
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSwitch-on near infrared emission in albumin behind dark fabric: toward application in forensic latent bloodstain detection†
Matthew A. Saucier ,
Nicholas A. Kruse,
Timothy A. Lewis,
Nathan I. Hammer and
Jared H. Delcamp*
,
Nicholas A. Kruse,
Timothy A. Lewis,
Nathan I. Hammer and
Jared H. Delcamp*
Department of Chemistry and Biochemistry, University of Mississippi, University, MS 38677, USA. E-mail: delcamp@olemiss.edu
First published on 19th March 2024
Abstract
Latent bloodstain detection remains imperative for crime scene investigators. Widely used luminol offers high sensitivity to human blood, but can produce untrustworthy results from a bleach-cleaned crime scene or in a room not dark enough. Furthermore, dark pigments impede imaging bloodstains covered by dark materials with previously reported bloodstain detection agents. A novel on/off human albumin–sensing dye (SO3C7) is reported herein with a longer emission wavelength (942 nm) than previous materials that allows imaging behind ∼5 mm of black fabric. The switch-on emission of SO3C7 is selective and sensitive to human albumin and lasts longer than luminol (24–48 hours). Emission studies, transient absorption spectra (TAS), and near-infrared (NIR) photographs herein describe the albumin sensing properties of the dye.
1 Introduction
Since the late 1980s when rapid and accurate forensic analysis of deoxyribonucleic acid (DNA) via polymerase chain reaction (PCR) became available, crime lab forensic teams have relied heavily on bloodstain detection and analysis to solve cases.1 Luminol remains the most common presumptive latent blood stain detection agent.2,3 Luminol, like most other blood detection agents, uses the peroxidase-like property of hemoglobin present in red blood cells to transform the molecule into a light emitting species.2,4 While luminol has high sensitivity to blood, it has low selectivity due to promiscuity with strong oxidants like hypochlorite (bleach) or plant-based peroxidases (e.g., horseradish sauce) leading to false positives.3–5 Some latent blood stain detection agents can even damage the DNA in a blood sample, which renders detection moot.2,6,7 Additional drawbacks include requiring additional activation (usually with base and peroxide or perborate),4,8 short-lived chemiluminescence (on the order of seconds to minutes),9 instability to light and heat (requires cold and dark storage due to the innate instability of the molecule),2,10 and usually requiring a dark room for analysis all leading to false negatives.3,4Our group has previously reported an indolizine squaraine dye (SO3SQ, Scheme 1) that demonstrates “ultra-bright” switch-on near infrared (NIR, 700–1000 nm) emission upon binding to albumin in fetal bovine serum (FBS), human serum albumin (HSA), and on latent blood stains.11–13 Albumin has previously been shown to enhance dye emission for sensors and for in vivo biological imaging; however, these reports have not been found to be applicable in forensic imaging (Table S2†).9,14–25 The albumin–SO3SQ complex displays longer lived emission (on the order of days to weeks), more selectivity for blood (since albumin is specific to and ubiquitous in blood),26,27 and leaves DNA less perturbed than with oxidizing solutions or ultraviolet (UV, 100–400 nm) light.13 Upon binding to albumin, the dye most likely assumes a restricted conformation with fewer avenues for non-radiative decay and thus a brighter emission compared to water alone.12,13,28 The NIR emission of SO3SQ (λemismax = 722 nm) extends beyond the absorption of UV and visible light (400–700 nm) by molecules like hemoglobin in the blood.29 For this reason, NIR emission is preferred over the blue emission from luminol at 425 nm. Nevertheless, dark pigments absorb light ≤850 nm, which can absorb the emitted photons from both luminol and SO3SQ on or behind dark fabrics.3,30,31 Furthermore, black fabric can significantly obscure visibly detecting bloodstains with an investigator's eyes, visible-wavelength camera, or NIR camera.32,33 Therefore, a longer wavelength (>850 nm) albumin–sensing emitter is needed for optimal detection on all surfaces and background colors.
 | ||
| Scheme 1 Synthesis of SO3C7 and structure of SO3SQ. | ||
Removing the IR-blocking filter (hot mirror) from an inexpensive point-and-shoot digital camera allows the silicon photodetector to detect NIR light up to ∼1000 nm.34 An optimal imaging window from 850–1000 nm then forms between black pigment absorption and the limit of silicon photodetectors.33–36 Imaging beyond 1000 nm would require a different photodetector (often indium gallium arsenide, InGaAs), which is multiple orders of magnitude more expensive and can require cryogenic cooling (as low as −80 °C).34,37–42
2 Results and discussion
2.1 Synthetic design
Previously, sulfonate indolizine dyes (SO3C5, Fig. S9†, and SO3SQ) were rigorously studied via computational protein docking.12 In that study, the dyes were found to bind primarily to the heme cleft of HSA. A significant contributor in binding was the sulfonate indolizine moiety where both the indolizine heterocycle and sulfonate group appended to the heterocycle had distinct protein group binding. In fact, a symmetric cyanine dye (SO3C5) binds almost exclusively with one of the two indolizine donors while the cyanine chain and second indolizine remain unbound. The prior study shows turn-on fluorescence when bound to albumin with high sensitivity. However, the spectral region of the prior work precludes the ability to see through black materials such as inks, paints, or fabrics that can be used to conceal latent blood stains (Table S2†). In order to maintain albumin–sensing properties, we chose to retain the sulfonate-indolizine moiety used in the prior designs. The cyanine chain can be modified, however, so a longer π-bridge with increased conjugation was chosen to allow longer wavelength absorption and emission than previous materials. In 2021, Xu, et al.,43 reported access to a C7 spacer (2 in Scheme 1), which importantly expanded earlier work on similar compounds by demonstrating the synthetic conditions to access it.44–46 That molecule was used in this study to produce the first heptamethine indolizine-cyanine in literature (SO3C7, Scheme 1). We hypothesized this extended π-bridge dye would allow for the detection of albumin in latent blood stains through NIR light use even when albumin is covered by black materials.To prepare SO3C7, starting materials 111 and 243 were synthesized according to literature precedent. Then 1 and 2 were combined in a condensation reaction with perchloric acid in acetic anhydride and methanol followed by triethylamine to afford the final dye SO3C7. The dye was then precipitated with diethyl ether and purified via reversed-phase column chromatography using ethanol and water.
