
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTumor microenvironment-regulated drug delivery system combined with sonodynamic therapy for the synergistic treatment of breast cancer
Chao Qian ab,
Guoliang Zhaoc,
Mengping Huob,
Meixia Sud,
Xuexue Hue,
Qiang Liu*ad and
Lei Wang*b
ab,
Guoliang Zhaoc,
Mengping Huob,
Meixia Sud,
Xuexue Hue,
Qiang Liu*ad and
Lei Wang*b
aShandong Provincial Hospital, Shandong University, Jinan, 250000, China. E-mail: 2002md@163.com
bDepartment of Ultrasound, Shandong Provincial Hospital Affiliated to Shandong First Medical University, Jinan, 250021, China. E-mail: sdslwanglei@163.com
cDepartment of Gastroenterology, The First Affiliated Hospital of Shandong First Medical University, Jinan, 250014, China
dDepartment of Radiology, Shandong Provincial Hospital Affiliated to Shandong First Medical University, Jinan, 250021, China
eSchool of Clinical Medicine, Shandong First Medical University, Jinan, 250117, China
First published on 31st May 2024
Abstract
Co-loading of sonosensitizers and chemotherapeutic drugs into nanocarriers can improve the biocompatibilities, stabilities, and targeting of drugs and reduce the adverse reactions of drugs, providing a robust platform to orchestrate the synergistic interplay between chemotherapy and sonodynamic therapy (SDT) in cancer treatment. In this regard, biodegradable manganese dioxide (MnO2) has attracted widespread attention because of its unique properties in the tumor microenvironment (TME). Accordingly, herein, MnO2 nanoshells with hollow mesoporous structures (H-MnO2) were etched to co-load hematoporphyrin monomethyl ether (HMME) and doxorubicin (DOX), and DOX/HMME-HMnO2@bovine serum albumin (BSA) obtained after simple BSA modification of DOX/HMME-HMnO2 exhibited excellent hydrophilicity and dispersibility. H-MnO2 rapidly degraded in the weakly acidic TME, releasing loaded HMME and DOX, and catalysed the decomposition of H2O2 abundantly present in TME, producing oxygen (O2) in situ, significantly increasing O2 concentration and downregulating the hypoxia-inducible factor 1α (HIF-1α). After irradiation of the tumor area with low-frequency ultrasound, the drug delivery efficiency of DOX/HMME-HMnO2@BSA substantially increased, and the excited HMME generated a large amount of reactive oxygen species (ROS), which caused irreversible damage to tumor cells. Moreover, the cell death rate exceeded 60% after synergistic SDT-chemotherapy. Therefore, the pH-responsive nanoshells designed in this study can realize drug accumulation in tumor regions by responding to TME and augment SDT-chemotherapy potency for breast cancer treatment by improving hypoxia in tumors. Thus, this study provides theoretical support for the development of multifunctional nanocarriers and scientific evidence for further exploration of safer and more efficient breast cancer treatments.
Introduction
Breast cancer stands out as the predominant malignancy affecting women, marked by elevated morbidity and mortality. Conventional therapeutic modalities, encompassing surgical interventions, radiation therapy, chemotherapy, and hormonal therapy, often cause significant damage to normal tissues and exhibit strong toxicities and adverse reactions. Furthermore, non-specific drug resistance in tumor cells is typical during drug treatment, decreasing treatment efficacy.1 Sonodynamic therapy (SDT) generates reactive oxygen species (ROS) and induces cavitation effects by activating sonosensitizers via ultrasound (US) irradiation, which can cause apoptosis and necrosis, thereby killing tumor cells.2,3 SDT demonstrates the potential for targeted therapy as it offers the ability to focus US on small tissue areas in the body. Therefore, SDT is considered a non-invasive, precise, safe, efficient, and repeatable treatment.4,5HMME is a second-generation hematoporphyrin (HP) sonosensitizer, which has the advantages of single composition, stable performance and high tumor selectivity, and can induce cell apoptosis through the mitochondrial apoptosis pathway, making it widely used in clinical practice.6–8 However, conventional sonosensitizers suffer from limitations including inferior biocompatibility, rapid degradation, and low ROS production, leading to low SDT efficiency.9,10 To address these issues, researchers have explored the use of nanocarriers for delivering sonosensitizers to enhance their therapeutic efficacy. Owing to their biological characteristics, for example, small particle sizes and large surface-to-body ratios, nanomaterials can efficiently carry small drugs to tumor areas. They also possess characteristics such as size tunability, high stability, high drug-loading capacity (LC), and the capability to encapsulate hydrophobic and hydrophilic substances, rendering them particularly alluring in the oncological domain. Due to the dense tumor stroma and inadequate vascular permeability, current nanodrugs based on enhanced permeability and retention (EPR) effects mainly accumulate around tumor blood vessels, resulting in limited passive penetrations of these nanodrugs into the tumor.11 However, when low-frequency US is used to irradiate the tumor area, vascular permeabilities of tumors significantly increase, and the generation of acoustic radiation, microfluidizer, and shock waves facilitate the intratumoral accumulations of nanodrugs.12–14 Unlike the normal tissue microenvironment, the tumor microenvironment (TME) exhibits hypoxia (below 30–40 mm Hg) and acidity (pH of 6.5–6.8), with elevated levels of hydrogen peroxide (H2O2) (concentration of 10–100 μM) and glutathione (GSH).15–17 The antitumor effectiveness of oxygen-dependent SDT is significantly hindered by the prevailing hypoxia in tumors.18,19 Additionally, oxygen consumption during SDT further aggravates tumor growth and metastasis.20–22 Therefore, the synthesis and development of nanodrug delivery systems (NDDS) with the abilities to modulate TME have become research hotspots in SDT.23
Accordingly, herein, a pH-responsive TME-regulated NDDS, hollow mesoporous manganese dioxide (H-MnO2, HM) nano-shells, is established. HM can efficiently load both chemotherapeutic drugs and sonosensitizers.18,24,25 Bovine serum albumin (BSA), a hydrophilic protein, is used to modify the surface of HM via hydrogen bonding to maintain the stability of HM during its circulation in the body, reduce its aggregation, and improve its water solubility and stability. Furthermore, BSA-mediated drug transport via albumin receptors (gp60) can increase drug accumulation in tumors.26,27 HM degrades in the weakly acidic TME and releases drugs, thereby minimizing premature drug leakage under physiological conditions.28 HM can also catalyse the decomposition of H2O2 abundantly present in TME, producing oxygen in situ, thereby alleviating hypoxia in TME and enhancing the synergistic efficacy of SDT and chemotherapy.29,30 Moreover, after degradation of HM under acidic conditions, Mn2+ from HM can undergo a Fenton-like reaction with H2O2, yielding highly reactive hydroxyl radicals (˙OH).31,32 Furthermore, HM can convert GSH present in high levels in TME into glutathione disulfide (GSSG), thereby reducing the scavenging effect of GSH on ˙OH and eliminating other potential adverse effects of high GSH levels.33 Therefore, HM exhibits excellent chemical properties that match those of TME. When HM is efficiently loaded with drugs and appropriately modified, it can realize TME-dependent drug release and precise combination therapy, providing theoretical support for the development of multifunctional NDDS for breast cancer treatment.34
Experimental
Reagents and materials
Hematoporphyrin monomethyl ether (HMME) was obtained from Shanghai Macklin Biochemical Co., Ltd. Doxorubicin (DOX) was acquired from Shanghai Aladdin Biochemical Technology Co., Ltd. Calcein-AM/PI live/dead cell double staining kit, BSA, PBS with pH = 7.4, and a penicillin–streptomycin double antibody mixture were obtained from Beijing Solarbio Science & Technology Co., Ltd. Singlet oxygen sensor green fluorescent probe (SOSG) was procured from Dalian Meilun Biotech Co., Ltd. Cell counting kit-8 (CCK-8) was obtained from APExBIO Technology, USA. 4′,6-Diamidino-2-phenylindole (DAPI) and 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA) were acquired from Shanghai Beyotime Biotechnology Co., Ltd. Phosphate buffered saline (PBS) solutions with pH = 6.5 and 5.5 were provided by Shanghai Yuanye Biotechnology Co., Ltd. Trypsin was procured from Gibco, USA.Synthesis and characterization of HM
HM was synthesized following a previously reported method.35 First, monodispersed silica nanoparticles (SiO2 NPs) were fabricated via the hydrolysis of tetraethyl orthosilicate (TEOS) (5 mL). Subsequently, potassium permanganate solution (KMnO4) (25 mmol L−1, 75 mL) was added to SiO2 NPs (50 mmol L−1, 50 mL), and the resulting SiO2 NPs were used as hard templates to synthesize SiO2@MnO2 NPs with core/shell structures. Finally, Na2CO3 (2 mol L−1) was employed to etch SiO2 coated with MnO2 to obtain HM (50 mg). The elemental composition and morphology of HM (50 μg mL−1) were determined utilizing FEI Tecnai F20 transmission electron microscopy (TEM) and Zeiss Sigma 300 scanning electron microscopy (SEM). Zeta potential and particle size of HM were measured using Malvern Zetasizer (ZS90, Malvern, UK). Shell thickness and pore size were roughly measured using Image J.Preparations and characterizations of DOX/HMME-HMnO2 (DH-HM)@BSA and HMME-HMnO2 (H-HM)@BSA
For DOX and HMME loading, HM (10 mg mL−1, 100 μL), DOX (10 mg mL−1, 600 μL or more) and HMME (10 mg mL−1, 600 μL or more) were added into the 10 mL mixed solution of ultrapure water and anhydrous ethanol (volume ratio = 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6). After ultrasonic mixing, the mixture was stirred at 500 rpm for 24 h using a magnetic stirrer under light-deprived conditions at 26 °C. And then DH-HM was obtained through the process of centrifugation (12000 rpm, 10 min × 3 times) and utilized for further experiments. At the same time, the supernatant was collected for further determination. H-HM was prepared via the same abovementioned procedure except that only HMME was loaded in this case. BSA was introduced into the DH-HM solution at a feed weight ratio (BSA:MnO2) of 8:1 under stirring (500 rpm, 3 h) to achieve DH-HM@BSA. Furthermore, H-HM@BSA was fabricated using the same abovementioned procedure.
