
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aryl diazonium salts and benzene derivatives bearing two electron-donor substituents: chemical and mechanistic behaviours†
Gabriele Micheletti * and
Carla Boga
* and
Carla Boga
Department of Industrial Chemistry, Alma Mater Studiorum – Università di Bologna, Via Gobetti 85, 40129 Bologna, Italy. E-mail: gabriele.micheletti3@unibo.it
First published on 14th March 2024
Abstract
The reaction between benzene derivatives 1–4 and p-substituted benzenediazonium tetrafluoroborates 5a–c provided novel azo-coupling products in high yields under mild conditions. The monitoring of the reaction progress using 1H-NMR provided mechanistic information on both the relative reactivity of the reagents and the possibility to detect novel reaction intermediates.
Introduction
Electrophilic aromatic substitution (SEAr) is one of the most widely studied types of reactions in organic chemistry. Numerous studies on this reaction have been carried out to obtain both new compounds and mechanistic information. In particular, a class of compounds used for mechanistic studies is that of 1,3,5-tris(N,N-dialkylamino)-benzenes belonging to neutral aromatic substrates rich in electrons due to the presence of three electron-donating groups on the ring. These compounds are able to react as carbon nucleophiles with a plethora of electrophiles such as, halogens,1,2 protons, and3–12 alkyl-,13 acyl-,14–16 and aryl-halides.17–19Moreover, the reaction of 1,3,5-tris(N,N-dialkylamino)-benzenes with charged20 and neutral carbon electrophiles has recently been studied, providing interesting information on the relevant reaction intermediates.21–23
From long time our interest lies in the reaction between 1,3,5-tris(N,N-dialkylamino)-benzene derivatives and aryl diazonium salts from which it was possible to obtain evidence for the reversibility of the azo-coupling reaction and to detect the related Wheland-like reaction intermediates. Moreover, the reaction gave access to novel products of interest in applied chemistry.24–26 For example, the reaction between 1,3,5-tris(N,N-dialkylamino)-benzenes and 2 equivalents of p-substituted benzenediazonium salts provided dicationic species, which collapsed to new benzimidazole derivatives with the expulsion of p-substituted anilines.27
Unlike 1,3,5-tris(N,N-dialkylamino)-benzene derivatives, just a few examples are reported for the electrophilic aromatic substitution involving 1,3-N,N-dialkylamino benzenes.28–31
Based on these, we decided to study the azo-coupling reaction between these nucleophiles and para-substituted benzenediazonium salts to possibly obtain new compounds of interest for application in fields such as optoelectronics32,33 and dyes34 together with further mechanistic information on this kind of reaction.
Results and discussion
A new series of azo-coupling compounds 6a–c, 7a–c, 8a–c, and 9a–c were synthesized via the reaction of 1,3-di-substituted benzenes 1–4, chosen as nucleophiles, and benzenediazonium salts 2a–c, chosen as electrophiles (Scheme 1).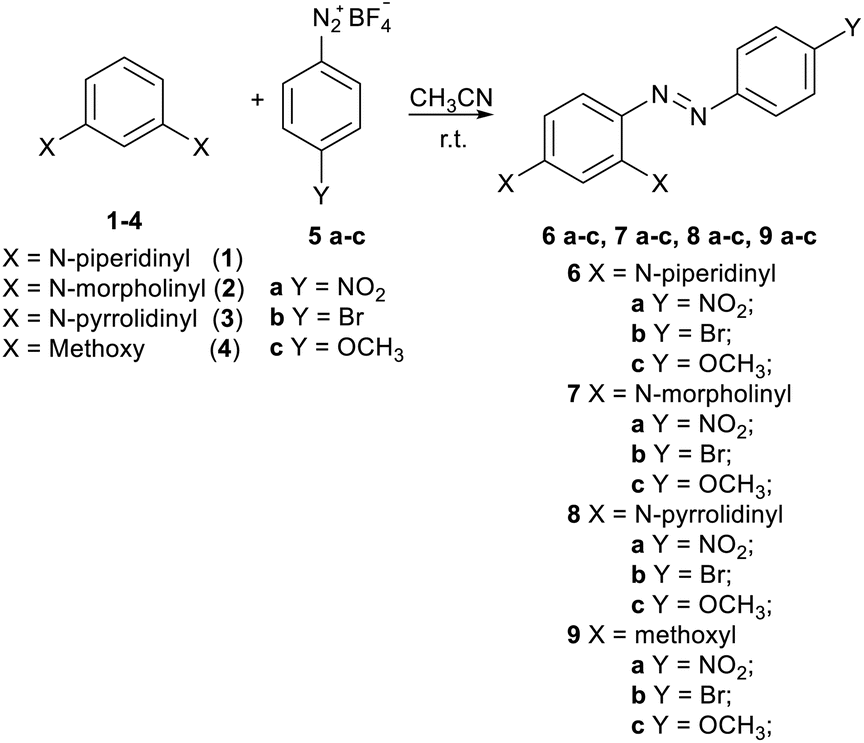 | ||
| Scheme 1 Reaction between di-substituted benzenes 1–4 and p-substituted benzenediazonium salts 5a–c. | ||
The reactions were conducted at room temperature in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of reagents, and the final products were purified by chromatography on silica gel and fully characterized.
:1 molar ratio of reagents, and the final products were purified by chromatography on silica gel and fully characterized.
The reaction can lead to the formation of two distinct isomeric products (A and B, Scheme 2), resulting from the attack of the diazonium salt in ortho–ortho (A) or in ortho–para (B) position with respect to the two substituents present on the nucleophilic reagent.
