
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDispersive micro solid phase extraction of glibenclamide from plasma, urine, and wastewater using a magnetic molecularly imprinted polymer followed by its determination by a high-performance liquid chromatography-photodiode array detector†
Najmeh Sabbaghia,
Shayessteh Dadfarnia *a,
Ali Mohammad Haji Shabani*a and
Majid Farsadroohb
*a,
Ali Mohammad Haji Shabani*a and
Majid Farsadroohb
aDepartment of Chemistry, Faculty of Science, Yazd University, 89195-741, Yazd, Iran. E-mail: sdadfarnia@yazd.ac.ir; hshabani@yazd.ac.ir
bRenewable Energies Research Laboratory, Department of Chemistry, Faculty of Science, University of Sistan and Baluchestan, P.O. Box 98135 674, Zahedan, Iran
First published on 23rd April 2024
Abstract
The present study describes the development of a simple and selective analytical method for dispersive micro solid phase extraction and determination of glibenclamide (GLB) using magnetic molecularly imprinted polymer (MMIP) as a sorbent. MMIP was fabricated by the non-covalent method on the surface of silicated Fe3O4 and had a high affinity for glibenclamide; dual monomers, itaconic acid and allylamine, were used for this. Polymerization was achieved by the precipitation method in the presence of glibenclamide as the template and ethylene glycol dimethacrylate as the cross-linker. The morphology and structural properties of the MMIP were characterized by different analytical methods. To achieve maximum extraction efficiency, influencing parameters were optimized. The linearity range was 1–2000 and 12–2000 μg L−1 by high-performance liquid chromatography-photodiode array detector (HPLC-PDA) and UV-vis spectroscopy, respectively. The detection and quantification limits with UV-vis and HPLC-PDA analyses were 4 and 12 μg L−1 and 0.3 and 1 μg L−1, respectively. Under optimized conditions, recovery of glibenclamide spiked in plasma, human urine, and wastewater was between 89.4 and 102.9% at the concentration levels of 25, 250, and 500 μg L−1; relative standard deviations were below 3.7% by HPLC-PDA. The developed method has a favorable pre-concentration factor of 140.0. Equilibrium data and sorption isotherms fitted well with the Langmuir model. A maximum sorption capacity of 24.260 mg g−1 was acquired based on the Langmuir model. The synthesized sorbent with high selectivity was used to separate GLB from complex biological systems and wastewater before measurement with UV-vis or HPLC-PDA.
1. Introduction
Glibenclamide (GLB), 5-chloro-N-[2-[4-[[[(cyclohexylamino)carbonyl]amino]sulphonyl]phenyl]ethyl]-2-methoxybenzamide (Fig. S1†), is a relatively water-insoluble, second-generation product of sulfonylurea with log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Kow of 4.79 and pKa of 5.3. It is a drug administered for the treatment of type 2 diabetes mellitus.1–3 Glibenclamide has an acute hypoglycemic influence as it acts on pancreatic beta cells and stimulates insulin secretion, leading to the cells of the organism to enhance their glucose consumption.3 It is one of the two important oral antidiabetic medications (together with metformin) that have been recognized by the World Health Organization.4 Glibenclamide is quickly absorbed in the gastrointestinal tract and remains effective in the body for up to 24 hours and has a half-life of 10 hours. Its peak response occurs with the secretion of insulin within 2 to 3 hours after oral consumption. However, the increased concentrations of glibenclamide in the body caused by excessive consumption can have different adverse effects on the kidneys and pancreas. Furthermore, unmetabolized GLB residue in the body is excreted and enters wastewater, which is not fully removed by sewage treatment plants, allowing GLB to enter natural and groundwater and harm human health and entire ecosystems. For instance, the presence of GLB in wastewater discharged into rivers has been observed to cause alterations in the sex of fish and a decline in their ability to reproduce, ultimately having a detrimental impact on their survival.5 Consequently, it is essential to develop sensitive, simple, and inexpensive analytical methods to monitor and determine the levels of glibenclamide in biological fluids and wastewater to prevent its adverse effects on humans and natural ecosystems.
Kow of 4.79 and pKa of 5.3. It is a drug administered for the treatment of type 2 diabetes mellitus.1–3 Glibenclamide has an acute hypoglycemic influence as it acts on pancreatic beta cells and stimulates insulin secretion, leading to the cells of the organism to enhance their glucose consumption.3 It is one of the two important oral antidiabetic medications (together with metformin) that have been recognized by the World Health Organization.4 Glibenclamide is quickly absorbed in the gastrointestinal tract and remains effective in the body for up to 24 hours and has a half-life of 10 hours. Its peak response occurs with the secretion of insulin within 2 to 3 hours after oral consumption. However, the increased concentrations of glibenclamide in the body caused by excessive consumption can have different adverse effects on the kidneys and pancreas. Furthermore, unmetabolized GLB residue in the body is excreted and enters wastewater, which is not fully removed by sewage treatment plants, allowing GLB to enter natural and groundwater and harm human health and entire ecosystems. For instance, the presence of GLB in wastewater discharged into rivers has been observed to cause alterations in the sex of fish and a decline in their ability to reproduce, ultimately having a detrimental impact on their survival.5 Consequently, it is essential to develop sensitive, simple, and inexpensive analytical methods to monitor and determine the levels of glibenclamide in biological fluids and wastewater to prevent its adverse effects on humans and natural ecosystems.