:6). After ultrasonic mixing, the mixture was stirred at 500 rpm for 24 h using a magnetic stirrer under light-deprived conditions at 26 °C. And then DH-HM was obtained through the process of centrifugation (12000 rpm, 10 min × 3 times) and utilized for further experiments. At the same time, the supernatant was collected for further determination. H-HM was prepared via the same abovementioned procedure except that only HMME was loaded in this case. BSA was introduced into the DH-HM solution at a feed weight ratio (BSA:MnO2) of 8:1 under stirring (500 rpm, 3 h) to achieve DH-HM@BSA. Furthermore, H-HM@BSA was fabricated using the same abovementioned procedure.
The content of HMME and DOX in the nanoparticles (DH-HM@BSA, H-HM@BSA) can be deduced to determine by calculating the mass of free HMME and DOX in the recovered supernatant. The specific procedure is as follows: at the end of the preparation of nanoparticles, when the precipitate was collected the centrifuged supernatant was recovered. High-performance liquid chromatography (HPLC) (Agilent, USA) with a mobile phase of ethanol and ultrapure water (v/v = 70:30) was used to separate HMME and DOX from the recovered supernatant. To determine the content of HMME in H-HM@BSA, this step is not necessary. Then, the absorbance of DOX, HMME, and BSA in the supernatant was recorded separately using ultraviolet spectrophotometer (Biotek Epoch 2). Finally, the concentrations of DOX, HMME, and BSA were calculated according to the standard curve of DOX, HMME, and BSA, followed by the calculation of drug loading capacity (LC), encapsulation efficiency (EE), and BSA modification efficiency. LC (%w/w) = (WFed drug − WDrug in supernatant)/Wnanoparticles × 100%. EE (%w/w) = (WFed drug − WDrug in supernatant)/WFed drug × 100%.
In addition, ultraviolet (UV)-visible (vis) spectra of full wavelength (300–800 nm) of all the substances involved in drug-loading were obtained using ultraviolet spectrophotometer. Particle sizes and zeta potentials of HM, DH-HM, and DH-HM@BSA during the sequential preparing process were determined using the Malvern Zetasizer. To analyze the stabilities of as-prepared DH-HM@BSA and DH-HM, these materials were separately added to PBS (pH 7.4) and fetal bovine serum (FBS). Measurements of particle sizes in the resultant mixtures were conducted both immediately and one week after incubation using the Malvern Zetasizer.
Catalase-like property of HM
To detect the catalase-like property of HM for catalysing the decomposition of H2O2 to produce oxygen (O2), HM at different concentrations was introduced into H2O2 solution (100 μM), and measurement of the concentration of dissolved O2 produced was carried out with the aid of a dissolved oxygen meter (WLDO-300 portable fluorescence dissolved oxygen meter, Shanghai Shuiyi Technology Co., Ltd).Degradation of carriers and release of drugs from DH-HM@BSA
To examine the reactivity of the nanocarrier HM, PBS solutions with pH = 6.5 and 5.5 were employed to simulate the mildly acidic TME. HM was separately incubated in 10 mL PBS at pH values of 5.5, 6.5, and 7.4 for varying durations. At specific time points, the degradation of HM was characterized using TEM and UV-vis spectroscopy. Specifically, the morphology of HM nano-shells was observed by TEM. And the retained rates of HM were characterized by the decrease of HM absorbance obtained by UV-vis spectrophotometer.Subsequently, the release of DOX and HMME loaded into DH-HM@BSA were investigated in simulated TME. DH-HM@BSA was separately dispersed in 20 mL PBS at various pH levels (6.5 and 7.4) with or without US irradiation (WED-100, Shenzhen, China) (1.5 W cm−2, 1 MHz, duty cycle: 50%, and 3 min) at room temperature. The dispersions were stirred in the dark, samples (1 mL from each experiment) were taken out at certain time points (0, 10, 20, 30, 60, 120, 240, 480, 720, and 1440 min) and immediately centrifuged to collect the supernatant. And then HMME and DOX from the supernatant were separated by HPLC and their absorbance was recorded separately at λ = 490 nm and 396 nm using a UV-vis spectrophotometer, thus determining the content of DOX and HMME. Finally, the drug cumulative release rates of DOX and HMME were calculated according to the following equation: drug cumulative release rates = (weight of released drug/weight of total loaded drug) × 100%.