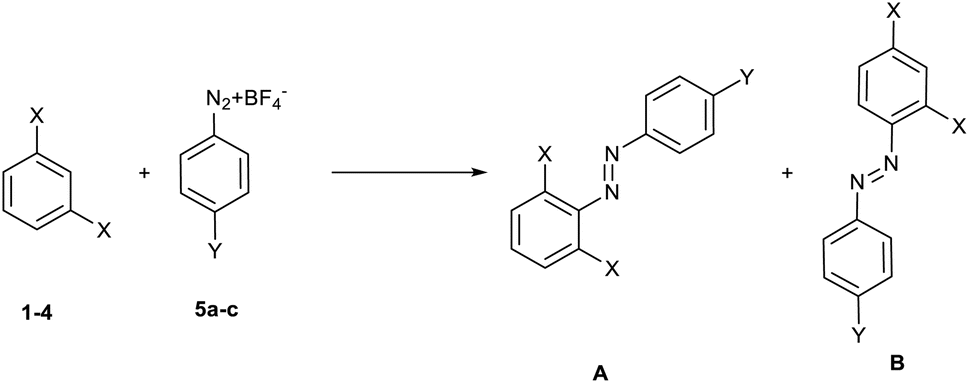 | ||
| Scheme 2 Possible regioisomers from the reaction between 1,3-disubstituted benzenes 1–4 and diazonium salts 5a–c. | ||
From the study of the 1H-NMR spectra of the purified final compounds, it has been possible to note (see 1H-NMR spectra in ESI†) that the nucleophilic moiety of the products shows three different signals. This behaviour is possible only for the product resulting from the attack of diazonium salt in the ortho–para position (case B), as it was expected on the basis of steric hindrance considerations.
From the data reported in Table 1, it is possible to note that the syntheses with 1,3-dimethoxybenzene (4) (entry 10–12) have required a very longer reaction time than that of all other reactions. Moreover, in the reaction with a para-methoxy benzene diazonium salt (5c), any product was obtained after a longer reaction time (72 h). These behaviours are due to the lower activation of the benzene ring towards a nucleophilic attack caused by the two methoxy substituents, compared with the amino groups and it is also similar to those reported previously.35–37
| Entry | Nucleophile | Electrophile | Reaction time | Product | Yielda |
|---|---|---|---|---|---|
| a Yield after purification by chromatography on silica gel. | |||||
| 1 | 1 | 5a | 20 min | 6a | 98% |
| 2 | 1 | 5b | 20 min | 6b | 97% |
| 3 | 1 | 5c | 20 min | 6c | 78% |
| 4 | 2 | 5a | 30 min | 7a | 94% |
| 5 | 2 | 5b | 30 min | 7b | 80% |
| 6 | 2 | 5c | 30 min | 7c | 75% |
| 7 | 3 | 5a | 10 min | 8a | 95% |
| 8 | 3 | 5b | 10 min | 8b | 77% |
| 9 | 3 | 5c | 10 min | 8c | 73% |
| 10 | 4 | 5a | 24 h | 9a | 77% |
| 11 | 4 | 5b | 48 h | 9b | 26% |
| 12 | 4 | 5c | 72 h | 9c | 0% |
The reactions with di(pyrrolidinyl)benzene 3 (entries 7–9) show a lower reaction time, due to the strong activation of the benzene ring caused by the two pyrrolidinyl substituents, which makes reagent 3 the stronger nucleophile.
Moreover, it is possible to note, particularly comparing the results reported in entries 10–12 of Table 1, that the electrophilicity of the diazonium salts also influences the progress of the reaction. In fact, as reported in Mayr's electrophilicity scale,38 the values for 5a–c are −5.1, −6.6 and −8.4, respectively.
It is important to note that all reactions were conducted in an equimolar amount between the two reagents, and given the formation of tetrafluoroboric acid, we should have obtained a maximum of 50% yield in the reactions, since the released acid would have to react with the nucleophilic reagent deactivating it. On the contrary, except to case 11 and 12 in Table 1, the yields obtained were greater than 50%. A possible explanation for this finding is that the final product is more basic than the starting diaminobenzene reagent. If this were the case, only the protonated product would be found in the reaction crude and the protonated diaminobenzene reagent would be absent.
Given that no data are reported in the literature for the protonation of dialkylaminobenzenes, we decided to carry out the reaction between 1,3-di(pyrrolidinyl) benzene (3) and tetrafluoroboric acid directly in the NMR spectroscopy tube.
It is reported in the literature3,12,15 that the protonation of 1,3,5-tris(N,N-dialkylamino) benzenes manifests the formation of both Wheland intermediates from the C-attack and ammonium salts from the proton attack on the nitrogen atom. Only in the case of protonation on 1,3,5-tripyrrolidinyl benzene, the attack on the carbon atom was detected.
In the present case involving dialkylamino benzene derivatives, there are three possibilities for the attack of the proton (Scheme 3): (i) in the ortho–ortho position (W-1), (ii) in the ortho-para position (W-2), and (iii) on one of the nitrogen atoms (ammonium salt NH).
