
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnalyzing atomic scale structural details and nuclear spin dynamics of four macrolide antibiotics: erythromycin, clarithromycin, azithromycin, and roxithromycin†
Bijay Laxmi Pradhanab,
Lekhan Lodhic,
Krishna Kishor Dey*d and
Manasi Ghosh *a
*a
aPhysics Section, Mahila Maha Vidyalaya, Banaras Hindu University, Varanasi-221005, Uttar-Pradesh, India. E-mail: manasi.ghosh@bhu.ac.in; vijaylaxmi@bhu.ac.in; manasi.ghosh@gmail.com
bDepartment of Physics, Institute of Science, Banaras Hindu University, Varanasi-221005, Uttar-Pradesh, India
cDepartment of Zoology, Dr Harisingh Gour Central University, Sagar-470003, Madhya-Pradesh, India. E-mail: lekhanlodhi5@gmail.com
dDepartment of Physics, Dr Harisingh Gour Central University, Sagar-470003, Madhya-Pradesh, India. E-mail: dey.krishna@gmail.com; kkdey@dhsgsu.edu.in
First published on 3rd June 2024
Abstract
The current investigation centers on elucidating the intricate molecular architecture and dynamic behavior of four macrolide antibiotics, specifically erythromycin, clarithromycin, azithromycin, and roxithromycin, through the application of sophisticated solid-state nuclear magnetic resonance (SSNMR) methodologies. We have measured the principal components of chemical shift anisotropy (CSA) parameters, and the site-specific spin-lattice relaxation time at carbon nuclei sites. To extract the principal components of CSA parameters, we have employed 13C 2DPASS CP-MAS SSNMR experiments at two different values of magic angle spinning (MAS) frequencies, namely 2 kHz and 600 Hz. Additionally, the spatial correlation between 13C and 1H nuclei has been investigated using 1H–13C frequency switched Lee-Goldburg heteronuclear correlation (FSLGHETCOR) experiment at a MAS frequency of 24 kHz. Our findings demonstrate that the incorporation of diverse functional groups, such as the ketone group and oxime group with the lactone ring, exerts notable influences on the structure and dynamics of the macrolide antibiotic. In particular, we have observed a significant decrease in the spin-lattice relaxation time of carbon nuclei residing on the lactone ring, desosamine, and cladinose in roxithromycin, compared to erythromycin. Overall, our findings provide detailed insight into the relationship between the structure and dynamics of macrolide antibiotics, which is eventually correlated with their biological activity. This knowledge can be utilized to develop new and more effective drugs by providing a rational basis for drug discovery and design.
1 Introduction
Macrolide antibiotics are composed of a macrocyclic lactone ring connected to two deoxysugars, cladinose and desosamine, as depicted in Fig. 1–3 (Fig. S5–S8† of ESI†). They belong to the polyketide class of natural compounds. Erythromycin, roxithromycin, clarithromycin, and azithromycin are widely utilized macrolide antibiotics for treating infections caused by both Gram-positive and Gram-negative bacteria. These antibiotics effectively target various bacteria including Streptococci, Staphylococcus aureus, Propionibacterium acnes, Listeria monocytogenes, Corynobacteria species, Legionella pneumophilia, and Haemophilus species. Compared to erythromycin, the newer macrolides (roxithromycin, clarithromycin, and azithromycin) offer improved chemical stability and better tolerability. Moreover, they exhibit a broader spectrum of activity against Mycobacterium avium complex (MAC), Haemophilus influenzae, nontuberculous mycobacteria, and Chlamydia trachomatis. Macrolides exhibit remarkable effectiveness in combating atypical respiratory pathogens like C. pneumoniae, Legionella species, and Mycoplasma species. Azithromycin and clarithromycin, due to their pharmacokinetic properties and sustained tissue levels, enable shorter dosing intervals.1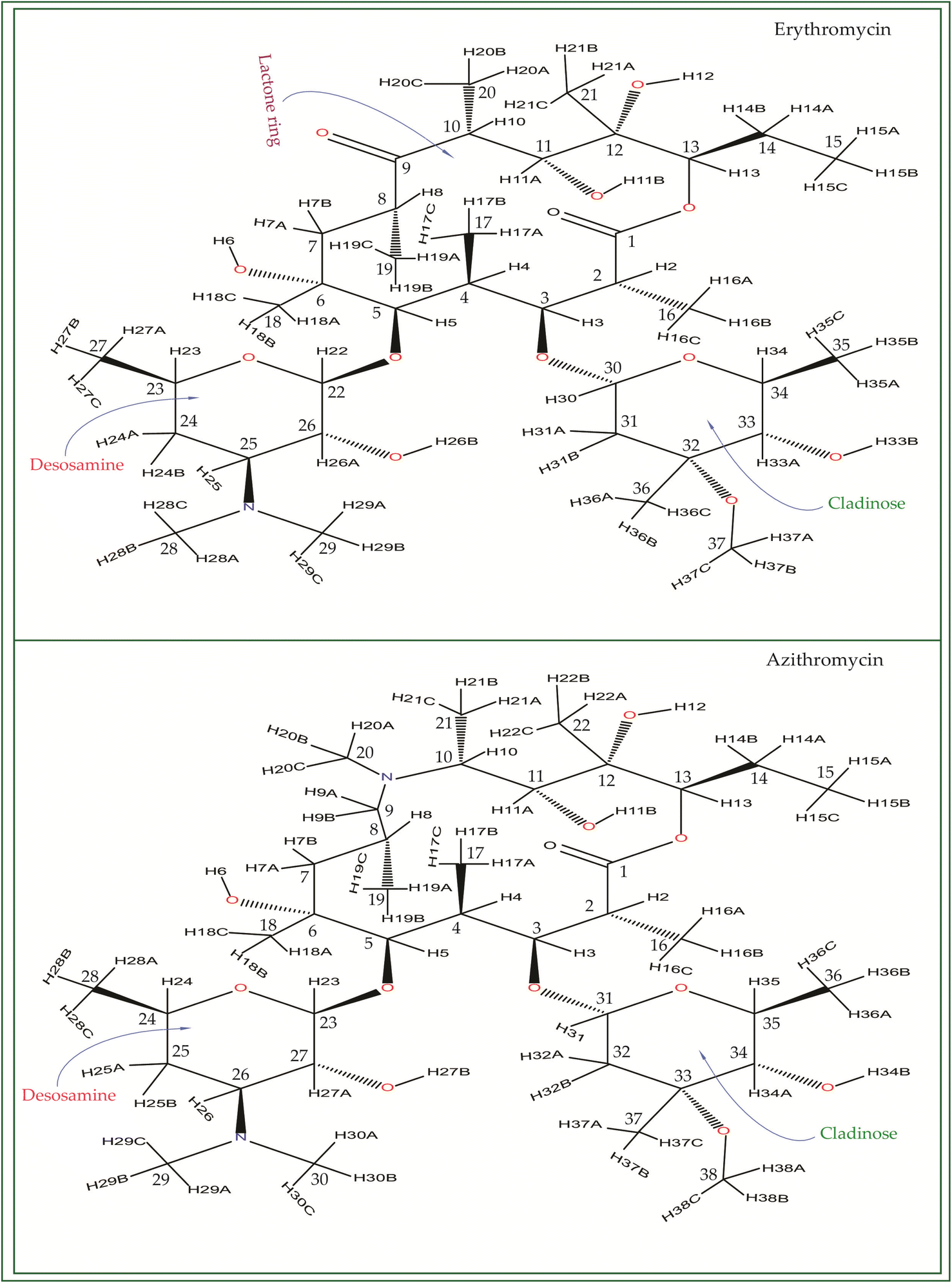 | ||
| Fig. 1 Chemical structure of erythromycin, and azithromycin. By inserting a methyl-substituted nitrogen at position C9 in the lactone ring, azithromycin forms a 15-membered macrocycle, distinguishing it structurally from erythromycin. | ||
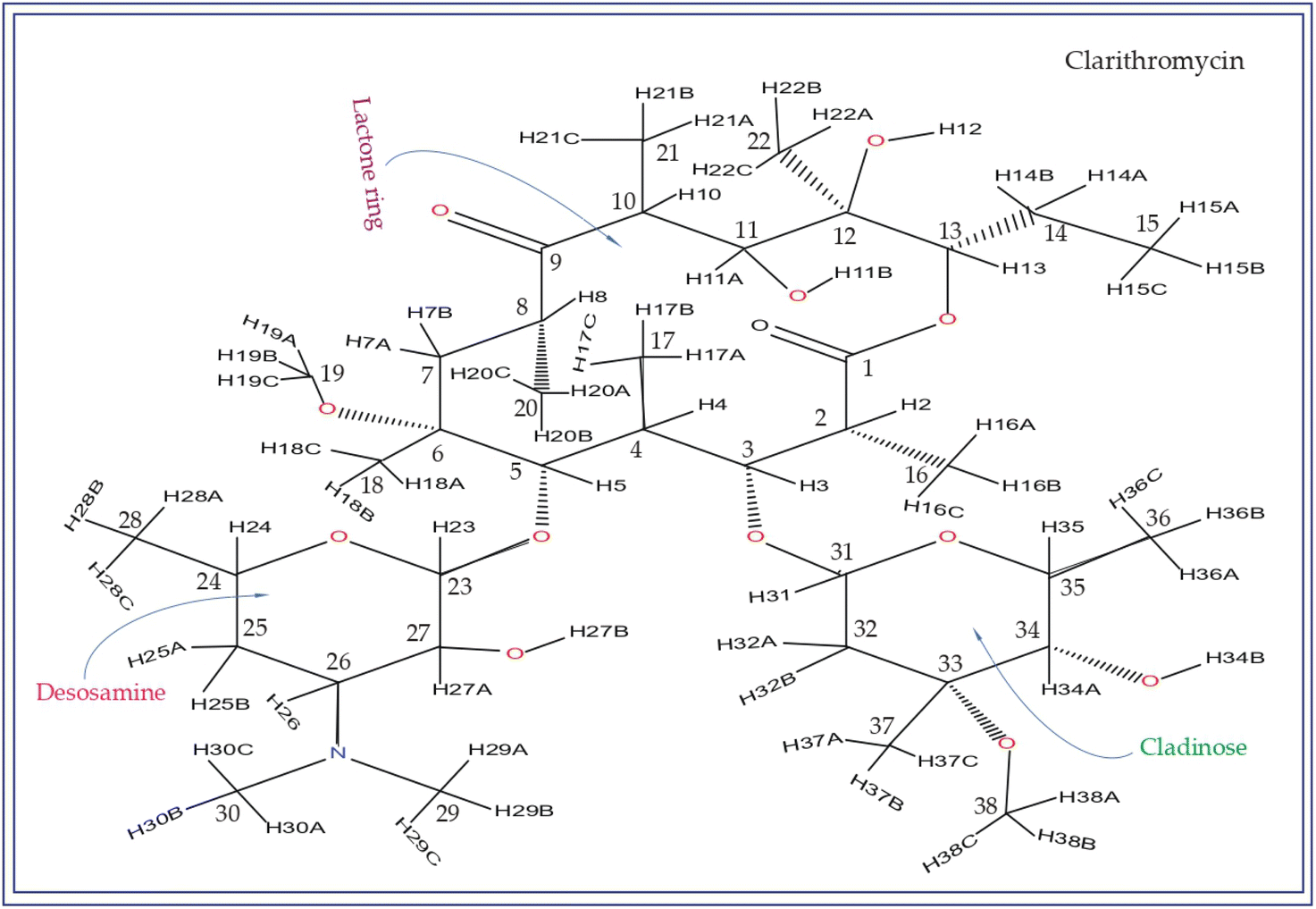 | ||
| Fig. 2 The chemical structure of clarithromycin closely resembles that of erythromycin, with the only difference being the attachment of the OCH3 group to the C6 atom of the lactone ring in clarithromycin, whereas the OH group is attached to C6 in erythromycin. | ||
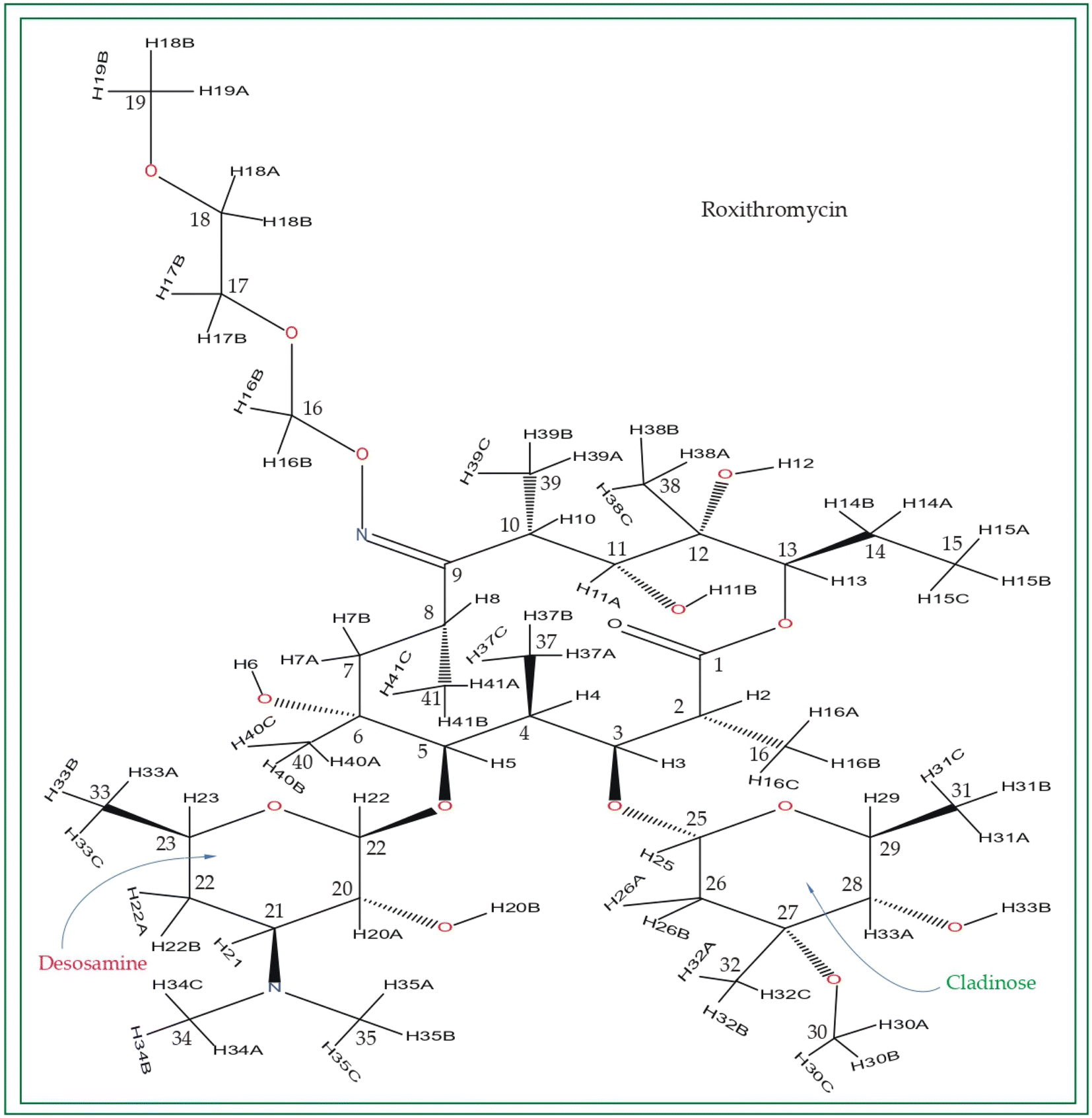 | ||
| Fig. 3 Chemical structure of roxithromycin. N-oxime side chain is attached with the lactone ring. | ||
Roxithromycin is a modified version of erythromycin, specifically designed to resist inactivation caused by acidic conditions. This modification involves converting the C9 carbonyl group into an oxime called 9-[O-(2-methoxyethoxy)methyl]oxime (28), which enhances its stability. In contrast to erythromycin, roxithromycin possesses greater lipophilicity.2–6 It remains unchanged into its parent compound in physiological conditions. Roxithromycin demonstrates similar antibacterial activity to erythromycin in vitro, effectively targeting macrolide-susceptible bacteria such as S. aureus, S. pyogenes, and S. pneumoniae. However, its effectiveness against H. influenzae is diminished due to its larger size, which hinders penetration through the outer membrane porins. Notably, roxithromycin exhibits exceptional bioavailability (72–85%), leading to high concentrations in the bloodstream and a prolonged half-life in humans.3,4 Metabolism of roxithromycin is minimal.
Azithromycin possesses enhanced tissue penetration capability and a prolonged terminal half-life due to the incorporation of a nitrogen atom, expanding the lactone ring and forming an azalide. Notably, clarithromycin demonstrates significantly higher activity against Gram-positive organisms in in vitro settings when compared to erythromycin. Both clarithromycin and azithromycin offer improved tolerability compared to erythromycin. Clarithromycin shares similarities with erythromycin in pharmacokinetic aspects such as half-life, tissue distribution, and drug interactions.7–17 Roxithromycin, a semi-synthetic macrolide antibiotic, is utilized for treating urinary tract, respiratory tract, and soft tissue infections. Similar to erythromycin, it is a derivative that incorporates an N-oxime side chain attached to the C9 carbon atom of a 14-membered lactone ring, as depicted in Fig. 3. Additionally, a keto function has been introduced to create a ketolide. In both in vitro and in vivo settings, macrolide antibiotics facilitate the emergence of macrolide-resistant strains of Streptococcus pyogenes, Streptococcus aureus, and Streptococcus pneumoniae by inhibiting the production of pneumolysin, the pneumococcal toxin.1–17 Their primary mechanism involves reversibly binding to 50S ribosomal subunits of susceptible microorganisms, inhibiting the biosynthesis of bacterial proteins and preventing the translocation of peptidyl-tRNA.
It is revealed by biochemical investigations that the binding of drugs to phosphatidylinositol-containing liposomes leads to the inhibition of lysosomal phospholipase A1 activity. The inhibition primarily targets phosphatidylcholine, a component of the bilayer, and the degree of inhibition strongly correlates with the number of cationic groups present in the drugs. Among the tested drugs, the order of increasing inhibitory activity is erythromycin < roxithromycin < azithromycin.14 In experiments conducted on cultured fibroblast cells, it was observed that phospholipidosis occurs after 3 days of exposure to these drugs. This condition affects all major phospholipids except sphingomyelin, with the following ranking in terms of severity: erythromycin, roxithromycin, and azithromycin.9 Furthermore, employing a robust and validated approach to explore drug–membrane interactions, the conformational analyses unveiled that erythromycin, roxithromycin, and azithromycin traverse the hydrophobic region of a phosphatidylinositol monolayer by means of their desosamine and cladinose moieties, while their macrocycle maintains close proximity to the interfacial region.
Erythromycin and roxithromycin are both monocationic macrolides, whereas azithromycin is dicationic. Roxithromycin, distinguished by a bulky side chain attached to C9 of the lactone ring instead of the keto group present in erythromycin, represents one of the acid-stable macrolides specifically developed for human use. In terms of the sugars attached to the macrocyclic lactone ring, all four drugs (azithromycin, erythromycin, clarithromycin, and roxithromycin) carry an amino function on one of the two sugars (desosamine). However, azithromycin possesses an additional aminofunction on its macrocyclic lactone ring as shown in Fig. 1 and S6 of the ESI.† Significantly, azithromycin has achieved worldwide recognition as a highly effective drug for numerous macrolide indications, including its use in long-term prophylaxis against intracellular infections in AIDS patients.9
The isotropic chemical shift (δiso) proves valuable in distinguishing nuclei within distinct electronic and coordination environments; however, its capacity to unveil non-bonded interactions, conformation and dynamics of molecule remains constrained. The anisotropic interactions like chemical shift anisotropy, quadrupolar and dipole–dipole interactions are primarily accessible through solid-state NMR, offering abundance information about detail molecular conformations as well as dynamics.18–32 The determination of CSA (Chemical Shift Anisotropy) parameters can be readily accomplished through the examination of powder patterns from a solid sample, wherein experiments are conducted in a static mode. However, this method faces challenges due to spectral overlap among non-equivalent sites, restricting its application to simpler spin systems. To overcome this limitation, the magic-angle spinning (MAS) experiment is commonly employed, which effectively reduces most anisotropic interactions, including CSA, thereby enhancing site resolution.
The primary objective of this study is to comprehensively investigate the detailed structure and dynamics of four significant macrolide antibiotics: erythromycin, azithromycin, clarithromycin, and roxithromycin. To achieve this, an NMR crystallographic approach is employed, utilizing advanced high-resolution solid-state NMR techniques to gather crystallographic information. Unlike X-ray crystallography, which solely provides the structure of the compound, NMR crystallography offers precise insights into the local structure and electronic environment surrounding a nucleus, along with its local dynamics. The study relies on solid-state NMR methodologies to determine crucial parameters such as isotropic chemical shift, site-specific spin-lattice relaxation time, and the principal components of the chemical shift (CS) tensor. The spin-lattice relaxation time for 13C nuclei is mainly influenced by heteronuclear dipole–dipole coupling and chemical shift anisotropy interactions. Consequently, the principal components of the CS tensor for 13C nuclei are instrumental in revealing the local structure and dynamics of each distinct carbon nucleus in the crystal. These principal components of the CS tensor are of great significance in understanding the structure and dynamics of the molecule.34–54
2 Experimental
2.1 Solid-state NMR measurements
For this investigation, the Active Pharmaceutical Ingredients (APIs) comprising erythromycin, clarithromycin, azithromycin, and roxithromycin were procured from Sigma Aldrich. The experimental procedures employed state-of-the-art Nuclear Magnetic Resonance (NMR) techniques, namely 13C Cross-Polarization Magic Angle Spinning (CP-MAS) SSNMR, and 1H–13C Frequency Switched Lee-Goldburg Heteronuclear Correlation (FSLGHETCOR),88 which were meticulously executed on a BRUKER AVANCE NEO 600 MHz NMR spectrometer featuring a 3.2 mm double-resonance MAS probe. During the course of the investigations, the MAS frequency employed for both 13C CP-MAS SSNMR and 1H–13C FSLGHETCOR experiments was set at 24 kHz. Furthermore, the 13C spin-lattice relaxation time was meticulously measured using the well-established approach described by D. C. Torchia,87 utilizing a JEOL ECX 500 MHz NMR spectrometer equipped with a 3.2 mm JEOL double-resonance MAS probe, operating at a MAS frequency of 10 kHz. It is important to note that the specific conditions pertaining to the 13C CP-MAS SSNMR experiment and the spin-lattice relaxation experiment were previously detailed in a published article, thereby upholding transparency and scientific rigor.81,82In the 1H–13C FSLGHETCOR experiment, a precise CP contact time of 200 microseconds was employed to facilitate efficient magnetization transfer between the hydrogen and carbon nuclei. During t1 increments, the proton channel utilized FSLG homo-nuclear decoupling, employing a notably short block size of 10 microseconds, which is significantly smaller than the MAS period. To guarantee the acquisition of accurate data, TPPM hetero-nuclear decoupling was judiciously employed during the acquisition process. To ensure sufficient relaxation time for the nuclei to return to their equilibrium states between scans, a relaxation delay of 15 seconds was implemented. Furthermore, the intricacies of the 13C CP-MAS 2DPASS SSNMR experiment have been extensively discussed in previously published articles.65–84
The 2D PASS CP-MAS SSNMR experiment employs a pulse sequence comprising five π pulses, with a fixed total duration and time intervals among five π pulses were determined by PASS-equations, as elucidated by Antzutkin et al.64 Initially, the 13C nucleus undergoes a precisely calibrated 90° pulse lasting 3.3 μs, followed by a relaxation delay of 15 seconds. The thirteen steps of cogwheel phase cycling COG13 (0, 1, 0, 1, 0, 1; 6) were applied, culminating 4030 scans, which is an integral multiple of 13.89,90 The coherence transfer pathway for this experiment was previously documented by Ghosh et al.65 In the indirect dimension, the acquisition of sixteen data points becomes necessary, as the number of sidebands is fewer than sixteen. The intensity of the sidebands, utilizing a graphical method, allowed the determination of the CSA tensor. The 13C 2DPASS CP-MAS SSNMR experiments were conducted at two distinct MAS frequencies: 600 Hz and 2 kHz. The precision of the MAS frequencies was upheld using the JEOL commercial MAS controller, which effectively stabilized the frequencies at (600 ± 4) Hz and (2000 ± 4) Hz, ensuring reliability and accuracy. To ascertain the principal components of the CSA parameters, sophisticated HBA (https://anorganik.uni-tuebingen.de/klaus/soft/index.php?p=hba/hba)91 and RMN (https://www.physyapps.com/rmn-intuitive-signal-processing-physical-sciences) software were employed.
Chemical shift anisotropy (CSA) interaction constitutes a valuable tool for gaining profound insights into molecular structure and dynamics.33,35,85 In order to surmount the obstacles encountered in determining CSA parameters of static powder sample, arising from low sensitivity and spectral overlap, numerous ingenious approaches have been devised, including the two-dimensional MAS/CSA NMR experiment,55 SUPER (separation of undistorted powder patterns by effortless recoupling),56 ROCSA (recoupling of chemical shift anisotropy),57 RNCSA (γ-encoded R Nνn-symmetry based chemical shift anisotropy),58 2DMAF (Two-dimensional magic angle flipping) experiment,59–61 2DMAT (two-dimensional magic angle turning) experiment,62 and the 2DPASS CP-MAS (two-dimensional phase-adjusted spinning sideband cross-polarization magic angle spinning) SSNMR experiment.63,64
While numerous recoupling techniques rely on the analysis of spinning-sideband or powder patterns for obtaining CSA parameters, 2DPASS experiments are found to be less suitable for CSA measurements in strongly coupled systems due to their limited efficacy in suppressing homonuclear dipolar interactions. However, in our specific research, our focus lies on measuring CSA parameters of 13C carbon nuclei with natural abundance (∼1.1%), rendering the homonuclear dipole–dipole interaction inconsequential to the observed spinning sideband pattern. Nonetheless, it remains crucial to decouple heteronuclear dipole–dipole interactions, particularly the 1H–13C heteronuclear dipolar coupling, which holds significant relevance for our study. To effectively achieve this, we employ the SPINAL 64 decoupling sequence, ensuring accurate and precise measurement of the CSA parameters while eliminating undesirable interferences.
Several sources of error may arise when employing the 2DPASS sequence, such as an unoptimized heteronuclear decoupling sequence, instability in the spinning frequency, and temperature fluctuations during the experiment. Careful consideration and mitigation of these factors are imperative to ensure the validity and accuracy of the obtained results. The 2DPASS experiment is utilized to ascertain the principal components of CSA parameters for various substances, including biopolymers, drug molecules,65–70 glass,83 and charge-transferred cocrystals.84
The Torchia CP (T1CP) experiment,87 pioneered by Torchia in 1978, aimed to gauge the 13C spin-lattice relaxation time under specific conditions: a magic angle spinning (MAS) frequency of 10 kHz and SPINAL-64 proton decoupling. This method, renowned for its advantages in 13C relaxation investigations, amplifies 13C nucleus signals via cross-polarization of 1H spins. Notably, it requires a relaxation delay five times that of the 1H relaxation time, considerably shorter than the 13C relaxation time, thus trimming down experimental duration compared to the inversion recovery method for solid samples. However, the efficacy of the Torchia scheme heavily hinges on the 1H–13C heteronuclear dipolar coupling constant (DCH) and proton density, given its initial preparation through 1H → 13C cross-polarization. Consequently, systems characterized by high molecular local motion, which tend to diminish DCH values, may not be apt for Torchia measurements. The referencing of 13C nuclei was conducted against the tetramethylsilane resonance peak at 0 ppm.
