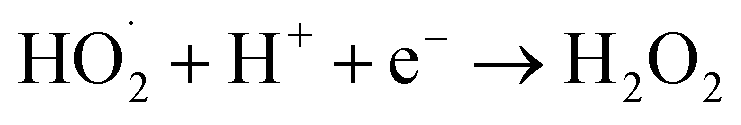DOI:
10.1039/D4RA00366G
(Paper)
RSC Adv., 2024,
14, 14784-14792
Kill two birds with one stone: simultaneous removal of volatile organic compounds and ozone secondary pollution by a novel photocatalytic process†
Received
14th January 2024
, Accepted 2nd May 2024
First published on 7th May 2024
Abstract
Volatile organic compounds (VOCs) originating from diverse sources with complex compositions pose threats to both environmental safety and human health. Photocatalytic treatment of VOCs has garnered attention due to its high efficacy at room temperature. However, the intricate photochemical reaction generates ozone (O3), causing secondary pollution. Herein, our work developed a novel “synergistic effect” system for photocatalytic co-treatment of VOCs and O3 secondary pollution. Under the optimized reactor conditions simulated with computational fluid dynamics (CFD), MgO-loaded g-C3N4 composites (MgO/g-C3N4) were synthesized as efficient catalysts for the photocatalytic synergistic treatment process. Density functional theory (DFT) calculations, characterization, and electron paramagnetic resonance (EPR) tests revealed that the addition of MgO reduced the band gap of g-C3N4, and increased O3 molecule adsorption in the composites, efficiently harnessing the synergistic effect of O3 to generate a significant quantity of reactive oxygen radicals, thereby facilitating the removal of VOCs and O3. This study provides new insights for simultaneous elimination of VOCs and O3 secondary pollution by a photocatalytic process.
1. Introduction
In recent years, the emission of volatile organic pollutants (VOCs), precursors to fine particulate matter and photochemical smog, has surged significantly due to the extensive combustion of fuels, coupled with rapid industrial and transportation growth.1,2 VOCs defined by the World Health Organization (WHO) as gaseous air pollutants with a boiling point of 50–260 °C, include acids, ketones, benzene, and nitrogen-containing, and oxygen-containing organics.3,4 These compounds, known for their volatility, toxicity, and uncontrolled emissions, constitute a menace to the well-being of the general population.5–8 For these rationales, the eradication of these pollutants is regarded as an imperative practical research subject. The main control strategies for VOCs generally consist of recovery methods such as condensation, absorption, and membrane separation.9–11 Various elimination procedures, including catalytic combustion, low-temperature plasma, and biodegradation, are also utilized.12–16 However, these methods frequently encounter disadvantages including the generation of additional contaminants, exorbitant expenses, heightened energy usage, and intricate operational protocols. Hence, there is a significant demand for the exploration of an efficient and straightforward method to eliminate VOCs under facile and mild conditions.
Photocatalytic technology, a component of the advanced oxidation processes (AOPs), stands out as a highly promising method for efficiently degrading VOCs under ambient conditions by generating reactive oxygen species (ROS), such as hydroxyl radicals (˙OH) and superoxide radicals (˙O2−), which possess strong oxidizing properties and facilitate the transformation of VOCs into carbon dioxide and water.17–20 Nevertheless, the utilization of this technique is greatly limited by problems related to the non-properly designed reactor, deactivation of the catalyst and its low effectiveness. The commonly used 185 nm UV irradiation generates high-energy photons that commence the breakdown of oxygen and water, leading to the formation of reactive oxygen and ˙OH.21–25 This process efficiently resolves the trade-off between efficiency and catalyst deactivation. However, the utilization of 185 nm UV irradiation in photocatalysis, is more prone to induce the production of ozone (O3), potentially leading to secondary pollution.26,27 Hence, it is imperative to tackle O3 generation during 185 nm UV photocatalysis by implementing in situ catalytic or post-treatment measures.28,29 Moreover, owing to its robust oxidizing capability, O3 finds widespread application in the mitigation of diverse pollutants, encompassing wastewater treatment, exhaust gas purification, and microbial inactivation.30–32 Consequently, the prospect of achieving synergistic removal of O3 and VOCs through the utilization of in situ generated O3 in the photocatalytic process is anticipated.
Developing appropriate photocatalysts is crucial for achieving the simultaneous elimination of VOCs and O3 secondary pollution through synergistic processes. Due to its narrow band gap of 2.7 eV, appropriate redox potential, and strong stability, graphite-carbon nitride (g-C3N4) is extensively employed in the field of photocatalysis. Nevertheless, the limited surface area and rapid formation of photogenerated carrier complexes in pure g-C3N4 impede its broader use in photocatalysis.33,34 Moreover, pure g-C3N4 cannot be directly utilized for the production of active substances from O3. To tackle these issues, specialists have improved the application of g-C3N4 in photocatalysis by making changes to its shape and structure, incorporating different elements, and creating heterojunctions.35,36 These enhancements aim to reduce the rate at which photogenerated carriers form complexes, thereby improving the practical use of g-C3N4. Muniandy et al. employed a catalyst consisting of copper-modified graphite carbon nitride nanosheets (Cu-g-C3N4) to successfully carry out the hydroxylation of benzene under mild conditions, utilizing H2O2 as an oxidizing agent.37 Li et al. synthesized g-C3N4@CsxWO3 nanocomposites that showed exceptional VOCs breakdown capabilities when exposed to a wide range of light wavelengths, including UV, visible, and near infrared ray (NIR).38 Nevertheless, the previous researches have primarily concentrated on investigating the exceptional characteristics and reaction mechanisms of the created substances, while giving less attention to the possible ramifications of O3 secondary pollution throughout the reaction procedure and its associated functions. Furthermore, multiple investigations have shown that the synergistic effect of combining the oxidizing and adsorbing properties of magnesium oxide (MgO) with O3 leads to an enhanced conversion of O3 into ˙OH, hence increasing the catalytic activity of O3.39 Agarkoti et al. employed the characteristics of MgO to expedite the breakdown of O3, proved successful in treating CETP effluents.40 MgO possesses favourable characteristics including strong surface reactivity, affordability, and environmental compatibility, rendering it a promising contender.41
Hence, this study explored a novel photocatalytic system with “synergistic effect” for co-treatment of VOCs and O3 secondary pollution. Computational fluid dynamics (CFD) simulation tools were employed to model airflow rates and the distribution of UV lamp radiation within the photocatalytic reaction unit, offering a theoretical basis for rational reactor design. Exploiting the respective advantages of MgO and g-C3N4, the composite catalyst (MgO/g-C3N4) was synthesized, and subsequently fixed onto a cordierite carrier for the purpose of catalyzing the removal of toluene and in situ secondary generated O3 at ambient temperature. Systematic adjustments of factors such as relative humidity, residence time, initial concentrations, and UV radiation were made to investigate optimal conditions. The reaction mechanism of the synergistic treatment of VOCs and O3 using UV/MgO/g-C3N4 photocatalytic process was explored using material characterization tests, free radical determination, and density functional theory (DFT) calculations.