Currently, different techniques, including high-performance liquid chromatography-mass spectrometry (HPLC-MS),6 HPLC-fluorescence,7 HPLC-UV,8 atmospheric pressure chemical ionization/MS–MS detection,9 micellar electrokinetic chromatography with non-ionic surfactants,10 gas chromatography with electron capture detector,11 and UV-vis spectroscopy,12 have been used to measure glibenclamide in biological fluids. However, because of complex matrices, trace levels of the analyte, and low sensitivity of some analytical techniques, a quick and simple method for isolation and pre-concentration of analytes is often essential. For glibenclamide, extraction and pre-concentration methods have been reported, such as solid-phase extraction (SPE),8 solid-phase microextraction (SPME),13 liquid–liquid extraction (LLE),14 liquid–liquid microextraction,15 and magnetic solid-phase extraction (MSPE).16 Magnetic solid-phase extraction (MSPE) has recently received popularity owing to its unique characteristics of ease of separation, the possibility of obtaining a high pre-concentration factor, environmental safety, consumption of small volumes of organic solvent, and high efficiency.17–20 Dispersive magnetic solid phase extraction (DMSPE) is an MSPE mode in which the magnetic sorbent is directly dispersed into sample solutions, resulting in enhanced effective interfacial area between the analytes and sorbent and better mass transfer, which alleviates the possibility of high pressure and frequent column blocking encountered in SPE.21 Furthermore, magnetic sorbents can be easily reused and recycled, making them both cost-effective and eco-friendly.22,23 Until now, different non-magnetic or magnetic sorbents, such as S@SnO2-NPs,13 Fe3O4@PPy NPs,24 CoFe2O4/MCM-48/chitosan,25 and SiO2-coated Fe3O4,26 have been utilized for SPE of glibenclamide from various media.
Molecular imprinting polymers (MIPs) are tailored synthetic receptors with cavities that are chemically or geometrically complementary to the analytes, and thus, they have a great potential for selective or specific binding and extraction of target molecules. However, the MIPs made from traditional methods suffer from broad size distributions, deeply embedded template molecules, low binding rate, slow mass transfer, and challenging elution.27 These limitations can be overcome by the surface imprinting technique.27 Surface imprinted polymers have recognition sites on the support surface that allow them to enjoy the benefits of the ease of accessibility of the cavity by the template molecule, fast mass transfer, ease of elution of the extracted analyte, high binding capacity, and kinetics.28 Different supports, including magnetic nanoparticles,22 polymeric supports,29 titanium dioxide particles,30 carbon nanotubes,31 and silica particles,32 have been used for the synthesis of surface MIPs. Among these supports, the MIPs synthesized on magnetic materials (such as Fe3O4) have the aforementioned advantages typical of the MIPs, as well as the possibility of dispersion of the sorbent in solution, followed by its facile separation with a potent magnetic field.33,34 Magnetic MIPs have been widely utilized as selective recognition materials for targeted analyte separation and extraction in different matrices.
So far, several MIPs with magnetic and non-magnetic natures have been synthesized using single monomers for the extraction of glibenclamide from different media,35–38 but the synthesis of the MIPs with dual monomers has not been reported. Based on recent studies, the use of multiple monomers for MIP synthesis has beneficial effects on its sorption capacity, efficiency, stability, and imprinting factor, as well as its selective performance.39,40
The aim of the present study is the fabrication of a magnetic molecularly imprinted polymer (MMIP) sorbent with high selectivity and stability for the extraction of GLB using two functional monomers of itaconic acid and allylamine. The synthesized MMIP has been characterized in this study by analytical methods of field emission scanning electron microscopy (FESEM), Brunauer–Emmett–Teller (BET) analysis, Fourier transform infrared (FT-IR) spectroscopy, vibrating sample magnetometer (VSM) and X-ray diffraction (XRD) spectroscopy. A dispersive magnetic-micro solid phase extraction (DM-μ-SPE) based on the synthesized MMIP is designed and combined with HPLC or a UV-visible instrument for the separation, extraction, and quantification of GLB. The effectiveness of specific cavities for the extraction of GLB in the designed MMIP was demonstrated by comparing its extraction performance with that of the magnetic non-molecular imprinted polymer (MNIP). To achieve high efficiency of extraction, parameters important for sorption, such as sample volume, solution pH, contact time, analyte concentration, and temperature, are also optimized. Finally, the practical applicability of the designed method for GLB extraction from biological fluids and wastewater is tested. To describe the extraction process, the equilibrium isotherms of the GLB extraction process on the fabricated MMIP are examined.
2. Materials and methods
2.1. Reagents
GLB (99%) was donated by a pharmaceutical company (Tehran Darou Pharmaceutical Company, Iran). Analytical reagent-grade iron(III) chloride, iron(II) sulfate, sodium hydroxide, trisodium citrate, ethanol, ammonium hydroxide, tetraethyl orthosilicate (TEOS), 3-methacryl oxypropyl trimethoxy silane (MPS), monomers of allylamine, and itaconic acid, azobisisobutyronitrile (AIBN), ethylene glycol dimethacrylate (EGDMA), methanol, acetonitrile, tetrahydrofuran, and acetic acid were purchased from Merck Company (Darmstadt, Germany). The solvents used for HPLC analysis were HPLC-grade, and the other chemicals were analytical reagent-grade.2.2. Apparatus
The MMIP and crystallography, morphology, and structure of other nanoparticles were studied by X-ray diffraction (XRD) spectroscopy (Philips-X'pertpro diffractometer, X'pert pro, Tokyo, Japan) with a Cu-Kα radiation source (PerkinElmer) (Model Lambda 25). Other techniques applied were spectrum GX Fourier transforms infrared spectroscopy (FT-IR 410, Jasco Inc., Easton, Maryland, USA) and field emission scanning electron microscopy (FE-SEM) (SIGMA VP-500, ZEISS Company, Germany). The MMIP pore diameter and surface area were studied by the Brunauer–Emmett–Teller (BET) technique using the BELSORP MINI II instrument (BEL Company, Japan). The magnetic character of the synthesized MMIP was determined by a vibrating sample magnetometer (VSM) (LBKFB Meghnatise Kavir, Iran). GLB was quantified by an HPLC with a photodiode array (HPLC-PDA) detector (Waters HPLC separation system, USA) equipped with a Waters 1525 binary pump and C18 column (4.6 × 250 mm; 5 μm, Agilent, USA), with a run time of 8 minutes. The Waters 996 photodiode array (PDA) detector operates in a range of 200 to 600 nm, and the absorbance was measured at a wavelength of 229 nm. Isocratic elution (water:acetonitrile at a ratio of 30:70 v/v and a flow rate of 1 mL min−1) was used for HPLC separation. A Shimadzu (1800, Japan) spectrophotometer was used to record sample absorbance. A magnet of neodymium (15 × 15 × 2.5 cm, 1.5 T) was used to collect the magnetic sorbent from the solution. Water from the Millipore water purification system (Bedford, Massachusetts, USA) was used to prepare the solutions throughout the study unless otherwise stated. The Memert oven (Memert Company, Germany), Heidolph stirrer (Heidolph Company, Germany), Methrohm pH meter (Metrohm, model 691, Switzerland), and ultrasonic cleaning bath (Tecno GAZ SPA; 130 W; 40 kHz frequency, Italy) were utilized throughout experiments.