2.2 PXRD measurements
The Powder X-ray diffraction (PXRD) patterns for the four antibiotics were acquired using a Malvern Panalytical Empyrean diffractometer at the Department of Physics, Institute of Science, Banaras Hindu University, Varanasi, India. The entire diffraction experiment was conducted at room temperature, employing CuKα radiation (λ = 1.54 Å). The 2θ angle ranged from 5° to 80° with a step size of 0.013°. The obtained diffraction patterns underwent Rietveld refinement using the General Structure and Analysis Software (GSAS)-II, developed by Brian Toby and Robert Von Dreele at Argonne National Laboratory.982.3 DSC and TGA measurements
Thermogravimetric measurements were performed on the active pharmaceutical ingredients (API) of azithromycin, clarithromycin, erythromycin, and roxithromycin using a PerkinElmer STA 6000 thermal analyzer. The measurements took place in a platinum measuring cell under a nitrogen atmosphere, employing a heating rate of 10 °C min−1. The temperature range extended from room temperature up to 700 °C. Calorimetric assessments were carried out using a Mettler-Toledo 822E apparatus, featuring a liquid nitrogen cooling accessory and ceramic sensor. The sensor incorporated a heat flux sensor with thermocouples. Precise temperature and enthalpy calibration were achieved using indium and zinc standards, while sapphire disk calibration was applied for heat capacity. The samples underwent scanning at a rate of 10 °C min−1, covering a temperature range from 298 K to well beyond their respective transition points.3 Results and discussion
3.1 Powder XRD data analysis
Fig. 4 shows PXRD pattern of erythromycin, azithromycin, clarithromycin, and roxithromycin. The XRD pattern of erythromycin closely resembled that of erythromycin dihydrate.99 The refined parameters for erythromycin were determined as a = 9.15714 Å, b = 9.61654 Å, c = 47.07366 Å, α = 90°, β = 90°, γ = 90°, with a cell volume of 4145.306 Å3. The unit cell structure exhibited orthorhombic symmetry (P21P21P21), and the final χ2 (the ratio of the weighted profile residual and expected profile residual) value was 2.3, with a weighted R factor (wR) of 12.6%.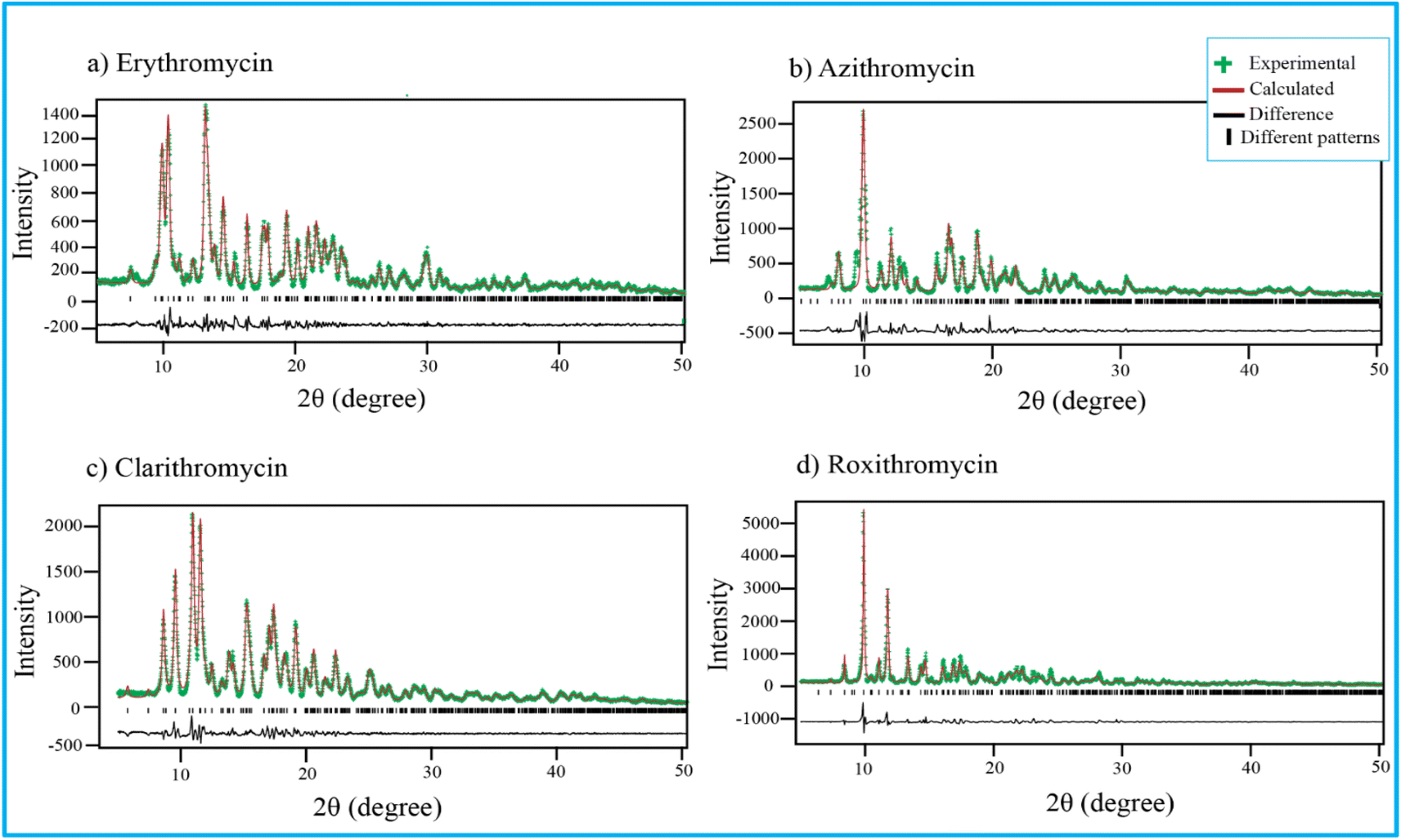 | ||
| Fig. 4 Rietveld refinement of four antibiotics: (a) erythromycin, (b) azithromycin, (c) clarithromycin, and (d) roxithromycin. | ||
Similarly, the XRD pattern of azithromycin closely matched that of azithromycin monohydrate.100 The refined cell parameters for azithromycin were a = 16.05245 Å, b = 16.1023 Å, c = 18.41222 Å, α = 90°, β = 109.151°, γ = 90°, resulting in a cell volume (V) of 4495.816 Å3. The unit cell structure was monoclinic with space group P21. The final χ2 value after Rietveld refinement was reduced to 7.6, with a wR of 23.8%.
The crystal structure of clarithromycin was found to be nearly identical to clarithromycin hemihydrate.101 The orthorhombic unit cell parameters were refined as a = 8.80922 Å, b = 20.04765 Å, c = 23.88248 Å, α = 90°, β = 90°, γ = 90°, resulting in a volume (V) of 4217.747 Å3. The final reduced χ2 value was 2.7, and wR was 13.3%.
For roxithromycin, the XRD pattern best matched that of roxithromycin semihydrate.102 The refined unit cell parameters were a = 11.69711 Å, b = 16.86219 Å, c = 16.86219 Å, α = 90°, β = 90°, γ = 90°, resulting in a volume (V) of 4762.400 Å3. The orthorhombic unit cells belonged to the P21P21P21 space group. The final reduced χ2 value was 11.0, with a wR of 28.3%.
3.2 DSC and TGA analysis of erythromycin, azithromycin, clarithromycin and roxithromycin
The TGA and DSC curves for azithromycin, clarithromycin, erythromycin, and roxithromycin are depicted in Fig. 5 and 6 respectively. These figures illustrate that azithromycin, clarithromycin, roxithromycin, and erythromycin exhibit stability up to 220 °C, 270 °C, 220 °C, and 230 °C, respectively. Beyond these temperatures, thermal decomposition initiates, concluding at 400 °C for azithromycin, 340 °C for clarithromycin, 340 °C for roxithromycin, and 340 °C for erythromycin. The findings align with the TGA analysis of azithromycin, clarithromycin, and roxithromycin conducted by K. Adrjanowicz and colleagues.103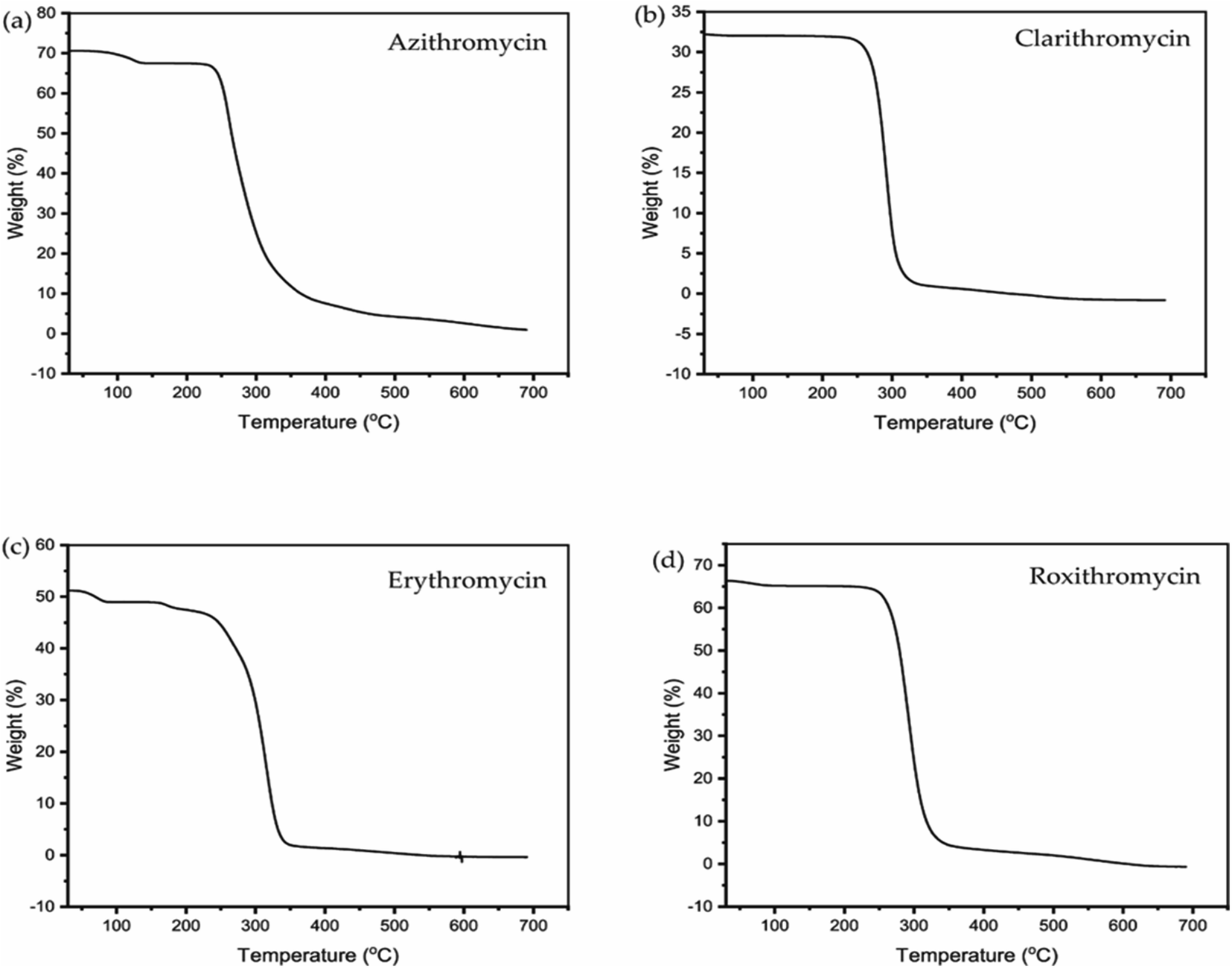 | ||
| Fig. 5 TGA curve of (a) azithromycin, (b) clarithromycin, (c) erythromycin, (d) roxithromycin. | ||
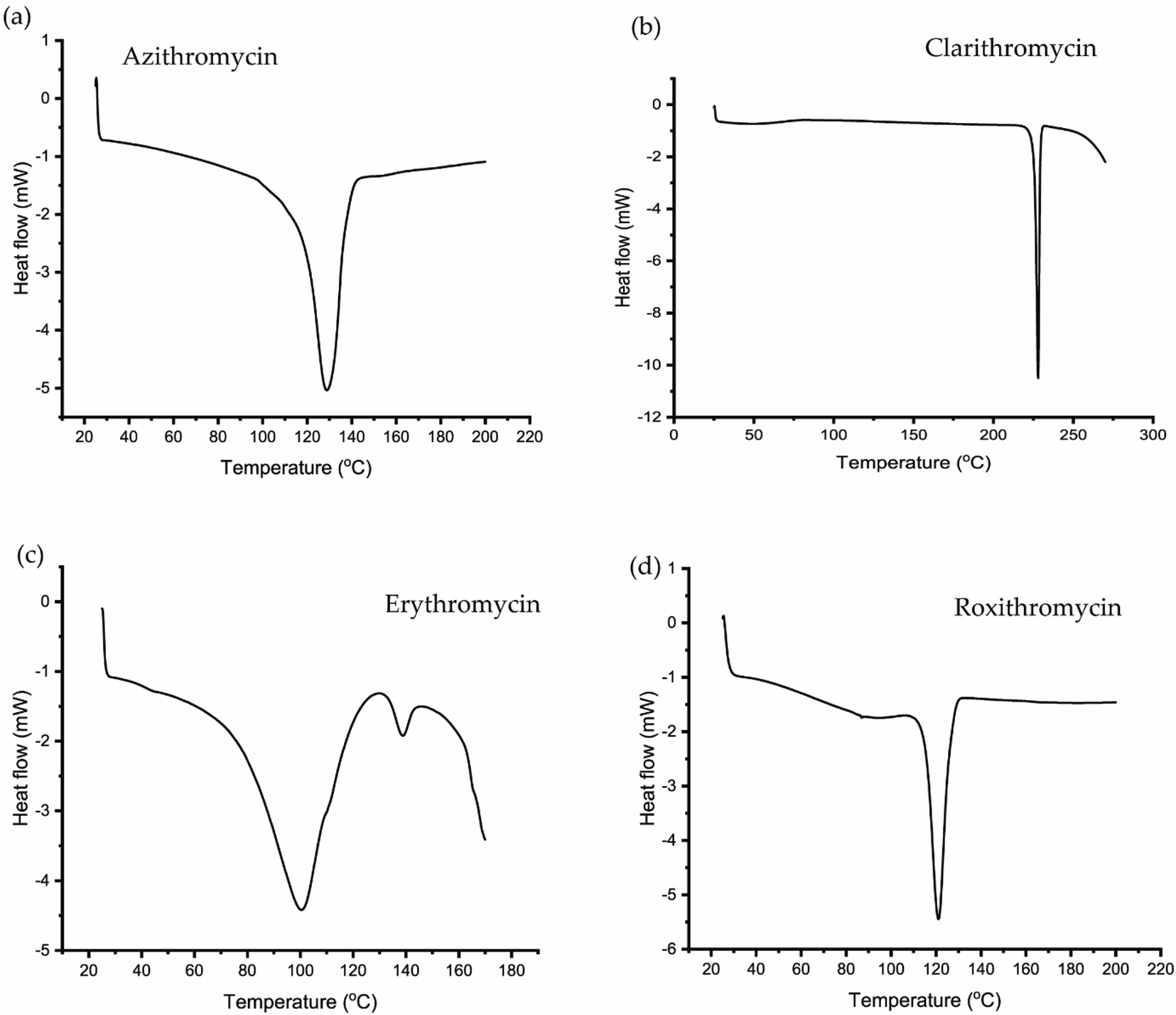 | ||
| Fig. 6 DSC curve of (a) azithromycin, (b) clarithromycin, (c) erythromycin, (d) roxithromycin. | ||
In DSC analysis, erythromycin exhibits two endothermic peaks at 100.34 °C and 138.59 °C. This result corresponds to a previous study on erythromycin dihydrate, which also displayed two similar endothermic peaks.104 Thus, combining PXRD and DSC data confirms that the erythromycin used in the study exists in dihydrate form.
For azithromycin, a single endothermic peak was observed in the DSC analysis, consistent with findings by R. Gandhi and co-authors in their analysis of azithromycin hydrates.105 The experimental results from TGA also match those of azithromycin monohydrate. Therefore, analysis of PXRD, DSC, and TGA collectively indicates that azithromycin is in monohydrate form by following the article by Bakheit et al.109
In the DSC analysis of roxithromycin and clarithromycin, both plots exhibit single endothermic peaks. The results from DSC and TGA for clarithromycin and roxithromycin parallel those found in studies of roxithromycin semihydrate and clarithromycin hemihydrates.103,106,107
3.3 13C CP-MAS NMR spectral analysis
Fig. 7–10 show the 13C CP-MAS SSNMR spectra of erythromycin, azithromycin, clarithromycin, and roxithromycin respectively. The resonance lines of 13C CP-MAS spectrum of roxithromycin is assigned by following Gharbi-Benarous et al.92 The resonance lines of 13C CP-MAS spectrum of azithromycin is assigned by following Brennan and Barber,94 and Bakheit et al.109 The resonance lines of 13C CP-MAS spectrum of clarithromycin is assigned by following Amaw et al.96 and I. I. Salem.107 The resonance lines of 13C CP-MAS spectrum of erythromycin is assigned by following Everett and Tyler.97 Carbon nuclei connected to polar bonds exhibit the highest isotropic chemical shift. The significant influence on the secondary magnetic field arises from the polarization caused by the polar bond and its effect on the surrounding electron cloud. As a result, the nucleus experiences an effective magnetic field that varies with the orientation of the molecule concerning the external magnetic field. As a result, carbon nuclei associated with polar bonds have a large isotropic and anisotropic chemical shift. The presence of electronegative atoms leads to electron cloud attraction around the carbon nuclei, resulting in a shift of the isotropic chemical shift towards higher frequency values.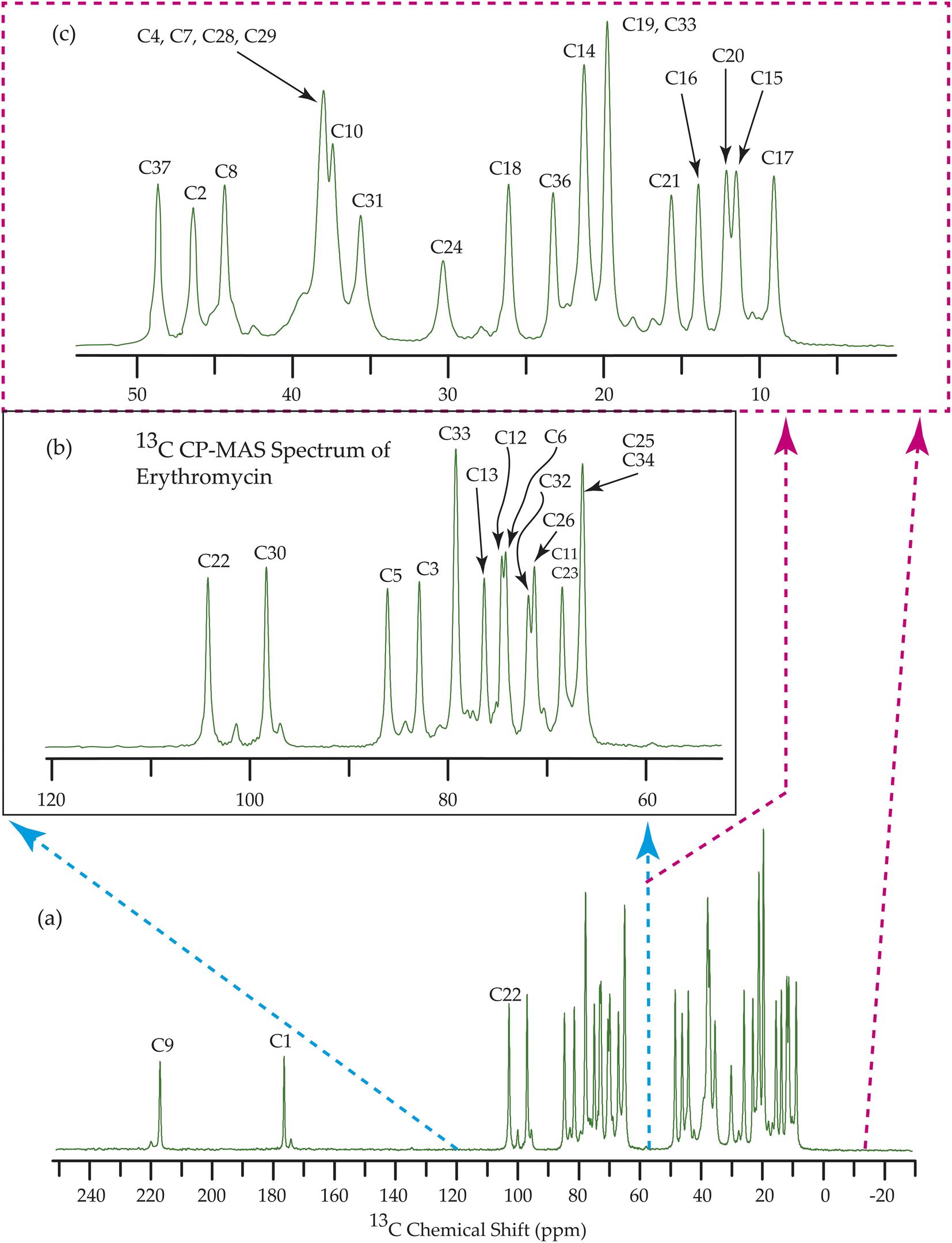 | ||
| Fig. 7 13C CP-MAS SSNMR spectrum of erythromycin at MAS frequency 24 kHz. The experiments were performed at 600 MHz BRUKER AVANCE NEO Spectrometer with contact time 2 ms, recycle delay 15 s, and number of scan was 1024. | ||
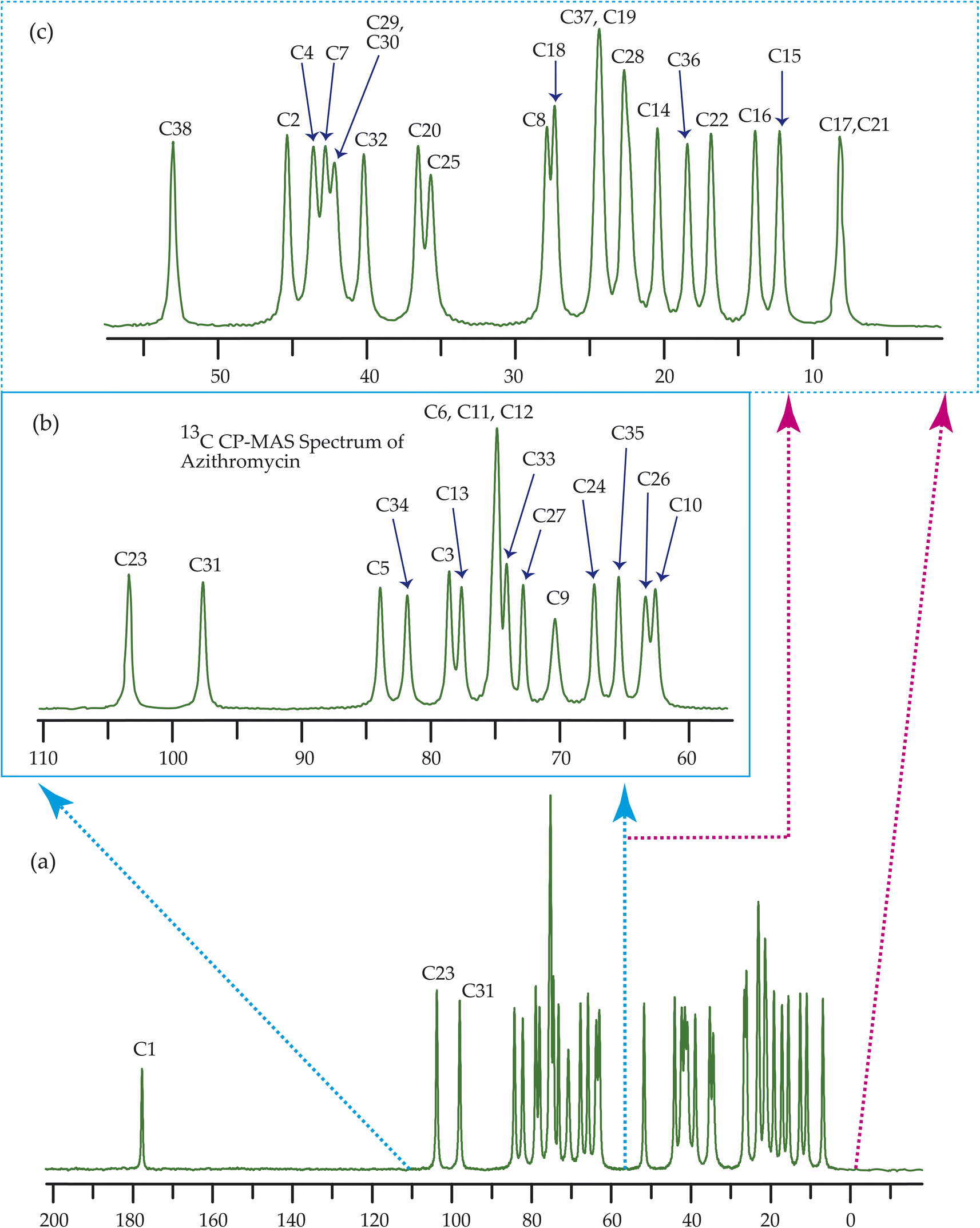 | ||
| Fig. 8 13C CP-MAS SSNMR spectrum of azithromycin at MAS frequency 24 kHz. The experiments were performed at 600 MHz BRUKER AVANCE NEO Spectrometer with contact time 2 ms, recycle delay 15 s, and number of scan was 1024. | ||
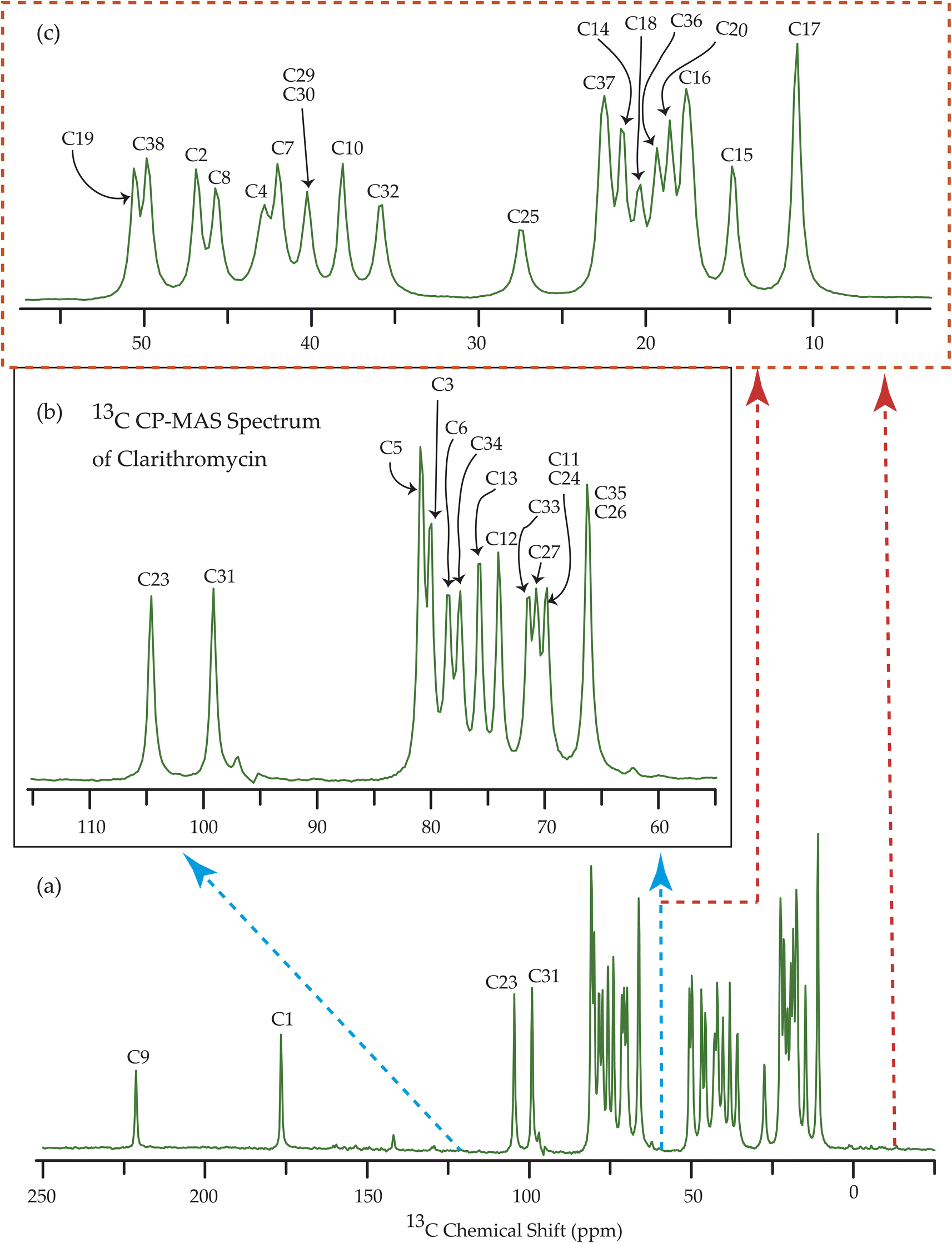 | ||
| Fig. 9 13C CP-MAS SSNMR spectrum of clarithromycin at MAS frequency 10 kHz. The experiments were performed at 500 MHz JEOL Spectrometer with contact time 2 ms, recycle delay 15 s, and number of scan was 1024. | ||
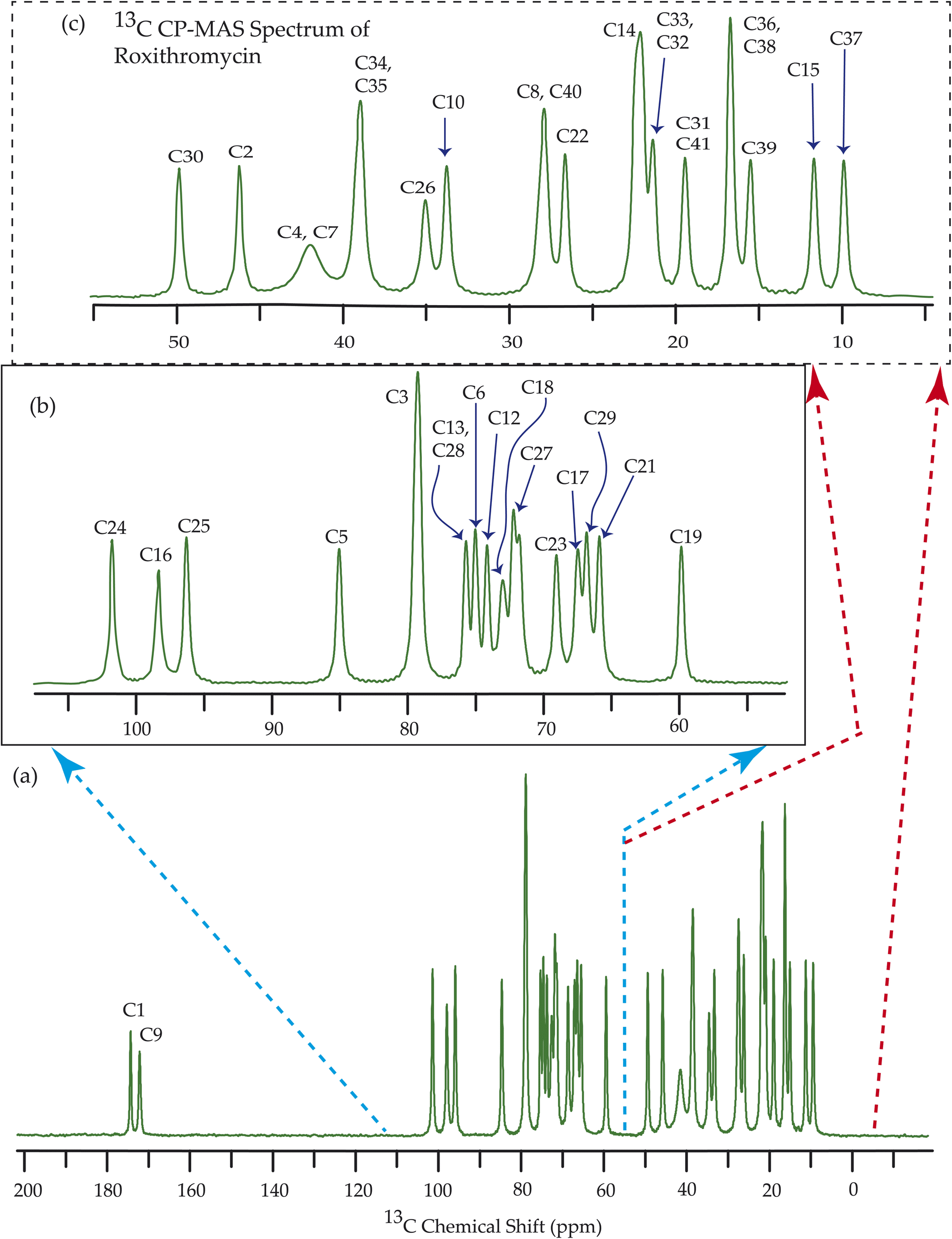 | ||
| Fig. 10 13C CP-MAS SSNMR spectrum of roxithromycin at MAS frequency 24 kHz. The experiments were performed at 600 MHz BRUKER AVANCE NEO Spectrometer with contact time 2 ms, recycle delay 15 s, and number of scan was 1024. | ||
3.4 Determination of principal components of CSA tensor
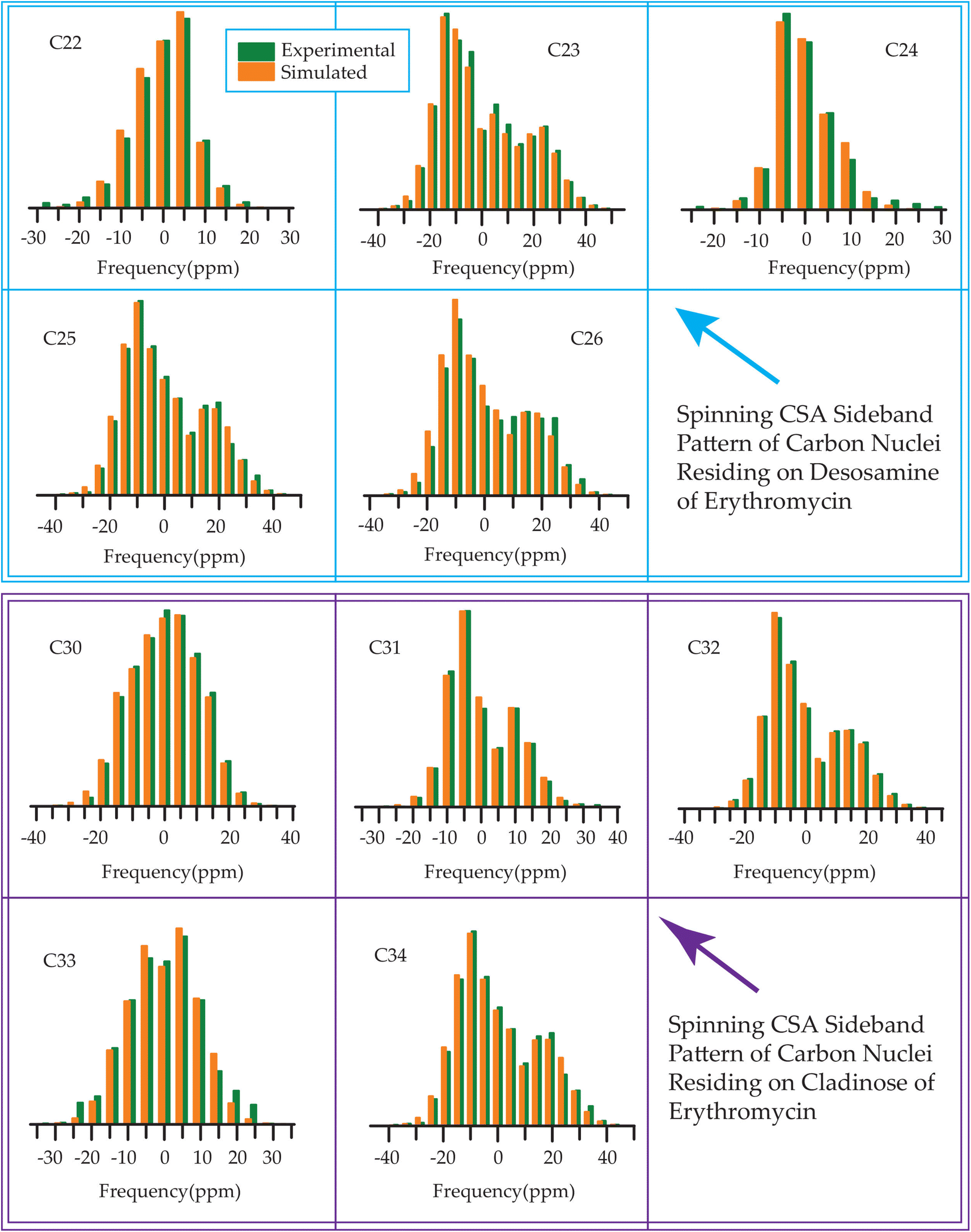
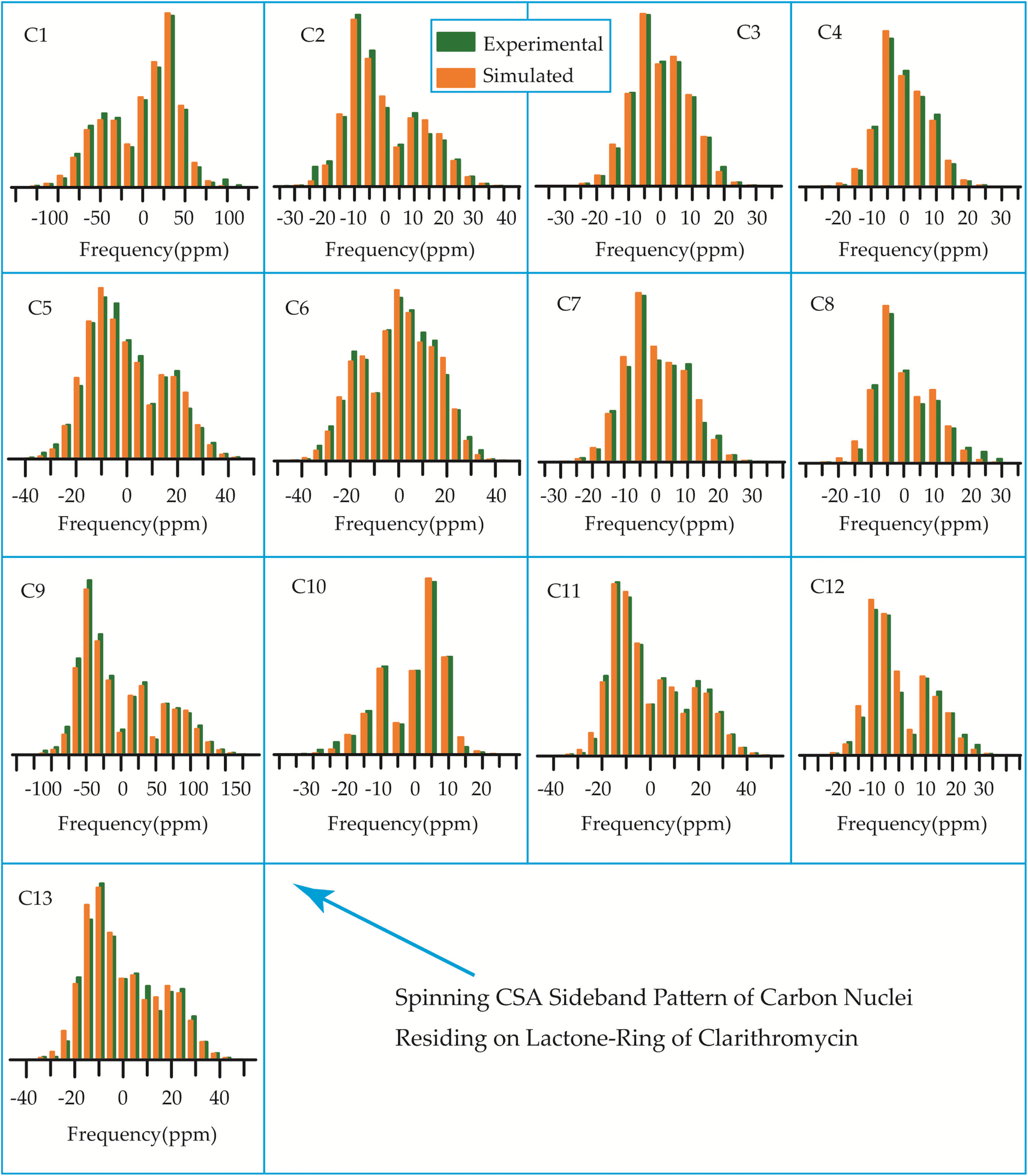
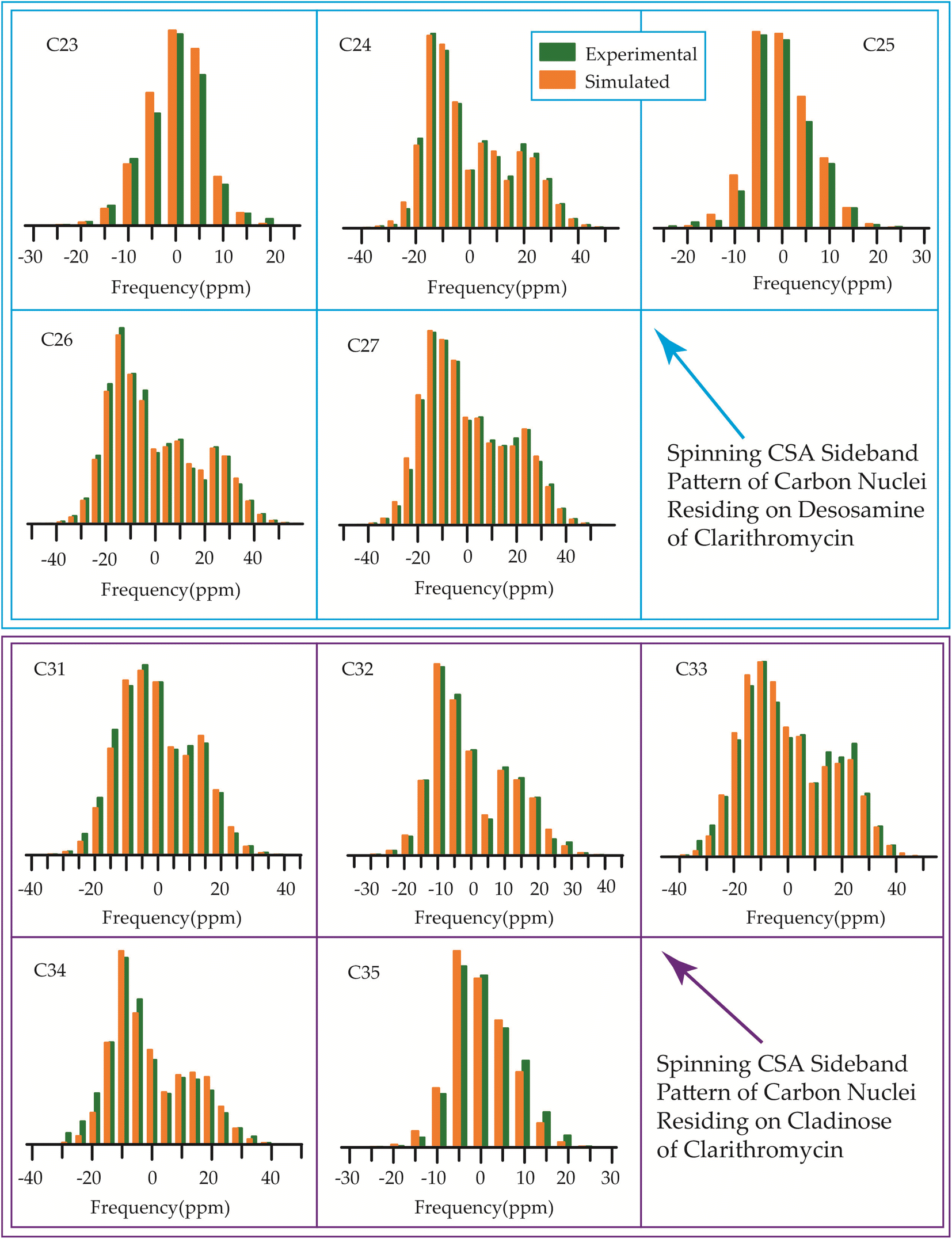
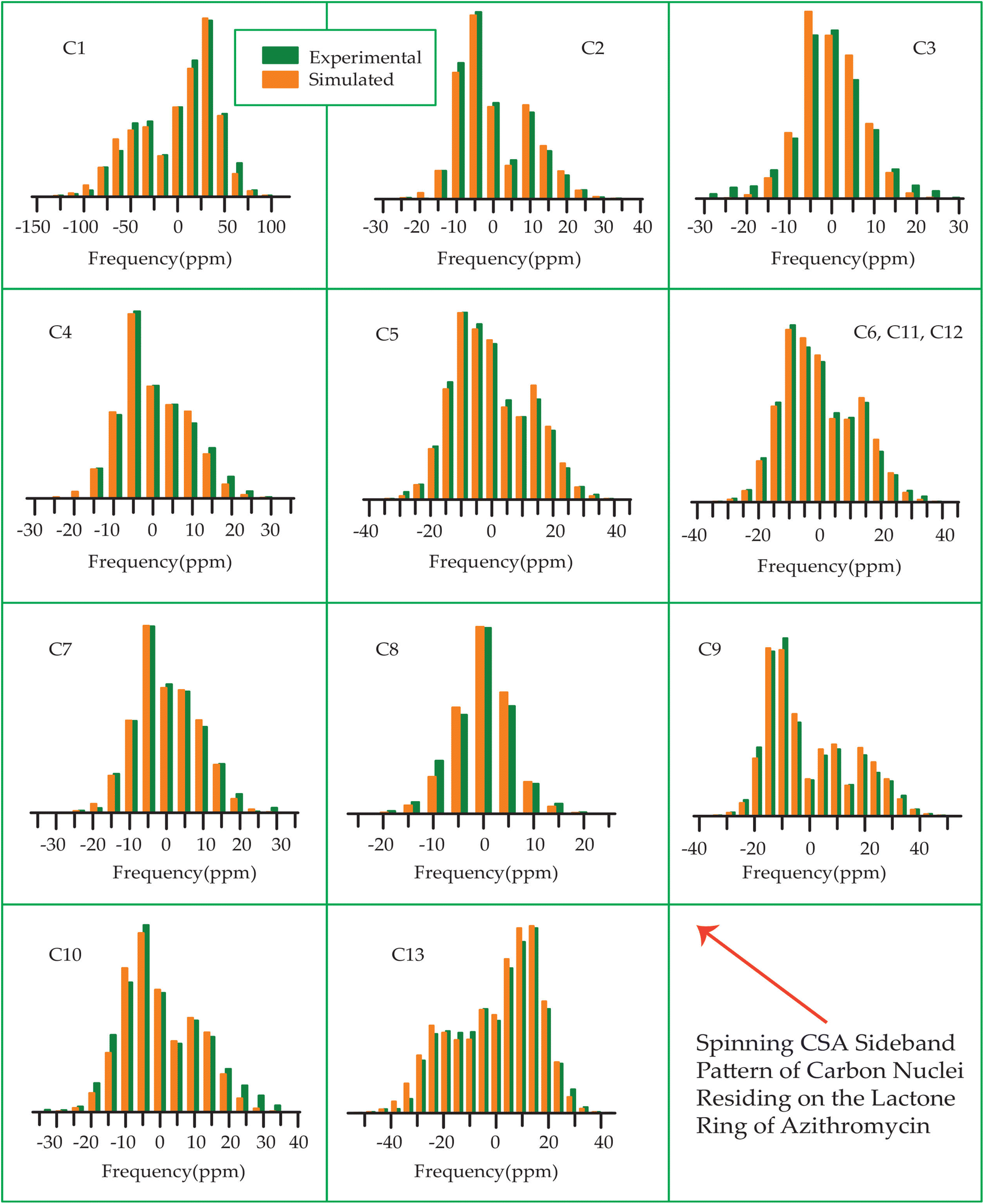
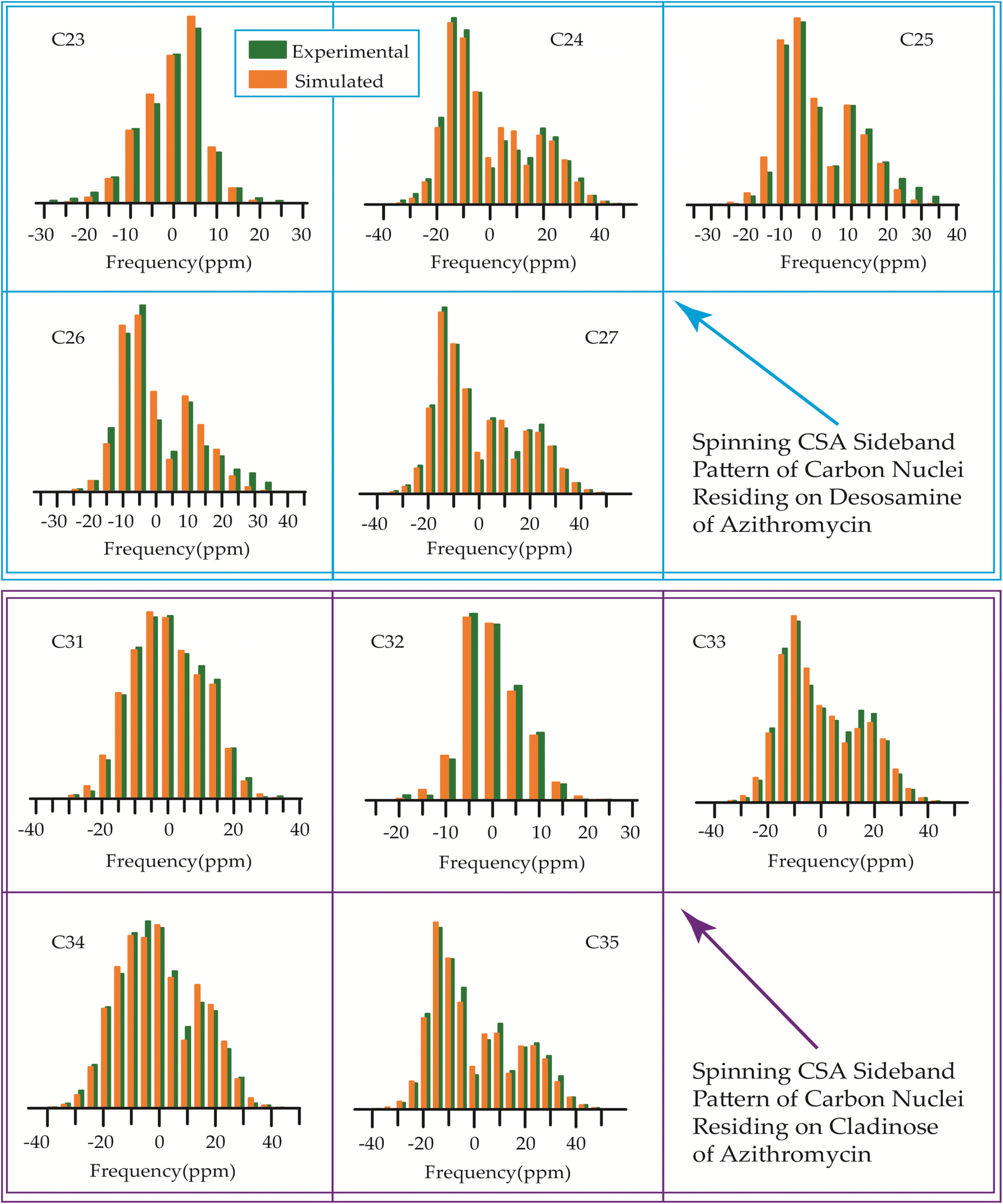
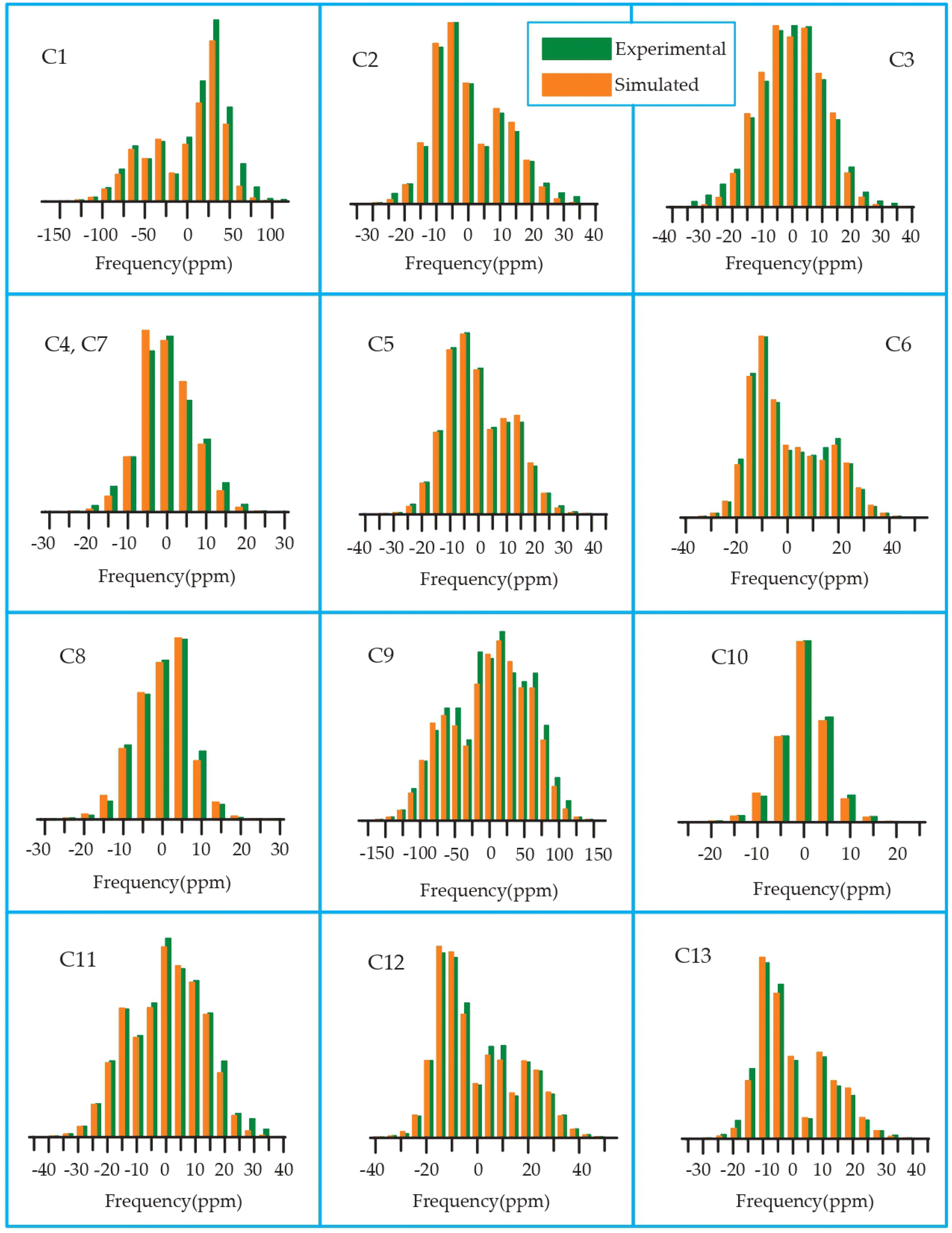
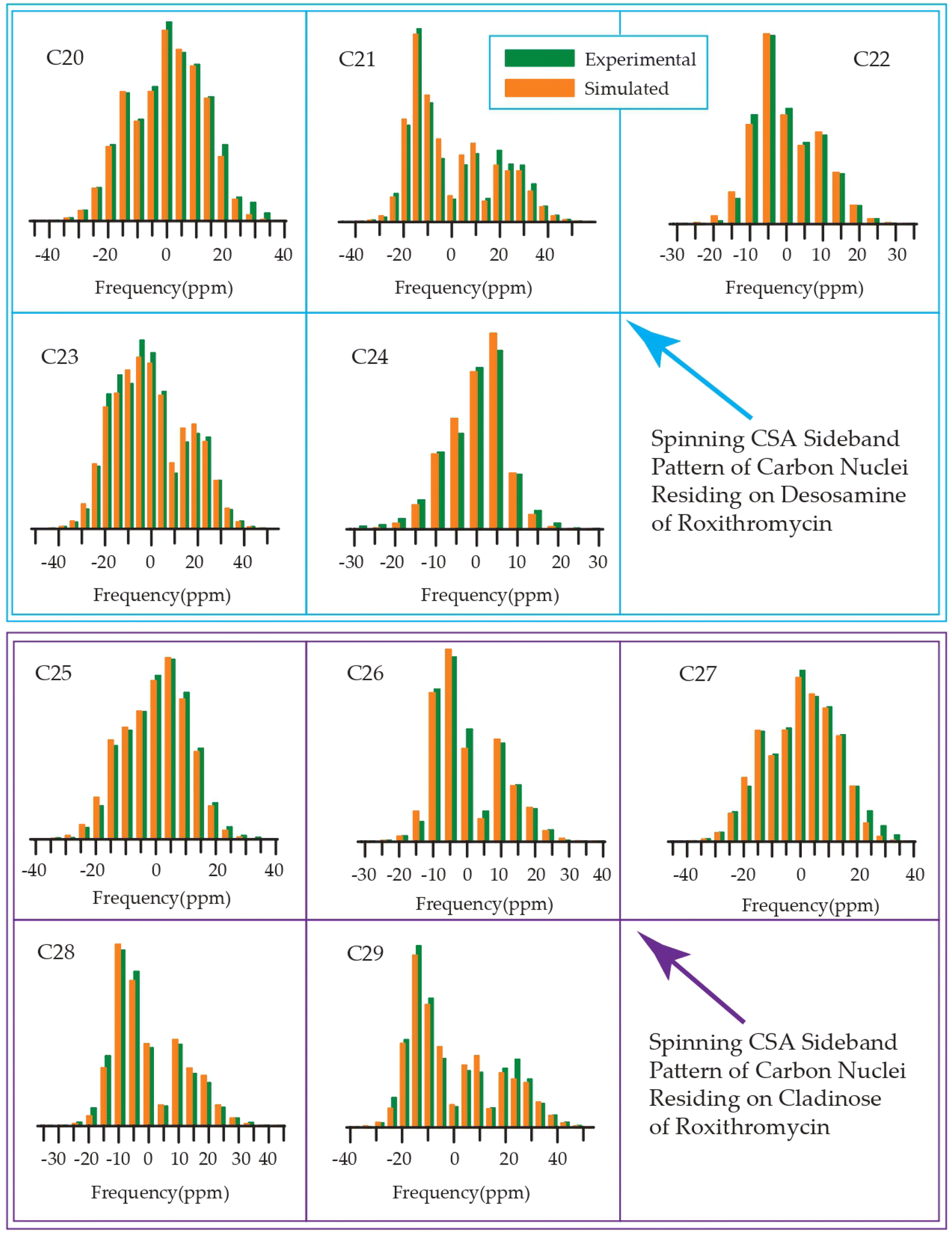