Detection of singlet oxygen (SO)
Singlet oxygen is produced by the sonosensitizers upon exposure to US irradiation. The singlet oxygen generated can be detected using SOSG. Herein, 8 groups (PBS, HMME, US, H-HM@BSA, HMME+US, H-HM@BSA+US, HMME(+H2O2)+US, and H-HM@BSA(+H2O2)+US) were designed, and each group was incubated with SOSG (10 μM) with or without US irradiation (1.5 W cm−2, 1 MHz, duty cycle: 50%, and 3 min). Production of 1O2 by H-HM@BSA (HMME = 9 μg mL−1) under different conditions was explored by measuring the fluorescence intensity of SOSG using a fluorescence microplate reader (SpectraMax i3, Molecular Devices, USA) at 494 nm excitation. In the abovementioned experiments, HM degraded and released drugs in a slightly acidic environment; therefore, the pH values of all reagents were adjusted to 6.5 for this experiment.Cell culture
MCF-7 human and 4T1 murine breast cancer cells were acquired from Wuhan Procell Life Science & Technology Co., Ltd. MCF-7 cells were grown in Dulbecco's Modified Eagle Medium supplemented with 10% FBS and 1% penicillin/streptomycin, whereas 4T1 cells were grown in Roswell Park Memorial Institute-1640 medium with the same supplements. These cells were incubated at 37 °C in a humidified incubator with 5% CO2. Furthermore, for subsequent investigations, cells in their optimal proliferative state were utilized.In vitro cytotoxicity of HM
Cytotoxicity of HM at different concentrations were assessed using CCK-8 (ApexBio, USA). Briefly, MCF-7 cells were transferred in a 96-well plate (2 × 104 cells per well) for seeding until adherent followed by incubation with HM at various concentrations. Herein, three replicate wells were set for each concentration. Following a 24 h incubation period, the CCK-8 solution was introduced into each well, and the viability of MCF-7 cells was assessed by their optical density (OD) at 450 nm using a microplate reader (Multiskan GO, Thermo Fisher Scientific, USA), in comparison to those of the untreated cells.Cellular uptake of DH-HM@BSA
MCF-7 cells were seeded in glass-bottom culture dishes (2 × 105 cells per dish) until adherent followed by incubation with DH-HM@BSA (DOX = 8 μg mL−1 and HMME = 9 μg mL−1) under light-deprived conditions for several periods (0.5, 1, and 4 h). The cells were then washed thrice using PBS. Thereafter, they were fixed with paraformaldehyde, stained with DAPI (Beyotime, China) (Ex = 364 nm, Em = 454 nm), and observed using a confocal laser scanning fluorescence microscope (SP5, Leica, USA). Similarly, MCF-7 cells were incubated with free DOX (Ex = 485 nm, Em = 593 nm) and HMME (Ex = 395 nm, Em = 613 nm) (DOX = 8 μg mL−1 and HMME = 9 μg mL−1) for 1 h and examined. To avoid potential false positives caused by overlapping fluorescence emissions, the samples were irradiated with different wavelength of lasers sequentially, while the fluorescence signals were collected through corresponding detection channels alternately, and confocal images of each fluorescence were displayed.In vitro ROS generation and SDT
To investigate intracellular ROS production, DCFH-DA was utilized as a fluorescent probe. Initially, the influences of various US powers on ROS generation and cell viability were compared to determine the appropriate US power for successive experiments. Thereafter, six groups were designed to compare ROS generation under different conditions. Specifically, MCF-7 cells were seeded in six-well plates (2 × 105 cells per well) until adherent followed by separate incubation with free HMME (18 μg mL−1) and H-HM@BSA (HMME = 18 μg mL−1) for 4 h and further incubation in a medium containing DCFH-DA (10 μmol L−1) for 0.5 h. Subsequently, they were irradiated with US at selected power (1 MHz, duty cycle: 50%, and 3 min). Finally, intracellular ROS fluorescence images were acquired with a fluorescence microscope (Axio Observer, Zeiss, Germany). To further evaluate the efficacy of SDT in the six groups, determination of the cell viability for each group was conducted via the CCK-8 assay.In vitro combination therapy
Qualitative experiment: MCF-7 cells seeded in 12-well plates (1 × 105 cells per well) were separately incubated with PBS, free DOX, H-HM@BSA, and DH-HM@BSA (DOX = 16 μg mL−1 and HMME = 18 μg mL−1) for 4 h followed by no treatment or treatment with US irradiation (0.5 W cm−2, 1 MHz, 50%, and 3 min) and incubation for another 2 h. Following the addition of Calcein-AM/PI, the cells were further incubated for 0.5 h under light-deprived conditions at 37 °C. The fluorescence microscope was used to capture images of live/dead cells.Quantitative experiment: the cells prepared in 96-well plates (2 × 104 cells per well) were similarly treated, and the cell viability was then evaluated via the CCK-8 assay.
Establishment of animal model
Female BALB/c nude mice (approximately 5 weeks old) were acquired from Beijing SPF Biotechnology Co., Ltd and raised in a controlled environment (20–26 °C, 40–70% humidity, and artificial lighting with a 12 hours light–dark alternation) at the Experimental Animal Research Institute of Shandong Provincial Hospital Affiliated to Shandong First Medical University. To establish a subcutaneous breast cancer xenograft model in nude mice, 1 × 106 4T1 cells suspended in 100 μL PBS were subcutaneously administered into the right forelimb armpit of BALB/c nude mice. Mice with suitable tumor volume were used for subsequent experiments after approximately 7–14 days.In vivo biodistribution
For mouse xenografts, the following formula was used to determine the tumor volume: V = (L × W2)/2. The animals with tumor volumes of around 80 mm3 were randomized into one of two groups, each consisting of three specimens. Biodistributions of free DOX and DH-HM@BSA in tumor-bearing mice were assessed using a small-animal in vivo imaging system (IVIS) (Spectrum, PerkinElmer, USA). Typically, 100 μL of free DOX and DH-HM@BSA (DOX = 4 mg kg−1) were intravenously injected. At specific time intervals (1, 4, 8, 12 and 24 h) post-injection, the distribution and intensity of red fluorescence were examined using the imaging system. After 24 h, the kidneys, lungs, spleen, liver, heart, and tumor tissues were separated from both groups of mice, and the red fluorescence intensity was quantitatively analyzed using the imaging system.In vivo antitumor efficacy
Mice harboring tumors with volumes of around 60 mm3 were randomized into one of six groups, each consisting of five specimens. And they were given 100 μL respective treatment solutions via the tail veins. After 1, 8, and 24 h of injection, the tumor was locally irradiated (1.5 W cm−2, 1 MHz, duty cycle: 50%, and 3 min) using the therapeutic US device. The treatment plans are presented in Table 1. Five injection–irradiation cycles were conducted every three days.| Groups | Injection | US | |
|---|---|---|---|
| 1 | Control | PBS, 100 μL | No |
| 2 | US | PBS, 100 μL | Yes |
| 3 | Free DOX | DOX is 4 mg kg−1, 100 μL | No |
| 4 | DH-HM@BSA | DH-HM@BSA (DOX is 4 mg kg−1), 100 μL | No |
| 5 | H-HM@BSA+US | H-HM@BSA (HMME is 4.5 mg kg−1), 100 μL | Yes |
| 6 | DH-HM@BSA+US | DH-HM@BSA (DOX is 4 mg kg−1 and HMME is 4.5 mg kg−1), 100 μL | Yes |
Mice were provided with a normal diet during treatment. To determine the trend curve of the tumor size and the body weight of mice at the end of treatment, these parameters were monitored every three days throughout treatment. Changes in tumors were also visually assessed and imaged. After five treatments, the tumors in each group were dissected, imaged, and weighed. Furthermore, tumor weights in each group were compared and statistically analyzed.
To further evaluate the antitumor effectiveness of the treatment and explore the treatment mechanism, the tumor tissues of each group were sectioned. In this regard, hematoxylin and eosin (H&E) staining, immunofluorescent (IF) staining of hypoxia-inducible factor-1α (HIF-1α), dihydroethidium (DHE) and immunohistochemical (IHC) staining of caspase-3, proliferating cell nuclear antigen (PCNA), terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) assay were conducted. The stained tumor sections were examined using an optical microscope (Axio Observer, Zeiss, Germany).