2. Experimental
Detailed information on materials, CFD simulation, catalysts synthesis, photocatalytic degradation processes, characterization and DFT calculation can be found in the ESI file.†
3. Results and discussion
3.1. Reactor optimization
To optimize the structure of the UV reactor, a more widely utilized model from existing engineering actual models was chosen for enhanced simulation. As shown in Fig. 1a, the airflow enters the chamber through the intake indentation in traditional equipment. While some of the airflow moves in the radial direction, it is mostly focused in the horizontal direction. Consequently, the airflow is not uniformly dispersed across the whole chamber. To solve this problem, a gas equalizer was introduced into the inlet indentation of the reactor (Fig. 1b) to disperse the gas flow at the entrance of the reaction system. This dispersion, facilitated by the gas equalizer, ensures a more even distribution of the gas flow within the reaction chamber. When viewed in a three-dimensions (Fig. 1c & d), the gas distribution is consistent with that described above. The inlet gas stream in the enhanced equipment exhibits a more uniform distribution within the chamber, leading to a reduction in the lateral velocity of the gas stream. This decrease in lateral flow velocity helps mitigate eddy currents around the catalyst carrier. To comprehend the dispersion of UV light within the chamber, an examination of the optical radiation emitted by the reactor was performed in a supplementary simulation. A part of it was intercepted for visualization to facilitate the analysis. The UV radiation is evenly distributed across the reaction chamber, as depicted in Fig. 1e & f, where the intensity of radiation from the UV lamp remains strong even at a certain distance from the UV lamp. Hence, the photocatalytic reactor possessing a gas equalizer was constructed to perform the following experiments. The model schematic and actual photos of the photoreactor (including the gas equalizer) are shown in Fig. S2.†
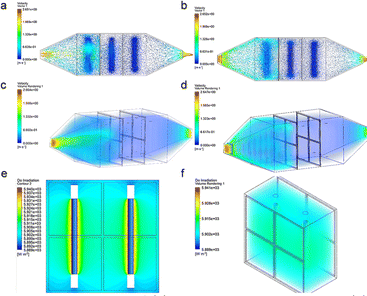 |
| | Fig. 1 Fluid trajectory of (a) conventional equipment and (b) improved equipment, spatial distribution of fluids in (c) conventional equipment and (d) improved equipment, (e and f) reactor radiation simulation. | |
3.2. Catalyst characterizations
Morphologies of the samples were analyzed by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). As shown in Fig. 2a and S3,† g-C3N4 exhibits a clear layered structure and is in the form of thin sheets. The pure MgO exhibits typical granular shape (Fig. 2b). From the SEM of 0.75MgO/g-C3N4, it can be seen that the MgO particles are loaded on the surface of g-C3N4 (Fig. 2c), ensuring the intimate electrical contact between MgO and g-C3N4. In addition, the TEM image of 0.75MgO/g-C3N4 (Fig. 2d) indicates that with the introduction of MgO, the basic structure of g-C3N4 remains unchanged and remains lamellar structure. In order to confirm the successful loading of MgO onto g-C3N4, the EDS characterization of the 0.75MgO/g-C3N4 was conducted, as depicted in Fig. 2f–i. The plots in Fig. 2h and i demonstrate the even distribution of O and Mg on the material surface, suggesting that MgO has effectively bonded with g-C3N4.
 |
| | Fig. 2 SEM images of (a) g-C3N4, (b) MgO, (c) 0.75MgO/g-C3N4, (d) TEM images of 0.75MgO/g-C3N4, (e) EDS mapping positions and EDS elemental profiles (f) C, (g) N, (h) O and (i) Mg of 0.75MgO/g-C3N4. | |
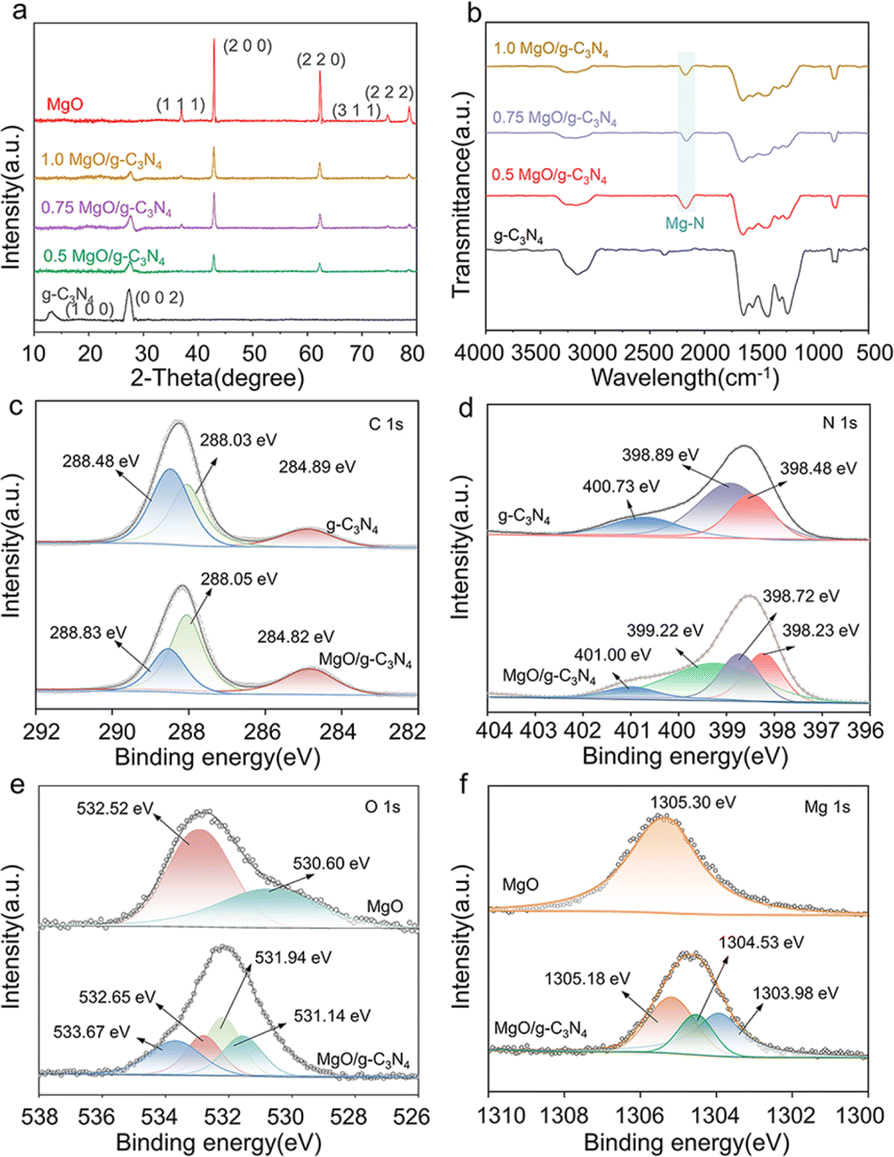
X-ray diffraction (XRD) examination was conducted on MgO, g-C3N4, and MgO/g-C3N4 composites, as depicted in Fig. 3a. The g-C3N4 exhibits distinct peaks at 13.1° and 27.4°, corresponding to the (100) and (002) crystallographic planes, respectively. The presence of these peaks suggests that g-C3N4 exhibits planar interlayer stacking and a conjugated aromatic system, which aligns with the characteristics described in the standard card (JCPDS 87-1526) for g-C3N4.42 The X-ray diffraction pattern of MgO has distinct peaks at 36.94°, 42.92°, and 62.30°, which correspond to the crystallographic planes (111), (200), and (220), respectively. All samples of MgO/g-C3N4 exhibit distinct diffraction peaks corresponding to both g-C3N4 and MgO, as shown in Fig. 3a. Furthermore, Fig. 3b displays the FTIR spectra of g-C3N4 and the composites of MgO/g-C3N4.
