
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of an imidazole-based N,N-bidentate ligand for the manganese catalyzed direct coupling of nitriles with alcohols†
Qian Tangac,
Dingguo Songb,
Kali Zhangb,
Wenhao Maob,
Xianghua Zhaob,
Ding Du *a,
Fei Ling*b and
Weihui Zhong*b
*a,
Fei Ling*b and
Weihui Zhong*b
aDepartment of Chemistry, State Key Laboratory of Natural Medicines, China Pharmaceutical University, Nan Jing, 210009, P. R. China. E-mail: ddmn9999@cpu.edu.cn
bCollege of Pharmaceutical Sciences, Zhejiang University of Technology, Hangzhou 310014, P. R. China. E-mail: lingfei@zjut.edu.cn; weihuizhong@zjut.edu.cn
cZhejiang Center for Drug &Cosmetic Evaluation, Hangzhou, 310012, P. R. China
First published on 22nd April 2024
Abstract
3d-Metal catalyzed borrowing hydrogen (BH) reactions represent powerful and environmentally friendly approaches for the direct coupling of alcohols with nitriles to assemble various important branched nitriles. The development of simple and efficient ligands is a crucial issue in this field. In this study, we designed a series of readily available N,N-bidentate ligands that demonstrated good efficiency in the Mn-catalyzed BH reaction of alcohols and nitrile derivatives, yielding the targeted nitriles in moderate to good yields. Remarkably, the mildness and practicality of this protocol were further demonstrated by the successful synthesis of anipamil via a two-cascade borrowing hydrogen procedure.
Introduction
α-Alkylated nitriles are not only widely present in numerous biologically active molecules, pharmaceuticals, and light-emitting diodes but also serve as versatile building blocks for the synthesis of diols, lactones, lactams, amino alcohols, and cyclic amine derivatives.1–4 Considering the significant importance of these scaffolds, tremendous efforts have been dedicated to developing efficient methodologies for α-alkylated nitriles. Traditionally, a general approach for accessing these compounds involved the condensation of nitriles with ketones, aldehydes, or alkyl halides under strong base-promoted conditions.5,6 However, this traditional strategy faced a significant challenge in overcoming multiple side reactions, such as self-condensation of the nitriles, the aldol reaction, or the Cannizzaro reaction. It also needed to expand the functional group tolerance towards stoichiometric strong bases while avoiding the generation of copious wastes. Alternatively, the borrowing hydrogen (BH) strategy, using alcohol as an efficient alkylating agent, offers an atom-economical and environmentally benign tool for constructing new C–C bonds with water as the sole byproduct.7 A breakthrough in this field was made by Grigg and co-workers in 1981, who reported a ruthenium-catalyzed alkylation of nitriles with alcohols to deliver α-alkylated nitriles.8 Since then, several other noble metals such as Ir,9 Rh,10 Os11 and Ru12 have been successfully employed as efficient catalysts in the direct coupling of nitriles and alcohols. However, considering the toxicity, cost, and limited availability of noble metals, the development of abundant first-row-transition metal catalysts (Fe,13 Co,14 Ni,15 Mn16) has become a highly attractive strategy to reduce the use of noble metals in homogeneous catalysis.Manganese, the third most abundant transition metal in the earth's crust, has emerged as an efficient catalyst in borrowing hydrogen (BH) reactions, replacing precious metals (Scheme 1). In 2018, Maji and co-workers introduced a phosphine-free bidentate ligand derived from the condensation of thiophen-2-yl ketone and pyridine-containing hydrazine.16a They applied this ligand in the first Mn-catalyzed α-alkylation of aryl nitriles with alcohols. Soon after, the groups of Reuping and El-Sepelgy demonstrated the utility of the Mn-PNP complex for the same reaction, synthesizing a diverse range of substituted alkylated nitriles.16b Notably, the PNP ligand's instability due to the easy oxidation of P(III) to P(V) was recognized. Recently, Mukherjee's group identified a simple Mn catalyst, generated in situ from Mn(CO)5Br and 2,2′-bipyridine, for this reaction, albeit with high catalyst loading and a narrow substrate scope.17 Despite the significant advances in Mn-based catalytic systems for α-alkylation of aryl nitriles, the discovery of a cheap and efficient Mn catalyst remains a great challenge.