In vivo biosafety
After the five treatments, blood was acquired from anesthetized animals for routine blood and functional tests, such as serum creatinine (CREA), aspartate aminotransferase (AST), red blood cells (RBC), white blood cells (WBC), platelets (PLT), serum alanine aminotransferase (ALT) and urea nitrogen (BUN), of the liver and kidney. Then, all test subjects from all groups were euthanized, and their vital organs (namely, kidneys, lungs, heart, liver, and spleen) were dissected and stained with H&E to assess the biosafety of the treatment.Statistical analysis
Data are presented as the mean of three separate experiments ± standard deviation (SD). Data analysis was performed using Origin 8.0 and GraphPad Prism 8.0. When the two groups were compared, statistical significance was evaluated using Student's t-test. *p < 0.05, **p < 0.01, and ***p < 0.001 were considered statistically significant.Results and discussion
Synthesis and characterization of HM
HM was successfully fabricated using the three steps mentioned in Materials and methods (Fig. 1a). TEM and SEM images of HM clearly demonstrated spherical morphologies, hollow structures, and excellent dispersibilities of HM (Fig. 1b and c). Internal and external diameters were approximately 130 and 150–190 nm, respectively, with a shell thickness of about 20–30 nm and pore size of several nanometers, as measured by Image J. Energy-dispersive X-ray spectroscopy (EDS)-high-angle annular dark-field (HAADF) images confirmed the presence of Mn and O and further verified the hollow structures of HM (Fig. 1e). Dynamic light scattering (DLS) indicated normal distributions of particle sizes, with peak diameters of 165.5 ± 3.26 nm and zeta potentials of −34.7 ± 0.52 mV (Fig. 1d). | ||
| Fig. 1 Synthesis and characterization of HM. (a) Schematic depicting the sequential fabrication of HM and DH-HM@BSA. (b) SEM and (c) TEM images of HM. (d) Distributions of particle size and zeta potential of HM. (e) Elemental mapping results of HM. | ||
Preparations and characterizations of DH-HM@BSA and H-HM@BSA
The anticancer drug DOX and the sonosensitizer HMME were simultaneously loaded into HM under stirring to obtain DH-HM. To enhance the physiological stability and water solubility of DH-HM, the surface of HM was modified with BSA to achieve DH-HM@BSA for further experiments. DOX entered the hollow shell via charge attraction, hydrophobic HMME was connected to MnO2 via coordination bonds (Mn–O), and BSA was attached to MnO2 via hydrogen bonding.36 H-HM was effectively prepared by a similar method. UV-vis spectra of HMME and DOX demonstrated characteristic peaks at 396 and 490 nm, respectively, with no significant interference; therefore, herein, these peaks were selected to detect HMME and DOX (Fig. 2a). In the spectrum of DH-HM@BSA, the characteristic peaks of HMME and DOX (indicated by black arrows) were acquired, revealing the successful loading of HMME and DOX into HM. Similarly, the characteristic peak of HMME (indicated by a black arrow) was detected in the UV-vis spectrum of H-HM@BSA, confirming the effective loading of HMME into HM (Fig. 2b). | ||
| Fig. 2 Preparations and characterizations of drug-loaded nanoparticles. (a) and (b) UV-vis spectra of free HMME, free DOX, HM, H-HM@BSA, and DH-HM@BSA. (c) Particle sizes and zeta potentials of HM, DH-HM and DH-HM@BSA during the sequential preparing process. (d) LCs and EEs of HMME in H-HM@BSA at various feed weight ratios of drugs to MnO2. (e) EEs of HMME and DOX in H-HM@BSA or DH-HM@BSA at various feed weight ratios of drugs to MnO2. (f) Stabilities of DH-HM and DH-HM@BSA in PBS and FBS reflected by changes in particle size. The date are expressed as means ± standard deviation (SD) (n = 3). (***p < 0.001, **p < 0.01, and *p < 0.05). | ||
DLS was employed to determine the particle sizes and zeta potentials of NDDS. Gradual changes in the zeta potential after each step further verified the successful loadings of HMME, DOX, and BSA (Fig. 2c). The initial zeta potential of HM was −34.7 ± 0.52 mV, and it increased to −15.6 ± 1.11 mV after HMME and DOX loading. After the modification of DH-HM with BSA, the zeta potential slightly decreased to −20.2 ± 1.06 mV. Negative surface charge can reduce the adhesion of plasma proteins and clearance of the reticuloendothelial system, which is beneficial for maintaining the circulation stability and prolonging the circulation time of NDDS in blood vessels. After HMME, DOX, and BSA loading, no considerable changes were noticed in the morphology of HM, and the hydrodynamic size of NDDS slightly increased. Final particle size of DH-HM@BSA was 171.7 ± 7.13 nm (Fig. 2c). Generally, NDDS with particle sizes in the 10–200 nm range are suitable for in vivo drug delivery and conducive to the EPR effect. Therefore, the particle sizes of the drug-loaded nanoparticles fabricated herein are ideal.37 Monodispersity of DH-HM@BSA in solution was higher than that of DH-HM, which was beneficial for subsequent drug delivery.
LCs and EEs of HMME and DOX in nanoparticles (H-HM@BSA, DH-HM@BSA) were examined by UV-vis spectroscopy after centrifugation to remove excess HMME and DOX (Fig. 2d and e). With an increase in the feed weight ratio (HMME:MnO2), LC and EE of HMME in H-HM@BSA increased, and a LC of 89% and an EE of 82% were achieved at a 10:1 ratio (Fig. 2d). In this system, when both DOX and HMME were incorporated into the hollow shell of HM at the same time, the simultaneous loadings of DOX and HMME did not significantly affect EE of HMME. When the weight ratio of both drugs to the carrier was 6:1, EEs of DOX and HMME were 74 and 81%, respectively (Fig. 2e). Due to the ample pore size of HM and the electrostatic adsorption between the negatively charged HM and positively charged DOX, the loading efficiency was exceptionally high. The same method was used to determine the modification efficiency of BSA in the nano-system. When the BSA:MnO2 weight ratio was 8:1, the modification efficiency of BSA was approximately 87.8%. DOX demonstrated no substantial detachment, whereas HMME exhibited minimal detachment after BSA modification, indicating that BSA modification was simple and reliable.
Protein adsorption can lead to the aggregation or dispersion of DH-HM@BSA, causing rapid clearance of DH-HM@BSA from the serum. Therefore, the stability of DH-HM@BSA in the serum is a critical parameter. Minor aggregation was observed for DH-HM stored in PBS and FBS on day 7, and a significant statistical difference was noticed between the particle sizes measured on days 1 and 7 (Fig. 2f). Under the same conditions, no apparent sedimentation or aggregation was observed for DH-HM@BSA in 7 days; particularly, no significant statistical difference was noticed between the particle sizes of DH-HM@BSA in FBS on days 1 and 7, and the change in particle size of DH-HM@BSA in PBS was considerably less than that of DH-HM in PBS. These results confirm that addition of BSA significantly enhances the physiological stability of NDDS, which can prevent damage to normal tissues and organs caused by premature leakage of drugs from the nano-system during in vivo drug delivery, prolong the blood circulation time of NDDS, promote efficient accumulation of drugs in tumor cells, and thereby improve the EPR effect, facilitating passive targeting of tumors.
Catalase-like property of HM
Previous studies have demonstrated that hypoxia in TME is an important reason for the limited efficacies of solid tumor treatments such as SDT and chemotherapy, and endogenous H2O2 is commonly present in most solid tumors, with concentrations ranging from 10 to 100 μM.15,17 To explore the ability of HM to catalyse the decomposition of H2O2, HM at different concentrations was introduced into H2O2 (100 μM) solution, and the concentration of the produced dissolved O2 was measured. Before the addition of HM, the dissolved O2 concentration in the H2O2 solution was stable. After the introduction of HM, H2O2 was effectively triggered to rapidly decompose and generate O2, and the rate of oxygen production and concentration of generated oxygen increased with an increase in the HM concentration (Fig. 3a), which is consistent with the findings of previous studies.12 | ||
| Fig. 3 Performance testing of HM in vitro. (a) Catalase-like property of HM: variations of the concentration of O2 in H2O2 solutions (100 μm) with the addition of HM at various concentrations. (b) Degradation of HM: TEM images of HM incubated in PBS at pH levels of 6.5 and 7.4 for varying durations. (c) Percentages of retained MnO2 after HM degradation over time in the presence of PBS at pH levels of 5.5, 6.5 and 7.4. (d) Percentages of HMME and DOX released from DH-HM@BSA over time in the presence of PBS at varying pH levels (7.4 and 6.5) with or without us irradiation. (e) Fluorescence intensities of so in different groups detected by SOSG. The date are expressed as means ± SD (n = 3) (***p < 0.001, **p < 0.01, and *p < 0.05). | ||
Degradation of carriers and release of drugs in DH-HM@BSA
An ideal drug delivery system can release drugs in a controlled manner in vivo. Most solid tumors possess slightly acidic TMEs, with pH = 6.5–6.8.38 To assess the acid responsiveness of HM, it was added to PBS at different pH values. TEM was employed to obtain images of HM incubated in PBS at pH levels of 6.5 and 7.4 for varying durations (Fig. 3b). Physical appearance of HM mainly remained unchanged after the incubation of HM in PBS at pH = 7.4 for 4 h, indicating stability of HM in neutral environments. However, degradation of HM was observed in PBS at pH = 6.5, as evidenced by the collapse of the hollow mesoporous structure and transformation of the spherical morphology to irregular morphology; HM disintegrated into very small particles of several nanometers in 4 h. This behavior demonstrates the time-dependent degradation of HM in acidic solutions.Additionally, UV-vis spectra revealed that the absorbance of HM remained stable in PBS with pH = 7.4, whereas it rapidly decreased in PBS solutions with pH = 6.5 and 5.5, implying acidity- and time-dependent degradation of HM (Fig. 3c). This acid responsiveness of HM can protect the potential activities of the loaded drugs before release, ensuring their subsequent release in tumors.