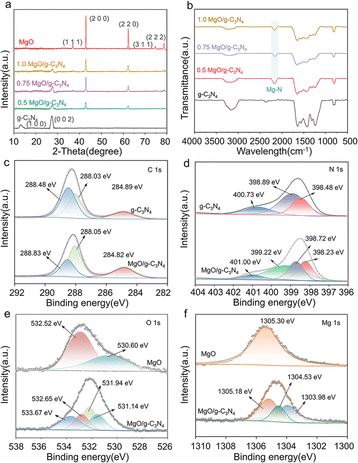 |
| | Fig. 3 (a) XRD pattern, (b) FTIR spectrum of the material, XPS patterns of (c) C 1s, (d) N 1s, (e) O 1s and (f) Mg 1s. | |
The FTIR spectra of g-C3N4 displays a distinct peak within the 1200–1650 cm−1 region. This peak corresponds to the characteristic stretching vibration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond and the resonance of the C–N heterocycle.43 The peak observed at 807 cm−1 is associated with the distinct vibrating of the triazine molecule. Furthermore, the spectral range of 3000–3400 cm−1 corresponds to a distinct peak in the N–H stretching vibration, which can be attributed to the existence of amino groups (–NH2 or NH) on the heptazine ring of g-C3N4.44,45 The g-C3N4 exhibits a prominent peak within the 1200–1650 cm−1 range, which corresponds to the characteristic stretching vibration of the CN bond and the vibration of the C–N heterocycle. The distinctive peaks of g-C3N4 and MgO/g-C3N4 composites exhibit a notable similarity. However, the peaks within the 1240–1640 cm−1 range are comparatively weaker in the composites than those in g-C3N4. This observation can be attributed to the potential creation of a chemical link between Mg and N.46 In addition, the newly observed signal at 2165 cm−1 corresponds to the vibration of the N–C–N bond, providing additional evidence for the presence of the Mg–N link between MgO and g-C3N4.47 While the peak observed at 2360 cm−1, which corresponds to the distinctive peak of CN in pure g-C3N4, gradually diminishes with the addition of MgO, suggesting the production of Mg–N bonds.48
N bond and the resonance of the C–N heterocycle.43 The peak observed at 807 cm−1 is associated with the distinct vibrating of the triazine molecule. Furthermore, the spectral range of 3000–3400 cm−1 corresponds to a distinct peak in the N–H stretching vibration, which can be attributed to the existence of amino groups (–NH2 or NH) on the heptazine ring of g-C3N4.44,45 The g-C3N4 exhibits a prominent peak within the 1200–1650 cm−1 range, which corresponds to the characteristic stretching vibration of the CN bond and the vibration of the C–N heterocycle. The distinctive peaks of g-C3N4 and MgO/g-C3N4 composites exhibit a notable similarity. However, the peaks within the 1240–1640 cm−1 range are comparatively weaker in the composites than those in g-C3N4. This observation can be attributed to the potential creation of a chemical link between Mg and N.46 In addition, the newly observed signal at 2165 cm−1 corresponds to the vibration of the N–C–N bond, providing additional evidence for the presence of the Mg–N link between MgO and g-C3N4.47 While the peak observed at 2360 cm−1, which corresponds to the distinctive peak of CN in pure g-C3N4, gradually diminishes with the addition of MgO, suggesting the production of Mg–N bonds.48
The XPS analysis was used to characterize the successful loading of MgO on g-C3N4. As illustrated in Fig. S4,† the presence of the Mg 1s peak at about 1304 eV and the enhanced intensity of the O 1s peak at roughly 530 eV in the complete spectrum of 0.75MgO/g-C3N4, as compared to that of g-C3N4, suggest the effective integration of MgO onto g-C3N4. The appearance of O 1s peaks on pure g-C3N4 can be attributed to the incorporation of oxygen during the calcination process.49 The C 1s XPS spectrum (Fig. 3c) shows three different peaks detected at 284.82, 288.05 and 288.83 eV, corresponding to C–C, C–O and C–N bonds of MgO/g-C3N4, respectively.50,51 The C–O and C–N bonds of MgO/g-C3N4 exhibit slightly higher binding energies than those of g-C3N4, which may be attributed to the electron-absorbing effect of MgO that alters the electronic environment of the C atoms in C–O and C–N. Fig. 3d displays three distinct peaks in the g-C3N4 material at energy levels of 398.48 eV, 398.89 eV, and 400.73 eV. These peaks correspond to the presence of sp2 C–NC, N–(C)3, and C–N–H, respectively.47 The newly observed signal at 399.22 eV in the MgO/g-C3N4 material can be attributed to the presence of the Mg–N bond.52 In the high-resolution O 1s XPS spectrum of the MgO/g-C3N4 composite (Fig. 3e), a peak at 533.67 eV indicates the appearance of C–O coordination bonds. While, the peaks at 532.65 eV, 531.94 eV, and 531.14 eV correspond to adsorbed ˙OH, surface adsorbed H2O, and the introduction of oxygen from air during calcination.53 As shown in Fig. 3f, the high-resolution Mg 1s spectrum of MgO/g-C3N4 has two more peaks (1303.98 and 1304.53 eV) than that of pure MgO, which correspond to N–Mg–N and N–Mg–O bonds, respectively.54 The negative shift of the peaks of Mg 1s and O 1s compared to pure MgO may be due to the charge transfer from g-C3N4 to MgO, reflecting the strong interaction between g-C3N4 and MgO.49,55
The UV-visible spectra of g-C3N4 and various series of MgO/g-C3N4 are presented in Fig. 4a. It is observed that MgO/g-C3N4 demonstrates a higher light absorption intensity compared to g-C3N4 in both the UV and visible regions. This suggests that the successful incorporation of MgO onto g-C3N4 enhances the efficiency of light absorption.56 To study the change in band gap of the material before and after loading MgO, the band gap values of g-C3N4 and MgO/g-C3N4 materials were calculated based on the measured UV-vis plots. The band gap values of g-C3N4 and MgO/g-C3N4 materials were calculated in this study based on the measured UV-vis plots. Fig. 4b demonstrates a decrease in the band gap values of the materials upon the introduction of MgO to g-C3N4. This suggests that MgO/g-C3N4 produces a greater number of photogenerated carriers when exposed to light, thereby enhancing the catalytic efficiency.57
