
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
First total synthesis of caerulomycin K: a case study on selective, multiple C–H functionalizations of pyridines†
Alessandro Dimasi‡
 a,
Mattia Failla‡a,
Arianna Montolia,
Andrea Citarellaa,
Paolo Ronchib,
Daniele Passarellaa and
Valerio Fasano*a
a,
Mattia Failla‡a,
Arianna Montolia,
Andrea Citarellaa,
Paolo Ronchib,
Daniele Passarellaa and
Valerio Fasano*a
aDepartment of Chemistry, Università degli Studi di Milano, Via Camillo Golgi, 19, 20133 Milano, Italy. E-mail: valerio.fasano@unimi.it; Web: https://www.fasanolab.com
bMedicinal Chemistry and Drug Design Technologies Department, Global Research and Preclinical Development, Chiesi Farmaceutici S.p.A, Largo Francesco Belloli 11/a, 43126 Parma, Italy
First published on 13th February 2024
Abstract
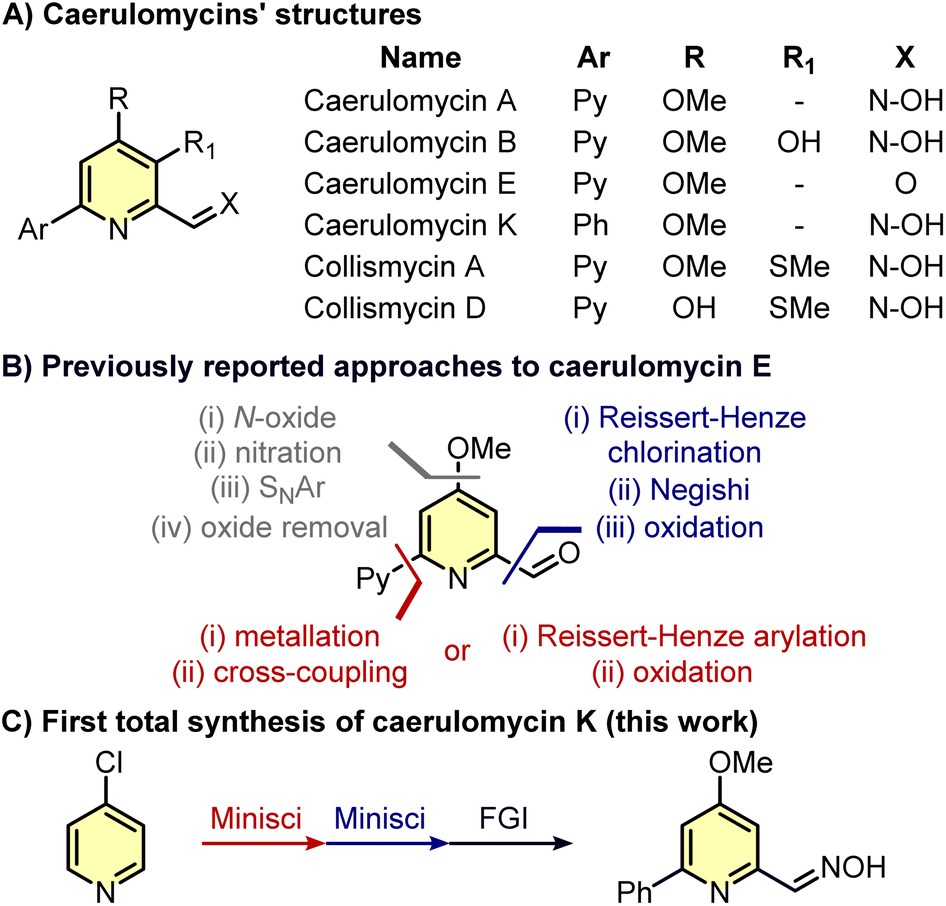
Caerulomycins, natural alkaloids with antimicrobial properties, have been previously synthesized starting with highly pre-functionalized building blocks or requiring many functional group manipulations. In this work, we report the first total synthesis of caerulomycin K, a diversely trifunctionalized pyridine readily assembled in three steps exploiting the recent advancements in the C–H activation of N-heterocycles.