The 13C 2DPASS CP-MAS spectrum of azithromycin, erythromycin, roxithromycin, and clarithromycin are shown in Fig. 11 and 12. The principal components of chemical shift anisotropy parameters of erythromycin, clarithromycin, azithromycin and roxithromycin, are displayed in Tables 1–4 respectively. The spinning CSA sideband pattern of carbon nuclei residing on the lactone ring of erythromycin, clarithromycin, azithromycin and roxithromycin are shown in Fig. 13, 15, 17, and 19 respectively. On the hand, the spinning CSA sideband pattern of the carbon nuclei residing on desosamine and cladinose sugar units attached with lactone ring are shown in Fig. 14, 16, 18 and 20 for erythromycin, clarithromycin, azithromycin and roxithromycin respectively.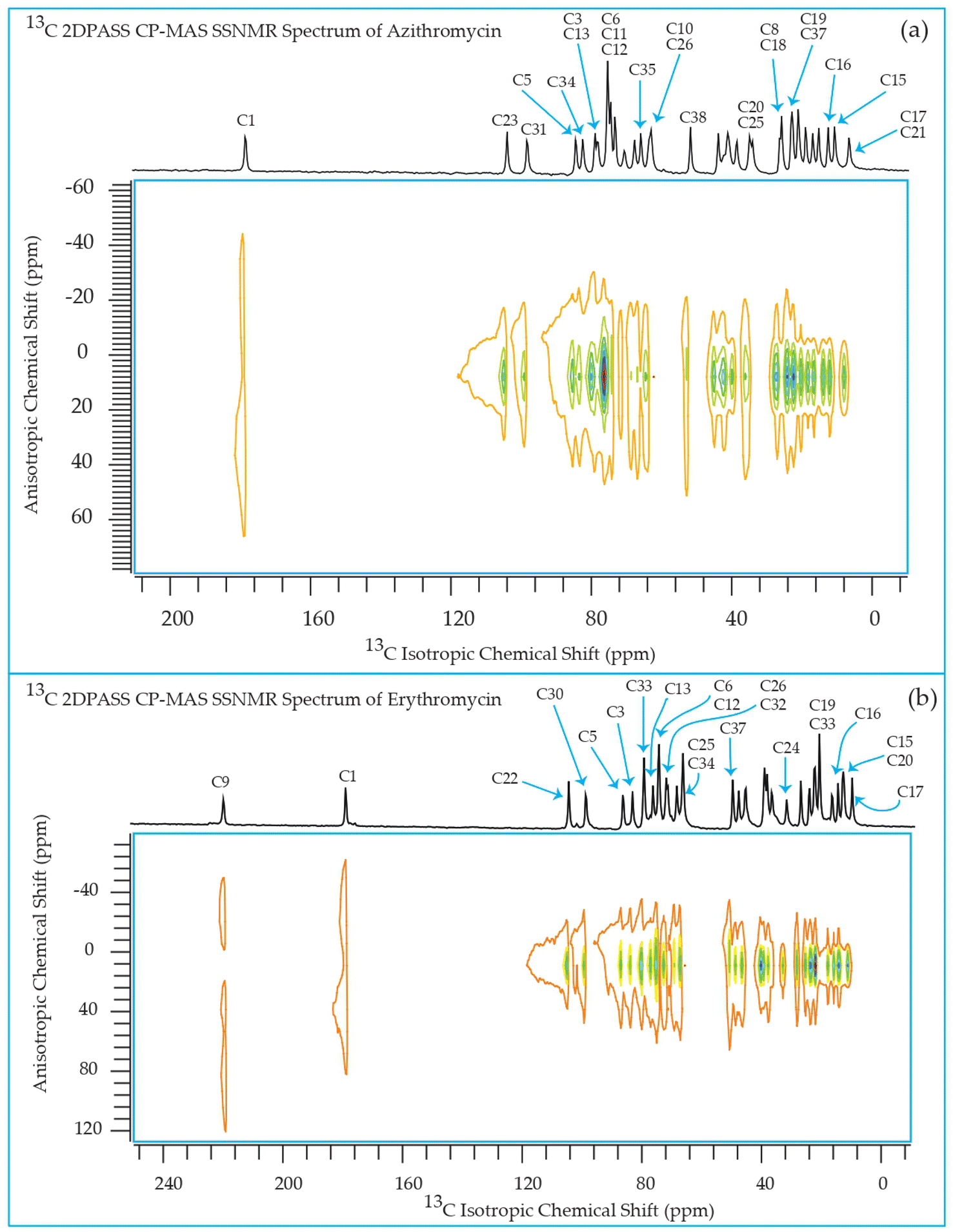 | ||
| Fig. 11 Two-dimensional phase-adjusted spinning sideband (2DPASS) cross-polarization magic angle spinning (CP-MAS) SSNMR spectrum of (a) azithromycin, and (b) erythromycin. The experiments were performed at 500 MHz JEOL Spectrometer with contact time 2 ms, recycle delay 15 s, and number of scan was 4030. The MAS frequency were 2 kHz and 600 Hz. | ||
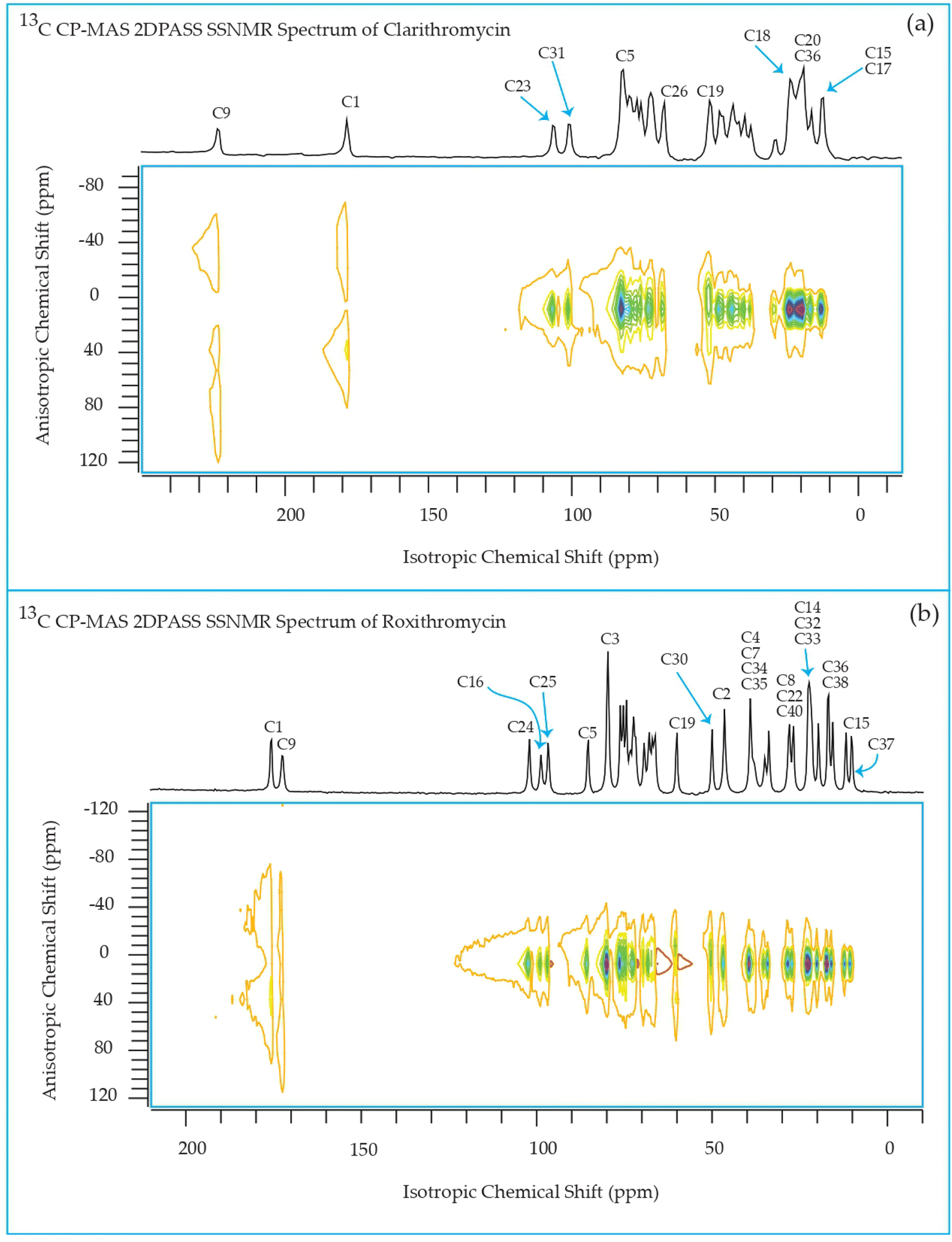 | ||
| Fig. 12 Two-dimensional phase-adjusted spinning sideband (2DPASS) cross-polarization magic angle spinning (CP-MAS) SSNMR spectrum of (a) clarithromycin, and (b) roxithromycin. The experiments were performed at 500 MHz JEOL Spectrometer with contact time 2 ms, recycle delay 15 s, and number of scan was 4030. The MAS frequency were 2 kHz and 600 Hz. | ||
| Principal components of 13C CSA tensor of erythromycin | ||||||||
|---|---|---|---|---|---|---|---|---|
| Carbon nuclei | δiso (ppm) | δ11 (ppm) | δ22 (ppm) | δ33 (ppm) | Span (ppm) | Skew | Anisotropy (ppm) | Asymmetry |
| C1 | 177.8 | 242.2 ± 1.7 | 192.1 ± 1.3 | 99.0 ± 1.1 | 143.2 ± 1.8 | 0.3 | −118.1 ± 1.7 | 0.6 |
| C2 | 47.3 | 72.7 ± 0.1 | 34.6 ± 0.1 | 34.6 ± 0.08 | 38.2 ± 0.1 | −1.0 | 38.2 ± 0.2 | 0.0 |
| C3 | 82.9 | 112.6 ± 0.2 | 75.9 ± 0.1 | 60.1 ± 0.1 | 52.5 ± 0.2 | −0.4 | 44.6 ± 0.3 | 0.5 |
| C4 | 38.8 | 53.6 ± 0.1 | 37.8 ± 0.1 | 24.9 ± 0.1 | 28.6 ± 0.2 | −0.1 | 22.2 ± 0.2 | 0.9 |
| C5 | 86.1 | 113.9 ± 0.2 | 78.1 ± 0.2 | 66.2 ± 0.1 | 47.7 ± 0.1 | −0.5 | 41.7 ± 0.3 | 0.4 |
| C6 | 73.8 | 109.1 ± 0.3 | 60.4 ± 0.2 | 51.8 ± 0.1 | 57.3 ± 0.2 | −0.7 | 52.9 ± 0.4 | 0.2 |
| C7 | 38.8 | 53.6 ± 0.1 | 37.8 ± 0.1 | 24.9 ± 0.1 | 28.6 ± 0.2 | −0.1 | 22.2 ± 0.2 | 0.9 |
| C8 | 45.2 | 64.7 ± 0.1 | 39.6 ± 0.1 | 31.3 ± 0.1 | 33.4 ± 0.1 | −0.5 | 29.2 ± 0.2 | 0.4 |
| C9 | 218.2 | 332.7 ± 2.0 | 180.0 ± 1.6 | 141.8 ± 1.1 | 190.9 ± 1.6 | −0.6 | 171.8 ± 3.0 | 0.3 |
| C10 | 37.9 | 50.8 ± 0.1 | 40.8 ± 0.1 | 22.1 ± 0.1 | 28.6 ± 0.2 | 0.3 | −23.6 ± 0.2 | 0.6 |
| C11 | 67.8 | 104.0 ± 0.2 | 57.5 ± 0.2 | 41.9 ± 0.1 | 62.0 ± 0.2 | −0.5 | 54.3 ± 0.4 | 0.4 |
| C12 | 74.2 | 105.7 ± 0.2 | 63.7 ± 0.1 | 53.2 ± 0.1 | 52.5 ± 0.2 | 0.6 | 47.2 ± 0.3 | 0.3 |
| C13 | 75.9 | 102.4 ± 0.1 | 65.9 ± 0.1 | 59.4 ± 0.1 | 42.9 ± 0.1 | −0.7 | 39.7 ± 0.3 | 0.2 |
| C14 | 22.4 | 39.6 ± 0.3 | 16.7 ± 0.2 | 10.9 ± 0.2 | 28.6 ± 0.4 | −0.6 | 25.8 ± 0.5 | 0.3 |
| C15 | 12.4 | 21.3 ± 0.2 | 13.7 ± 0.08 | 2.2 ± 0.2 | 19.1 ± 0.4 | 0.2 | −15.3 ± 0.3 | 0.7 |
| C16 | 14.4 | 24.6 ± 0.06 | 13.1 ± 0.04 | 5.5 ± 0.04 | 19.1 ± 0.07 | −0.2 | 15.2 ± 0.09 | 0.7 |
| C17 | 9.7 | 13.8 ± 0.04 | 10.9 ± 0.02 | 4.3 ± 0.03 | 9.5 ± 0.06 | 0.4 | −8.1 ± 0.05 | 0.5 |
| C18 | 26.7 | 49.0 ± 0.2 | 20.3 ± 0.1 | 10.8 ± 0.1 | 38.2 ± 0.3 | −0.5 | 33.4 ± 0.3 | 0.4 |
| C19 | 20.6 | 36.8 ± 0.1 | 16.8 ± 0.1 | 8.2 ± 0.09 | 28.6 ± 0.1 | −0.4 | 24.3 ± 0.2 | 0.5 |
| C20 | 13.0 | 23.8 ± 0.1 | 10.4 ± 0.05 | 4.7 ± 0.1 | 19.1 ± 0.2 | −0.4 | 16.2 ± 0.2 | 0.5 |
| C21 | 16.5 | 32.2 ± 0.1 | 13.6 ± 0.09 | 3.6 ± 0.08 | 28.6 ± 0.1 | −0.3 | 23.6 ± 0.2 | 0.6 |
| C22 | 103.8 | 117.2 ± 0.2 | 105.7 ± 0.1 | 88.5 ± 0.1 | 28.6 ± 0.2 | 0.2 | −22.9 ± 0.2 | 0.7 |
| C23 | 67.8 | 104.0 ± 0.2 | 57.5 ± 0.2 | 41.9 ± 0.1 | 62.0 ± 0.2 | −0.5 | 54.3 ± 0.4 | 0.4 |
| C24 | 31.4 | 45.3 ± 0.1 | 27.4 ± 0.08 | 21.4 ± 0.08 | 23.8 ± 0.1 | −0.5 | 20.9 ± 0.2 | 0.4 |
| C25 | 66.0 | 98.4 ± 0.6 | 58.3 ± 0.5 | 41.2 ± 0.4 | 57.3 ± 0.5 | −0.4 | 48.7 ± 0.9 | 0.5 |
| C26 | 70.7 | 101.3 ± 0.4 | 61.9 ± 0.3 | 48.8 ± 0.2 | 52.5 ± 0.3 | −0.5 | 45.9 ± 0.6 | 0.4 |
| C27 | 20.6 | 36.8 ± 0.1 | 16.8 ± 0.1 | 8.2 ± 0.09 | 28.6 ± 0.1 | −0.4 | 24.3 ± 0.2 | 0.5 |
| C28,C29 | 38.8 | 53.6 ± 0.1 | 37.8 ± 0.1 | 24.9 ± 0.1 | 28.6 ± 0.2 | −0.1 | 22.2 ± 0.2 | 0.9 |
| C30 | 98.2 | 119.7 ± 0.3 | 98.2 ± 0.2 | 76.7 ± 0.1 | 42.9 ± 0.2 | 0.0 | −32.2 ± 0.2 | 1.0 |
| C31 | 36.4 | 58.7 ± 0.3 | 30.0 ± 0.2 | 20.5 ± 0.2 | 38.2 ± 0.3 | −0.5 | 33.4 ± 0.5 | 0.4 |
| C32 | 71.3 | 99.9 ± 0.3 | 61.7 ± 0.2 | 52.2 ± 0.1 | 47.7 ± 0.2 | −0.6 | 42.9 ± 0.4 | 0.3 |
| C33 | 79.1 | 98.2 ± 0.4 | 79.1 ± 0.3 | 60.0 ± 0.2 | 38.2 ± 0.4 | 0.0 | −28.6 ± 0.4 | 1.0 |
| C34 | 66.0 | 98.4 ± 0.6 | 58.3 ± 0.5 | 41.2 ± 0.4 | 57.3 ± 0.5 | −0.4 | 48.7 ± 0.9 | 0.5 |
| C35 | 20.6 | 36.8 ± 0.1 | 16.8 ± 0.1 | 8.2 ± 0.09 | 28.6 ± 0.1 | −0.4 | 24.3 ± 0.2 | 0.5 |
| C36 | 24.1 | 43.0 ± 0.1 | 19.6 ± 0.1 | 9.6 ± 0.09 | 33.4 ± 0.1 | −0.4 | 28.4 ± 0.2 | 0.5 |
| C37 | 49.3 | 87.5 ± 0.5 | 34.8 ± 0.4 | 25.5 ± 0.3 | 62.0 ± 0.4 | −0.7 | 57.4 ± 0.8 | 0.2 |
| Principal components of 13C CSA tensor of clarithromycin | ||||||||
|---|---|---|---|---|---|---|---|---|
| Carbon nuclei | δiso (ppm) | δ11 (ppm) | δ22 (ppm) | δ33 (ppm) | Span (ppm) | Skew | Anisotropy (ppm) | Asymmetry |
| a The information regarding the anisotropy and the asymmetry of the shielding tensor are Δδ = δ33 − 1/2(δ11 + δ22) and η = (δ22 − δ11)/(δ33 − δiso) respectively. The magnitude of the anisotropy defines the distance of the largest separation of the spinning CSA sideband pattern from the position of the center of gravity (δiso = δ11 + δ22 + δ33/3)- of the sideband pattern, and the sign of the anisotropy represents on which side of the center of gravity the distance of separation is maximum. The ‘asymmetry’ parameter says the deviation of the spinning CSA sideband pattern from its axially symmetric shape. The ‘span’ (Ω = δ33 − δ11)monitors the breadth of the spinning CSA sideband pattern, and ‘skew’ (k = (δiso − δ22)/(δ33 − δ11)) measures the amount of orientation of the asymmetry pattern. When k = ±1, the spinning CSA pattern is axially symmetric, and the CS tensor is non-axial when k = +0.3. | ||||||||
| C1 | 177.0 | 240.6 ± 1.1 | 208.8 ± 0.9 | 81.5 ± 0.6 | 159.1 ± 0.9 | 0.6 | −143.2 ± 0.9 | 0.3 |
| C2 | 46.8 | 65.7 ± 0.2 | 42.3 ± 0.1 | 32.3 ± 0.1 | 33.4 ± 0.2 | −0.4 | 28.4 ± 0.4 | 0.5 |
| C3 | 80.0 | 97.3 ± 0.4 | 78.9 ± 0.3 | 63.8 ± 0.3 | 33.4 ± 0.5 | −0.1 | 25.9 ± 0.6 | 0.9 |
| C4 | 43.0 | 58.7 ± 0.1 | 40.1 ± 0.09 | 30.1 ± 0.08 | 28.6 ± 0.1 | −0.3 | 23.6 ± 0.2 | 0.6 |
| C5 | 81.0 | 113.4 ± 0.4 | 73.3 ± 0.3 | 56.2 ± 0.2 | 57.3 ± 0.3 | −0.4 | 48.7 ± 0.6 | 0.5 |
| C6 | 78.5 | 106.2 ± 0.8 | 80.4 ± 0.6 | 48.9 ± 0.4 | 57.3 ± 0.6 | 0.1 | −44.4 ± 0.7 | 0.9 |
| C7 | 42.0 | 62.3 ± 0.4 | 39.4 ± 0.3 | 24.2 ± 0.3 | 38.2 ± 0.4 | −0.2 | 30.5 ± 0.6 | 0.7 |
| C8 | 45.7 | 71.5 ± 0.4 | 37.1 ± 0.3 | 28.5 ± 0.3 | 42.9 ± 0.4 | −0.6 | 38.6 ± 0.7 | 0.3 |
| C9 | 221.9 | 349.2 ± 2.7 | 158.3 ± 1.6 | 158.3 ± 1.5 | 190.9 ± 1.6 | −1.0 | 190.9 ± 3.5 | 0.0 |
| C10 | 38.1 | 50.9 ± 1.5 | 45.9 ± 1.0 | 17.5 ± 1.0 | 33.4 ± 1.7 | 0.7 | −30.9 ± 1.5 | 0.2 |
| C11 | 69.0 | 104.3 ± 0.7 | 55.6 ± 0.6 | 47.0 ± 0.4 | 57.3 ± 0.6 | −0.7 | 53.0 ± 1.1 | 0.2 |
| C12 | 74.0 | 98.8 ± 0.3 | 62.5 ± 0.2 | 60.6 ± 0.1 | 38.2 ± 0.3 | −0.9 | 37.2 ± 0.4 | 0.08 |
| C13 | 75.7 | 109.1 ± 0.4 | 66.1 ± 0.3 | 51.8 ± 0.2 | 57.3 ± 0.3 | −0.5 | 50.1 ± 0.6 | 0.4 |
| C14 | 21.4 | 33.1 ± 0.1 | 16.9 ± 0.1 | 14.1 ± 0.1 | 19.1 ± 0.2 | −0.7 | 17.6 ± 0.2 | 0.2 |
| C15 | 10.9 | 19.7 ± 0.1 | 7.5 ± 0.06 | 5.4 ± 0.09 | 14.3 ± 0.1 | −0.7 | 13.2 ± 0.1 | 0.2 |
| C16 | 14.7 | 23.3 ± 0.1 | 16.6 ± 0.08 | 4.2 ± 0.1 | 19.1 ± 0.3 | 0.3 | −15.7 ± 0.2 | 0.6 |
| C17 | 10.9 | 19.7 ± 0.1 | 7.5 ± 0.06 | 5.4 ± 0.09 | 14.3 ± 0.1 | −0.7 | 13.2 ± 0.1 | 0.2 |
| C18 | 20.4 | 35.7 ± 0.2 | 18.5 ± 0.1 | 7.0 ± 0.1 | 28.6 ± 0.2 | −0.2 | 22.9 ± 0.3 | 0.7 |
| C19 | 50.6 | 91.8 ± 0.4 | 35.0 ± 0.3 | 25.0 ± 0.2 | 66.8 ± 0.2 | −0.7 | 61.8 ± 0.5 | 0.2 |
| C20 | 17.6 | 41.2 ± 0.9 | 13.3 ± 0.7 | −1.7 ± 0.6 | 42.9 ± 0.9 | −0.3 | 35.4 ± 1.4 | 0.6 |
| C21 | 18.5 | 32.0 ± 0.1 | 15.3 ± 0.1 | 8.1 ± 0.1 | 23.8 ± 0.2 | −0.4 | 20.3 ± 0.2 | 0.5 |
| C22 | 27.4 | 40.5 ± 0.2 | 25.0 ± 0.1 | 16.7 ± 0.2 | 23.8 ± 0.3 | −0.3 | 19.7 ± 0.3 | 0.6 |
| C23 | 104.7 | 115.8 ± 0.2 | 106.3 ± 0.1 | 92.0 ± 0.1 | 23.8 ± 0.2 | 0.2 | −19.0 ± 0.2 | 0.7 |
| C24 | 69.0 | 104.3 ± 0.7 | 55.6 ± 0.6 | 47.0 ± 0.4 | 57.3 ± 0.6 | −0.7 | 53.0 ± 1.1 | 0.2 |
| C25 | 27.4 | 40.5 ± 0.2 | 25.0 ± 0.1 | 16.7 ± 0.2 | 23.8 ± 0.3 | −0.3 | 19.7 ± 0.3 | 0.6 |