 |
| | Fig. 4 (a) UV-diffuse reflectance spectra of the materials, (b) the band gap of g-C3N4 and 0.75MgO/g-C3N4, (c) photocurrent response diagram and (d) electrochemical impedance diagram of the material. | |
Fig. 4c demonstrates a notable enhancement in the photocurrent intensity of the MgO/g-C3N4 composites in comparison to that of g-C3N4. In particular, the 0.75 ratio of MgO/g-C3N4 has the strongest photocurrent response intensity. The correlation between higher photocurrent intensity and faster charge separation efficiency indicates that the presence of MgO enhances the charge separation efficiency of g-C3N4.58 Electrochemical impedance analysis is commonly used to study the transfer of charge carriers at interfaces. As shown in Fig. 4d, the 0.75MgO/g-C3N4 material shows the smallest semicircle in all samples, which indicates that its interfacial charge carrier transfer is the fastest and more efficient for the separation and transport of photogenerated carriers.59 In conclusion, the introduction of MgO can effectively reduce the band gap of g-C3N4, which is favourable for electron leaps and rearrangements. The formation of C–O and Mg–N bonds changes the electronic environments of C and N atoms in g-C3N4, which in turn promotes the transfer and separation of photogenerated carriers through Mg–N and C–O chemical bonds, which is expected to improve the photocatalytic performance of MgO/g-C3N4 in degrading pollutants.60,61
3.3. Catalytic performance
To investigate more comprehensively the degradation effect and the mechanism of synergistic effect in the MgO/g-C3N4/UV system, this study selected 0.75MgO/g-C3N4 with optimal optical properties as catalyst and investigated the optimal degradation conditions by controlling univariate variables. Firstly, the removal of toluene and O3 by g-C3N4, MgO and MgO/g-C3N4 was compared under light conditions (Fig. S5†). The removal of toluene by MgO/g-C3N4 was significantly enhanced compared to g-C3N4 or MgO. In addition, the loading of MgO, which has excellent oxidation and adsorption properties with O3, improved the O3 removal ability of MgO/g-C3N4.39,40 According to the data presented in Fig. S6a,† the rate of degradation reduces as the concentration increases. The reaction system achieves its optimum efficiency at a concentration of 113.06 mg m−3. This is mainly due to the fact that the capacity of the catalyst to treat pollutants per unit area is certain, and the degradation efficiency decreases due to the excessive number of pollutants not being degraded in time per unit time. Although lower concentrations of toluene exhibit higher degradation rates, higher concentrations favour O3 elimination and utilization (Fig. S6b†). This is mostly attributed to the larger concentrations, which allow for greater contact between pollutants and O3 on a per unit area and per unit time basis, thus the rate of O3 removal is increased.62
Currently, many investigations indicate that the adsorption of organic pollutant molecules on the catalyst surface and the synthesis of active chemicals in the catalytic oxidation process are influenced by the relative humidity (RH) in the catalytic oxidation of VOCs, which has an impact on the degradation effect.63–65 As shown in Fig. S6c,† the optimal toluene degradation rate occurs when the RH of the process system is approximately 60%; however, the toluene degradation efficiency decreases to a certain extent when the RH is too high or too low. This is mainly due to the fact that under high RH conditions, moisture in the gas stream will be adsorbed on the catalyst surface, which results in competitive adsorption with pollutant molecules,66 while low RH will reduce the yield of active substances in the system, which will also reduce the degradation efficiency of toluene.67 As shown in Fig. S6d,† there are also some differences in the synergistic elimination of O3 under different RH conditions. This is possibly due to the fact that the presence of moisture in the reaction system affects the formation of reactive substances from O3, which in turn affects the degradation of pollutants.68
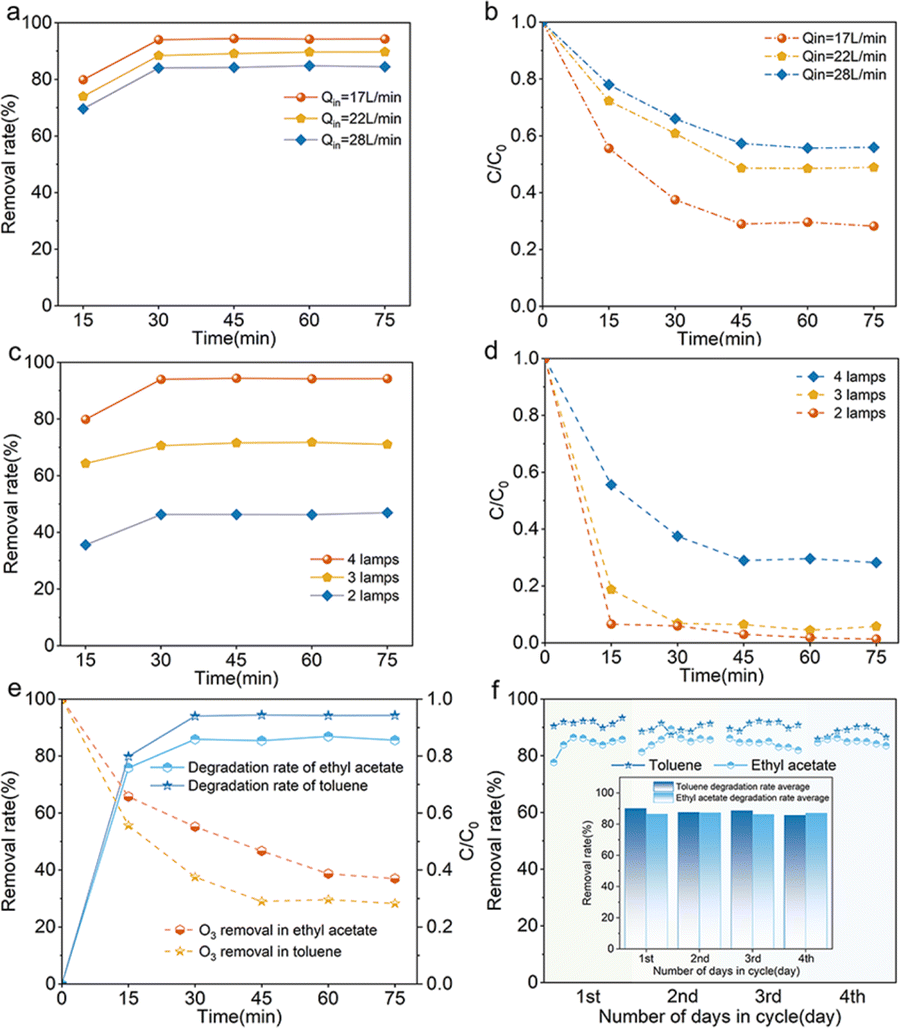
The residence time determines the contact area and time of a certain amount of catalyst and pollutant per unit of time in the reaction system. Fig. 5a demonstrates that the degradation rate of the pollutants increases with decreasing inlet flow rate and toluene can be degraded with an efficiency of 95%. In addition, the concentration of O3 is low under this condition and the removal rate is about 75% (Fig. 5b). This is possibly due to the fact that the slower flow rate allows greater contact between the pollutant and the catalyst in the reaction chamber, allowing the pollutant molecules to fully react with the active substances produced by the photocatalysis, resulting in a higher degradation efficiency of the pollutant. As shown in Fig. 5c, the decrease in the number of O3-producing UV lamps results in a varying degree of reduction in the effectiveness of pollutant breakdown, and the concentration of terminal O3 also decreases with the reduction of the generated amount. As shown in Fig. 5d, when the number of O3-producing UV lamps is 3, the remaining O3 concentration of the whole reaction system is nearly 0, which indicates that the system can effectively degrade organic pollutants and eliminate the secondary pollution brought by excess O3 at the same time under the premise of certain O3 concentration.