2.2 Photophysical studies
The photophysical properties of SO3C7 were studied in organic and aqueous environments. SO3C7 exhibits typical π–π* type absorption and emission peaks (Fig. 1) in organic solvents methanol (MeOH) and dimethylsulfoxide (DMSO) with λabsmax at 900 nm and 916 nm, respectively (Table 1). In an aqueous medium (saline, 0.85% NaCl in H2O), however, the absorption curve suggests aggregation due to its λabsmax shifting to higher energy at 719 nm. Emission in this environment is essentially quenched—displaying only a minimal increase compared to a blank sample (Fig. S1†). Upon introduction of HSA in the saline solution, π–π* behavior is restored and emission at 942 nm increases nearly 17-fold compared to saline. The emissive quantum yield (Φ) increases after adding HSA from 0.28% to 0.36%, which does not completely reflect the increase in emission observed since the absorption value used in quantum yield calculations is much lower for saline (absorbance at the excitation wavelength, 882 nm) thus inflating the value for saline. | ||
| Fig. 1 Normalized absorption (solid) and emission (dashed) spectra of SO3C7 in saline, HSA, MeOH, and DMSO with optimal imaging window indicated in grey from 850–1000 nm. Note: the saline emission curve is proportional to HSA since no significant emission peak was observed to normalize. | ||
| Solventa | λabsmax (nm) | λemismax (nm) | ε (M−1 cm−1) | Φ (%) |
|---|---|---|---|---|
| a Saline = 0.85% NaCl solution in H2O, HSA = human serum albumin in water, BSA = bovine serum albumin in water.b sh = shoulder. | ||||
| Saline | 719, 934 (sh)b | — | 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
0.28 |
| HSA | 923 | 942 | 16500 |
0.36 |
| BSA | 938 | 984, 950 (sh)b | 12000 |
0.24 |
| MeOH | 900 | 953 | 19500 |
0.40 |
| DMSO | 916 | 967 | 11000 |
1.30 |
Importantly, the λemismax in HSA at 942 nm falls within the aforementioned optimal imaging window for forensic imaging from 850–1000 nm (Fig. 1). The emission spectra reported in Fig. 1 were collected on an InGaAs photodetector due to its photodetectivity range capturing the entire emission spectrum of SO3C7. To support its use with inexpensive silicon photodetector-based cameras, emission was also collected using a silicon charge-coupled device (CCD) photodetector. The dye in saline remains negligibly emissive, while the dye in HSA forms a distinct emission peak easily capcieof these unique propery range of the silicon photodetector (Fig. S2†). Emission in saline remains minimal at all excitation wavelengths, including those near its λabsmax, and emission in the presence of HSA is strongest at wavelengths ≥882 nm, corresponding to its λabsmax (Fig. S3†). Human and bovine albumin were also compared along with other sources of albumin (Table 1, Fig. 3a and S4†). Dye peak emission in human albumin (HSA) is nearly triple that in bovine serum albumin (BSA), displaying enhanced specificity to human blood for forensic application.
