Manila, Metro Manila 1000, Philippines
First published on 26th March 2024
Abstract
African swine fever virus (ASFV) has emerged as a serious threat to the pork industry resulting in significant economic losses and heightened concerns about food security. With no known cure presently available, existing control measures center on animal quarantine and culling. Considering the severity and challenges posed by ASFV, it is imperative to discover new treatment strategies and implement additional measures to prevent its further spread. This study recognized the potential of 1830 fungal metabolites from medicinal fungi as antiviral compounds against base excision repair (BER) proteins of ASFV, specifically ASFVAP, ASFVPolX, and ASFVLig. A wide array of computer-aided drug discovery techniques were employed to carry out the virtual screening process: ADMET profiling revealed 319 molecules with excellent bioavailability and toxicity properties; consensus docking identified the 10 best-scoring ligands against all targets; molecular dynamics simulation elucidated the stability of the protein-ligand complexes; and MM/PB(GB)SA energy calculations predicted the binding energies of the compounds as well as the key residues integral to binding. Through in silico methods, we identified two theoretical lead candidates against ASFVAP, four against ASFVLig, and five against ASFVPolX. Two compounds, methyl ganoderate E and antcamphin M, exhibited potential multi-target inhibitory characteristics against ASFVPolX and ASFVLig, while compound cochlactone A showed promising antagonistic results against all three BER proteins. It is recommended to prioritize these hit compounds in future in vitro and in vivo studies to validate their potential as antiviral drugs against ASFV.
Introduction
African swine fever (ASF) is a highly infectious haemorrhagic disease that affects both domestic pigs and wild boars caused by the African swine fever virus (ASFV).1,2 It was initially reported in Kenya in 1921, and since its discovery, ASFV has spread unprecedentedly in Europe, Latin America, and Asia.3 Globally, China bears the brunt of the impact of ASFV. It produces half of the world's total pork output, with its pig sector valued at $4.04 billion.4 The massive outbreak in China that began in 2018 has led to the deaths of approximately 699 million pigs, resulting in a 27% drop in pig meat production and a direct economic loss of $141 billion.4–6 Between January 2021 and December 2023, 27 277 ASF outbreaks have been reported in 53 countries with over 1.9 million associated pig deaths.7 Due to its high morbidity and mortality rates, the disease has resulted in substantial economic losses and presents an ongoing threat to food security, particularly in developing countries.8,9 There is neither a cure nor vaccine commercially available for ASF,2,10 and its control relies solely on biosecurity protocols, such as animal quarantine and culling of infected swine.2,11 However, these methods entail negative economic implications. Therefore, the development of an antiviral compound stands as an attractive option to combat ASF.The ASFV genome encodes over 150 proteins with varying functions.12 However, only the base excision repair (BER) proteins—specifically, the apurinic/apyrimidinic (AP) endonuclease (ASFVAP), the repair DNA polymerase (ASFVPolX), and the DNA ligase (ASFVLig)—are involved in the correction of a damaged DNA.13 When a damaged nucleotide base is detected, DNA glycosylase excises the damaged base and creates an abasic site. ASFVAP cleaves the sugar-phosphate bond of the abasic site, generating a break in the DNA strand and leaving a 3′-OH and a 5′-deoxyribose phosphate (5′dRP) end. ASFVPolX then inserts the correct nucleotide base to fill the gap in the DNA while simultaneously removing the 5′ dRP end. Finally, ASFVLig seals the nick in the DNA backbone through the formation of phosphodiester bonds between 3′-OH and 5′-phosphate of adjacent nucleotides.13 These proteins serve as the sole repair system of the virus to mitigate the oxidative damage induced by free oxygen radicals. The inhibition of these proteins could disrupt the repair process and maintenance of the ASFV,14 leading to a damaged and non-functional genome, thereby arresting viral replication. Additionally, due to the low fidelity and error-prone nature of ASFVPolX and ASFVLig, any damage to viral DNA can potentially lead to lethal mutations and contribute to the virus's suceptibility.14 Owing to their functional importance, BER proteins have been extensively studied,15 highlighting their potential as therapeutic targets (Fig. 1).
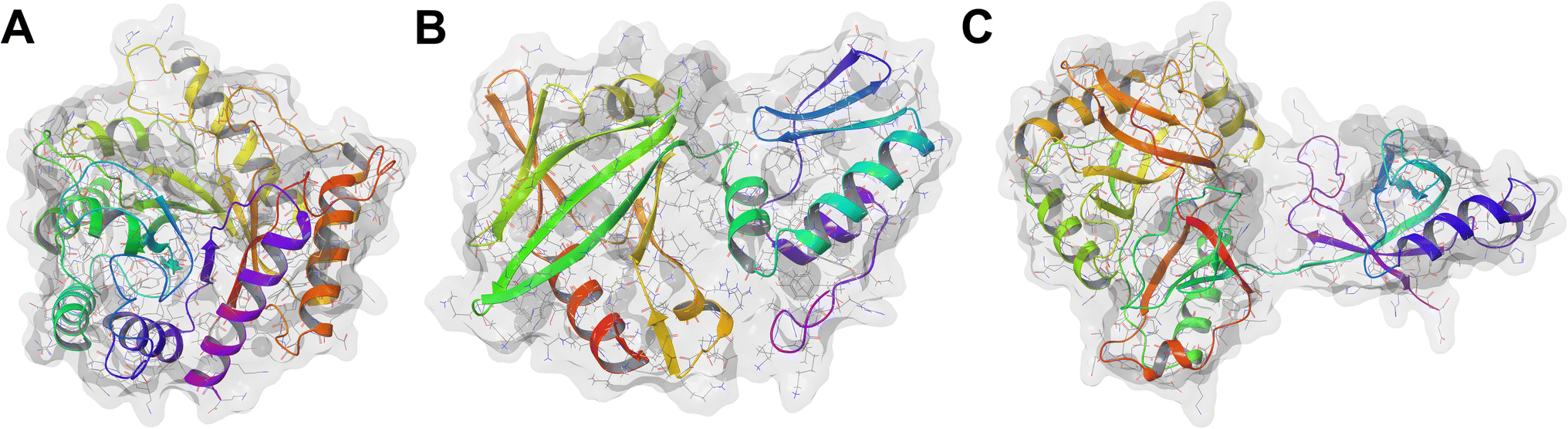 | ||
| Fig. 1 Structures of the BER pathway proteins used as targets. (A) ASFVAP, PDB ID: 6KI3 (B) ASFVPolX, PDB ID: 2M2U (C) ASFVLig, PDB ID: 6IMK. | ||
Numerous in vitro studies have investigated natural products from plants as antiviral agents to inhibit ASFV;16–20 however, their specific targets and underlying mechanisms remain unclear. Fungi are rich sources of natural compounds that have gained considerable attention in the development of new drug compounds.21 They have played a significant role in the development of some of today's most important medicines, such as penicillin. Recent studies have tested fungal-derived metabolites, such as paclitaxel, muscimol, and psilocybin, in clinical trials for the treatment of chronic diseases, including cancer, epilepsy, and treatment-resistant depression, respectively.22,23 However, despite their potential, fungal secondary metabolites are still underutilized as a source of bioactive drug molecules to address veterinary diseases, including ASF.24–26 Furthermore, considering the extensive biodiversity of secondary fungal metabolites, the likelihood of discovering bioactive antiviral compounds against ASFV remains substantial.
With recent technological advancements in the in silico drug development process, enhanced computing capabilities, and new tools in computer-aided drug design (CADD), there has been renewed interest in exploring natural products as potential drug candidates.27 CADD approaches employ various molecular modelling techniques, such as ADMET profiling, molecular docking, and molecular dynamics simulations (MD), enabling a comprehensive understanding of the predicted bioavailability and structural behavior of the candidate compounds against their targets. It allows an atomistic view of the interaction between the ligand and the receptor even before bioassays, thereby reducing both the time and costs associated in the drug development pipeline.28
In this study, compounds derived from medicinal fungi were explored as potential antiviral inhibitors of ASFV. The primary goal of this study is to identify fungal metabolites with ASFV BER multi-target inhibitory potential using a wide array of CADD techniques. Specifically, this work aims to find compounds that adopt a stable conformation with the target proteins, exhibits strong binding affinity to the proteins' binding pocket, and interacts with known binding site residues.
Materials and methods
Curation and ADMET profiling of fungi metabolites
The 3D chemical structures in structured data format (SDF) of the 1830 fungal secondary metabolites were downloaded from MeFSAT (https://cb.imsc.res.in/mefsat/)29 and subsequently optimized in Maestro 12.9 using the OPLS_2005 force field. The metabolites were then subjected to absorption, distribution, metabolism, excretion, and toxicity (ADMET) profiling to predict their safety and efficacy. The SDF files were uploaded to ADMETLab2.0 (https://admetmesh.scbdd.com/)30 where seven parameters were used to assess the drug-likeness of the compounds: (1) Caco-2 permeability, commonly used to estimate in vivo drug permeability, (2) Madin–Darby Canine Kidney cells (MDCK) permeability, considered as one of the most reliable methods in assessing drug uptake efficiency, (3) plasma protein binding, to measure the binding of the drug to its target in plasma, which has a significant influence on the compound's bioavailability, (4) clearance of drug, (5) ames toxicity test, the most widely used assay for testing the mutagenicity of compounds, (6) acute toxicity rule, which identifies molecules that contain toxic moieties or functional groups, (7) hepatoxicity, and (8) Lipinski's Rule of Five (Ro5), one of the most well-known drug-likeness scoring tool.30 Lipinski's rule states that for a drug to be orally bioavailable, it must have a molecular weight of less than 500 g mol−1, less than ten hydrogen bond acceptors, less than five hydrogen donors, and its octanol–water partition coefficient (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) must be less than five. More violations in any of these rules decrease the likelihood of good permeability and solubility. However, this rule cannot predict a drug's potency or whether a drug is pharmacologically active.31 Only the compounds that passed all seven ADMET descriptors were considered in the next steps.
P) must be less than five. More violations in any of these rules decrease the likelihood of good permeability and solubility. However, this rule cannot predict a drug's potency or whether a drug is pharmacologically active.31 Only the compounds that passed all seven ADMET descriptors were considered in the next steps.