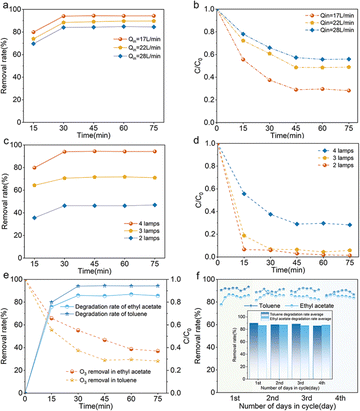 |
| | Fig. 5 Degradation of (a) toluene and (b) O3 at different flow rates, degradation of (c) toluene and (d) O3 at different radiation levels, (e) degradation efficiency of ethyl acetate and (f) degradation of toluene and ethyl acetate by catalyst recycling (inset shows the average degradation rate per cycle). Conditions: Qin = 17 L min−1 except for (a and b), C0 = 113.06 mg m−3, RH = 61.2%. | |
The system's versatility and cyclic stability were demonstrated by evaluating the degradation efficiency of ethyl acetate as well as long-term running and cycling experiments with toluene and ethyl acetate. As shown in Fig. 5e, the breakdown rate of ethyl acetate in this system can exceed 85%, meeting regional emission standards (DB 31/933—2023). And the concentration of O3 also decreases in this process, which indicates that this system still has a good degradation effect for other VOCs. As seen in Fig. 5f, the degradation rates of toluene and ethyl acetate were basically above 88% and 85% in each cycle, which indicated that the system could maintain a high degradation efficiency even under the long-time working conditions, and thus the reaction system had a good cyclic stability.
3.4. Mechanism discussion
With the successful introduction of MgO, the MgO/g-C3N4 forms Mg–N and C–O chemical bonds, and its band gap is smaller than that of pure g-C3N4, which improves the charge separation efficiency of g-C3N4, and enhances the separation and transport efficiency of photogenerated carriers. To further investigate the electronic behaviour and energy level changes upon loading, the energy band structure and density of states of g-C3N4 and MgO/g-C3N4 composites were analyzed by DFT calculations. The energy band structure and density of states for g-C3N4 and MgO/g-C3N4 are displayed in Fig. S7 and S8,† respectively, indicating that the energy band gap of the composites is significantly reduced due to the successful introduction of MgO. The density of states plot in Fig. S7† reveals that the energy band of g-C3N4 mostly comprises the s and p orbitals of C and N. Furthermore, the conduction band is predominantly composed of the p orbitals of C and N. Compared to g-C3N4, the density of states plots of MgO/g-C3N4 in Fig. S8† shows an upward shift in the valence band energy and a downward shift in the conduction band energy, which results in a decrease in the band gap. Additionally, the s and p orbitals of the composite material contribute to both the valence and conduction bands. This indicates that the loading of MgO onto g-C3N4 facilitates the formation of band gap and more photogenerated carriers.3
To further investigate the synergistic mechanism of O3 utilization by catalyst materials before and after MgO loading, the adsorption energy of catalysts for O3, oxygen, and toluene was investigated in this paper. The change in adsorption energy of catalyst materials before and after MgO loading was calculated using DFT. The adsorption sites and adsorption energies of g-C3N4 and MgO/g-C3N4 on O3, oxygen, and toluene are shown in Fig. 6. From the results, it is clear that the adsorption of O3, O2, and toluene is enhanced more obviously after the loading of MgO on g-C3N4, which indicates that the introduction of MgO is beneficial to the adsorption and utilization of O3, O2, and toluene. That is verified by adsorption experiments under light-free conditions (Fig. S9†), where the adsorption of toluene by MgO/g-C3N4 is about 20% higher than that of g-C3N4 in the first 30 min of the reaction, and the concentration of toluene tends to stabilize as the adsorption continues to be saturated. To further clarify the active compounds generated in the photocatalytic-O3 synergistic system, auxiliary electron paramagnetic resonance (EPR) tests were conducted.69 As shown in Fig. S10,† in the absence of light there is no production of active substances, in contrast, when the light is turned on a more pronounced ˙OH (Fig. S10a†), ˙O2− (Fig. S10b†) and 1O2 (Fig. S10c†) signal peaks were produced. The addition of O3 greatly amplified the signal peaks of the MgO/g-C3N4 system, suggesting the existence of a synergistic interaction between MgO/g-C3N4 and O3.