Pyridines are ubiquitous in many natural products and drugs, often with a wide selection of functionalities decorating these aromatic rings.1,2 While classical pyridine syntheses (e.g. Bohlmann–Rahtz reaction, Hantzsch condensation, etc.) allow the introduction of substituents in the final ring, the functionalization of existing pyridines using C–H activation is usually a better option to avoid the de novo synthesis of complex pyridines.3 However, despite pyridines exhibiting a clear similarity to benzenes, they present distinct challenges when it comes to their C–H functionalization.4,5 As a result, relatively simple pyridines may require several steps to be synthesized, especially if the substituents around the aromatic ring are different in nature. This is the case for caerulomycins (and related collismycins), a class of natural alkaloids produced by Streptomyces caeruleus and endowed with antimicrobial properties (Scheme 1A).6–11 For instance, taking caerulomycin E as the prototype of this type of bioactive compounds, different routes have been designed to decorate the core pyridine ring with common substituents such as a carbonyl group (ortho-aldehyde), an alkoxide (para-MeO), and an aromatic ring (ortho-pyridine).12–21 Yet, the installation of these functionalities via C–H activation is not straightforward since it requires several functional group interconversions (Scheme 1B).
 | ||
| Scheme 1 Structures and synthetic routes to caerulomycins. | ||
Specifically, the installation of the methoxy group can require four steps: formation of the N-oxide with an oxidant, nitration with concentrated H2SO4, nucleophilic aromatic substitution (SNAr) with MeONa, and removal of the oxide with Ac2O.18,19 The insertion of an ortho-pyridine group is usually more rapid but requires the use of Grignard reagents or prefunctionalized 2-bromopyridines.15,16 Finally, the insertion of the carbonyl group is achieved by oxidation of a methyl group whose installation has been obtained only with a halogen (Cl or Br) already placed in ortho-position.18,21 Given the recent advancements in selective C–H functionalizations of pyridines,4,5 it would be expected that alternative strategies should now allow a faster synthesis of caerulomycins. Herein, we report our efforts to rapidly convert a cheap monosubstituted pyridine into caerulomycin K, a recently isolated alkaloid whose total synthesis has never been reported before.
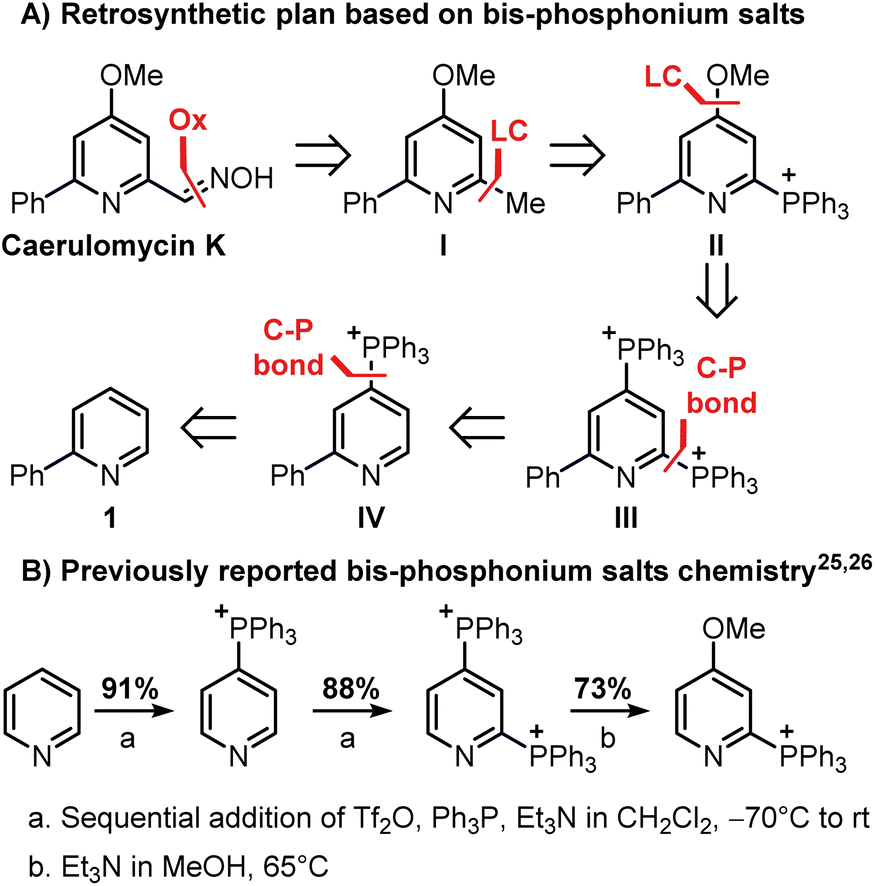
Our investigation began with the design of a synthetic route that would furnish caerulomycin K in a few steps using two C–H activations, thus avoiding highly pre-functionalized starting materials. In an initial retrosynthetic approach, we imagined that the aldoxime group could be derived from a methyl group, as reported by Quéguiner and co-workers, thus leading to trifunctionalized pyridine I (Scheme 2A).18 At this point, we envisaged that I could be accessed by a selective difunctionalization of pyridyl bis-phosphonium salt III via two sequential ligand-coupling (LC) reactions, that is formal SNAr reactions where a phosphonium group is replaced by opportune nucleophiles.22–24 Salt III could then be obtained from 2-phenylpyridine 1 via two consecutive C–P bond formation reactions, in analogy with a rare example of a pyridyl bis-phosphonium salt (Scheme 2B).25,26