| C26 | 66.3 | 106.4 ± 0.6 | 52.9 ± 0.5 | 39.6 ± 0.3 | 66.8 ± 0.4 | −0.6 | 60.1 ± 0.9 | 0.3 |
| C27 | 70.7 | 106.9 ± 0.7 | 60.4 ± 0.6 | 44.8 ± 0.4 | 62.0 ± 0.6 | −0.5 | 54.3 ± 1.1 | 0.4 |
| C28 | 18.5 | 32.0 ± 0.1 | 15.3 ± 0.1 | 8.1 ± 0.1 | 23.8 ± 0.2 | −0.4 | 20.3 ± 0.2 | 0.5 |
| C29, C30 | 40.2 | 59.9 ± 0.2 | 38.9 ± 0.1 | 21.7 ± 0.1 | 38.2 ± 0.2 | −0.1 | 29.6 ± 0.3 | 0.9 |
| C31 | 99.4 | 122.3 ± 0.3 | 96.5 ± 0.2 | 79.3 ± 0.2 | 42.9 ± 0.4 | −0.2 | 34.4 ± 0.5 | 0.7 |
| C32 | 35.9 | 61.7 ± 0.8 | 27.3 ± 0.6 | 18.7 ± 0.5 | 42.9 ± 0.8 | −0.6 | 38.6 ± 1.1 | 0.3 |
| C33 | 71.5 | 106.6 ± 0.4 | 63.2 ± 0.3 | 44.6 ± 0.2 | 62.0 ± 0.3 | −0.4 | 52.7 ± 0.7 | 0.5 |
| C34 | 77.4 | 106.0 ± 0.3 | 67.8 ± 0.2 | 58.3 ± 0.2 | 47.7 ± 0.3 | −0.6 | 42.9 ± 0.5 | 0.3 |
| C35 | 66.3 | 106.4 ± 0.6 | 52.9 ± 0.5 | 39.6 ± 0.3 | 66.8 ± 0.4 | −0.6 | 60.1 ± 0.9 | 0.3 |
| C36 | 18.5 | 32.0 ± 0.1 | 15.3 ± 0.1 | 8.1 ± 0.1 | 23.8 ± 0.2 | −0.4 | 20.3 ± 0.2 | 0.5 |
| C37 | 22.5 | 39.7 ± 0.4 | 21.4 ± 0.3 | 6.3 ± 0.3 | 33.4 ± 0.5 | −0.1 | 25.9 ± 0.6 | 0.9 |
| C38 | 49.8 | 88.8 ± 0.6 | 38.7 ± 0.5 | 21.9 ± 0.3 | 66.8 ± 0.4 | −0.5 | 58.4 ± 0.8 | 0.4 |
| Principal components of CSA tensor of azithromycin | ||||||||
|---|---|---|---|---|---|---|---|---|
| Carbon nuclei | δiso(ppm) | δ11(ppm) | δ22(ppm) | δ33(ppm) | Span (ppm) | skew | Anisotropy (ppm) | Asymmetry |
| C1 | 178.7 | 242.3 ± 2.4 | 210.5 ± 1.9 | 83.2 ± 1.4 | 159.1 ± 2.1 | 0.6 | −143.2 ± 2.1 | 0.3 |
| C2 | 44.6 | 65.7 ± 0.1 | 35.7 ± 0.1 | 32.3 ± 0.1 | 33.4 ± 0.2 | −0.8 | 31.7 ± 0.2 | 0.1 |
| C3 | 79.4 | 94.2 ± 0.2 | 78.4 ± 0.1 | 65.5 ± 0.2 | 28.6 ± 0.3 | −0.1 | 22.2 ± 0.3 | 0.9 |
| C4 | 43.0 | 60.5 ± 0.09 | 41.6 ± 0.07 | 27.0 ± 0.06 | 33.4 ± 0.1 | −0.2 | 26.7 ± 0.1 | 0.7 |
| C5 | 84.6 | 110.8 ± 0.2 | 79.8 ± 0.1 | 63.1 ± 0.1 | 47.7 ± 0.2 | −0.3 | 39.4 ± 0.2 | 0.6 |
| C6 | 75.8 | 102.8 ± 0.5 | 69.4 ± 0.4 | 55.1 ± 0.3 | 47.7 ± 0.4 | −0.4 | 40.5 ± 0.7 | 0.5 |
| C7 | 42.1 | 59.9 ± 0.09 | 39.9 ± 0.07 | 26.5 ± 0.06 | 33.4 ± 0.1 | −0.2 | 26.7 ± 0.1 | 0.7 |
| C8 | 26.9 | 38.4 ± 0.3 | 27.7 ± 0.2 | 14.6 ± 0.2 | 23.8 ± 0.4 | 0.1 | −18.5 ± 0.4 | 0.9 |
| C9 | 71.2 | 105.3 ± 0.5 | 55.4 ± 0.3 | 52.8 ± 0.2 | 52.5 ± 0.4 | −0.9 | 51.2 ± 0.7 | 0.08 |
| C10 | 63.4 | 85.0 ± 0.3 | 58.3 ± 0.2 | 46.8 ± 0.2 | 38.2 ± 0.4 | −0.4 | 32.4 ± 0.5 | 0.5 |
| C11, C12 | 75.8 | 102.8 ± 0.5 | 69.4 ± 0.4 | 55.1 ± 0.3 | 47.7 ± 0.4 | −0.4 | 40.5 ± 0.7 | 0.5 |
| C13 | 78.5 | 104.3 ± 0.7 | 88.8 ± 0.6 | 42.3 ± 0.4 | 62.0 ± 0.5 | 0.5 | −54.3 ± 0.6 | 0.4 |
| C14 | 19.5 | 35.4 ± 0.1 | 11.5 ± 0.08 | 11.5 ± 0.1 | 23.8 ± 0.1 | −1.0 | 23.8 ± 0.2 | 0.0 |
| C15 | 11.3 | 22.8 ± 0.1 | 12.1 ± 0.1 | −1.0 ± 0.08 | 23.9 ± 0.1 | 0.1 | −18.5 ± 0.1 | 0.9 |
| C16 | 13.2 | 14.8 ± 1.4 | 14.8 ± 0.7 | 10.0 ± 1.1 | 4.8 ± 2.2 | 1.0 | −4.8 ± 1.7 | 0.0 |
| C17 | 7.3 | 13.5 ± 0.09 | 9.2 ± 0.06 | −0.8 ± 0.07 | 14.3 ± 0.1 | 0.4 | −12.2 ± 0.1 | 0.5 |
| C18 | 26.3 | 45.2 ± 0.2 | 21.8 ± 0.1 | 11.8 ± 0.1 | 33.4 ± 0.3 | −0.4 | 28.4 ± 0.4 | 0.5 |
| C19 | 23.6 | 45.8 ± 0.1 | 22.1 ± 0.1 | 2.8 ± 0.1 | 42.9 ± 0.1 | −0.1 | 33.3 ± 0.2 | 0.9 |
| C20 | 35.5 | 60.9 ± 0.4 | 32.3 ± 0.3 | 13.2 ± 0.2 | 47.7 ± 0.4 | −0.2 | 38.2 ± 0.6 | 0.7 |
| C21 | 7.3 | 13.5 ± 0.09 | 9.2 ± 0.06 | −0.8 ± 0.07 | 14.3 ± 0.1 | 0.4 | −12.2 ± 0.1 | 0.5 |
| C22 | 15.8 | 27.9 ± 0.06 | 10.7 ± 0.04 | 8.8 ± 0.05 | 19.1 ± 0.09 | −0.8 | 18.1 ± 0.1 | 0.1 |
| C23 | 104.3 | 117.2 ± 0.2 | 107.2 ± 0.2 | 88.5 ± 0.1 | 28.6 ± 0.3 | 0.3 | −23.6 ± 0.3 | 0.6 |
| C24 | 68.2 | 103.5 ± 0.2 | 54.8 ± 0.2 | 46.2 ± 0.1 | 57.2 ± 0.2 | −0.7 | 52.9 ± 0.4 | 0.2 |
| C25 | 34.6 | 57.5 ± 0.2 | 26.9 ± 0.1 | 19.3 ± 0.1 | 38.2 ± 0.2 | −0.6 | 34.4 ± 0.3 | 0.3 |
| C26 | 64.2 | 85.9 ± 0.3 | 59.2 ± 0.2 | 47.5 ± 0.2 | 38.2 ± 0.4 | −0.4 | 32.4 ± 0.5 | 0.5 |
| C27 | 73.5 | 109.8 ± 0.2 | 58.2 ± 0.2 | 52.5 ± 0.1 | 57.3 ± 0.2 | −0.8 | 54.4 ± 0.3 | 0.1 |
| C28 | 21.5 | 38.2 ± 0.2 | 16.7 ± 0.1 | 9.5 ± 0.2 | 28.6 ± 0.4 | −0.5 | 25.0 ± 0.4 | 0.4 |
| C29,C30 | 41.5 | 58.7 ± 0.07 | 40.4 ± 0.05 | 25.3 ± 0.04 | 33.4 ± 0.08 | −0.1 | 25.9 ± 0.1 | 0.9 |
| C31 | 98.4 | 120.6 ± 0.1 | 96.9 ± 0.09 | 77.6 ± 0.07 | 42.9 ± 0.1 | −0.1 | 33.3 ± 0.1 | 0.9 |
| C32 | 39.3 | 52.4 ± 0.08 | 36.9 ± 0.04 | 28.5 ± 0.07 | 23.9 ± 0.1 | −0.3 | 19.7 ± 0.1 | 0.6 |
| C33 | 75.0 | 108.4 ± 0.3 | 65.4 ± 0.2 | 51.1 ± 0.1 | 57.3 ± 0.2 | −0.5 | 50.1 ± 0.5 | 0.4 |
| C34 | 82.9 | 110.9 ± 0.3 | 79.4 ± 0.2 | 58.4 ± 0.2 | 52.5 ± 0.3 | −0.2 | 42.0 ± 0.4 | 0.7 |
| C35 | 66.6 | 104.8 ± 0.3 | 52.1 ± 0.3 | 42.8 ± 0.2 | 62.0 ± 0.2 | −0.7 | 57.4 ± 0.5 | 0.2 |
| C36 | 17.5 | 29.6 ± 0.1 | 12.4 ± 0.08 | 10.5 ± 0.1 | 19.1 ± 0.2 | −0.8 | 18.1 ± 0.2 | 0.1 |
| C37 | 23.6 | 45.8 ± 0.1 | 22.1 ± 0.1 | 2.8 ± 0.1 | 42.9 ± 0.1 | −0.1 | 33.3 ± 0.2 | 0.9 |
| C38 | 52.4 | 93.6 ± 0.3 | 36.8 ± 0.3 | 26.8 ± 0.2 | 66.8 ± 0.2 | −0.7 | 61.8 ± 0.5 | 0.2 |
| Principal components of 13C CSA tensor of roxithromycin | ||||||||
|---|---|---|---|---|---|---|---|---|
| Carbon nuclei | δiso (ppm) | δ11 (ppm) | δ22 (ppm) | δ33 (ppm) | Span (ppm) | Skew | Anisotropy (ppm) | Asymmetry |
| C1 | 175.2 | 222.9 ± 1.9 | 222.9 ± 1.5 | 79.7 ± 1.2 | 143.2 ± 1.9 | 1.0 | −143.2 ± 1.8 | 0.0 |
| C2 | 46.0 | 68.3 ± 0.2 | 39.6 ± 0.1 | 30.1 ± 0.1 | 38.2 ± 0.3 | −0.5 | 33.4 ± 0.3 | 0.4 |
| C3 | 79.3 | 98.4 ± 0.2 | 79.3 ± 0.2 | 60.2 ± 0.1 | 38.2 ± 0.2 | 0.0 | −28.6 ± 0.2 | 1.0 |
| C4 | 42.2 | 54.9 ± 0.2 | 40.6 ± 0.1 | 31.1 ± 0.2 | 23.8 ± 0.3 | −0.2 | 19.1 ± 0.3 | 0.7 |
| C5 | 85.2 | 108.8 ± 0.09 | 80.9 ± 0.07 | 65.9 ± 0.07 | 42.9 ± 0.1 | −0.3 | 35.4 ± 0.1 | 0.6 |
| C6 | 74.8 | 106.3 ± 0.2 | 64.3 ± 0.1 | 53.8 ± 0.1 | 52.5 ± 0.2 | −0.6 | 47.2 ± 0.3 | 0.3 |
| C7 | 42.2 | 54.9 ± 0.2 | 40.6 ± 0.1 | 31.1 ± 0.2 | 23.8 ± 0.3 | −0.2 | 19.1 ± 0.3 | 0.7 |
| C8 | 26.5 | 45.4 ± 0.1 | 22.0 ± 0.08 | 12.0 ± 0.08 | 33.4 ± 0.1 | −0.4 | 28.4 ± 0.2 | 0.5 |
| C9 | 172.0 | 268.5 ± 2.4 | 185.8 ± 2.0 | 61.7 ± 1.4 | 206.8 ± 1.8 | 0.2 | −165.4 ± 2.0 | 0.7 |
| C10 | 33.5 | 42.4 ± 0.1 | 34.8 ± 0.05 | 23.3 ± 0.1 | 19.1 ± 0.3 | 0.2 | −15.2 ± 0.2 | 0.7 |
| C11 | 71.4 | 93.7 ± 0.6 | 74.6 ± 0.4 | 45.9 ± 0.3 | 47.7 ± 0.6 | 0.2 | −38.2 ± 0.6 | 0.7 |
| C12 | 73.9 | 109.2 ± 0.2 | 60.5 ± 0.1 | 51.9 ± 0.1 | 57.3 ± 0.1 | −0.7 | 52.9 ± 0.3 | 0.2 |
| C13 | 75.6 | 101.0 ± 0.1 | 62.9 ± 0.1 | 62.9 ± 0.1 | 38.2 ± 0.2 | −1.0 | 38.2 ± 0.2 | 0.0 |
| C14 | 22.2 | 37.9 ± 0.3 | 19.3 ± 0.2 | 9.3 ± 0.2 | 28.6 ± 0.5 | −0.3 | 23.6 ± 0.5 | 0.6 |
| C15 | 11.5 | 19.8 ± 0.07 | 14.0 ± 0.05 | 0.7 ± 0.05 | 19.1 ± 0.08 | 0.4 | −16.2 ± 0.07 | 0.5 |
| C16 | 98.5 | 117.8 ± 0.3 | 102.8 ± 0.2 | 74.8 ± 0.2 | 42.9 ± 0.3 | 0.3 | −35.4 ± 0.3 | 0.6 |
| C17 | 67.2 | 87.6 ± 0.5 | 64.6 ± 0.4 | 49.4 ± 0.3 | 38.2 ± 0.6 | −0.2 | 30.5 ± 0.8 | 0.7 |
| C18 | 72.7 | 101.3 ± 0.4 | 63.1 ± 0.3 | 53.6 ± 0.2 | 47.7 ± 0.4 | −0.6 | 42.9 ± 0.7 | 0.3 |
| C19 | 59.7 | 107.4 ± 0.9 | 35.8 ± 0.7 | 35.8 ± 0.5 | 71.6 ± 0.6 | −1.0 | 71.6 ± 1.3 | 0.0 |
| C20 | 71.4 | 93.7 ± 0.6 | 74.6 ± 0.4 | 45.9 ± 0.3 | 47.7 ± 0.6 | 0.2 | −38.2 ± 0.6 | 0.7 |
| C21 | 65.8 | 105.0 ± 0.6 | 49.2 ± 0.5 | 43.0 ± 0.3 | 62.0 ± 0.5 | −0.8 | 58.9 ± 0.9 | 0.1 |
| C22 | 27.5 | 40.8 ± 0.2 | 29.4 ± 0.1 | 12.2 ± 0.1 | 28.6 ± 0.3 | 0.2 | −22.9 ± 0.2 | 0.7 |
| C23 | 68.8 | 101.9 ± 0.3 | 64.6 ± 0.3 | 39.8 ± 0.2 | 62.0 ± 0.3 | −0.2 | 49.6 ± 0.6 | 0.7 |
| C24 | 101.7 | 114.6 ± 0.3 | 104.6 ± 0.2 | 85.9 ± 0.2 | 28.6 ± 0.4 | 0.3 | −23.6 ± 0.3 | 0.6 |
| C25 | 96.4 | 116.4 ± 0.2 | 99.3 ± 0.1 | 73.5 ± 0.1 | 42.9 ± 0.2 | 0.2 | −34.4 ± 0.2 | 0.7 |
| C26 | 34.6 | 56.9 ± 0.1 | 23.4 ± 0.1 | 23.4 ± 0.1 | 33.4 ± 0.1 | −1.0 | 33.4 ± 0.2 | 0.0 |
| C27 | 72.0 | 95.5 ± 0.1 | 63.1 ± 0.08 | 57.3 ± 0.06 | 38.2 ± 0.1 | −0.7 | 35.3 ± 0.1 | 0.2 |
| C28 | 75.6 | 101.0 ± 0.1 | 62.9 ± 0.1 | 62.9 ± 0.1 | 38.2 ± 0.2 | −1.0 | 38.2 ± 0.2 | 0.0 |
| C29 | 66.5 | 104.7 ± 0.4 | 47.4 ± 0.3 | 47.4 ± 0.2 | 57.3 ± 0.3 | −1.0 | 57.3 ± 0.7 | 0.0 |
| C30 | 49.5 | 90.7 ± 0.4 | 33.9 ± 0.3 | 23.9 ± 0.2 | 66.8 ± 0.3 | −0.7 | 61.8 ± 0.6 | 0.2 |
| C31 | 19.2 | 34.7 ± 0.07 | 12.0 ± 0.05 | 10.8 ± 0.04 | 23.8 ± 0.08 | −0.9 | 23.3 ± 0.1 | 0.07 |
| C32 | 21.2 | 36.5 ± 0.2 | 19.3 ± 0.1 | 7.8 ± 0.2 | 28.6 ± 0.3 | −0.2 | 22.9 ± 0.3 | 0.7 |
| C33 | 21.8 | 37.5 ± 0.4 | 18.9 ± 0.3 | 8.9 ± 0.3 | 28.6 ± 0.5 | −0.3 | 23.6 ± 0.6 | 0.6 |
| C34, C35 | 38.7 | 51.4 ± 0.2 | 37.1 ± 0.1 | 27.5 ± 0.1 | 23.9 ± 0.3 | −0.2 | 19.1 ± 0.3 | 0.7 |
| C36 | 16.5 | 27.6 ± 0.09 | 13.3 ± 0.04 | 8.5 ± 0.08 | 19.1 ± 0.1 | −0.5 | 16.7 ± 0.1 | 0.4 |
| C37 | 9.8 | 16.2 ± 0.1 | 6.6 ± 0.09 | 6.6 ± 0.1 | 9.5 ± 0.2 | −1.0 | 9.5 ± 0.2 | 0.0 |
| C38 | 16.5 | 27.6 ± 0.09 | 13.3 ± 0.04 | 8.5 ± 0.08 | 19.1 ± 0.1 | −0.5 | 16.7 ± 0.1 | 0.4 |