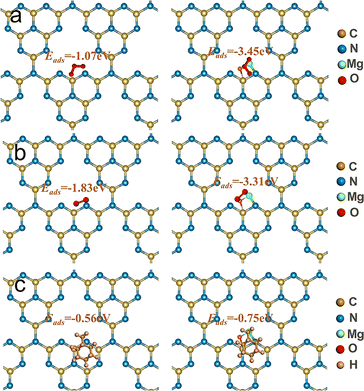 |
| | Fig. 6 Adsorption sites of (a) O3, (b) O2 and (c) toluene. | |
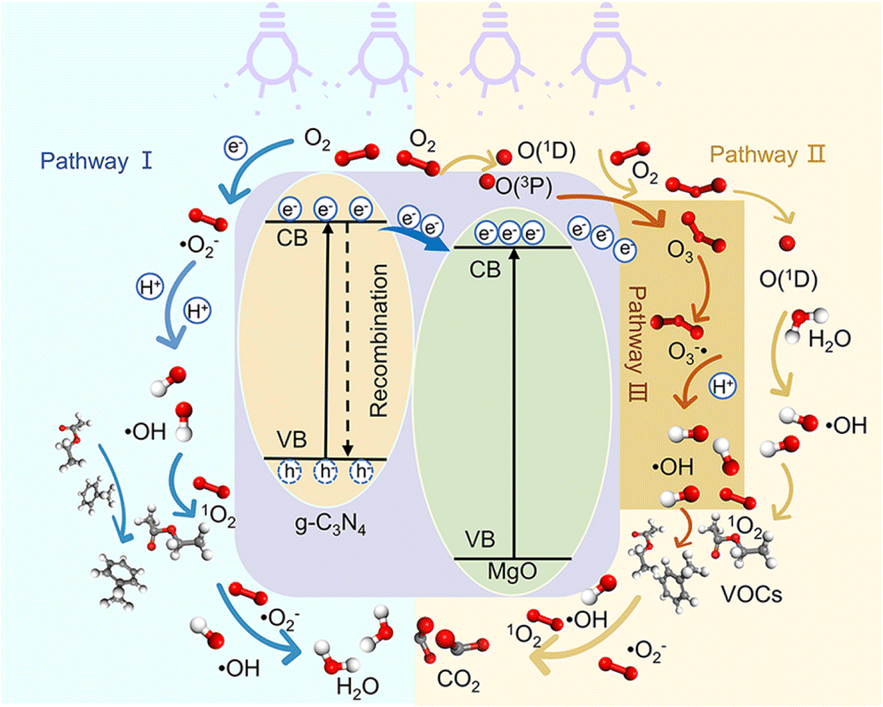
Combining the characterization of materials, the detection of active substances during the reaction, the main active substances in the present system are ˙OH, ˙O2− and 1O2. In the present system, the dual role of MgO is crucial. Firstly, in the photocatalytic O3 oxidation system, the chemical bond between MgO and g-C3N4 establishes the charge transfer channel, so MgO significantly speeds up the separation of photogenerated charges in g-C3N4 and boosts the production of ˙OH. In addition, MgO can improve the active site, increase the utilization of O3, and actively decompose O3, thus generating more ˙OH, which in turn creates an additional catalytic O3 oxidation system. Therefore, there are three main ways to degrade pollutants in this system, as shown in Fig. 7: (I) a portion of the O2 will undergo a direct reaction with electrons, resulting in the formation of ˙O2−. The generated ˙O2− will then combine with H2O to generate additional ˙OH, thereby intensifying the degradation of pollutants.70 (II) O2 is photolyzed in this system to produce O3, and the photocatalytic material can be used to generate ˙O2−, which will further generate ˙OH to degrade organic pollutants. The detailed process is as follows: high-energy UV light first cracks the oxygen molecules in the air to form (O(1D)) and triplet state oxygen atoms (O(3P)). Then, they react with oxygen molecules in the air to form O3, and this reaction can be expressed by the eqn (1)–(3).71 Subsequently, in the presence of ultraviolet light, the generated O3 continues to form O(1D), which then reacts with water molecules in the air to form ˙OH, with the reaction eqn (4) and (5).68 (III) The O3 generated in the MgO/g-C3N4 synergistic system can be used as an electron trapping agent to accelerate the separation of photogenerated carriers should thus generate ˙OH, and eqn (6)–(8) demonstrate the generation process, while the generation of superoxide radicals and the final generation of ˙OH can be expressed by eqn (9)–(12).72,73 Due to the short lifespan of radical ˙OH radicals, the generated radical ˙OH radicals would transform into 1O2 on the MgO/g-C3N4 surface by the disproportionation reaction (eqn (13)).74
| | |
O2 + hv(<243 nm) → O(1D) + O(3P)
| (1) |
| | |
O(1D) + M → O(3P) + M (M = O2 or N2)
| (2) |
| | |
O(3P) + O2 + M → O3 + M
| (3) |
| | |
O3 + hv(<310 nm) → O(1D) + O2
| (4) |
| | |
hv(185 nm) + H2O → ˙OH + H+
| (6) |
| |
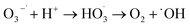 | (8) |
| |
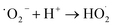 | (10) |
| |
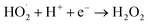 | (11) |
| | |
H2O2 + e− → ˙OH + OH
| (12) |
| | |
˙OH + ˙OH → 1O2 + H2O
| (13) |
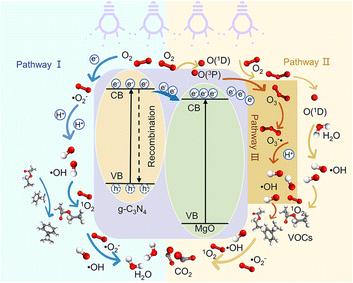 |
| | Fig. 7 Degradation mechanism diagram. | |
To insight the degradation products of gaseous toluene, the gas chromatography-mass spectrometry (GC-MS) is often used to analyse the composition of the treated gas. In addition, in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) allows the analysis of adsorbed substances on the catalyst during the reaction process, ultimately revealing the degradation intermediates.75,76 Due to the limitations of the photoreactor, the study used GC-MS to explore the degradation products of toluene. As shown in Fig. S11,† the main intermediates in the photocatalytic degradation of toluene are the long-chain products produced by the ring-opening reaction, and a series of short-chain compounds such as alcohols, aldehydes, ketones, olefins, and so on, formed by further degradation.77–79 Finally, these short-chain products were degraded to CO2 and H2O.
4. Conclusion
A novel photocatalytic system holding “kills two birds with one stone” was explored, i.e. towards simultaneous elimination of VOCs and O3 secondary pollution. The distribution of airflow field and UV lamp radiation in the photocatalytic reaction device was simulated by CFD, which optimized the reactor design in this system. MgO/g-C3N4 synthesized as efficient catalysts for the photocatalytic oxidation process exhibit satisfied removal efficiencies for VOCs and O3 under optimized conditions, which meets the industrial emission standard. On the other hand, the long-term degradation experiments of toluene and ethyl acetate indicate that the system possesses good stability and wide applicability. Based on DFT calculations and EPR experiments, the reaction mechanism for the effective degradation of pollutants by MgO/g-C3N4 was elucidated. These results might provide a new insight for the application of photocatalytic technology to remove VOCs and O3 pollution in the industry.
Author contributions
Yifan Sui: investigation, visualization, writing original draft. Xiaohu Sun: methodology, and formal analysis. Jie Guan: resources and funding acquisition. Zeqiu Chen: writing – review & editing, and supervision. Xinjie Zhu: methodology, and investigation. Xiaoyi Lou: resources, and investigation. Xiuli Li: methodology, and investigation. Jiaowen Shen: methodology. Xiaomei Liu: methodology, and investigation. Xiaojiao Zhang: methodology. Yaoguang Guo: funding acquisition, writing – review & editing, and supervision. Gangfeng Zhang: methodology, and investigation. Rui-Qin Zhang: methodology, resources, and investigation.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
The present work was financially supported by Natural Science Foundation of China (52270129, 52070127 and 52370142), Shanghai Natural Science Foundation (20ZR1421100), Oriental Talent Youth Program, Shanghai Shuguang Program (23SG52), Guizhou Provincial Key Technology R&D Program (QKHZC(2024)153) and Fundamental Research Funds of Shanghai Polytechnic University (EGD24DS15). Dr Guo also thanks the financial support of Science and Technology Development Fund of Pudong New Area (PKJ2022-C07, and PKJ2022-C10).
References
- Z. Zhang, H. Wang, D. Chen, Q. Li, P. Thai, D. Gong, Y. Li, C. Zhang, Y. Gu, L. Zhou, L. Morawska and B. Wang, Sci. Total Environ., 2017, 584–585, 1162–1174 CrossRef CAS PubMed.
- Q. Zhang, B. Yuan, M. Shao, X. Wang, S. Lu, K. Lu, M. Wang, L. Chen, C. C. Chang and S. C. Liu, Atmos. Chem. Phys., 2014, 14, 6089–6101 CrossRef.