 | ||
| Scheme 2 Reactivity of bis-phosphonium salts and application in the planned retrosynthesis of caerulomycin K. The triflate anion is not reported for clarity. | ||
This strategy would provide the desired product in 5 steps, whereas the ionic nature of most intermediates would reduce the need for column chromatography. Moreover, considering the wide versatility of ligand-coupling reactions,25,26 III would be a strategic intermediate for the synthesis of libraries of trifunctionalized pyridines by simply changing the order and the nature of the added nucleophiles. In the laboratory, 1 was dissolved in dichloromethane and cooled down to −78 °C, before sequentially adding Tf2O, Ph3P, and DBU (Scheme 3). In agreement with McNally's work,27 upon workup, 2 was easily precipitated out as a white powder from cold ether (88% isolated yield). Notably, the Ph3P addition occurs almost exclusively at the para-position (due to stereoelectronic reasons), thus no regioselectivity problems are encountered during this reaction.28 This was also confirmed by 31P NMR, with only a sharp singlet observed at 23.01 ppm. Repeating the procedure using now 2 as the starting material, the reaction crude revealed two new signals of similar intensity at 23.55 ppm and 15.37 ppm.
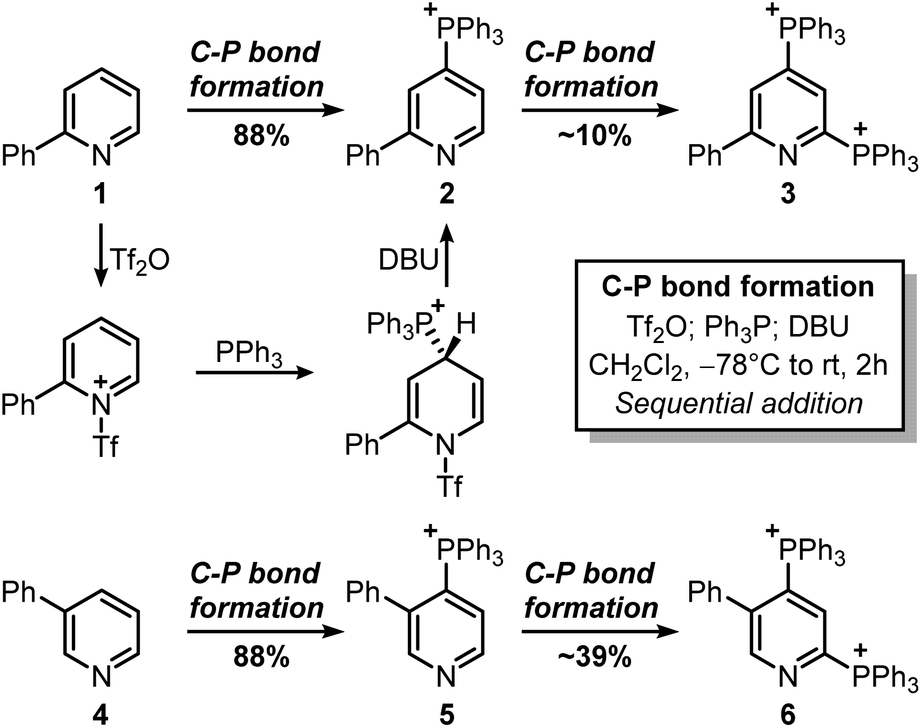 | ||
| Scheme 3 Attempted bis-phosphonium salts synthesis. The triflate anion is not reported for clarity. | ||
These signals were respectively assigned to the para- and ortho-phosphine of bis-phosphonium bis-triflate 3. However, the conversion was only modest by 31P NMR, with significant unreacted 2 and Ph3PO observed in the reaction mixture. The failure of the second C–P bond formation was attributed to a problematic N-activation since 2 should be less nucleophilic than 1 due to its cationic nature. This was confirmed using phosphonium 5, obtained in good yield from 3-phenylpyridine 4: moving away the phenyl ring from the ortho- to the meta-position improved the second C–P bond formation (5 less sterically encumbered than 2), yet not to a significant extent due to electronic reasons. Indeed, bis-phosphonium 6 was found as a minor component in 31P NMR spectrum of the reaction crude (signals at 21.93 ppm and 17.08 ppm, with a 3JP–P = 6.0 Hz). Deuterodephosphination29 of this reaction mixture further confirmed the poor conversion, with the isolated pyridine showing almost quantitative d-incorporation in para-position but a limited deuteration on the C6-site (Scheme 4).