| C39 | 15.2 | 28.7 ± 0.07 | 12.0 ± 0.04 | 4.8 ± 0.05 | 23.8 ± 0.08 | −0.4 | 20.3 ± 0.1 | 0.5 |
| C40 | 26.5 | 45.4 ± 0.1 | 22.0 ± 0.08 | 12.0 ± 0.08 | 33.4 ± 0.1 | −0.4 | 28.4 ± 0.2 | 0.5 |
| C41 | 19.2 | 34.7 ± 0.07 | 12.0 ± 0.05 | 10.8 ± 0.04 | 23.8 ± 0.08 | −0.9 | 23.3 ± 0.1 | 0.07 |
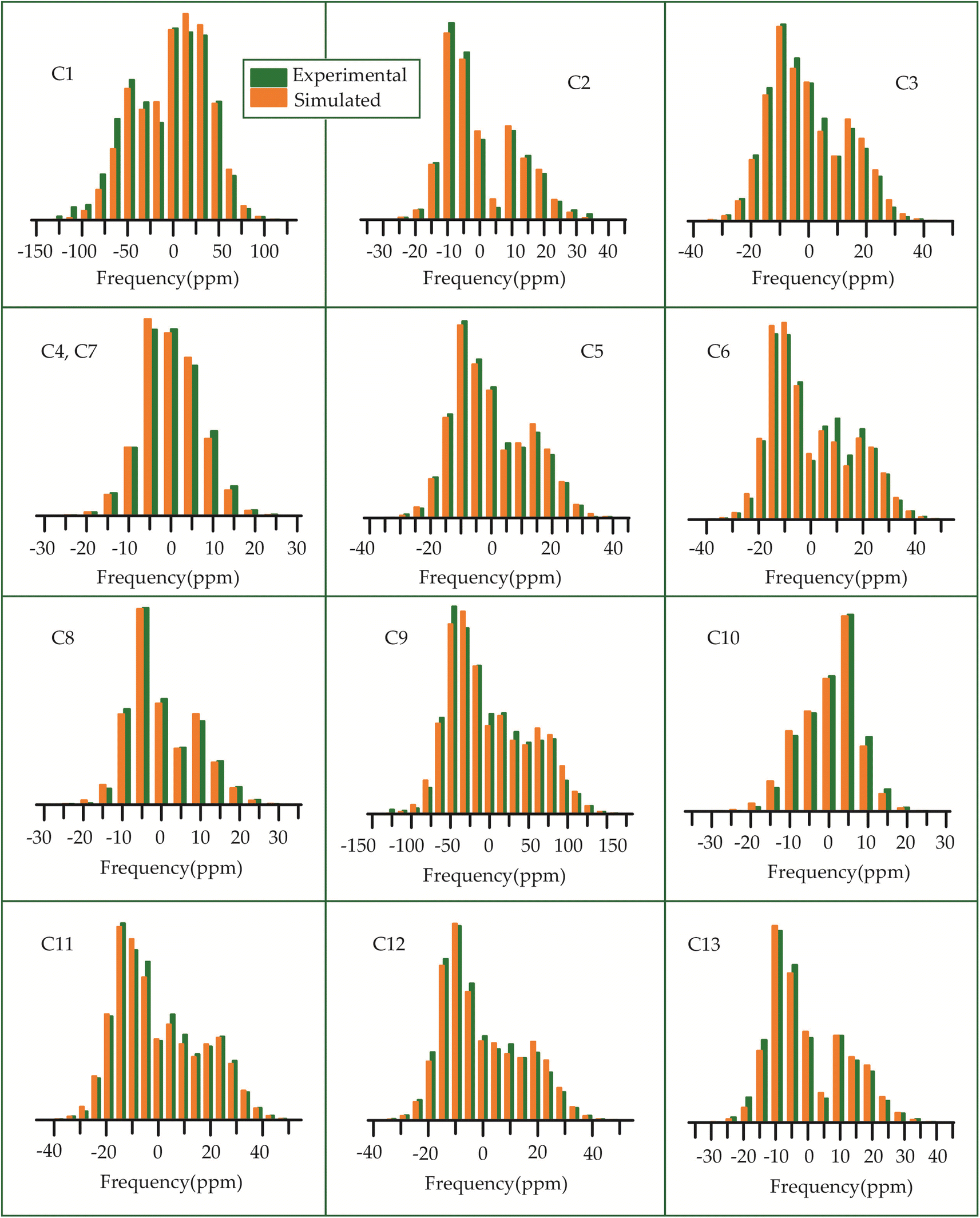 | ||
| Fig. 13 The spinning CSA sideband pattern of the carbon nuclei residing on the lactone ring of erythromycin. | ||
 | ||
| Fig. 14 The spinning CSA sideband pattern of the carbon nuclei residing on desosamine and cladinose of erythromycin. | ||
 | ||
| Fig. 15 The spinning CSA sideband pattern of the carbon nuclei residing on lactone ring of clarithromycin. | ||
 | ||
| Fig. 16 The spinning CSA sideband pattern of the carbon nuclei residing on desosamine and cladinose of clarithromycin. | ||
 | ||
| Fig. 17 Spinning CSA sideband pattern of crystallographically distinct carbon nuclei residing on lactone ring of azithromycin. | ||
 | ||
| Fig. 18 Spinning CSA sideband pattern of crystallographically distinct carbon nuclei residing on desosamine and cladinose of azithromycin. | ||
 | ||
| Fig. 19 Spinning CSA sideband pattern of crystallographically distinct carbon nuclei residing on lactone ring of roxithromycin. | ||
 | ||
| Fig. 20 Spinning CSA sideband pattern of crystallographically distinct carbon nuclei residing on desosamine and cladinose of roxithromycin. | ||
Table 3 demonstrates that for azithromycin, the principal component values of CSA parameters such as ‘span’ (159.1 ppm) and ‘anisotropy’ (−143.2 ppm) for C1 nuclei are higher than those for erythromycin, as evidenced by Table 1 where the ‘span’ and ‘anisotropy’ values are 143.2 ppm and −118.1 ppm, respectively. Furthermore, the value of principal component of chemical shift anisotropy tensor (δ22) for the C1 nucleus in azithromycin is recorded as 210.5 ppm (Table 3), whereas for roxithromycin, it is noted as 222.9 ppm (Table 4), both showing a shift towards higher frequency side compared to erythromycin (δ22 = 192.1 ppm, Table 1). The δ22 value serves as an indicator of hydrogen bonding strength associated with the keto group carbon, with a higher frequency indicating weaker hydrogen bonding.85,86 Thus, the higher value of δ22 in azithromycin and roxithromycin suggest a reduction in hydrogen bond strength associated with the C1 nucleus compared to erythromycin. Table 1 presents notable differences in the principal component values of chemical shift anisotropy (CSA) parameters for the C9 nucleus between erythromycin and azithromycin. For erythromycin, the parameters exhibit higher values: δ11 = 332.7 ppm, δ22 = 180.0 ppm, δ33 = 141.8 ppm, ‘span’ = 190.9 ppm, and ‘anisotropy’ = 171.8 ppm. In contrast, Table 3 illustrates substantially lower values for azithromycin: δ11 = 105.3 ppm, δ22 = 55.4 ppm, δ33 = 52.8 ppm, ‘span’ = 52.5 ppm, and ‘anisotropy’ = 51.2 ppm. As it is shown in Fig. 1 that a polar bond is associated with C9 in erythromycin, contributing to the elevated isotropic and anisotropic chemical shift values observed. The substantially large values of CSA parameters associated with the (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) group result from the shielding/deshielding effect induced by magnetic anisotropy. Lacking a symmetry axis, the carbonyl group exhibits three distinct magnetic susceptibilities (χx, χy, χz) along the axes of the principal axes system (PAS). Consequently, two anisotropic susceptibilities arise: one parallel (Δχ‖ = χz − χx) and the other perpendicular (Δχ⊥ = χy − χx) to the magnetic field.110 The McConnell equation111 describes the magnetic anisotropy for non-symmetric carbonyl groups as: δanis = {Δχ‖(3cos2θ1 − 1) + Δχ⊥(3cos2θ2 − 1)}/3R3, where θ1 denotes the angle between the radius vector and the x-axis, while θ2 represents the angle between the radius vector and the z-axis.110 This configuration generates a substantial directional-dependent magnetic field owing to the highly anisotropic magnetic susceptibility. Consequently, this phenomenon manifests as a significant anisotropic chemical shift at the carbonyl site. The electrostatic effect significantly influences carbonyl groups containing polar bonds with nonspherical charge distributions.86 The interaction of the central molecule with other molecules induces substantial polarization in the electronic density surrounding the central molecule. In the presence of an external magnetic field, this polarization alters the strength of the induced magnetic field around the C9 nucleus. This phenomenon contributes to the large anisotropy observed in carbonyl group carbon.
O) group result from the shielding/deshielding effect induced by magnetic anisotropy. Lacking a symmetry axis, the carbonyl group exhibits three distinct magnetic susceptibilities (χx, χy, χz) along the axes of the principal axes system (PAS). Consequently, two anisotropic susceptibilities arise: one parallel (Δχ‖ = χz − χx) and the other perpendicular (Δχ⊥ = χy − χx) to the magnetic field.110 The McConnell equation111 describes the magnetic anisotropy for non-symmetric carbonyl groups as: δanis = {Δχ‖(3cos2θ1 − 1) + Δχ⊥(3cos2θ2 − 1)}/3R3, where θ1 denotes the angle between the radius vector and the x-axis, while θ2 represents the angle between the radius vector and the z-axis.110 This configuration generates a substantial directional-dependent magnetic field owing to the highly anisotropic magnetic susceptibility. Consequently, this phenomenon manifests as a significant anisotropic chemical shift at the carbonyl site. The electrostatic effect significantly influences carbonyl groups containing polar bonds with nonspherical charge distributions.86 The interaction of the central molecule with other molecules induces substantial polarization in the electronic density surrounding the central molecule. In the presence of an external magnetic field, this polarization alters the strength of the induced magnetic field around the C9 nucleus. This phenomenon contributes to the large anisotropy observed in carbonyl group carbon.
By introducing a methyl-substituted nitrogen at position C9 in the lactone ring, azithromycin forms a larger macrocycle consisting of 15 members. By making this modification, the drug's efficacy against bacteria is enhanced, its stability in acidic environments is improved, and it leads to notably longer elimination half-lives and higher concentrations in tissues as compared to erythromycin.93
Although structurally erythromycin and clarithromycin are similar, but the spinning CSA sideband pattern of C9 nuclei of both drugs are different. For clarithromycin it is axially symmetric with ‘skew’ −1, but for roxithromycin (as shown in Table 4) it is nearly axially symmetric.
The spinning CSA sideband pattern (as shown in Figure 19 and Table 4) exhibits axial symmetry for C1 (asymmetry parameter η = (δ22 − δ11)/(δ33 − δiso) = 0) nuclei in roxithromycin, while it is nearly axial symmetry in clarithromycin and azithromycin (η = (δ22 − δ11)/(δ33 − δiso)=0.3) as shown in Tables 2 and 3. Roxithromycin, in contrast to erythromycin, displays a change in the sign of the ‘anisotropy’ parameter. For roxithromycin Δδ = δ33 − 1/2(δ11 + δ22) = −165.4 ppm (as it is shown in Table 4), while it is Δδ = δ33 − 1/2(δ11 + δ22) = 171.8 ppm for erythromycin (as it is shown in Table 1).
Notably, the local electronic distribution surrounding C9 nuclei shows a clear indication of change. Despite having a similar basic configuration (refer to Fig. 1 and 3), erythromycin and roxithromycin differ only the attachment of the N-oxime side chain to the C9 atom of the 14-membered lactone ring. In roxithromycin, the N-oxime side chain replaces the keto group of erythromycin.15 The ketone group in erythromycin is protonated under acidic conditions, converting it to anhydroerythromycin by forming an intramolecular cyclic acetal, owing to the presence of alcohols at C6 and C11.12 Consequently, the structural modification of erythromycin through the replacement of the ketone group with the oxime side chain enhances the stability of the macrolide antibiotic in acidic environments (it is discussed in Section 4 by Molecular dynamic simulation that the biological activity is enhanced in roxithromycin, compared to erythromycin). In the following section, we will delve into how this structural modification significantly impacts the spin-dynamics of the C9 nuclei.