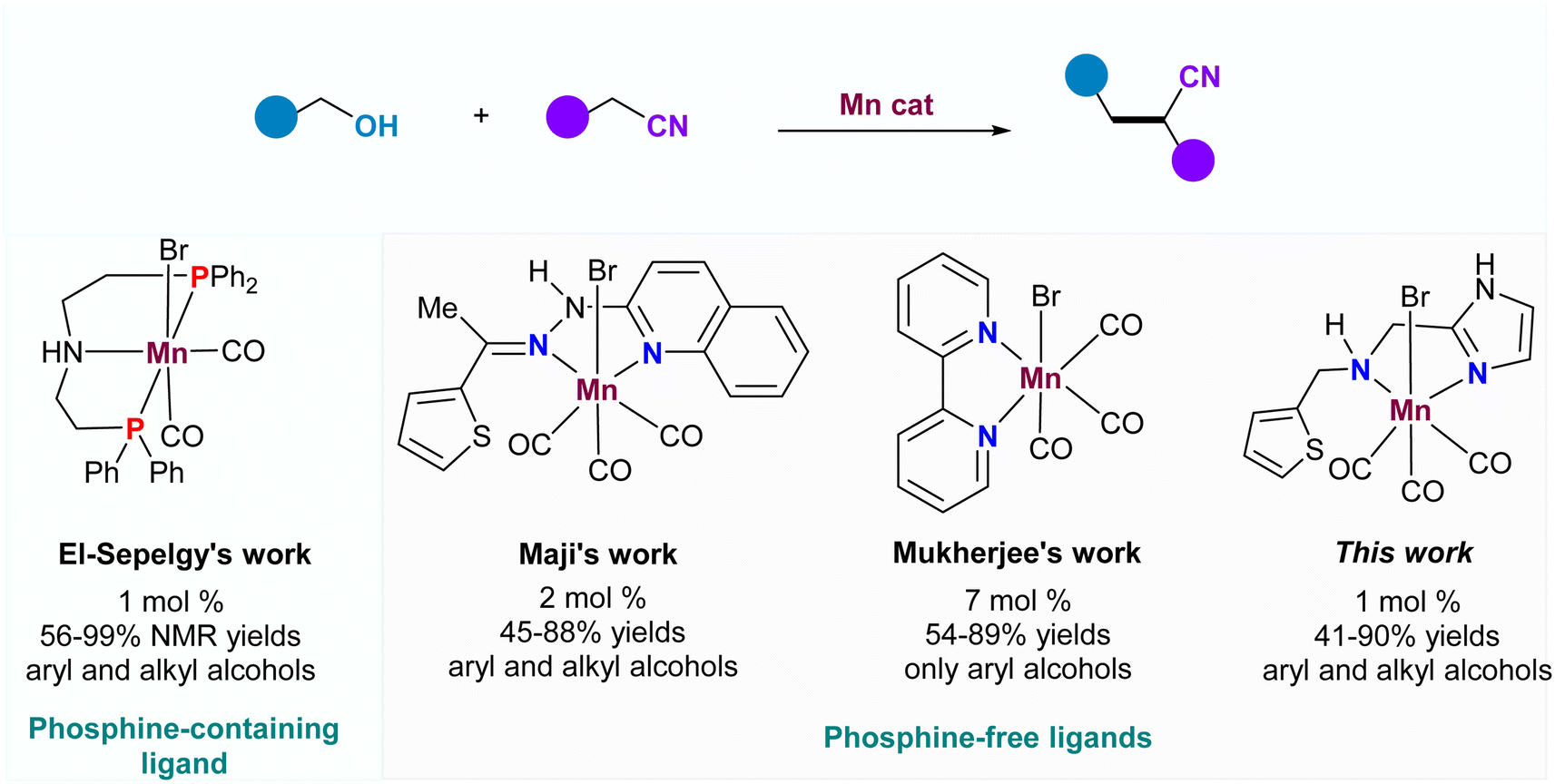 | ||
| Scheme 1 Manganese catalyzed α-alkylation of nitriles with alcohols. | ||
In our previous work, we developed several powerful imidazole-based tridentate ligands for Mn-catalyzed asymmetrical hydrogenation of various ketones, leading to highly enantioenriched alcohols.18 Recognizing the crucial role of the imidazole moiety in these ligands, we envisioned that combining the imidazole group with another nitrogen component could yield a readily available and efficient ligand for Mn-catalyzed α-alkylation of aryl nitriles with alcohols. Herein, in our continuous interest in this field,19 we report a simple imidazole-based N,N-bidentate ligand-assisted Mn-catalyzed efficient synthesis of α-alkylated nitriles through a BH process. This protocol demonstrates a broad substrate scope with good functional tolerance at low catalyst loading (26 examples, 41–90% yields) and is applicable in the total synthesis of anipamil.