Protein optimization
The 3D structure of the target proteins was obtained from the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank32 (https://www.rcsb.org/). The PDB IDs of the receptors used are as follows: 6KI3 (ref. 33) for ASFVAP, 2M2U34 for ASFVPolX, and 6IMK14 for ASFVLig. Water molecules, co-crystallized ligands, and other unnecessary atoms were manually removed using Maestro 12.9.35The protein structures were then processed in Modeller 9.23, where a set of Python scripts was used to fill in missing residues and refine the new loops generated. A 5000-step steepest descent minimization was performed until a tolerance of 10 kJ mol−1 is achieved to ensure that the protein structure was relaxed before proceeding to the docking experiments. For ASFVLig, the protein is composed of three domains: the adenylation domain (AD), the OB-fold domain (OB), and the N-terminal domain (NTD). However, since the nick site residues in the DNA-binding surface are found between the AD and OB domains, the NTD domain is excluded from the PDB structure of ASFVLig.14
Molecular docking
Two docking programs, Autodock 4.2 (AD4)36 and Autodock Vina (Vina),37 were utilized to predict the most probable binding poses of the ligands against their respective targets. Consensus docking was then implemented to consolidate the results from the two software. Since AD4 and Vina employ different scoring functions, integrating their docking scores leads to an improved accuracy, as each program can compensate for the weaknesses of the other scoring function. Studies have further reported that consensus docking improves the reliability of docking experiments by increasing the likelihood of obtaining the correct binding pose for the ligand.38 In consensus docking, if the RMSD between the top poses from AD4 and Vina is less than 2 Å, the binding pose of the ligand is accepted; otherwise, it is dropped from the list of potential inhibitors. The consensus docking score was determined by getting the sum of the scores from AD4 and Vina. Afterward, the consensus docking scores for the ligands from each protein target are totalled and re-ranked to determine the top ten ligands with the highest potential to inhibit all three proteins. AutodockTools-1.5.7 (ref. 36) was used to compute the Root Mean Square Deviations (RMSD) of the ligands. Since the binding site residues of the proteins are already known,14,16,33 the grid box was placed around the centroid of the known residues (Table 1). In the case of ASFVAP, three binding sites were investigated in the docking experiments, corresponding to three DNA binding regions.| Target protein | Binding site residues | Grid box center coordinates |
|---|---|---|
| ASFVAP_R1 | His 8, Ser 14, Cys 16, Cys 20 (ref. 33) | x = −10.0, y = 23.0, z = 5.944 |
| ASFVAP_R2 | His 148, His 145, His 149 (ref. 33) | x = −3.056, y = 25.0, z = −26.14 |
| ASFVAP_R3 | Arg 272, Asn 273, Glu 287, Leu 219 (ref. 33) | x = −4.028, y = 19.0, z = −9.112 |
| ASFVPolX | His 115, Arg 127, Val 120, Leu 123 (ref. 16) | x = 9.0, y = −1.0, z = 7.0 |
| ASFVLig | Asn 153, Leu 211, Leu 402, Gln 403 (ref. 14) | x = 0.285, y = 45.816, z = 107.357 |
Polar hydrogens were added to the protein, and the structure was saved in a coordinate file (PDBQT), which contained the atom types, partial charges, and position of the atoms. In AD4, a 40 × 40 × 40 box was employed, while Vina used a 15 × 15 × 15 box to constrain the identified residues and ensure that the ligands remained within the specified region. Protein residues were maintained in a rigid state, and only translational and rotational movements were allowed for the ligands. The Lamarckian genetic algorithm (LGA) with 1000 iterations was utilized in AD4 to explore ligand conformations and select the best binding pose with the most negative docking scores. Python scripts prepare_gp4.py and prepare_dp4.py were used to generate the grid parameter files (GPF) and docking parameter files (DPF), respectively. For the Vina parameters, the exhaustiveness was set to 20, num_modes to 1000, and energy_range to 1. All other parameters were set to their default values.
Molecular dynamics (MD) simulations
Molecular dynamics simulations were conducted in GROMACS 2023.1 (ref. 39) on the top ten highest-scoring ligands to assess the stability and dynamic interactions between the ligands and the proteins. The topology of the protein was built using the Chemistry at Harvard Macromolecular Mechanics (CHARMM) 36 force field.40 Simultaneously, the CGenFF server (https://cgenff.umaryland.edu/) of the University of Maryland—Baltimore was used to generate the topology of the ligands. The topology files provide information regarding the system being simulated. It contains the details about the molecular structure, atom types, and both bonded and non-bonded parameters needed for calculating forces and updating atom positions.41The protein residues were protonated using the pdb2gmx command of GROMACS with the assumption of canonical pKa values and a pH of 7. The system was then enclosed in a cubic box with periodic boundary conditions to maintain a minimum distance of at least 20.0 Å between any two periodic images of the protein. The box was filled with TIP3P water molecules, and sodium and chlorine ions were added to neutralize any excess charge. Following neutralization, more salt ions were introduced to attain a salt concentration of 0.15 M. Once the solvated and ionized protein-ligand complex is assembled, the system is relaxed through a 5000-steps energy minimization using the steepest descent algorithm to eliminate any steric clashes between atoms. Next, the solvent and ions were equilibrated around the protein under NVT and NPT ensembles. NVT equilibration was performed under constant numbers of particles, volume, and temperature until the system reached the target temperature of 312 K, which corresponds to the normal body temperature of pigs. After temperature stabilization, NPT equilibration at a constant number of particles, temperature, and pressure was performed to reach the desired pressure of 1 bar. The system temperature and pressure were regulated using the velocity rescaling thermostat algorithm and the Berendsen barostat, respectively. Finally, upon achieving the target temperature and pressure during equilibration, the systems underwent a 100 ns molecular dynamics (MD) production run under the NPT ensemble. The 100 ns MD simulation required 50000000 time steps, with each step corresponding to 2 fs. A snapshot was saved every 100 ps, producing a total of 1000 frames.
Molecular mechanics Poisson–Boltzmann (generalized Born)/surface area (MM/PB(GB)SA) calculations
The molecular mechanics/Poisson–Boltzmann surface area (MM/PBSA) and molecular mechanics/generalized Born surface area (MM/GBSA) were used to predict the binding affinity between the protein and ligand. Both models integrate molecular mechanics calculations with continuum solvation models, allowing for a more accurate assessment of the intermolecular interactions. In this approach, explicit water molecules are replaced with a continuum water model to optimize calculation efficiency while preserving the balance between computational speed and accuracy.42The MM/PB(GB)SA score was calculated from the total energy difference between the bound and unbound states of the protein–ligand complex (eqn (1)). Gprotein+ligand is the total free energy of the complex, while Gprotein and Gligand are the total free energies of the unbound protein and ligand in a solvent, respectively. Each term can be decomposed as the sum of energies in a vacuum and a solution (eqn (2)). Evacuum is the average molecular mechanics potential energy in the gas phase, which includes the bonded (i.e., bond stretching, bond angles, dihedral angles, etc.) and non-bonded interactions (i.e., electrostatic and van der Waals interactions). An entropic term is also included in Evacuum to account for the entropic contribution in vacuum. In calculating the molecular mechanics potential energy, it is assumed that the bonded interactions of the protein and ligand in their bound and unbound states are identical, so the total Ebonded is zero. Therefore, Evacuum is solely determined by the sum of the electrostatic and van der Waals energies, and the entropic term. Gsolvation is the energy required to transfer a solute from the gas phase medium into the solvent. It can be expressed as the sum of the electrostatic and non-polar interactions between the solute and the implicit solvent (eqn (6)). Gpolar calculates the electrostatic interactions of the ligand with the continuum solvent, whereas Gnonpolar accounts for both the energy cost associated with creating a cavity in the continuum solvent and the attractive van der Waals interactions between the solute and the solvent.42–44
| ΔGblind = Gprotein+ligand − Gprotein − Gligand | (1) |
| Gx = Evacuum + Gsolvation | (2) |
| Evacuum = Ebonded + Enonbonded − TΔS | (3) |
| Ebonded = Estretch + Eangles + Edihedrals + Eimproper | (4) |
| Enonbonded = EvdW + Eelec | (5) |
| Gsolvation = Gpolar + Gnonpolar | (6) |
| ΔGblind = EvdW + Eelec + Gpolar + Gnonpolar − TΔS | (7) |
The electrostatic and van der Waals interaction energies were calculated using the Coulomb and Lennard-Jones potential functions, respectively, based on the molecular mechanics force field parameters. The main difference between the PB and GB models is the continuum solvation model used for estimating the Gpolar term. MM/PBSA employs the Poisson–Boltzmann (PB) equation to calculate polar solvation energies, while MM/GBSA relies on the generalized Born (GB) model. The GB approach is significantly faster and less computationally intensive compared to the PB model. The GB model is preferred when ranking the relative binding energies of candidate inhibitors, whereas PB provides more accurate predictions for absolute free energies.45 Studies have compared the performance of MM/PBSA and MM/GBSA, demonstrating that the accuracy of their results can vary considerably depending on the system being studied.46 For the non-polar contribution of the solvation energy, it is assumed that the non-polar solvation energy due to cavity formation is directly proportional to the surface accessible surface area (SASA).43
The MM/PBSA binding energies were predicted using two GROMACS compatible packages, g_mmpbsa,43 and gmx_MMPBSA,44 while MM/GBSA was estimated through gmx_MMPBSA. The dielectric constant values for the solute and the solvent were kept 2.0 and 80.0, respectively. For both packages, the entropy term from eqn (3) is omitted due to its high computational costs and a tendency to introduce a higher degree of error compared to other binding energy terms.43,47 Additionally, entropic energies often yield small values that do not significantly improve the accuracy of the binding energy. As a result, they are often ignored in many MM/PBSA studies.43,46 After incorporating eqn (1)–(6) and considering the assumptions mentioned above, the binding strength between the ligand and protein can be estimated using eqn (8). Only the last 10 ns or the stable phase of the complex trajectory was considered for the evaluation of binding energies.
| ΔGblind = ΔEvdW + ΔEelec + ΔGPB + ΔGSASA | (8) |
Results and discussion
ADMET profiling
The fungal metabolites were subjected to initial ADMET profiling to predict their bioavailability and safety when administered to pigs. Only those with predicted values within the recommended range were retained for additional screening, while those that failed in at least one parameter were omitted. To date, there is no ADMET server capable of predicting the pharmacokinetic properties of swine; therefore, the ADMET parameters used in this study were carefully selected to ensure their applicability for use in organisms other than humans. Physiologically, the gastrointestinal tract (GIT) of pigs demonstrates greater similarity to the human GIT than rats, thus the absorption parameters derived from humans can reasonably estimate porcine intestinal permeability.48 Out of the 1830 compounds tested, only 319 metabolites met all eight ADMET criteria (Table 2). The compounds that scored favorably for the selected ADMET parameters are listed in Table S1.†| ADMET property | Passed | Failed |
|---|---|---|
| Caco-2 permeability | 854 | 976 |
| Madin–Darby Canine Kidney cells (MDCK) permeability | 1824 | 6 |
| Plasma protein binding | 983 | 847 |
| Clearance of drug | 1145 | 685 |
| Ames toxicity | 1728 | 102 |
| Hepatoxicity | 1539 | 291 |
| Acute toxicity rule | 1830 | 0 |
| Lipinski's rule of five | 1617 | 213 |
| All eight properties | 319 | 1511 |
Among the two permeability tests, Caco-2 was more stringent than MDCK. The Caco-2 test resulted in the elimination of over half of the metabolites, whereas MDCK excluded only six compounds. Compounds that successfully passed both tests were more likely to exhibit favorable drug permeability in in vivo methods. 983 metabolites were predicted to not bind to proteins in the blood plasma, which could potentially enhance their distribution to target sites. Moreover, 1145 out of the 1830 metabolites demonstrated effective drug elimination.
For toxicity assessment, we conducted the Ames toxicity test to predict mutagenicity and genetic toxicity, and the Hepatoxicity test to assess potential liver damage.30 The Ames toxicity test is particularly important in ADMET profiling, considering the ligand's proximity to the DNA molecule. Through this test, the tendency of compounds to cause genetic toxicity to DNA can be predicted. In the Ames toxicity test, 1728 compounds yielded promising results, while 1539 passed the Hepatotoxicity tests. After applying the Acute toxicity rule, it was confirmed that none of the metabolites contained any toxic moieties. Finally, Lipinski's rule of five revealed 1617 compounds with favorable absorption and permeation characteristics, emphasizing their viability as orally active drugs. Considering all parameters, 319 compounds successfully passed all specified ADMET criteria, thereby enhancing their potential as orally bioavailable and safe inhibitor drugs.