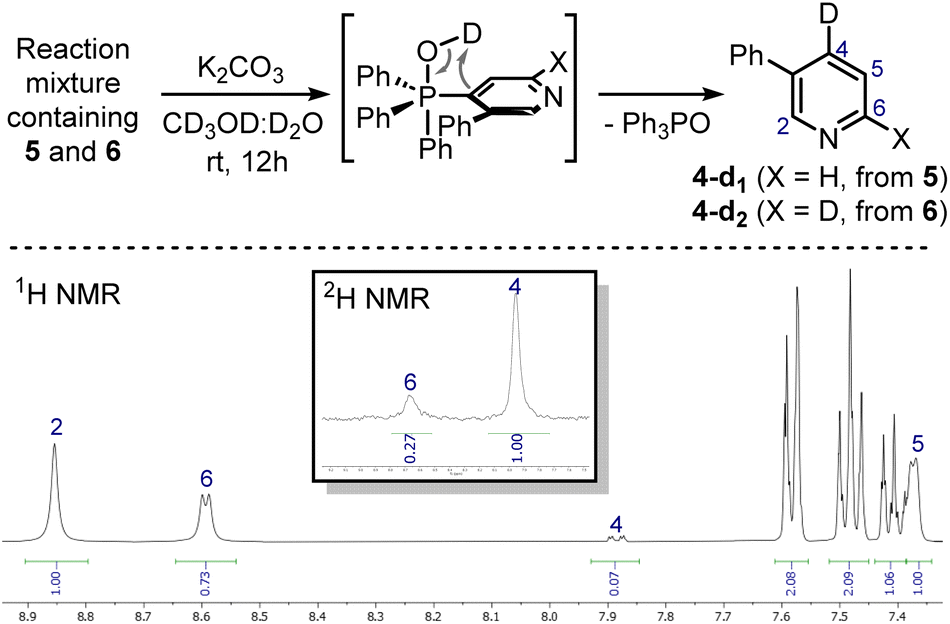 | ||
| Scheme 4 Deuterodephosphination of the 5/6 mixture (top) and 1H NMR and 2H NMR spectra (CDCl3, 400 MHz) of the isolated mixture of 4-d1/4-d2 (bottom). Deuterium incorporation was determined by the relative integration of the signals of hydrogens on the pyridine ring. | ||
Finally, attempts to use a more nucleophilic phosphine (i.e. (4-anisyl)3P) did not improve the C–P bond formation, and neither did the use of it as the first installed phosphine (see ESI†). Indeed, the use of (4-anisyl)3P mainly resulted in the formation of the corresponding phosphine oxide, as expected for electron-rich phosphines. Given the problematic separation of salts 2 and 3 and the modest conversion observed in the second step, we decided to perform one C–P bond formation at a time. Treatment of 2 with MeONa in dichloromethane gave disubstituted pyridine 7 in 53% 1H NMR yield (Scheme 5), although its isolation was complicated by co-eluting Ph3PO (the product, together with 1, of competitive protodephosphination). Before performing the second C–P bond formation on 7, the installation of a methyl group using 2 as a model compound was attempted.