Results and discussion
The currently developed imidazole-based N,N-bidentate ligands L1–L4 can be easily obtained in yields ranging from 47% to 65% by treating imidazole aldehydes or ketones with amines in MeOH under reductive conditions (Scheme 2). Subsequently, the Mn catalysts Mn-1–Mn-4 are prepared by coordinating Mn(CO)5Br with L1–L4 in toluene at reflux. With these catalysts in hand, our studies commenced by screening them for the α-alkylation of benzonitrile with benzyl alcohol (Table 1). Initially, the screening of Mn catalysts revealed that NH unprotected imidazole ligands provided better yields compared to those protected by a methyl group, and Mn-1 emerged as the optimal catalyst for this reaction (Table 1, entries 1–4). A subsequent examination of solvents showed that tAmOH yielded better results than others (Table 1, entries 5–9). Moreover, bases containing potassium resulted in higher yields than those with lithium and sodium, with KOtBu proving to be the best base, offering 3a in a 75% yield (Table 1, entries 10–13). Additionally, a slight decrease in yield was observed when reducing the catalyst loading from 2 mol% to 1 mol% (Table 1, entry 14). Fortunately, extending the reaction time to 48 h with 1 mol% of Mn-1 showed improved conversion, yielding the desired 3a in 83% (Table 1, entry 15). As expected, the reaction failed to yield α-alkylated nitrile without the presence of Mn-1 (Table 1, entry 16). Notably, the reaction could be successfully conducted on a 10 mmol scale, yielding 3a in 78% (Table 1, entry 17).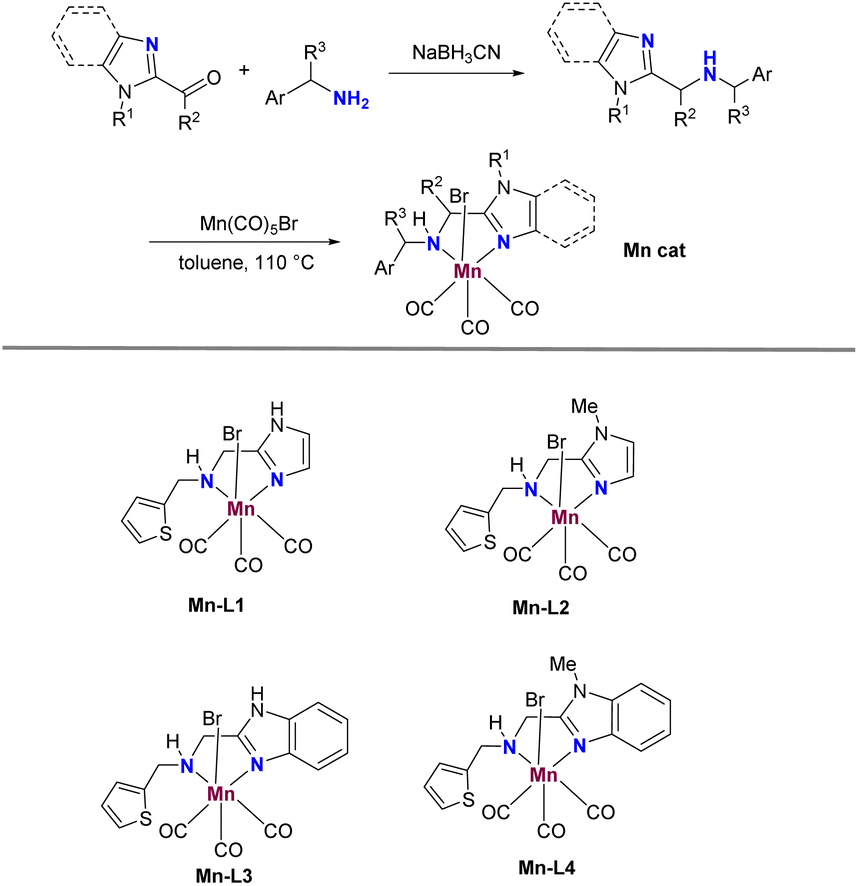 | ||
| Scheme 2 Synthetic route for the novel Mn catalyst. | ||
|
||||
|---|---|---|---|---|
| Entry | Cat. | Base | Solvent | Yieldb (%) |
| a General conditions: 1a (1.0 mmol), 2a (0.5 mmol), Mn cat. (2 mol%), tBuOK (20 mol%) were dissolved in t-AmOH (1.5 mL) and heated to 140 °C for 36 h.b Isolated yield.c 1 mol% of Mn-1 was used.d Stirred for 48 h.e 10 mmol scale. | ||||
| 1 | Mn-1 | KOtBu | tAmOH | 75 |
| 2 | Mn-2 | KOtBu | tAmOH | 35 |
| 3 | Mn-3 | KOtBu | tAmOH | 70 |
| 4 | Mn-4 | KOtBu | tAmOH | 41 |
| 5 | Mn-1 | KOtBu | Toluene | 62 |
| 6 | Mn-1 | KOtBu | Xylenes | 64 |
| 7 | Mn-1 | KOtBu | tBuOH | 63 |
| 8 | Mn-1 | KOtBu | THF | 21 |
| 9 | Mn-1 | KOtBu | 1,4-Dioxane | 43 |
| 10 | Mn-1 | tBuOLi | tAmOH | 15 |
| 11 | Mn-1 | tBuONa | tAmOH | 46 |
| 12 | Mn-1 | AcONa | tAmOH | 30 |
| 13 | Mn-1 | KOH | tAmOH | 59 |
| 14c | Mn-1 | KOtBu | tAmOH | 72 |
| 15c,d | Mn-1 | KOtBu | tAmOH | 83 |
| 16 | — | KOtBu | tAmOH | Trace |
| 17c,d,e | Mn-1 | KOtBu | tAmOH | 78 |
With the optimized reaction conditions established, our focus shifted to exploring the substrate scope for accessing α-alkylated nitriles (Scheme 3). Initial examination of aryl rings adjacent to the nitrile group revealed that electron-donating substituents on benzene rings (Me-, CH3O-, and tBu-) provided higher yields (3b, 86%; 3c, 89%; 3d, 83%) compared to the electron-withdrawing group (3f, 41%). Importantly, halide groups like chlorine were retained under the reaction conditions, yielding product 3e in an acceptable yield. The substituent position had minimal impact on the reaction outcomes; ortho-substituted aryl nitrile showed lower yield than those with meta- or para-substituents (3a, 83% vs. 3g, 76% vs. 3h, 74%). Additionally, heteroaryl-substituted nitriles were compatible, delivering products 3i, 3j, and 3k in 69–84% yields. A variety of aromatic primary alcohols were screened, showing no significant electron and steric effects and producing the targeted products 3l-3p in moderate to good yields. Fluorenyl and naphthyl groups were also tolerated, generating the desired 3q–3s in 67–74% yields. The reaction proceeded smoothly with primary alkyl alcohols, yielding 3t–3z in moderate to good yields. Unfortunately, when the primary alcohol is changed to a secondary alcohol, the hydrogenation product cannot be obtained, but the dehydrogenation product 3za can be obtained in 82% yield.