Consensus docking
Docking experiments were employed to predict the preferred binding conformation of a ligand, estimate its associated binding score, and determine residue interactions. However, most docking software, including AD4 and Vina, assume a rigid protein structure and do not incorporate explicit water molecules in their scoring functions.49,50 In reality, protein mobility and water molecules both play significant roles in determining binding affinity. Thus, the docking score does not correspond to the actual binding energy; it only provides a rough approximation of the binding affinity of a ligand to a protein. Therefore, the total docking score presented in this study will only be used to rank the compounds and will not serve as a measure of the actual binding strength of the ligand.Two docking software programs were used to generate the binding conformations and rank the docking scores of the ligands against the receptors. Since both programs employ different search algorithms and scoring functions, it is expected that the docking scores and rankings will also differ. Fig. 2 illustrates the distribution of the docking scores of Vina and AD4 across all 1830 metabolites. Autodock 4.2 can better differentiate the affinity of the compounds as the scores exhibited a broader distribution along the x-axis, whereas Vina tended to yield similar scores resulting in a concentration of scores around a single docking value. The differences in scoring patterns from employing multiple docking software highlight the need for validation to generate the final list of promising compounds against each protein target.
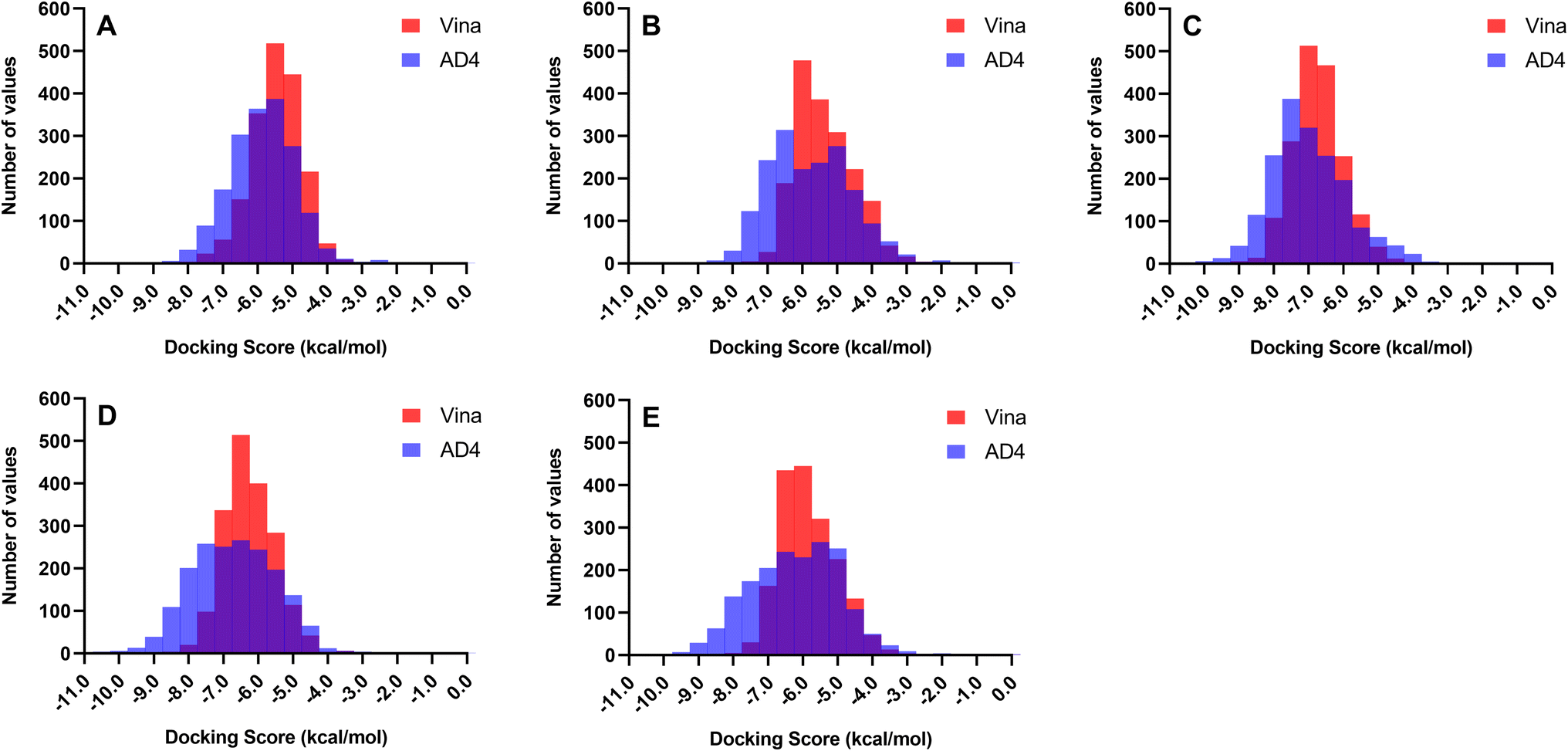 | ||
| Fig. 2 Distribution of docking scores between Autodock 4.2 (AD4) and Autodock Vina (Vina) across all 1830 metabolites. Autodock 4.2 better differentiates compound affinity with scores showing a broader distribution along the x-axis. In contrast, Vina tends to produce similar scores for most ligands. (A) ASFVAP_R1. (B) ASFVAP_R2. (C) ASFVAP_R3. (D) ASFVPolX. (E) ASFVLig. | ||
To consolidate the docking poses and scores obtained from the two software tools, consensus docking was implemented, considering only the docking poses created by the two docking software with RMSD within 2 Å. The final rankings of the top ten compounds with the best consolidated docking scores are listed in Table 3. These ten compounds exhibited the highest potential to inhibit all protein targets, including the three binding regions of ASFVAP, so they will undergo further tests to verify their binding stability and affinity. Again, given that docking scores are solely employed for ranking and selecting the final candidate compounds and do not precisely reflect the potency of the ligands, all ten ligands will still be considered in the subsequent stage of the virtual screening process. A more accurate estimation of the binding energy was performed in the MM/PB(GB)SA experiments. The docking scores for all ligands are available in Table S2 of the ESI,† the structures of the top ten ligands and their interactions with their respective targets are shown in Fig. S1 to S5,† and the summary of the ligand–protein interaction are presented in Table 4 to Table 8.
| Ligand | ASFVAP (kcal mol−1) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | ||||||||||
| Vina | AD4 | RMSD | Score | Vina | AD4 | RMSD | Score | Vina | AD4 | RMSD | Score | |
| F1385 | −7.8 | −7.42 | 1.899 | −15.22 | −7.7 | −6.93 | 1.991 | −14.63 | −9.5 | −8.92 | 0.542 | −18.42 |
| F1384 | −6.8 | −7.54 | 1.555 | −14.34 | −6.3 | −7.33 | 0.866 | −13.63 | −8.8 | −8.86 | 0.798 | −17.66 |
| F1187 | −6.9 | −7.31 | 1.485 | −14.21 | −6.5 | −8.21 | 1.037 | −14.71 | −7.0 | −6.98 | 1.662 | −13.98 |
| F1540 | −6.4 | −7.63 | 1.028 | −14.03 | −6.0 | −7.35 | 1.389 | −13.35 | −7.8 | −7.95 | 0.93 | −15.75 |
| F1792 | −5.9 | −6.42 | 0.448 | −12.32 | −7.3 | −7.36 | 1.048 | −14.66 | −7.6 | −7.01 | 0.833 | −14.61 |
| F0329 | −6.0 | −6.97 | 1.066 | −12.97 | −6.2 | −7.75 | 1.311 | −13.95 | −6.9 | −7.66 | 1.052 | −14.56 |
| F1342 | −5.9 | −7.27 | 1.147 | −13.17 | −6.2 | −7.62 | 1.605 | −13.82 | −7.9 | −8.24 | 1.383 | −16.14 |
| F1771 | −5.2 | −6.59 | 1.407 | −11.79 | −6.4 | −8.01 | 1.605 | −14.41 | −7.5 | −7.92 | 1.525 | −15.42 |
| F0059 | −6.0 | −6.52 | 1.208 | −12.52 | −6.1 | −6.57 | 0.757 | −12.67 | −8.2 | −8.17 | 0.713 | −16.37 |
| F1740 | −5.4 | −7.19 | 1.836 | −12.59 | −6.0 | −6.87 | 1.712 | −12.87 | −7.3 | −8.05 | 1.505 | −15.35 |
| Ligand | ASFVPolX (kcal mol−1) | ASFVLig (kcal mol−1) | Total docking score | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Vina | AD4 | RMSD | Score | Vina | AD4 | RMSD | Score | ||
| PEN | −6.2 | −8.78 | 1.974 | −14.98 | NA | NA | NA | NA | NA |
| ATP | NA | NA | NA | NA | −5.6 | −5.32 | 1.290 | −10.92 | NA |
| F1385 | −8.6 | −7.86 | 1.651 | −16.46 | −6.6 | −7.11 | 1.773 | −13.71 | −78.44 |
| F1384 | −8.0 | −7.50 | 1.12 | −15.50 | −7.0 | −6.05 | 1.132 | −13.05 | −74.18 |
| F1187 | −6.4 | −6.73 | 1.751 | −13.13 | −6.8 | −8.40 | 1.046 | −15.20 | −71.23 |
| F1540 | −6.8 | −7.20 | 1.08 | −14.00 | −7.2 | −6.36 | 1.472 | −13.56 | −70.69 |
| F1792 | −7.1 | −6.06 | 1.224 | −13.16 | −8.0 | −7.64 | 0.664 | −15.64 | −70.39 |
| F0329 | −6.4 | −7.77 | 1.326 | −14.17 | −7.2 | −7.26 | 1.572 | −14.46 | −70.11 |
| F1342 | −6.5 | −6.63 | 1.824 | −13.13 | −5.9 | −7.90 | 0.985 | −13.80 | −70.06 |
| F1771 | −6.6 | −8.10 | 1.909 | −14.70 | −5.8 | −7.20 | 1.731 | −13.00 | −69.32 |
| F0059 | −7.7 | −7.06 | 1.448 | −14.76 | −6.3 | −6.66 | 1.486 | −12.96 | −69.28 |
| F1740 | −6.8 | −8.10 | 1.071 | −14.90 | −6.3 | −7.00 | 1.219 | −13.30 | −69.01 |
| F0329 | F1187 | F1342 | F1384 | F1385 | F1540 | F1792 | F0059 | F1740 | F1771 | |
|---|---|---|---|---|---|---|---|---|---|---|
| ARG 9 | X | X | X | X | X | X | X | |||
| ASL 60 | X | X | X | X | X | X | X | X | X | |
| ASN 55 | X | X | X | X | X | X | X | X | X | |
| ASP 13 | X | X | X | X | X | X | ||||
| CYS 16 | X | X | X | X | X | X | X | X | ||
| CYS 20 | X | X | X | X | X | X | X | X | X | X |
| GLN 57 | X | X | X | X | X | X | X | X | X | |
| GLY 15 | X | X | X | X | X | X | X | X | ||
| GLY 48 | X | X | X | |||||||
| HIS 8 | X | X | X | X | X | X | X | X | X | X |
| ILE 21 | X | X | X | X | X | X | X | X | X | |
| ILE 46 | X | X | X | |||||||
| ILE 56 | X | X | X | X | X | X | X | X | X | |
| PHE 45 | X | X | X | |||||||
| PRO 49 | X | X | X | |||||||
| SER 7 | X | X | X | |||||||
| SER 12 | X | X | X | X | X | X | ||||
| SER 14 | X | X | X | X | X | X | X | X | ||
| THR 17 | X | X | X | X | X | X | X | X | X | X |
| THR 18 | X | X | X | X | X | X | X | X | X | |
| THR 19 | X | X | X | X | X | X | ||||
| VAL 44 | X | X |
Generally, the structures of the ten promising ligands consist of a polar group functionalized with hydroxyl groups at one end, and a linear or cyclic aliphatic group at the opposite end (Fig. S6 to S10†). Compound F1385 registered the highest consolidated score of −78.44 kcal mol−1 followed by compounds F1384 and F1187. Pentagastrin (PEN) is chosen as the control compound for ASFVPolX, which has been shown to exhibit inhibitory characteristics against ASFVPolX in both in silico and in vitro methods.51 As for ASFVLig, its natural substrate, adenosine triphosphate (ATP), served as the control compound.14 To the best of our knowledge, no compounds have been tested against ASFVAP using either in silico or in vitro methods, and thus, no control compound has been examined for comparison. Nevertheless, the negative docking score signifies affinity towards the protein. Compound F1385 also scored the highest against the R1 and R3 regions of ASFVAP and ASFVPolX. Two metabolites scored better than PEN, while all candidate ligands registered more negative scores than ATP. Despite variations in the rankings of the top ten compounds for each target, these ten compounds consistently ranked among the highest-scoring ligands out of all the docked ligands. The candidate ligands adopted similar docking conformations within the three binding domains of the ASFVAP protein (Fig. S6 to S8 of the ESI†). The binding site residues were predominantly polar, with only a few nonpolar and charged residues. Among the three regions, R2 region had the highest concentration of polar amino acids. Interactions with polar residues can help stabilize the ligand through hydrogen bonds and other non-covalent interactions, especially when the ligand is also highly functionalized with polar groups. However, the presence of polar residues inside the cavity can also attract mobile water molecules, which may compete for interactions with the ligand and potentially displace it from the pocket. Thus, the high number of polar residues in all three binding regions of ASFVAP poses a double-edged sword, potentially contributing to instability in most systems during MD simulations. From the study of Chen et al.33 the three binding regions of the ASFVAP establish an extensive hydrogen bond network during DNA interaction. H-bonding with the DNA molecule occurs through His 8 and Ser 14 in the R1 region, implying that inhibitor interactions with these amino acids may potentially decrease DNA binding efficiency. Our results showed that His 8, Thr 17, and Cys 20 in the R1 region formed interactions with all ten candidate ligands, while Ser 14 interacted with eight of the ten ligands (Table 4). Additionally, hydrogen bond networks were observed among several residues, including His 8, Cys 16, Thr 17, Cys 20, and Gln 57 (Fig. S1†).