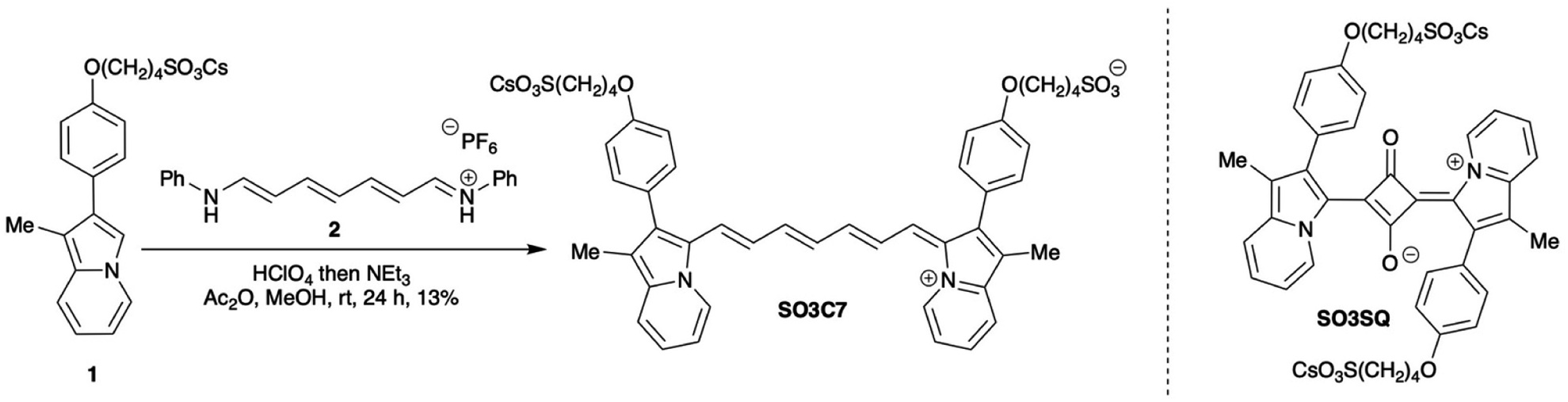
Excited state dynamics were determined for SO3C7 via transient absorption spectroscopy (TAS). A stimulated emission (SE) band at 950 nm was observed (Fig. S5a and b†), which occurs at the same wavelength as the dye fluorescence suggesting the same transition. The signal is both more intense and longer lasting (7-fold increase in lifetime, 10.68 to 68.03 ps) in HSA compared to saline, which can be attributed to restricted motion of the dye by albumin (Fig. S5c and d†). In the visible region, an excited state absorption (ESA) band can be seen for SO3C7 in HSA at 600 nm, suggesting the energy gap between excited states increases with each excited state. The lifetime of that ESA peak was determined to be 56.48 ps and no ESA was observed for SO3C7 in saline (Fig. 2c–e). The relatively long lifetimes observed for this spectral region could improve visibility in NIR photography.47–51
 | ||
| Fig. 2 (a) Emission spectra of SO3C7 in HSA kept in the dark over 96 h and (b) emission spectra of SO3C7 in saline kept in the dark for 96 h, then HSA was added at 96 h, with emission of a fresh HSA sample to compare. Excited state absorption spectrum at the maximum signal in time (c) and in wavelength (d) and three-dimensional differential absorption spectrum (e) of SO3C7 in HSA. | ||
An important aspect of applicability in field work is stability of the dye or dye–albumin complex. Two samples were prepared with SO3C7 in HSA: one was kept in a dark box and one was kept in ambient indoor lighting for 5 days and emission was collected each day. In the dark, the emission peak intensity maintains 96% of the fresh sample emission intensity over 24 hours, then by 96 hours the emission drops to 24% of the original value with a clear peak maintained (Fig. 2a). In the light, however, the dye degrades faster. After 24 hours in the light, the peak emission intensity drops to 49% of the original value, then to 21% after 48 hours and the emission peak shape begins to change (Fig. S6†). These samples were kept under ambient temperature and atmosphere, which indicates that the dye–albumin emission is maintained for at least a day and up to a week. Additionally, to test storage conditions, a sample of SO3C7 was prepared in saline and stored in the dark under ambient atmosphere and temperature for 96 hours, at which point HSA was added. The dye in saline displays minimal emission, as expected. After HSA was added at 96 hours, the emission increased to 79% of the peak emission value of a fresh HSA sample, demonstrating the capability of storing a stock solution of SO3C7 in saline for use within a week at ambient temperature and atmosphere in the dark (Fig. 2b). More concentrated stock solutions of dye were kept in DMSO at 4 °C in the dark under ambient atmosphere. Samples prepared from these stock solutions displayed similar emission intensities even after being stored for >4 months.
The effect of several biologically relevant environments on the emission intensity of SO3C7 both with and without albumin present were probed (Fig. 3a). Saline (NaCl), acidic (pH 5), basic (pH 9), and nucleophilic (reduced L-glutathione, GSH) conditions were used to see if the switch-on emission of SO3C7 with HSA is interrupted by additives that might be present in a forensic blood stain sample. Emission in all these environments without HSA remains negligible (<4% of HSA + NaCl), leading to no false-positives detected. Conversely, the samples with HSA all show substantial switch-on emission, with minimal effect in the presence of NaCl (control, 100%), pH 5 (91%), and GSH (74%). The emission spectrum changes more dramatically in pH 9, with the emission peak shifting to higher energy (Fig. S7†). The peak intensity (39%) is comparable to the emission intensity in BSA (34%).
 | ||
| Fig. 3 (a) Relative emission peak intensity of SO3C7 in the presence of several biologically relevant environments (0.85% NaCl, 0.1 M pH 5 acetate buffer, 0.1 M pH 9 carbonate buffer, 1 × 10−4 M GSH) both with and without albumin (HSA or BSA). (b) Hill plot of HSA concentration versus peak emission intensity of SO3C7 with dissociation constant (KD), binding association constant (KA), and binding free energy (ΔG). | ||
Lastly, concentration dependent emission studies were conducted with SO3C7 in order to understand the binding affinity of the dye to albumin. Normalized emission intensities fit nicely to the Hill–Langmuir equation (Fig. 3b). The dissociation constant (KD) was determined from the fit as the concentration of albumin at half the maximum emission intensity. With KD (9.42 × 10−6 M), KA can be determined by the equation: KD = 1/KA. The binding affinity (KA = 1.06 × 105 M−1) of SO3C7 to HSA is comparable to previous materials.12 The binding free energy can also be determined with the equation: ΔG = −RTln(KA), where R is the gas constant (1.987 × 10−3 kcal K−1 mol−1) and T is the temperature in Kelvin (room temperature, 298 K). The binding free energy for the SO3C7–albumin binding reaction is −6.85 kcal mol−1. These binding affinity factors display the sensitivity of SO3C7 to HSA with a KD at ∼37× more dilute protein concentration than whole human blood.