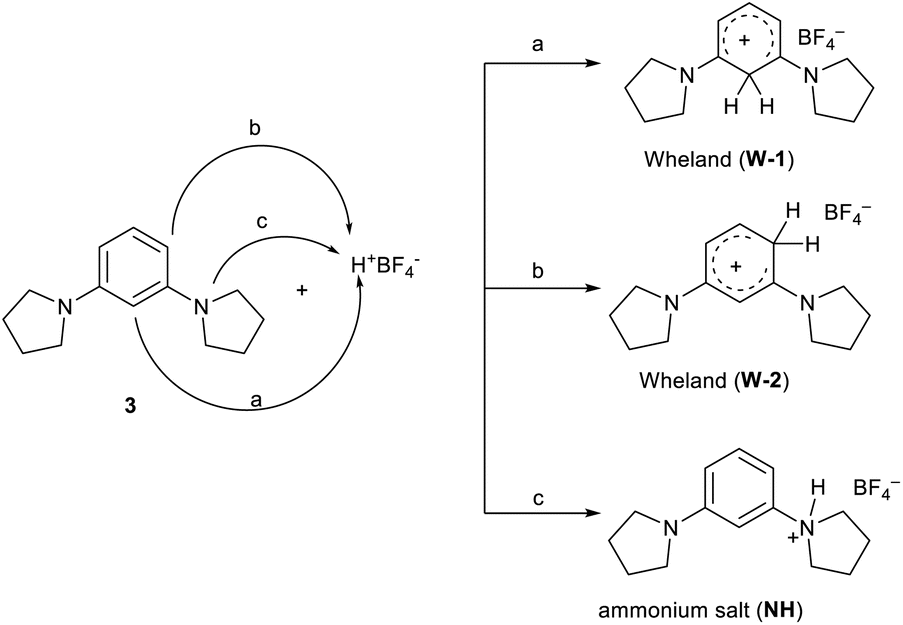 | ||
| Scheme 3 Possible attacks in the protonation reaction of compound 3. | ||
The reaction was carried out in CD3CN by adding an equimolar amount of a solution of HBF4 in Et2O to a solution of 3. The mixture was analysed by 1H-NMR and 2D-NMR (g-COSY and g-HSQC) experiments. The 1H-NMR spectrum (Fig. 1) shows the presence of two sets of signals. In particular, by studying the g-COSY spectrum, it is possible to observe that the signals at 7.43, 6.90 and 6.81 ppm are attributed to the same species.
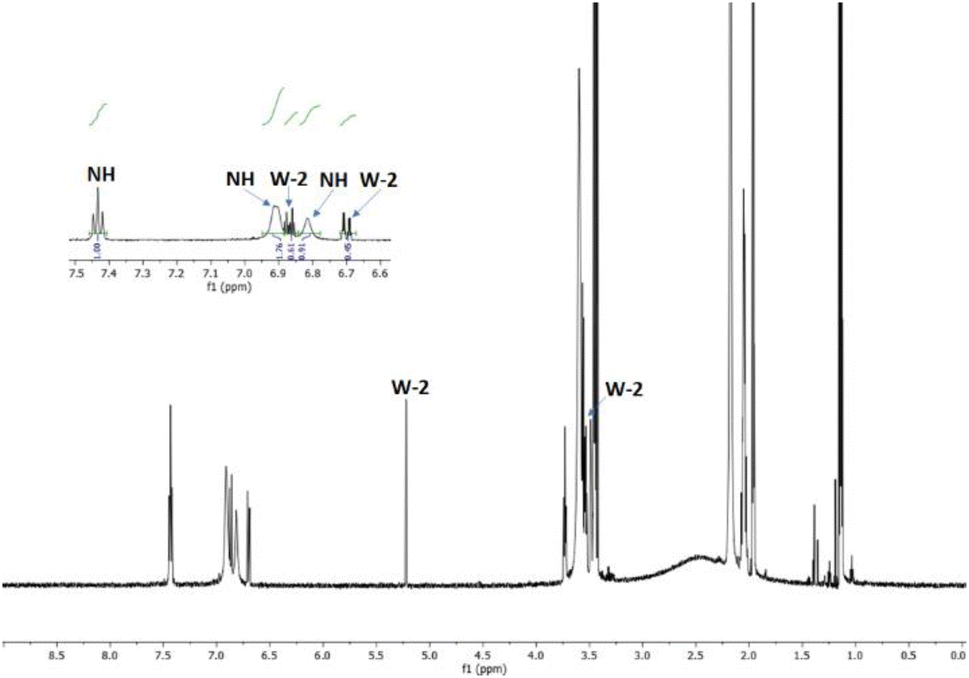 | ||
| Fig. 1 1H-NMR spectrum of the product mixture resulting from the protonation reaction of 3, with enlarged view. | ||
These signals are shifted at a lower field than those of compound 3 (6.98, 5.91 and 5.75, see Fig. S35†); this finding confirms the presence of ammonium salt NH. Theoretically, for the salt NH, the number of signals should be four, but the rapid movement of the proton from one nitrogen atom to another makes the molecule appear symmetrical, in the NMR scale time. This agrees with the relative integration 1:2:1 of the signals, analogously to what has been previously observed for the protonation of triaminobenzenes.12 Furthermore, from the g-COSY experiment, the signals at 6.87, 6.70, 5.21 and 3.50 ppm are correlated with each other. These signals are compatible with the formation of the Wheland-like intermediate. In particular, the signal at 3.50 ppm is very diagnostic, because it shifted in the sp3 region of the spectrum. This is also confirmed by the HSQC experiment, which indicates that the signals at 3.50 ppm in the 1H-MNR spectrum are related to the signal at about 50 ppm in the 13C-NMR spectrum. The presence of four signals for this molecule is consistent with the structure of the W-2 intermediate.
This experiment furnished us the possibility to verify if compounds W-2 and/or NH are present in the 1H-NMR spectra of the raw reaction mixture between equimolar amounts of 3 and 5c, which implies the absence of the signals belonging to the protonated spices of 3 (Fig. 2).