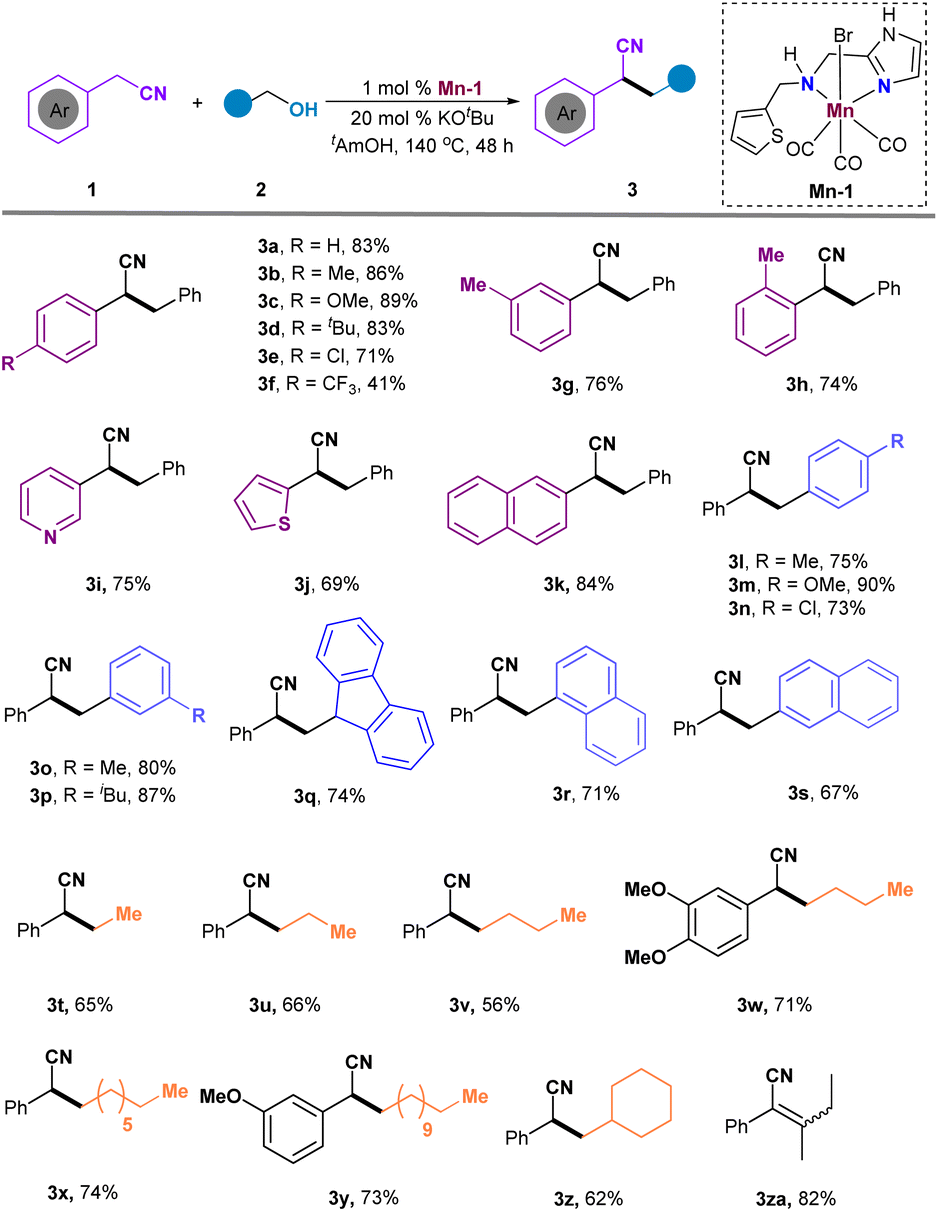 | ||
| Scheme 3 Substrate scope. | ||
To showcase the scalability and utility of this method, we applied it to the synthesis of the calcium channel blocker anipamil (Scheme 4). Initially, a 5 mmol scale of 3-methoxyphenylacetonitrile 1y underwent a Mn-1-catalyzed BH reaction with 1-dodecanol to yield secondary nitrile 3y in 72% yield after 48 h. Subsequently, the second BH coupling reaction of 3y with propylene alcohol, enabled by a Ru-MACHO catalyst, proceeded smoothly to generate tertiary nitrile 4y in 95% yield. The bromination of nitrile 4y with PBr3 led to the formation of 5y in 78% yield. Finally, the nucleophilic attack of 6 on 5y resulted in the generation of the calcium channel blocker anipamil in an 80% yield.