2.3 Near infrared photography
To visually display the emissive properties of SO3C7 for application in forensic imaging, NIR photographs were taken using a digital point-and-shoot camera (Kolari Vision) whose internal IR-blocking filter (hot mirror) was removed. A 900 nm longpass filter was affixed to the front of the camera lens and samples were irradiated with 850 nm light. The light source was positioned directly above the camera and photographs were taken under ambient indoor lighting. In saline, SO3C7 appears dark in the photograph, almost identical in appearance to a blank sample with only water. However, upon introduction of HSA, the vial appears bright in the photograph, demonstrating the switch-on emission seen in the emission spectra (Fig. 4a). | ||
| Fig. 4 NIR photographs of: (a) blank (water with no dye), SO3C7 in saline, and SO3C7 in HSA irradiated with 850 nm light; (b) SO3SQ in HSA versus SO3C7 in HSA bare, with 4 layers of Kimwipe, or 1 layer of black fabric wrapped around the vials. Samples were irradiated with 660 nm and 850 nm light, respectively. Vials with Kimwipe and black fabric imaged with visible light included in the box. | ||
In order to probe fabric penetration, several layers of Kimwipe (white fabric) or black fabric were wrapped around vials containing SO3C7 in HSA and SO3SQ in HSA. For photographs of SO3SQ, the camera was equipped with a 720 nm longpass filter in front of the lens and the sample was irradiated with 660 nm light. SO3SQ is very bright when the vial is bare, and even remains emissive with 4 layers (64% of the bare vial brightness) and 8 layers (41%) of Kimwipe wrapped around the vial (Fig. 4b, S10, S13 and Table S1†). Similarly, SO3C7 emission is bright when the vial is bare and can be detected through 4 layers (38% of the bare vial brightness) and 8 layers (17%) of Kimwipe. The contrast (signal-to-noise ratio, SNR) is higher in the image of bare SO3C7 (4.62 versus 3.23 for SO3SQ) most likely due to less background light being captured by the camera equipped with the longer wavelength filter. Once layers of Kimwipe are wrapped around the vial, the contrast is only slightly higher for SO3C7 compared to SO3SQ.
Black fabric, however, blocks the higher energy excitation and emission light for SO3SQ (Fig. 4b, S11, S13 and Table S1†). Just 1 layer of black fabric wrapped around the vial inhibits detection of the dye emission (10% of the bare vial brightness with 1 layer of black fabric and 7% with 2 layers). In fact, the vial of SO3SQ with 1 layer of black fabric appears identical to the blank sample and the SNR is nearly the same as the blank sample (1.00 meaning the entire vial is uniform in brightness). Impressively, taking advantage of the longer wavelength excitation and emission, SO3C7 can emit strongly behind 1 layer (56% of the bare vial brightness) and up to 4 layers (14%) of black fabric. In a direct comparison, SO3C7 is noticeably brighter than SO3SQ with 1 layer of black fabric, retaining 56% of the bare vial brightness (versus 10% for SO3SQ). Furthermore, the images of SO3C7 with black fabric retain high contrast (SNR = 2.34 with 1 layer, 1.38 with 4 layers) enabling easier detection against background noise, which is a key aspect of diagnostic forensic imaging. This experiment reflects the importance of extending both the excitation and emission wavelengths beyond the intense absorption of black pigment, allowing light to penetrate through fabric more easily.