2.3. Synthesis of the MMIP as a sorbent
Magnetic iron nanoparticles (Fe3O4), SiO2@Fe3O4 nanoparticles, SiO2-MPS@Fe3O4, MMIP, and MNIP were fabricated using the precipitation polymerization method according to previous works with some modification.22,23 The schematic of MMIP synthesis is shown in Fig. 1. The synthesis details are provided in the ESI.† | ||
| Fig. 1 Schematic of the synthesis route of the MMIP. | ||
2.4. MIP-based DM-μ-SPE procedure for the extraction of GLB
The pH of the standard or sample solution of GLB (14.0 mL) with a concentration of less than 2000 μg L−1 was adjusted to 8.0 by diluted NaOH and HCl solutions. Then, 8.0 mg of MMIP was added to it, and the mixture was vortexed for 5 minutes. The MMIP was isolated utilizing a magnet, and the aqueous phase was discarded. The GLB sorbed onto the MMIP was eluted with 500 μL of a methanol and acetic acid (8:2 v/v) mixture while vortexing for 5 minutes. The MMIP was finally isolated by an external magnet. The GLB concentration in the eluent was quantified either by a UV-vis spectrophotometer or HPLC-PDA. For spectrophotometric analysis, the eluent was transferred to a quartz cell, and its absorbance was measured at a wavelength of 229 nm. For analysis with HPLC-PDA, the eluent was dried under an N2 stream, followed by the dissolution of the residue into 100 μL of acetonitrile:water (70:30 v/v). Twenty microliters of it were then injected into the HPLC-PDA system. The extraction efficiency was calculated by the following equation:22
 | (1) |
2.5. Study of sorption isotherms
The sorption mechanism and sorbent capacity were determined by considering the sorption isotherms and comparing determination coefficients (R2). In this regard, under optimal conditions, 8 mg of MMIP was added to 14 mL of GLB solution, with concentrations ranging from 2 to 25 mg L−1 at room temperature. GLB was then extracted by the designed DM-μ-SPE. The obtained data was analyzed by different isotherm models.2.6. Preparation of samples
Human plasma and urine samples were collected from healthy volunteers (aged 30 and 35, respectively) who had not consumed any drugs for the last three months (Kimiya Medical Laboratory, Tehran, Iran). The samples were stored at −20 °C before use. The method was also applied to pharmaceutical wastewater (Tehranchem Pharmaceutical Company, Tehran, Iran) and urban wastewater (Tehran, Iran). The plasma samples were thawed, followed by the addition of 14 mL of acetonitrile and centrifugation for 10 minutes at 10000 rpm. The aqueous layer was then filtered through a 0.45 μm filter, diluted with water (pH = 8.0) in a 1:4 ratio, and treated according to the procedure given in Section 2.4.41 The wastewater and urine samples (14 mL) were centrifuged for 10 minutes at 10000 rpm and filtered through a 0.45 μm filter. The pH was then adjusted to 8.0 and subjected to extraction and analysis.22,23
3. Results and discussion
3.1. Characterization
The FTIR spectra of Fe3O4, SiO2@Fe3O4, SiO2-MPS@Fe3O4, and MMIP were recorded in the range of 400 to 4000 cm−1 (Fig. 2A). The band at 574 cm−1 was attributed to the Fe–O vibration and confirmed the synthesis of Fe3O4 nanoparticles (Fig. 2a). The FTIR spectrum of SiO2@Fe3O4 (Fig. 2b) contained bands at 1098 and 3391 cm−1 as well, which were related to the vibrations of Si–O–Si and –OH groups, respectively, confirming the existence of silica on the Fe3O4 surface, such as the coating of Fe3O4 with silica.42 In Fig. 2c, the appearance of a band at 1630 cm−1 is attributed to the stretching vibration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, indicating the successful graft of MPS on SiO2-MPS@Fe3O4. Furthermore, in Fig. 2d, the presence of bands at 2943 and 3440 cm−1 are related to the C–H group of methyl and N–H stretching vibrations, respectively; they confirm that the MIP layer was formed on the SiO2@Fe3O4 surface.
C bond, indicating the successful graft of MPS on SiO2-MPS@Fe3O4. Furthermore, in Fig. 2d, the presence of bands at 2943 and 3440 cm−1 are related to the C–H group of methyl and N–H stretching vibrations, respectively; they confirm that the MIP layer was formed on the SiO2@Fe3O4 surface.
 | ||
| Fig. 2 (A) FTIR spectra of Fe3O4 (a), SiO2@Fe3O4 (b), SiO2-MPS@Fe3O4 (c), and the MMIP (d); (B) XRD spectra of Fe3O4 (a), SiO2@Fe3O4 (b), and the MMIP (c). | ||
The XRD patterns of Fe3O4, SiO2@Fe3O4, and MMIP are shown in Fig. 2B. The peaks at 2θ of 30.3, 35.5, 43.1, 53.4, 57.0, and 62.6° in the Fe3O4 spectrum correspond to (220), (311), (400), (422), (511) and (440) peaks according to the JCPDS card (19-0629), respectively. The broad diffraction peak in the 2θ range from 20 to 28° is due to the presence of SiO2, affirming the successful grafting of SiO2 on Fe3O4. Moreover, after polymerization and the modification of Fe3O4 with SiO2, its peak shape remained unchanged, confirming that the anti-spinel structure of Fe3O4 was not destroyed by successive synthetic processes.
