
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual active pyrimidine-based carbocyclic nucleoside derivatives: synthesis, and in silico and in vitro anti-diabetic and anti-microbial studies†
Kalyani Mallidia,
Rambabu Gundla*a,
Parameshwar Makamb,
Naresh Kumar Katari *ac and
Sreekantha Babu Jonnalagaddac
*ac and
Sreekantha Babu Jonnalagaddac
aDepartment of Chemistry, GITAM School of Science, GITAM Deemed to be University, Hyderabad, Telangana 502329, India. E-mail: nkatari@gitam.edu
bDepartment of Chemistry, School of Applied and Life Sciences, Uttaranchal University, Arcadia Grant, P.O. Chandanwari, Premnagar, Dehradun, Uttarakhand 248007, India
cSchool of Chemistry & Physics, College of Agriculture, Engineering & Science, WestvilleCampus, University of KwaZulu-Natal, P Bag X 54001, Durban 4000, South Africa
First published on 21st March 2024
Abstract
Diabetes mellitus (DM) is a chronic metabolic disorder marked by high blood glucose levels, impairing glucose production in the body. Its prevalence has steadily risen over the past decades, leading to compromised immunity and heightened susceptibility to microbial infections. Immune dysfunction associated with diabetes raises vulnerability, while neuropathy dulls sensation in the extremities, reducing injury awareness. Hence, the development of novel chemical compounds for anti-diabetic and anti-infective treatments is imperative to mitigate adverse effects. In this study, we designed and synthesized pyrimidine-based carbocyclic nucleoside derivatives with C-4 substitution to assess their potential in inhibiting α-glucosidase for managing diabetes mellitus (DM) and microbial infections. Compounds 8b and 10a displayed promising IC50 values against α-glucosidase (43.292 nmol and 48.638 nmol, respectively) and noteworthy docking energies (−9.4 kcal mol−1 and −10.3 kcal mol−1, respectively). Additionally, compounds 10a and 10b exhibited better antimicrobial activity against Bacillus cereus, with the zone of inhibition values of 2.2 ± 0.25 mm and 1.4 ± 0.1 mm at a 100 μl concentration, respectively. Compound 10a also exhibited a modest zone of inhibition of 1.2 ± 0.15 mm against Escherichia coli at 100 μl.
1 Introduction
Diabetes Mellitus (DM) is a chronic metabolic disorder1 characterized by elevated blood glucose levels, which impairs the body's ability to produce glucose.2,3 Diabetes is directly accountable for causing the deaths of 1.5 million people annually. Over the past few decades, there has been a steady increase in both the occurrence and the overall number of cases of diabetes. Type 1 diabetes is caused by the autoimmune destruction of pancreatic beta cells, requiring lifelong insulin therapy,4,5 Type 2 diabetes is caused by poor diet, lack of exercise, and obesity, leading to increased insulin resistance.6–8 α-Glucosidase inhibitors are used to treat type 2 diabetes by slowing the absorption of carbohydrates in the small intestine.9 These inhibitors have multiple advantages in controlling diabetes, including delayed digestion of complex carbohydrates, slowing glucose release, reducing postprandial hyperglycemia, and regulating postprandial glucose levels.3,6 Typically used alongside other anti-diabetic medications or insulin therapy, α-glucosidase inhibitors can enhance a diabetes management plan and are rarely used as initial treatments (Fig. 1).3,10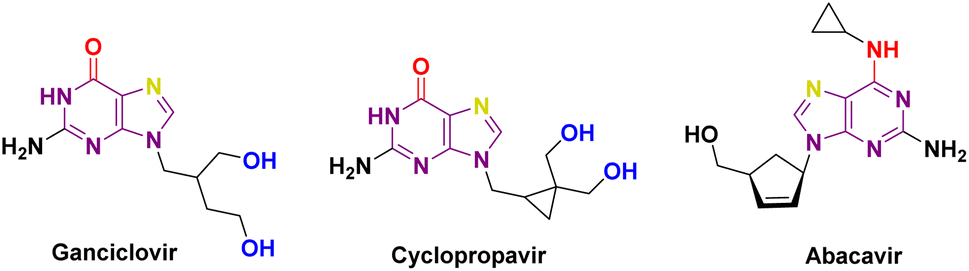 | ||
| Fig. 1 Carbocyclic nucleoside analogues. | ||
Diabetes can lead to reduced immunity and increased risk of microbial infections.11 Chronic hyperglycemia can impair neutrophil and macrophage infection defence, while wound healing is impaired due to inadequate circulation and nerve damage.12 Elevated blood glucose levels promote the proliferation of microorganisms that facilitate the accelerated growth of pathogens such as bacteria and fungi.13 Diabetes-related immune dysfunction increases vulnerability, and neuropathy numbs extremities, reducing awareness of injuries and wounds. Undiagnosed infections worsen, and urinary tract infections increase.13 Diabetes-related neuropathy affects bladder