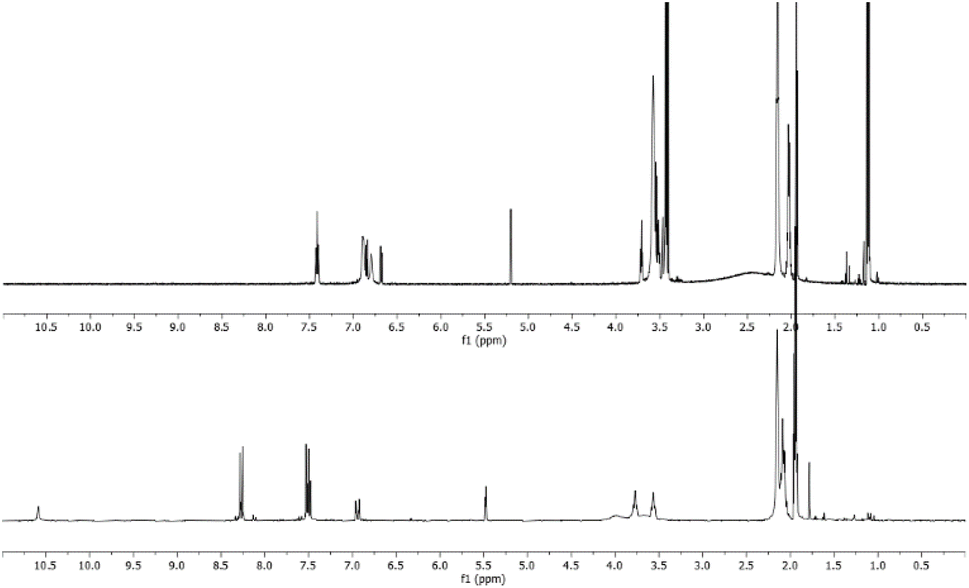 | ||
| Fig. 2 Comparison between the raw spectrum for the reaction between 3 and 5c (lower) and the protonation reaction spectrum of 3 (upper). | ||
Furthermore, comparison of the spectra of the raw reaction and that of the purified product indicates that the broad signals of the crude reaction mixture shifted to lower fields than those of the purified products (Fig. 3). This implies that the obtained products are in salt form. A similar behaviour was reported in the literature in the reaction between tris-dialkylamino benzene derivatives and electrophiles such as benzofuroxan19 and benzofurazan18 derivatives.
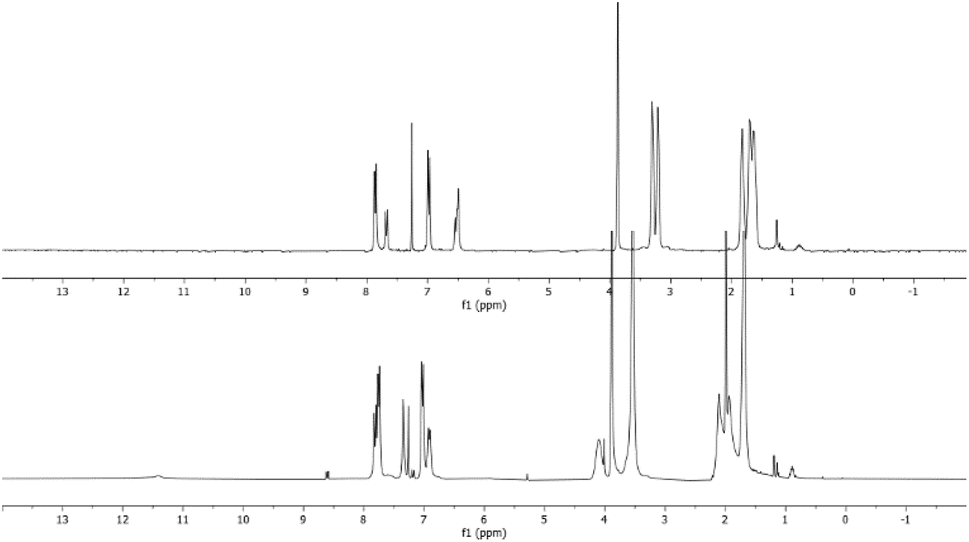 | ||
| Fig. 3 Comparison between the raw reaction spectrum (lower) and the spectrum of purified product (upper) for the reaction between 1 and 5c. | ||
The only explanation for this is that the neutralization process might have occurred during the chromatographic purification, since this was the only work-up step to which the reactions were subjected.
It was reported in the literature24–26 that during the reaction between 1,3,5-tris(N,N-dialkylamino)-benzene derivatives and benzenediazonium salts, the formation of the relevant Wheland intermediates can be confirmed by NMR analyses at low temperatures. For this reason, we performed some reactions directly in the NMR tube, using deuterated acetonitrile at −35 °C or deuterated dichloromethane at −80 °C, but the 1H-NMR spectra did not confirm the presence of the Wheland intermediate, as only the signals belonging to the products in the salt form were present.
These results allow us to affirm that Wheland intermediates, in the azo-coupling reaction, are sufficiently stable, in the NMR time scale, to allow their identification by NMR spectroscopy, only when three strong electron-donor substituents are present on the benzene ring, such as in the case of 1,3,5-tris(N,N-dialkylamino)-benzenes.
Conclusion
Novel azo-compounds have been synthesized from benzenediazonium tetrafluoroborates and 1,3-disubstituted benzenes 1–4 in good yields under mild reaction conditions.The data enabled us to obtain information on the relative reactivity of the studied 1,3-disubstituted benzenes, that is: 1,3-di(pyrrolidinyl)benzene (3) > 1,3-dipiperidinyl benzene (1) > 1,3-di(morpholinyl)benzene (2) > 1,3-di(methoxybenzene) (4).