In the R2 region, the ten candidate ligands displayed common interactions with nine residues, namely Ala 118, His 145, His 146, Lys 147, His 148, His 149, Ser 228, Gly 229, and Ile 230 (Table 5). Studies suggest that the primary driving force for DNA binding in the R2 region arises from His 145, His 148, and His 149, which is attributed to the formation of hydrogen bonds.33 Thus, the interaction of the ten ligands with these three residues may lead to a decreased affinity of the DNA for the protein. Additionally, ligands F0059, F0329, F1187, F1384, F1740, and F1792 formed hydrogen bonds with the three histidine residues, thereby further enhancing the binding strength (Fig. S2†).
| F0329 | F1187 | F1342 | F1384 | F1385 | F1540 | F1792 | F0059 | F1771 | F1740 | |
|---|---|---|---|---|---|---|---|---|---|---|
| ALA 118 | X | X | X | X | X | X | X | X | X | X |
| ASN 146 | X | X | X | X | X | X | X | X | X | X |
| GLY 117 | X | X | X | X | X | X | X | X | X | |
| GLY 229 | X | X | X | X | X | X | X | X | X | X |
| HIS 115 | X | X | X | X | X | X | X | X | ||
| HIS 145 | X | X | X | X | X | X | X | X | X | X |
| HIS 148 | X | X | X | X | X | X | X | X | X | X |
| HIS 149 | X | X | X | X | X | X | X | X | X | X |
| ILE 230 | X | X | X | X | X | X | X | X | X | X |
| LEU 116 | X | X | X | X | X | X | X | X | X | |
| LYS 147 | X | X | X | X | X | X | X | X | X | X |
| LYS 152 | X | X | ||||||||
| PRO 144 | X | X | X | X | X | X | X | X | X | |
| SER 228 | X | X | X | X | X | X | X | X | X | X |
The R3 region of ASFVAP features a narrower nucleotide-binding pocket compared to the other two regions, where two residues, Asn 273 and Tyr 81, are known to directly form hydrogen bond interactions with DNA molecules.33 From the ligand interaction table (Table 6), these two residues, along with His 115, Asp 231, and Glu 271, generated interactions with all ten final candidate ligands. Hydrogen bond pairs were also formed between Asn 273 and Tyr 81, and ligands F0059 and F1740 (Fig. S3†). Studies have reported the unique structural features of the three binding regions of ASFVAP, which significantly differ from other homologous proteins and AP endonucleases.33 This suggests that ASFVAP adopts a novel DNA-binding mode, making it an interesting target for drug design and development.
| F0329 | F1187 | F1342 | F1384 | F1385 | F1540 | F1792 | F0059 | F1771 | F1792 | |
|---|---|---|---|---|---|---|---|---|---|---|
| ALA 234 | X | X | X | |||||||
| ARG 9 | X | X | X | |||||||
| ARG 50 | X | X | X | X | ||||||
| ARG 232 | X | X | X | X | X | X | X | X | X | |
| ARG 272 | X | X | X | X | X | X | X | |||
| ASN 273 | X | X | X | X | X | X | X | X | X | X |
| ASP 179 | X | X | ||||||||
| ASP 231 | X | X | X | X | X | X | X | X | X | X |
| GLN 43 | X | X | X | X | X | X | X | |||
| GLU 142 | X | X | X | X | ||||||
| GLU 271 | X | X | X | X | X | X | X | X | X | X |
| HIS 8 | X | X | X | |||||||
| HIS 78 | X | X | X | X | X | X | X | |||
| HIS 115 | X | X | X | X | X | X | X | X | X | X |
| HIS 145 | X | X | X | |||||||
| HIS 182 | X | X | X | X | X | X | X | X | ||
| HIS 218 | X | X | X | |||||||
| HIS 233 | X | X | X | X | X | X | X | X | X | |
| LEU 274 | X | X | X | X | X | X | X | X | ||
| PHE 5 | X | X | X | X | X | |||||
| PHE 45 | X | X | X | X | X | X | X | X | ||
| PRO 49 | X | X | X | |||||||
| PRO 144 | X | X | ||||||||
| SER 7 | X | X | X | |||||||
| TYR 81 | X | X | X | X | X | X | X | X | X | X |
For the ASFVPolX complexes, the top ten ligands settled into a localized region within the binding site and presented analogous binding poses (Fig. S9†). The ligands are strategically docked, with polar groups situated near hydrophilic residues, while nonpolar groups are adjacent to hydrophobic residues. Site interactions were primarily hydrophobic. Electrostatic and charged residues were also observed, along with a few polar interactions. Five residues interacted with all ten compounds: Arg 84, Lys 85, Phe 102, His 115, and Arg 127 (Table 7). Notably, His 115 and Arg 127 play crucial roles in the misincorporation of deoxyguanosine triphosphate (dGTP) into the growing DNA strand, leading to mutations and increased survival of the virus.15 Establishing strong connections with these two residues can prevent dGTP mispair stabilization, but more importantly, it can inhibit the activity of ASFVPolX and disrupt the entire replication process of the polymerase. Compounds F0059, F1187, F1385, F1740, and F1771 formed hydrogen bonds with these two residues (Fig. S4†). Additionally, F0059 formed pi–pi stacking and pi–cation interactions, enhancing its binding affinity. Although Lys 85 is not listed as a crucial residue of ASFVPolX, it formed hydrogen bond connections with six candidate ligands, potentially contributing to the stability of the protein–ligand complexes.
| PEN | F0329 | F1187 | F1342 | F1384 | F1385 | F1540 | F1792 | F0059 | F1771 | F1740 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ALA 104 | X | X | X | X | X | X | X | X | X | ||
| ALA 112 | X | X | X | X | X | X | X | X | X | ||
| ARG 84 | X | X | x | X | X | X | X | X | X | X | X |
| ARG 127 | X | X | X | X | X | X | X | X | X | X | X |
| ASN 138 | X | X | X | X | |||||||
| GLN 139 | X | X | X | X | X | X | X | X | X | X | |
| GLU 83 | X | X | X | X | X | ||||||
| GLU 108 | X | X | X | X | |||||||
| GLY 82 | X | X | X | ||||||||
| HIS 115 | X | X | X | X | X | X | X | X | X | X | X |
| ILE 124 | X | X | X | X | X | X | X | X | |||
| LEU 53 | X | X | X | X | X | X | X | X | |||
| LEU 123 | X | X | X | X | X | X | X | ||||
| LEU 137 | X | X | X | ||||||||
| LEU 159 | X | X | |||||||||
| LYS 85 | X | X | X | X | X | X | X | X | X | X | X |
| PHE 102 | X | X | X | X | X | X | X | X | X | X | X |
| PHE 114 | X | ||||||||||
| PHE 116 | X | X | X | X | X | X | X | X | X | X | |
| THR 103 | X | X | X | X | X | X | X | X | X | ||
| TYR 111 | X | X | |||||||||
| TYR 140 | X | X | X | X | X | X | X | X | X | ||
| VAL 120 | X | X | X | X | X | X | X | X | X |
The top-scoring candidate ligands against ASFVLig also adopted similar binding poses when docked into the protein's binding pocket (Fig. S10†). The nick site residues targeted in this study were sandwiched between the OB-fold domain (OB) and the adenylation domain (AD). However, between the two domains, the hydroxyl groups of the ligands were directed towards the OB domain rather than the AD domain (Fig. S12†). Most interactions were hydrophobic and charged, while only a few polar connections were present (Fig. S5†). F0329 formed the fewest interactions as the ligand exhibited a preference for binding to the AD domain only, limiting its number of contacts. In contrast, the remaining candidate ligands demonstrated shared interactions between the residues of the OB and AD domains. Only one residue, Gln 403 from the AD domain, showed a binding preference for all candidate ligands (Table 8). However, excluding F0329, which exclusively docked against the OB domain, Asn 153 also presented polar interactions with the remaining nine candidate ligands. Gln 403 and Asn 153 have been identified in in vitro studies as crucial for the catalytic efficiency of ASFVLig. Moreover, as these residues are unique to ASFVLig, drugs targeting them will not interfere with other DNA ligases in pigs which makes them excellent targets for small inhibitors.14 Compared to ASFVPolX, fewer hydrogen bonds were observed as the ligands docked in the region between the OB and AD domains. Consequently, the number of adjacent residues capable of forming hydrogen bonds with the ligands was limited. Only F0059, F1342, F1540, and F1771 generated one to two hydrogen bonds with the binding site residues.