The SEM images of the MMIP and its components are presented in Fig. 3. As can be seen, the morphologies of the Fe3O4 nanoparticles, SiO2@Fe3O4, and MMIP reveal a relatively uniform size distribution across a nanometric diameter.
 | ||
| Fig. 3 FESEM images of SiO2@Fe3O4 (A), SiO2-MPS@Fe3O4 (B), and the MMIP (C). | ||
N2 adsorption–desorption analysis was used to evaluate surface areas, pore volumes, and pore sizes of MMIP and MNIP (Fig. 4A and B). Pore size distribution and isotherms of N2 adsorption–desorption for MMIP and MNIP are provided in Table S1.† The surface areas were determined by the multipoint Brunauer–Emmett–Teller (BET) technique, utilizing adsorption data as a function of relative pressure. The N2 adsorption analysis presented an isotherm of type (IV) with an H3-type hysteresis loop, suggesting a highly ordered mesoporous MMIP according to the IUPAC. The pore size distribution of the MMIP based on Barrett–Joyner–Halenda (BJH) analysis was 1.21 nm, signifying a narrow pore size distribution. The specific surface area, total pore volume, and average pore diameter of the MMIP were 116.85 m2 g−1, 0.458 cm3 g−1, and 15.673 nm, respectively, while those of the MNIP were 67.64 m2 g−1, 0.292 cm3 g−1, and 12.299 nm. Thus, it is assumed that the MMIP possesses an adequate surface area for utilization as a sorbent in GLB extraction. Furthermore, the utilization of the porous MMIP enables polymeric networks to sorb the target template more quickly and attain equilibrium rapidly.
 | ||
| Fig. 4 N2 adsorption–desorption isotherm analysis of the MMIP (A) and MNIP (B); VSM of Fe3O4 and the MMIP (C). | ||
The magnetic properties of Fe3O4 and the MMIP were investigated by a vibrating sample magnetometer (VSM) at room temperature (Fig. 4C). The values of saturation magnetizations were 91.84 and 26.06 emu g−1 for Fe3O4 and the MMIP, respectively. The value of saturation magnetization of the MMIP was lower than that of Fe3O4 due to the coating of the Fe3O4 surface with a SiO2 layer, MPS, and the molecularly imprinted polymer. Although the deposition of an imprinted polymer layer on the Fe3O4 surface led to a reduction in its saturation magnetization value, the MMIP could still be rapidly isolated by a magnet within 30 seconds.
The value of pHpzc for the MMIP was determined by mixing 0.01 g of the prepared MMIP with 2 mL of NaCl solution (0.1 mol L−1) in the pH ranges of 2.0–10.0. The mixture was kept at room temperature for 24 hours, and the final pH (pHf) was measured. The pHpzc of the sorbent was found to be 4.76 based on the plot of the initial pH of the solutions (pHi) against the corresponding ΔpH = (pHi − pHf) (Fig. S2†).43
3.2. Optimization of extraction conditions
The parameters affecting the DM-μ-SPE procedure and recovery of GLB were optimized using the one-at-a-time method, which was repeated thrice. For optimization, the extracted analyte was quantified by a UV-vis spectrophotometer. To study the analytical performance and applicability of the method, the extracted GLB was quantified by HPLC-PDA, as well as a UV-visible spectrophotometer.:1, 8:2, and 7:3 v/v) were investigated on the elution of GLB from the MMIP. As illustrated in Fig. S4B,† the highest GLB recovery was obtained with a mixture of methanol and acetic acid (8:2 (v/v)). It was observed that methanol cannot effectively elute GLB from the MMIP, and acetic acid plays a crucial role in GLB desorption. Specifically, acetic acid protonates GLB and helps break hydrogen bonds between GLB molecules and the amino-functional group of MMIP. Hence, a mixture of methanol and acetic acid (8:2 v/v) was selected as the ideal eluent in subsequent experiments.The influence of eluent volume within a range of 300 to 700 μL was investigated on the recovery of GLB. The results (Fig. S4C†) show that 500 μL of the mixture of methanol and acetic acid (8:2 v/v) is sufficient for the complete desorption of GLB from the sorbent. At higher volumes, signals decrease due to the increase in dilution of the analyte, but the extraction percentage remains almost constant. Thus, 500 μL was selected as the optimal volume for desorption.
 | ||
| Fig. 5 Effect of sorption time (A), desorption time (B), solution pH (C), and temperature on the recovery of glibenclamide (D). Conditions: MMIP mass, 8 mg; eluent, 500 μL; methanol/acetic acid (8:2 v/v); concentration of GLB, 250 μg L−1; and sample volume, 10 mL. | ||
In DM-μ-SPE, desorption time is an important factor affecting the recovery of the analytes from the sorbent. To find optimal desorption time, the eluent was added to the sorbent containing the analyte, and the mixture was vortexed for 1 to 7 minutes. Fig. 5B confirms that a 5 minute vortex is enough for the complete recovery of GLB from the MMIP sorbent.