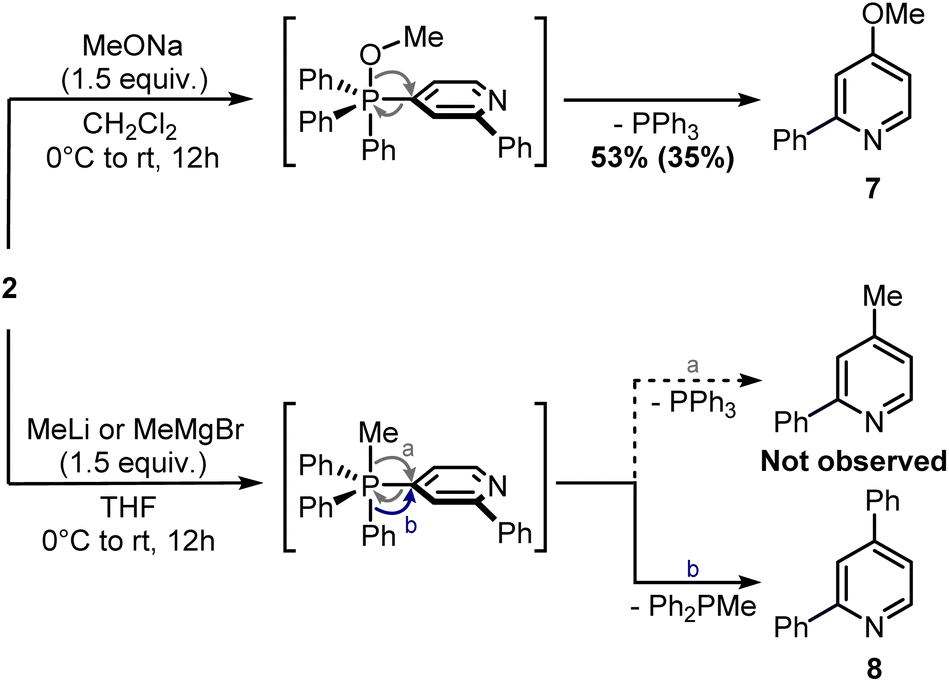 | ||
| Scheme 5 Ligand-coupling reactions with phosphonium salt 2. 1H NMR yield determined using CH2Br2 as internal standard (in brackets, isolated yield). | ||
Indeed, while the replacement of Ph3P with chalcogens/pnictogen nucleophiles (–OR, –SR, –NR2) is relatively straightforward,27,30,31 the installation of alkyl or aryl groups via ligand-coupling requires additional manipulations.32–34 However, the direct use of organolithium has been shown successful in a couple of cases (e.g. ArLi), hence we hoped the use of MeLi or MeMgBr would avoid extra steps.27 Unfortunately, treatment of 2 with these organometallics provided equimolar amounts of 2,4-diphenylpyridine 8 and Ph2P(O)Me (Scheme 5), the latter observable in the 1H NMR spectrum (2.01 ppm, d, 2JP–H = 13.2 Hz, 3H, Me). This result highlights how, in contrast to alkoxides, the phenyl ring has a higher migration aptitude than a methyl group during the ligand-coupling of the phosphorane intermediate,35,36 thus leading to 8 and Ph2PMe (then oxidized during the workup). An alternative approach to the use of phosphonium salts would be an ortho-halogenation followed by Negishi coupling with MeZnCl, in analogy with the reported synthesis of caerulomycin E.18 These halogenations (Reissert–Henze reactions) require the use of N-oxides, easily made upon treatment of pyridines with an oxidant such as H2O2 or m-chloroperbenzoic acid (mCPBA).37 Initially, using 1-O as a model substrate, activation with Tf2O and bromination with tetra-n-butylammonium bromide (TBAB) was attempted, in analogy with the ortho-bromination of quinolines reported by Baran and co-workers (Scheme 6).38 However, no desired product was observed, but only a mixture of brominated pyridines, probably due to some Br2 formed from the residual oxidant (mCPBA) still present in 1-O. In contrast to pyridines, the successful ortho-bromination observed by Baran for quinolines reflects the lower loss in resonance stabilization typical of bicyclic aromatics (i.e. naphthalene vs. benzene). Chlorination of 1-O with POCl3 gave better results (9 isolated in 55% yield), but the need for harsh conditions (neat POCl3 refluxing at 106 °C) somehow defeated our original purpose for a short and mild synthesis, thus a completely different strategy was considered.
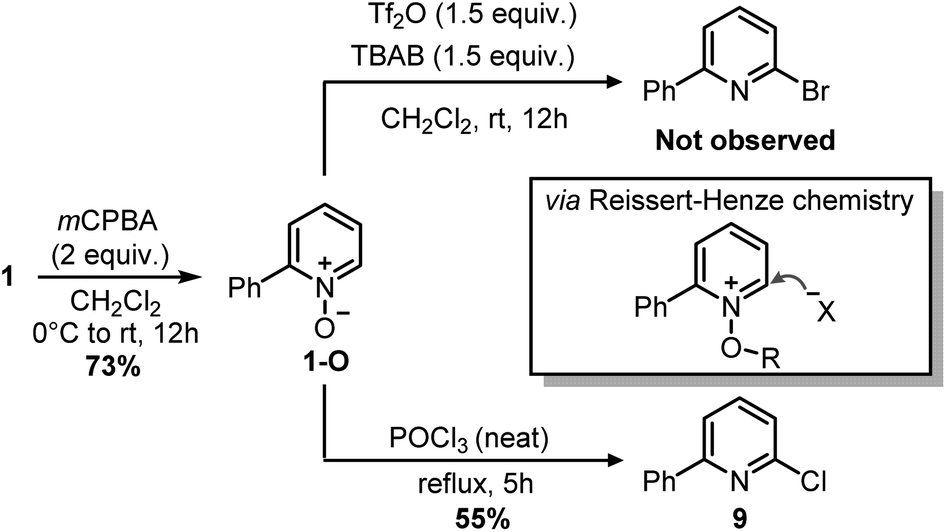 | ||
| Scheme 6 ortho-Halogenations with 1-O. | ||