| ATP | F0329 | F1187 | F1342 | F1384 | F1385 | F1540 | F1792 | F0059 | F1740 | F1771 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ALA 215 | X | X | |||||||||
| ALA 404 | X | X | X | X | X | X | X | X | |||
| ARG 152 | X | X | X | X | X | ||||||
| ARG 156 | X | X | X | X | X | X | |||||
| ARG 172 | X | X | X | X | X | X | X | X | X | ||
| ASN 153 | X | X | X | X | X | X | X | X | X | X | |
| GLN 403 | X | X | X | X | X | X | X | X | X | X | X |
| GLU 203 | X | X | X | X | X | X | X | ||||
| GLU 291 | X | X | X | X | X | X | X | X | |||
| GLU 393 | X | X | X | X | X | X | X | X | |||
| GLY 154 | X | X | X | X | X | X | X | X | X | ||
| ILE 400 | X | X | X | X | X | X | X | ||||
| LEU 211 | X | X | X | X | X | X | X | X | |||
| LEU 402 | X | X | X | X | X | X | X | X | X | X | |
| LYS 316 | X | X | X | X | X | X | X | ||||
| LYS 318 | X | X | X | X | X | ||||||
| LYS 397 | X | X | |||||||||
| LYS 151 | X | X | X | X | X | X | |||||
| PHE 354 | X | X | X | X | X | ||||||
| PRO 212 | X | X | X | X | X | X | X | X | X | X | |
| PRO 396 | X | X | |||||||||
| PRO 401 | X | X | X | X | X | X | X | ||||
| SER 395 | X | X | |||||||||
| THR 357 | X | X | |||||||||
| THR 398 | X | X | |||||||||
| VAL 155 | X | X | X | X | X | X | X | X | |||
| VAL 355 | X | X | X | X | X | X | X | ||||
| VAL 356 | X | X | X | X | X | X |
Molecular dynamics (MD) simulation
The ten complexes from each protein target were tested for stability using 100 ns MD simulations. Fig. S11† shows the complex RMSD of all candidate ligands against all protein targets, including the three regions of ASFVAP. Among the ten candidates, only one ligand demonstrated a stable behavior with ASFVAP_R1, none with ASFVAP_R2, two with ASFVAP_R3, five with ASFVPolX, and four with ASFVLig (Fig. 3). The stable complexes required approximately 20–30 ns after equilibration before achieving a constant RMSD trajectory and reaching their most stable conformation. Sample ESI Videos S1 to S4† presents how the stable ligands adjusted to their environment during first few nanoseconds and achieved their final binding pose upon reaching a steady state.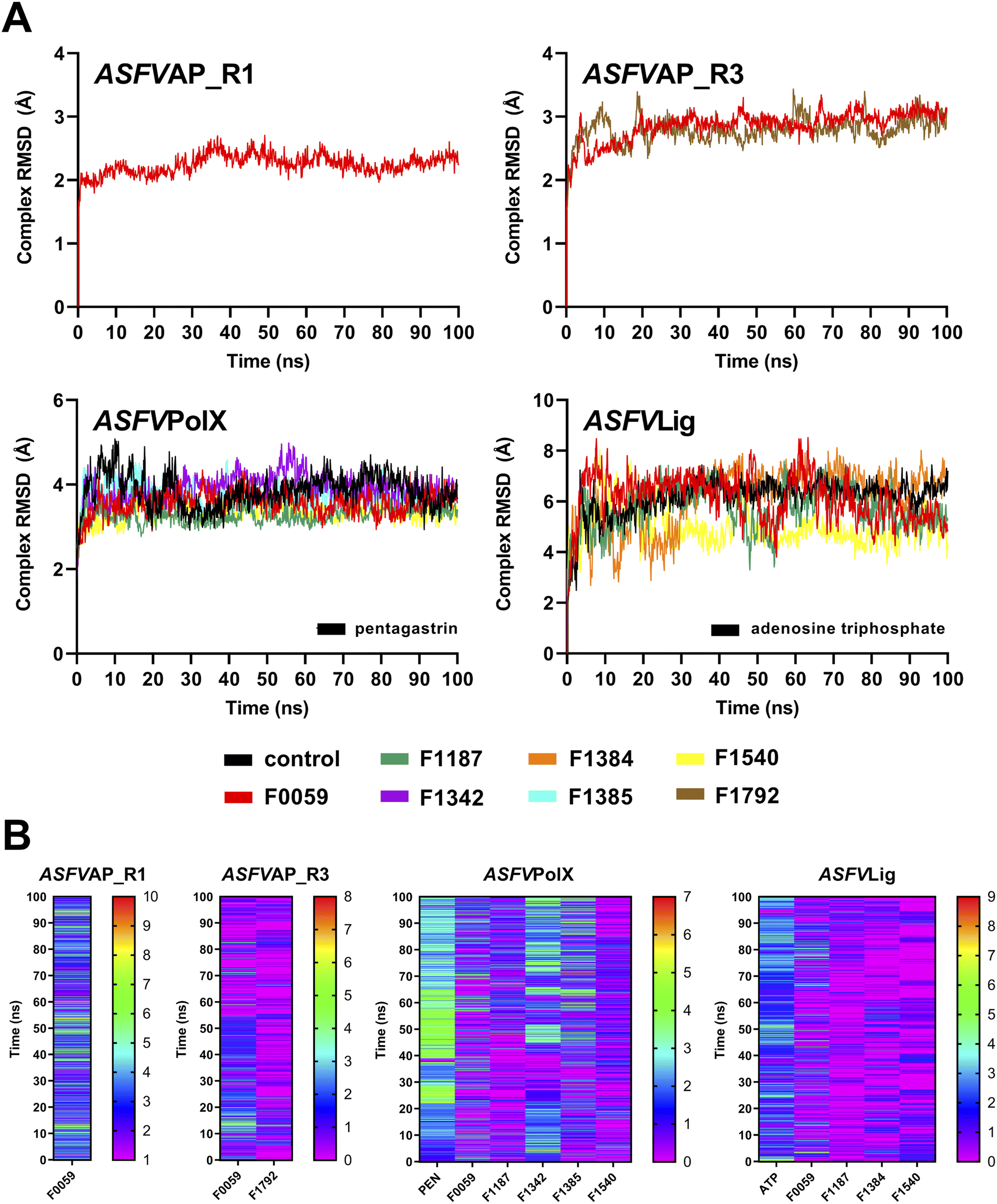 | ||
| Fig. 3 (A) Complex RMSD and (B) H-bonds diagram of the stable complexes. Only one candidate ligand presented stability against ASFVAP_R1, two against ASFVAP_R3, five against ASFVPolX, and four against ASFVLig. In the H-bond diagram, a color closer to red suggests more frequent H-bonding, whereas a color closer to purple indicates fewer H-bond connections. | ||
Due to the highly polar nature of the pockets in the ASFVAP enzyme, most ligands detached from the cavity and moved into the solvent. With the increased concentration of the solvent inside the pocket, the mobile water molecules likely formed strong interactions with the polar residues, which competed with the ligands. Moreover, the mobile water molecules might have also interacted with the ligands, overpowering the noncovalent interaction between the ligand and the protein, ultimately resulting in the ligand being pulled out of the binding site. ASFVAP_R2, which has the most polar binding site among the three regions, prevented any ligands from displaying a stable behavior. Only F0059 against ASFVAP_R1 and ASFVAP_R3, and F1792 against ASFVAP_R3, maintained their structural stability with the protein throughout the entire simulation. The RMSD values for the stable ASFVAP complexes averaged between 2.247 Å and 2.859 Å. The relatively low RMSD values suggest that the initial conformation of the complex closely resembles the stable conformation after the 100 ns simulation. Additionally, an increased occurrence of H-bond formation was observed in the R1 region of ASFVAP compared to the R3 region (Fig. 3). However, even with a few hydrogen bonds, F0059 and F1792, docked in the R3 region still exhibited a stable behavior with the protein. Since the functional groups of F1792 are ketones and ethers, which only serve as hydrogen bond acceptors, and most of the side chains of the residues within the binding pocket of R3 also only act as hydrogen bond acceptors, this limits the formation of hydrogen bonds. In contrast, the binding pocket of the R1 region contains residues with side chains capable of both donating (e.g., cysteine, tryptophan, histidine, and glutamine) and accepting (e.g., aspartic acid) hydrogen bonds, which allows the participation of more hydrogen bond pairs.
As shown in Fig. S4,† the binding pocket of the ASFVPolX protein contains positively charged residues capable of forming strong hydrogen bonds and electrostatic interactions with the polar groups of the ligand. These positively charged residues appeared to play a pivotal role in establishing stability, as evidenced by the highest number of stable complexes in the ASFVPolX system. Among the ten compounds, five became stable with the target proteins: F0059, F1187, F1342, F1385, and F1540. Hydrogen bonds were more frequent with the charged residues, specifically arginine and lysine. Compared to neutral atoms, hydrogen bonds formed with at least one charged partner are considerably stronger owing to a larger coulombic or electrostatic attraction.52 F1342 generated the most H-bonds, while F1540 obtained the least number of H-bonds because of its lower number of oxygen-containing functional groups.
Similar to ASFVPolX, the binding site of ASFVLig also contains positively charged residues, potentially contributing to the stability of the formed complexes. However, due to its larger size, the average RMSD was significantly higher (5.750 Å) and the trajectories fluctuated more compared to the other protein systems (2.247 Å to 3.566 Å). Since the protein is composed of two distinct domains (AB and OD), and the ligand is sandwiched between them, there is a tendency for the ligand to shift its position between the two domains, leading to oscillations in the binding interactions. The two domains also exhibited significant movements during the simulations, but the four stable ligands remained securely bound within the binding pocket. The ASFVLig complexes presented a modest number of hydrogen bonds, with a maximum of only two hydrogen bond pairs. The shifting of ligands' binding positions between the two domains might have prevented them from establishing a consistent number of hydrogen bonds owing to the constant breaking and formation of new hydrogen bonds, even with a stable complex conformation.
The RMSF and RMSD values of the ligands are shown in Fig. S12 and S13 of the ESI,† respectively. Peaks in the RMSF plots represent residues that are constantly moving throughout the simulation. Most of these residues are located in protein loops with no defined patterns, and hence, the constant and random movements. Additionally, unstable complexes generally incurred higher RMSF values compared to complexes that demonstrated stable behaviours. Lower RMSF values signify restricted movements and fewer conformational changes, indicating stability. Meanwhile, ligands with a higher number of rotatable bonds, such as F1771 and the control compound PEN, recorded higher ligand-RMSD values, which was attributed to their increased freedom of movement. In contrast, compounds with a more compact structure, such as F1792, consistently exhibited lower ligand-RMSD values.