3 Conclusions
In conclusion, a novel heptamethine indolizine-cyanine dye, SO3C7, appended with sulfonate groups to enable albumin–sensing was synthesized and shown to have a longer wavelength emission than previous materials in the literature. The increase in its emission upon sensing albumin—compared to a relatively non-emissive aqueous solution—was described using both photophysical emission spectra and NIR photography. Since longer wavelengths penetrate dark fabrics more easily, the emission from the dye–albumin complex can be observed through up to 4 layers (∼5 mm) of black fabric, while an “ultra-bright” dye in literature with higher energy emission cannot be seen through a single layer of black fabric. This imaging depth is relevant in the field of forensic imaging considering the distance that blood travels during fabric wicking.52,53 The dye–albumin complex emission lies in a unique forensic imaging window that enables inexpensive imaging with a silicon-photodetector-based camera behind dark fabric. To our knowledge, SO3C7 is unique in emitting in this forensic imaging window and displaying the switch-on/off and SNR properties described herein (Table S2†). Because of these unique properties, this work unveils a potential path toward enhanced forensic imaging with selectivity and sensitivity to human blood and the ability to penetrate deep into materials that might otherwise obscure currently used bloodstain detection methods. Furthermore, the properties of this dye–albumin complex enable imaging over longer periods of time and more convenient storage conditions compared to commonly used luminol. Importantly, the overall cost to implement this method is relatively low. The camera ($350), filters ($300), and the LED light source ($400) with accessories ($700) results in about $1750 in total imaging costs for a simple setup, which is less expensive than 1 digital single-lens reflex (DSLR) camera commonly used by forensic crime labs (up to $2500).4 Experimental details
All reagents, solvents, and starting materials were purchased from commercial vendors and were used without further purification. SO3SQ was prepared according to literature precedent.11 HSA (albumin solution human, A9080-10 ML), HS (human serum from human male AB plasma, USA origin, sterile-filtered, H4522-20 ML), and BSA (bovine serum albumin solution, A9205-10 ML) were purchased from Sigma-Aldrich (St. Louis, MO, USA). FBS (premium grade fetal bovine serum, 100% US origin, 89510–194 50 mL) was purchased from VWR Seradigm Life Science (Randor, PA, USA). Reversed-phase thin-layer silica gel chromatography (TLC) was conducted with Sorbent Technologies, Inc. (Atlanta, GA, USA) glass-backed 250 μm C18-W silica TLC plates and visualized under a UV (254 nm) lamp or ambient light. Flash column chromatography was performed on a Teledyne CombiFlash Rf+ with prepacked Teledyne RediSep Rf Gold C18 Reversed-Phase 15.5–30 g cartridges. The 1H and 13C NMR spectra were recorded on a Bruker Avance-400 (400 MHz) spectrometer, and the chemical shifts were reported in ppm using residual solvent signals as internal standards (CD3OD δ = 3.31 ppm for 1H NMR and δ = 49.00 ppm for 13C NMR). Data were reported as s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet, br = broad; coupling constants, J, were in Hz. For electrospray ionization (ESI) high-resolution mass spectrometry (HRMS), quadruple-TOF with an Orbitrap Exploris 240 to obtain the data in positive mode with a spray voltage of 3600 V, a resolution of 240000, the ion transfer tube temperature set at 300 °C, and the mass analyzer set to the 200–2000 Da range. ATR-IR was taken using a Bruker Alpha Platinum-ATR FTIR spectrometer, and spectra were processed on OPUS 6.5 software. All IR samples were taken as a solid/neat, unless otherwise noted. NIR digital photography was recorded using a Kolari Pocket Full-Spectrum Converted Infrared Photography Camera (Kolari Vision, New Jersey, USA, model C2282) equipped with a 900 nm (Thorlabs FELH0900, for SO3C7) or a 720 nm (Kolari 37mmK720-01, for SO3SQ) cut-on filter affixed in front of the lens. Photographed samples were irradiated with an 850 nm LED (Thorlabs M850LP1) with an 850 nm bandpass filter (Thorlabs FBH850-40) in front of the light source (for SO3C7) or a 660 nm LED (Aukvi from Amazon) with a visible light bandpass filter (blocks light >700 nm, Thorlabs FGB37M) in front of the light source (for SO3SQ). All photographed samples were prepared at the same concentration of dye and albumin as the emission samples. Camera settings include long shutter mode, macro focus range, 1600 ISO, and 15′′ (15 seconds) exposure time for SO3C7 or 1′′ (1 second) exposure time for SO3SQ, unless otherwise specified.Exposure time was selected to hold the brightness of the photographed vials of SO3C7 and SO3SQ near constant to allow for direct comparison of SNRs. Also, ambient lighting conditions were used to illustrate the practicality of SO3C7. However, there is significantly more ambient light (noise) at the wavelengths SO3SQ must be imaged at which requires reduced exposure time in order for reasonable image contrast. For grayscale NIR photography, the raw images were imported to a computer and opened with Microsoft PowerPoint, after which the images were cropped and the color saturation was set to zero to convert the pictures into grayscale. The dropper tool was used to determine the grayscale brightness value given in the grayscale slider page by picking a pixel in the middle of the emissive area of the vial pictured.
UV-vis-NIR absorption spectra were measured using an Avantes/AvaSpecULS2048-USB2-50 spectrometer (Pine Research part RRAVSP3) with an Avantes/AvaSpec light source (Pine Research part RRAVSP) and AvaSoft8 software program or using a Cary 5000 UV-vis-NIR spectrophotometer. All absorption, emission, and NIR photography samples were prepared with 1.5 × 10−5 M dye from a 1 × 10−3 M stock solution in DMSO. Unless otherwise noted, a concentration of 6 × 10−5 M albumin was used for albumin containing solutions. All albumin sources were diluted with 0.85% NaCl(aq.) to that albumin concentration based on average albumin content of commercial serums. FBS was used as purchased due to the albumin content being ∼4 × 10−5 M. All albumin solutions were equilibrated at 37 °C for 30 min before use.