Minisci-type chemistry is an excellent method for ortho-functionalizations of pyridines, especially employing nucleophilic carbon-based radicals (ideal for the synthesis of caerulomycin K).39,40 Moreover, starting with a 4-substituted pyridine would prevent regioselectivity issues (C2 vs. C6) typical of unsymmetrical starting materials. For the ortho-arylation of pyridines, Baran and others have shown how aryl boronic esters, in combination with AgNO3, Na2S2O8, and TFA, are excellent aryl radical precursors.41,42 To install a carbonyl group, Angeles, Yeung, and colleagues have used 1,3,5-trioxanes as an aldehyde equivalent in Minisci-type carbonylation of pyridines.43 Based on this precedent, a Minisci arylation of 4-chloropyridine 10 was performed (Scheme 7). In this case, an excess of phenylboronic acid (1.5 equiv.) was needed to compensate for competitive protodeboronation, whereas a higher loading of AgNO3 allowed the isolation of product 11 in 56% yield. A second Minisci reaction was then performed on this pyridine using 1,3,5-trioxane in the presence of (nBu4N)2S2O8. A successful ortho-alkylation gave product 12 in 50% yield (a value in agreement with previous reports),44–47 whereas a subsequent nucleophilic aromatic substitution allowed to access trifunctionalized pyridine 13 almost quantitatively. It has to be noted that starting from 4-methoxypyridine 14 would shorten the synthesis, but the electron-donating effect of the methoxy group will negatively affect both steps since Minisci reactions are based on the addition of nucleophilic radicals. The final conversion of 13 into caerulomycin K was achieved in a one-pot procedure by treatment with HCl (to reveal the aldehyde functionality), followed by condensation with NH2OH. Importantly, 12 could be directly converted into caerulomycin K without the need for isolation of 13, further simplifying the synthesis (route in green). Therefore, this three-step total synthesis (overall yield of 10%) represents the first synthesis of caerulomycin K as well as a potential alternative to the synthesis of caerulomycins.
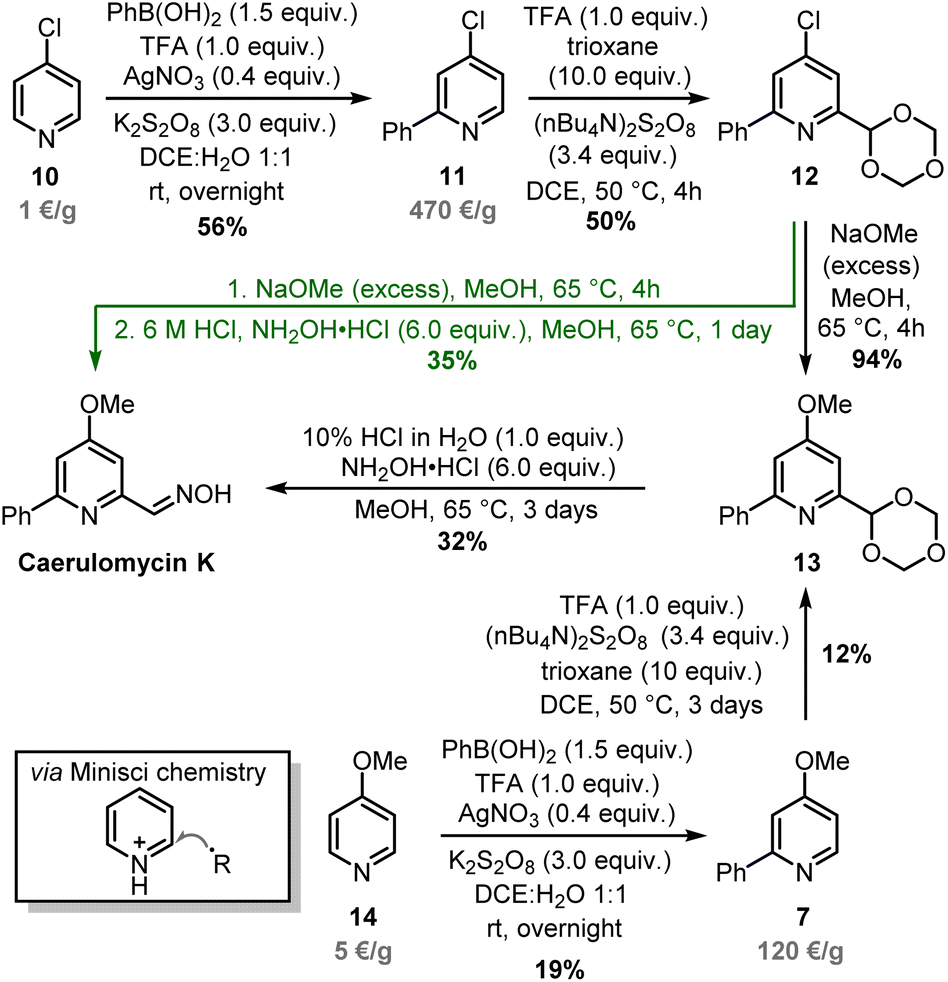 | ||
| Scheme 7 ortho-Functionalization of 10 and total synthesis of caerulomycin K. | ||
In conclusion, the first total synthesis of caerulomycin K has been reported. Starting from monofunctionalized pyridines, the first strategy looked at a double C–H activation by means of phosphonium chemistry. However, a poor conversion of the second C–P bond formation and a problematic ortho-methylation, including via halogenation, prompted the search for a better alternative. This was achieved by sequential Minisci ortho-arylation and