Binding energy calculations
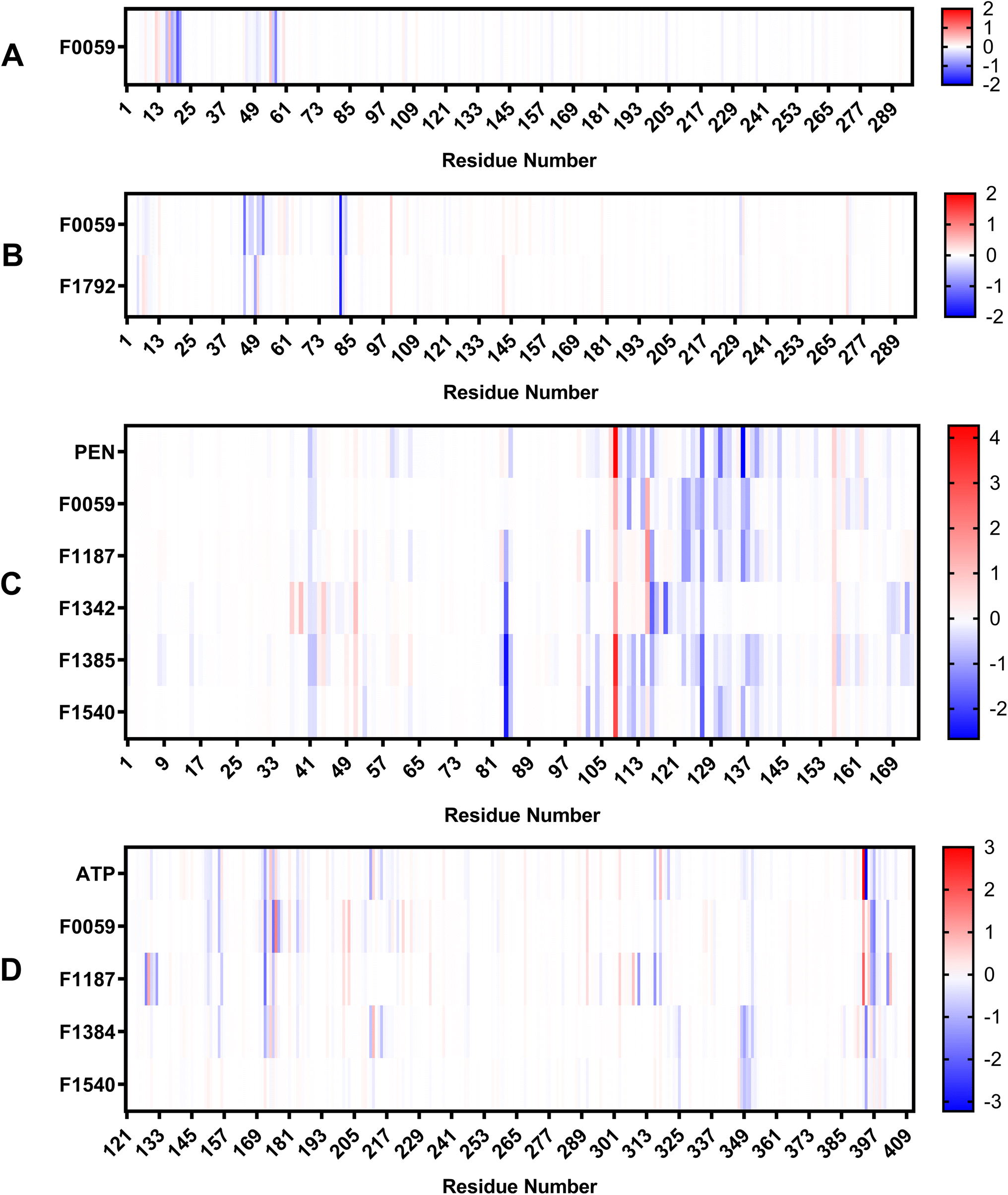
The estimated binding energies of stable complexes were determined using three open-source tools: gmx_MMPBSA, gmx_MMGBSA, and g_mmpbsa. The van der Waals and electrostatic energies were comparable for all three packages. However, because different equations and constants were employed for polar an non-polar solvation energies, there were variations in the results among the three packages.Compound F0059 registered a negative score in both the R1 (−21.81 kcal mol−1, −23.420 kcal mol−1, and −14.916 kcal mol−1) and R3 (−18.750 kcal mol−1, −18.260 kcal mol−1, and −17.064 kcal mol−1) regions using all three tools (Fig. 4). Similarly, F1792 also demonstrated a negative binding energy towards R3 (−14.470 kcal mol−1, −15.720 kcal mol−1, and −11.179 kcal mol−1), albeit considerably weaker compared to F0059. Despite the absence of control compounds for comparison, the negative binding energies displayed by the compounds suggested susceptibility to complex formation. This hinders the binding of viral DNA to the enzyme and impedes the repair process. The electrostatic contribution in the R1 region was significantly stronger than in the R3 region, indicating that the polar residues within the R1 region played a more substantial role in the ligand's affinity. The residues Cys 20, Gln 57, Ile 21, Thr 18, and Cys 16 (Fig. 5) were among the highest contributing residues in the R1 region, which is consistent with the observations of previous studies. It has been reported that mutations of Cys 20 and Cys 16 can lead to up to a 6-fold weaker DNA binding activity.33 Therefore, the strong interaction of F0059 with Cys16 and Cys 20 is an indicator of possible potency against the ASFVAP enzyme. In the R3 region, the Tyr 81 residue imparted the strongest binding energy to both F0059 and F1792. Tyr 81 plays a crucial role in the protein assembly and catalytic function of ASFVAP through the formation of an H-bond pair with the C11 base of the DNA,33 so the strong interaction of the candidate ligands with Tyr 81 weakens the residue interaction with the DNA molecule. F1792 also presented a strong affinity towards Asn 272, which plays a key role in maintaining the structural conformation of Asn 273, another crucial residue of the R3 region. Asn 272 forms extensive hydrogen-bond networks with its adjacent residues, reinforcing the structural integrity of Asn 273. An in vitro study also revealed that mutations in Asn 272 can lead to a 4.5-fold decrease in DNA binding affinity which establishes its importance in the function of ASFVAP.33 Thus, the added interaction of F1792 with Asn 272 can improve the ligand's inhibitory potential against ASFVAP. Other residues with significant energy contribution include Phe 45, Leu 82, Lys 51, Gly 48, and Asp 231.
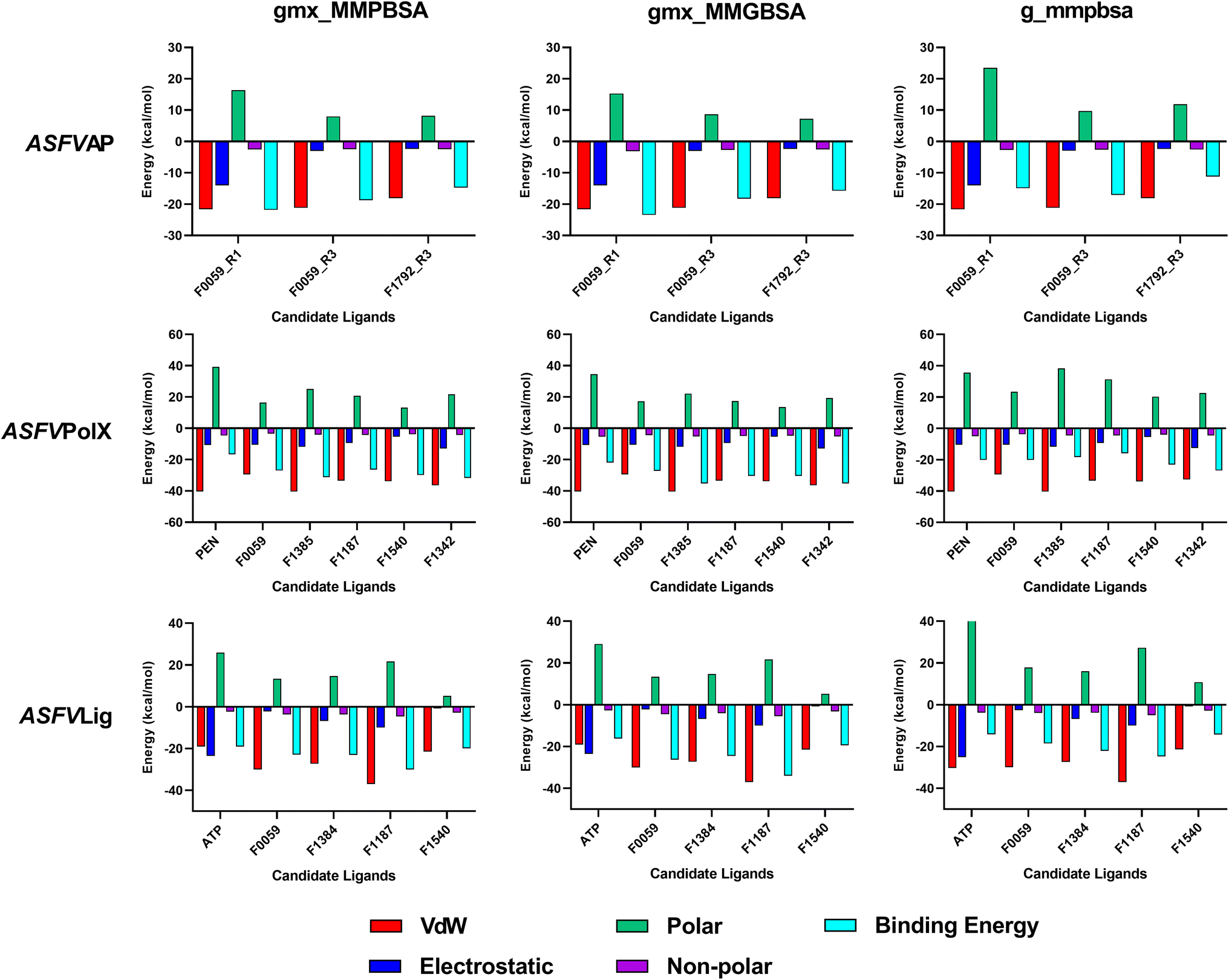 | ||
| Fig. 4 Average binding energies of the final candidate ligands estimated from three binding energy tools: gmx_MMPBSA, gmx_MMGBSA, and g_mmpbsa. All three tools share similar van der Waals and electrostatic forces but differ in their polar and non-polar solvation energies due to the utilization of distinct equations and parameters. The binding energy was calculated as the sum of van der Waals, electrostatic, polar, and non-polar solvation energies. The tabulated values of the binding energies are presented in Table S4 to S6 of the ESI.† | ||
 | ||
| Fig. 5 Binding energy contribution of the protein residues. Values are in kcal mol−1 (A) ASFVAP_R1 (B) ASFVAP_R3 (C) ASFVPolX (D) ASFVLig. Blue streaks represent attractive interactions, while red streaks represent repulsive interactions. | ||
The positive control, PEN, recorded binding energy scores of −16.52 kcal mol−1, −21.86 kcal mol−1, and −20.10 kcal mol−1 against ASFVPolX in all binding energy tools used. F1342, F1385, and F1540 ranked among the highest-scoring compounds, with the remaining ligands exhibiting comparable energies to the control. van der Waals interactions served as the primary driving force for the binding activity, effectively countering the positive polar solvation energies. The energy breakdown in Fig. 6 shows that the control compound, PEN, shares similar residues contributing to the attractive forces that stabilized the ligand. In particular, the residues Arg 127, Leu 123, Phe 116, and Arg 84 exerted the strongest binding strength within the protein's binding pocket. Arg 84 is an important residue of ASFVPolX, located in the main binding site, while Arg 127 and Leu 123 are unique structural features of the protein, distinct from other polymerases. In vitro studies have also indicated that mutations in Arg 127 may lead to a reduced catalytic activity.15 Therefore, the strong interaction of the final candidate ligands with these residues emphasizes the potential potency and improved selectivity of the five compounds as inhibitor drugs of ASFVPolX.