3.3. Comparison of GLB extraction with the MMIP and MNIP
To compare the performance of synthesized MMIP and MNIP polymers, GLB was extracted in both polymers under optimal conditions. The results showed that extraction percentages for GLB by the MMIP and MNIP were 93.5 and 15.5%, respectively. The MMIP is imprinted well and can selectively extract GLB. The low GLB sorption by MNIP can be due to the non-selective sorption by the polymer.3.4. Selectivity test
The main advantage of MIPs is their selectivity. The selectivity of the MIP is dependent on the shape, size, and functional groups of the target analyte that are considered during the synthesis of the polymer structure.36 The selectivity of the synthesized MMIP for GLB in comparison to other antidiabetic drugs, such as pioglitazone, glipizide, and rosiglitazone, was assessed, and extraction recoveries for all were found to be 93.60, 7.10, 13.02, and 8.14%, respectively. These results confirm the high selectivity of the prepared MMIP and the method developed here for the determination of GLB.3.5. Evaluation of MMIP reusability
The reusability of the MMIP without losing its performance efficiency was evaluated from an economic standpoint. The reusability of an MIP depends on the attachment of incoming monomers to the polymer network and degree of polymer cross-linking. To investigate this aspect, the MMIP was collected after each extraction cycle, rinsed, dried, and used again in the next cycle. It was found that after 7 cycles, the extraction efficiency decreased slightly (93.5 to 92.0%), indicating the good chemical stability and appropriate quality of MMIP.3.6. Non-linear isotherm studies
Isotherms play a very important role in predicting and modeling absorption systems containing a sorbate and sorbent. To investigate the mechanism of sorption, 8 mg of MMIP was added to 14 mL of GLB solution, with concentrations ranging from 2 to 25 mg L−1 at room temperature. The resulting data were fitted to Langmuir, Freundlich, and Temkin isotherm models.The non-linear form of the Langmuir isotherm model can be described as follows:46
| qe = qmaxKLCe/(1 + KLCe) | (2) |
Freundlich's sorption isotherm non-linear form is:46
| qe = KFCe1/n | (3) |
The Temkin isotherm non-linear form is represented as follows:46
|
qe = RT/bln(ACe)
| (4) |
The data from the non-linear plot of above-mentioned sorption isotherms are presented in Fig. 6 and Table 1. Based on the regression coefficient (R2) and error analysis function (sum of squares of errors  ), the Langmuir isotherm model shows a high R2 (0.9947) with a low SSE value (0.614), indicating that GLB sorbs onto the MMIP surface as a homogenous monolayer.46 Based on the Langmuir isotherm model, the maximum sorption capacity of the sorbent was found to be qmax = 24.260 mg g−1. In addition, the 1/n value for GLB sorption was less than 1 (1/n = 0.360), indicating a favorable sorption process.
), the Langmuir isotherm model shows a high R2 (0.9947) with a low SSE value (0.614), indicating that GLB sorbs onto the MMIP surface as a homogenous monolayer.46 Based on the Langmuir isotherm model, the maximum sorption capacity of the sorbent was found to be qmax = 24.260 mg g−1. In addition, the 1/n value for GLB sorption was less than 1 (1/n = 0.360), indicating a favorable sorption process.
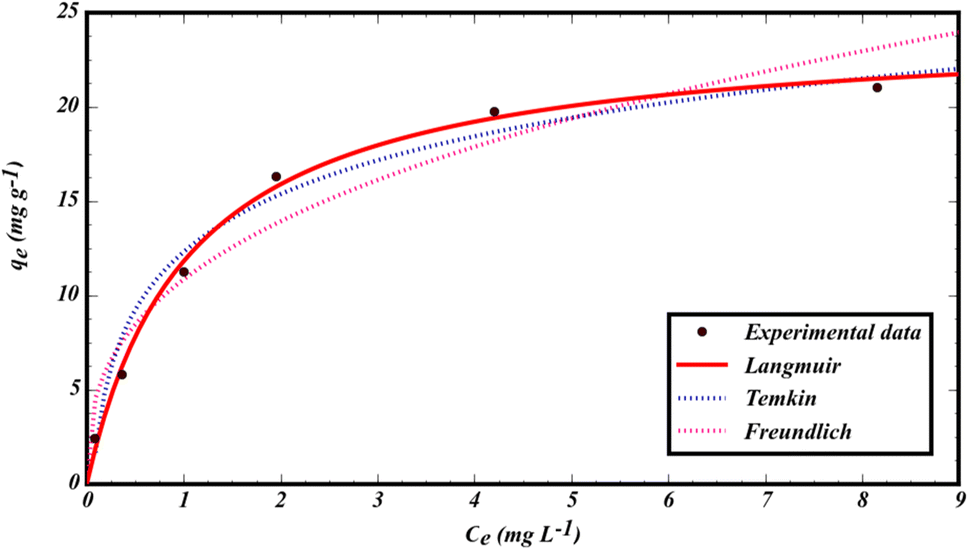 | ||
| Fig. 6 Non-linear isotherm plots for the sorption of GLB onto the MMIP. | ||
| Model | Parameter | Value |
|---|---|---|
| Langmuir | qmax (mg g−1) | 24.260 |
| KL (L mg−1) | 0.946 | |
| R2 | 0.9947 | |
| SSE | 0.614 | |
| Freudlich | N | 2.774 |
| KF (L1/n mg1−1/n g−1) | 1.083 | |
| R2 | 0.9315 | |
| SSE | 2.221 | |
| Temkin | A (L g−1) | 16.127 |
| B | 4.420 | |
| R2 | 0.9688 | |
| SSE | 1.499 |
The empirical sorption capacity is obtained using the following equation:47
 | (5) |
In this equation, Ce, C0, V, and W, are the equilibrium, and initial GLB concentrations (mg L−1), volume of solution (L), and sorbent mass (g), respectively. Based on this equation, the experimental capacity was found to be 21.05 mg g−1. The closeness of qmax (24.260 mg g−1) to qexp (21.05 mg g−1) implies the best adaptation of the sorption process to the Langmuir model. Thus, the rate-controlling step is sorption to the surface of the MMIP.48
The imprinting factor (IF) describes the tendency of the MMIP for selective identification and sorption affinity of recognition sites toward the imprinted GLB molecule. It is calculated by the following equation:22
 | (6) |
3.7. Thermodynamic studies
Thermodynamic studies were performed at four temperatures of 10, 20, 30, and 40 °C under optimal conditions to determine the Gibbs free energy (ΔG), enthalpy (ΔH), and entropy (ΔS). These were determined using following equations:23
 | (7) |
|
ΔG° = −RTlnKC
| (8) |
 | (9) |
According to results presented in Table 2, negative values of ΔG° at all temperatures and positive values of the enthalpy indicate that the sorption process was spontaneous and endothermic. Additionally, positive entropy changes in sorption indicate an increase in disorder at the sorbent–solution interface during sorption.