Degradation of the nanocarrier could also coincide with the release of the loaded drugs DOX and HMME. Therefore, we investigated the release behaviors of HMME and DOX from DH-HM@BSA in solutions at pH levels of 6.5 and 7.4 (Fig. 3d). Unlike the gradual release pattern observed for DH-HM@BSA in neutral environments (pH = 7.4), HMME and DOX exhibited notably increased release rates in the mildly acidic environment (pH = 6.5) and the rates were further accelerated under US irradiation. The application of US led to increased mechanical and thermal energy, contributing to the breakdown of the HM nano-shells, and the release and diffusion of loaded DOX and HMME.39 Therefore, DH-HM@BSA can be considered an acidic pH- and US-responsive drug delivery system suitable for the targeted deliveries of DOX and HMME to tumors. Thus, our designed nano-delivery system, DH-HM@BSA, can remain stable during systemic circulation, minimizing premature drug leakage, and lead to rapid vector degradation and site-specific drug release in tumors when combined with US irradiation. Therefore, it offers an exceptional foundation for in vivo antitumor activity.
Detection of SO
To detect the production of 1O2 by H-HM@BSA after US irradiation, we employed highly selective. The probe emits green fluorescence, similar to that of fluorescein, in the presence of 1O2. No significant green fluorescence was noticed for free HMME and H-HM@BSA without US irradiation. In contrast, both H-HM@BSA+US and HMME+US groups exhibited green fluorescence, and no substantial difference was observed between the fluorescence intensities of the two groups. Nevertheless, the addition of acidic H2O2 (100 μM, pH = 6.5) significantly increased the green fluorescence intensity in the case of the HM@BSA(+H2O2)+US group as compared to that in the case of the HMME(+H2O2)+US group (Fig. 3e). Therefore, under ultrasound irradiation, H-HM@BSA can generate 1O2, and the production of 1O2 can be further increased after adding H2O2 because of increased oxygen through the decomposition of H2O2 catalysed by the carrier HM.In vitro cytotoxicity of HM
For further in vivo applications of HM, we assessed its biosafety and biocompatibility. HM at different concentrations was co-incubated with MCF-7 human breast cancer cells and its cytotoxicity was indirectly reflected by the cell viability (Fig. 4a). Results showed no considerable cytotoxicity of HM even when the concentrations were increased to 100 μg mL−1, showing outstanding biosafety and biocompatibility of HM.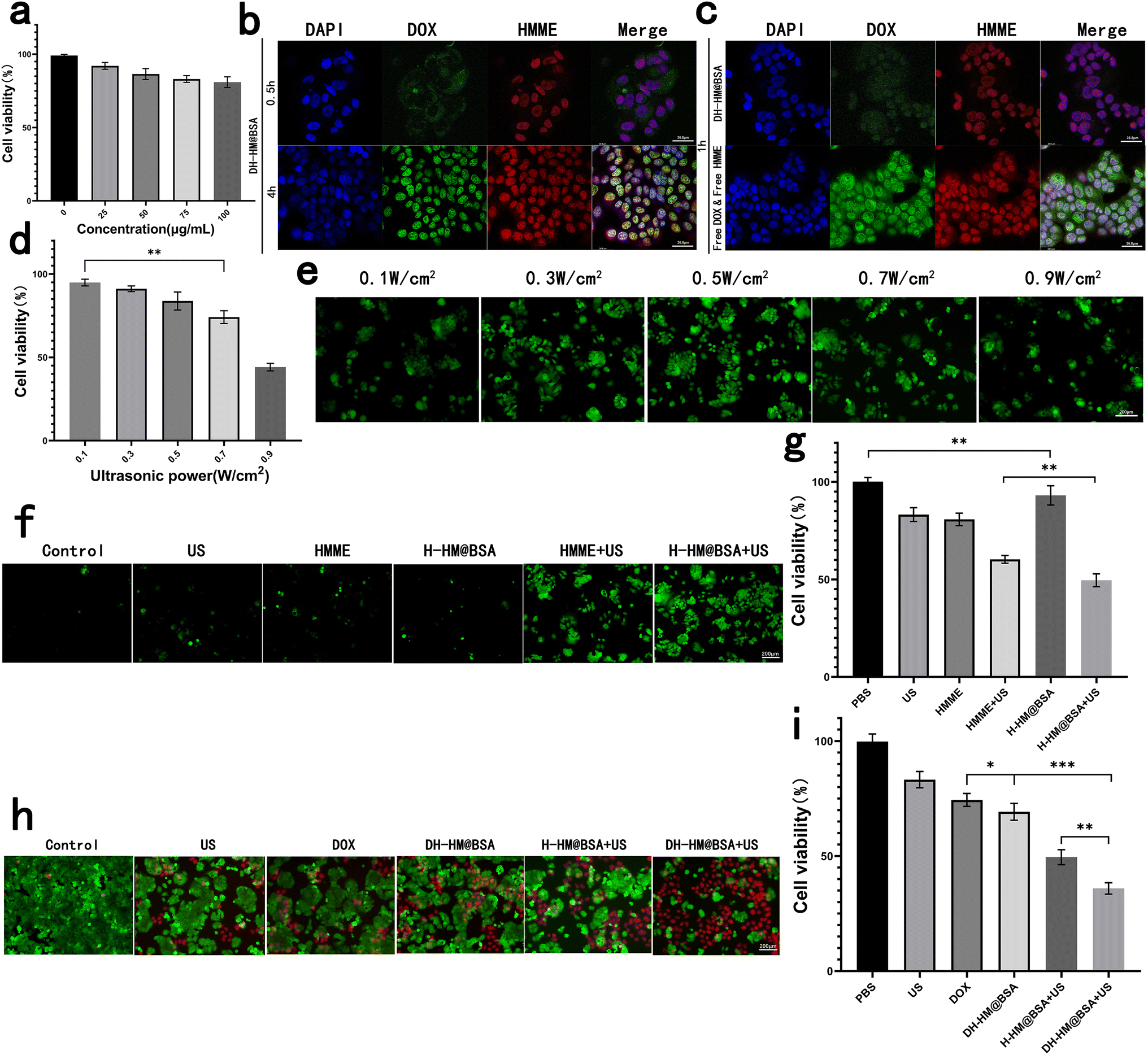 | ||
| Fig. 4 In vitro experiments with HM, H-HM@BSA and DH-HM@BSA. (a) In vitro cytotoxicity of HM: CCK-8 results for the MCF-7 human breast cancer cells co-incubated with HM at different concentrations. (b) Confocal images of the uptake of DH-HM@BSA by the MCF-7 cells at various time points (red, green, and blue represent emissions from HMME, DOX, and DAPI, respectively). (c) Confocal images of the uptake of free DOX & HMME and DH-HM@BSA by the MCF-7 cells at 1 h (white scale bars denote 36.8 μm). (d) CCK-8 results for MCF-7 cells irradiated with simple us at different powers. (e) ROS green fluorescence for H-HM@BSA irradiated with us at different powers. (f) ROS green fluorescence and (g) CCK-8 results of different groups. (h) Fluorescent images of live/dead cell and (i) CCK-8 results of the MCF-7 cells acquired after various treatments (white scale bar denotes 200 μm). The date are expressed as means ± SD (n = 3). (***p < 0.001, **p < 0.01, and *p < 0.05). | ||
Cellular uptake of DH-HM@BSA
Before in vitro combination therapy with DH-HM@BSA, we imaged the MCF-7 human breast cancer cells incubated with DH-HM@BSA for several durations via confocal fluorescence microscopy (Fig. 4b). As the incubation period extends, the fluorescence intensities of both HMME and DOX inside the cells significantly enhanced. Fluorescence of lipophilic HMME appeared in the nucleus at 0.5 h, whereas most of the fluorescence of DOX was noticed around the nucleus at this time. However, at approximately 4 h, considerable accumulation of DOX also appeared in the nucleus.In this experiment, free DOX and free HMME were also taken up by MCF-7 cells (Fig. 4c). Compared with the brighter fluorescence of free DOX and free HMME in MCF-7 cells at 1 h, fluorescence of DH-HM@BSA in MCF-7 cells was weaker at the same time point. As after cells uptake equal amounts of DOX and HMME, the loaded DOX and HMME had to undergo vector degradation in the acidic environment and loaded-drugs release, and then displayed fluorescence, while the free DOX and HMME could display fluorescence but didn't need to go through such processes.22 This implies that DH-HM@BSA can achieve acid-responsive drug release within tumors.