In their study, Gharbi-Benarous et al.95 observed that the hydroxyl protons attached to the C6 and C11 carbon atoms of the lactone ring in roxithromycin establish intra-molecular hydrogen bonds with the oxime chain (it is shown in Fig. S8 of ESI†). Conversely, in erythromycin, the hydroxyl groups of the lactone ring form intermolecular hydrogen bonds with other molecules or water molecules. Consequently, roxithromycin lacks free hydroxyl groups, leading to increased hydrophobicity. The involvement of C6–OH and C11–OH in hydrogen bonding with the oxime chain gives rise to a globular molecular structure. In contrast, hydroxyl groups C6–OH and C11–OH of erythromycin exhibit a less rigid conformation, allowing them to interact with polar solvents or associate with other units.95
3.5 Determination of 13C spin-lattice relaxation time of macrolide antibiotic
The bar-diagram of the spin-lattice relaxation time of the carbon nuclei residing on lactone ring, cladinose, and desosamine for erythromycin, roxithromycin, and azithromycin are shown in Fig. 21. Similarly, the bar-diagram of the spin-lattice relaxation time for erythromycin and clarithromycin are shown in Fig. 22 for the carbon nuclei residing on lactone ring, cladinose, desosamine. The larger molecules exhibit higher conformational flexibility due to intermolecular and intramolecular interactions. The substitutional group determines the spin-lattice relaxation time of various carbon nuclei on the lactone ring, and a substantial difference in relaxation time exists among 13 carbon nuclei (C1 to C13 as depicted in Fig. 1 and 2) in four macrolide antibiotics. The relaxation time is comparatively longer for C5 and C3 carbon (attached to two deoxysugars desosamine and cladinose respectively) than C2 (attached to a methyl group). In roxithromycin, the spin-lattice relaxation time of C9 nucleus is significantly shortened as the ketone group is replaced by the oxime group.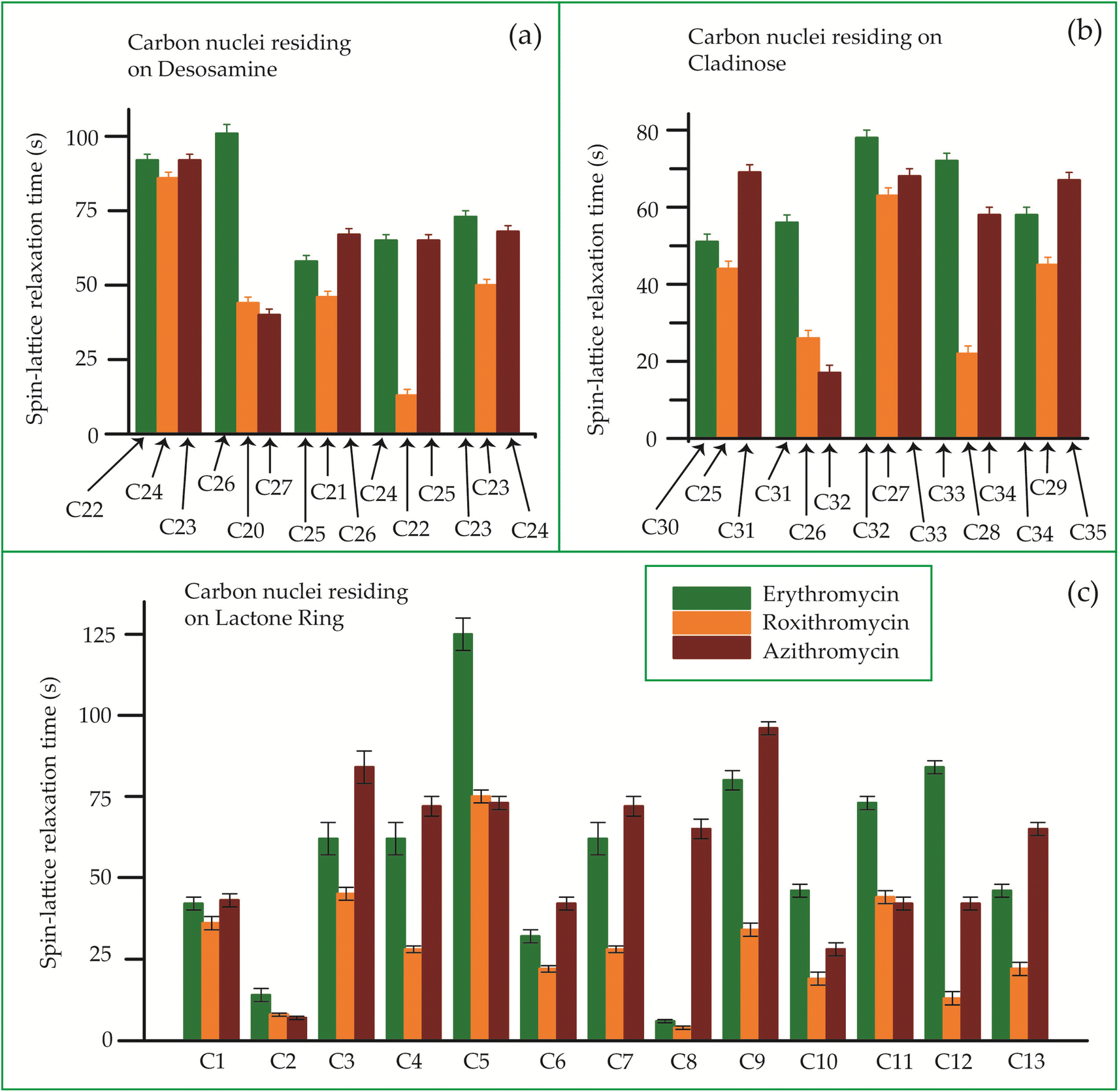 | ||
| Fig. 21 Bar-diagram of the spin-lattice relaxation time of the carbon nuclei residing on (c) lactone ring, (b) cladinose, (a) desosamine for erythromycin, roxithromycin, and azithromycin. | ||
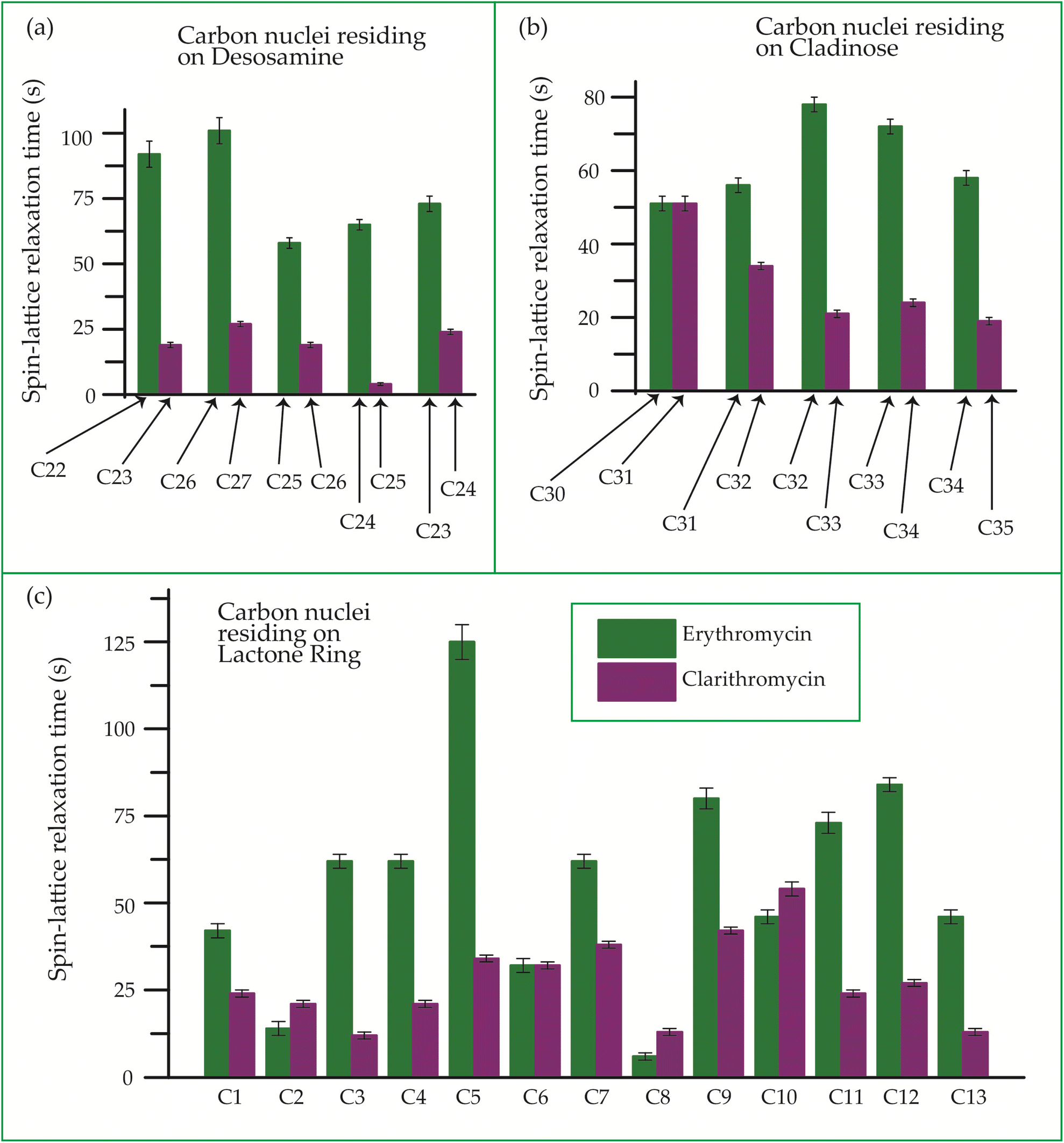 | ||
| Fig. 22 Bar-diagram of spin-lattice relaxation time of the carbon nuclei residing on (c) lactone ring, (b) cladinose, (a) desosamine for erythromycin, and clarithromycin. | ||
In their study, Benarous et al.95 examined the comparative conformation of erythromycin and roxithromycin. They found that while the overall three-dimensional structures of both molecules are quite similar, there are slight differences in torsion angles in certain regions. Specifically, the C6–C10 and Cll–C13 portions of the aglycons share similar torsion angles, whereas the Cl–C3, C4–C6, C10–C11, and C13–C14 regions exhibit slight variations. Additionally, the researchers noted that the orientation of the hydroxyl group and lactone ring differs between erythromycin and roxithromycin, and the orientation of the sugar rings with respect to each other and with respect to the lactone ring is also different for these two molecules. It was reported by in vitro simulations of steady-state pharmacokinetics in human tonsils that the antistreptococcal effect of azithromycin on Streptococcus pyogenes and streptococcus pneumonia is more than roxithromycin.12 Notably, Table 5 and Fig. 21 demonstrate that the spin-lattice relaxation time of carbon nuclei in the lactone ring, desosamine, and cladinose is shorter in roxithromycin compared to erythromycin. This distinction may contribute to the superior pharmacokinetic properties observed in roxithromycin when compared to erythromycin.
| Erythromycin | Roxithromycin | Azithromycin | Clarithromycin | ||||
|---|---|---|---|---|---|---|---|
| Carbon nuclei | Relaxation time (s) | Carbon nuclei | Relaxation time (s) | Carbon nuclei | Relaxation time (s) | Carbon nuclei | Relaxation time |
| Relaxation time of the carbon nuclei residing on lactone ring | |||||||
| C1 | 42 ± 2 | C1 | 36 ± 2 | C1 | 43 ± 2 | C1 | 24 ± 1 |
| C2 | 14 ± 2 | C2 | 8 ± 0.5 | C2 | 7 ± 0.5 | C2 | 21 ± 1 |
| C3 | 62 ± 2 | C3 | 45 ± 2 | C3 | 84 ± 5 | C3 | 12 ± 1 |
| C4 | 62 ± 2 | C4 | 28 ± 2 | C4 | 72 ± 2 | C4 | 21 ± 1 |
| C5 | 125 ± 5 | C5 | 75 ± 2 | C5 | 73 ± 2 | C5 | 34 ± 1 |
| C6 | 32 ± 2 | C6 | 22 ± 2 | C6 | 42 ± 2 | C6 | 32 ± 1 |
| C7 | 62 ± 2 | C7 | 28 ± 2 | C7 | 72 ± 2 | C7 | 38 ± 1 |
| C8 | 6 ± 1 | C8 | 4 ± 1 | C8 | 65 ± 2 | C8 | 13 ± 1 |
| C9 | 80 ± 3 | C9 | 34 ± 2 | C9 | 96 ± 2 | C9 | 42 ± 1 |
| C10 | 46 ± 2 | C10 | 19 ± 2 | C10 | 28 ± 2 | C10 | 54 ± 2 |
| C11 | 73 ± 3 | C11 | 44 ± 2 | C11 | 42 ± 2 | C11 | 24 ± 1 |
| C12 | 84 ± 2 | C12 | 13 ± 1 | C12 | 42 ± 2 | C12 | 27 ± 1 |
| C13 | 46 ± 2 | C13 | 22 ± 2 | C13 | 65 ± 2 | C13 | 13 ± 1 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||||
| Relaxation time of the carbon nuclei residing on desosamine | |||||||
| C22 | 92 ± 5 | C24 | 86 ± 5 | C23 | 92 ± 5 | C23 | 19 ± 1 |
| C26 | 101 ± 5 | C20 | 44 ± 2 | C27 | 40 ± 2 | C27 | 27 ± 1 |
| C25 | 58 ± 2 | C21 | 46 ± 2 | C26 | 67 ± 2 | C26 | 19 ± 1 |
| C24 | 65 ± 2 | C22 | 13 ± 2 | C25 | 65 ± 2 | C25 | 4 ± 0.5 |
| C23 | 73 ± 3 | C23 | 50 ± 2 | C24 | 68 ± 2 | C24 | 24 ± 1 |
|
|||||||
| Relaxation time of the carbon nuclei residing on cladinose | |||||||
| C30 | 51 ± 2 | C25 | 44 ± 2 | C31 | 69 ± 2 | C31 | 51 ± 2 |
| C31 | 56 ± 2 | C26 | 26 ± 1 | C32 | 17 ± 1 | C32 | 34 ± 1 |
| C32 | 78 ± 2 | C27 | 63 ± 2 | C33 | 68 ± 2 | C33 | 21 ± 1 |
| C33 | 72 ± 2 | C28 | 22 ± 1 | C34 | 58 ± 2 | C34 | 24 ± 1 |
| C34 | 58 ± 2 | C29 | 45 ± 2 | C35 | 67 ± 2 | C35 | 19 ± 1 |
Clarithromycin, a semisynthetic 14-membered macrolide antibiotic with antimicrobial properties, differs structurally from erythromycin solely by the substitution of an O-methyl group for the hydroxyl group attached to the C6 carbon of the lactone ring. Despite this seemingly minor alteration, its impact is significant. This modification enhances the tissue penetration capability of clarithromycin when compared to erythromycin, thereby contributing to an improved pharmacokinetic profile for this particular macrolide.107 Conformational analysis has revealed that macrolide antibiotics ingress into the hydrophobic domain of a phosphatidylinositol monolayer via their desosamine and cladinose constituents.14 Table 5 and Fig. 22 demonstrate that the spin-lattice relaxation time of carbon nuclei residing on lactone ring (except C2 and C10), desosamine and cladinose of clarithromycin is shorter compared to erythromycin. This distinction may contribute to the higher bioavailability observed in clarithromycin.
Azithromycin features a methyl-substituted nitrogen atom integrated into its lactone ring, whereas roxithromycin incorporates an N-oxime side chain attached to the lactone ring.103 These small chemical modification results in increased acid stability in both roxithromycin and azithromycin compared to erythromycin.108 The enhancement of spin-lattice relaxation rate is evident in carbon nuclei located on the lactone ring, desosamine ring, and cladinose ring of both roxithromycin and azithromycin, as depicted in Fig. 21 and Table 5. This enhanced relaxation rate is found to be correlated with the improved acid stability observed in these two macrolide antibiotics—roxithromycin and azithromycin.
We have observed the phenomena of the correlation between the enhancements of the spin-lattice relaxation rate and bioactivity in ofloxacin and its levo-isomer levofloxacin. Levofloxacin is more active antibiotic than ofloxacin and we have observed by NMR relaxometry measurements that the spin-lattice relaxation time is shortened for each nucleus of levofloxacin except C8. Bioavailability closely ties into a drug molecule's bioactivity. Hence, it's plausible to propose a connection between nuclear spin-lattice relaxation and drug bioactivity. The gradual shortening in spin-lattice relaxation time of levofloxacin compared to ofloxacin might underpin the heightened antibacterial effectiveness of levofloxacin.121
Quinine (1S, 3R, 4S, 8S, 9R) and its pseudoenantiomer quinidine (1S, 3R, 4S, 8R, 9S) differ in configuration at the C8 and C9 positions, leading to significant differences in their motional dynamics. For the three independent molecules of quinine, the relaxation times for C8 and C9 are nearly same and the values are 537 s and 555 s, respectively. In contrast, for quinidine, these values are 330 s and 370 s, respectively. Among Cinchona alkaloids, quinidine is more effective than quinine, as antimalarial activity is highly correlated with the orientation of the amine and hydroxyl groups (8R, 9S). Relaxation measurements indicate that the spin-lattice relaxation time of the C8 and C9 nuclei correlates with the efficiency of Cinchona alkaloids as antimalarial drugs. The rotation around the C8–C9 bond significantly influences the conformational behavior of these alkaloids. The higher motional dynamics of the C8 and C9 nuclei in quinidine, which connects the aromatic quinoline ring with the aliphatic bicyclic quinuclidine ring of Cinchona alkaloids, can be considered as the origin of the greater efficiency of quinidine compared to quinine.81 C6′ is one of the stereogenic centers of Cinchona alkaloids, due to the rotation of the methoxy group (CH3O) around the C6′–O bond. The spin-lattice relaxation times of the C6′ nuclei in the ‘a’, ‘b’, and ‘c’ molecules of quinine are 383 s, 285 s, and 290 s, respectively, while for quinidine, it is 170 s. Therefore, the motional dynamics of the stereogenic centers in quinidine are faster than those in quinine, which can be considered as a reason of the higher efficacy of quinidine as antimalarial drug.81
In case of five glucocorticoid we also have observed the same phenomena of the interrelation between spin-lattice relaxation time and bioactivity. From prednisone to prednisolone to methylprednisolone, and from prednisolone to prednisolone acetate to methylprednisolone acetate, both the spin-lattice relaxation time and the local correlation time decrease. In silico analysis further supports this trend, indicating enhanced bioactivity from prednisone to prednisolone to methylprednisolone, and from prednisolone to prednisolone acetate to methylprednisolone acetate.82 There is no such concrete theory correlating drug activity with the enhancement of nuclear spin-dynamics, but our observations on various drug molecules will definitely stimulate scientists to investigate this important aspect of the drug discovery.
3.6 13C–1H frequency-switched Lee-Goldburg heteronuclear correlation (FSLGHETCOR) spectrum of erythromycin, clarithromycin, azithromycin, and roxithromycin
The motional dynamics of the molecule profoundly influence the 1H–13C heteronuclear dipolar coupling, a phenomenon amenable to investigation through 2D 1H–13C FSLG HETCOR (Heteronuclear Correlation) experiments. Employing a contact time of 0.2 ms, the cross-peaks discernible in the HETCOR spectrum (Fig. 23–25) predominantly originate from direct 1H–13C bonding interactions. The presence of pronounced 1H–13C dipolar couplings indicates reduced molecular mobility, with the notable abundance of cross-peaks in Fig. 23–25 suggesting restricted nuclear motions.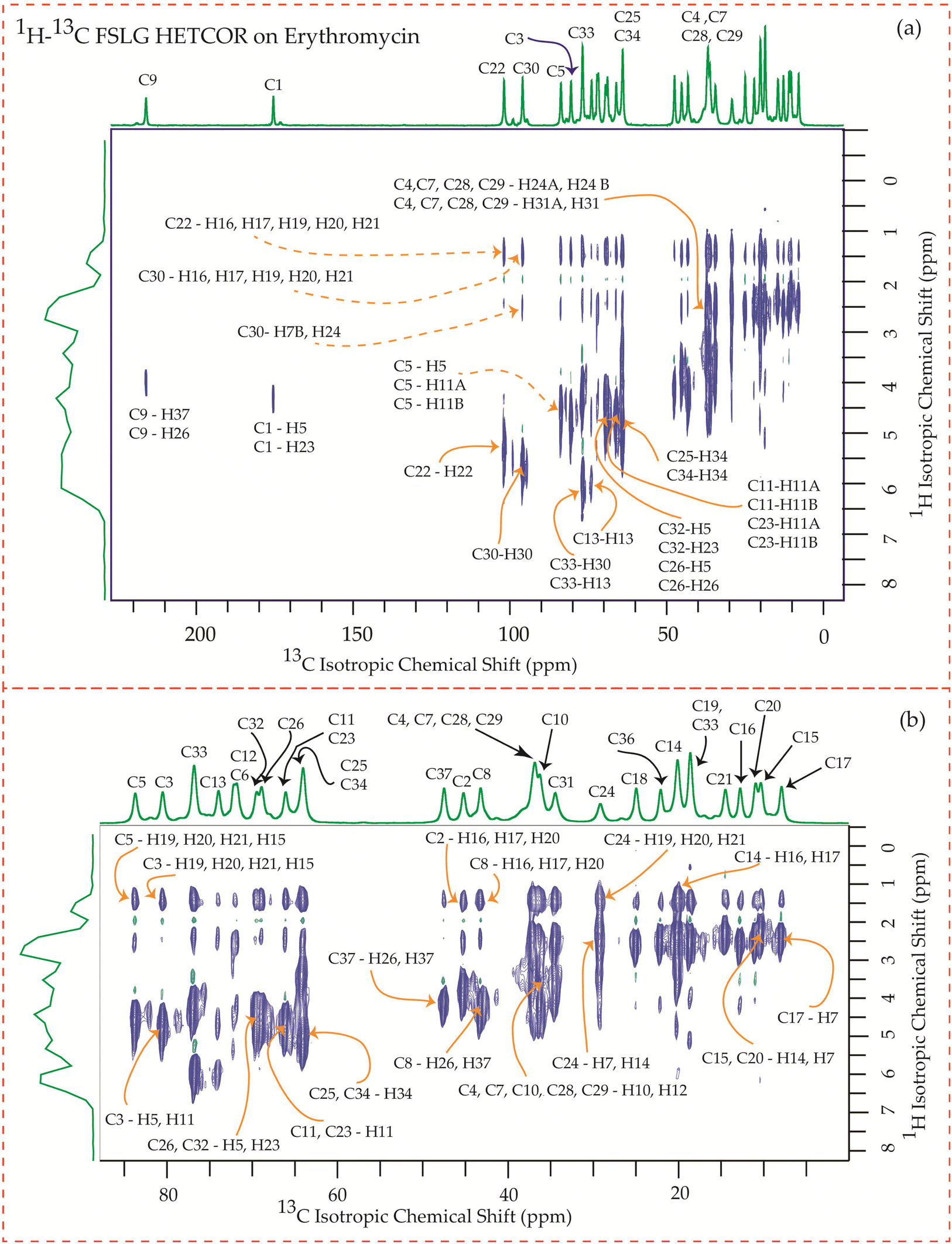 | ||
| Fig. 23 (a) 1H–13C FSLG HETCOR spectrum of erythromycin. (b) Enlarged version of 1H–13C FSLG HETCOR spectrum from C5 to C17. | ||
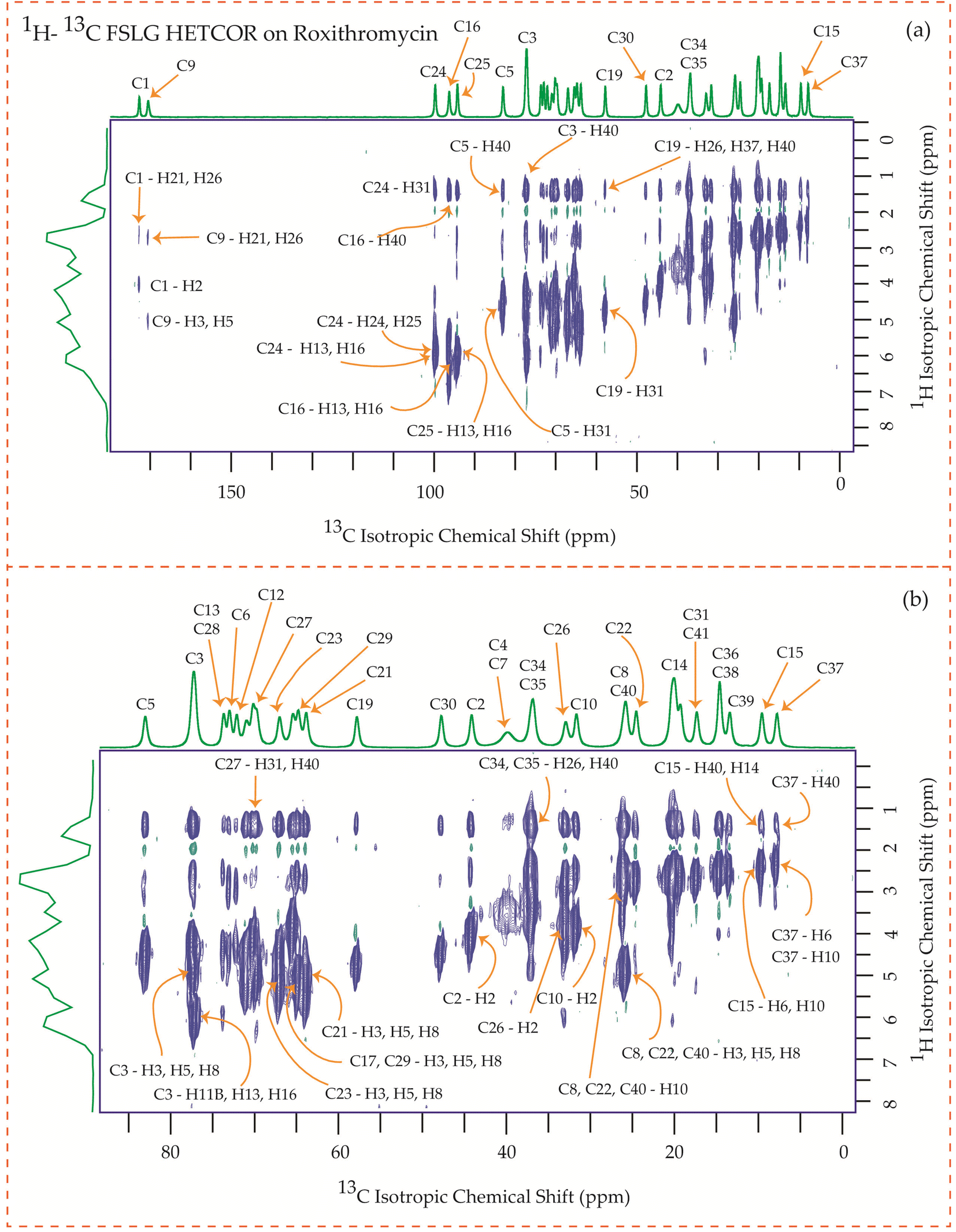 | ||
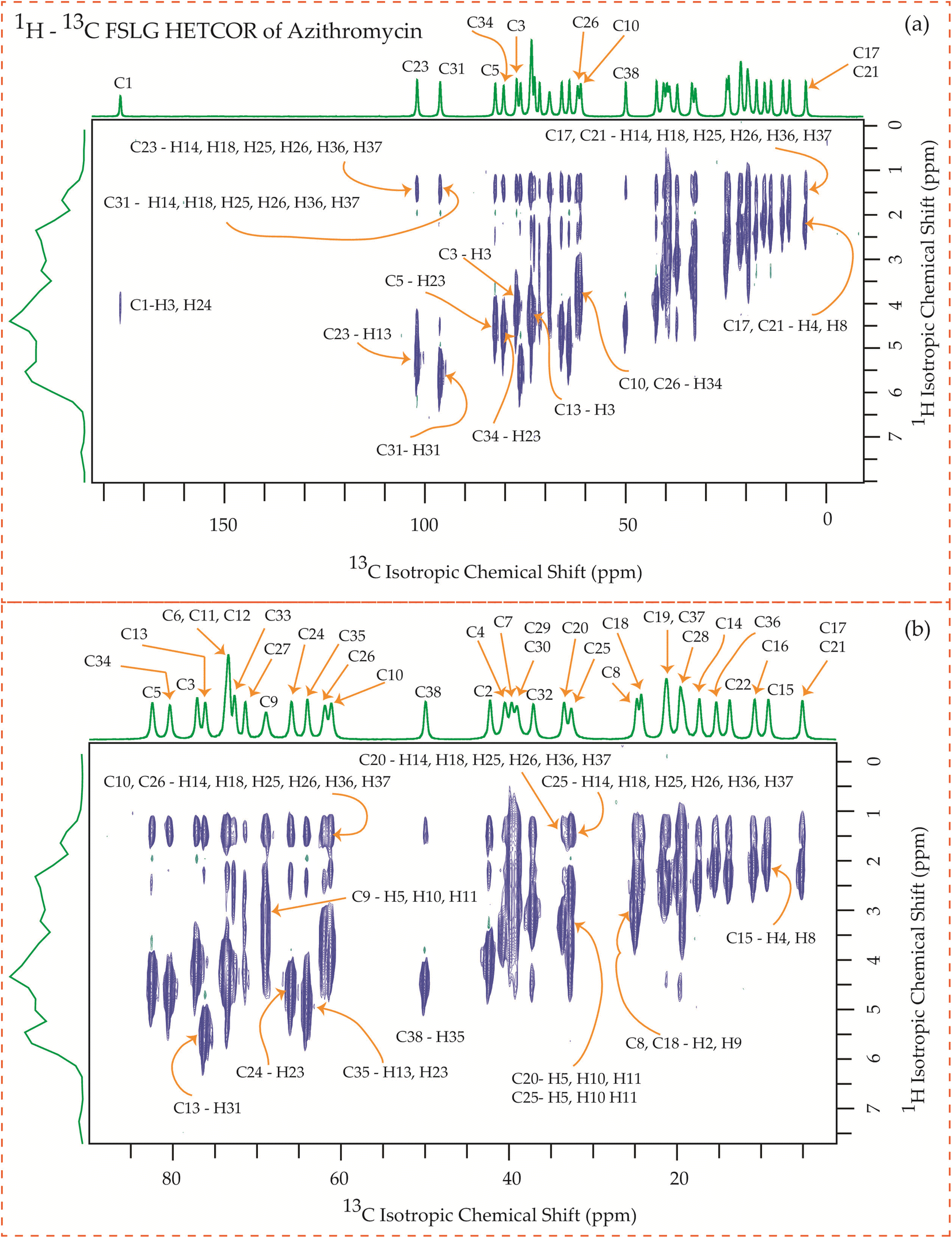
| Fig. 24 1H–13C FSLG HETCOR spectrum of roxithromycin. (b) Enlarged version of 1H–13C FSLG HETCOR spectrum from C5 to C37. | ||
 | ||
| Fig. 25 1H–13C FSLG HETCOR spectrum of azithromycin. (b) Enlarged version of 1H–13C FSLG HETCOR spectrum from C5 to C17. | ||
The 1H–13C heteronuclear correlation spectrum of erythromycin, depicted in Fig. 23, reveals several spatial correlations between carbon and hydrogen atoms in different rings. In particular, the C9 carbon of the lactone ring exhibits spatial proximity with the H37 hydrogen atom of cladinose and the H26 hydrogen atom of desosamine. Additionally, the C1 carbon of the lactone ring is spatially correlated with the H23 atom of desosamine. The C4, C7, and C10 carbon atoms of the lactone ring show spatial correlation with the H28 and H29 protons of desosamine, as well as the H31A and H33B protons of cladinose. Furthermore, the C8 carbon atom of the lactone ring is correlated with the H26A and H26B protons of desosamine, as well as the C37 protons of cladinose. The C32 carbon of cladinose exhibits spatial correlation with the H5 protons of the lactone ring and the H23 protons of desosamine. Similarly, the C26 carbon atoms of desosamine are correlated with the H5 proton of the lactone ring. Moreover, the C22 carbon of the desosamine ring shows correlation with the H20 and H21 protons of the lactone ring. The C30 carbon of the cladinose ring demonstrates spatial correlation with the H7B, H14B, H16, H18, H19, H20, H21 protons of the lactone ring, as well as the H24 protons of desosamine. These spatial proximities between various carbons and protons in the three rings are observed through the 1H–13C FSLG HETCOR spectrum of erythromycin.