- X. Liu, J. Zheng, K. Peng, G. Qin, Y. Yang and Z. Huang, J. Environ. Chem. Eng., 2022, 10, 107390 CrossRef CAS.
- Y. Guo, M. Wen, G. Li and T. An, Appl. Catal., B, 2021, 281, 119447 CrossRef CAS.
- S. Weon and W. Choi, Environ. Sci. Technol., 2016, 50, 2556–2563 CrossRef CAS PubMed.
- J. Lee, J. Jang, J. Kim and S.-H. Lim, Chem. Eng. J., 2022, 430, 132891 CrossRef CAS.
- Y.-W. Li and W.-L. Ma, Chemosphere, 2021, 280, 130667 CrossRef CAS PubMed.
- E. Cetin, M. Odabasi and R. Seyfioglu, Sci. Total Environ., 2003, 312, 103–112 CrossRef CAS PubMed.
- K. Kante, M. Florent, A. Temirgaliyeva, B. Lesbayev and T. J. Bandosz, Carbon, 2019, 146, 568–571 CrossRef CAS.
- G. Zheng, K. Wei, X. Kang, W. Fan, N. L. Ma, M. Verma, H. S. Ng and S. Ge, Environ. Pollut., 2023, 336, 122451 CrossRef CAS PubMed.
- B. Shen, S. Zhao, X. Yang, M. Carta, H. Zhou and W. Jin, Sep. Purif. Technol., 2022, 280, 119974 CrossRef CAS.
- H. Huang, Y. Xu, Q. Feng and D. Y. C. Leung, Catal. Sci. Technol., 2015, 5, 2649–2669 RSC.
- M. S. Kamal, S. A. Razzak and M. M. Hossain, Atmos. Environ., 2016, 140, 117–134 CrossRef CAS.
- S. Rouhani and F. Taghipour, Chem. Eng. Sci., 2023, 272, 118617 CrossRef CAS.
- X. Wu, Y. Lin, Y. Wang, S. Wu and C. Yang, Chem. Eng. J., 2023, 471, 144420 CrossRef CAS.
- J. R. Silva, D. S. Ascensão and L. M. Castro, Energy Rep., 2020, 6, 250–255 CrossRef.
- J. Sun, X. Yan, K. Lv, S. Sun, K. Deng and D. Du, J. Mol. Catal. A: Chem., 2013, 367, 31–37 CrossRef CAS.
- W. Dechapanya, A. Eusebi, Y. Kimura and D. T. Allen, Environ. Sci. Technol., 2003, 37, 3662–3670 CrossRef CAS PubMed.
- J. Li, W. Cui, P. Chen, X. a. Dong, Y. Chu, J. Sheng, Y. Zhang, Z. Wang and F. Dong, Appl. Catal., B, 2020, 260, 118130 CrossRef CAS.
- A. H. Mamaghani, F. Haghighat and C.-S. Lee, Appl. Catal., B, 2017, 203, 247–269 CrossRef CAS.
- J. Ji, Y. Xu, H. Huang, M. He, S. Liu, G. Liu, R. Xie, Q. Feng, Y. Shu, Y. Zhan, R. Fang, X. Ye and D. Y. C. Leung, Chem. Eng. J., 2017, 327, 490–499 CrossRef CAS.
- J. Kim, P. Zhang, J. Li, J. Wang and P. Fu, Chem. Eng. J., 2014, 252, 337–345 CrossRef CAS.
- M. Li, Z. Qiang, P. Hou, J. R. Bolton, J. Qu, P. Li and C. Wang, Environ. Sci. Technol., 2016, 50, 5849–5856 CrossRef CAS PubMed.
- J. Jeong, K. Sekiguchi and K. Sakamoto, Chemosphere, 2004, 57, 663–671 CrossRef CAS PubMed.
- P. Fu, P. Zhang and J. Li, Int. J. Photoenergy, 2012, 2012, 174862 CrossRef.
- K. Zoschke, H. Börnick and E. Worch, Water Res., 2014, 52, 131–145 CrossRef CAS PubMed.
- H. Claus, Photochem. Photobiol., 2021, 97, 471–476 CrossRef CAS PubMed.
- Y. Liu and P. Zhang, J. Phys. Chem. C, 2017, 121, 23488–23497 CrossRef CAS.
- J. Ji, Y. Fang, L. He and H. Huang, Catal. Sci. Technol., 2019, 9, 4036–4046 RSC.
- S. Lim, J. L. Shi, U. von Gunten and D. L. McCurry, Water Res., 2022, 213, 118053 CrossRef CAS PubMed.
- L. Xiang, L. Zhang, J. Shao, F. Lin, Z. Wang, B. Yan and G. Chen, J. Hazard. Mater., 2023, 441, 129997 CrossRef CAS.
- L. Z. Deng, A. S. Mujumdar, Z. Pan, S. K. Vidyarthi, J. Xu, M. Zielinska and H. W. Xiao, Crit. Rev. Food Sci. Nutr., 2020, 60, 2481–2508 CrossRef CAS PubMed.
- L. Chen, M. A. Maigbay, M. Li and X. Qiu, Adv. Powder Mater., 2023, 1, 100150 Search PubMed.
- R. Cheng, J. Wen, J. Xia, L. Shen, M. Kang, L. Shi and X. Zheng, Catal. Today, 2022, 388–389, 26–35 CAS.
- F. Fu, J. Yang, H. Xu, Y. Li, S. Cao, K. Da, W. Chen and X. Fan, Chem. Eng. Sci., 2023, 276, 118793 CrossRef CAS.
- X. Li, Y. Huang, W. Ho, S. Han, P. Wang, S. Lee and Z. Zhang, Appl. Catal., B, 2023, 338, 123048 CrossRef CAS.
- L. Muniandy, F. Adam, A. R. Mohamed, A. Iqbal and N. R. A. Rahman, Appl. Surf. Sci., 2017, 398, 43–55 CrossRef CAS.
- Y. Li, X. Wu, J. Li, K. Wang and G. Zhang, Appl. Catal., B, 2018, 229, 218–226 CrossRef CAS.
- G. Moussavi, A. Yazdanbakhsh and M. Heidarizad, J. Hazard. Mater., 2009, 171, 907–913 CrossRef CAS PubMed.
- C. Agarkoti, P. R. Gogate and A. B. Pandit, Sep. Purif. Technol., 2022, 284, 120281 CrossRef CAS.
- L. Du, W. Gao, Z. Li, W. Jiao and Y. Liu, Chem. Eng. Process., 2020, 155, 108053 CrossRef CAS.
- W. An, L. Tian, J. Hu, L. Liu, W. Cui and Y. Liang, Appl. Surf. Sci., 2020, 534, 147518 CrossRef CAS.
- Q. Chen, S. Li, H. Xu, G. Wang, Y. Qu, P. Zhu and D. Wang, Chin. J. Catal., 2020, 41, 514–523 CrossRef CAS.
- Y. Liang, R. Shang, J. Lu, L. Liu, J. Hu and W. Cui, ACS Appl. Mater. Interfaces, 2018, 10, 8758–8769 CrossRef CAS PubMed.