Moreover, from 0.5 to 4 h, the red fluorescence intensity of HMME in the cells gradually increased, ultimately overlapping with the blue fluorescence of DAPI in the cell nucleus, indicating successful cellular uptake of HMME. Therefore, 4 h was chosen as the incubation period of cells with DH-HM@BSA in further in vitro experiments and then US irradiation was applied.
In vitro ROS generation and SDT
At the extracellular level, with the intensity of US applied to HMME increasing, the presence of 1O2 became more apparent. Nevertheless, at the cellular level, with the intensity of US increasing, cell viability and ROS production decreased (Fig. 4d and e). The cell viability of MCF-7 cells was evaluated after exposure to US at various intensities. With a gradual increase in the US power to 0.5 W cm−2, the cell viability remained as high as 80%. However, with a continuous increase in the US power, cell viability decreased to below 80% (Fig. 4d); hence, a US power of 0.5 W cm−2 was chosen for subsequent SDT of cells.After addition of DCFH-DA solution, the green fluorescence of ROS generated inside the cells upon US irradiation was observed using the fluorescence microscope. Images demonstrated that in the absence of HMME, extremely weak green fluorescence appeared in the cells after US irradiation. After MCF-7 cells separately incubated with free HMME and H-HM@BSA were irradiated with US, the fluorescence intensity inside the cells considerably increased over time, indicating an increase in ROS production (Fig. 4f). The amount of ROS generated by the H-HM@BSA+US group was the highest among the six groups, revealing the most significant US-responsive ROS generation ability of H-HM@BSA. This might be ascribed to the catalysis of endogenous H2O2 inside the tumor cells by HM to decompose, providing additional oxygen to promote ROS production.
Further quantitative assessment using the CCK-8 assay revealed that compared to the free HMME+US group, the H-HM@BSA+US group was more lethal to the MCF-7 cells (Fig. 4g). Moreover, the higher the ROS level, the lower the cell viability (Fig. 4f and g). Therefore, under ultrasound irradiation, H-HM@BSA can generate more ROS than that produced by free HMME combined with US, thereby improving the SDT efficacy.
In vitro combined therapy
To further analyze the antitumor properties of this treatment in vitro, the cells were stained using Calcein-AM/PI live/dead cell staining Kit after different treatments and then observed under a fluorescence microscope. Live cells were characterized by green fluorescence, whereas dead cells displayed red fluorescence (Fig. 4h). More than half of the cells in the DH-HM@BSA+US group exhibited strong red fluorescence, indicating widespread apoptosis and necrosis after chemo-SDT. Further quantitative assessment using the CCK-8 assay (Fig. 4i) revealed that the cell survival rate in the case of combination therapy (DH-HM@BSA+US group) was the lowest (approximately 35.9%) as compared to those in the cases of SDT (H-HM@BSA+US group) or chemotherapy (DH-HM@BSA group) alone. This result demonstrates that DH-HM@BSA+US exhibits a significant chemo-SDT effect.In vivo biodistribution
To explore the tumor-targeted delivery of DH-HM@BSA in vivo and observe drug accumulation in tumors and potential toxicity of DH-HM@BSA to normal organs and tissues, biological distribution of DH-HM@BSA in mice harboring 4T1 tumors was investigated using a small-animal IVIS (Fig. 5a), and the mean fluorescence intensity of the images was quantified using IVIS Spectrum (Fig. 5b). Only weak fluorescence signals were obtained from the tumors of the free DOX group, which quickly disappeared, indicating that free DOX exhibited a short retention time in tumors after injection and was quickly cleared from the blood. In contrast, after injecting DH-HM@BSA, time-dependent gradually growing fluorescence signals were found in the tumors of the mice, and significant fluorescence signals were noticed at the tumor site after 8 h of injection, which persisted for up to 24 h. These findings reveal that DH-HM@BSA facilitates passive accumulation of DOX at the tumor site, demonstrating better tumor-targeting ability. These findings also indicate that the optimal US irradiation time of SDT in vivo is 8 h post-administration of DH-HM@BSA (Fig. 5b). | ||
| Fig. 5 In vivo and ex vivo imaging after DH-HM@BSA and free DOX injections. (a) In vivo fluorescence images of mice harboring 4T1 tumors acquired at various intervals of time after separately intravenous injections of DH-HM@BSA and free DOX (three mice in each group). (b) Semi-quantitative analysis results of in vivo fluorescence images for the various groups in (a). (c) Ex vivo fluorescence images of the vital organs and tumors 24 h after injection. (d) Semi-quantitative analysis results of ex vivo fluorescence images for the various groups in (c). The date are expressed as means ± SD (n = 3). (***p < 0.001, **p < 0.01, and *p < 0.05). | ||
In addition, ex vivo fluorescence imaging was carried out on tumor tissues and major organs (kidneys, lungs, spleen, liver and heart) 24 h after injection (Fig. 5c), and the mean fluorescence intensity of the images was quantified (Fig. 5d). The DH-HM@BSA group exhibited a greater intensity of fluorescence at the tumor site in comparison to the free DOX group. Therefore, relative to free DOX, DH-HM@BSA exhibited a higher capability of enrichment in tumors and substantially higher drug accumulation in tumors as compared to those in other organs, revealing excellent tumor-targeting ability of DH-HM@BSA.40 Overall, DH-HM@BSA can be retained in tumors for at least 24 h, demonstrating EPR effects.
In vivo antitumor efficacy
Based on the excellent therapeutic effects of DH-HM@BSA under US irradiation in vitro and its ability to penetrate and accumulate in tumors in vivo, we further investigated its antitumor effects in 4T1 tumor-bearing mice. Fig. 6a depicts a schematic for this treatment. Antitumor efficacies of different treatments were evaluated by observing and measuring the tumor volume (Fig. 6b and d). Tumor growth curves revealed that compared with the PBS group, the US, the free DOX, the DH-HM@BSA, the H-HM@BSA+US, and the DH-HM@BSA+US groups slowed tumor growth, and the tumor inhibition rates were 16.79%, 35.69%, 56.51%, 69.62% and 78.62%, respectively. Compared to the limited tumor suppression of the free DOX group, the DH-HM@BSA group exhibited more effective tumor suppression. Among all groups, the DH-HM@BSA+US group demonstrated the most significant inhibition of tumor growth (Fig. 6d). | ||
| Fig. 6 In vivo antitumor efficacy. (a) Schematic of the treatments in vivo. (b) Images of the mice harboring tumors throughout 15 days of treatments. (c) Weight change and (d) tumor growth curves of the mice in various groups after several treatments. (e) Typical images and (f) average weights of the tumors dissected from mice of various groups on the 16th day. The date are expressed as means ± SD (n = 5) (***p < 0.001, **p < 0.01, and *p < 0.05). | ||
Average weights of the tumor tissues acquired after 15 days of various treatments were 1225, 1017, 790, 480, 365, and 267 mg. Compared to the other groups, the DH-HM@BSA+US group only exhibited a three-fold rise in the final tumor volume, which was comparable with the results of the in vitro cell experiment, suggesting superior synergistic antitumor effects of DH-HM@BSA+US (Fig. 6e and f). Therefore, the combination treatment DH-HMD@BSA+US can effectively hinder the progression of tumors. This can be related to the improvement of hypoxia in TME and delivery of chemotherapeutic drugs and sonosensitizers by HM effectively coupled with US irradiation, enhancing drug penetration and accumulation in tumors, thereby realizing a perfect combination of SDT and chemotherapy.