Femtosecond transient absorption spectra and kinetics were acquired using a Helios Fire Femtosecond Transient Absorption Spectrometer from Ultrafast Systems. Excitation pulses (882 nm) were generated by routing the 800 nm (<100 fs FWHM, 1 kHz repetition rate) fundamental output from a femtosecond amplifier (Coherent Astrella) into an optical parametric oscillator (Light Conversion Topas). Probe beams were generated by taking a portion of the 800 nm fundamental and focusing them into CaF2 and YAG crystals for the visible and NIR regions, respectively. By generating the probe in this manner, the temporal characteristics of the fundamental beam are preserved in the broadband continua. Pump and probe beams are spatially overlapped in the sample, while the time delay between the actinic and probe beams is controlled by a mechanical optical delay line (0.02 ps step size). Excited state lifetimes are found via a standard single exponential decay function. Unless otherwise noted, the emission data for SO3C7 were collected using a Horiba PTI QuantaMaster QM-8075-21 fluorometer with a liquid nitrogen cooled InGaAs detector that used an 882 nm excitation source (unless otherwise specified, using a Xenon lamp and monochromator), an excitation slit width of 5 mm, and an emission slit width of 5 mm, with IR-1061 in CH2Cl2 as the reference dye (Φ = 0.32% (ref. 54)). Rectangular 10 mm path cuvettes were used for all fluorescence measurements under ambient atmosphere. For any silicon-based CCD emission spectra reported, the spectra were recorded on a Horiba LabRAM HR evolution Raman spectrometer with a 785 nm excitation laser with a hole size of 100 μm and a 600 g mm−1 grating. Unless otherwise noted, all emission spectra and intensities subtract the emission intensity value of a blank sample (solvent with no dye) at each wavelength in order to account for instrumental noise. Absorption and emission spectra reported were smoothed with the locally estimated scatterplot smoothing (LOESS) function in the 1st order at <0.1 smoothing value. The photoluminescent quantum yields (Φ) were determined using the integrated emission intensity values (summation of all Y-value data points using Microsoft Excel) by using the relative quantum yield equation below,55 where Φ denotes the quantum yield; E refers to integrated emission intensity; S is equal to 1 − 10−A, with the superscript A being the absorbance value at the excitation wavelength; η is the refractive index of the solvent; sample is the dye studied herein; and reference is the reference standard chosen for quantum yield studies.

Synthesis of SO3C7: to a flame dried 25 mL round bottom flask backfilled with nitrogen was added cesium 4-(4-(1-methylindolizin-2-yl)phenoxy)butane-1-sulfonate (1, 117 mg, 0.238 mmol), N-((1E,2E,4E,6E)-7-(phenylamino)hepta-2,4,6-trien-1-ylidene)benzenaminium hexafluorophosphate (2, 50 mg, 0.119 mmol), acetic anhydride (8.0 mL) and methanol (1.5 mL). The mixture was then sonicated and stirred for 5 min. Subsequently, concentrated perchloric acid (21 μL, 0.238 mmol) was added dropwise and the mixture was stirred at rt for 1.5 h. Then, triethylamine (21 μL, 0.286 mmol) was added dropwise and the mixture was stirred at rt for 22 h. Once the starting materials were consumed by NMR, the reaction mixture was poured into diethyl ether to precipitate the crude product, which was filtered and dissolved into 20% EtOH/H2O. The solution was passed through a 0.45 μm syringe filter, then purified via reverse-phase flash column chromatography using a gradient from 20% EtOH/H2O to 40% EtOH/H2O to elute the product. The column fractions containing pure dye were combined and the solvent was removed via flow of nitrogen at 35 °C to afford the product SO3C7 as a dark green solid (13%, 15 mg, 0.016 mmol). 1H NMR (400 MHz, CD3OD, Fig. S16†) δ 8.85 (d, J = 6.7 Hz, 2H), 7.64 (d, J = 8.6 Hz, 2H), 7.51–7.37 (m, 4H), 7.21 (d, J = 8.3 Hz, 4H), 7.08 (d, J = 8.6 Hz, 4H), 7.06–6.99 (m, 1H), 6.93 (d, J = 8.5 Hz, 1H), 6.59 (t, J = 13.1 Hz, 2H), 6.22 (t, J = 13.0 Hz, 1H), 4.12 (t, J = 5.8 Hz, 4H), 2.93 (t, J = 8.2 Hz, 4H), 2.16 (s, 3H), 2.10–1.94 (m, 12H). 13C NMR (101 MHz, CD3OD, Fig. S17†) δ 161.9, 160.8, 155.3, 143.7, 139.4, 133.7, 132.6, 130.6, 129.7, 127.6, 126.4, 123.9, 119.3, 117.8, 116.0, 68.8, 52.4, 29.4, 23.1, 9.3. Note that one aromatic peak is not observed in the carbon NMR spectrum due to limitations in concentration in methanol, even with an extended number of scans. IR (neat, cm−1) 3082, 3042, 2939, 2923, 2868, 1605. HRMS (ESI) m/z calc'd for C45H46N2O8S2 [M + H]+: 806.2701, found: 806.2729.
Author contributions
Conceptualization: JHD & MAS; investigation: MAS, NAK, & TAL; funding acquisition: JHD & NIH; writing – original draft: MAS; writing – review & editing: JHD & NAK.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge and thank the University of Mississippi School of Pharmacy GlyCORE mass spectrometry facility. Mass spectrometry research reported in this publication was supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institute of Health under award number P20GM130460. This work was funded by the National Science Foundation grant number OIA-1757220 and the National Institute of Justice grant number 15PNIJ-22-GG-04427-RESS. All authors have read and agreed to the manuscript.Notes and references
- Crime Scene and DNA Basics for Forensic Analysts Online Training Course, https://nij.ojp.gov/nij-hosted-online-training-courses/crime-scene-and-dna-basics-forensic-analysts/evidence-crime-scene/collection-techniques, accessed October 18, 2023.
- F. Barni, S. W. Lewis, A. Berti, G. M. Miskelly and G. Lago, Talanta, 2007, 72, 896–913 CrossRef CAS PubMed.