- C. Zhou, P. Xu, C. Lai, C. Zhang, G. Zeng, D. Huang, M. Cheng, L. Hu, W. Xiong, X. Wen, L. Qin, J. Yuan and W. Wang, Chem. Eng. J., 2019, 359, 186–196 CrossRef CAS.
- L. Ge, Z. Peng, W. Wang, F. Tan, X. Wang, B. Su, X. Qiao and P. K. Wong, J. Mater. Chem. A, 2018, 6, 16421–16429 RSC.
- P. Ramacharyulu, S. J. Abbas, S. R. Sahoo and S.-C. Ke, Catal. Sci. Technol., 2018, 8, 2825–2834 RSC.
- J. Zhou, X. Ji, X. Zhou, J. Guo, J. Sun and Y. Liu, Sep. Sci. Technol., 2019, 54, 2817–2829 CrossRef CAS.
- N. Mao and J.-X. Jiang, Appl. Surf. Sci., 2019, 476, 144–150 CrossRef CAS.
- M. Zhu, S. Kim, L. Mao, M. Fujitsuka, J. Zhang, X. Wang and T. Majima, J. Am. Chem. Soc., 2017, 139, 13234–13242 CrossRef CAS PubMed.
- X. An, X. Xu, W. Guo, Z. Chen, Z. Miao, J. Yuan and Z. Wu, J. Environ. Manage., 2023, 344, 118489 CrossRef CAS PubMed.
- W. Gao, Graphene Oxide: Reduction Recipes, Spectroscopy, and Applications, 2015, pp. 61–95 Search PubMed.
- C. Wang, H. Fan, X. Ren, J. Ma, J. Fang and W. Wang, ChemSusChem, 2017, 11, 700–708 CrossRef.
- J. Sun, H. Wu, C. Fu, C. Zhang, Z. Hu and M. Zhou, Appl. Catal., B, 2024, 351, 123976 CrossRef CAS.
- Z. Zhang, R. Ji, Q. Sun, J. He, D. Chen, N. Li, H. Li, A. Marcomini, Q. Xu and J. Lu, Appl. Catal., B, 2023, 324, 122276 CrossRef CAS.
- W. He, L. Liu, T. Ma, H. Han, J. Zhu, Y. Liu, Z. Fang, Z. Yang and K. Guo, Appl. Catal., B, 2022, 306, 121107 CrossRef CAS.
- W. Wang, Z. Zeng, G. Zeng, C. Zhang, R. Xiao, C. Zhou, W. Xiong, Y. Yang, L. Lei and Y. Liu, Chem. Eng. J., 2019, 378, 122132 CrossRef CAS.
- I. Khan, S. Ali, M. Mansha and A. Qurashi, Ultrason. Sonochem., 2017, 36, 386–392 CrossRef CAS PubMed.
- S. Baqi, B. Deng, Y. Guo and R.-Q. Zhang, J. Phys. Chem. C, 2021, 125, 25207–25216 CrossRef CAS.
- E. Giamello, D. Murphy, E. Garrone and A. Zecchina, Spectrochim. Acta, Part A, 1993, 49, 1323–1330 CrossRef.
- Y.-j. Hao, B. Liu, L.-g. Tian, F.-t. Li, J. Ren, S.-j. Liu, Y. Liu, J. Zhao and X.-j. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 12687–12693 CrossRef CAS PubMed.
- B. M. da Costa Filho, G. V. Silva, R. A. Boaventura, M. M. Dias, J. C. Lopes and V. J. Vilar, Sci. Total Environ., 2019, 687, 1357–1368 CrossRef CAS PubMed.
- F. Almomani, R. Bhosale and M. Shawaqfah, Chemosphere, 2020, 255, 126878 CrossRef CAS PubMed.
- T. Gopi, G. Swetha, S. C. Shekar, R. Krishna, C. Ramakrishna, B. Saini and P. Rao, Arabian J. Chem., 2019, 12, 4502–4513 CrossRef CAS.
- C. H. A. Tsang, K. Li, Y. Zeng, W. Zhao, T. Zhang, Y. Zhan, R. Xie, D. Y. Leung and H. Huang, Environ. Int., 2019, 125, 200–228 CrossRef CAS PubMed.
- H. Gu, J. Lan, Y. Liu, C. Ling, K. Wei, G. Zhan, F. Guo, F. Jia, Z. Ai and L. Zhang, ACS Catal., 2022, 12, 11272–11280 CrossRef CAS.
- R. Radhakrishnan and S. T. Oyama, J. Catal., 2001, 199, 282–290 CrossRef CAS.
- J. Xiao, Y. Xie, F. Nawaz, S. Jin, F. Duan, M. Li and H. Cao, Appl. Catal., B, 2016, 181, 420–428 CrossRef CAS.
- L.-S. Zhang, X.-H. Jiang, Z.-A. Zhong, L. Tian, Q. Sun, Y.-T. Cui, X. Lu, J.-P. Zou and S.-L. Luo, Angew. Chem., 2021, 133, 21919–21923 CrossRef.
- J. Lei, B. Chen, L. Zhou, N. Ding, Z. Cai, L. Wang, S.-I. In, C. Cui, Y. Zhou and Y. Liu, Chem. Eng. J., 2020, 400, 125902 CrossRef CAS.
- J. Jeong, K. Sekiguchi, W. Lee and K. Sakamoto, J. Photochem. Photobiol., A, 2005, 169, 279–287 CrossRef CAS.
- J. Xiao, J. Rabeah, J. Yang, Y. Xie, H. Cao and A. Brückner, ACS Catal., 2017, 7, 6198–6206 CrossRef CAS.
- P. Chen, Y. Mu, Y. Chen, L. Tian, X.-H. Jiang, J.-P. Zou and S.-L. Luo, Chemosphere, 2022, 291, 132817 CrossRef CAS PubMed.
- M. Tian, X. Ren, S. Ding, N. Fu, Y. Wei, Z. Yang and X. Yao, Environ. Res., 2024, 243, 117848 CrossRef CAS PubMed.
- Q. Cheng, Z. Wang, X. Wang, J. Li, Y. Li and G. Zhang, Nano Res., 2023, 16, 2133–2141 CrossRef CAS.
- S. Fan, S. Luo, Y. Wang, X. Yue, D. Zheng, Z. Zhang, X. Fu and W. Dai, Sep. Purif. Technol., 2024, 336, 126256 CrossRef CAS.
- J. Rao, X. Chen, X. Zheng and C. Du, Atmos. Pollut. Res., 2023, 14, 101854 CrossRef CAS.
- S. Cao, L. Wu, J. Li, Y. Li, K. Da, W. Chen, R. Xue, J. Yang and X. Fan, J. Environ. Chem. Eng., 2024, 12, 112321 CrossRef CAS.
- L. Guo, J. Zhang, X. Zhang, R. Wang, Y. Jia and H. Long, Mol. Catal., 2023, 550, 113603 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Gangfeng Zhangd and
Rui-Qin Zhang
*a,
Gangfeng Zhangd and
Rui-Qin Zhang