Control and US-only groups demonstrated regular tumor tissue. In contrast, various degrees of tissue damage and cell necrosis were detected in the other treatment groups, among which the DH-HM@BSA+US group exhibited exceptional antitumor efficacy (Fig. 7a). Nuclear lysis, fragmentation, and condensation were observed in the majority of the tumor cells, according to histological analysis of the DH-HM@BSA+US group stained with H&E. Capase-3 apoptosis staining and TUNEL assay revealed widespread brown apoptotic cells. PCNA staining implied a considerable reduction in the number of proliferation-positive tumor cells. Combination treatment (DH-HM@BSA+US) led to extensive tumor cell necrosis, apoptosis, and deoxyribonucleic acid damage, significantly reducing proliferative activities of tumor cells. This suggests that chemo-SDT can reduce tumor cell proliferation and induce apoptosis, hence inhibiting tumor growth.
 | ||
| Fig. 7 Typical histological images of the tumor tissue sections from the mice harboring 4T1 tumors after different treatments. (a) Images of H&E, TUNEL, capase-3 and PCNA immunohistochemical staining. (b) and (c) Immunofluorescence staining images of DHE and HIF-1α (scale bars are 100 μm). | ||
DHE immunofluorescence staining revealed strong red fluorescence of ROS in both the H-HM@BSA+US and DH-HM@BSA+US groups (Fig. 7b). This indicates successful delivery of the sonosensitizer to the tumors and production of ROS by US irradiation. In combination with the aforementioned staining results, this finding suggests that an increase in ROS levels in tumors can promote necrosis and apoptosis of tumor cells.
HIF-1α is an essential factor in tumor growth, metastasis, and chemotherapy resistance. The HIF-1α signaling pathway is hypothesized to provide an indirect indication of potential alterations in oxygen levels in tumors.41,42 In this investigation, immunofluorescence staining was utilized to examine HIF-1α expression in tumors, which was displayed with red fluorescence (Fig. 7c). The findings demonstrated that, due to chemotherapy aggravating tumor hypoxia, the red fluorescence intensity was highest in all group. Compared to the cases of the control, US-only, and free DOX groups, lower HIF-1α expressions were detected in the DH-HM@BSA, H-HM@BSA+US, and DH-HM@BSA+US groups. These results are ascribed to the improvement of intratumoral hypoxia caused by HM. These findings, when combined with the aforementioned effects on tumor suppression, indicate that approaches to improve hypoxia in tumors and downregulate HIF-1α expression can be advantageous for improving the synergistic SDT-chemotherapy.
In vivo biosafety
To assess the biosafety of DH-HM@BSA+US in vivo, weight changes of the mice were recorded throughout treatment, and H&E staining and laboratory tests of main organs were conducted after the treatment. Because of the severe adverse effects of free DOX, the mice in the free DOX group had a considerable loss in body weight, according to the results (Fig. 6c). No substantial weight changes were observed in the cases of other groups, indicating the safety of DH-HM@BSA+US in vivo.This result was in agreement with the major organ H&E staining results (Fig. 8a). H&E staining of the myocardial cells in the free DOX group demonstrated degeneration, aggregation and uneven distribution, whereas no evident histopathological abnormalities were noticed in the myocardium of the DH-HM@BSA+US group. Renal cortical damage, such as significant glomerular consolidation, degeneration, and expansion of Bowman's space, was observed in the free DOX group; nevertheless, no considerable renal cortical injury was noticed in the DH-HM@BSA+US group. This suggests that toxicity to the heart and kidneys may result from free DOX. The H&E staining images of other main tissues in the DH-HM@BSA+US group did not reveal any significant damage or pathological alterations, implying that DH-HM@BSA avoided the severe systemic side effects of free DOX.
 | ||
| Fig. 8 In vivo biosafety evaluation of chemo-SDT based on DH-HM@BSA. (a) H&E staining images of the main organs dissected from the mice of various groups on day 16 after various treatments (scale bars are 50 μm). (b) Hematological tests, such as regular blood indicators (WBC, PLT, PMN, LYM) and functions of liver and kidney (ALT, AST, CREA, BUN), of different groups on day 16 after various treatments. The date are expressed as means ± SD (n = 3) (***p < 0.001, **p < 0.01, and *p < 0.05). | ||
Furthermore, routine blood and functional tests of the liver and kidney (Fig. 8b) revealed excluding RBC, abnormalities in WBC, PLT, polymorphonuclear neutrophils, lymphocytes, ALT, AST, CREA, and BUN in the free DOX group as compared to the case of the PBS group, whereas no significant differences were noticed between the laboratory test results of the DH-HM@BSA+US and PBS groups, indicating that DH-HM@BSA+US exerted no considerable adverse effects on the blood, liver, and kidneys. These results further demonstrate that DH-HM@BSA exhibits no significant toxicity in vivo and demonstrates high biosafety when combined with low-power US for antitumor treatment in vivo.
Conclusions
pH-responsive DH-HM prepared herein exhibited excellent hydrophilicity and dispersion after simple modification with BSA, where HM improved the cyclic stabilities of HMME and DOX, facilitated drug accumulation at tumor sites, improved the EPR effect, and minimized premature drug leakage. After low-frequency US irradiation, the micro-jet and micro-impulse generated via the “cavitation effect” enhanced the permeabilities of blood vessels and cell membranes of tumor, and the concentration of drug that infiltrated from blood vessels was substantially increased in tumor cells and extracellular matrix. Notably, owing to the highly matched chemical activity of MnO2 with that of TME, hypoxia in the tumor was effectively improved, which enhanced the SDT efficacy. Moreover, activated HMME transferred energy from low-frequency US to oxygen molecules, resulting in highly oxidative SO, which increased intracellular oxidative stress and promoted tumor cells apoptosis. The combination treatment of DH-HMD@BSA+US reduced the systemic toxicities and side effects of chemotherapeutic drugs, reversed chemotherapy resistance, and achieved precise tumor targeting, eventually leading to enhanced tumor growth inhibition in tumor-bearing mice without significant adverse reactions. This HM-based nano-delivery system is a simple, efficient, and safe multifunctional integrated nanotherapeutic platform. This study provides a promising strategy for improving immune microenvironment in tumor and inhibiting drug resistance in tumor therapy in the future and proposes HM with considerable potential as a nanocarrier for clinical application.Ethical statement
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Shandong Provincial Hospital Affiliated to Shandong First Medical University and approved by the Animal Ethics Committee of Shandong Provincial Hospital Affiliated to Shandong First Medical University.Author contributions
Chao Qian: methodology, investigation, data curation, formal analysis, writing – original draft. Guoliang Zhao: methodology, investigation, data curation, formal analysis. Mengping Huo: validation, writing – original draft preparation. Meixia Su: data curation. Xuexue Hu: formal analysis. Qiang Liu: project administration, conceptualization, funding acquisition. Lei Wang: methodology, conceptualization, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of Shandong Province (ZR2019MH106), Youth Science Fund Cultivation Program of Shandong First Medical University of China (202201-064) and Transverse Research of Shandong First Medical University (342564).Notes and references
- E. F. Blackley and S. Loi, Breast, 2019, 48, S44–S48 CrossRef PubMed
.