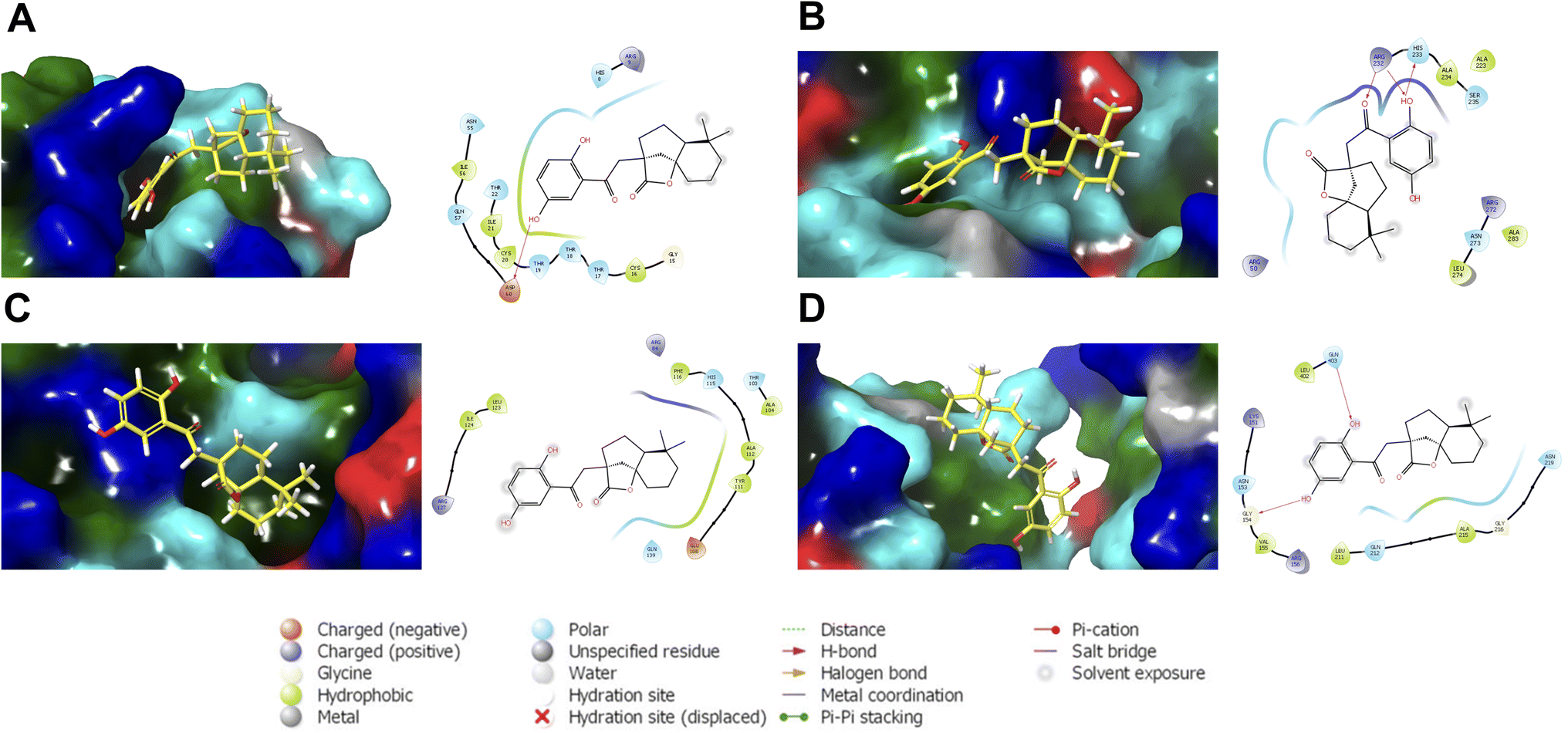 | ||
| Fig. 6 Representative structures of cochlactone A against the binding regions of (A) ASFVAP_R1, (B) ASFVAP_R3, (C) ASFVPolX, and (D) ASFVLig. Surface residues were colored based on their type: cyan for polar, green for hydrophobic, red for negatively charged, and blue for positively charged residues. Also shown are the interacting residues within 4.0 Å. The hydroxyl groups of the molecules were situated near the polar residues. Additionally, the hydroquinone of cochlactone A interacts the most, forming hydrogen bonds with adjacent amino acids. | ||
All final four candidate ligands for ASFVLig received more negative scores (−14.292 kcal mol−1 to −34.010 kcal mol−1) than the positive control ATP, which only recorded binding energy scores between −14.077 kcal mol−1 to −19.030 kcal mol−1. Compound F1187 consistently obtained the strongest MMPBSA scores for all three packages. Similar to previous protein systems, the dominant contribution to the binding energy comes from van der Waals interactions. However, for the control, ATP, electrostatic contributions were also as significant due to the highly polar nature of the compounds. The favorable interactions with the ligands primarily occurred in the residues Arg 172, Lys 175, Lys 316, Pro 396, Lys 397, and Leu 402 (Fig. 5). Leu 402 is a unique residue of the ASFVLig enzyme that has been identified in bioassays to influence the catalytic efficiency of the protein. Additionally, replacing Leu 402 with different residues resulted in decreased ligation rates by approximately 20-fold, which highlights its importance in the activity of ASFVLig.14 Although the majority of residues with high energy contributions are not recognized as crucial residues of the protein, these residues still impart significant binding affinity that helps to establish the stability of the complex.
It is also worth noting that all systems obtained positive polar solvation energies (Fig. 4), indicating that the mobile water molecules favor the separation of the ligand from the protein. Polar solvation energy accounts for the electrostatic contribution of the solvation free energy resulting from the interactions between the partial charges of the polar solute molecules and the surrounding solvent molecules.42,45 The hydrophilic and polar R-groups exposed on the surface of the enzyme interact with water molecules, resulting in a negative electrostatic energy for the unbound protein. Similarly, when unbound ligands interact with the bulk solvent through polar groups, a negative electrostatic energy is expected. As for the bound protein–ligand, since the ligand is docked inside the binding pocket, only a portion of the compound interacts freely with the solvent. In addition, the docked ligand blocked the connections of some protein residues to the bulk water phase.
Consequently, the electrostatic interaction of the complex with the continuum solvent was similar to that of the unbound protein. When calculating the difference in polar solvation energy between the bound and unbound states of the system protein. When calculating the difference in polar solvation (i.e., Gcomplex − (Gligand + Gprotein)), the overall polar solvation energy, or electrostatic contribution to the solvation energy becomes positive, which indicates a preference for complex dissociation. Thus, when a ligand has more polar groups or contains atoms with high electronegativity in its structure, it results in a more negative electrostatic interaction with water in its unbound state, but a more positive overall polar solvation energy.
Cochlactone A as a multi-target inhibitor against all BER proteins
Table 9 presents the list of fungal metabolites that have the potential to inhibit the BER pathway proteins and disrupt the repair process of ASFV. These compounds displayed structural stability and exhibited favorable binding affinities with their respective targets. Furthermore, their excellent ADMET properties have enhanced their potential for development as safe, effective, and potent antiviral drugs for ASF.| ASFVAP_R1 | ASFVAP_R3 | ASFVPolX | ASFVLig | ||||
|---|---|---|---|---|---|---|---|
| F0059 | Cochlactone A | F0059 | Cochlactone A | F0059 | Cochlactone A | F0059 | Cochlactone A |
| F1792 | Lepiotaprocerin G | F1187 | Methyl ganoderate E | F1187 | Methyl ganoderate E | ||
| F1342 | Resinacein F | F1384 | Schizine A | ||||
| F1385 | Schizine B | F1540 | Antcamphin M | ||||
| F1540 | Antcamphin M | ||||||
Four of the identified compounds, namely cochlactone A, methyl ganoderate E, resinacein F, and antcamphin M, belong to the terpenoids class of compounds, while lepiotaprocerin G, schizine B, and schizine A are classified as lactones. It is also interesting to note that two compounds, methyl ganoderate E and antcamphin M, possess the potential to inhibit two BER proteins. Both exhibited antagonistic characteristics in targeting ASFVPolX and ASFVLig. The triterpenoid cochlactone A was the only ligand that bound stably to all three BER pathway proteins.
Methyl ganoderate E is derived from Ganoderma lucidum, while antcamphin M is isolated from Antrodia cinnamomea.29 Ganoderma lucidum has a long history of traditional use in Chinese medicine, known for its antioxidant, anti-inflammatory, and immune-boosting effects.53 Conversely, Antrodia cinnamomea is also recognized for its various medicinal benefits, including hepatoprotection, anti-allergy, antihypertension, and antioxidant properties.54 To the best of our knowledge, this is the first time that methyl ganoderate E and antcamphin M are investigated as antiviral compounds. Previous studies have only reported methyl ganoderate E as a potential inhibitor for cholinisterate, making it a candidate for the treatment of Alzheimer's disease,55 while antcamphin M has only been shown to exhibit potent anti-inflammatory activity.56
Meanwhile, cochlactone A is isolated from Ganoderma cochlear, an edible medicinal fungus used in traditional medicine for the treatment of inflammation-associated disorders.57 Cochlactone A has also been specifically investigated in a study, revealing its significant anti-inflammatory property. Furthermore, its structure can serve as a scaffold for designing new antibacterial and anti-inflammatory agents.58 However, no studies have explored the antiviral effects of cochlactone A against any pathogenic diseases.
The most probable structures of cochlactone A against the BER pathway proteins are presented in Fig. 6. These structures, derived from the most populated clusters in the last ten nanoseconds of the MD trajectories, represent the most probable ligand binding conformations within the binding pocket. The hydroxyl groups of cochlactone A are situated near charged and polar residues. For the R1 region of ASFVAP, a hydroxyl group formed an H-bond pair with Asp 60. The molecule was also able to preserve its interactions with some of the crucial residues of the binding site, including His 8. In the R3 region, a hydroxyl and a carbonyl functional group are involved in hydrogen bonding with residues Arg 232 and His 233. Additionally, Asn 273, a crucial residue in the R3 region of ASFVAP,33 is also located in nearby residues. No special types of interactions were found in the representative structure of cochlactone A against ASFVPolX. However, the critical residues described in literature, such as Arg 84, His 115, Arg 127, and Leu 123,15 are within the binding domain. Finally, for ASFVLig, both hydroxyl groups of hydroquinone formed hydrogen bonds with residues Gly 154 and Gln 403. Interestingly, Gln 403 is among the crucial residues of ASFVLig;14 therefore, forming a special interaction with this residue would likely improve the potency of cochlactone A as an inhibitor drug. Aside from Gln 403, other important residues that interact with the ligand include Gln 402, Asn 153, and Leu 211. In contrast, the nonpolar segments of the molecule were in proximity to hydrophobic residues. While van der Waals interactions between nonpolar pairs are generally weak, they produce a powerful adhesion effect between the ligand and the protein when considered collectively across all atom pairs. This was evident in the binding energy results, where van der Waals interactions consistently promoted the most substantial energy for binding.
Conclusion
Various computer-aided drug screening tools were used to investigate the inhibitory potential of fungal metabolites against base excision repair pathway proteins of ASFV, including ASFVAP, ASFVPolX, and ASFVLig. Out of 1830 metabolites screened, 319 showed favorable ADMET profiles and proceeded to molecular docking. Two docking software were used to rank the binding poses, and consensus docking was employed to validate the results. The remaining 319 ligands were given docking scores, but only the top ten best-scoring ligands were considered for further evaluation. The final ten candidate ligands were then tested for structural stability using 100 ns MD simulation. RMSD, RMSF, and H-bonds diagrams elucidated the stability of the complexes while MM/PB(GB)SA energy calculations confirmed the strong binding affinities of the compounds. Through our in silico methods, we have identified the best theoretical lead compounds with antiviral potential against the BER pathway proteins: cochlactone A against the R1 region of ASFVAP; cochlactone A and lepiotaprocerin G against the R3 region of ASFVAP; cochlactone A, methyl ganoderate E, resinacein F, schizine B, and antcamphin M against ASFVPolX; and cochlactone A, methyl ganoderate E, schizine A, and antcamphin M against ASFVLig. Detailed interaction analysis and energy contribution breakdown also revealed the key residues integral to the binding activity: Cys 20, Gln 57, Ile 21, Thr 18, and Cys 16 for the R1 region of ASFVAP; Tyr 81, Phe 45, Leu 82, Lys 51, Gly 48, and Asp 231 for the R3 region of ASFVAP; Arg 127, Leu 123, Phe 116, and Arg 84 for ASFVPolX; and Arg 172, Lys 175, Lys 316, Pro 396, Lys 397, and Leu 402 for ASFVLig. H-bonds helped establish stability, particularly in compounds with polar moieties, as reflected by the stronger electrostatic interactions. However, van der Waals energies in hydrophobic interactions provided the largest energy contribution for promoting complex formation. Meanwhile, two compounds, methyl ganoderate E and antcamphin M, targeted two repair proteins, ASFVPolX and ASFVLig. Cochlactone A was the only fungal metabolite capable of inhibiting all three targets. These compounds have not been explored for their inhibitory effects against ASFV. Thus, they are recommended to be investigated in the next stage of the drug discovery process to verify their viability as multi-target drugs for ASF.Conflicts of interest
There are no conflicts of interest to report.Acknowledgements
The authors want to thank the Department of Science and Technology – Philippine Council for Agriculture, Aquatic and Natural Resources Research and Development (PCAARRD), the Department of Science and Technology - S&T Fellows Program, the Department of Science and Technology - Science Education Institute (DOST-SEI) Career Incentive Program, the Department of Science and Technology - Industrial Technology Development Institute (DOST-ITDI), and the Department of Science and Technology – Advanced Science and Technology Institute (DOST-ASTI) Computing and Archiving Research Environment (COARE) for their support to this research.References
- S. Blome, K. Franzke and M. Beer, Virus Res., 2020, 287, 198099 CrossRef CAS PubMed.
- F.L. Orosco, Vet. Integr. Sci., 2023, 21, 751–781 Search PubMed.
- S. Yang, C. Miao, W. Liu, G. Zhang, J. Shao and H. Chang, Front. Microbiol., 2023, 14, 1043129 CrossRef PubMed.
- The global economic impact of ASF, https://bulletin.woah.org/?panorama=02-2-2-2020-1-economic, accessed 7 November 2023.
- G. Wang, ASF in China: Current Status, Control Measures and Experience, https://www.woah.org/app/uploads/2023/04/12-4-gongmin-wang-slides.pdf, accessed 18 October 2023.
- C. Frezal, S. H. Gay and C. Nenert, The impact of the African swine fever outbreak in China on global agricultural markets, https://www.oecd.org/publications/the-impact-of-the-african-swine-fever-outbreak-in-china-on-global-agricultural-markets-96d0410d-en.htm, accessed 7 November 2023.
- African swine fever (ASF) – Situation Report 44, https://www.woah.org/en/document/african-swine-fever-asf-situation-report-44/, accessed 25 January 2024.
- K. Schulz, F. J. Conraths, S. Blome, C. Staubach and C. Sauter-Louis, Viruses, 2019, 11, 866 CrossRef PubMed.
- G. Wang, M. Xie, W. Wu and Z. Chen, Viruses, 2021, 13, 2124 CrossRef CAS PubMed.
- A. M. Simbulan, E. C. Banico, E. M. J. S. Sira, N. M. O. Odchimar and F. L. Orosco, Sci. Rep., 2024, 14, 1354 CrossRef CAS PubMed.
- S. Costard, B. Wieland, W. de Glanville, F. Jori, R. Rowlands, W. Vosloo, F. Roger, D. U. Pfeiffer and L. K. Dixon, Philos. Trans. R. Soc., B, 2009, 364, 2683 CrossRef PubMed.
- F. L. Orosco, Open Vet. J., 2024, 13, 1517 CrossRef PubMed.
- M. Redrejo-Rodríguez, J. M. Rodríguez, J. Salas and M. L. Salas, DNA Repair–On the Pathways to Fixing DNA Damage and Errors, ed. F. Storici, IntechOpen, London, 2011, ch. 5 Search PubMed.
- Y. Chen, H. Liu, C. Yang, Y. Gao, X. Yu, X. Chen, R. Cui, L. Zheng, S. Li, X. Li, J. Ma, Z. Huang, J. Li and J. Gan, Nat. Commun., 2019, 10, 387 CrossRef CAS PubMed.
- Y. Chen, J. Zhang, H. Liu, Y. Gao, X. Li, L. Zheng, R. Cui, Q. Yao, L. Rong and J. Li, PLoS Biol., 2017, 15, e1002599 CrossRef PubMed.
- J. Choi, D. Tark, Y.-S. Lim and S. B. Hwang, Int. J. Mol. Sci., 2021, 22, 13414 CrossRef CAS PubMed.
- M. Juszkiewicz, M. Walczak, G. Woźniakowski and A. Szczotka-Bochniarz, Pathogens, 2021, 10, 1357 CrossRef CAS PubMed.
- Q. L. Truong, L. T. Nguyen, H. Y. Babikian, R. K. Jha, H. T. Nguyen and T. L. To, Vet. World, 2021, 14, 794–802 CAS.
- S. Jo, S. Kim, D. H. Shin and M.-S. Kim, J. Enzyme Inhib. Med. Chem., 2020, 35, 1045–1049 CrossRef CAS PubMed.
- F. L. Orosco, Vet. Integr. Sci., 2024, 22, 969–991 Search PubMed.
- R. M. Chugh, P. Mittal, N. MP, T. Arora, T. Bhattacharya, H. Chopra, S. Cavalu and R. K. Gautam, Front. Pharmacol, 2022, 13, 925387 CrossRef CAS PubMed.
- T. A. K. Prescott, R. Hill, E. Mas-Claret, E. Gaya and E. Burns, Biomolecules, 2023, 13, 986 CrossRef CAS PubMed.
- A. H. Aly, A. Debbab and P. Proksch, Fungal Diversity, 2011, 50, 3–19 CrossRef.
- T. Rämä and C. A. Quandt, Front. Microbiol., 2021, 12, 706044 CrossRef PubMed.
- A. Bhambri, M. Srivastava, V. G. Mahale, S. Mahale and S. K. Karn, Front. Microbiol., 2022, 13, 837266 CrossRef PubMed.
- M. G. Roth, N. M. Westrick and T. T. Baldwin, Front. Fungal Biol., 2023, 4, 1135263 CrossRef PubMed.
- A. L. Harvey, R. Edrada-Ebel and R. J. Quinn, Nat. Rev. Drug Discovery, 2015, 14, 111–129 CrossRef CAS PubMed.
- E. H. B. Maia, L. C. Assis, T. A. de Oliveira, A. M. da Silva and A. G. Taranto, Front. Chem., 2020, 8, 481382 Search PubMed.
- R. P. Vivek-Ananth, A. K. Sahoo, K. Kumaravel, K. Mohanraj and A. Samal, RSC Adv., 2021, 11, 2596–2607 RSC.
- G. Xiong, Z. Wu, J. Yi, L. Fu, Z. Yang, C. Hsieh, M. Yin, X. Zeng, C. Wu, A. Lu, X. Chen, T. Hou and D. Cao, Nucleic Acids Res., 2021, 49, W5–W14 CrossRef CAS PubMed.
- C. A. Lipinski, Drug Discovery Today: Technol., 2004, 1, 337–341 CrossRef CAS PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- Y. Chen, X. Chen, Q. Huang, Z. Shao, Y. Gao, Y. Li, C. Yang, H. Liu, J. Li and Q. Wang, Cell Discovery, 2020, 6, 13 CrossRef CAS PubMed.
- W.-J. Wu, M.-I. Su, J.-L. Wu, S. Kumar, L. Lim, C.-W. E. Wang, F. H. T. Nelissen, M.-C. C. Chen, J. F. Doreleijers, S. S. Wijmenga and M.-D. Tsai, J. Am. Chem. Soc., 2014, 136, 4927–4937 CrossRef CAS PubMed.
- Schrödinger Release 2023-4, Maestro Schrödinger, LLC, New York, NY, 2023 Search PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CrossRef CAS PubMed.
- D. R. Houston and M. D. Walkinshaw, J. Chem. Inf. Model., 2013, 53, 384–390 CrossRef CAS PubMed.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- K. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu, S. Zhong, J. Shim, E. Darian, O. Guvench, P. Lopes, I. Vorobyov and A. D. Mackerell, J. Comput. Chem., 2010, 31, 671–690 CrossRef CAS PubMed.
- E. R. Lindahl, in Molecular Modeling of Proteins, ed. A. Kukol, Humana Press, Totowa, NJ, 2008, vol. 443, pp. 3–23 Search PubMed.
- L. Li, L. Wang and E. Alexov, Front. Mol. Biosci., 2015, 2, 5 Search PubMed.
- R. Kumari, R. Kumar, Open Source Drug Discovery Consortium and A. Lynn, J. Chem. Inf. Model., 2014, 54, 1951–1962 CrossRef CAS PubMed.
- M. S. Valdés-Tresanco, M. E. Valdés-Tresanco, P. A. Valiente and E. Moreno, J. Chem. Theory Comput., 2021, 17, 6281–6291 CrossRef PubMed.
- E. Wang, H. Sun, J. Wang, Z. Wang, H. Liu, J. Z. H. Zhang and T. Hou, Chem. Rev., 2019, 119, 9478–9508 CrossRef CAS PubMed.
- S. Genheden and U. Ryde, Expert Opin. Drug Discovery, 2015, 10, 449–461 CrossRef CAS PubMed.
- R. Izairi and H. Kamberaj, J. Chem. Inf. Model., 2017, 57, 2539–2553 CrossRef CAS PubMed.
- Q. Sciascia, G. Daş and C. C. Metges, J. Anim. Sci., 2016, 94, 441–452 CrossRef CAS.
- L. G. Ferreira, R. N. Dos Santos, G. Oliva and A. D. Andricopulo, Molecules, 2015, 20, 13384–13421 CrossRef CAS PubMed.
- A. Sethi, K. Joshi, K. Sasikala, M. Alvala, A. Sethi, K. Joshi, K. Sasikala and M. Alvala, in Drug Discovery and Development - New Advances, IntechOpen, 2019 Search PubMed.
- J. Choi, J. S. Yun, H. Song, Y.-K. Shin, Y.-H. Kang, P. R. Munashingha, J. Yoon, N. H. Kim, H. S. Kim, J. I. Yook, D. Tark, Y.-S. Lim and S. B. Hwang, Molecules, 2021, 26, 3592 CrossRef CAS PubMed.
- E. V. Anslyn and D. A. Dougherty, Modern Physical Organic Chemistry, University science books, 2006 Search PubMed.
- E. Ekiz, E. Oz, A. M. Abd El-Aty, C. Proestos, C. Brennan, M. Zeng, I. Tomasevic, T. Elobeid, K. Çadırcı, M. Bayrak and F. Oz, Foods, 2023, 12, 1512 CrossRef CAS PubMed.
- Y.-W. Liu, K.-H. Lu, C.-T. Ho and L.-Y. Sheen, J. Tradit. Complementary Med., 2012, 2, 284–294 CrossRef PubMed.
- I. Lee, B. Ahn, J. Choi, M. Hattori, B. Min and K. Bae, Bioorg. Med. Chem. Lett., 2011, 21, 6603–6607 CrossRef CAS PubMed.
- W.-H. Liu, L.-S. Shi, M.-C. Chung, T.-C. Chang and S.-Y. Lee, Am. J. Chin. Med., 2019, 47, 1611–1626 CrossRef CAS PubMed.
- F.-Y. Qin, H.-X. Zhang, Q.-Q. Di, Y. Wang, Y.-M. Yan, W.-L. Chen and Y.-X. Cheng, Org. Lett., 2020, 22, 2574–2578 CrossRef CAS PubMed.
- X. Peng, X. Wang, L. Chen, H. Yang, L. Li, S. Lu, L. Zhou and M. Qiu, Fitoterapia, 2018, 127, 286–292 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra00819g |
| This journal is © The Royal Society of Chemistry 2024 |