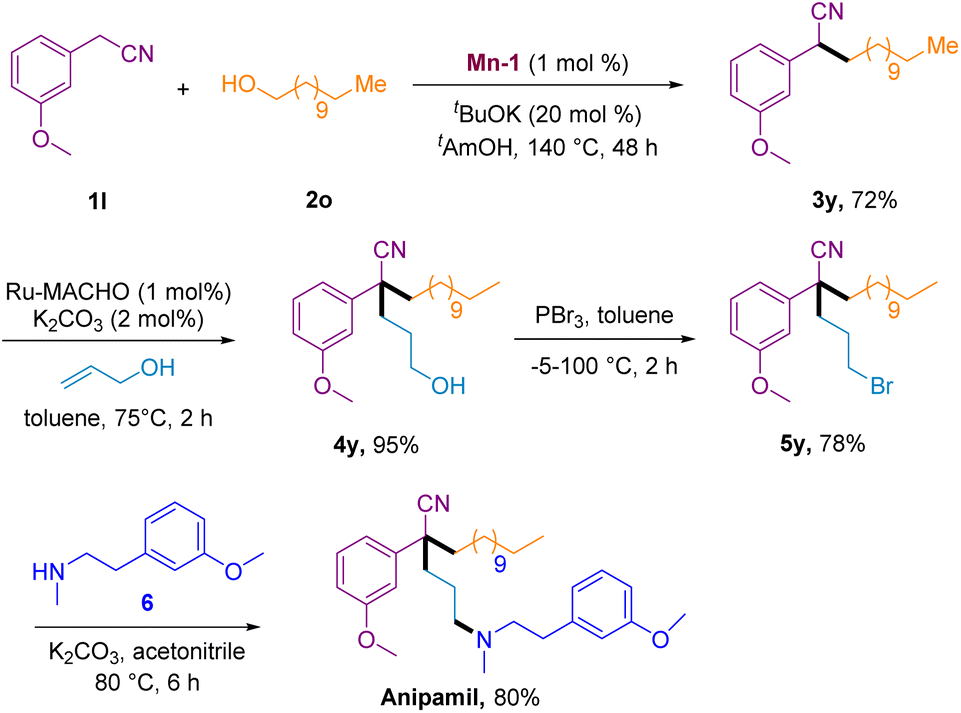 | ||
| Scheme 4 Total synthesis of anipamil. | ||
A series of control experiments were conducted to gain a preliminary understanding of the reaction mechanism, as depicted in Scheme 5. Firstly, the reaction of 1a with deuterated benzyl alcohol 2a-d2 resulted in the formation of deuterated product 3a-d in 78% yield, with a 63% deuteration at the β-position of the nitrile group. This implies that the hydride originated from the alcohol 2a-d2 (Scheme 5(1)). Subsequently, the condensation of 1a with benzaldehyde led to the formation of unsaturated aryl nitrile 8a in 90% yield. This intermediate was successfully reduced to 3a-d in 82% yield, with 91% deuteration under standard conditions using deuterated benzyl alcohol 2a-d2 as a hydrogen transfer reagent (Scheme 5(2) and (3)). These experiments suggested that the aldehyde may be the key intermediate in this reaction, further confirming that the hydrogen source comes from the alcohol.
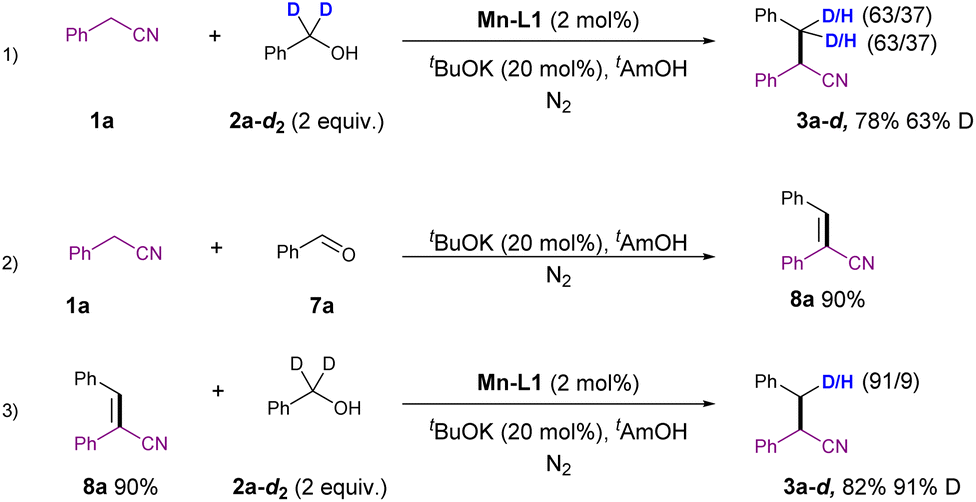 | ||
| Scheme 5 Control experiments. | ||
Based on these observations and previous literature,20 we propose a plausible mechanism illustrated in Fig. 1. Initially, an active Mn catalyst I is formed with the assistance of a base, initiating the dehydrogenative process of alcohol 2a to produce aldehyde 7a, along with Mn-D species III through a six-membered Mn complex II. Subsequently, aldehyde 7a and phenylacetonitrile 1a undergo a Knoevenagel-type condensation to yield α,β-substituted acrylonitriles 8a. Finally, the Mn-D species III facilitates the reduction of acrylonitriles 8a via a transient state IV, leading to the formation of nitrile 3a and the regeneration of active Mn catalyst I.