| Temperature (K) | lnKc |
ΔG° (kJ mol−1) | ΔH° (kJ mol−1) | ΔS° (kJ mol−1 K−1) |
|---|---|---|---|---|
| 283.15 | 0.11 | −0.26 | +84.09 | +0.2991 |
| 293.15 | 1.71 | −4.18 | ||
| 303.15 | 2.59 | −6.54 | ||
| 313.15 | 3.61 | −9.40 |
3.8. Analytical performance of the DM-μ-SPE method developed
The analytical performance of the method was investigated under optimal conditions. The calibration equation for the UV-vis spectrophotometric method was A = 0.219C + 0.0059, with a linear range of 12 to 2000 μg L−1, a regression coefficient of 0.9965, and a detection limit of 4 μg L−1. For the HPLC-PDA analysis, the calibration equation was A = 299519C + 7043.9 in the range of 1–2000 μg L−1 with a detection limit of 0.3 μg L−1. In these equations, A and C (μg L−1) denote the solution absorbance and GLB concentration in the aqueous phase, respectively. The limit of quantification (LOQ) and limit of detection (LOD) for GLB were calculated using formulas LOQ = 10Sb/m and LOD = 3Sb/m, respectively. Here, Sb represents the standard deviation of five replicates of blank measurement, and m refers to the slope of the calibration curve after extraction. The determined LOD and LOQ were 0.3 and 1.0 μg L−1, respectively. Moreover, relative intra and inter-standard deviations for five consecutive measurements at 500 μg L−1, using the designed DM-μ-SPE method, followed by UV-vis analysis were 5.1 and 5.7, respectively. These values were 3.2 and 4.1%, respectively, for the HPLC-PDA analysis. The pre-concentration factor, defined as the volume ratio of the sample (14.0 mL) to the eluent volume (100 μL) was 140.0. The enhancement factor (i.e., the ratio of the slope of calibration curves with and without pre-concentration) was 135.1, indicating that the recovery was more than 96%.3.9. Analysis of real samples
The synthesized MMIP was tested for the selective sorption of GLB by the developed method in plasma, urine, and wastewater samples. The accuracy of the developed method was studied by a recovery experiment, whereby samples were spiked with GLB at three levels: 25, 50, and 500 μg L−1 of GLB. As presented in Table 3, recoveries were in the acceptable ranges of 89.4–102.9%. These results show that the developed method has a good ability for selective extraction in complex real samples. The chromatograms for wastewater, urine, and plasma samples before and after spiking are shown in Fig. S5.†| Sample | Added (μg L−1) | Found (μg L−1) ± SDa (%) | ER (%) | RSDb (%) |
|---|---|---|---|---|
| a Average and standard deviation of three independent measurements.b Relative standard deviation. | ||||
| Plasma 1 | — | Not detected | — | — |
| 25 | 23.9 ± 0.2 | 95.6 | 0.8 | |
| 250 | 223.5 ± 3.0 | 89.4 | 1.3 | |
| 500 | 461.5 ± 17.1 | 92.3 | 3.7 | |
| Plasma 2 | — | Not detected | — | — |
| 25 | 24.1 ± 0.2 | 96.4 | 0.8 | |
| 250 | 230.2 ± 3.6 | 92.0 | 1.5 | |
| 500 | 489.5 ± 16.1 | 97.9 | 3.2 | |
| Urine 1 | — | Not detected | — | — |
| 25 | 24.6 ± 0.1 | 98.4 | 0.4 | |
| 250 | 246.2 ± 3.0 | 98.4 | 1.2 | |
| 500 | 490.0 ± 12.8 | 98.0 | 2.6 | |
| Urine 2 | — | Not detected | — | — |
| 25 | 23.6 ± 0.12 | 94.4 | 0.8 | |
| 250 | 239.0 ± 2.4 | 95.6 | 1.0 | |
| 500 | 465.5 ± 9.8 | 93.1 | 2.1 | |
| Pharmaceutical wastewater | — | 13.5 ± 0.1 | — | 0.7 |
| 25 | 37.5 ± 0.5 | 97.4 | 1.3 | |
| 250 | 265.1 ± 6.9 | 100.6 | 2.6 | |
| 500 | 528.5 ± 17.0 | 102.9 | 3.2 | |
| Urban wastewater | — | Not detected | — | — |
| 25 | 23.5 ± 0.2 | 94.0 | 0.8 | |
| 250 | 239.0 ± 2.4 | 95.6 | 1.0 | |
| 500 | 460.0 ± 6.5 | 92.0 | 1.4 | |
3.10. Comparison with similar methods for GLB extraction
The analytical characteristics of the DM-μ-SPE-HPLC/PDA method were compared to methods previously reported in the literature for the pre-concentration and extraction of glibenclamide in different sample matrices (Table 4). The present method demonstrated excellent extraction efficiencies. LODs achieved in this study were lower or comparable to those achieved by previously reported methods. In terms of extraction time, the suggested method reaches sorption equilibrium faster than other comparable methods. Additionally, the sample volume required, recovery, and RSD of this method are comparable to other methods reported in Table 4.| Sorbent | Extraction technique | Determination technique | Matrix | PFa | Desorption time (min) | Sample volume (mL) | LRb (μg L−1) | LODc (μg L−1) | Recovery (%) | RSDd (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Pre-concentration factor.b Linear range.c Limit of detection.d Relative standard deviation.e Polymerization Fe3O4@GO@SiO2-OH.f Polypyrrole modified magnetic nanoparticles.g Hollow porous molecularly imprinted polymer dispersive solid phase extraction.h Glibenclamide-surface molecularly imprinted polymers. | |||||||||||