ortho-alkylation, with the latter converted in one pot into the desired oxime. Compared to previously reported caerulomycin syntheses, this novel approach does not require highly pre-functionalized starting materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Miriana Guarnaccia and Marissa Panethiere for preliminary results as well as Dr Elisa Moretti and Dr Francesco Castagnetti for mass analyses. The authors acknowledge Chiesi Farmaceutici S.p.A for funding and the support of the APC central fund of the University of Milan.Notes and references
- S. De, A. Kumar, S. K. Shah, S. Kazi, N. Sarkar, S. Banerjee and S. Dey, RSC Adv., 2022, 12, 15385–15406 RSC.
- Y. Ling, Z. Y. Hao, D. Liang, C. L. Zhang, Y. F. Liu and Y. Wang, Drug Des., Dev. Ther., 2021, 15, 4289–4338 CrossRef CAS PubMed.
- S. Maity, A. Bera, A. Bhattacharjya and P. Maity, Org. Biomol. Chem., 2023, 21, 5671–5690 RSC.
- M. Failla, G. W. Lombardo, P. Orlando, D. Fiorito, E. Bombonato, P. Ronchi, D. Passarella and V. Fasano, Eur. J. Org Chem., 2023, e202300074 CrossRef CAS.
- C. M. Josephitis, H. M. H. Nguyen and A. McNally, Chem. Rev., 2023, 123, 7655–7691 CrossRef CAS PubMed.
- L. C. Vining, A. G. McInnes, A. W. McCulloch, D. G. Smith and J. A. Walter, Can. J. Chem., 1988, 66, 191–194 CrossRef CAS.
- A. G. Mcinnes, D. G. Smith, J. A. Walter, L. C. Vining and J. L. C. Wright, Can. J. Chem., 1979, 57, 3200–3204 CrossRef CAS.
- D. K. Chatterjee, W. Raether, N. Iyer and B. N. Ganguli, Z. Parasitenkd, 1984, 70, 569–573 CrossRef CAS PubMed.
- D. Chen, Q. Zhao and W. Liu, J. Ind. Microbiol. Biotechnol., 2019, 46, 459–468 CrossRef CAS PubMed.
- B. Pang, R. Liao, Z. Tang, S. Guo, Z. Wu and W. Liu, Nat. Commun., 2021, 12, 3124 CrossRef CAS PubMed.
- P. Fu, S. Wang, K. Hong, X. Li, P. Liu, Y. Wang and W. Zhu, J. Nat. Prod., 2011, 74, 1751–1756 CrossRef CAS PubMed.
- B. D. Alreja, S. L. Kattige, B. La and N. J. de Souza, Heterocycles, 1986, 24, 1637–1640 CrossRef CAS.
- J. Dash and H. U. Reissig, Chem.–Eur. J., 2009, 15, 6811–6814 CrossRef CAS PubMed.
- D. N. Bobrov and V. Tyvorskii, Tetrahedron, 2010, 66, 5432–5434 CrossRef CAS.
- F. Zhang and X.-F. Duan, Org. Lett., 2011, 13, 6102–6105 CrossRef CAS PubMed.
- A. M. Horan, V. K. Duong and E. M. McGarrigle, Org. Lett., 2021, 23, 9089–9093 CrossRef CAS PubMed.
- F. Mongin, F. Trécourt, B. Gervais, O. Mongin and G. Quéguiner, J. Org. Chem., 2002, 67, 3272–3276 CrossRef CAS PubMed.
- F. Trécourt, B. Gervais, O. Mongin, C. Le Gal, F. Mongin and G. Quéguiner, J. Org. Chem., 1998, 63, 2892–2897 CrossRef.