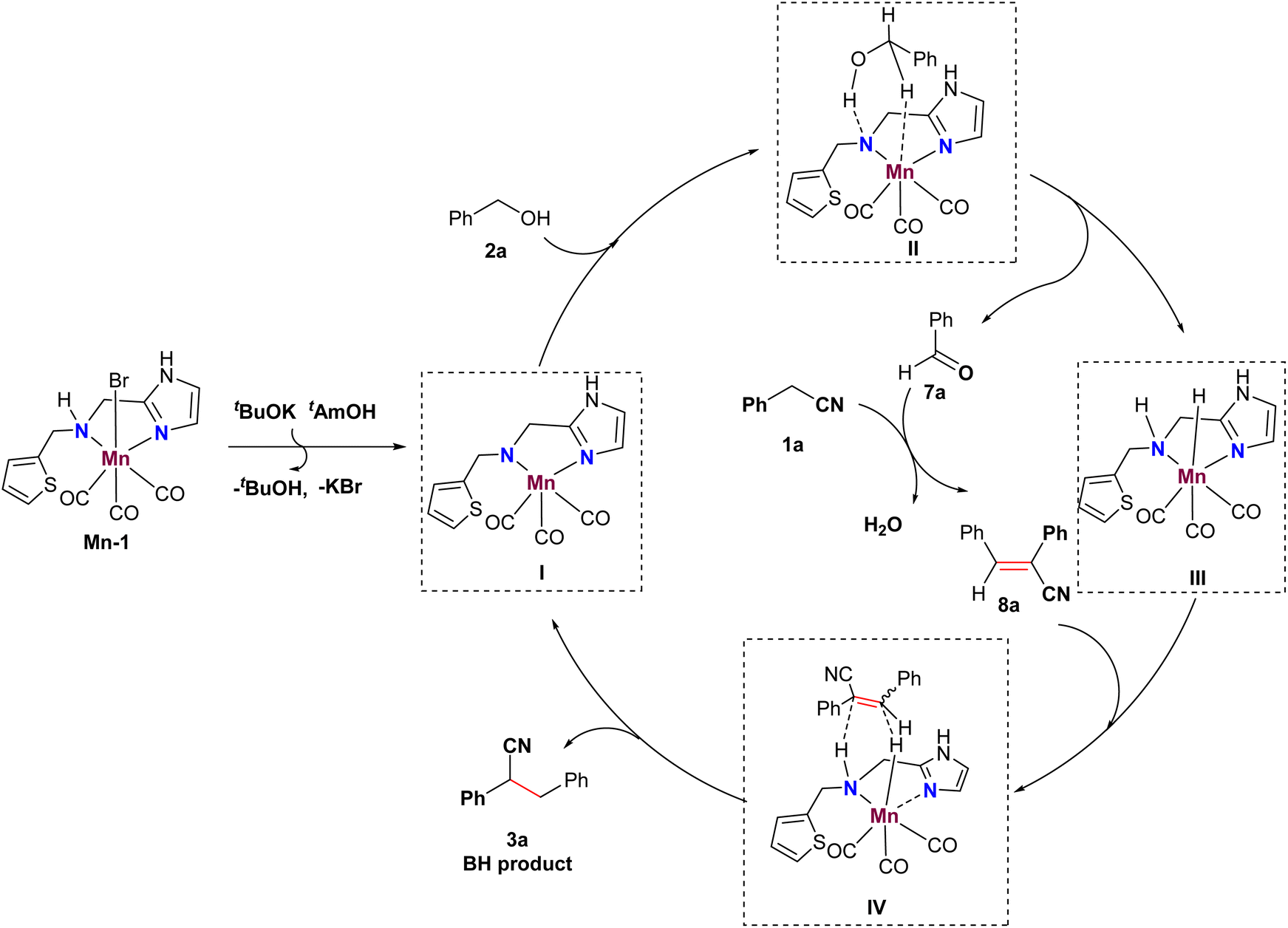 | ||
| Fig. 1 Plausible reaction mechanism. | ||
Conclusions
In conclusion, we have developed a series of novel N,N-bidentate ligands for the Mn catalyzed borrowing hydrogenation of alcohols with nitriles. This procedure featured a broad substrate scope with a good functional tolerance under simple conditions to provide the secondary nitriles in moderate to good yields (26 examples, 41–90% yields). Remarkably, the mildness and practicality of this protocol was further demonstrated by the synthesises of anipamil via two-cascade borrowing hydrogen procedure. Mechanistic studies were also conducted, which confirmed that the hydride came from the alcohol and aldehyde was the key intermediate. The present cheap metal-catalyzed mild, general strategies are expected to be of high interest to scientists in academia and industry.Experimental
General procedure for the synthesis of α-alkylated nitrile
To a mixture of Mn-1 catalyst (1 mol%), KOtBu (0.2 eq.), nitrile (1.0 mmol) and primary alcohol (0.5 mmol), 1.5 mL of tAmOH was added. Then, the reaction was stirred under Ar in a pressure tube (ACE pressure tube, 15 mL). The reaction was stirred at 140 °C for 48 hours. After cooling to room temperature, the reaction was diluted with ethyl acetate (10 mL) and water (10 mL). The organic layer was separated, and the aqueous layer was extracted with ethyl acetate (10 mL) for three times. The combined organic layers were washed by brine and dried over magnesium sulfate and the volatiles were removed under reduced pressure. The residue was purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1–10:1) to give the desired product.