The study of the progress of the reaction at low temperatures using NMR spectroscopy did not highlight the presence of Wheland intermediates, in contrast to the azo-coupling reaction with 1,3,5-tris(dialkylamino)benzenes. This suggests that the Wheland intermediates are stable enough to be detected by NMR spectroscopy, only when three strong electron-donating substituents are present on the aromatic ring.
Experimental section
Material and methods
The reagents used, unless stated otherwise, were purchased from Sigma-Aldrich. Chromatographic purifications (FC) were carried out using glass columns packed with silica gel Geduran Si 60, 0.063–0.200 mm) or neutral alumina at a medium pressure. Thin-layer chromatography (TLC) was performed on silica gel 60 F254-coated aluminium foils. 1H-NMR spectra were recorded at 25 °C using Varian spectrometers Inova 300, Mercury 400 or Inova 600 operating at 300, 400 or 600 MHz, respectively. 13C-NMR spectra were recorded at 25 °C using Varian spectrometers Mercury 400 or Inova 600 operating at 100.56 or 150.80 MHz, respectively. Chemical shifts were measured in ppm with reference to the solvent CDCl3 = 7.26 ppm for 1H-NMR and 77.0 ppm for 13C-NMR. The J-values are given in Hz. Electrospray ionization (ESI)-MS spectra and ESI high-resolution HRMS spectra were recorded using a Waters ZQ 4000 and a Xevo instrument, respectively. For mass analyses, the samples were dissolved in methanol. UV/Vis spectra were recorded using a PerkinElmer Lambda 12 spectrophotometer, at 20 °C in CHCl3 and quartz cells (path length of the cell: 1 cm). Melting points (m.p.) were measured using a Büchi 535 apparatus and were uncorrected. 1H-NMR, 13C-NMR, ESI-MS, and UV/Vis spectra of the synthesized compounds are reported in the ESI.† Syntheses of the dialkylamino benzene derivatives were reported in the literature.31 Compounds 5a–c and tetrafluoroboric acid diethyl ether complex are commercially available.General procedure
To a solution of 1,3 di-substituted benzene derivative (0.1 mmol, corresponding to 24.4 mg of 1, 24.8 mg of 2, 21.6 mg of 3 and 13.8 mg of 4) in CH3CN (5 mL), the selected benzenediazonium salt (0.1 mmol, corresponding to 23.7 mg of 5a, 27.1 mg of 5b and 22.2 mg of 5c) was added at room temperature. The mixture was magnetically stirred, and the reaction progress was monitored through TLC analysis. After solvent removal, the reaction products were purified by column chromatography on silica gel (FC) using appropriate eluents (reported in the characterization).:2; yield = 98% (38.5 mg); 1H-NMR (600 MHz, CDCl3, 25 °C) δ, ppm: 8.30 (d, J = 8.9 Hz, 2H), 7.89 (d, J = 8.9 Hz, 2H), 7.80 (d, J = 9.2 Hz, 1H), 6.94 (dd, J1 = 9.2 Hz, J2 = 2.3 Hz 1H), 6.36 (d, J = 2.3 Hz, 1H), 3.43–3.39 (m, 4H), 3.31 (t, J = 4.6 Hz, 4H), 1.82 (q, J = 4.6 Hz, 4H), 1.74–1.63 (m, 8H, three signals overlapped); 13C-NMR (150 MHz, CDCl3, 25 °C) δ, ppm: 157.3 (C), 155.5 (C, two signals overlapped), 146.7 (C), 136.6 (C), 124.8 (CH), 122.4 (CH), 118.8 (CH), 108.2 (CH), 102.4 (CH), 54.5 (CH2), 48.7 (CH2), 26.4 (CH2), 25.5 (CH2), 24.4 (CH2), 24.3 (CH2); ESI MS (ES+) m/z: 394 [M + H]+, 416 [M + Na]+; HRMS (ES+) m/z: [M + H]+ calculated for C22H28N5O2 394.2243; found 394.2246.:1; yield = 97% (41.4 mg); 1H-NMR (600 MHz, CDCl3, 25 °C) δ, ppm: 7.72 (m, 3H, Two signals overlapped), 7.58 (d, J = 8.1 Hz, 2H), 6.52 (s, 1H), 6.43 (s, 1H), 3.35 (s, 4H), 3.24 (s, 4H), 1.81 (s, 4H), 1.73–1.67 (m, 4H), 1.67–1.59 (m, 4H); 13C-NMR (150 MHz, CDCl3, 25 °C) δ, ppm: 154.8 (C), 154.0 (C), 152.3 (C), 136.5 (C), 132.1 (CH), 123.8 (CH), 122.8 (C), 118.2 (CH), 108.5 (CH), 103.6 (CH), 54.6 (CH2), 49.1 (CH2), 26.4 (CH2), 25.5 (CH2), 24.3 (CH2), 22.7 (CH2); ESI MS (ES+) m/z: 427 (79Br) [M + H]+, 429 (81Br) [M + H]+, 449 (79Br) [M + Na]+, 451 (81Br) [M + Na]+; HRMS (ES+) m/z: [M + H]+ calculated for C22H28BrN4 427.1497 (79Br) and 429.1477 (81Br); found 427.1498 (79Br) and 429.1479 (81Br).:2; yield = 78% (29.5 mg); 1H-NMR (300 MHz, CDCl3, 25 °C) δ, ppm: 7.86 (d, J = 8.4 Hz, 2H), 7.68 (d, J = 8.4 Hz, 1H), 6.98 (d, J = 8.8 Hz, 2H), 6.58–6.45 (m, 2H, two signals overlapped), 3.87 (s, 3H), 3.30 (br. s, 4H), 3.21 (br. s, 4H), 1.88–1.77 (m, 4H), 1.77–1.55 (m, 8H); 13C-NMR (150 MHz, CDCl3, 25 °C) δ, ppm: 160.8 (C), 154.3 (C), 153.0 (C), 147.8 (C), 136.9 (C), 124.0 (CH), 117.8 (CH), 114.1 (CH), 108.7 (CH), 104.3 (CH), 55.5 (CH3), 54.6 (CH2), 49.4 (CH2), 26.4 (CH2), 25.6 (CH CH2, two signals overlapped), 24.3; ESI MS (ES+) m/z: 379 [M + H]+, 401 [M + Na]+; HRMS (ES+) m/z: [M + H]+ calculated for C23H31N4O 379.2498; found 379.2500.:4; yield = 97% (38.5 mg); 1H-NMR (400 MHz, CDCl3, 25 °C) δ, ppm: 8.33 (d, J = 8.9 Hz, 2H), 7.88 (d, J = 8.9 Hz, 2H), 7.82 (s, 1H), 6.55 (dd, J1 = 9.6 Hz, J2 = 2.6 Hz 1H), 6.39 (d, J = 2.6 Hz, 1H), 3.98–3.92 (m, 4H), 3.90–3.84 (m, 4H), 3.41–3.32 (m, 8H, two signals overlapped); 13C-NMR (150 MHz, CDCl3, 25 °C) δ, ppm: 156.8 (C), 155.3 (C), 153.8 (C), 147.4 (C), 137.4 (C), 124.8 (CH), 122.6 (CH), 118.9 (CH), 108.3 (CH), 102.5 (CH), 67.1 (CH2), 66.5 (CH2), 53.4 (CH2), 47.5 (C CH2H2); ESI MS (ES+) m/z: 398 [M + H]+, 420 [M + Na]+; HRMS (ES+) m/z: [M + H]+ calculated for C20H24N5O4 398.1828; found 398.1829.:4; yield = 96% (41.5 mg); 1H-NMR (300 MHz, CDCl3, 25 °C) δ, ppm: 7.76 (d, J = 9.1 Hz, 1H), 7.69 (d, J = 9.1 Hz, 2H), 7.59 (d, J = 8.5 Hz, 2H), 6.56 (d, J = 8.5 Hz, 1H), 6.43 (s, 1H), 3.95 (s, 4H), 3.87 (t, J = 5.05, 4H), 3.35–3.25 (m, 8H, two signals overlapped); 13C-NMR (150 MHz, CDCl3, 25 °C) δ, ppm: 154.4 (C), 152.5 (C), 151.9 (C), 137.2 (C), 132.3 (CH), 123.8 (CH), 123.7 (C), 118.5 (CH), 108.6 (CH), 103.2 (CH), 67.1 (CH2), 66.6 (CH2), 53.4 (CH2), 47.9 (CH2); ESI MS (ES+) m/z: 431 (79Br) [M + H]+, 433 (81Br) [M + H]+, 453 (79Br) [M + Na]+, 455 (81Br) [M + Na]+; HRMS (ES+) m/z: [M + H]+ calculated for C20H24BrN4O2 431.1083 (79Br) and 433.1062 (81Br); found 31.1086 (79Br) and 433.1064 (81Br).:3; 1H-NMR (600 MHz, CDCl3, 25 °C) δ, ppm: 7.82 (d, J = 8.8 Hz, 2H), 7.71 (d, J = 8.8 Hz, 1H), 6.99 (d, J = 8.5 Hz, 2H), 6.57 (d, J = 8.5 Hz, 1H), 6.47 (s,1H), 3.95 (t, J = 4.44, 4H), 3.88–3.86 (m, 7H, two signals overlapped), 3.30–3.26 (m, 8H, Two signals overlapped): 13C-NMR (150 MHz, CDCl3, 25 °C) δ, ppm: 161.1 (C), 153.7 (C), 151.6 (C), 147.6 (C), 137.7 (C), 124.1 (CH), 118.1 (CH), 114.2 (CH), 108.8 (CH), 103.6 (CH), 67.2 (CH2), 66.7 (CH2), 55.5 (CH3), 53.4 (CH2), 48.3 (CH2); ESI MS (ES+) m/z: 383 [M + H]+, 405 [M + Na]+; HRMS (ES+) m/z: [M + H]+ calculated for C21H27N4O3 383.2083; found 383.2085.:2; yield = 95% (34.7 mg); 1H-NMR (600 MHz, CDCl3, 25 °C) δ, ppm: 8.24 (d, J = 8.2 Hz, 2H), 7.99 (s, 1H), 7.69 (s, 2H), 6.14 (s, 1H), 5.63 (s, 1H), 3.72 (s, 4H), 3.43 (s, 4H), 2.09–2.01 (m, 8H, two signals overlapped); 13C-NMR (150 MHz, CDCl3, 25 °C) δ, ppm: 158.4 (C), 152.1 (C), 150.5 (C), 145.0 (C), 134.5 (C), 124.8 (CH), 121.4 (CH), 119.0 (CH), 105.5 (CH), 94.5 (CH), 52.6 (CH2), 47.8 (CH2), 25.9 (CH2), 