Fig. 24 illustrates the 1H–13C FSLG HETCOR spectrum of roxithromycin, revealing the spatial proximity between carbon atoms in the oxime chain with the protons in the lactone ring, and cladinose, not with the protons of desosamine. Specifically, the C18 carbon of the oxime chain shows correlation with the H3, H5, H8, H37, and H40 protons of the lactone ring, as well as the H31 proton of cladinose. Similarly, the C16 carbon of the oxime chain correlates with the H11B, H13, and H40 protons of the lactone ring, and the H31 proton of cladinose. The C17 carbon is connected to the H3, H5, H8, H37, and H40 protons of the lactone ring, along with the H31 proton of cladinose. Moreover, the C19 carbon is connected to the H37 and H40 protons of the lactone ring, as well as the H26 and H31 protons of the cladinose ring. It is notable that the C24 carbon of desosamine is spatially correlated with H16 proton, bonded with C16 carbon, of oxime chain. In addition, the C1 and C9 carbons of the lactone ring exhibit spatial correlation with the H21 proton of desosamine and the H26 proton of cladinose. C2, C3, C5, C8, C10 carbons are spatially correlated with H21 proton of desosamine and H26 proton of cladinose. The C24 carbon of desosamine is correlated with the H25 and H31 protons of cladinose, as well as the H40 proton of the lactone ring.
Fig. 25 showcases the 1H–13C FSLG HETCOR spectrum of azithromycin, highlighting the spatial proximity between various carbon nuclei in the lactone ring of the macrolide antibiotic and the protons bonded to the carbon nuclei in the desosamine and cladinose sugar units. Likewise, certain carbon nuclei in the desosamine and cladinose units exhibit spatial proximity with specific protons attached to the lactone ring. The figure includes designated cross-peaks, such as the correlation between C1 of the lactone ring and H24 of desosamine. Furthermore, the C2 carbon of the lactone ring is spatially connected to the H24, H25, and H26 protons of desosamine, as well as the H36 and H37 protons of the methyl groups bonded with cladinose. Similarly, the C5 carbon is spatially correlated with the H23, H25, and H26 protons of desosamine, along with the H36 and H37 protons of the methyl groups bonded with cladinose. Additionally, the C3 and C13 carbons exhibit spatial correlation with the H24, H25, and H26 protons of desosamine, as well as the H36 and H37 protons of cladinose. Likewise, the C6, C11, and C12 carbons are spatially correlated with the H23, H24, H25, and H26 protons of desosamine, and the H36 and H37 protons of cladinose. The C10 carbon shows spatial correlation with the H34, H36, and H37 protons of cladinose, as well as the H25 and H26 protons of desosamine. The C8 carbon of the lactone ring exhibits spatial correlation with the H25 and H26 protons of desosamine, and the H36 and H37 protons of the methyl groups bonded with cladinose.
The C4 and C7 carbons of the lactone ring in azithromycin exhibit spatial correlation with the H25 and H26 protons of desosamine, as well as the H32 protons of cladinose and the H36 and H37 protons of the methyl groups bonded with cladinose. Similarly, the C9 carbon of the lactone ring is spatially correlated with the H25 and H26 protons of desosamine, the H32 protons of cladinose, and the H36 and H37 protons of the methyl groups bonded with cladinose. Consequently, through 1H–13C HETCOR experiments, it is observed that all thirteen carbon nuclei in the lactone ring display spatial correlation with specific protons from the two sugar units, desosamine and cladinose, that are connected to the lactone ring in azithromycin. In both erythromycin and azithromycin, the C1 carbon is spatially correlated with desosamine. However, for roxithromycin, it is correlated with both desosamine and cladinose.
The C33 carbon of azithromycin located in cladinose shows spatial correlation with the H14 and H18 protons of the methyl and ethyl groups connected to the lactone ring, as well as the H25 and H26 protons of desosamine. Similarly, the C23 carbon in desosamine exhibits spatial correlation with the H14 and H18 protons of the methyl and ethyl groups connected to the lactone ring, along with the H13 proton of the lactone ring. Additionally, it is correlated with the H36 and H37 protons of the methyl group connected to cladinose. Furthermore, the C24 carbon in desosamine displays spatial correlation with the H4 and H8 protons of the lactone ring, the H14 and H18 protons of the methyl and ethyl groups connected to the lactone ring, and the H36 and H37 protons of the methyl group connected to cladinose.
In these four macrolide antibiotics the cladinose and desosamine sugar units are connected with lactone ring via C3 and C5 carbons. It is worth noting from Fig. 25 that the C3 and C5 carbons exhibit spatial correlation with specific protons from the desosamine and cladinose units in azithromycin. However, in the case of erythromycin, as shown in Fig. 23, the C3 and C5 carbons are spatially correlated with the H11, H15, H19, H20, and H21 protons, which are either bonded to carbon atoms within the lactone ring or to methyl groups attached to the lactone ring. Hence, no spatial correlation is observed among C3, C5 carbons of erythromycin with any protons residing on cladinose and desosamine units. Conversely, in roxithromycin, both the C3 and C5 carbons are spatially correlated with the H31 and H26 protons of cladinose, as well as the H21 proton of desosamine.
4 Molecular dynamics simulation of two macrolide antibiotics erythromycin and roxithromycin in complex with ribosomal protein L4
Molecular dynamics simulation serves as a sophisticated computational method for exploring and understanding the intricate movements and behaviors of atoms and molecules within a given system over a defined period.112,113 In this investigation, we embarked on molecular dynamics simulations involving erythromycin and roxithromycin when bound to ribosomal protein L4. Our primary aim was to delve into the structural dynamics and interactions between these antibiotics and protein L4. Erythromycin and its derivatives belong to the macrolide antibiotic class, known for their ability to bind to the 50S ribosomal subunit of bacteria, thereby impeding bacterial protein synthesis. Notably, within this ribosomal subunit, both erythromycin and its derivatives engage in specific interactions with protein L4. These interactions play a pivotal role in disrupting bacterial protein synthesis machinery, consequently inhibiting bacterial growth.114–116 Thus, through the analysis of these molecular dynamics simulations, we aim to elucidate how structural alterations in erythromycin influence its biological activity. This research offers valuable insights into the diverse efficacies of these antibiotics and their potential implications for pharmacology.For MD simulation, first we have obtained crystal structure of erythromycin, roxithromycin and ribosomal protein L4 in the form of complexes from RCSB protein data bank (https://www.rcsb.org/). These structures were then processed and prepared using Discovery Studio Visualizer version 2021 (https://www.3ds.com/products/biovia/discovery-studio/visualization) to ensure they were clean and suitable for MD simulation. The entire MD simulation was performed by using GROMACS-2023 (https://www.gromacs.org/), a widely used molecular dynamics simulation software. We followed the recommended steps outlines in the GROMACS tutorial for simulating protein–ligand complexes (https://www.mdtutorials.com/gmx/complex/index.html) to set up and execute our simulations effectively. For our molecular dynamic simulations, we utilized the Charmm force field. Prior to initiating MD simulation, we prepared corresponding topology files for both the ligand and the L4 protein. Subsequently, a unit cell was defined and filled with water and ions subjected to replicate a physiological environment resembling that of the human body as it is shown in Fig. S1 of ESI.† To stabilize the system, the entire setup underwent energy minimization. Following this, the system was subjected to equilibrium using NVT (canonical ensemble) and NPT (isothermal-isobaric ensemble) statistics to ensure proper temperature and pressure conditions. After achieving equilibrium, the system was prepared for the MD simulation, where it was stimulated at a desired temperature of 300 K and pressure of 1 bar. Each complex, consisting of antibiotics-L4 protein complex, water molecules, and ions, underwent a 100 ns MD simulation to study their dynamic behaviour and interactions over an extended period. Upon completion of MD simulation, the coordinates of the system were rewrapped and recentred to prepare the data for further analysis and interpretation. This comprehensive process enabled us to simulate and study the behaviour of the antibiotics-L4 protein complexes under realistic physiological conditions, providing valuable insights into their structural dynamics and stability.
5 Results obtained from MD simulations
5.1 The root mean square deviation (RMSD) analysis
The root mean square deviation gives the idea about the dynamics of a molecule in a system over a particular timeframe. The root mean square deviation of the atoms is represented by the equation,
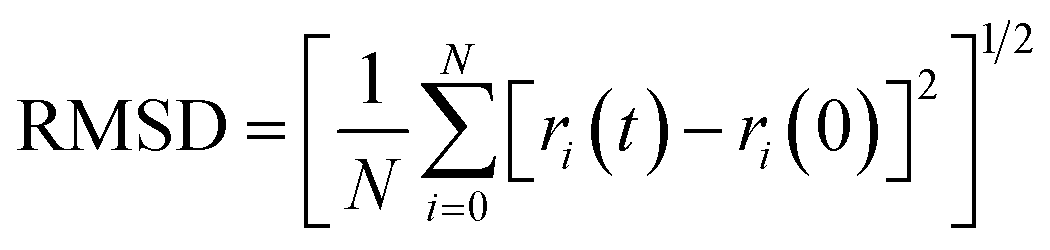 | (1) |
In Fig. 26(a), we observe the root mean square deviations (RMSD) of atoms within protein L4 during their interactions with erythromycin and roxithromycin across a 100 ns timeframe. When bound to erythromycin, protein L4 displayed RMSD values ranging between 1.5 Å and 2.8 Å throughout the initial 20 ns of simulation. Between 20 ns and 60 ns, these values continued to fluctuate, stabilizing around 2.0 Å on average. Subsequently, after 60 ns, the RMSD values increased further, reaching a range of 2.0 Å to 3.2 Å. Conversely, when protein L4 interacted with roxithromycin, the RMSD values initially remained lower than those observed with erythromycin, ranging from 1.5 Å to 2.4 Å over the first 20 ns. However, after 20 ns, the RMSD value peaked at 3.5 Å before stabilizing. Overall, roxithromycin induced greater RMSD values and stability compared to erythromycin, indicating its influence on the dynamic behavior of protein L4.
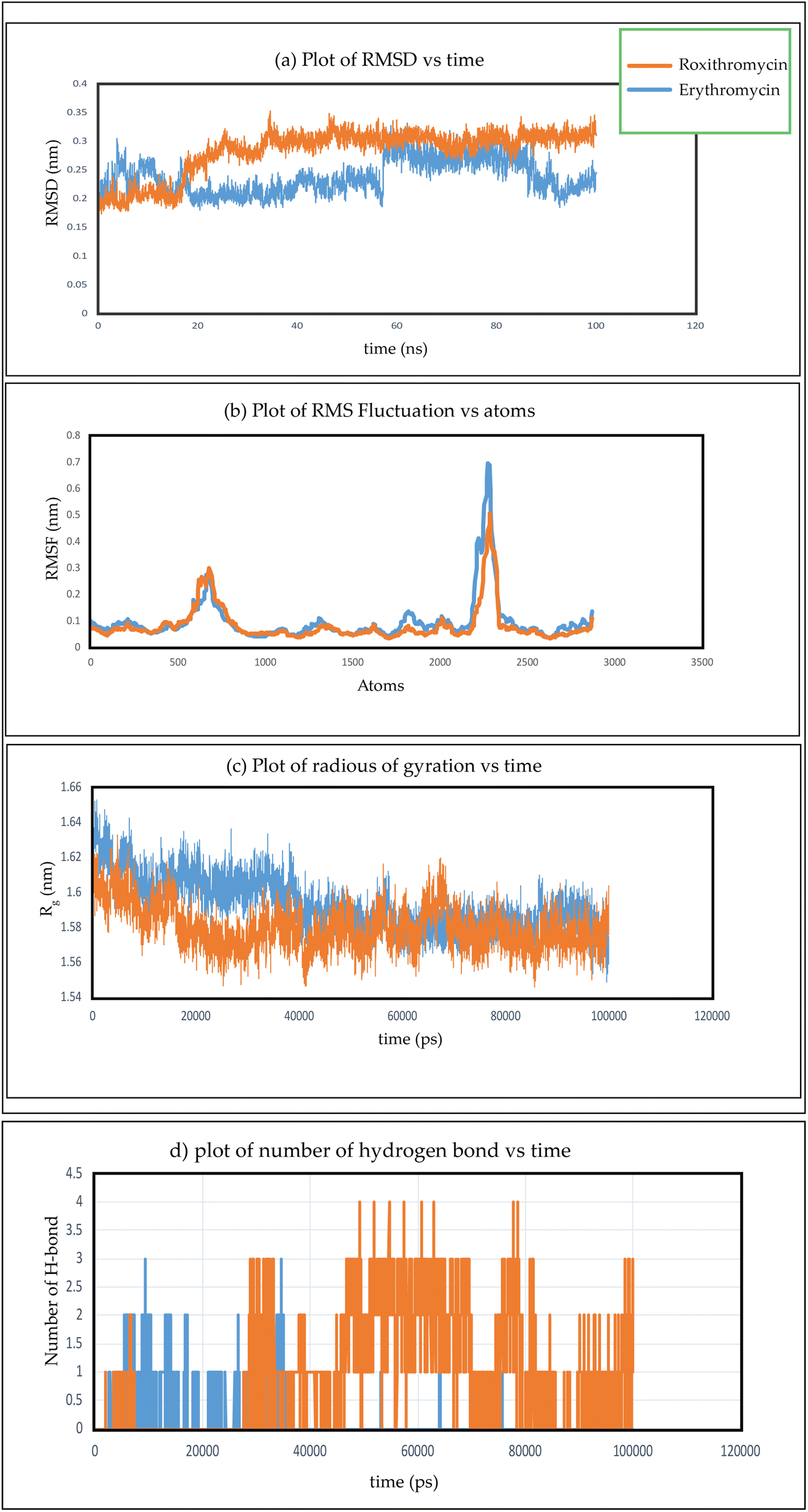 | ||
| Fig. 26 (a) Plot of RMSD verses time, (b) plot of RMSF verses atoms, (c) plot of radius of gyration verses time, (d) plot of number of hydrogen bond verses time for two complex of erythromycin–protein complex and roxithromycin–protein complex. | ||
5.2 RMSF analysis
To investigate the conformational changes of protein during molecular dynamics simulations, we analysed the fluctuations in the mean structure of protein, specifically focusing on the fluctuations in mean atomic quadratic deviations (RMSF). The displacement and stability of individual residues for each system throughout the simulation trajectory are quantified and further analysed by using the equation,113
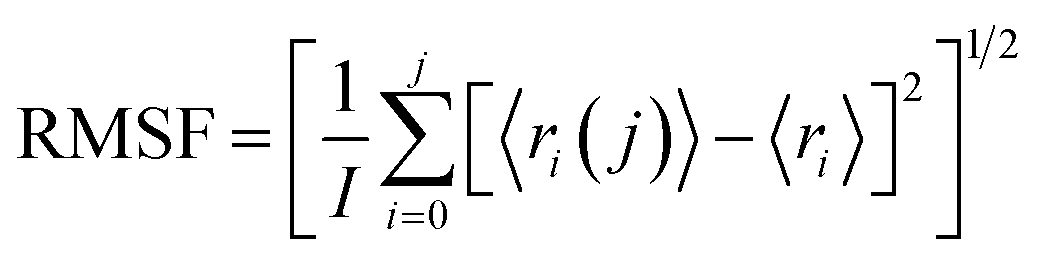 | (2) |
From the plot depicting root mean square fluctuation (RMSF) against protein atoms as shown in Fig. 26(b), it is evident that all atoms within protein L4 exhibits lightly higher fluctuation when interacting with erythromycin than that of roxithromycin. However, residues numbered 2022 to 2024 of the protein show noticeable higher fluctuations when interacting with erythromycin compared to roxithromycin. This suggests that overall, protein L4 displays lower fluctuation when interacting with roxithromycin, indicating that roxithromycin may be biologically more effective than erythromycin.
5.3 Radius of gyration analysis
The radius of gyration (Rg) verses time plot in molecular dynamics simulations provides valuable insights into the compactness and stability of protein-ligand complex over the course of simulation.120 In Fig. 26(c), we can observe how the compactness of the protein influenced by interacting with erythromycin and its derivative roxithromycin. The radius of gyration is calculated from the relation,
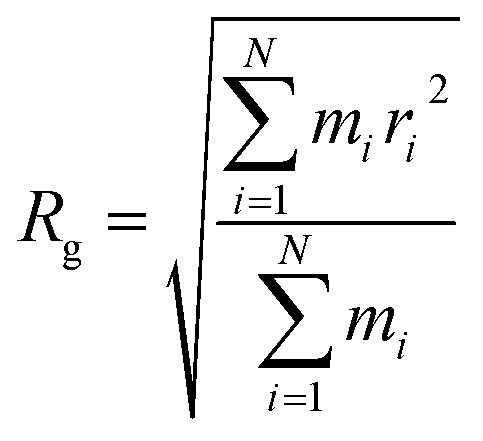 | (3) |
From the result of radius of gyration verses time plot, it is observed that the protein L4 exhibits lower radius of gyration and maintain a more stable structure when bound to roxithromycin compared to erythromycin, except for a specific period between approximately 65 ns and 75 ns in the simulation. Specifically, the lower radius of gyration observed in the roxithromycin–protein complex indicates a more compact and tightly folded structure throughout most of the simulation duration. A lower radius of gyration typically signifies reduced structural flexibility and extension within the protein–ligand complex. This stability suggests that roxithromycin interacts with protein L4 in a way that maintains a consistent and favourable binding conformation.
5.4 Hydrogen bond analysis
The presence of intermolecular hydrogen bonds plays an important role in maintaining the stability of protein–ligand complex, as highlighted in the previous studies.118,119 Fig. 26(d) gives the information about the formation of hydrogen bonds networks between each protein–ligand complex over extended production steps resulting from molecular dynamic simulations. Throughout the simulation, noticeable fluctuations in hydrogen bond networks were observed, with the number of interactions varying between one and four. In the initial phase of simulation, spanning up to 28 ns, erythromycin was found to form more hydrogen bond networks with protein L4 compared to roxithromycin. However, from 28 ns to 100 ns, roxithromycin exhibited a more extensive hydrogen bond network with protein L4 than erythromycin. The shift in dominance of hydrogen bonding towards roxithromycin after the initial phase indicates a potential stronger and more stable interaction between roxithromycin and protein L4, highlighting its superior binding capacity over erythromycin within the given molecular dynamics context.The molecular dynamic simulation of azithromycin and clarithromycin is discussed in the ESI Section S1.†
6 Conclusion
It is observed by 13C 2D PASS CP-MAS SSNMR experiments that the spinning CSA sideband pattern of C1 nuclei in roxithromycin displays axial symmetry, in contrast to the asymmetric pattern found for erythromycin for the same nuclei. Additionally, a significant alteration has been detected in the sign of anisotropy parameter of C9 nuclei in roxithromycin, as compared to erythromycin. This alteration has been attributed to the presence of an N-oxime side chain attached to the C9 atom of the 14-membered lactone ring. The analysis of hydrogen bonds between roxithromycin and erythromycin indicates that roxithromycin exhibits a stronger binding affinity with protein L4 compared to erythromycin. Additionally, the radius of gyration analysis reveals that roxithromycin maintains a favorable binding conformation with protein L4. Furthermore, the root mean square fluctuation (RMSF) analysis demonstrates that protein L4 experiences less fluctuation when interacting with roxithromycin than with erythromycin. Roxithromycin also induces greater root mean square deviation (RMSD) values, suggesting increased stability compared to erythromycin, thereby influencing the dynamic behavior of protein L4. In summary, roxithromycin exhibits higher bioactivity than erythromycin. The increased spin-lattice relaxation rate of each carbon nucleus in roxithromycin, compared to erythromycin, indicates that the motional dynamics and degrees of freedom are higher in roxithromycin. Hence, it can be predicted that the enhancement of the bioactivity of this macrolide antibiotic is somehow correlated with the enhancement of relaxation rate (1/T1).Clarithromycin exhibits structural similarity to erythromycin; however, it surpasses erythromycin in terms of its pharmacokinetic profile due to its increased bioavailability. Conformational analysis demonstrates that macrolide antibiotics effectively permeate the hydrophobic region of a phosphatidylinositol monolayer by utilizing their desosamine and cladinose components. In Table 5, it is evident that the spin-lattice relaxation time for the carbon nuclei present in desosamine and cladinose moieties of clarithromycin is shorter compared to that of erythromycin. This disparity may contribute to the enhanced bioavailability observed in clarithromycin.
The spatial proximities among various carbons and protons in the lactone, desosamine, and cladinose rings are observed in four macrolide antibiotics through the 1H–13C FSLG HETCOR spectrum. It is notable that all thirteen carbon nuclei in the lactone ring display spatial correlation with specific protons from the two sugar units, desosamine and cladinose, that are connected to the lactone ring in azithromycin. In both erythromycin and azithromycin, the C1 carbon is spatially correlated with desosamine. However, for roxithromycin, C1 carbon has spatial proximity with both desosamine and cladinose. For roxithromycin and azithromycin, C3 and C5 carbons are spatially correlated with some protons of desosamine and cladinose, whereas for erythromycin there is no spatial proximity of C3 and C5 carbon with any protons residing on desosamine and cladinose.
A minor alteration in the chemical composition of the lactone ring, such as substituting an O-methyl group for a hydroxyl group, attaching an N-oxime side chain, or incorporating a methyl-substituted nitrogen atom, induces significant changes in the acid stability, pharmacokinetic profile, and tissue penetration capabilities of the macrolide antibiotic erythromycin. This interplay between chemical structure and activity also extends to a crucial microscopic property of these antibiotics, namely, the nuclear spin-lattice relaxation time. While there is currently no direct evidence establishing a correlation between relaxation time and antibiotic activity in the existing literature, these findings serve as a compelling incentive for further exploration and investigation into this intriguing connection. These types of detailed analysis of four macrolide antibiotics will enlighten the route of drug discovery and drug-design.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author Manasi Ghosh is grateful to Science and Engineering Research Board (SERB)-POWER research grant (file no. SPG/2021/000303), Department of Science and Technology (DST), Government of India, and IoE-BHU Seed Grant-II (Dev. Scheme No. 6031(B), for financial support. The authors are thankful to DST sponsored SATHI-BHU scheme No. 6025 for providing 600 MHz solid-state NMR facility. The authors are thankful to Prof. Philip Grandinetti for RMN software. The authors are also thankful to Dr Harisingh Gour Central University, Sagar, Madhya-Pradesh for providing 500 MHz solid-state NMR facility.References
- T. Kaneko, T. J. Dougherty and T. V. Magee, Macrolide Antibiotics, Compr. Med. Chem. II, 2007, 7, 519–566 Search PubMed.
- S. K. Puri and H. B. Lassman, Roxithromycin: a pharmacokinetic review of a macrolide, J. Antimicrob. Chemother., 1987, 20, 89–100 CrossRef CAS PubMed.
- C. J. Dunn and L. B. Barradell, Azithromycin: a review of its pharmacological properties and use as 3-day therapy in respiratory tract infections, Drugs, 1996, 51, 483–505 CrossRef CAS PubMed.
- A. Markham and D. Faulds, Roxithromycin: an update of its antimicrobial activity, pharmacokinetic properties and therapeutic use, Drugs, 1994, 48, 297–326 CrossRef CAS PubMed.
- R. Jain and L. H. Danziger, The Macrolide Antibiotics: A Pharmacokinetic and Pharmacodynamic Overview, Curr. Pharm. Des., 2004, 10, 3045–3053 CrossRef CAS PubMed.
- J. M. Herron, Roxithromycin in the therapy of Streptococcus pyogenes throat infections, J. Antimicrob. Chemother., 1987, 20, 139–144 CrossRef PubMed.
- G. W. Amsden, Erythromycin, clarithromycin, and azithromycin: are the differences real?, Clin. Ther., 1996, 18, 56–72 CrossRef CAS PubMed.
- M. S. Whiteman and A. R. Tunkel, Azithromycin, and Clarithromycin: overview and comparison with erythromycin, Infect. Control Hosp. Epidemiol., 1992, 13, 357–368 CrossRef.
- J. D. Williams and A. M. Sefton, Comparison of macrolide antibiotics, J. Antimicrob. Chemother., 1993, 31, 11–26 CrossRef CAS PubMed.
- S. Kanoh and B. K. Rubin, Mechanisms of action and clinical application of macrolides as immunomodulatory medications, Clin. Microbiol. Rev., 2010, 23, 590–615 CrossRef CAS PubMed.
- K. Lagrou, W. A. Peetermans, M. Jorrisen, J. Verhaegen, J. Van Damme and J. Van Eldere, Subinhibitory concentrations of erythromycin reduce pneumococcal adherence to respiratory epithelial cells in vitro, J. Antimicrob. Chemother., 2000, 46, 717–723 CrossRef CAS PubMed.
- Y. Fukuda, K. Yanagihara, Y. Higashiyama, Y. Miyazaki, Y. Hirakata, H. Mukae, K. Tomono, Y. Mizuta, K. Tsukamoto and S. Kohno, Effects of macrolides on pneumolysin of macrolide-resistant Streptococcus pneumonia, Eur. Respir. J., 2006, 27, 1020–1025 CrossRef CAS PubMed.