:1–10:1) to give the desired product.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (No. 22078298, 22178315 and 22378363), Fundamental Research Funds for the Provincial Universities of Zhejiang (No. RF-B2023008), Natural Science Foundation of Zhejiang Province (No. LQ24B060008) and key research and development program of Zhejiang Province (No. 2023C03117) for financial support.Notes and references
- F. F. Fleming, L. Yao, P. C. Ravikumar, L. Funk and B. C Shook, J. Med. Chem., 2010, 53, 7902–7917 CrossRef CAS PubMed.
- F. F. Fleming, Nat. Prod. Rep., 1999, 16, 597–606 RSC.
- X. Bao, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2019, 58, 2139–2143 CrossRef CAS PubMed.
- A. D. Dorsey, J. E. Barbarow and D. Trauner, Org. Lett., 2003, 5, 3237–3239 CrossRef CAS PubMed.
- (a) D. F. Taber and S. J. Kong, J. Org. Chem., 1997, 62, 8575–8576 CrossRef CAS PubMed; (b) D. Savoia, E. Tagliavini, C. Trombini and A. Umani-Ronchi, J. Org. Chem., 1980, 45, 3227–3229 CrossRef CAS; (c) F. L. Cook, C. W. Bowers and C. L. Liotta, J. Org. Chem., 1974, 39, 3416–3418 CrossRef CAS; (d) S. Caron, E. Vazquez and J. M. Wojcik, J. Am. Chem. Soc., 2000, 122, 712–713 CrossRef CAS; (e) B. A. D’Sa, P. Kisanga and J. G. Verkade, J. Org. Chem., 1998, 63, 3961–3967 CrossRef; (f) R. Guillot, A. Loupy, A. Meddour, M. Pellet and A. Petit, Tetrahedron, 2005, 61, 10129–10137 CrossRef CAS.
- (a) B. A. D’Sa, P. Kisanga and J. G. Verkade, J. Org. Chem., 1998, 63, 3961–3967 CrossRef; (b) R. Guillot, A. Loupy, A. Meddour, M. Pellet and A. Petit, Tetrahedron, 2005, 61, 10129–10137 CrossRef CAS.
- (a) B. G. Reed-Berendt, D. E. Latham, M. B. Dambatta and L. C. Morrill, ACS Cent. Sci., 2021, 7, 570–585 CrossRef CAS PubMed; (b) T. Irrgang and R. Kempe, Chem. Rev., 2019, 119, 2524–2549 CrossRef CAS PubMed; (c) A. Corma, J. Navas and M. J. Sabater, Chem. Rev., 2018, 118, 1410–1459 CrossRef CAS PubMed; (d) R. H. Crabtree, Chem. Rev., 2017, 117, 9228–9246 CrossRef CAS PubMed; (e) Q. Yang, Q. Wang and Z. Yu, Chem. Soc. Rev., 2015, 44, 2305–2329 RSC; (f) G. Guillena, D. J. Ramón and M. Yus, Angew. Chem., Int. Ed., 2007, 46, 2358–2364 (Angew. Chem., 2007, 119, 2410–2424) CrossRef CAS PubMed.
- R. Grigg, T. R. B. Mitchell, S. Sutthivaiyakit and N. Tongpenyai, Tetrahedron Lett., 1981, 22, 4107–4110 CrossRef CAS.
- (a) C. Löfberg, R. Grigg, M. A. Whittaker, A. Keep and A. Derrick, J. Org. Chem., 2006, 71, 8023–8027 CrossRef PubMed; (b) B. Anxionnat, D. Gomez Pardo, G. Ricci and J. Cossy, Org. Lett., 2011, 13, 4084–4087 CrossRef CAS PubMed; (c) T. Sawaguchi and Y. Obora, Chem. Lett., 2011, 40, 1055–1057 CrossRef CAS; (d) M. Morita, Y. Obora and Y. Ishii, Chem. Commun., 2007, 2850–2852 RSC; (e) B. Anxionnat, D. Gomez Pardo, G. Ricci and J. Cossy, Eur. J. Org Chem., 2012, 4453–4456 CrossRef CAS.