25.4 (CH2); ESI MS (ES+) m/z: 366 [M + H]+, 388 [M + Na]+; HRMS (ES+) m/z: [M + H]+ calculated for C20H24N5O2 366.1930; found 366.1932.:1; yield = 77% (30.7 mg); 1H-NMR (300 MHz, CDCl3, 25 °C) δ, ppm: 7.92 (d, J = 8.7 Hz, 1H), 7.57 (d, J = 8.6 Hz, 2H), 7.52 (d, J = 8.6 Hz, 2H), 6.09 (d, J = 8.7 Hz, 1H), 5.72 (s, 1H), 3.69 (t, J = 6.3 Hz, 4H), 3.39 (t, J = 6.3 Hz, 4H), 2.04–1.98 (m, 8H); 13C-NMR (150 MHz, CDCl3, 25 °C) δ, ppm: 153.0 (C), 151.2 (C), 149.5 (C), 133.0 (C), 131.8 (CH), 123.1 (CH), 120.4 (C), 118.5 (CH), 104.0 (CH), 95.1 (CH), 52.5 (CH2), 47.7 (C CH2H2), 25.9 (CH2), 25.4 (CH2); ESI MS (ES+) m/z: 399 (79Br) [M + H]+, 401 (81Br) [M + H]+, 421 (79Br) [M + Na]+, 423 (81Br) [M + Na]+; HRMS (ES+) m/z: [M + H]+ calculated for C20H24BrN4 399.1184 (79Br) and 401.1164 (81Br); found 399.1187 (79Br) and 401.1166 (81Br).:1; yield = 73% (25.5 mg); 1H-NMR (600 MHz, CDCl3, 25 °C) δ, ppm: 7.90 (s, 1H), 7.71 (d, J = 8.9 Hz, 2H), 6.96 (d, J = 8.9 Hz, 2H), 6.09 (s, 1H), 5.76 (s, 1H), 3.85 (s, 3H), 3.69 (t, J = 6.6 Hz, 4H), 3.38 (t, J = 6.6 Hz, 4H), 2.03–1.98 (m, 8H); 13C-NMR (150 MHz, CDCl3, 25 °C) δ, ppm: 159.2 (C), 150.6 (C), 149.0 (C), 148.3 (C), 132.8 (C), 123.0 (CH), 118.3 (CH), 114.0 (CH), 103.4 (CH), 95.5 (CH), 55.4 (CH3), 52.4 (CH2), 47.6 (CH2), 25.9 (CH2), 25.4 (CH2); ESI MS (ES+) m/z: 351 [M + H]+; HRMS (ES+) m/z: [M + H]+ calculated for C21H27N4O 351.2185; found 351.2187.:1; yield = 77% (22.1 mg); 1H-NMR (600 MHz, CDCl3, 25 °C) δ, ppm: 8.34 (d, J = 8.4 Hz, 2H), 7.95 (d, J = 8.4 Hz, 2H), 7.81 (d, J = 8.7 Hz, 1H), 6.61 (s, 1H), 6.58 (d, J = 8.7 Hz, 1H), 4.04 (s, 3H), 3.92 (s, 3H); 13C-NMR (150 MHz, CDCl3, 25 °C) δ, ppm: 165.2 (C), 159.9 (C), 156.5 (C), 147.9 (C), 136.9 (C), 124.7 (CH), 123.1 (CH), 118.3 (CH), 106.2 (CH), 98.9 (CH), 56.4 (CH3), 55.7 (CH3); ESI MS (ES+) m/z: 288 [M + H]+, 310 [M + Na]+; HRMS (ES+) m/z: [M + H]+ calculated for C14H14N3O4 288.0984; found 288.0985.:1; yield = 26% (8.4 mg); 1H-NMR (400 MHz, CDCl3, 25 °C) δ, ppm: 7.77 (d, J = 8.9 Hz, 1H), 7.75 (d, J = 8.8 Hz, 2H) 7.60 (d, J = 8.8 Hz, 2H), 6.59 (d, J = 2.4 Hz, 1H), 6.55 (dd, J1 = 8.8 Hz, J2 = 2.4 Hz, 1H), 4.02 (s, 3H), 3.89 (s, 3H); 13C-NMR (150 MHz, CDCl3, 25 °C) δ, ppm: 164.1 (C), 159.0 (C), 151.9 (C), 136.7 (C), 132.2 (CH), 124.2 (C), 124.1(CH), 118.2 (CH), 105.8 (CH), 99.0 (CH), 56.4 (CH3), 55.7 (CH3); ESI MS (ES+) m/z: 321 (79Br) [M + H]+, 323 (81Br) [M + H]+, 343 (79Br) [M + Na]+, 345 (81Br) [M + Na]+, 359 (79Br) [M + K]+, 361 (81Br) [M + K]+; HRMS (ES+) m/z: [M + H]+ calculated for C14H14BrN2O2 321.0239 (79Br) and 323.0218 (81Br); found 321.0241 (79Br) and 323.0220 (81Br).Variable temperature NMR experiments
Author contributions
Gabriele Micheletti: conceptualization, investigation, and writing – original draft. Carla Boga: conceptualization, funding acquisition, supervision, and writing - original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Work supported by Alma Mater Studiorum – Università di Bologna (RFO funds). Authors thank Dr Martina Casadei and Silvia Cino.Notes and references
- P. Menzel and F. Effenberger, Angew Chem. Int. Ed. Engl., 1972, 11, 922–923 CrossRef CAS.
- F. Effenberger, P. Menzel and W. Seufert, Chem. Ber., 1979, 112, 1660–1669 CrossRef CAS.
- F. Effenberger and R. Niess, Angew Chem. Int. Ed. Engl., 1967, 6, 1067 CrossRef CAS.
- F. Effenberger, Acc. Chem. Res., 1989, 22, 27–35 CrossRef CAS.
- F. Effenberger and R. Niess, Chem. Ber., 1968, 101, 3787–3793 CrossRef CAS.