- C. R. Indalecio-Céspedes, D. Hernández-Romero, I. Legaz, M. F. Sánchez Rodríguez and E. Osuna, Forensic Chem., 2021, 26, 100368 CrossRef.
- B. A. Stoica, S. Bunescu, A. Neamtu, D. Bulgaru-Iliescu, L. Foia and E. G. Botnariu, J. Forensic Sci., 2016, 61, 1331–1336 CrossRef CAS PubMed.
- M. N. Hochmeister, B. Budowle and F. S. Baechtel, J. Forensic Sci., 1991, 36, 656–661 CrossRef CAS.
- J. P. de Almeida, N. Glesse and C. Bonorino, Forensic Sci. Int., 2011, 206, 58–61 CrossRef PubMed.
- J. Sloots, W. Lalonde, B. Reid and J. Millman, Forensic Sci. Int., 2017, 281, 141–146 CrossRef CAS PubMed.
- R. A. Zweidinger, L. T. Lytle and C. G. Pitt, J. Forensic Sci., 1973, 18, 10031J Search PubMed.
- Z. Wang, P. Zhang, H. Liu, Z. Zhao, L. Xiong, W. He, R. T. K. Kwok, J. W. Y. Lam, R. Ye and B. Z. Tang, ACS Appl. Mater. Interfaces, 2019, 11, 17306–17312 CrossRef CAS PubMed.
- R. A. W. Stott and L. J. Kricka, J. Proc. Int. Biolumin. Chemilumin. Symp., 4th, Meeting, ed. J. Schoelmerich, John Wiley & Sons Ltd, Chichester, 1987 Search PubMed.
- W. E. Meador, S. A. Autry, R. N. Bessetti, J. N. Gayton, A. S. Flynt, N. I. Hammer and J. H. Delcamp, J. Org. Chem., 2020, 85, 4089–4095 CrossRef CAS PubMed.
- W. E. Meador, K. Kapusta, I. Owolabi, S. A. Autry, J. Saloni, W. Kolodziejczyk, N. I. Hammer, A. S. Flynt, G. A. Hill and J. H. Delcamp, ChemPhotoChem, 2022, 6, e202200127 CrossRef CAS.
- J. Qu, W. Meador, P. Cheah, E. E. L. Tanner, J. Delcamp and Y. Zhao, RSC Adv., 2023, 13, 27549–27557 RSC.
- B. Li, L. Lu, M. Zhao, Z. Lei and F. Zhang, Angew. Chem., Int. Ed., 2018, 57, 7483–7487 CrossRef CAS PubMed.
- L. P. Jameson, N. W. Smith, O. Annunziata and S. V. Dzyuba, Phys. Chem. Chem. Phys., 2016, 18, 14182–14185 RSC.
- A. L. Antaris, H. Chen, S. Diao, Z. Ma, Z. Zhang, S. Zhu, J. Wang, A. X. Lozano, Q. Fan, L. Chew, M. Zhu, K. Cheng, X. Hong, H. Dai and Z. Cheng, Nat. Commun., 2017, 8, 15269 CrossRef CAS PubMed.
- Y. Fang, J. Shang, D. Liu, W. Shi, X. Li and H. Ma, J. Am. Chem. Soc., 2020, 142, 15271–15275 CrossRef CAS PubMed.
- R. Tian, Q. Zeng, S. Zhu, J. Lau, S. Chandra, R. Ertsey, K. S. Hettie, T. Teraphongphom, Z. Hu, G. Niu, D. O. Kiesewetter, H. Sun, X. Zhang, A. L. Antaris, B. R. Brooks and X. Chen, Sci. Adv., 2019, 5, eaaw0672 CrossRef CAS.
- J. A. Carr, D. Franke, J. R. Caram, C. F. Perkinson, M. Saif, V. Askoxylakis, M. Datta, D. Fukumura, R. K. Jain, M. G. Bawendi and O. T. Bruns, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 4465–4470 CrossRef CAS PubMed.
- B. Li, M. Zhao, L. Feng, C. Dou, S. Ding, G. Zhou, L. Lu, H. Zhang, F. Chen, X. Li, G. Li, S. Zhao, C. Jiang, Y. Wang, D. Zhao, Y. Cheng and F. Zhang, Nat. Commun., 2020, 11, 3102 CrossRef CAS PubMed.
- Y. Wang, M. Wang, G. Xia, Y. Yang, L. Si, H. Wang and H. Wang, Chem. Commun., 2023, 59, 3598–3601 RSC.
- X. Zhang, Y. Fu, G. Qian, R. Zhang and Z. P. Xu, J. Mater. Chem. B, 2020, 8, 5420–5424 RSC.
- K. Umezawa, D. Citterio and K. Suzuki, Anal. Sci., 2008, 24, 213–217 CrossRef CAS.
- Y. Zhang, X. Yue, B. Kim, S. Yao, M. V. Bondar and K. D. Belfield, ACS Appl. Mater. Interfaces, 2013, 5, 8710–8717 CrossRef CAS PubMed.
- N. Barbero, C. Butnarasu, S. Visentin and C. Barolo, Chem.–Asian J., 2019, 14, 896–903 CrossRef CAS PubMed.