| MGTPIIe | MSPE | HPLC-UV | Granules, tablets, and teas | — | 30 | 10 | 500–800 | 50.0 | 80.02–87.28 | ≤1.97 | 16 |
| Fe3O4@PPy NPsf | MSPE | HPLC-UV | Human serum and urine | 75–122 | 5 | 20 | 0.2–700 | 0.1 | 92.0–102.5 | 3.9 | 24 |
| HPMIP-d-SPEg | SPE | HPLC-UV | Human urine | — | 15 | 10 | 10.0–3000.0 | 3.5 | 87.7–104.3 | 2.3–4.4 | 36 |
| MIP | SPE | HPLC-UV | Water | 155 | 30 | 50 | 500–2500 | 40.0 | 63.4–87.1 | 1.76–4.98 | 37 |
| Gb-SMIPsh | DSPE | HPLC-UV | Health foods | — | <60 | 5 | 5–2000 | 1.56 | 81.46–93.53 | <4.07 | 38 |
| RP-18 and RP-8 cartridges | SPE | HPLC-UV | Plasma | — | 8 | 1 | 10–400 | 5.0 | 97.0–104.0 | <9.5 | 8 |
| Oasis® HLB cartridges | SPE | HPLC-UV | Plasma | — | 6.2 | 1 | 25–1000 | 4.0 | 81.8–87.3 | 0.99 | 49 |
| Magnetic CoFe2O4/MCM-48/chitosan | D-μ-SPE | HPLC-PDA | Urine, plasma, and water | — | 7 | 30 | 0.7–520 | 0.21 | 89.6–95.8 | 4.4–6.2 | 50 |
| MMIP | DM-μ-SPE | HPLC-PDA | Urine, plasma, and wastewater | 140.0 | 5 | 14 | 1–2000 | 0.3 | 89.4–102.9 | <3.7 | This work |
| UV-vis | 28.0 | 12–2000 | 4.0 | <5.3 | |||||||
4. Conclusions
In this research, a new MMIP was successfully synthesized using two functional monomers (itaconic acid and allylamine). It was used as an effective sorbent in DM-μ-SPE for the separation/pre-concentration and determination of GLB in trace amounts in plasma, wastewater, and urine samples. The MMIP was characterized by various methods. Under optimized conditions, the MMIP featured high selectivity and good sorption capacity for GLB. The maximum amount of GLB is extracted at pH = 8.0 at 40 °C within 5 minutes. The suggested method accurately determines GLB and offers a broad linear range; quick extraction time; and acceptable LOD, PF, and RSD. The impressive extraction efficiency of the synthesized sorbent can be attributed to multiple mechanisms, such as hydrogen bonding and electrostatic interactions between the MMIP and GLB molecules. It has been demonstrated that the GLB sorption process fits well to the Langmuir model, and thermodynamic calculations corroborated that the GLB sorption onto the MMIP occurs by an endothermic, spontaneous process. It was also demonstrated that the extracted GLB can be accurately determined by the simple method of UV-vis spectrophotometry, provided its concentration is in the range of 12–2000 μg L−1. Furthermore, the suggested technique enjoys good efficiency, simplicity, cost-effectiveness, and swiftness and can be used to enrich and determine the content of GLB in various real samples.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors appreciate the financial support from Yazd University, Iran.References
- T. N. Nanovskaya, I. Nekhayeva, G. D. Hankins and M. S. Ahmed, Biochem. Pharmacol., 2006, 72, 632–639 CrossRef CAS PubMed.
- F. Akhtar, S. Gul, S. Ashfaq, I. Rehman and A. Z. Mirza, J. Anal. Test., 2020, 4, 281–290 CrossRef.
- L. Azharshekoufeh, J. Shokri, M. Barzegar-Jalali and Y. Javadzadeh, Bioimpacts, 2017, 7, 5 CrossRef CAS PubMed.
- M. Gazizadeh, G. Dehghan and M. Amjadi, Luminescence, 2019, 34, 297–303 CrossRef CAS PubMed.
- N. J. Niemuth and R. D. Klaper, Chemosphere, 2015, 135, 38–45 CrossRef CAS PubMed.
- B.-M. Chen, Y.-Z. Liang, F.-Q. Guo, L.-F. Huang, F.-L. Deng, X. Chen and Y.-L. Wang, Anal. Chim. Acta, 2004, 514, 185–191 CrossRef CAS.
- N. González, S. P. L. Corral, A. G. Lista and C. C. Acebal, Microchem. J., 2018, 142, 288–296 CrossRef.
- J. V. Santurio and E. G. Porto, J. Chromatogr. B, 1996, 682, 364–370 CrossRef PubMed.
- F. Albu, C. Georgiţă, V. David and A. Medvedovici, J. Chromatogr. B, 2007, 846, 222–229 CrossRef CAS PubMed.
- V. Maier, J. Znaleziona, D. Jirovský, J. Skopalová, J. Petr and J. Ševčík, J. Chromatogr. A, 2009, 1216, 4492–4498 CrossRef CAS PubMed.
- P. Hartvig, C. Fagerlund and O. Gyllenhaal, J. Chromatogr. B, 1980, 181, 17–24 CrossRef CAS PubMed.
- M. R. Sohrabi, N. Kamali and M. Khakpour, Anal. Sci., 2011, 27, 1037–1041 CrossRef CAS PubMed.
- O. Eilami, A. Asfaram, M. Ghaedi and A. Goudarzi, Anal. Methods, 2019, 11, 627–634 RSC.
- I. Niopas and A. C. Daftsios, J. Pharm. Biomed. Anal., 2002, 28, 653–657 CrossRef CAS PubMed.
- C. M. Monzón, C. M. Teglia, M. R. Delfino and H. C. Goicoechea, Microchem. J., 2016, 127, 113–119 CrossRef.
- Z.-y. Wu, X.-y. Li, M. Liu, F. Wang, L. Liu, X.-c. Tan and F.-h. Lei, New J. Chem., 2017, 41, 12535–12540 RSC.
- M. Safari, Y. Yamini, A. Mani-Varnosfaderani and H. Asiabi, J. Iran. Chem. Soc., 2017, 14, 623–634 CrossRef CAS.
- W. Zhang, Y. Zhang, Q. Jiang, W. Zhao, A. Yu, H. Chang, X. Lu, F. Xie, B. Ye and S. Zhang, Anal. Chem., 2016, 88, 10523–10532 CrossRef CAS PubMed.