- (a) F. Li, X. Zou and N. Wang, Adv. Synth. Catal., 2015, 357, 1405–1415 CrossRef CAS; (b) J. Li, Y. Liu, W. Tang, D. Xue, C. Li, J. Xiao and C. Wang, Chem.–Eur. J., 2017, 23, 14445 CrossRef CAS PubMed.
- M. L. Buil, M. A. Esteruelas, J. Herrero, S. Izquierdo, I. M. Pastor and M. Yus, ACS Catal., 2013, 3, 2072–2075 CrossRef CAS.
- (a) S. Huang, X. Hong, Y. Sun, H.-Z. Cui, Q. Zhou, Y.-J. Lin and X.-F. Hou, Appl. Organomet. Chem., 2020, 34, e5451 CrossRef CAS; (b) T. Kuwahara, T. Fukuyama and I. Ryu, Chem. Lett., 2013, 42, 1163–1165 CrossRef CAS; (c) S. Thiyagarajan and C. Gunanathan, ACS Catal., 2017, 7, 5483–5490 CrossRef CAS.
- W. Ma, S. Cui, H. Sun, W. Tang, D. Xue, C. Li, J. Fan, J. Xiao and C. Wang, Chem.–Eur. J., 2018, 24, 13118 CrossRef CAS PubMed.
- (a) P. Chakraborty, N. Garg, E. Manoury, R. Poli and B. Sundararaju, ACS Catal., 2020, 10, 8023–8031 CrossRef CAS; (b) K. Paudel, S. Xu and K. Ding, J. Org. Chem., 2020, 85, 14980–14988 CrossRef CAS PubMed; (c) A. Singh and M. Findlater, Organometallics, 2022, 41, 3145–3151 CrossRef CAS.
- (a) R. Saha, S. Panda, A. Nanda and B. Bagh, J. Org. Chem., 2023 DOI:10.1021/acs.joc.3c00859; (b) S. Bera, A. Bera and D. Banerjee, Chem. Commun., 2020, 56, 6850–6853 RSC.
- (a) A. Jana, C. B. Reddy and B. Maji, ACS Catal., 2018, 8, 9226–9231 CrossRef CAS; (b) J. C. Borghs, M. A. Tran, J. Sklyaruk, M. Rueping and O. El-Sepelgy, J. Org. Chem., 2019, 84, 7927–7935 CrossRef CAS PubMed.
- B. Krishanu and A. Mukherjee, Chem.–Asian J., 2023, 18, e202300157 CrossRef PubMed.
- (a) F. Ling, H. Hou, J. Chen, S. Nian, X. Yi, Z. Wang, D. Song and W. Zhong, Org. Lett., 2019, 21, 3937–3941 CrossRef CAS PubMed; (b) F. Ling, J. Chen, S. Nian, H. Hou, X. Yi, F. Wu, M. Xu and W. Zhong, Synlett, 2020, 31, 285–289 CrossRef CAS; (c) Z. Wang, X. Zhao, A. Huang, Z. Yang, Y. Cheng, J. Chen, F. Ling and W. Zhong, Tetrahedron Lett., 2021, 82, 153389 CrossRef CAS; (d) J. He, W. Mao, J. Lin, Y. Wu, L. Chen, P. Yang, D. Song, P. Zhu, W. Zhong and F. Ling, Org. Chem. Front., 2023, 10, 3321–3327 RSC.
- (a) D. Song, L. Chen, Y. Li, T. Liu, X. Yi, L. Liu, F. Ling and W. Zhong, Org. Chem. Front., 2021, 8, 120–126 RSC; (b) X. Yi, Y. Chen, A. Huang, D. Song, J. He, F. Ling and W. Zhong, Org. Chem. Front., 2021, 8, 6830–6836 RSC; (c) S. Wang, D. Song, F. Shen, R. Chen, Y. Cheng, C. Zhao, Q. Shen, S. Yin, F. Ling and W. Zhong, Green Chem., 2023, 25, 357–364 RSC; (d) T. Liu, W. Zhang, C. Xu, Z. Xu, D. Song, W. Qian, G. Lu, C.-J. Zhang, W. Zhong and F. Ling, Green Chem., 2023, 25, 3606–3614 RSC; (e) D. Song, S. Wang, W. Huang, R. Chen, F. Hu, L. Chen, X. Zhao, F. Ling and W. Zhong, Org. Chem. Front., 2023, 10, 5908–5915 RSC.
- (a) R. Ferrari, R. Raddino, C. Ceconi, S. Curello, S. Ghielmi and O. Visioli, Cardiovasc. Drugs Ther., 1989, 3, 403–412 CrossRef CAS PubMed; (b) D. Lenke and C. D. Mueller, Use of Anipamil, US Pat., US4777183A, 1988 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data of all the compounds. See DOI: https://doi.org/10.1039/d4ra00817k |
| This journal is © The Royal Society of Chemistry 2024 |