- R. Anderson, H. C. Steel, R. Cockeran, A. M. Smith, A. von Gottberg, L. de Gouveia, A. Brink, K. P. Klugman, T. J. Mitchell and C. Feldman, Clarithromycin alone and in combination with ceftriaxone inhibits the production of pneumolysin by both macrolide-susceptible and macrolide-resistant strains of Streptococcus pneumonia, J. Antimicrob. Chemother., 2007, 59, 224–229 CrossRef CAS PubMed.
- R. Anderson, H. C. Steel, R. Cockeran, A. von Gottberg, L. de Gouveia, K. P. Klugman, T. J. Mitchell and C. Feldman, Comparison of the effects of macrolides, amoxicillin, ceftriaxone, doxycycline, tobramycin and fluoroquinolones, on the production of pneumolysin by Streotococcus pneumonia in vitro, J. Antimicrob. Chemother., 2007, 60, 1155–1158 CrossRef CAS PubMed.
- J. J. Holstein, P. Luger, R. Kalinowski, S. Mebs, C. Paulman and B. Dittrich, Validation of experimental charge densities: refinement of the macrolide antibiotic roxithromycin, Acta Crystallogr., Sect. B: Struct. Sci., 2010, 66, 568–577 CrossRef CAS PubMed.
- A. A. Firsov, S. H. Zinner, S. N. Vostrov, O. V. Kononenko, Y. A. Portnoy, L. V. Shustova and I. B. Kadenatsi, Comparative pharmacodynamics of azithromycin and roxithromycin with S. pyogenes and S. pheumoniae in a model that simulates in vitro pharmacokinetics in human tonsils, J. Antimicrob. Chemother., 2002, 49, 113–119 CrossRef CAS PubMed.
- J. P. Montenez, F. Van Bambeke, J. Piret, R. Brasseur, P. M. Tulkens and M. P. Mingeot-Leclercq, Interactions of Macrolide Antibiotics (Erythromycin A, Roxithromycin, Erythromycylamine [Dirithromycin], and Azithromycin) with Phospholipids: Computer-Aided Conformational Analysis and Studies on Acellular and Cell Culture Models, Toxicol. Appl. Pharmacol., 1999, 156, 129–140 CrossRef CAS PubMed.
- H. T. Edzes and J. P. C. Bernards, Two-dimensional exchange NMR in static powders -interchain C-13 spin exchange in crystalline polyethylene, J. Am. Chem. Soc., 1984, 106, 1515–1517 CrossRef CAS.
- P. M. Henrichs and M. C. Linder, 13 spin diffusion in the determination of intermolecular structure in solids, J. Magn. Reson., 1984, 58, 458–461 CAS.
- W. S. Veeman, C-13 chemical-shift anisotropy, Prog. Nucl. Magn. Reson. Spectrosc., 1984, 16, 193–235 CrossRef CAS.
- C. J. Hartzell, M. Whitfield, T. G. Oas and G. P. Drobny, Determination of the N-15 and C-13 chemical-shift tensors of L-C-13-Alanyl-L-N-15 alanine from the dipole-coupled powder patterns, J. Am. Chem. Soc., 1987, 109, 5966–5969 CrossRef CAS.
- R. Tycko and G. Dabbagh, Nuclear-magnetic-resonance crystallography – molecular orientational ordering in 3 forms of solid methanol, J. Am. Chem. Soc., 1991, 113, 3592–3593 CrossRef CAS.
- R. H. Havlin, H. B. Le, D. D. Laws, A. C. de Dios and E. Oldfield, An ab initio quantum chemical investigation of carbon-13 NMR shielding tensors in glycine, alanine, valine, isoleucine, serine, and threonine: comparisons between helical and sheet tensors, and the effects of χ1 on shielding, J. Am. Chem. Soc., 1997, 119, 11951–11958 CrossRef CAS.
- J. Heller, D. D. Laws, M. Tomaselli, D. S. King, D. E. Wemmer, A. Pines, R. H. Havlin and E. Oldfield, Determination of dihedral angles in peptides through experimental and theoretical studies of alpha-carbon chemical shielding tensors, J. Am. Chem. Soc., 1997, 119, 7827–7831 CrossRef CAS.
- S. Wi, H. H. Sun, E. Oldfield and M. Hong, Solid-state NMR and quantum chemical investigations of C-13(alpha) shielding tensor magnitudes and orientations in peptides: determining phi and psi torsion angles, J. Am. Chem. Soc., 2005, 127, 6451–6458 CrossRef CAS PubMed.
- G. J. Hou, I. J. L. Byeon, J. Ahn, A. M. Gronenborn and T. Polenova, Recoupling of chemical shift anisotropy by R-symmetry sequences in magic angle spinning NMR spectroscopy, J. Chem. Phys., 2012, 137, 134201 CrossRef PubMed.
- G. J. Hou, S. Paramasivam, S. Yan, T. Polenova and A. J. Vega, Multidimensional magic angle spinning NMR spectroscopy for site-resolved measurement of proton chemical shift anisotropy in biological solids, J. Am. Chem. Soc., 2013, 135, 1358–1368 CrossRef CAS PubMed.
- J. T. Damron, K. M. Kersten, M. K. Pandey, K. H. Mroue, J. R. Yarava, Y. Nishiyama, A. J. Matzger and A. Ramamoorthy, Electrostatic constraints assessed by H-1 MAS NMR illuminate differences in crystalline polymorphs, J. Phys. Chem. Lett., 2017, 8, 4253–4257 CrossRef CAS PubMed.
- M. K. Pandey, J. T. Damron, A. Ramamoorthy and Y. Nishiyama, Proton-detected 3d H-1 anisotropic/N-14/H-1 isotropic chemical shifts correlation NMR under fast magic angle spinning on solid samples without isotopic enrichment, Solid State Nucl. Magn. Reson., 2019, 97, 40–45 CrossRef CAS PubMed.
- Z. T. Gu, R. Zambrano and A. McDermott, Hydrogen-bonding of carboxyl groups in solid-state amino-acids and peptides - comparison of carbon chemical shielding, infrared frequencies, and structures, J. Am. Chem. Soc., 1994, 116, 6368–6372 CrossRef CAS.
- H. Saito, I. Ando and A. Ramamoorthy, Chemical shift tensor - the heart of NMR: insights into biological aspects of proteins, Prog. Nucl. Magn. Reson. Spectrosc., 2010, 57, 181–228 CrossRef CAS PubMed.
- Y. F. Wei, A. C. de Dios and A. E. McDermott, Solid-state N-15 NMR chemical shift anisotropy of histidines: experimental and theoretical studies of hydrogen bonding, J. Am. Chem. Soc., 1999, 121, 10389–10394 CrossRef CAS.
- B. J. Wylie and C. M. Rienstra, Multidimensional solid state NMR of anisotropic interactions in peptides and proteins, J. Chem. Phys., 2008, 128, 052207 CrossRef PubMed.
- A. Shoji, S. Ando, S. Kuroki, I. Ando and G. A. Webb, Structural studies of peptides and polypeptides in the solid state by nitrogen-15 NMR, Annu. Rep. NMR Spectrosc., 1993, 26, 55–98 CrossRef CAS.
- W. S. Veeman, Carbon-13 Chemical Shift Anisotropy, Prog. Nucl. Magn. Reson. Spectrosc., 1984, 16, 193–235 CrossRef CAS.
- L. Shao and J. J. Titman, Chemical Shift Anisotropy Amplification, Prog. Nucl. Magn. Reson. Spectrosc., 2007, 51, 103–137 CrossRef CAS.
- G. Wu, Solid-state 17O NMR studies of organic and biological molecules, Prog. Nucl. Magn. Reson. Spectrosc., 2008, 52, 118–169 CrossRef CAS.
- Solid State NMR of Polymers, ed. I. Ando and T. Asakura, Elsevier Science, Amsterdam, 1998 Search PubMed.
- Solid-state NMR Spectroscopy: Principles and Applications, ed. O. N. Antzutkin and M. J. Duer, Blackwell Sciences, Oxford, 2002, p. 280 Search PubMed.
- NMR Spectroscopy of Biological Solids, A. Ramamoorthy, CRC Press, Cleveland, 2005 Search PubMed.
- B. C. Gerstein and C. R. Dybowski, Transient Techniques in NMR of Solids, an Introduction to Theory and Practice, Academic Press, Orlando, 1985 Search PubMed.
- O. N. Antzutkin, Sideband manipulation in magic-angle-spinning nuclear magnetic resonance, Prog. Nucl. Magn. Reson. Spectrosc., 1999, 35, 203–266 CrossRef CAS.
- T. M. Duncan, A Compilation of Chemical Shift Anisotropics, The Farragut Press, Chicago, 1990 Search PubMed.
- A. C. de Dios, Ab initio calculations of the NMR chemical shift, Prog. Nucl. Magn. Reson. Spectrosc., 1996, 29, 229–278 CrossRef CAS.
- S. Sen, Dynamics in Inorganic Glass-forming Liquids by NMR Spectroscopy, Prog. Nucl. Magn. Reson. Spectrosc., 2019, 116, 155–176 CrossRef PubMed.
- D. D. Laws, L. B. Hans-Marcus and A. Jerschow, Solid-State NMR Spectroscopic Methods in Chemistry, Angew. Chem., Int. Ed., 2002, 41, 3096–3129 CrossRef CAS PubMed.
- M. J. Duer, Solid State NMR Spectroscopy Principles and Applications, Blackwell science, 2007 Search PubMed.
- C. M. Widdifield and R. W. Schurko, Understanding Chemical Shielding Tensors Using Group Theory, MO Analysis, and Modern Density-Functional Theory, Concepts Magn. Reson., Part A, 2009, 34(2), 91–123 CrossRef.
- N. F. Ramsey, Magnetic shielding of nuclei in molecules, Phys. Rev., 1950, 78, 699–703 CrossRef CAS.
- N. F. Ramsey, Dependence of magnetic shielding of nuclei upon molecular orientation, Phys. Rev., 1951, 83, 540–541 CrossRef CAS.
- N. F. Ramsey, Chemical effects in nuclear magnetic resonance and in diamagnetic susceptibility, Phys. Rev., 1952, 86, 243–246 CrossRef CAS.
- J. A. Pople, The theory of chemical shifts in nuclear magnetic resonance. I. Induced current densities, Proc. R. Soc. A: Math. Phys. Eng. Sci., 1957, A239, 541–549 Search PubMed.
- J. A. Pople, Nuclear magnetic resonance in diamagnetic materials. Theory of chemical shifts, Discuss. Faraday Soc., 1962, 34, 7–14 RSC.
- C. M. Widdifield, I. Moudrakovski and D. L. Bryce, Calcium-43 chemical shift and electric field gradient tensor interplay: a sensitive probe of structure, polymorphism, and hydration, Phys. Chem. Chem. Phys., 2014, 16, 13340–13359 RSC.
- R. Tycko, G. Dabbagh and P. A. Mirau, Determination of chemical shift anisotropy lineshapes in a two-dimensional magic angle spinning NMR experiment, J. Magn. Reson., 1989, 85, 265–274 CAS.
- S. F. Liu, J. D. Mao and K. Schmidt-Rohr, A robust technique for two-dimensional separation of undistorted chemical shift anisotropy powder patterns in magic angle spinning NMR, J. Magn. Reson., 2002, 155, 15–28 CrossRef CAS PubMed.
- J. C. C. Chan and R. Tycko, R. Recoupling of chemical shift anisotropies in solid state NMR under high speed magic angle spinning and in uniformly 13C labelled systems, J. Chem. Phys., 2003, 118, 8378–8389 CrossRef CAS.
- G. Hou, L. Byeon In-Ja, J. Ahn, A. M. Gronenborn and T. Polenova, Recoupling of chemical shift anisotropy by R-symmetry sequences in magic angle spinning NMR spectroscopy, J. Chem. Phys., 2012, 137, 134201–134210 CrossRef PubMed.
- A. D. Bax, N. M. Szeverenyi and G. E. Maciel, Chemical shift anisotropy in powdered solids studied by 2D FT NMR with flipping of the spinning axis, J. Magn. Reson., 1983, 55, 494–497 CAS.
- A. D. Bax, N. M. Szeverenyi and G. E. Maciel, Correlation of isotropic shifts and chemical shift anisotropies by two-dimensional Fourier-transform magic angle hopping NMR spectroscopy, J. Magn. Reson., 1983, 52, 147–152 CAS.
- A. D. Bax, N. M. Szeverenyi and G. E. Maciel, Chemical shift anisotropy in powdered solids studied by 2D FT CP/MAS NMR, J. Magn. Reson., 1983, 51, 400–408 CAS.
- Z. Gan, High-resolution chemical shift and chemical shift anisotropy correlation in solids using slow magic angle spinning, J. Am. Chem. Soc., 1992, 114, 8307–8309 CrossRef CAS.
- W. T. Dixon, Spinning-sideband-free and spinning-sideband-only NMR spectra in spinning samples, J. Chem. Phys., 1982, 77, 1800–1809 CrossRef CAS.
- O. N. Antzutkin, S. C. Shekar and M. H. Levitt, Two-dimensional sideband separation in magic angle spinning NMR, J. Magn. Reson., Ser. A, 1995, 115, 7–19 CrossRef CAS.
- M. Ghosh, S. Sadhukhan and K. K. Dey, Elucidating the internal structure and dynamics of α-chitin by 2DPASS-MAS-NMR and spin-lattice relaxation measurements, Solid State Nucl. Magn. Reson., 2019, 97, 7–16 CrossRef CAS PubMed.
- M. Ghosh, B. P. Prajapati, N. Kango and K. K. Dey, A comprehensive and comparative study of the internal structure and dynamics of natural β-keratin and regenerated β-keratin by solid state NMR spectroscopy, Solid State Nucl. Magn. Reson., 2019, 101, 1–11 CrossRef CAS PubMed.
- M. Ghosh, N. Kango and K. K. Dey, Investigation of the internal structure and dynamics of cellulose by 13C-NMR relaxometry and 2DPASS-MAS-NMR measurements, J. Biomol. NMR, 2019, 73, 601–616 CrossRef CAS PubMed.
- K. K. Dey and M. Ghosh, Understanding the effect of an anionic side-chain on the nuclear spin dynamics of a polysaccharide, Cellulose, 2022, 29, 1381–1392 CrossRef CAS.
- K. K. Dey and M. Ghosh, Understanding the effect of deacetylation on chitin by measuring chemical shift anisotropy tensor and spin lattice relaxation time, Chem. Phys. Lett., 2020, 738, 136782 CrossRef CAS.
- K. K. Dey, S. Gayen and M. Ghosh, Structure and dynamics of sodium alginate as elucidated by chemical shift anisotropy and site-specific spin–lattice relaxation time measurements, Eur. Biophys. J., 2021, 50(7), 963–977 CrossRef CAS PubMed.
- K. K. Dey, M. Deshmukh and M. Ghosh, A Description of the Local Structure and Dynamics of Ketoconazole Molecule by Solid-State NMR Measurements and DFT Calculations: Proposition for NMR Crystallography, ChemistrySelect, 2021, 6, 10208–10220 CrossRef CAS.
- K. K. Dey, L. Lodhi and M. Ghosh, Study of the Variation of the Electronic Distribution and Motional Dynamics of Two Independent Molecules of an Asymmetric Unit of Atorvastatin Calcium by Solid-State NMR Measurements, ACS Omega, 2021, 6, 22752–22764 CrossRef CAS PubMed.
- K. K. Dey and M. Ghosh, Study of the Structure and Dynamics at Various Parts of the Antibacterial Drug Molecule Cefpodoxime proxetil, Solid State Nucl. Magn. Reson., 2021, 115, 101752 CrossRef CAS PubMed.
- K. K. Dey and M. Ghosh, Investigation of the Structure and Dynamics of Antiviral Drug Adefovir Dipivoxil by Site-Specific Spin–Lattice Relaxation Time Measurements and Chemical Shift Anisotropy Tensor Measurements, ACS Omega, 2020, 5, 29373 CrossRef CAS PubMed.
- K. K. Dey and M. Ghosh, Determination of Chemical Shift Anisotropy Tensor and Molecular Correlation Time of Proton Pump Inhibitor Omeprazole by Solid State NMR Measurements, New J. Chem., 2020, 44, 19393 RSC.
- K. K. Dey and M. Ghosh, Determination of the Correlation between the Structure and Dynamics of Deflazacort by solid state NMR measurements, New J. Chem., 2020, 44, 18419 RSC.
- K. K. Dey and M. Ghosh, Understanding the Structure and Dynamics of Anti-inflammatory Corticosteroid Dexamethasone by solid state NMR Spectroscopy, RSC Adv., 2020, 10, 37564 RSC.
- K. K. Dey, S. Gayen and M. Ghosh, Understanding the correlation between structure and dynamics of clocortolonepivalate by solid state NMR measurement, RSC Adv., 2020, 10, 4310 RSC.
- K. K. Dey, S. Gayen and M. Ghosh, An atomic resolution description of folic acid using solid state NMR measurements, RSC Adv., 2020, 10, 24973 RSC.
- K. K. Dey, S. Gayen and M. Ghosh, Investigation of the Detailed Internal Structure and Dynamics of Itraconazole by Solid-State NMR Measurements, ACS Omega, 2019, 4, 21627–21635 CrossRef CAS PubMed.
- L. Lodhi, J. P. Yadav, T. Yamazaki, N. T. Duong, S. L. Poojary, K. K. Dey, Y. Nishiyama and M. Ghosh, NMR Crystallographic Approach to Study the Variation of the Dynamics of Quinine and its Quasienantimer Quinidine, J. Phys. Chem. C, 2022, 126, 17291–17305 CrossRef CAS.
- J. P. Yadav, L. Lodhi, T. Fatma, K. K. Dey and M. Ghosh, Investigation of the Influence of Various Functional Groups on the Dynamics of Glucocorticoids, ACS Omega, 2022, 7, 43190–43209 CrossRef CAS PubMed.
- I. Mandal, S. R. Keshri, L. Lodhi, K. K. Dey, M. Ghosh, A. Ghosh and A. R. Allu, Correlation of structure and ionic-conductivity in phosphate glass using MAS-NMR and impedance spectroscopy: Influence of sodium salt, Phys. Rev. Mater., 2022, 6, 115403 CrossRef CAS.
- R. Bhowal, A. Balaraman, M. Ghosh, S. Dutta, K. K. Dey and D. Chopra, Probing Atomistic Behavior to Unravel Dielectric Phenomena in Charge Transfer Cocrystals, J. Am. Chem. Soc., 2021, 143, 1024–1037 CrossRef CAS PubMed.
- H. Saito, I. Ando and A. Ramamoorthy, Chemical shift tensor-the heart of NMR: Insight into biological aspects of proteins, Prog. Nucl. Magn. Reson. Spectrosc., 2010, 57, 181–228 CrossRef CAS PubMed.
- N. Asakawa, T. Kameda, S. Kuroki, H. Kurosu, S. Ando, I. Ando and A. Shoji, Structural Studies of Hydrogen-bonded Peptides and Polypeptides by Solid-state NMR, Annu. Rep. NMR Spectrosc., 1998, 35, 55–137 CrossRef CAS.
- D. A. Torchia, The measurement of proton-enhanced carbon-13 T1 values by method which suppresses artifacts, J. Magn. Reson., 1978, 30, 613–616 CAS.
- B. J. Van Rossum, H. Forster and H. J. M. De Groot, High-Field and High-Speed CP-MAS 13C NMR Heteronuclear Dipolar Correlation Spectroscopy of Solids with Frequency-Switched Lee–Goldburg Homonuclear Decoupling, J. Magn. Reson., 1997, 124, 516–519 CrossRef CAS.
- M. H. Levitt, P. K. Madhu and C. E. Hughes, Cogwheel phase cycling, J. Magn. Reson., 2002, 155, 300–306 CrossRef CAS PubMed.
- N. Ivchenko, C. E. Hughes and M. H. Levitt, Application of cogwheel phase cycling to sideband manipulation experiments in solid-state NMR, J. Magn. Reson., 2003, 164, 286–293 CrossRef CAS PubMed.
- J. Herzfeld and A. E. Berger, Sideband Intensities in NMR Spectra of Samples Spinning at the Magic Angle, J. Chem. Phys., 1980, 73, 6021–6030 CrossRef CAS.
- J. Gharbi-Benarous, M. Delaforge, I. Artaud and J. P. Girault, Analysis of the 1H and 13C NMR Spectra of the Novel Macrolide Antibiotic Roxithrornycin. Structure and Conformation in Solution, Magn. Reson. Chem., 1990, 28, 846855 CrossRef.
- G. Lazarevski, M. Vinkovc, G. Kobrehel and S. Dokic, Conformational Analysis of Azithromycin by Nuclear Magnetic Resonance Spectroscopy and Molecular Modelling, Tetrahedron, 1993, 49, 721–730 CrossRef CAS.
- R. J. Brennan and J. Barber, Full Assignments of the 13C and lH NMR Spectra of Azithromycin in Buffered D20 and DMSO-d6, Magn. Reson. Chem., 1992, 30, 327–333 CrossRef CAS.
- J. Gharbi-Benarous, M. Delaforge, C. K. Jankowski and J. P. Girault, A Comparative NMR Study between the Macrolide Antibiotic Roxithromycin and Erythromycin A with Different Biological Properties, J. Med. Chem., 1991, 34, 1117–1125 CrossRef CAS PubMed.
- A. Awan, J. Barber, R. Brennan, J. John and A. Parkinson, Structural studies on clarithromycin (6-O-methylerythromycin A): Assignments of the 1H and 13C NMR spectra in organic and aqueous solutions, Magn. Reson. Chem., 1992, 30, 1241–1246 CrossRef CAS.
- J. R. Everett and J. W. Tyler, An analysis of the 1H and 13C NMR spectra of erythromycin A using two-dimensional methods, J. Chem. Soc., Perkin Trans. 1, 1985, 2599–2603 RSC.
- B. H. Toby and R. B. Von Dreele, GSAS-II: the genesis of a modern open-source allpurpose crystallography software package, J. Appl. Crystallogr., 2013, 46, 544–549 CrossRef CAS.
- X. G. A. Stephenson, O. G. Stowell, P. H. Toma, R. R. Pfeiffer and S. R. Byrn, Solid-state investigations of erythromycin A dihydrate: structure, NMR spectroscopy, and hygroscopicity, J. Pharm. Sci., 1997, 86(11), 1239–1244 CrossRef PubMed.
- J. M. Montejo-Bernardo, S. García-Grande, M. S. Bayod-Jasanada, L. Lavona-Díaz and I. Llorente, X-ray study of the pseudopolymorphism of the azithromycin monohydrate, Z. fur Krist. – Cryst. Mater., 2003, 218(10), 703–707 CrossRef CAS.
- J. Tian, P. K. Thallapally, S. J. Dalgarno and J. L. Atwood, Free transport of water and CO2 in nonporous hydrophobic clarithromycin form II crystals, J. Am. Chem. Soc., 2009, 131(37), 13216–13217 CrossRef CAS PubMed.
- J. J. Hos Holstein, P. Luger, R. Kalinowski, S. Mebs, C. Paulman and B. Dittrich, Validation of experimental charge densities: refinement of the macrolide antibiotic roxithromycin, Acta Crystallogr., Sect. B: Struct. Sci., 2010, 66(5), 568–577 CrossRef PubMed.
- K. Adrjanowicz, D. Zakowiecki, K. Kaminski, L. Hawelek, K. Grzybowska, M. Tarnacka, M. Paluch and K. Cal, Molecular dynamics in supercooled liquid and glassy states of antibiotics: azithromycin, clarithromycin and roxithromycin studied by dielectric spectroscopy. Advantages given by the amorphous state, Mol. Pharmaceutics, 2012, 9(6), 1748–1763 CrossRef CAS PubMed.
- S. Mirza, I. Miroshnyk, J. Heinämäki, L. Christiansen, M. Karjalainen and J. Yliruusi, Influence of solvents on the variety of crystalline forms of erythromycin, American Association of Pharmaceutical Scientists, 2003, 5, 39–47 Search PubMed.
- R. Gandhi, O. Pillai, R. Thilagavathi, B. Gopalakrishnan, C. L. Kaul and R. Panchagnula, Characterization of azithromycin hydrates, Eur. J. Pharm. Sci., 2002, 16(3), 175–184 CrossRef CAS PubMed.
- Y. T. Sohn, J. K. Rhee and W. B. Im, Polymorphism of clarithromycin, Arch. Pharmacal Res., 2000, 23, 381–384 CrossRef CAS PubMed.
- I. I. Salem, Clarithromycin, Anal. Profiles Drug Subst. Excipients, 1996, 24, 45–85 CAS.
- P. Kurath, P. H. Jones, R. S. Egan and T. Perun, Acid degradation of erythromycin A and erythromycin B, Experientia, 1971, 27, 362 CrossRef CAS PubMed.
- A. H. H. Bakheit, B. M. H. Al-Hadiya and A. A. Abd-Elgalil, Chapter One- Azithromycin, Profiles Drug Subst., Excipients, Relat. Methodol., 2014, 39, 1–40 CAS.
- R. J. Abraham, M. Mobli and R. J. Smith, 1H chemical shifts in NMR : Carbonyl anisotropies and steric effect in aromatic aldehydes and ketones, Magn. Reson. Chem., 2003, 41, 26–36 CrossRef CAS.
- H. M. McConnell, Theory of Nuclear Magnetic Shielding in Molecules: Long-Range Dipolar Shielding of protons, J. Chem. Phys., 1957, 27, 226 CrossRef CAS.
- M. R. Housaindokht, M. R. Bozorgmehr and M. Bahrololoom, Analysis of ligand binding to proteins using molecular dynamics simulations, J. Theor. Biol., 2008, 254(2), 294–300 CrossRef CAS PubMed.
- A. M. da Fonseca, B. J. Caluaco, J. M. C. Madureira, S. Q. Cabongo, E. M. Gaieta, F. Djata, R. P. Colares, M. M. Neto, C. F. C. Fernandes, G. S. Marinho, H. S. D. Santos and E. S. Marinho, Screening of potential inhibitors targeting the main protease structure of SARS-CoV-2 via molecular docking, and approach with molecular dynamics, RMSD, RMSF, H-bond, SASA and MMGBSA, Mol. Biotechnol., 2023, 1–15 CAS.
- B. Arsic, J. Barber and P. Novak. The macrolide antibiotics and their semi-synthetic derivatives, Macrolides: Properties, Synthesis and Applications, ed. B. Arsic, 2018, pp. 1-30 Search PubMed.
- G. G. Zhanel, M. Dueck, D. J. Hoban, L. M. Vercaigne, J. M. Embil, A. S. Gin and J. A. &Karlowsky, Review of macrolides and ketolides: focus on respiratory tract infections, Drugs, 2001, 61, 443–498 CrossRef CAS PubMed.
- D. Jelić and R. &Antolović, From erythromycin to azithromycin and new potential ribosome-binding antimicrobials, Antibiotics, 2016, 5(3), 29 CrossRef PubMed.
- M. Han, P. Chen and X. Yang, Molecular dynamics simulation of PAMAM dendrimer in aqueous solution, Polymer, 2005, 46(10), 3481–3488 CrossRef CAS.
- A. Ragunathan, K. Malathi and A. &Anbarasu, MurB as a target in an alternative approach to tackle the Vibrio cholerae resistance using molecular docking and simulation study, J. Cell. Biochem., 2018, 119(2), 1726–1732 CrossRef CAS PubMed.
- V. Mascoli, N. Liguori, L. Cupellini, E. Elias, B. Mennucci and R. Croce, Uncovering the interactions driving carotenoid binding in light-harvesting complexes, Chem. Sci., 2021, 12(14), 5113–5122 RSC.
- M. Enayatkhani, M. Salimi, K. Azadmanesh and L. &Teimoori-Toolabi, In-silico identification of new inhibitors for Low-density lipoprotein receptor-related protein6 (LRP6), J. Biomol. Struct. Dyn., 2022, 40(10), 4440–4450 CrossRef CAS PubMed.
- B. L. Pradhan, J. P. Yadav, L. Lodhi, P. Sen, K. K. Dey and M. Ghosh, Atomic-Scale Resolution Insights into Structural and Dynamic Differences between Ofloxacin and Levofloxacin, ACS Omega, 2023, 8(26), 24093–24105 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: S1. Exploring the interactions of ribosomal protein L4 with erythromycin, azithromycin, and clarithromycin through molecular dynamics simulations; S2. Docking of three antibiotics erythromycin, azithromycin, and roxithromycin with motilin receptor. See DOI: https://doi.org/10.1039/d4ra00718b |
| This journal is © The Royal Society of Chemistry 2024 |