- X. F. Duan, Z. Q. Ma, F. Zhang and Z. B. Zhang, J. Org. Chem., 2009, 74, 939–942 CrossRef CAS PubMed.
- T. Sammakia, E. L. Stangeland and M. C. Whitcomb, Org. Lett., 2002, 4, 2385–2388 CrossRef CAS PubMed.
- F. Mongin, F. Trécourt, B. Gervais, O. Mongin and G. Quéguiner, J. Org. Chem., 2002, 67, 3272–3276 CrossRef CAS PubMed.
- R. G. Anderson, B. M. Jett and A. McNally, Angew. Chem., Int. Ed., 2018, 57, 12514–12518 CrossRef CAS PubMed.
- R. D. Dolewski, M. C. Hilton and A. McNally, Synlett, 2018, 29, 8–14 CrossRef CAS.
- J.-P. Finet, Tetrahedron Org. Chem. Ser., 1998, 18, 9–46 Search PubMed.
- E. Anders and F. Markus, Chem. Ber., 1989, 122, 119–122 CrossRef CAS.
- E. Anders and F. Markus, Chem. Ber., 1989, 122, 113–118 CrossRef CAS.
- M. C. Hilton, R. D. Dolewski and A. McNally, J. Am. Chem. Soc., 2016, 138, 13806–13809 CrossRef CAS PubMed.
- P. Du, Y. Yin, D. Shi, K. Mao, Q. Yu and J. Zhao, Molecules, 2022, 27, 5694 CrossRef CAS PubMed.
- J. L. Koniarczyk, D. Hesk, A. Overgard, I. W. Davies and A. McNally, J. Am. Chem. Soc., 2018, 140, 1990–1993 CrossRef CAS PubMed.
- R. G. Anderson, B. M. Jett and A. McNally, Tetrahedron, 2018, 74, 3129–3136 CrossRef CAS PubMed.
- C. Patel, M. Mohnike, M. C. Hilton and A. McNally, Org. Lett., 2018, 20, 2607–2610 CrossRef CAS PubMed.
- X. Zhang and A. McNally, Angew. Chem., Int. Ed., 2017, 56, 9833–9836 CrossRef CAS PubMed.
- M. C. Hilton, X. Zhang, B. T. Boyle, J. V. Alegre-Requena, R. S. Paton and A. McNally, Science, 2018, 362, 799–804 CrossRef CAS PubMed.
- X. Zhang and A. McNally, ACS Catal., 2019, 9, 4862–4866 CrossRef CAS PubMed.
- S. Oae and Y. Uchida, Acc. Chem. Res., 1991, 24, 202–208 CrossRef CAS.
- R. Hoffmann, J. M. Howell and E. L. Muetterties, J. Am. Chem. Soc., 1972, 94, 3047–3058 CrossRef CAS.
- D. Wang, L. Désaubry, G. Li, M. Huang and S. Zheng, Adv. Synth. Catal., 2021, 363, 2–39 CrossRef CAS.
- S. E. Wengryniuk, A. Weickgenannt, C. Reiher, N. A. Strotman, K. Chen, M. D. Eastgate and P. S. Baran, Org. Lett., 2013, 15, 792–795 CrossRef CAS PubMed.
- R. S. J. Proctor and R. J. Phipps, Angew. Chem., Int. Ed., 2019, 58, 13666–13699 CrossRef CAS PubMed.
- M. A. J. Duncton, MedChemComm, 2011, 2, 1135–1161 RSC.
- I. B. Seiple, S. Su, R. A. Rodriguez, R. Gianatassio, Y. Fujiwara, A. L. Sobel and P. S. Baran, J. Am. Chem. Soc., 2010, 132, 13194–13196 CrossRef CAS PubMed.
- P. P. Singh, S. K. Aithagani, M. Yadav, V. P. Singh and R. A. Vishwakarma, J. Org. Chem., 2013, 78, 2639–2648 CrossRef CAS PubMed.
- J. M. Ganley, M. Christensen, Y. H. Lam, Z. Peng, A. R. Angeles and C. S. Yeung, Org. Lett., 2018, 20, 5752–5756 CrossRef CAS PubMed.
- H. Tian, H. Yang, C. Tian, G. An and G. Li, Org. Lett., 2020, 22, 7709–7715 CrossRef CAS PubMed.
- H. Zhao and J. Jin, Org. Lett., 2019, 21, 6179–6184 CrossRef CAS PubMed.
- H. Zhao, Z. Li and J. Jin, New J. Chem., 2019, 43, 12533–12537 RSC.
- G. P. Gardini, Tetrahedron Lett., 1972, 13, 4113–4116 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra00589a |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |