
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Access to C(sp3) borylated and silylated cyclic molecules: hydrogenation of corresponding arenes and heteroarenes†
Arzoo Chhabra ,
Sabrina Reich,
Timothy M. Shannon,
Robert E. Maleczka Jr* and
Milton R. Smith III
,
Sabrina Reich,
Timothy M. Shannon,
Robert E. Maleczka Jr* and
Milton R. Smith III
Department of Chemistry, Michigan State University, 578 S Shaw Lane, East Lansing, Michigan 48824, USA. E-mail: maleczka@chemistry.msu.edu
First published on 2nd April 2024
Abstract
This paper presents a simple and cost-effective hydrogenation method for synthesizing a myriad of cycloalkanes and saturated heterocycles bearing boryl or silyl substituents. The catalyst used are heterogeneous, readily available, bench stable, and recyclable. Also demonstrated is the application of the method to compounds that possess both boryl and silyl groups. When combined with Ir-catalyzed sp2 C–H borylation, such hydrogenations offer a two-step complementary alternative to direct sp3 C–H borylations that can suffer selectivity and reactivity issues. Of practical value to the community, complete stereochemical analyses of reported borylated compounds that were never fully characterized are reported herein.
Introduction
Organoboron compounds are often pivotal intermediates in the synthesis of natural products, biologically relevant molecules and compounds used in material science.1 Such broad applications come from the ability of C–B bonds to be readily transformed to C–OH, C–NH2, C–C bonds, etc. Additionally, boryl groups present on sp3 carbons can undergo couplings with retention or inversion of stereochemistry, Matteson homologation reactions, etc.2 Owing to their synthetic utility, the pursuit of new methods and strategies for making borylated compounds is an active area of research.Among the various borylation methods, catalytic sp2 C–H borylations are widely used for the preparation of borylated arenes and heteroarenes. In contrast, catalytic sp3 C–H borylations to afford borylated cycloalkanes and saturated heterocycles are relatively underdeveloped. Many sp3 C–H borylation methods are highly substrate limited requiring the presence of a directing group (e.g. 2-alkyl pyridines, alkyl amines, benzylic compounds, cyclopropyl amides) and/or demand excess starting materials or reagents.3–6
In recent years, some of these restraints have been eased. The Hartwig group has established directing group free sp3 C–H borylation chemistry that encompasses a broader substrate scope, including alkanes, ethers, protected amines, alcohols, and carbocycles. Their method affords good selectivity and when executed with an excess of boron reagent good reactivity. In addition, all reported examples were run in cyclooctane. As the authors note, this restricted functionalization of polar molecules due to poor solubility.7 Schley and coworkers were able to overcome the limitation of excess of boron reagent through the thoughtful application of dipyridylarylmethane as ligand.8 In an alternative strategy, sp3 C–H borylated compounds can be accessed by hydrogenating corresponding borylated arenes, which themselves can be obtained via iridium catalyzed sp2 C–H borylations. Hydrogenation of arenes and heteroarenes have been carried out in the past via transition metal (Pd, Ru, Pt and Rh) catalysis.9–20 However, none of these reports demonstrated the hydrogenation on borylated or silylated arenes or heteroarenes. On this front, Glorius and coworkers pioneered the catalytic heterogeneous hydrogenation of boryl- and silyl-arenes/heteroarenes by a cyclic (alkyl)(amino)carbene rhodium complexes (Fig. 1).21–25 In similar work, Zeng showed hydrogenation of borylated arenes and heteroarenes using a related Rh-catalyst (Fig. 1).26 Glorius' catalyst was utilized by Bach's group to hydrogenate one borylated 2-oxindole among other benzofused N-heterocycles, 2,5-diketopiperazine, and 3,4-dihydroquinolones.27–29
 | ||
| Fig. 1 Strategy to access sp3 C–H borylated and silylated substrates via arene/heteroarene hydrogenation using Rh catalysts. | ||
Glorius also reported hydrogenations using Rh/C in their optimization studies on six borylated substrates (five benzenes and one pyridine).21 Catalysis with Rh/C showed full conversion with TBS protected 4-Bpin phenol, but as the authors noted the other arenes tested and the pyridine gave diminished yields (17–33%). Separately,10 Rh/C catalyzed hydrogenation of six silylated benzenes were examined. It was reported that two of the six gave 0% of the corresponding cyclohexanes, but three substrates were saturated in 25, 35, and 40% yields respectively. In contrast, the hydrogenation of n-hexyl ether of 4-TMS-phenol was achieved in 90% yield with Rh/C in hexane (vs. 16% in EtOH) and could be further optimized up to 97% using Rh/Al2O3.
Other recent reports on saturating arenes and heteroarenes that do not bear boryl or silyl substituents include the work of Handa, who used [Ir(cod)Cl]2 to hydrogenate phosphine oxide scaffolds with Ir nanoparticles being the active catalyst.30 A cooperative heterogeneous and homogeneous strategy for asymmetric hydrogenations of arenes and heteroarenes has also been developed, which has been applied to a single borylated substrate (benzofuran).31–33 The hydrogenation of fluoropyridines using Pd(OH)2, in acidic media has also been reported.34
While we considered all prior art cited above, Glorius' results motivated us to fully evaluate bench stable and commercially available Rh-catalysts, e.g. Rh/C or Rh/Al2O3, or other standard hydrogenation catalysts against a larger substrate set of arenes and heteroarenes with boryl or silyl substituents. We were further inspired by Glorius' recent report on hydrogenation of arenes that were bisfunctionalized with germyl and boryl, and germyl and silyl,35 and sought to hydrogenate previously unexplored heteroarenes bearing both boryl and silyl substituents.
Results and discussion
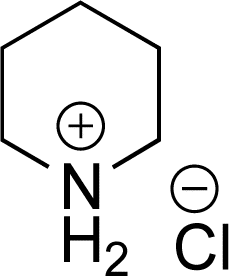
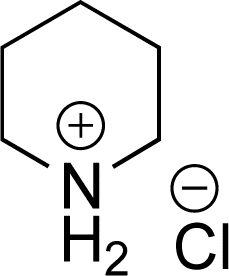
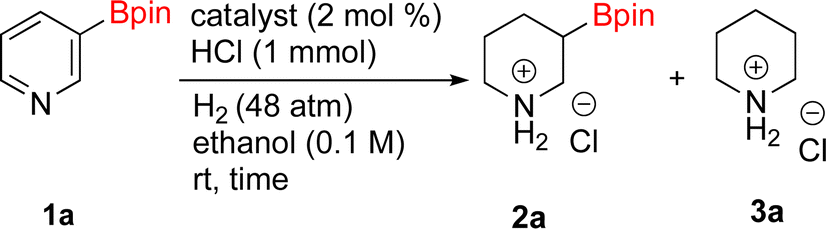
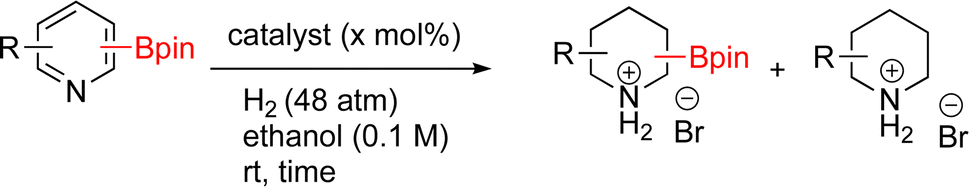
As the catalytic hydrogenations of pyridines using Rh/C is well established,36 we began our study by subjecting an ethanolic mixture of 3-borylated pyridine 1a to 5% Rh/C under a hydrogen atmosphere (Table 1; entry 1). As piperdines are known to poison Rh-catalysis36,37 and since pyridinium salts hydrogenate more readily than the free base,38 HCl was added to the reaction mixture. After 1 h, the pyridine ring was fully saturated to afford the desired borylated piperidine (2a) along with the deboronated product (3a). Rhodium on alumina (entry 2) afforded a similar mix of 2a and 3a, but the reaction was incomplete after 2 hours. Though platinum oxide has long been used to hydrogenate pyridines, reactions catalyzed by Pt2O and Pt/C met with an increased amount of deboronation (entries 3 and 4).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Starting material | Product 2a | Product 3a | Catalyst | Ratiob of 2a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3a:1a :3a:1a |
Time |
| a (4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine (0.25 mmol), hydrochloric acid (1 mmol), hydrogen gas (48 atm), ethanol (2 mL), 5% Rh/C (10 mg).b Relative composition based on 1H-NMR. | ||||||
| 1 | 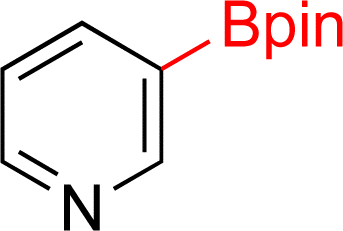 |
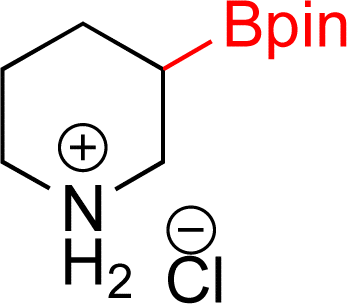 |
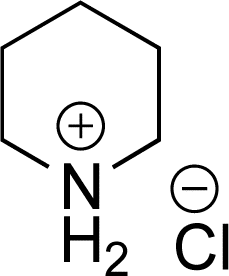 |
5% Rh/C (2 mol%) | 72:28:0 |
1 h |
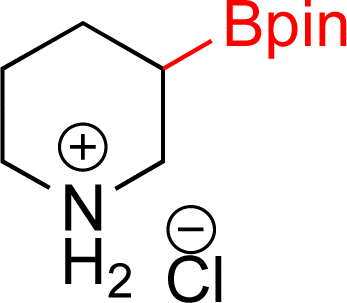
| 2 |  |
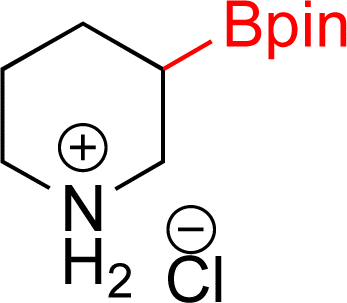 |
 |
5% Rh/Al2O3 (2 mol%) | 50:15:35 |
2 h |
| 3 |  |
 |
 |
PtO2 (2 mol%) | 32:32:36 |
2 h |
| 4 | 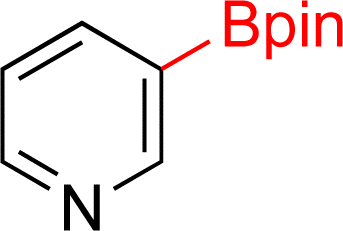 |
 |
 |
10% Pt/C (2 mol%) | 24:38:38 |
2 h |
| 5 |  |
 |
 |
10% Pd/C (2 mol%) | 0:0:100 |
2 h |
Catalytic hydrogenations of pyridines with palladium typically demand higher temperatures (70–80 °C) and higher catalyst loads,39 so it was not entirely surprising that 10% Pd/C failed to effect hydrogenation (entry 5). W2 RANEY® nickel40 and Ru-catalysts41 were not tested given the precedent for hydrogenations of pyridines with those catalysts requiring pressures > 1000 psi.
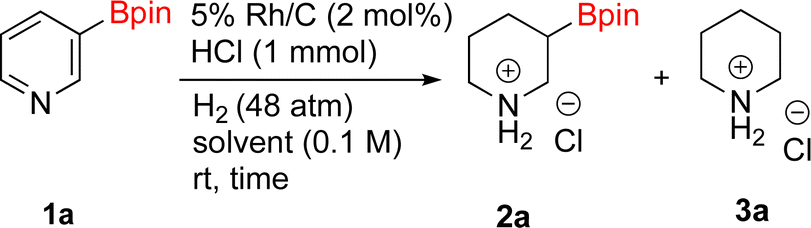
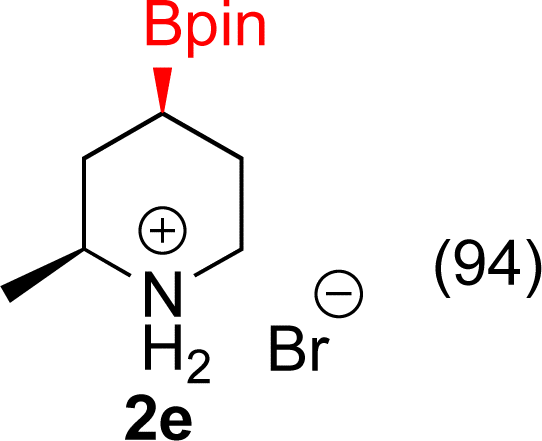
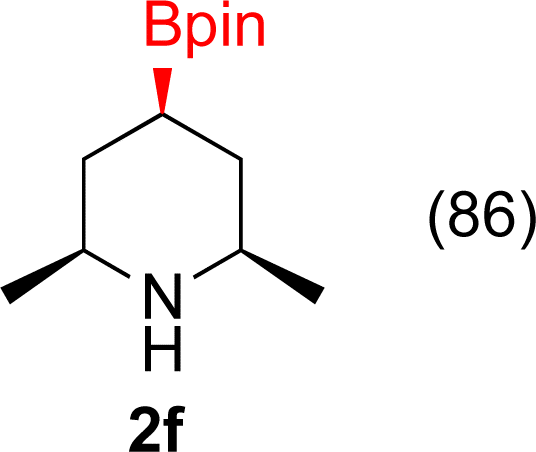
With Rh/C proving fastest at hydrogenating 1a, reactions with this catalyst were screened against different solvents (Table 2). In addition to ethanol, 1a could be hydrogenated in methanol, dichloromethane, dioxane, and THF. Yields of 2a were observed to be in order of ethanol/methanol > THF > dioxane > dichloromethane. This trend is consistent with previous reports on hydrogenations of non-polar substrates in polar solvents, where it has been shown that higher yields correlate to higher activity coefficients.42 Methanol was chosen as the reaction solvent because of the greenness of the solvent.43 Unfortunately, with all solvents deboronation remained a problem, with 3a being the only observed product when the reaction was run in dichloromethane for 36 hours. As metal mediated protiodeboronation by Brønsted acids and Lewis acids is well known,44 the reaction was run without HCl (Table 2; entry 2). This led to no reaction. Reducing the amount of conc. HCl in the reaction or using dry HCl (Table S1†) resulted in similar 2a:3a ratios as those observed in entry 1, Table 1. Other Brønsted and Lewis acids were also tested (Tables S1 and S2†), but none solved the deboronation problem. Lastly, we explored the idea that by starting with a halogenated 3-borylpyridine the reaction conditions would in situ generate 1 equiv. of HX, which would immediately form the pyridinium and not promote deboronation. Thus 3-bromo-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine (2b) was hydrogenated under the standard, albeit HCl free, conditions. Though debromination and hydrogenation occurred as predicted, deboronation was not eliminated. Unable to eliminate deboronation, we reevaluated the early catalyst screening data. Catalysis using Rh/Al2O3 (entry 2, Table 1) was slower than Rh/C, but afforded a better ratio of 2a to 3a. Therefore, as we looked at the hydrogenation of additional borylated pyridines, Rh/Al2O3 was employed as catalyst and reactions were run for 16 h. As shown in Table 3 (entries 2–5) 2c, 2d, 2e, and 2f were all formed >80% yield with no deboronation. Of note was the hydrogenation of 1f, which was carried out without the addition or in situ generation of a Brønsted acid. Presumably, the methyl groups at C2 and C6 inhibit poisoning of the catalyst. Furthermore, 2f was formed as single diastereomer, the stereochemistry of which as determined to be all cis by oxidizing the boronate ester to a hydroxy group and comparing that product to analogous literature compounds.45,46
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Time (h) | 2ab (%) | 3ab (%) | 1ab |
| a (4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine (0.25 mmol), HCl (1 mmol), H2 (48 atm), solvent (2 mL), 5% Rh/C (2 mol%), rt.b Relative composition determined by 1H-NMR.c Run without HCl. | |||||
| 1 | EtOH | 16 | 75 | 25 | 0 |
| 2 | EtOHc | 2 | 0 | 0 | 100 |
| 2 | DCM | 36 | 0 | 100 | 0 |
| 3 | Dioxane | 7 | 47 | 53 | 0 |
| 4 | THF | 7 | 65 | 35 | 0 |
| 5 | MeOH | 2 | 75 | 25 | 0 |
|
||||
|---|---|---|---|---|
| Entry | Starting material | Products (yield%) | Catalyst | Time |
| a Isolated yields and relative stereochemistry shown, starting material (0.5 mmol), ethanol (5 mL).b Isolated as a mixture.c Run at 0.3 mmol scale. | ||||
| 1 | 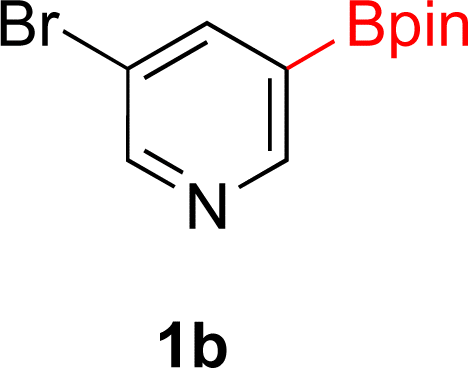 |
 |
5% Rh/C (2 mol%) | 16 h |
| 2 | 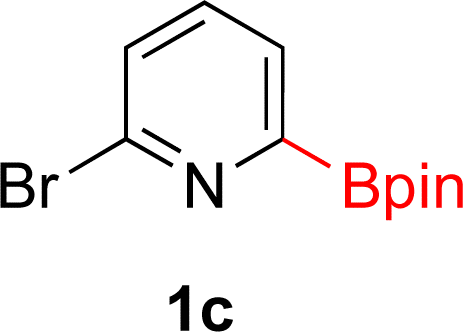 |
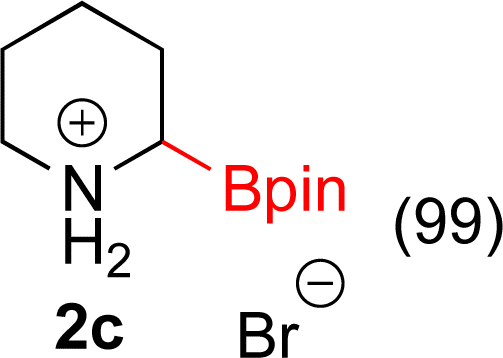 |
5% Rh/C (2 mol%) | 16 h |
| 3 | 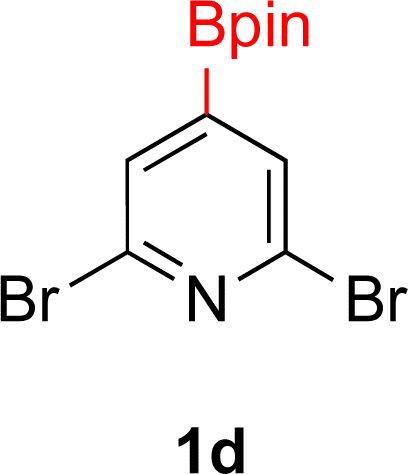 |
 |
5% Rh/Al2O3 (8 mol%) | 16 h |
| 4c | 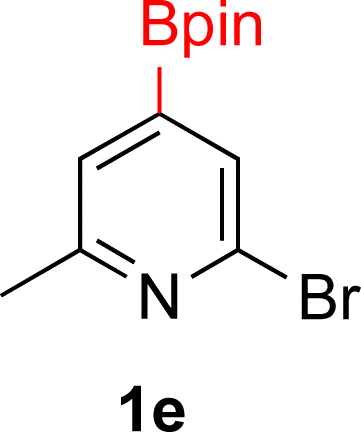 |
 |
5% Rh/Al2O3 (4 mol%) | 16 h |
| 5 | 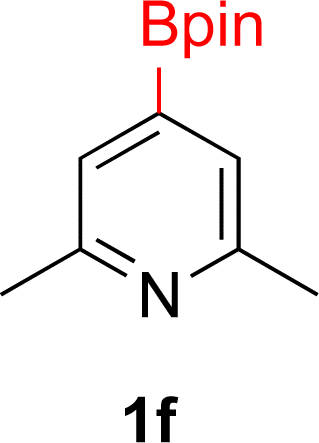 |
 |
5% Rh/Al2O3 (4 mol%) | 40 h |
Hydrogenation of different 5 membered borylated heterocycles were then investigated (Scheme 1). Under Rh/C catalysis, N-Boc-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-pyrrole (1g) was successfully hydrogenated (73% yield) on 1 gram scale without any observable loss of the Bpin. As Pd/C has been shown to selectively hydrogenate the pyrrole ring on nicotyrine,47 the catalytic hydrogenation of 1g was also run with 10% Pd/C and 2g was afforded in 82% yield. We also attempted a one pot Ir-catalyzed borylation/hydrogenation sequence using Boc-pyrrole as the starting substrate. In practice, running the hydrogenation step on the crude borylation mixture was not successful as only 1g was observed. Nonetheless, it should be noted that the CH borylation of Boc-pyrrole gives 1g in 90% yield. Thus, the 74% two-step combined yield of 2g from Boc-pyrrole compares favorably to 54% yield obtained in the Ir-catalyzed sp3 C–H borylation of Boc-pyrrolidine.7
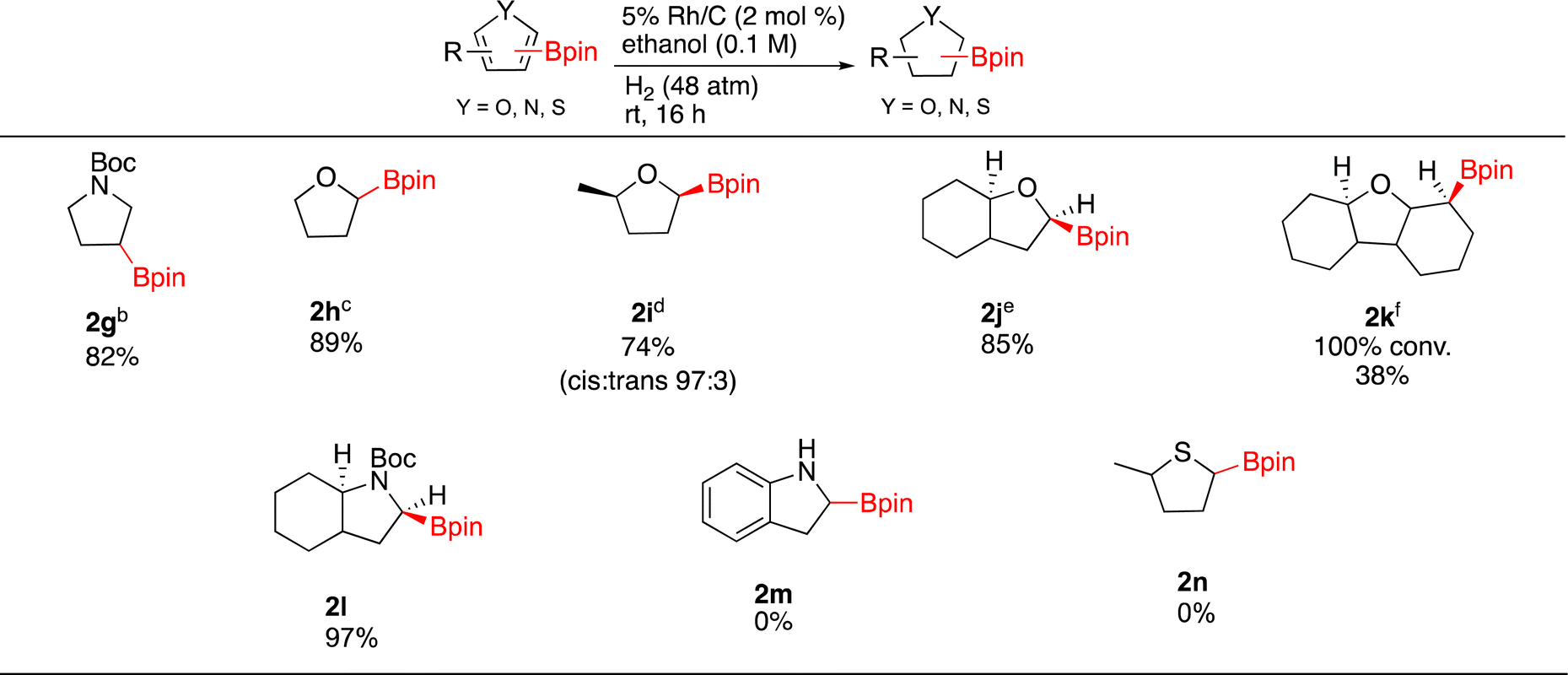 | ||
| Scheme 1 Hydrogenation of borylated heteroarenesa. aReaction conditions: 0.5 mmol substrate, relative stereochemistry shown, isolated yields shown. bN-Boc-3-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-pyrrole (1.0 g, 3.4 mmol), Rh/C (170 mg, 2 mol%) EtOH (10 mL, 0.3 M). c13 h, 8% OBpin monomer, d1 mmol substrate, Rh/C (1 mol%), 6.3% HOBpin byproduct esolvent = hexane, P = 54 atm, ∼20% OBpin monomer f8% deborylated product, solvent = hexane, P = 59 atm, T = 60 °C, t = 21 h. | ||
Moving on to other heterocycles, hydrogenation of 2-borylated furan (1h) occurred with 100% conversion to 2h. Isolation of 2h (89%) was complicated by the presence of HOBpin or oligomers thereof (∼8%). In this case, Rh/C is not as efficient as the Glorius catalyst, which affords 2h from 1h in 98% yield.21,26 It is worth nothing that when hydrogenating 1h at 2.5 mmol scale the catalyst could be recycled five times with reproducible results after each cycle. Direct sp3 Ir-catalyzed CH borylations of tetrahydrofuran gives the C-3 borylated product,7 in contrast, the sp2 Ir-catalyzed CH borylation of furan gives the C-2 borylated product. This highlights the potential for direct borylation and borylation/hydrogenation approaches to be complementary.
Hydrogenations of methyl substituted 1i, benzofuran 1j and dibenzofuran 1k were then carried out. Compound 2i was obtained in 74% yield as a 97:3 cis/trans mixture and as before a minor amount of boron byproduct (∼6%) was present in the isolated material. The cis stereochemistry of the major product was ascertained by COSY and 1D NOE NMR. Interestingly, hydrogenation of 1j generated the fully saturated borylated octahydrobenzofuran 2j again contaminated by the boron byproduct (78:22 by NMR). Compound 1k underwent full conversion affording 2k with 8% of the deboronated product. Though a minor product, the presence of the deboronated material made purification of 2k challenging and thus pristine 2k was isolated in only 38% yield. We were also able to generate 2l as an octahydroindole in 97% yield. The stereochemistry of hydrogen α to the nitrogen was found to be cis with the bridgehead hydrogen by 1D-NOE experiments. An unprotected borylated indole and a 2-borylated methylthiophene failed to produce saturated products 2m and 2n respectively, only starting material observed by 1H-NMR in both cases.
The hydrogenation of borylated arenes was also carried out (Scheme 2). All borylated arenes tested were easily hydrogenated with Rh/C and functional groups like esters, methoxy, alkyl, trifluoroalkane and alcohols were well tolerated. For compounds 2q, 2r, 2s, 2t, and 2u low ratios of cis and trans products were obtained with the cis diastereomers being major. The assignment for 2q was made by comparisons to a known silylated derivative.48 To do so for compounds 2r, 2t, and 2u, each of these products were oxidized to their alcohols,49 which were then compared to previously reported cis and trans alcohols. It is worth noting that when 2u was generated using caac-Rh-2 as the catalyst, a yield of 55% yield was observed vs. the 68% with Rh/C. Diastereoselectivity was similar in both cases (∼6:1).26 The major stereoisomer of 2s being cis was confirmed using 1D-NOE. The major stereochemistry of bisborylcyclohexane 2v was made by direct comparison to literature data.50
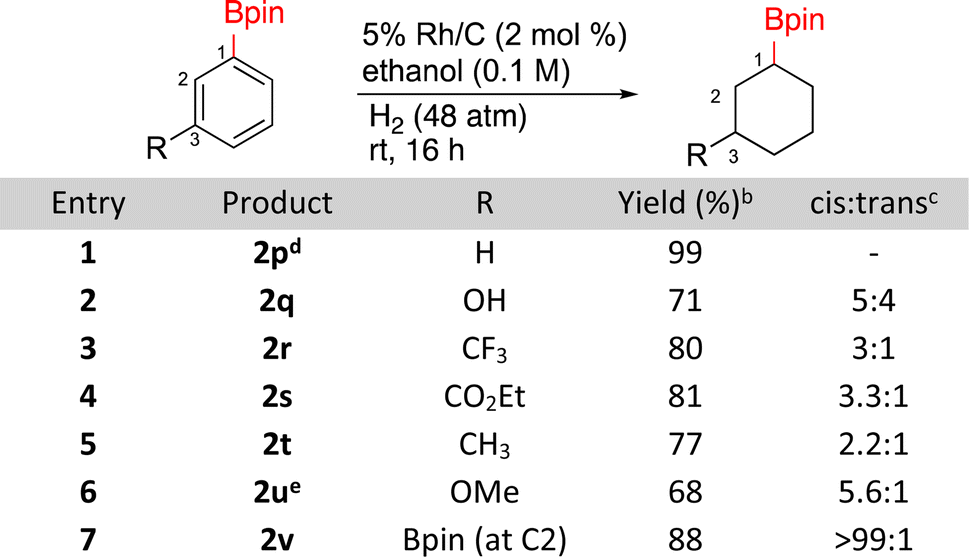 | ||
| Scheme 2 Hydrogenation of borylated benzenesa. aRun on 0.5 mmol substrate. bIsolated yields. cCis/trans ratios determined by 1H NMR on the crude reaction product. d1 mmol substrate, Rh/C (1 mol%). e22% demethoxylated byproduct. | ||
Carbon–silicon bonds, similar to carbon–boron bonds, are also versatile as they can be transformed to various functional groups such as hydroxy, amine, halogen and aryl.51 Thus, we sought to apply the same chemistry to organosilicon bearing heterocycles (Scheme 3). TMS-substituted pyridines 2-TMS 1w and 4-TMS 1x were easily hydrogenated with Rh/C and Rh/Al2O3 respectively, demonstrating the utility of both these catalysts for such substrates. Compound 1w was hydrogenated as a salt of camphor sulphonate. This was to see whether adding an optically active acid could induce chirality in the hydrogenated product. This proved not to be the case as a 1:1 mixture of enantiomers was obtained. Surprisingly, 1y, which only differs from 1x by the position of the TMS group gave 2y as a 1:5 mixture with the desilylated material being major as determined by LCMS and 19F-NMR. Two siloxanes, 1z and 1aa were also tested. Compound 1z presented the opportunity to probe complementary reactivity beyond just the ability to saturate siloxane containing hetero arenes.
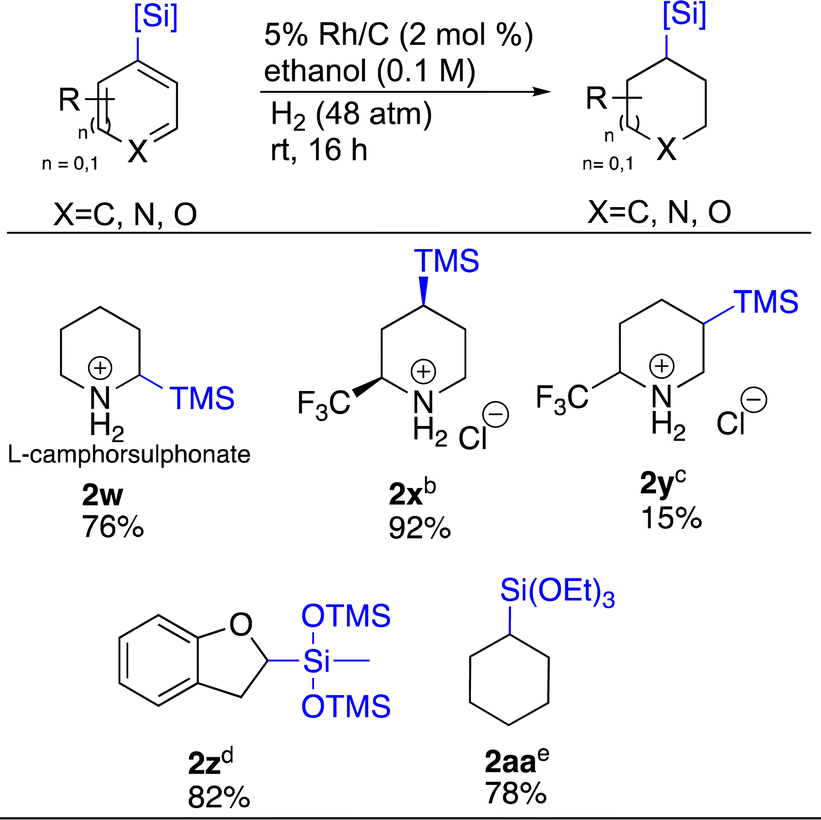 | ||
| Scheme 3 Hydrogenation of silylated arenesa. aReaction conditions: heterocycle (0.5 mmol), relative stereochemistry shown, all yields are isolated. bRh/Al2O3 (4.5 mol%), starting pyridine (0.2 mmol). cDesilylated product observed. Ratio of desilylated:silylated = 5:1. dPd/C (10 mol%). eRh/Al2O3 (2 mol%). | ||
As Rh/C led to the saturation of both rings of 1j, for 1z we employed Pd/C to see if 2,3-dihydrobenzofuran 2z could be formed selectively.25,52,53 We were delighted to see this hypothesis realized. Catalytic hydrogenation of arene 1aa showcased how catalyst choice matters. For this substrate, Pd/C only returned starting material. Rh/C saturated the ring, but the siloxane was lost. In contrast, Rh/Al2O3 successfully provided 2aa in 78% yield.54
Dual functionalized compounds that incorporate both silyl and boryl group have shown potential in facilitating diverse reaction pathways due to their orthogonal reactivity. An example of such reactivity was demonstrated by Hartwig where Bpin was converted to a Boc protected amine without compromising silyl groups. In another example, the silyl group was manipulated without compromising the Bpin group.55
Given such demonstrations of Si/B dual functionality, we prepared and then hydrogenated 1ab–1af under Rh/C catalysis (Scheme 4). To drive the hydrogenation of pyrrole 1ab to full conversion, increasing the reaction time to 48 h and doubling the catalyst loading to 4 mol% was required. This enabled isolation of 2ab in 93% yield with a high cis:trans ratio (96:4). The cis configuration was confirmed by NOE and 2D NMR experiments. Hydrogenation of 2-bromo-4-(Bpin)-6-(TMS)pyridine 1ac was also carried out. As seen earlier, ring saturation was accompanied by debromination affording 2ac as its HBr salt. Compound 2ac was isolated in 46% yield by precipitating out the product using ethyl acetate. The 100% cis stereochemistry was determined by 1D-NOE.
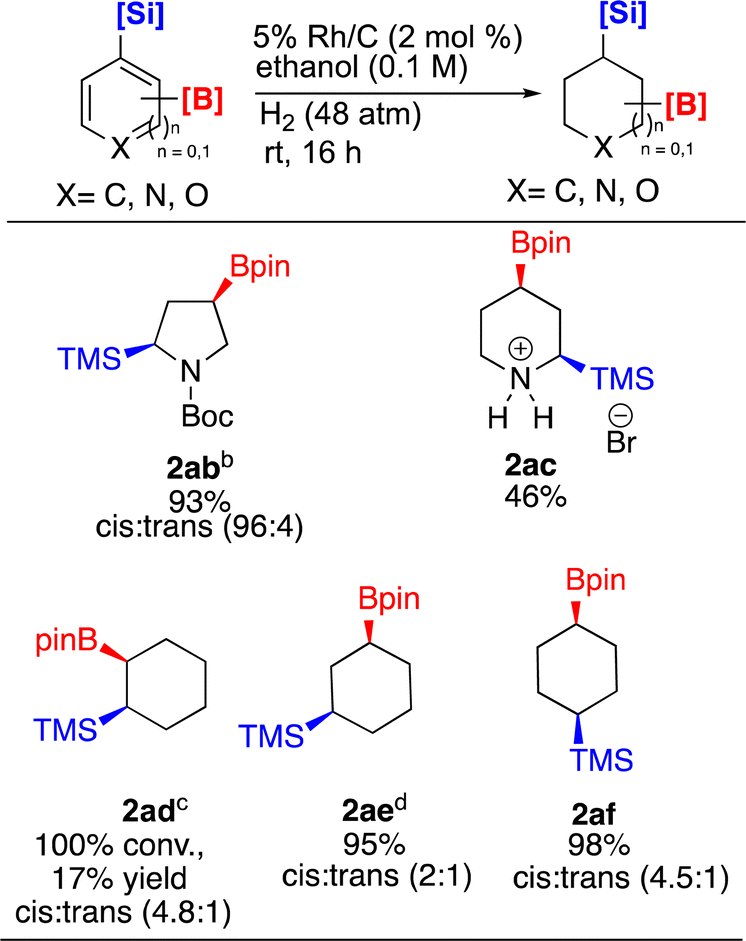 | ||
| Scheme 4 Hydrogenation of borylated and silylated arenes and heteroarenesa. aReaction conditions: substrate (0.5 mmol), relative stereochemistry shown, all yields are isolated. bt = 48 h, Rh/C (4 mol%). c12.5% desilylation observed. d93:7 (product:desilylated product ratio) observed. | ||
Lastly, we examined the three substituted benzenes bearing boryl and silyl group in ortho, meta, and para arrangements. All three converted to the corresponding cyclohexanes, albeit with some differences. The reaction to produce ortho 2ad showed 100% conversion to product (4.8:1 cis:trans) by 1H-NMR, however ∼12.5% of the material had desilylated. Difficulty in isolating 2ad by flash column chromatography was experienced. In contrast, 2ae (2:1 cis:trans) was isolated in higher yield, but some desilylation still occurred. As the amount of desilylation when forming 2ae was lower than that experienced with 2ad, we experimented to see if this side reaction could be eliminated. Unfortunately, we were unable to lessen desilylation by reducing the reaction time from 16 h to 8.5 h. Interestingly compound 2af (4.5:1 cis:trans) was isolated in 98% yield with no desilylation. We note the observed diastereoselectivities of 2ae and 2af were lower (2:1 vs. 4:1 for 2ae) and (4.5:1 vs. 7:1 for 2af) than those observed with Glorius' catalyst,22 whereas 2ad was formed with a comparable cis:trans ratio. We also synthesized tert-butyl 2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-5-(trimethylsilyl)-1H-pyrrole-1-carboxylate and trimethyl(6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)dibenzo[b,d]furan-4-yl)silane compounds of dual functionality however their hydrogenation reactions under several different reaction conditions did not yield the desired hydrogenated products. They gave largely a mixture of deboronation and desilylation side products.
Conclusion
In summary, Rh/C, Rh/Al2O3, and other readily available catalysts can affect the hydrogenation of borylated and silylated arenes and heteroarenes. Demonstrations of these catalysts to substrates bearing both boryl and silyl group were also shown to be viable. Catalyst selection is often key to successful hydrogenations, especially for 5- and 6-membered heterocycles. In some cases, minimizing unwanted loss of the boron or silicon substituent can also be achieved through catalyst choice. The scope, including limitations disclosed, and comparisons made to the pioneering work of Glorius and Zheng can help guide practitioners as to whether to employ more complex Rh-catalysts or those tested herein. Spectroscopic data reported herein for new molecules as well as previously described compounds where full characterization data were lacking may also prove valuable for those who find those compounds of interest.Experimentals
General remarks
Unless indicated otherwise all reactions were carried out in oven-dried glassware with magnetic stirring and monitored by GC-MS or 1H-NMR/19F-NMR. Tetrahydrofuran was freshly distilled from sodium/benzophenone under nitrogen. Diisopropylamine was freshly distilled from calcium hydride. n-BuLi was used as a 2.5 M solution in hexanes. Flash column chromatography was performed with silica gel (230–400 mesh). Spectra taken in CDCl3 were referenced to 7.26 ppm in 1H NMR and 77.2 ppm in 13C{1H} NMR, C6D6 was referenced to 7.16 ppm in 1H NMR and 128.4 ppm in 13C{1H} NMR, C7D8 was referenced to 7.17 ppm in 1H NMR and 128.9 ppm in 13C{1H} NMR, CD2Cl2 was referenced to 5.30 ppm in 1H NMR and 53.5 ppm in 13C{1H} NMR. Resonances for the boron-bearing carbon atom were not observed due to quadrupolar relaxation. All coupling constants are apparent J values measured at the indicated field strengths in Hertz (s = singlet, d = doublet, t = triplet, q = quartet, dd = doublet of doublets, ddd = doublet of doublet of doublets, bs = broad singlet).High-resolution mass spectra (HRMS) were obtained at the Michigan State University Mass Spectrometry Service Center using electrospray ionization (ESI+ or ESI−) on quadrupole time-of-flight (Q-TOF) instruments. Low resolution mass spectra were obtained on GCMS-QP2010 SE Shimadzu instrument. Melting points were measured in a capillary melting point apparatus and are uncorrected.
General procedure A for synthesis of starting materials via iridium catalysis (1d–1g and 1z)
In a nitrogen filled glove box in a vial (5 mL)/round bottom (50 mL) loaded with a stir bar was added bis(1,5-cyclooctadiene)di-μ-methoxydiiridium(I), bis(pinacolato)diboron or bis(trimethylsiloxy)methylsilane, di-tert-butylbipyridine followed by addition of substrate in THF. The vial/round bottom was closed, removed from the glove box, connected to a Schlenk line, and placed in an oil bath. The solution was stirred under nitrogen at 70–80 °C for 16–48 h. The reaction mixture was concentrated by rotary evaporation and purified by passing through a silica plug or flash column chromatography.:90 EtOAc/hexane). Fractions containing the desired product were combined and the volatiles evaporated to afford a white solid (2.5 g, 86% yield). 1H NMR (500 MHz, CDCl3) δ 7.62 (s, 1H), 7.43 (s, 1H), 2.52 (s, 3H), 1.33 (s, 12H). 13C{1H} NMR (126 MHz, CDCl3) δ 159.6, 141.6, 130.1, 127.2, 84.9, 25.0, 24.1. 11B NMR (160 MHz, CDCl3) δ 30.3 mp = 101–102 °C.:ethyl acetate 95:5). Fractions containing the desired product were evaporated and dried under high vacuum to yield a clear colorless oil (1.3 g, 76% yield). 1H NMR (500 MHz, CDCl3) δ 7.60 (d, J = 7.6 Hz, 1H), 7.51 (d, J = 8.3 Hz, 1H), 7.29 (t, J = 6.9 Hz, 1H), 7.21 (t, J = 7.4 Hz, 1H), 7.01 (s, 1H), 0.35 (s, 3H), 0.13 (s, 18H). 13C{1H} NMR (126 MHz, CDCl3) δ 160.8, 157.8, 127.8, 124.7, 122.5, 121.5, 116.5, 111.6, 1.9, 0.1. 29Si NMR (99 MHz, CDCl3) δ 10.0, −43.2. 1H and 13C{1H} NMR values were consistent with those previously reported.46General procedure for synthesis of starting materials via pinacol coupling (1j, 1l, 1q, 1ae, 1af)
To an oven dried round bottom flask (50 mL)/vial (5 mL) containing a stir bar was added pinacol (1 equiv.) and the corresponding boronic acid (1 equiv.) in hexane/DCM. The resulting solution was stirred for 2–48 hours at room temperature. The product was isolated by passing through a Celite plug or washing with water.Synthesis of starting material (1k) via Miyaura borylation
Synthesis of starting materials via lithiation, silylation and borylation (1y, 1x, 1ab, 1ac, 1ad)
General procedure B for hydrogenation
Arene/heteroarene (0.5 mmol), 5% Rh/C (25 mg, 2 mol%) or Rh/Al2O3 (50 mg, 4 mol%), and a stir bar were loaded into a 300 mL Parr reactor pressure vessel. Each run was flushed with hydrogen at least twice. Ethanol (5 mL) was added and the reactor was sealed and pressurized with hydrogen gas (48 atm). After 16 hours the reactor was opened and the reaction mixture filtered through Celite. The Celite was washed with methanol (3 × 5 mL) and the solvent was removed by rotary evaporation to yield the corresponding product. (Note: we chose 16 h for our reaction time owing to our preference for a common time point and related literature hydrogenations being typically run for 16–24 h. Reactions for individual substrates were not optimized.):27 molar mixture of 2b:3b (106 mg of 2b). 1H NMR (500 MHz, D2O) δ 3.42–3.29 (m, 2H), 3.13 (t, J = 5.8 Hz, 1H, deboronated piperidine), 3.04–2.86 (m, 2H), 1.96–1.80 (m, 2H), 1.80–1.59 (m, 3H, deboronated piperidine), 1.55–1.42 (m, 1H), 1.42–1.32 (m, 1H), 1.19 (s, 12H); 13C{1H} NMR (126 MHz, D2O) δ 75.6, 45.9, 44.4, 44.3, 23.6, 23.4, 22.8, 22.1, 21.4; 11B NMR (160 MHz, D2O) δ 30.2; HRMS (ESI) m/z calcd for C11H23BNO2 [M − Br]+˙ 212.1821; found. 212.1826.Recyclability test experiment
4,4,5,5-Tetramethyl-2-(5-methyltetrahydrofuran-2-yl)-1,3,2-dioxaborolane (2i). General procedure B was followed with 4,4,5,5-tetramethyl-2-(5-methylfuran-2-yl)-1,3,2-dioxaborolane 208 mg, 1.0 mmol), 5% Rh/C (25 mg, 1 mol%) for 16 h. After purification solvent was removed by rotary evaporation to yield a clear oil (165 mg (with 6.3% HOBpin) amounting to 157 mg of 2i (74% yield). A diastereomeric ratio of 97
:3 was measured by 1H NMR and GC. Cis stereochemistry was established by COSY and 1D NOE NMR experiments. In the NOE experiments, irradiating 3.63 ppm (H on C2) showed enhancement of the peak at 3.73 ppm (H on C5) and irradiating the peak 3.73 ppm (H on C5) showed enhancement of the peak at 3.63 ppm (H on C2). Byproduct HOBpin was confirmed by adding HOBpin to the NMR tube and observing the increase in intensity of the 11B peak for HOBpin. When Pd/C was used as a catalyst, 18% HOBpin was observed. Attempts to remove HOBpin by sublimation were unsuccessful. Hydrogenation using Rh/C gave only 6.3% HOBpin and reducing the time of reaction to 12.5 h further lowered the amount of HOBpin to 4%. 1H NMR (500 MHz, C6D6) δ 3.77 (dq, J = 12.8, 6.3 Hz, 1H), 3.67 (dd, J = 9.9, 7.9 Hz, 1H), 2.04–1.84 (m, 2H), 1.75–1.64 (m, 1H), 1.34–1.26 (m, 1H), 1.24 (d, J = 6.0 Hz, 3H), 1.028 (s, 6H, Bpin), 1.035 (s, 6H, Bpin); 13C{1H} NMR (126 MHz, C6D6) δ 83.4 (Bpin CH), 76.8 (Bpin CH), 34.0, 28.8, 24.9 (Bpin CH3), 24.8 (Bpin CH3), 21.2; 11B NMR (160 MHz, C6D6) δ 32.7; HRMS (ESI) m/z calcd for C11H21BO3Na [M + Na]+˙ 235.1481 found 235.1493.
4,4,5,5-Tetramethyl-2-(octahydrobenzofuran-2-yl)-1,3,2-dioxaborolane (2j). General procedure B was followed with 2-(benzofuran-2-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (1j) (122 mg, 0.5 mmol), 5% Rh/C (25 mg, 2 mol%) dissolved in hexane (5 mL) for 16 h at 55 atm pressure. This compound was purified by passing the reaction mixture in ethyl acetate through Celite and evaporating the mother liquor by rotary evaporation to yield a clear oil (124 mg (with ∼20 mol% OBpin monomer via 1H-NMR)) amounting to ∼107 mg of 2j (∼85% yield). 1H NMR (500 MHz, CDCl3) δ 3.73 (q, J = 4.1 Hz, 1H), 3.69 (dd, J = 10.9, 7.1 Hz, 1H), 2.22–2.12 (m, 1H), 2.05–1.97 (m, 1H), 1.97–1.89 (m, 1H), 1.61–1.46 (m, 5H), 1.40–1.31 (m, 1H), 1.27 (s, 6H), 1.25 (s, 6H), 1.25 (s, 3H, B byproduct) 1.23–1.17 (m, 2H); 13C{1H} NMR (126 MHz, CDCl3) δ 83.7, 83.1 (B byproduct), 78.3, 38.2, 34.5, 28.2, 28.21, 24.77 (Bpin CH3), 24.55 (B byproduct), 24.53 (Bpin CH3), 24.3, 21.0; 11B NMR (160 MHz, CDCl3) δ 32.7, 22.4 (B byproduct). The stereochemical assignment and NMR data were consistent with those previously reported.21
2-(Dodecahydrodibenzo[b,d]furan-4-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (2k). General procedure B was followed with 4,4,5-trimethyl-2-(tetrahydrodibenzofuran-4-yl)-1,3,2-dioxaborolane (147 mg, 0.5 mmol) (1k), 5% Rh/C (25 mg, 2 mol%), H2 at 60 atm, using hexane (5 mL) as a solvent at 60 °C for 21 h. The catalyst was removed by passing through Celite using diethyl ether as a solvent (3 × 5 mL). The solvent was removed by rotary evaporation to give a colorless oil that contained product (100% conversion by 1H NMR) and 8% deboronated material (tetrahydrodibenzofuran) as determined by GCMS analysis. The desired compound was purified by flash column chromatography eluting with an ethyl acetate/hexane gradient (2–10%) to give a colorless oil (58 mg, 38%). Only the major isomer was isolated. The stereochemistry was determined to be cis using 2-D NMRs and 1D NOE (see S49†). All spectral data matched those previously reported26 except for the 11B chemical shift that was reported to be at 22.40, which is inconsistent with a C–boronic ester peak and is more likely boron byproducts. 1H NMR (500 MHz, CDCl3) δ 3.88 (t, J = 3.7 Hz, 1H), 3.82 (td, J = 10.5, 5.9 Hz, 1H), 2.29–2.16 (m, 1H), 1.87–1.75 (m, 2H), 1.68 (dt, J = 12.9, 3.5 Hz, 1H), 1.64–1.53 (m, 2H), 1.47 (tt, J = 12.8, 4.7 Hz, 5H), 1.42–1.35 (m, 1H), 1.33–1.25 (m, 2H), 1.22 (s, 6H), 1.23 (s, 7H) 1.19 (s, J = 4.6 Hz, 1H), 1.07 (qt, J = 12.7, 3.0 Hz, 1H); 13C{1H} NMR (126 MHz, CDCl3) δ 82.9, 77.6, 76.4, 42.0, 41.4, 28.6, 25.7, 25.1, 24.8 (Bpin C), 24.5 (Bpin C), 22.1, 21.3, 21.2, 19.6; 11B NMR (160 MHz, CDCl3) δ 33.4; GCMS C18H31BO3 calcd [M]+˙ 306.24 found 306.25.
tert-Butyl 2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)octahydro-1H-indole-1-carboxylate (2l). General procedure B was followed with tert-butyl 2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole-1-carboxylate (1l) (174 mg, 0.5 mmol), 5% Rh/C (25 mg, 2 mol%) for 16 h. After purification solvent was removed by rotary evaporation to yield a colorless oil (170 mg, 97% yield). 1H NMR (500 MHz, CDCl3) δ 3.78 (dt, J = 11.6, 6.3 Hz, 1H), 3.74–3.68 (m, 1H), 3.62–3.50 (m, 1H), 3.18–3.01 (m, 2H), 2.33–2.18 (m, 2H), 2.07–1.97 (m, 2H), 1.89–1.77 (m, 2H), 1.76–1.52 (m, 8H), 1.46–1.39 (m, 19H, Boc of 2 rotamers), 1.37–1.28 (m, 2H), 1.27–1.18 (m, 25H, 2 Bpin rotamers), 1.18–1.07 (m, 3H); 13C{1H} NMR (151 MHz, toluene-d8, 80 °C) δ 154.4, 83.4, 78.2, 57.0, 28.9, 28.6, 26.8, 25.2, 24.8, 24.3, 21.4; 11B NMR (160 MHz, CDCl3) δ 32.5; GCMS C15H25BNO4 calcd [M − tBu]+˙ 294.19 found 294.20. 1H and 13C{1H} NMR data are consistent with those previously reported,9 however that report only tabulated data for one rotamer although both the rotamers are observed in the spectrum.21
2-Cyclohexyl-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (2p). General procedure B was followed with 4,4,5,5-tetramethyl-2-phenyl-1,3,2-dioxaborolane (210 mg, 1 mmol), 5% Rh/C (25 mg, 1 mol%) for 16 h. After purification solvent was removed by rotary evaporation to yield a clear oil (208 mg, 99% yield). 1H NMR (500 MHz, CDCl3) δ 1.69–1.62 (m, 2H), 1.62–1.53 (m, 3H), 1.37–1.26 (m, 5H), 1.23 (s, 12H), 1.01–0.92 (m, 1H); 13C{1H} NMR (126 MHz, CDCl3) δ 82.9, 28.1, 27.3, 26.9, 24.9; 11B NMR (160 MHz, CDCl3) δ 33.8. NMR data matched those previously reported.21
3-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)cyclohexan-1-ol (2q). General procedure B was followed with 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenol (1q) (110 mg, 0.5 mmol), 5% Rh/C (25 mg, 2 mol%) for 16 h. After purification solvent was removed by rotary evaporation to yield a clear oil (80 mg, 71% yield). A 1.2
:1 mixture of diastereomers was obtained. The major product was assumed to be the cis isomer based on a comparison of its 1H NMR to that of known48 TMS protected 2q. NMR data of the mixture are reported. 1H NMR67 (500 MHz, CDCl3) δ 3.79 (s, 1H), 3.59 (s, 1H), 1.96 (d, J = 13.0 Hz, 1H), 1.93–1.82 (m, 2H), 1.82–1.70 (m, 2H), 1.70–1.60 (m, 2H), 1.55–1.44 (m, 3H), 1.44–1.32 (m, 6H), 1.23 (s, 24H), 1.20–1.11 (m, 1H), 1.00 (t, J = 11.3 Hz, 1H), 0.91–0.79 (m, 1H); 13C{1H} NMR (126 MHz, CDCl3) δ 83.16, 83.13, 71.01, 68.29, 36.77, 35.99, 35.46, 34.50, 26.92, 26.78, 24.87, 24.83, 24.80; 11B NMR (160 MHz, CDCl3) δ 34.0. GCMS C11H20BO2 calcd [M − OH–Me]+˙ 195.16 found 195.05.
4,4,5,5-Tetramethyl-2-(-3-(trifluoromethyl)cyclohexyl)-1,3,2-dioxaborolane (2r). General procedure B was followed with 4,4,5,5-tetramethyl-2-(3-(trifluoromethyl)phenyl)-1,3,2-dioxaborolane (136 mg, 0.5 mmol), 5% Rh/C (25 mg, 2 mol%) for 16 h. After purification solvent was removed by rotary evaporation to yield a clear oil (109 mg, 80% yield). Cis and trans diastereomers were observed via 19F NMR in a 3
:1 cis to trans ratio. 1H NMR66 (500 MHz, CDCl3) δ 2.19–2.07 (m, 1H), 2.04–1.89 (m, 8H), 1.89–1.70 (m, 8H), 1.48–1.32 (m, 3H), 1.24 (s, 17H), 1.23 (s, 29H), 1.21–1.09 (m, 4H), 0.99–0.87 (m, 2H); 13C{1H} NMR (126 MHz, CDCl3) δ 129.1 (q), 83.4, 83.3, 42.7 (q, J = 26.1 Hz), 40.2 (q, J = 25.8 Hz), 27.0, 26.9, 26.2 (q, J = 2.5 Hz), 26.0, 25.2 (q, J = 2.6 Hz), 25.0 (q, J = 2.9 Hz), 24.9 (d, J = 4.3 Hz), 24.9 (d, J = 3.3 Hz), 24.0; 19F NMR (470 MHz, CDCl3) δ −73.61 (d, J = 8.8 Hz, 1H), −74.03 (d, J = 8.3 Hz, 3H); 11B NMR (160 MHz, CDCl3) δ 33.7; GCMS calcd for C12H19BO2F3 [M − Me]+˙ 263.14, found 263.20.Cis and trans isomers were assigned by oxidation of the Bpin and then comparing spectra of the alcohols to those previously reported.74 The proton peak on the OH bearing carbon appeared at 4.23 ppm (t, J = 3.29 Hz) (lit. 74 4.23 (brtt J = 3.5, 3.0 Hz) and for the cis isomer at 3.62 ppm (tt, J = 10.8, 4.2 Hz) (lit. 74 3.60 ppm (tt, J = 10.7, 4.2 Hz)). The ratios of cis and trans isomers after oxidation (2.6:1) (cis:trans) was comparable to the ratio before oxidation.
Ethyl-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)cyclohexane-1-carboxylate (2s). General procedure B was followed with ethyl 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate (116 mg, 0.42 mmol), 5% Rh/C (25 mg, 2 mol%) for 16 h. After purification solvent was removed by rotary evaporation to yield a clear oil (95 mg, 81% yield). 1H-NMR showed both cis and trans diastereomers that were isolated as a mixture in a 3.3
:1 ratio. The major isomer was determined using 2D NMR and 1D NOE. The proton α to carboxyl group and the proton α to Bpin both showed NOEs with each other indicating a cis orientation. 1H NMR81 (500 MHz, CDCl3) δ 4.15–4.02 (m, 7H), 2.44–2.34 (m, 1H), 2.26–2.12 (m, 4H), 1.99 (d, J = 13.3 Hz, 4H), 1.90 (d, J = 12.8 Hz, 5H), 1.83–1.75 (m, 5H), 1.73 (d, J = 13.0 Hz, 4H), 1.68–1.58 (m, 4H), 1.56–1.42 (m, 5H), 1.40–1.27 (m, 12H), 1.25–1.17 (m, 57H), 1.14 (dt, J = 12.8, 3.0 Hz, 3H), 1.13–1.06 (m, 1H), 0.92 (dt, J = 11.4, 3.2 Hz, 3H), 0.87–0.78 (m, 1H) 13C{1H} NMR (126 MHz, CDCl3) δ 176.4, 176.3, 83.1, 60.1, 44.3, 41.4, 30.4, 29.5, 29.1, 29.0, 27.2, 27.1, 26.5, 24.9, 24.9, 24.8, 24.8, 24.5, 14.4, 14.3; 11B NMR (160 MHz, CDCl3) δ 33.2; GCMS calcd for C14H24BO4 [M − Me]+˙ 267.18, found 267.20.
4,4,5,5-Tetramethyl-2-(3-methylcyclohexyl)-1,3,2-dioxaborolane (2t). General procedure B was followed with 4,4,5,5-tetramethyl-2-(m-tolyl)-1,3,2-dioxaborolane (109 mg, 0.5 mmol), 5% Rh/C (25 mg, 2 mol%) for 16 h. After purification, solvent was removed under vacuum to yield a clear oil (86 mg, 77% yield). The cis and trans diastereomeric ratio was 2.2
:1 via NMR and 2.6:1 via GC. 1H NMR81 (500 MHz, CDCl3) δ 2.17 (s, 1H), 1.77 (d, J = 13.0 Hz, 1H), 1.71 (d, J = 10.5 Hz, 8H), 1.67–1.55 (m, 5H), 1.43 (ddp, J = 10.1, 6.8, 3.3 Hz, 1H), 1.36–1.28 (m, 6H), 1.23 (s, 12H, Bpin, minor isomer), 1.22 (s, 26H, Bpin, major isomer), 1.19 (t, J = 2.8 Hz, 1H), 1.12–1.05 (m, 4H), 0.92 (tt, J = 12.8, 2.7 Hz, 4H), 0.87 (d, J = 6.6 Hz, 4H), 0.84 (d, J = 6.5 Hz, 9H); 13C{1H} NMR (126 MHz, CDCl3) δ 82.93, 82.87, 36.59, 36.06, 35.47, 35.14, 33.64, 30.98, 27.64, 27.58, 25.00, 24.93, 24.87, 24.86, 23.22, 22.46; 11B NMR (160 MHz, CDCl3) 33.8. GCMS calcd for C12H22BO2 [M − Me]+˙ 209.17 found 209.20. The NMR data of the mixture were consistent with those previously reported.21 Cis and trans isomer assignments were confirmed by oxidation of the Bpin and comparing the spectra of the alcohols to those previously reported.75 The proton on the OH bearing carbon appeared at 4.05 ppm (1H, m) (lit. 75 4.06 (m)) for the trans isomer and 3.57 ppm (lit. 75 3.57 (m)) for the cis isomer. The cis and trans ratio after oxidation (3.3:1) was comparable to that before oxidation ratio (2.6:1, via GC).
2-(3-Methoxycyclohexyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (2u). General procedure B was followed with 2-(3-methoxyphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (117 mg, 0.5 mmol), 5% Rh/C (25 mg, 2 mol%) for 16 h. After purification, solvent was removed by rotary evaporation to yield a clear oil (104 mg with 22% (by GCMS)) demethoxylated cyclohexane-Bpin, which corresponded to 82 mg of 2u as a 5.6
:1 cis/trans mixture (68% yield). For the cis isomer: 1H NMR (500 MHz, CDCl3) δ 3.33 (s, 3H), 3.08 (tt, J = 10.2, 4.0 Hz, 1H), 2.08 (d, J = 10.5 Hz, 1H), 1.97 (d, J = 13.8 Hz, 1H), 1.83–1.75 (m, 1H), 1.70–1.53 (m, 2H), 1.23 (s, 12H), 1.20–1.07 (m, 3H), 1.00–0.85 (m, 1H). (Note: the integration values reported were adjusted for the presence of overlapping signals from cyclohexane-Bpin.) 13C{1H} NMR (126 MHz, CDCl3) δ 83.0, 80.0, 55.6, 32.9, 32.4, 27.1, 25.7, 24.8; 11B NMR (160 MHz, CDCl3) δ 33.8; HRMS (ESI) m/z calcd for C13H25BO3Na [M + Na]+˙ 263.1794 found 263.1806. NMR data match those previously reported.26 Cis and trans isomer assignments were confirmed by oxidation of the Bpin and comparing the spectra of the alcohols to those previously reported.76 The proton on the OH bearing carbon appeared at 3.69 ppm (dp, J = 8.1, 3.6 Hz) (lit. 76 3.71 ppm (tt, J = 8.2, 3.9 Hz)) for the cis isomer and at 3.99 ppm (tt, J = 7.9, 3.7 Hz) (lit. 76 4.02 ppm (tt, J = 7.9, 3.8 Hz)) for the trans isomer. The cis and trans ratios after oxidation was found to be 3.3:1.
Cis-1,2-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)cyclohexane (2v). General procedure B was followed with 1,2-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzene (1v) (165 mg, 0.5 mmol), 5% Rh/C (25 mg, 2 mol%) for 6 h. After filtration solvent was removed under reduced pressure, mixture purified over flash column chromatography (hexane as eluent) and concentrated and dried over rotovap to give colorless oil (106 mg, 88% yield). 1H NMR (500 MHz, CDCl3) δ 1.69–1.49 (m, 5H), 1.49–1.35 (m, 5H), 1.22 (s, 24H); 13C{1H} NMR (126 MHz, CDCl3) δ 82.9, 28.2, 27.0, 25.0, 24.9; 11B NMR (160 MHz, CDCl3) δ 34.0. NMR data were consistent with those previously reported.50
2-(Trimethylsilyl)piperidin-1-ium-(+)-camphorsulfonate (2w). General procedure B was followed with 2-bromo-6-(trimethylsilyl)pyridine (115 mg, 0.5 mmol), 5% Rh/C (25 mg, 0.2 mol%), (+)-camphor sulfonic acid (CS) (116 mg, 0.5 mmol). After purification, solvent was removed under reduced pressure to give a colorless oil (181 mg, 76% yield). 1H NMR (500 MHz, D2O) δ 3.32–3.22 (m, 1H), 3.17 (d, J = 14.8 Hz, 1H, CS), 2.84 (td, J = 13.0, 2.4 Hz, 1H), 2.74 (d, J = 14.9 Hz, 1H, CS), 2.62–2.52 (m, 1H), 2.41–2.16 (m, 2H, CS), 2.06 (t, J = 4.5 Hz, 1H, CS), 1.99–1.90 (m, 1H, CS), 1.90–1.85 (m, 1H, CS), 1.85–1.73 (m, 3H), 1.66–1.28 (m, 5H, CS + compound), 0.94 (s, 3H, CS), 0.73 (s, 3H, CS), 0.04 (s, 9H); 13C{1H} NMR (126 MHz, D2O) δ 221.6 (CS), 58.3 (CS), 48.2, 48.0 (CS), 47.0 (CS), 46.4, 42.4 (CS), 42.1 (CS), 26.1 (CS), 24.3 (CS), 24.1, 22.8, 22.2, 18.8 (CS), 18.6 (CS), −4.9; 29Si NMR (99 MHz, D2O) δ 2.7; mp 122–124 °C; GCMS calcd for C8H19NSi [M − CS–H]+˙ 157.13 found 157.15.
2-(Trifluoromethyl)-4-(trimethylsilyl)piperidine (2x). General procedure B was followed with 2-chloro-6-(trifluoromethyl)-4-(trimethylsilyl)pyridine (1x) (44 mg, 0.17 mmol), 5% Rh/Al2O3 (20 mg, 4.5 mol%). After purification, the solvent was removed by rotary evaporation. Adding ethyl acetate formed a white solid precipitate which was dried under vacuum (44 mg, 92% yield). 1H NMR (500 MHz, D2O) δ 4.02–3.92 (m, 1H), 3.57 (dd, J = 12.5, 4.0 Hz, 1H), 3.07 (td, J = 13.0, 3.3 Hz, 1H), 2.19 (d, J = 14.0 Hz, 1H), 1.94 (d, J = 14.8 Hz, 1H), 1.67–1.45 (m, 2H), 1.00 (tt, J = 13.3, 3.4 Hz, 1H), 0.01 (s, 9H); 13C{1H} NMR (126 MHz, D2O) δ 123.1 (d, J = 280.4 Hz), 57.1 (q, J = 31.5 Hz), 46.1, 22.5, 22.1, 19.8, −5.1; 19F NMR (470 MHz, D2O) δ −75.47 (d, J = 6.6 Hz); 29Si NMR (99 MHz, D2O) δ 3.6; mp: decomposes at 237 °C; HRMS (ESI) m/z calcd for C9H19F3NSi [M − Cl]+˙ 226.1238 found 226.1239. Stereochemistry was ascertained by 2D NMR and the cis geometry determined by a 1D-NOE experiment. Irradiation of the proton at the carbon with a CF3 substituent showed enhancement of the proton at the carbon bearing TMS confirming the cis assignment.
2-(Trifluoromethyl)-5-(trimethylsilyl)piperidine (2y). General procedure B was followed with 2-chloro-5-(trifluoromethyl)-4-(trimethylsilyl)pyridine (1y) (126 mg, 0.5 mmol), 5% Rh/C (25 mg, 2 mol%). After purification, solvent was removed by rotary evaporation to yield a yellow oil (90 mg (mixture), 15% yield of 2y). A 5
:1 ratio of desilylated to silylated product was observed in the 1H NMR, which was confirmed by LCMS (226.3 m/z for silylated material and 154.1 m/z for desilylated material) and 19F NMR. Spectral data of the mixture (2y + desilylated 2y): 1H NMR (500 MHz, D2O) δ 4.46–4.14 (m, 1H), 4.09–3.97 (m, 5H), 3.53 (d, J = 12.96, 5H), 3.40 (dd, J = 13.0, 4.2 Hz, 1H), 3.21–3.01 (m, 6H), 2.22 (s, 1H), 2.21–2.12 (m, 5H), 2.03–1.85 (m, 12H), 1.82–1.50 (m, 18H), 1.27–1.09 (m, 1H), 0.01 (d, J = 1.2 Hz, 9H); 13C{1H} NMR (126 MHz, D2O) δ 123.75, 51.4, 51.2, 45.1, 43.6, 30.1, 21.4, 21.4, 21.3, 21.0, 20.6, 20.1, 17.7, −4.9; 19F NMR (470 MHz, D2O) δ −68.51 (d, J = 8.9 Hz), −75.55 (d, J = 6.8 Hz). 1H-NMR data for the desilylated compound matched those previously reported.77 1H NMR (500 MHz, D2O) 4.09–3.97 (m, 1H), 3.53 (d, J = 13.0, 1H), 3.21–3.01 (m, 1H), 2.21–2.12 (m, 1H), 2.03–1.85 (m, 2H), 1.82–1.50 (m, 3H);19F NMR (470 MHz, D2O) δ −75.6 (d, J = 6.7 Hz).
3-(2,3-Dihydrobenzofuran-2-yl)-1,1,1,3,5,5,5-heptamethyltrisiloxane (2z). General procedure B was followed with 3-(benzofuran-2-yl)-1,1,1,3,5,5,5-heptamethyltrisiloxane (1z) (169 mg, 0.5 mmol), 10% Pd/C (20 mg, 3 mol%), ethanol (5 mL) for 16 h. After purification, the solvent was removed by rotary evaporation to yield a yellowish oil (139 mg, 82% yield); 1H NMR (500 MHz, C6D6) δ 7.04 (d, J = 7.3 Hz, 1H), 7.01–6.94 (m, 1H), 6.87–6.81 (m, 1H), 6.77 (td, J = 7.4, 1.0 Hz, 1H), 4.14 (t, J = 11.1 Hz, 1H), 3.15 (dd, J = 15.0, 11.4 Hz, 1H), 3.03 (dd, J = 15.0, 10.8 Hz, 1H), 0.21 (s, 3H), 0.16 (s, 9H), 0.10 (s, 9H); 13C{1H} NMR (126 MHz, C6D6) δ 161.2, 124.7, 120.0, 109.6, 75.0, 31.1, 1.5, 1.4, −2.9; 29Si NMR (99 MHz, CDCl3) δ 9.1, 9.3, −31.62; HRMS (APCI) m/z calcd for C15H29O3Si3 [M]+˙ 341.1425 found 341.1428.
Cyclohexyltriethoxysilane (2aa)54. General procedure B was followed with triethoxy(phenyl)silane (120 mg, 0.5 mmol), 5% Rh/Al2O3 catalyst (25 mg, 2 mol%). After purification, the solvent was removed by rotary evaporation to yield a colorless oil (95.8 mg, 78% yield). Per Reaxys the only IR data were previously reported.78 1H NMR (500 MHz, CDCl3) δ 3.82 (q, J = 7.0 Hz, 6H), 1.77 (d, J = 11.4 Hz, 2H), 1.70 (d, J = 12.1 Hz, 2H), 1.67 (s, 1H), 1.32–1.25 (m, 2H), 1.22 (t, J = 7.0 Hz, 12H), 0.81 (tt, J = 12.5, 3.1 Hz, 1H); 13C{1H} NMR (126 MHz, CDCl3) δ 58.6, 27.9, 27.0, 26.9, 23.0, 18.5; 29Si NMR (99 MHz, CDCl3) δ 48.5; GCMS for C12H26O3Si calcd [M]+˙ 246.17 found 246.10.
tert-Butyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2(trimethylsilyl)pyrrolidine-1-carboxylate (2ab). General procedure B was followed with tert-butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2-(trimethylsilyl)-1H-pyrrole-1-carboxylate (1ab) (182 mg, 0.5 mmol), 5% Rh/C (25 mg, 2 mol%). After 16 h, the reaction had run to 40% conversion of 1ab and was worked up. After filtration, solvent was removed by rotary evaporation. The resulting mixture was purified by silica flash column chromatography. Fractions containing the desired product were combined and the volatiles evaporated to yield a white solid (66 mg, 36% yield, 96
:4 dr). Increasing the stirring speed from 300 rpm to 1000 rpm did not affect conversion. Increasing the pressure to 72 atm from 48 atm gave a 66% conversion and 60% yield. The reaction showed full conversion after 48 h with 4 mol% catalyst and 93–100% yields were obtained. The cis configuration of the compound was confirmed by 2D NMRs, COSY and 2D NOESY NMR. By 2D NOESY NMR, the peak at 2.93 ppm (H at C α to N with TMS) showed a stronger correlation with the neighboring proton at 1.96 ppm and a weaker correlation with the neighbor at 1.72 ppm. The peak at 1.96 ppm showed a correlation with the proton at 1.44 ppm whereas no correlation was observed with the proton at 1.72 ppm. Characterization of the major cis diastereomer: 1H NMR (500 MHz, C6D6) δ 3.85 (t, J = 9.6 Hz, 1H), 3.40 (t, J = 11.4 Hz, 1H), 2.97–2.87 (m, 1H), 2.00–1.89 (m, 1H), 1.72 (q, J = 12.6 Hz, 1H), 1.41 (s, 10H), 0.95 (s, 12H), 0.20 (s, 9H); 13C{1H} NMR (126 MHz, C6D6) δ 154.3, 82.8, 77.8, 50.1, 49.3, 32.3, 28.3, 24.4, −1.8; 11B NMR (160 MHz, C6D6) δ 33.4; 29Si NMR (99 MHz, C6D6) δ 1.7; mp 85–87 °C. HRMS (ESI) m/z calcd for C18H36BNO4Si [M]+˙ 369.2506 found 369.2510.
4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-2-(trimethylsilyl)piperidin-1-ium bromide (2ac). General procedure B was followed with 2-bromo-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-6-(trimethylsilyl)pyridine (1ac) (178 mg, 0.5 mmol), 5% Rh/C (25 mg, 2 mol%). After purification, solvent was concentrated by rotary evaporation and the product precipitated by adding ethyl acetate. After drying under vacuum a white solid was obtained (83 mg, 46% yield). Structure determination was made by 2D NMRs and the cis stereochemistry was ascertained by 1D NOE. 1H NMR (500 MHz, D2O) δ 3.22 (d, J = 12.4 Hz, 1H), 2.78 (dt, J = 12.79 Hz, 2.45 Hz, 1H), 2.52 (dd, J = 13.2 Hz, 2.45, 1H), 1.80 (d, J = 14.4 Hz, 2H), 1.56–1.33 (m, 2H), 1.12 (s, 12H), 1.09 (s, 1H), −0.02 (s, 9H); 13C{1H} NMR (126 MHz, D2O) δ 75.5, 48.6, 46.7, 25.7, 23.7, 23.6, −5.01; 11B NMR (160 MHz, D2O) δ 31.2; 29Si NMR (99 MHz, D2O) δ 2.7; mp: decomposes at 230 °C; HRMS (ESI) m/z calcd for C14H31BNSiO2 calcd [M − Br]+˙ 284.2217 found 284.2220.
Cis-trimethylsilyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)cyclohexane (2ad). General procedure B was followed with trimethyl(2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)silane (1ad) (138 mg, 0.5 mmol), 5% Rh/C (25 mg, 2 mol%) at 48 atm pressure for 6 h. After purification, solvent was removed under reduced pressure and the residue purified by flash column chromatography (diethyl ether/hexane 1–10% gradient). Fractions containing the desired product were combined and the volatiles evaporated to give a colorless oil (86% conversion, 17% yield). 1H NMR (500 MHz, CDCl3) δ 1.92–1.83 (m, 1H), 1.72 (dtt, J = 12.8, 3.6, 1.8 Hz, 1H), 1.65–1.58 (m, 1H), 1.58–1.50 (m, 1H), 1.48–1.25 (m, 4H), 1.24 (d, J = 3.6 Hz, 12H), 1.19 (dt, J = 12.3, 3.8 Hz, 1H), 0.71 (dt, J = 12.7, 3.3 Hz, 1H), −0.04 (s, 9H); 13C{1H} NMR (126 MHz, CDCl3) δ 82.9, 31.1, 28.9, 26.3, 25.8, 25.1, 25.0, −1.8; 11B NMR (160 MHz, CDCl3) δ 34.1; 29Si NMR (99 MHz, CDCl3) δ 1.9. GCMS calcd for C14H28BO2Si [M − Me]+˙ 267.20 found 267.20. NMR data and the stereochemical assignment were consistent with those previously reported.22
Trimethyl(3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)cyclohexyl)silane (2ae). General procedure B was followed with trimethyl(3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)silane (1ae) (138 mg, 0.5 mmol), 5% Rh/C (25 mg, 2 mol%). After purification, the solvent was removed by rotary evaporation and compound obtained as a colorless liquid (141 mg (with ∼7% desilylated material observed via GC-MS)), which corresponded to 133.5 mg of 2ae (95% yield). The major diastereomer was identified by oxidizing the boronic ester. The peak for the proton α to the OH in the cis isomer was observed at 3.55 ppm (sept) (lit. 79 3.42 ppm, sept) and the peak for proton α to OH was observed at 4.0 ppm (bs) (lit. 79 3.96 ppm (bs)) for the trans diastereomer. Cis/trans ratios were before (2.1
:1) and after (2.5:1) oxidation were comparable. Data of the mixture: 1H NMR (500 MHz, CDCl3) δ 1.91–1.53 (m, 6H), 1.42–1.27 (m, 2H), 1.27–1.20 (m, 14H), 1.19 (d, J = 10 Hz, 1H), 1.12–0.98 (m, 2H), 0.89 (t, J = 11.7 Hz, 1H), 0.53 (t, J = 12.6 Hz, 1H),–0.07 (s, 9H); 13C{1H} NMR (126 MHz, CDCl3) δ 82.9, 82.8, 28.7, 28.7, 28.3, 28.3, 28.1, 28.0, 27.5, 27.3, 27.1, 26.9, 25.2, 25.2, 24.9, 24.9, 24.8, −3.3, −3.4; 11B NMR (160 MHz, CDCl3) δ 34.0; 29Si NMR (99 MHz, CDCl3) δ 2.1, 2.3; GCMS for C14H28BO2Si [M − Me]+˙ calcd 267.20 found 267.20. The NMR data were consistent with those previously reported.22
Trimethyl(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)cyclohexyl)silane (2af). General procedure B was followed with trimethyl(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)silane (1af) (138 mg, 0.5 mmol), 5% Rh/C (25 mg, 2 mol%) at 48 atm pressure for 8.5 h. After purification, solvent was removed under reduced pressure to obtain a colorless oil (138 mg, 98% yield). A 4.5
:1 cis to trans ratio was observed by 1H NMR and 6:1 via GC. Data for the major cis diastereomer: 1H NMR (500 MHz, CDCl3) δ 1.95–1.86 (m, 2H), 1.64–1.55 (m, 2H), 1.41–1.30 (m, 3H), 1.25 (s, 12H), 1.15 (qd, J = 12.8, 3.3 Hz, 2H), 0.56–0.47 (m, 1H), −0.09 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3) δ 83.0, 29.2, 26.2, 25.9, 25.0, −3.5. 11B NMR (160 MHz, CDCl3) δ 34.6; 29Si NMR (99 MHz, CDCl3) δ 2.4, 2.2. 1H-NMR data were consistent with those previously reported.22 (Note: the major diastereomer was identified by oxidizing the boronic ester. The differentiable peak for the proton α to the OH in the cis isomer was observed at 4.05 ppm (s, 1H) (lit. 80 4.07 ppm (s, 1H))). The proton α to the OH in the trans isomer was observed at 3.54–3.46 ppm (m, 1H) (lit. 80 3.58–3.52 ppm (m, 1H)). By 1H NMR the cis:trans ratios were comparable before (4.5:1) and after (4.4:1) oxidation.
Data availability
All underlying data is available in the article itself and its ESI.†Conflicts of interest
The authors declare the following competing financial interest(s): M. R. S. and R. E. M. own a percentage of BoroPharm, Inc.Acknowledgements
We thank the NIH (GM63188) for financial support and BoroPharm, Inc. for a generous gift of bis(pinacolato)diboron (B2pin2). We acknowledge Ryan Fornwald for helpful discussions during the initial stages of this research.References
- J. F. Hartwig, Evolution of C-H Bond Functionalization from Methane to Methodology, J. Am. Chem. Soc., 2016, 138(1), 2–24 CrossRef CAS PubMed
.
- C. Sandford and V. K. Aggarwal, Stereospecific Functionalizations and Transformations of Secondary and Tertiary Boronic Esters, Chem. Commun., 2017, 53(40), 5481–5494 RSC
- S. Miyamura, M. Araki, T. Suzuki, J. Yamaguchi and K. Itami, Stereodivergent Synthesis of Arylcyclopropylamines by Sequential C-H Borylation and Suzuki-Miyaura Coupling, Angew. Chem., Int. Ed., 2015, 54(3), 846–851 CrossRef CAS PubMed
- S. Kawamorita, R. Murakami, T. Iwai and M. Sawamura, Synthesis of Primary and Secondary Alkylboronates through Site-Selective C(sp3)-H Activation with Silica-Supported Monophosphine-Ir Catalysts, J. Am. Chem. Soc., 2013, 135(8), 2947–2950 CrossRef CAS PubMed
- Q. Li, C. W. Liskey and J. F. Hartwig, Regioselective Borylation of the C-H Bonds in Alkylamines and Alkyl Ethers. Observation and Origin of High Reactivity of Primary C-H Bonds Beta to Nitrogen and Oxygen, J. Am. Chem. Soc., 2014, 136(24), 8755–8765 CrossRef CAS PubMed
- M. A. Larsen, C. V. Wilson and J. F. Hartwig, Iridium-Catalyzed Borylation of Primary Benzylic C-H Bonds without a Directing Group: Scope, Mechanism, and Origins of Selectivity, J. Am. Chem. Soc., 2015, 137(26), 8633–8643 CrossRef CAS PubMed
- R. Oeschger, B. Su, I. Yu, C. Ehinger, E. Romero, S. He and J. Hartwig, Diverse Functionalization of Strong Alkyl C-H Bonds by Undirected Borylation, Science, 2020, 368(6492), 736–741 CrossRef CAS PubMed
- M. R. Jones, C. D. Fast and N. D. Schley, Iridium-Catalyzed sp3 C–H Borylation in Hydrocarbon Solvent Enabled by 2,2′-Dipyridylarylmethane Ligands, J. Am. Chem. Soc., 2020, 142(14), 6488–6492 CrossRef CAS PubMed
- F. Bourriquen, J. Hervochon, R. Qu, S. Bartling, N. Rockstroh, K. Junge, C. Fischmeister and M. Beller, Diastereoselective Hydrogenation of Arenes and Pyridines Using Supported Ruthenium Nanoparticles under Mild Conditions, Chem. Commun., 2022, 58(63), 8842–8845 RSC
- S. Williams, L. Qi, R. J. Cox, P. Kumar and J. Xiao, Hydrogenation of Functionalised Pyridines with a Rhodium Oxide Catalyst under Mild Conditions, Org. Biomol. Chem., 2024, 22(5), 1010–1017 RSC
- M. Ibrahim, R. Poreddy, K. Philippot, A. Riisager and E. J. Garcia-Suarez, Chemoselective Hydrogenation of Arenes by PVP Supported Rh Nanoparticles, Dalton Trans., 2016, 45(48), 19368–19373 RSC
- C. Chaudhari, K. Sato, Y. Nishida, T. Yamamoto, T. Toriyama, S. Matsumura, Y. Ikeda, K. Terada, N. Abe, K. Kusuda, H. Kitagawa and K. Nagaoka, Chemoselective Hydrogenation of Heteroarenes and Arenes by Pd-Ru-PVP under Mild Conditions, RSC Adv., 2020, 10(72), 44191–44195 RSC
- T. Maegawa, A. Akashi, K. Yaguchi, Y. Iwasaki, M. Shigetsura, Y. Monguchi and H. Sajiki, Efficient and Practical Arene Hydrogenation by Heterogeneous Catalysts under Mild Conditions, Chemistry, 2009, 15(28), 6953–6963 CrossRef CAS PubMed
- X. Cui, A.-E. Surkus, K. Junge, C. Topf, J. Radnik, C. Kreyenschulte and M. Beller, Highly Selective Hydrogenation of Arenes Using Nanostructured Ruthenium Catalysts Modified with a Carbon–Nitrogen Matrix, Nat. Commun., 2016, 7, 11326 CrossRef PubMed
- A. Sánchez, M. Fang, A. Ahmed and R. A. Sánchez-Delgado, Hydrogenation of Arenes, N-Heteroaromatic Compounds, and Alkenes Catalyzed by Rhodium Nanoparticles Supported on Magnesium Oxide, Appl. Catal., A, 2014, 477, 117–124 CrossRef
- P. Kumar, L. Qi, S. Williams, R. A. Dop, Y. Liu, T. Zhang, C. Li and J. Xiao, Selective Hydrogenation of Lignin-Derived Aromatics to Give Cyclohexanes with a Rhodium-Pincer Precatalyst, J. Organomet. Chem., 2023, 997, 122795 CrossRef CAS
- M. Nasiruzzaman Shaikh, M. A. Aziz, A. N. Kalanthoden, A. Helal, A. S. Hakeem and M. Bououdina, Facile Hydrogenation of N-Heteroarenes by Magnetic Nanoparticle-Supported Sub-Nanometric Rh Catalysts in Aqueous Medium, Catal. Sci. Technol., 2018, 8(18), 4709–4717 RSC
- D.-S. Wang, Q.-A. Chen, S.-M. Lu and Y.-G. Zhou, Asymmetric Hydrogenation of Heteroarenes and Arenes, Chem. Rev., 2012, 112(4), 2557–2590 CrossRef CAS PubMed
- Z. Wei, F. Shao and J. Wang, Recent Advances in Heterogeneous Catalytic Hydrogenation and Dehydrogenation of N-Heterocycles, Chin. J. Catal., 2019, 40(7), 980–1002 CrossRef CAS
- M. P. Wiesenfeldt, Z. Nairoukh, T. Dalton and F. Glorius, Selective Arene Hydrogenation for Direct Access to Saturated Carbo- and Heterocycles, Angew Chem. Int. Ed. Engl., 2019, 58(31), 10460–10476 CrossRef CAS PubMed
- M. Wollenburg, D. Moock and F. Glorius, Hydrogenation of Borylated Arenes, Angew Chem. Int. Ed. Engl., 2019, 58(20), 6549–6553 CrossRef CAS PubMed
- M. P. Wiesenfeldt, T. Knecht, C. Schlepphorst and F. Glorius, Silylarene Hydrogenation: A Strategic Approach That Enables Direct Access to Versatile Silylated Saturated Carbo- and Heterocycles, Angew Chem. Int. Ed. Engl., 2018, 57(27), 8297–8300 CrossRef CAS PubMed
- M. Wollenburg, A. Heusler, K. Bergander and F. Glorius, Trans-Selective and Switchable Arene Hydrogenation of Phenol Derivatives, ACS Catal., 2020, 10(19), 11365–11370 CrossRef CAS PubMed
- D. Moock, M. P. Wiesenfeldt, M. Freitag, S. Muratsugu, S. Ikemoto, R. Knitsch, J. Schneidewind, W. Baumann, A. H. Schäfer, A. Timmer, M. Tada, M. R. Hansen and F. Glorius, Mechanistic Understanding of the Heterogeneous, Rhodium-Cyclic (Alkyl)(Amino)Carbene-Catalyzed (Fluoro-)Arene Hydrogenation, ACS Catal., 2020, 10(11), 6309–6317 CrossRef CAS PubMed
- D. Moock, T. Wagener, T. Hu, T. Gallagher and F. Glorius, Enantio- and Diastereoselective, Complete Hydrogenation of Benzofurans by Cascade Catalysis, Angew Chem. Int. Ed. Engl., 2021, 60(24), 13677–13681 CrossRef CAS PubMed
- L. Ling, Y. He, X. Zhang, M. Luo and X. Zeng, Hydrogenation of (Hetero)Aryl Boronate Esters with a Cyclic (Alkyl)(Amino)Carbene–Rhodium Complex: Direct Access to Cis-Substituted Borylated Cycloalkanes and Saturated Heterocycles, Angew. Chem., Int. Ed., 2019, 58, 6554 CrossRef CAS PubMed
- C. H. Schiwek, S. Stegbauer, T. Pickl and T. Bach, Rhodium(CAAC)-catalyzed Arene Hydrogenation of Benzo-fused N-heterocycles to Saturated Building Blocks with an All-Cis Configuration, Adv. Synth. Catal., 2022, 364(19), 3360–3365 CrossRef CAS
- C. H. Schiwek, C. Jandl and T. Bach, All-Cis Saturated 2,5-Diketopiperazines by a Diastereoselective Rhodium-Catalyzed Arene Hydrogenation, ACS Catal., 2022, 12(6), 3628–3633 CrossRef CAS
- C. H. Schiwek, C. Jandl and T. Bach, Diastereoselective Rhodium-Catalyzed Hydrogenation of 2-Oxindoles and 3,4-Dihydroquinolones, Org. Lett., 2020, 22(24), 9468–9472 CrossRef CAS PubMed
- G. Hedouin, S. Sharma, K. Kaur, R. H. Choudhary, J. B. Jasinski, F. Gallou and S. Handa, Ligand-Free Ultrasmall Recyclable Iridium(0) Nanoparticles for Regioselective Aromatic Hydrogenation of Phosphine Oxide Scaffolds: An Easy Access to New Phosphine Ligands, Angew Chem. Int. Ed. Engl., 2023, e202307139 CAS
- F. Zhang, H. S. Sasmal, C. G. Daniliuc and F. Glorius, Ru-NHC-Catalyzed Asymmetric, Complete Hydrogenation of Indoles and Benzofurans: One Catalyst with Dual Function, J. Am. Chem. Soc., 2023, 145(29), 15695–15701 CrossRef CAS PubMed
- L. Lückemeier, M. Pierau and F. Glorius, Asymmetric Arene Hydrogenation: Towards Sustainability and Application, Chem. Soc. Rev., 2023, 52(15), 4996–5012 RSC
- H. Wu, J. Yang, B. B. C. Peters, L. Massaro, J. Zheng and P. G. Andersson, Asymmetric Full Saturation of Vinylarenes with Cooperative Homogeneous and Heterogeneous Rhodium Catalysis, J. Am. Chem. Soc., 2021, 143(48), 20377–20383 CrossRef CAS PubMed
- T. Wagener, A. Heusler, Z. Nairoukh, K. Bergander, C. G. Daniliuc and F. Glorius, Accessing (Multi)Fluorinated Piperidines Using Heterogeneous Hydrogenation, ACS Catal., 2020, 10(20), 12052–12057 CrossRef CAS PubMed
- A. Kaithal, H. S. Sasmal, S. Dutta, F. Schäfer, L. Schlichter and F. Glorius, Cis-Selective Hydrogenation of Aryl Germanes: A Direct Approach to Access Saturated Carbo- and Heterocyclic Germanes, J. Am. Chem. Soc., 2023, 145(7), 4109–4118 CrossRef CAS PubMed
- M. Freifelder, R. M. Robinson and G. R. Stone, Hydrogenation of Substituted Pyridines with Rhodium on Carbon Catalyst, J. Org. Chem., 1962, 27(1), 284–286 CrossRef CAS
- J. Becica and G. E. Dobereiner, The Roles of Lewis Acidic Additives in Organotransition Metal Catalysis, Org. Biomol. Chem., 2019, 17(8), 2055–2069 RSC