- R. A. McPherson and M. R. Pincus, Henry's Clinical Diagnosis and Management by Laboratory Methods, Saunders, 22nd edn, 2011 Search PubMed.
- C. A. Burtis and E. R. Ashwood, Tietz Textbook of Clinical Chemistry, Third Edition, Saunders, 3rd edn, 1999 Search PubMed.
- A. S. Tatikolov and S. M. B. Costa, Biophys. Chem., 2004, 107, 33–49 CrossRef CAS.
- G. Hong, A. L. Antaris and H. Dai, Nat. Biomed. Eng., 2017, 1, 1–22 CrossRef.
- C. Aitken, Y.-T. Chang, P. Buzzini, G. Zadora and G. Massonnet, Forensic Sci. Int., 2019, 305, 110007 CrossRef CAS PubMed.
- P. M. P. García, S. L. Ibáñez-Calero and R. E. Vásquez, UPB-Investigación & Desarrollo, No. 17, 2017, vol. 1, pp. 43–53 Search PubMed.
- A. J. Hart, G. C. Barnes, F. Fuller, A. M. Cornwell, J. Gyula and N. P. Marsh, Forensic Sci. Int., 2022, 330, 111124 CrossRef.
- T. G. Schotman, A. A. Westen, J. van der Weerd and K. G. de Bruin, Forensic Sci. Int., 2015, 257, 214–219 CrossRef CAS PubMed.
- M. Vollmer, K.-P. Möllmann and J. A. Shaw, in Education and Training in Optics and Photonics: ETOP 2015, SPIE, 2015, vol. 9793, pp. 174–181 Search PubMed.
- R. L. Schuler, P. E. Kish and C. A. Plese, J. Forensic Sci., 2012, 57, 1562–1569 CrossRef PubMed.
- J. Albanese and R. Montes, J. Forensic Sci., 2011, 56, 1601–1603 CrossRef PubMed.
- The influence of temperature on image quality in SWIR cameras, https://www.alliedvision.com/en/news/detail/the-influence-of-temperature-on-image-quality-in-swir-cameras/, accessed October 24, 2023.
- Graphene for Short-wave infrared (SWIR) cameras, https://www.graphenea.com/blogs/graphene-news/graphene-for-short-wave-infrared-swir-cameras, accessed October 24, 2023.
- B. Chen, Y. Chen and Z. Deng, Photonics, 2021, 8, 14 CrossRef CAS.
- Y. Yang, Y. H. Zhang, W. Z. Shen and H. C. Liu, Prog. Quantum Electron., 2011, 35, 77–108 CrossRef.
- L. Keal, Rev. Sci. Instrum., 2021, 92, 079501 CrossRef CAS PubMed.
- S. Dong, Z. Xiong, R. Li, Y. Chen and H. Wang, Electronics, 2022, 11, 2001 CrossRef CAS.
- W. Xu, E. Leary, S. Sangtarash, M. Jirasek, M. T. González, K. E. Christensen, L. Abellán Vicente, N. Agraït, S. J. Higgins, R. J. Nichols, C. J. Lambert and H. L. Anderson, J. Am. Chem. Soc., 2021, 143, 20472–20481 CrossRef CAS.
- S. M. Makin, N. V. Monich, O. A. Shavrygina, M. I. Berezhnaya and S. A. Kheifets, Tetrahedron, 1969, 25, 4939–4948 CrossRef CAS.
- L. Dähne, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A, 1992, 216, 55–60 CrossRef.
- H. Bian, D. Ma, X. Zhang, K. Xin, Y. Yang, X. Peng and Y. Xiao, Small, 2021, 17, 2100398 CrossRef CAS.
- A. L. Dorris, J. Watson, J. J. Mosely, E. C. Lambert, G. S. Tschumper, J. H. Delcamp and N. I. Hammer, J. Phys. Chem. C, 2023, 127, 649–659 CrossRef CAS.
- N. K. Petrov, D. A. Ivanov and M. V. Alfimov, ACS Omega, 2019, 4, 11500–11507 CrossRef CAS PubMed.
- P. Singhal and H. N. Ghosh, Phys. Chem. Chem. Phys., 2014, 16, 16824–16831 RSC.
- A. Fürstenberg, M. D. Julliard, T. G. Deligeorgiev, N. I. Gadjev, A. A. Vasilev and E. Vauthey, J. Am. Chem. Soc., 2006, 128, 7661–7669 CrossRef.
- B. Dietzek, A. Yartsev and A. N. Tarnovsky, J. Phys. Chem. B, 2007, 111, 4520–4526 CrossRef CAS.
- S. Michielsen, M. Taylor, N. Parekh and F. Ji, Bloodstain Patterns on Textile Surfaces: A Fundamental Analysis, United States Department of Justice Office of Justice Programs, 2015 Search PubMed.
- R. Faflak and D. Attinger, Exp. Fluids, 2021, 62, 87 CrossRef.
- E. D. Cosco, J. R. Caram, O. T. Bruns, D. Franke, R. A. Day, E. P. Farr, M. G. Bawendi and E. M. Sletten, Angew. Chem., Int. Ed., 2017, 56, 13126–13129 CrossRef CAS.
- C. A. Parker and W. T. Rees, Analyst, 1960, 85, 587–600 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra00756e |
| This journal is © The Royal Society of Chemistry 2024 |