- Q. Cui, J.-Q. Wang, Y. G. Assaraf, L. Ren, P. Gupta, L. Wei, C. R. Ashby, D.-H. Yang and Z.-S. Chen, Drug Resist. Updates, 2018, 41, 1–25 CrossRef PubMed
- H.-Q. Dong, X.-F. Fu, M.-Y. Wang and J. Zhu, World J. Clin. Cases, 2023, 11, 5193–5203 CrossRef PubMed
- L. Rengeng, Z. Qianyu, L. Yuehong, P. Zhongzhong and L. Libo, Photodiagnosis Photodyn. Ther., 2017, 19, 159–166 CrossRef
- P. Wang, J. Chen, R. Zhong, Y. Xia, Z. Wu, C. Zhang and H. Yao, Eur. J. Pharm. Biopharm., 2024, 198, 114246–114263 CrossRef CAS PubMed
- X. Su, P. Wang, X. Wang, B. Cao, L. Li and Q. Liu, Cancer Biother. Rad., 2013, 28, 207–217 CAS
- E. Hwang, M. Yun and H. S. Jung, Front. Chem., 2023, 11, 1212193–1212206 CrossRef CAS PubMed
- F. Yang, M. Xu, X. Chen and Y. Luo, Biomed. Pharmacother., 2023, 164, 114933–114948 CrossRef CAS PubMed
- S. Son, J. H. Kim, X. Wang, C. Zhang, S. A. Yoon, J. Shin, A. Sharma, M. H. Lee, L. Cheng, J. Wu and J. S. Kim, Chem. Soc. Rev., 2020, 49, 3244–3261 RSC
- Z. Jiang, W. Xiao and Q. Fu, J. Controlled Release, 2023, 361, 547–567 CrossRef CAS PubMed
- Y. Lu, E. Zhang, J. Yang and Z. Cao, Nano Res., 2018, 11, 4985–4998 CrossRef PubMed
- Q. Li, W. Hou, M. Li, H. Ye, H. Li and Z. Wang, Int. J. Nanomed., 2020, 15, 4825–4845 CrossRef CAS
- E. Thomas, J. U. Menon, J. Owen, I. Skaripa-Koukelli, S. Wallington, M. Gray, C. Mannaris, V. Kersemans, D. Allen, P. Kinchesh, S. Smart, R. Carlisle and K. A. Vallis, Theranostics, 2019, 9, 5595–5609 CrossRef CAS PubMed
- M. Li, Q. Li, W. Hou, J. Zhang, H. Ye, H. Li, D. Zeng and J. Bai, RSC Adv., 2020, 10, 15252–15263 RSC
- P. Vaupel, M. Höckel and A. Mayer, Antioxid. Redox Signaling, 2007, 9, 1221–1236 CrossRef CAS PubMed
- B. A. Webb, M. Chimenti, M. P. Jacobson and D. L. Barber, Nat. Rev. Cancer, 2011, 11, 671–677 CrossRef CAS PubMed
- S. Peng, F. Xiao, M. Chen and H. Gao, Adv. Sci., 2021, 9, 2103836 CrossRef PubMed
- J. Cao, M. Zheng, Z. Sun, Z. Li, X. Qi and S. Shen, Int. J. Nanomed., 2022, 17, 2577–2591 CrossRef PubMed
- J. Li, Z. Yue, M. Tang, W. Wang, Y. Sun, T. Sun and C. Chen, Adv. Healthcare Mater., 2023, 13, 2302028 CrossRef PubMed
- X. Jing, F. Yang, C. Shao, K. Wei, M. Xie, H. Shen and Y. Shu, Mol. Cancer, 2019, 18, 157 CrossRef
- Y. Chao and Z. Liu, Nat. Rev. Bioeng., 2023, 1, 125–138 CrossRef
- Z. Chen, Z. Liu, Q. Zhang, S. Huang, Z. Zhang, X. Feng, L. Zeng, D. Lin, L. Wang and H. Song, Front. Pharmacol, 2023, 14, 1133011 CrossRef CAS PubMed
- B. Du, Q. Du, Y. Bai, L. Yu, Y. Wang, J. Huang, M. Zheng, G. Shen, J. Zhou and H. Yao, J. Mater. Chem. B, 2020, 8, 9139–9150 RSC
- M. Xu, L. Zhou, L. Zheng, Q. Zhou, K. Liu, Y. Mao and S. Song, Cancer Lett., 2021, 497, 229–242 CrossRef CAS PubMed
- G. Yang, J. Ji and Z. Liu, Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol., 2021, 13, 1720 CrossRef PubMed
- W. Sun, H. Xiao, J. Zhu, Z. Hao, J. Sun, D. Wang, X. Wang, M. Ramalingam, S. Xie and R. Wang, ACS Appl. Bio Mater., 2023, 6, 4998–5008 CrossRef CAS PubMed
- N. Desai, Nanomed. Nanotechnol. Biol. Med., 2007, 3, 337–346 CrossRef
- F. Pi, X. Deng, Q. Xue, L. Zheng, H. Liu, F. Yang and T. Chen, J. Nanobiotechnol., 2023, 21, 90 CrossRef CAS
- H. Wang, W. Wang, L. Liu, M. Wang, G. Li, H. Li, B. Li, S. Yu, D. Ma and W. Xue, ACS Appl. Mater. Interfaces, 2021, 13, 57009–57022 CrossRef CAS PubMed
- X. Zhang, C. He, Y. Sun, X. Liu, Y. Chen, C. Chen, R. Yan, T. Fan, T. Yang, Y. Lu, J. Luo, X. Ma and G. Xiang, Acta Pharm. Sin. B, 2021, 11, 3608–3621 CrossRef CAS PubMed
- H.-L. Zhang, Y. Wang, Q. Tang, B. Ren, S.-P. Yang and J.-G. Liu, J. Inorg. Biochem., 2023, 241, 112133 CrossRef CAS PubMed
- J. Duan, T. Liao, X. Xu, Y. Liu, Y. Kuang and C. Li, J. Colloid Interface Sci., 2023, 634, 836–851 CrossRef CAS
- Q. Song, B. Chi, H. Gao, J. Wang, M. Wu, Y. Xu, Y. Wang, Z. Xu, L. Li, J. Wang and R. Zhang, J. Mater. Chem. B, 2023, 11, 6889–6895 RSC
- Q. Hu, W. Sun, Y. Lu, H. N. Bomba, Y. Ye, T. Jiang, A. J. Isaacson and Z. Gu, Nano Lett., 2016, 16, 1118–1126 CrossRef CAS
- G. Yang, L. Xu, Y. Chao, J. Xu, X. Sun, Y. Wu, R. Peng and Z. Liu, Nat. Commun., 2017, 8, 902 CrossRef
- X. Liu, K. Tian, J. Zhang, M. Zhao, S. Liu, Q. Zhao and W. Huang, ACS Applied Bio Materials, 2019, 2, 1225–1232 CrossRef CAS PubMed
- J. C. Murray, The American Society for Pharmacology and Experimental Therapeutics, Pharmacol. Rev., 2001, 53, 283–318 Search PubMed
- R. A. Cairns, I. S. Harris and T. W. Mak, Nat. Rev. Cancer, 2011, 11, 85–95 CrossRef CAS PubMed
- K. W. Jang, D. Seol, L. Ding, D. N. Heo, S. J. Lee, J. A. Martin and I. K. Kwon, Int. J. Biol. Macromol., 2018, 106, 1211–1217 CrossRef CAS PubMed
- P. Zhang, J. Wang, H. Chen, L. Zhao, B. Chen, C. Chu, H. Liu, Z. Qin, J. Liu, Y. Tan, X. Chen and G. Liu, J. Am. Chem. Soc., 2018, 140, 14980–14989 CrossRef PubMed
- T. Liu, Q. Gao, B. Yang, C. Yin, J. Chang, H. Qian, G. Xing, S. Wang, F. Li, Y. Zhang, D. Chen, J. Cai, H. Shi, M. Aschner, K. Appiah-Kubi, D. He and R. Lu, Toxicol. Lett., 2020, 331, 82–91 CrossRef CAS PubMed
- Z. Zhang, R. Wang, X. Huang, R. Luo, J. Xue, J. Gao, W. Liu, F. Liu, F. Feng and W. Qu, ACS Appl. Mater. Interfaces, 2020, 12, 5680–5694 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2024 |