- L.-S. Qing, Y. Xue, Y.-M. Liu, J. Liang, J. Xie and X. Liao, J. Agric. Food Chem., 2013, 61, 8072–8078 CrossRef PubMed.
- Q. Gao, D. Luo, M. Bai, Z.-W. Chen and Y.-Q. Feng, J. Agric. Food Chem., 2011, 59, 8543–8549 CrossRef CAS PubMed.
- B. Socas-Rodríguez, A. V. Herrera-Herrera, M. Asensio-Ramos and J. Hernández-Borges, Analytical Separation Science, Wiley-VCH, Germany, 2015, pp. 1525–1570 Search PubMed.
- N. Sabbaghi, A. M. Haji Shabani, S. Dadfarnia and M. Farsadrooh, Mikrochim. Acta, 2023, 190, 164 CrossRef CAS PubMed.
- N. Azizi-Khereshki, H. Z. Mousavi, M. G. Dogaheh, M. Farsadrooh, N. Alizadeh and A. Mohammadi, Spectrochim. Acta, Part A, 2023, 296, 122656 CrossRef CAS PubMed.
- A. Tavousi, S. Ahmadi and P. Zohrabi, IET Nanobiotechnol., 2019, 13, 503–509 CrossRef.
- Z. Bagheri Zomoorodi, M. Masrournia and M. R. Abedi, J. Iran. Chem. Soc., 2022, 19, 3637–3647 CrossRef CAS.
- K. C. de Souza, G. F. Andrade, I. Vasconcelos, I. M. de Oliveira Viana, C. Fernandes and E. M. B. de Sousa, Mater. Sci. Eng., C, 2014, 40, 275–280 CrossRef CAS PubMed.
- Y. Yuan, Y. Liu, W. Teng, J. Tan, Y. Liang and Y. Tang, J. Chromatogr. A, 2016, 1462, 2–7 CrossRef CAS PubMed.
- T. Anirudhan, J. Christa and J. Deepa, Food Chem., 2017, 227, 85–92 CrossRef CAS PubMed.
- J. Pan, H. Yao, W. Guan, H. Ou, P. Huo, X. Wang, X. Zou and C. Li, Chem. Eng. J., 2011, 172, 847–855 CrossRef CAS.
- W.-z. Xu, W. Zhou, P.-p. Xu, J.-m. Pan, X.-y. Wu and Y.-s. Yan, Chem. Eng. J., 2011, 172, 191–198 CrossRef CAS.
- D. Xiao, P. Dramou, N. Xiong, H. He, D. Yuan, H. Dai, H. Li, X. He, J. Peng and N. Li, Analyst, 2013, 138, 3287–3296 RSC.
- J. Ma, L. Yuan, M. Ding, S. Wang, F. Ren, J. Zhang, S. Du, F. Li and X. Zhou, Biosens. Bioelectron., 2011, 26, 2791–2795 CrossRef CAS PubMed.
- H. Gholami, M. Arabi, M. Ghaedi, A. Ostovan and A. R. Bagheri, J. Chromatogr. A, 2019, 1594, 13–22 CrossRef CAS PubMed.
- X. Fu, D. Zhu, L. Huang, X. Yan, S. Liu and C. Wang, Microchem. J., 2019, 150, 104169 CrossRef CAS.
- X. Ren and X. Li, Anal. Methods, 2020, 12, 2858–2864 RSC.
- A. Ostovan, M. Ghaedi, M. Arabi and A. Asfaram, J. Chromatogr. A, 2017, 1520, 65–74 CrossRef CAS PubMed.
- R. Lahsini, M. R. Louhaichi, N. Adhoum and L. Monser, Acta Pharm., 2013, 63, 265–275 CAS.
- R. Wang, Y. Wang, C. Xue, T. Wen, J. Wu, J. Hong and X. Zhou, J. Sep. Sci., 2013, 36, 1015–1021 CrossRef CAS PubMed.
- U. D. Thach, H. H. Nguyen Thi, T. D. Pham, H. D. Mai and T.-T. Nhu-Trang, Polymers, 2021, 13, 2788 CrossRef CAS PubMed.
- A. A. Elaine, S. I. Krisyanto and A. N. Hasanah, Polymers, 2022, 14, 3498 CrossRef CAS PubMed.
- A. A. Pebdani, A. M. H. Shabani and S. Dadfarnia, J. Iran. Chem. Soc., 2016, 13, 155–164 CrossRef CAS.
- A. B. Shatan, K. Venclíková, B. A. Zasońska, V. Patsula, O. Pop-Georgievski, E. Petrovský and D. Horák, Pharm. Res., 2019, 36, 1–12 CrossRef CAS PubMed.
- K. Y. Urbain, K. Ollo, K. K. K. Gains and T. Albert, Am. J. Appl. Chem., 2021, 9, 207–214 CrossRef.
- X. Wu, X. Wang, W. Lu, X. Wang, J. Li, H. You, H. Xiong and L. Chen, J. Chromatogr. A, 2016, 1435, 30–38 CrossRef CAS PubMed.
- M. M. Moein, R. Said, F. Bassyouni and M. Abdel-Rehim, J. Anal. Methods Chem., 2014, 2014 Search PubMed.
- J. Wang and X. Guo, Chemosphere, 2020, 258, 127279 CrossRef CAS PubMed.
- R. M. Toudeshki, S. Dadfarnia and A. M. H. Shabani, Anal. Chim. Acta, 2019, 1089, 78–89 CrossRef CAS PubMed.
- N. Kataria and V. Garg, J. Environ. Chem. Eng., 2017, 5, 5420–5428 CrossRef CAS.
- S. AbuRuz, J. Millership and J. McElnay, J. Chromatogr. B, 2005, 817, 277–286 CrossRef CAS PubMed.
- Z. Bagheri Zomoorodi, M. Masrournia and M. R. Abedi, J. Iran. Chem. Soc., 2022, 19, 3637–3647 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra00452c |
| This journal is © The Royal Society of Chemistry 2024 |