- R. M. Skomoroski and A. Schreisheim, The Hydrogenation of Pyridine in Acid Media, J. Phys. Chem., 1961, 65, 1340 CrossRef CAS
- G. N. Walker, Palladium Catalyzed Hydrogenation of Pyridines, J. Org. Chem., 1962, 27, 2966 CrossRef CAS
- H. Adkins, L. F. Kuick, M. Farlow and B. Wojcik, Hydrogenation of Derivatives of Pyridine, J. Am. Chem. Soc., 1934, 56(11), 2425–2428 CrossRef CAS
- M. Freifelder and G. Stone, Reductions with Ruthenium. II. Its Use in the Hydrogenation of Pyridines, J. Org. Chem., 1961, 26, 3805 CrossRef CAS
- R. A. Rajadhyaksha and S. L. Karwa, Solvent Effects in Catalytic Hydrogenation, Chem. Eng. Sci., 1986, 41, 1765–1770 CrossRef CAS
- F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. Robert McElroy and J. Sherwood, Tools and Techniques for Solvent Selection: Green Solvent Selection Guides, Sustainable Chem. Processes, 2016, 4(1), 1–24 CrossRef
- V. A. Kallepalli, K. A. Gore, F. Shi, L. Sanchez, G. A. Chotana, S. L. Miller, R. E. Maleczka Jr and M. R. Smith 3rd, Harnessing C-H Borylation/Deborylation for Selective Deuteration, Synthesis of Boronate Esters, and Late Stage Functionalization, J. Org. Chem., 2015, 80(16), 8341–8353 CrossRef CAS PubMed
- V. A. Kallepalli, F. Shi, S. Paul, E. N. Onyeozili, R. E. Maleczka Jr and M. R. Smith 3rd, Boc Groups as Protectors and Directors for Ir-Catalyzed C-H Borylation of Heterocycles, J. Org. Chem., 2009, 74(23), 9199–9201 CrossRef CAS PubMed
- C. Cheng and J. F. Hartwig, Rhodium-Catalyzed Intermolecular C-H Silylation of Arenes with High Steric Regiocontrol, Science, 2014, 343(6173), 853–857 CrossRef CAS PubMed
- E. S. Kuffner, A Simplification of Pictect’s Nicotine Synthesis (Part II on Tobacco Bases), Ber. Dtsch. Chem. Ges., 1935, 68, 494–497 CrossRef
- L. M. Mori-Quiroz, K. W. Shimkin, S. Rezazadeh, R. A. Kozlowski and D. A. Watson, Copper-Catalyzed Amidation of Primary and Secondary Alkyl Boronic Esters, Chemistry, 2016, 22(44), 15654–15658 CrossRef CAS PubMed
- J. Wu, R. M. Bär, L. Guo, A. Noble and V. K. Aggarwal, Photoinduced Deoxygenative Borylations of Aliphatic Alcohols, Angew Chem. Int. Ed. Engl., 2019, 58(52), 18830–18834 CrossRef CAS PubMed
- M. Huang, J. Hu, S. Shi, A. Friedrich, J. Krebs, S. A. Westcott, U. Radius and T. B. S. Marder, Transition Metal-Free 1,2-Diboration of Alkyl Halides, Tosylates, and Alcohols, Chemistry, 2022, 28(24), e202200480 CrossRef CAS PubMed
- C. Karmel and J. F. Hartwig, Mechanism of the Iridium-Catalyzed Silylation of Aromatic C-H Bonds, J. Am. Chem. Soc., 2020, 142(23), 10494–10505 CrossRef CAS PubMed
- L. R. Augustine, Catalytic Hydrogenation, Marcel Dekker, Inc., New York, 1965 Search PubMed
- A. Coaviche-Yoval, E. Andrade-Jorge, C. Pérez-González, H. Luna, R. Tovar-Miranda and J. G. Trujillo-Ferrara, Quantum Reality in the Selective Reduction of a Benzofuran System, Molecules, 2019, 24(11), 2061–2078 CrossRef PubMed
- Y. Yasuhisa, H. Kazuhiro and M. Hiroshi, Method for Producing Alicyclic Substituted Silane Compound, Japanese Pat. Appl., JP2004307392A, 2004 Search PubMed
- M. A. Larsen, S. H. Cho and J. Hartwig, Iridium-Catalyzed, Hydrosilyl-Directed Borylation of Unactivated Alkyl C-H Bonds, J. Am. Chem. Soc., 2016, 138(3), 762–765 CrossRef CAS PubMed
- N. Saito, J. Takaya and N. Iwasawa, Stabilized Gallylene in a Pincer-Type Ligand: Synthesis, Structure, and Reactivity of PGaI P-Ir Complexes, Angew Chem. Int. Ed. Engl., 2019, 58(29), 9998–10002 CrossRef CAS PubMed
- M. S. Congreve, S. P. Andrews, J. S. Mason, C. M. Richardson and G. A. Brown, 1,2,4-Triazine-4-Amine Derivatives, WO2011095625A1, 2011
- H. Ren, Y.-P. Zhou, Y. Bai, C. Cui and M. Driess, Cobalt-Catalyzed Regioselective Borylation of Arenes: N-Heterocyclic Silylene as an Electron Donor in the Metal-Mediated Activation of C-H Bonds, Chemistry, 2017, 23(24), 5663–5667 CrossRef CAS PubMed
- M. Tobisu, T. Igarashi and N. Chatani, Iridium/N-Heterocyclic Carbene-Catalyzed C-H Borylation of Arenes by Diisopropylaminoborane, Beilstein J. Org. Chem., 2016, 12, 654–661 CrossRef CAS PubMed
- W. Xiong, Q. Shi and W. H. Liu, Simple and Practical Conversion of Benzoic Acids to Phenols at Room Temperature, J. Am. Chem. Soc., 2022, 144(34), 15894–15902 CrossRef CAS PubMed
- J. Xie, K. Sekine, S. Witzel, P. Krämer, M. Rudolph, F. Rominger and A. S. K. Hashmi, Light-Induced Gold-Catalyzed Hiyama Arylation: A Coupling Access to Biarylboronates, Angew Chem. Int. Ed. Engl., 2018, 57(51), 16648–16653 CrossRef CAS PubMed
- L. Britton, J. H. Docherty, G. S. Nichol, A. P. Dominey and S. P. Thomas, Iron-catalysed C(sp2)–H Borylation with Expanded Functional Group Tolerance, Chin. J. Chem., 2022, 40(24), 2875–2881 CrossRef CAS
- E. G. Lee, C. Si, M. H. Cho, G. Si, H. D. Oh, G. S. Kim, D. Gu, S. J. Kim and C. Si, Compound for Organic Electronic Element, Organic Electronic Element using Same, And Electronic Device Thereof, US Pat., 2020039993A1, February 2020
- L. Gong, C. Li, F. Yuan, S. Liu and X. Zeng, Chromium-Catalyzed Selective Borylation of Vinyl Triflates and Unactivated Aryl Carboxylic Esters with Pinacolborane, Org. Lett., 2022, 24(17), 3227–3231 CrossRef CAS PubMed
- F. Mongin, A. Tognini, F. Cottet and M. Schlosser, Halogen Shuffling in Pyridines: Site Selective Electrophilic Substitutions of 2-Chloro-6-(Trifluoromethyl)Pyridine, Tetrahedron Lett., 1998, 39, 1749–1752 CrossRef CAS
- E. M. Beck, R. Hatley and M. J. Gaunt, Synthesis of Rhazinicine by a Metal-Catalyzed C–H Bond Functionalization Strategy, Angew. Chem., Int. Ed., 2008, 47(16), 3004–3007 CrossRef CAS PubMed
- S. D. Bull, S. G. Davies, D. J. Fox, M. Gianotti, P. M. Kelly, C. Pierres, E. D. Savory and A. D. Smith, Asymmetric Synthesis of β-Pyridyl-β-Amino Acid Derivatives, J. Chem. Soc., Perkin Trans. 1, 2002,(16), 1858–1868 RSC
- M. Onoe, K. Baba, Y. Kim, Y. Kita, M. Tobisu and N. Chatani, Rhodium-Catalyzed Carbon–Silicon Bond Activation for Synthesis of Benzosilole Derivatives, J. Am. Chem. Soc., 2012, 134(47), 19477–19488 CrossRef CAS PubMed
- T. Niwa, H. Ochiai and T. Hosoya, Copper-Catalyzed Ipso-Borylation of Fluoroarenes, ACS Catal., 2017, 7(7), 4535–4541 CrossRef CAS
- R. H. Verschueren, P. Gilles, S. Van Mileghem and W. M. De Borggraeve, Solvent-Free N-Boc Deprotection by Ex Situ Generation of Hydrogen Chloride Gas, Org. Biomol. Chem., 2021, 19(26), 5782–5787 RSC
- A. H. Harkiss and A. Sutherland, Access to 2,6-Disubstituted 4-Oxopiperidines Using a 6-Endo-Trig Cyclization: Stereoselective Synthesis of Spruce Alkaloid and (+)-241D, J. Org. Chem., 2018, 83(1), 535–542 CrossRef CAS PubMed
- C. Gnamm, K. Brödner, C. M. Krauter and G. Helmchen, A Configurational Switch Based on Iridium-Catalyzed Allylic Cyclization: Application in Asymmetric Total Syntheses of Prosopis, Dendrobate, and Spruce Alkaloids, Chemistry, 2009, 15(40), 10514–10532 CrossRef CAS PubMed
- B. Wang, P. Peng, W. Ma, Z. Liu, C. Huang, Y. Cao, P. Hu, X. Qi and Q. Lu, Electrochemical Borylation of Alkyl Halides: Fast, Scalable Access to Alkyl Boronic Esters, J. Am. Chem. Soc., 2021, 143(33), 12985–12991 CrossRef CAS PubMed
- A. Arnone, R. Bernardi, F. Blasco, R. Cardillo, G. Resnati, I. I. Gerus and V. P. Kukhar, Trifluoromethyl vs. Methyl Ability to Direct Enantioselection in Microbial Reduction of Carbonyl Substrates, Tetrahedron, 1998, 54(12), 2809–2818 CrossRef CAS
- T. Hattori, T. Ida, A. Tsubone, Y. Sawama, Y. Monguchi and H. Sajiki, Facile Arene Hydrogenation under Flow Conditions Catalyzed by Rhodium or Ruthenium on Carbon, Eur. J. Org. Chem., 2015, 2015(11), 2492–2497 CrossRef CAS
- P. R. de Oliveira and R. Rittner, The Relevant Effect of an Intramolecular Hydrogen Bond on the Conformational Equilibrium of Cis-3-Methoxycyclohexanol Compared to Trans-3-Methoxycyclohexanol and Cis-1,3-Dimethoxycyclohexane, Spectrochim. Acta, Part A, 2005, 61(8), 1737–1745 CrossRef PubMed
- S. Gille, A. Ferry, T. Billard and B. R. Langlois, Synthesis of Alpha-Trifluoromethylated Nitrogen Heterocycles, J. Org. Chem., 2003, 68(23), 8932–8935 CrossRef CAS PubMed
- J. Pola, M. Jakoubková and V. Chvalovský, The Oxygen Basicity in Siloxanes. Variable Electronic Effect of Trimethylsiloxy Group, Collect. Czech. Chem. Commun., 1978, 43(12), 3373–3379 CrossRef CAS
- M.-D. Jesus, O. Rosario and G. L. Larson, The Hydroboration-Oxidation of the Isomeric Trimethylsilylcyclo-Hexenes and the Isomeric Trimethylsilylcyclopentenes, J. Organomet. Chem., 1977, 132(2), 301–320 CrossRef
- K. Guckian, G. Kumaravel, B. Ma, S. Mi, P. Hairuo, Z. Shao, L. Sun, A. Taveras, Z. Xin and L. Zhang, Compounds That Are S1P Modulating Agents and/or ATX Modulating Agents, WO/2014/018891A1, 2014
- Integration of the 1H NMR spectrum appeared higher than expected, possibly due to relaxation delay differences or impurities not detected by GCMS.
Footnote |
| † Electronic supplementary information (ESI) available: Compound characterization data. See DOI: https://doi.org/10.1039/d4ra00491d |
| This journal is © The Royal Society of Chemistry 2024 |