- T. Yamaoka, H. Hosoya and S. Nagakura, Tetrahedron, 1968, 24, 6203–6213 CrossRef.
- T. Yamaoka, H. Hosoya and S. Nagakura, Tetrahedron, 1970, 26, 4125–4130 CrossRef CAS.
- W. Knoche, W. Sachs and S. Vogel, Bull. Soc. Chim. Fr., 1988, 377–382 CAS.
- W. Knoche, W. W. Schoeller, R. Schomaecker and S. Vogel, J. Am. Chem. Soc., 1988, 110, 7484–7489 CrossRef CAS.
- W. Sachs, W. Knoche and S. Herrmann, J. Chem. Soc., Perkin Trans. 2, 1991, 701–710 RSC.
- D. T. Glatzhofer, D. Allen and R. W. Taylor, J. Org. Chem., 1990, 55, 6229–6231 CrossRef CAS.
- C. Boga, L. Forlani, S. Tozzi, E. Del Vecchio, A. Mazzanti, M. Monari and N. Zanna, Curr. Org. Chem., 2014, 18, 512–523 CrossRef CAS.
- P. Menzel and F. Effenberger, Angew Chem. Int. Ed. Engl., 1975, 14, 62–63 CrossRef.
- R. Niess, K. Nagel and F. Effenberger, Tetrahedron Lett., 1968, 4265–4268 CrossRef CAS.
- F. Effenberger, K. E. Mack, K. Nagel and R. Niess, Chem. Ber., 1977, 110, 165–180 CrossRef CAS.
- P. Fkher, K. E. Mack, E. Mhsner and F. Effenberger, Chem. Ber., 1977, 110, 181–198 CrossRef.
- F. Effenberger, W. Agster, P. Fischer, K. H. Jogun, J. J. Stezowski, E. Daltrozzo and G. Kollmannsberger-von Nell, J. Org. Chem., 1983, 48, 4649–4658 CrossRef CAS.
- G. Micheletti, C. Boga, M. Pafundi, S. Pollicino and N. Zanna, Org. Biomol. Chem., 2016, 14, 768–776 RSC.
- G. Micheletti, C. Boga, S. Cino, S. Bordoni and E. Chugunova, RSC Adv., 2018, 8, 41663–41674 RSC.
- G. Micheletti, R. J. Mayer, S. Cino, C. Boga, A. Mazzanti, A. R. Ofial and H. Mayr, Eur. J. Org Chem., 2021, 2021, 6347–6357 CrossRef CAS.
- C. Boga, E. Del Vecchio, L. Forlani, A. Mazzanti and P. E. Todesco, Angew. Chem., Int. Ed., 2005, 44, 3285–3289 CrossRef CAS.
- C. Boga, E. Del Vecchio, L. Forlani, A. Mazzanti, C. Menchen Lario, P. E. Todesco and S. Tozzi, J. Org. Chem., 2009, 74, 5568–5575 CrossRef CAS PubMed.
- C. Boga, G. Micheletti, S. Cino, S. Fazzini, L. Forlani, N. Zanna and D. Spinelli, Org. Biomol. Chem., 2016, 14, 4267 RSC.
- C. Boga, E. Del Vecchio and L. Forlani, Eur. J. Org Chem., 2004, 1567–1571 CrossRef CAS.
- C. Boga, E. Del Vecchio, L. Forlani and S. Tozzi, J. Org. Chem., 2007, 72, 8741–8747 CrossRef CAS.
- L. Forlani, C. Boga, E. Del Vecchio, A. Ngobo and S. Tozzi, J. Phys. Org. Chem., 2007, 20, 201–205 CrossRef CAS.
- E. Del Vecchio, C. Boga, L. Forlani, S. Tozzi, G. Micheletti and S. Cino, J. Org. Chem., 2015, 80, 2216–2222 CrossRef CAS PubMed.
- A. A. Tolmachev, S. P. Ivonin, A. V. Kharchanko and E. S. Kozlov, J. Gen. Chem. USSR, 1992, 62, 868–871 Search PubMed.
- F. Effenberger, R. Gleiter, L. Heider and R. Niess, Chem. Ber., 1968, 101, 502–511 CrossRef CAS.
- M. Hartnagel and K. Grimm, Liebigs Ann.-Recl., 1997, 55–69 Search PubMed.
- G. Micheletti, S. Bordoni, E. Chugunova and C. Boga, Molecules, 2017, 22, 684 CrossRef PubMed.
- Y. Zhou, S. Zhang, S. Li, J. Li, N. Li and L. Zhang, Dyes Pigm., 2023, 209, 110898 CrossRef CAS.
- M. F. Asif, R. Bano, R. Farooq, S. Muhammad, T. Mahmood, K. Ayub, S. Tabassum and M. A. Gilani, Dyes Pigments, 2022, 202, 110284 CrossRef CAS.
- A. R. Patel, G. Patel, A. Srivastava and S. Banerjee, Curr. Org. Chem., 2023, 27, 1611–1628 CrossRef CAS.
- J. Rosevear and J. F. K. Wilshire, Aus. J. Chem., 1987, 40, 1663–1673 CrossRef CAS.
- D. Kosynkin, T. M. Bockman and J. K. Kochi, J. Chem. Soc., Perkin Trans. 2, 1997, 2003–2012 RSC.
- R. Weiss and F. G. Pühlhofer, J. Am. Chem. Soc., 2007, 129, 547–553 CrossRef CAS.
- H. Mayr, M. Hartnagel and K. Grimm, Liebigs Ann.-Recl., 1997, 55–69 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra00652f |
| This journal is © The Royal Society of Chemistry 2024 |