function, causing incomplete emptying and infection risk. High urine glucose allows germs to grow, and diabetics with retinopathy can see poorly, increasing skin and other infections.14 High blood sugar glycates proteins and impedes infection-fighting proteins and immune cells.15 Therefore, developing novel chemical compounds for anti-diabetic and anti-infective medications is crucial to minimize side effects. Several pharmaceutical compounds and prospective compounds categorized as carbocyclic nucleosides have been developed for the management of diabetes. Multiple studies have confirmed the importance of carbocyclic pyrimidine and purine derivatives in the field of diabetes treatment. Carbocyclic nucleoside derivatives have a wide range of applications in the treatment of numerous disorders. Various instances can be given, such as Abacavir (ABC), also referred to as Ziagen or 1592U89, which is a carbocyclic nucleoside analogue that is chemically produced from guanosine. H2G, or (R)-9-[4-hydroxy-2-(hydroxymethyl)butyl]guanine, is a type of nucleoside analogue that has shown strong effectiveness against different types of herpes viruses. Amprenavir, the fifth nonpeptidic inhibitor of HIV-1 protease to be commercially accessible, was initially created for the treatment of AIDS patients in combination with authorized antiretroviral nucleoside analogues.16,17 Carbocyclic nucleosides have been predominantly investigated for their antiviral effects. However, they have also been examined for their ability to combat bacterial infections, particularly in the context of diabetes treatment. Diabetes mellitus increases the likelihood of different infections, such as bacterial infections, because it weakens the immune system and hampers the wound healing process.18 Certain carbocyclic nucleosides exhibit anti-inflammatory effects, perhaps aiding in the reduction of inflammation-related problems in diabetic patients and indirectly decreasing the likelihood of bacterial infections.19 Recent studies indicate that some carbocyclic nucleosides may demonstrate synergistic properties when used with traditional antibiotics to combat bacterial infections as well.20
2 Results and discussion
2.1 Design and evaluation of physico-chemical properties
Lower molecular weight and lipophilicity compounds have been shown to increase paracellular and transcellular absorption and clearance,21–23 resulting in increased renal excretion,24,25 and moderate toxicity.26 The “rule of 5” (Ro5) defines a drug-like molecule (DLM). The Ro5 criterion includes molecules having a molecular mass below 500, a log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P value below 5, and a hydrogen bond donor and acceptor count below 5 and 10 respectively.27–29 According to the Veber rule, compounds with polar surface areas, hydrogen bond donors, and acceptors of 140, 10, or 12 improve oral bioavailability.30 Leeson et al. found that NCEs approved from 1983 to 2002 had 16% to 23% higher molecular mass, rings, rotatable bonds, and hydrogen bonding groups. These qualities help build compounds with improved ADMET.31–33 Physico-chemical characteristics are increasingly important in determining a molecule's therapeutic potential and efficacy during medication development (Fig. 2).31,32
P value below 5, and a hydrogen bond donor and acceptor count below 5 and 10 respectively.27–29 According to the Veber rule, compounds with polar surface areas, hydrogen bond donors, and acceptors of 140, 10, or 12 improve oral bioavailability.30 Leeson et al. found that NCEs approved from 1983 to 2002 had 16% to 23% higher molecular mass, rings, rotatable bonds, and hydrogen bonding groups. These qualities help build compounds with improved ADMET.31–33 Physico-chemical characteristics are increasingly important in determining a molecule's therapeutic potential and efficacy during medication development (Fig. 2).31,32
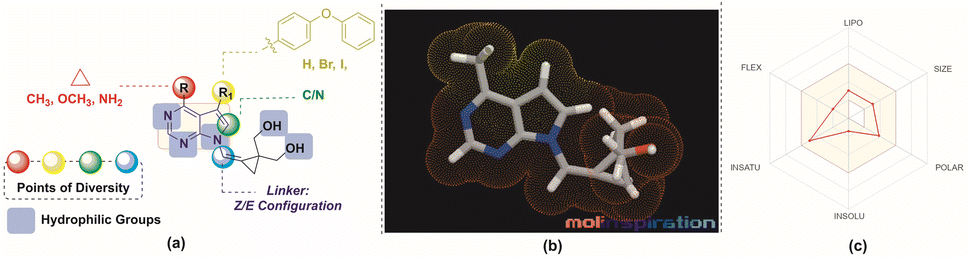 | ||
| Fig. 2 (a) Design of the molecules (b) MLP 3D representation of the molecule, 8a (c) web representation of the physico-chemical properties of the molecule, 8a. | ||
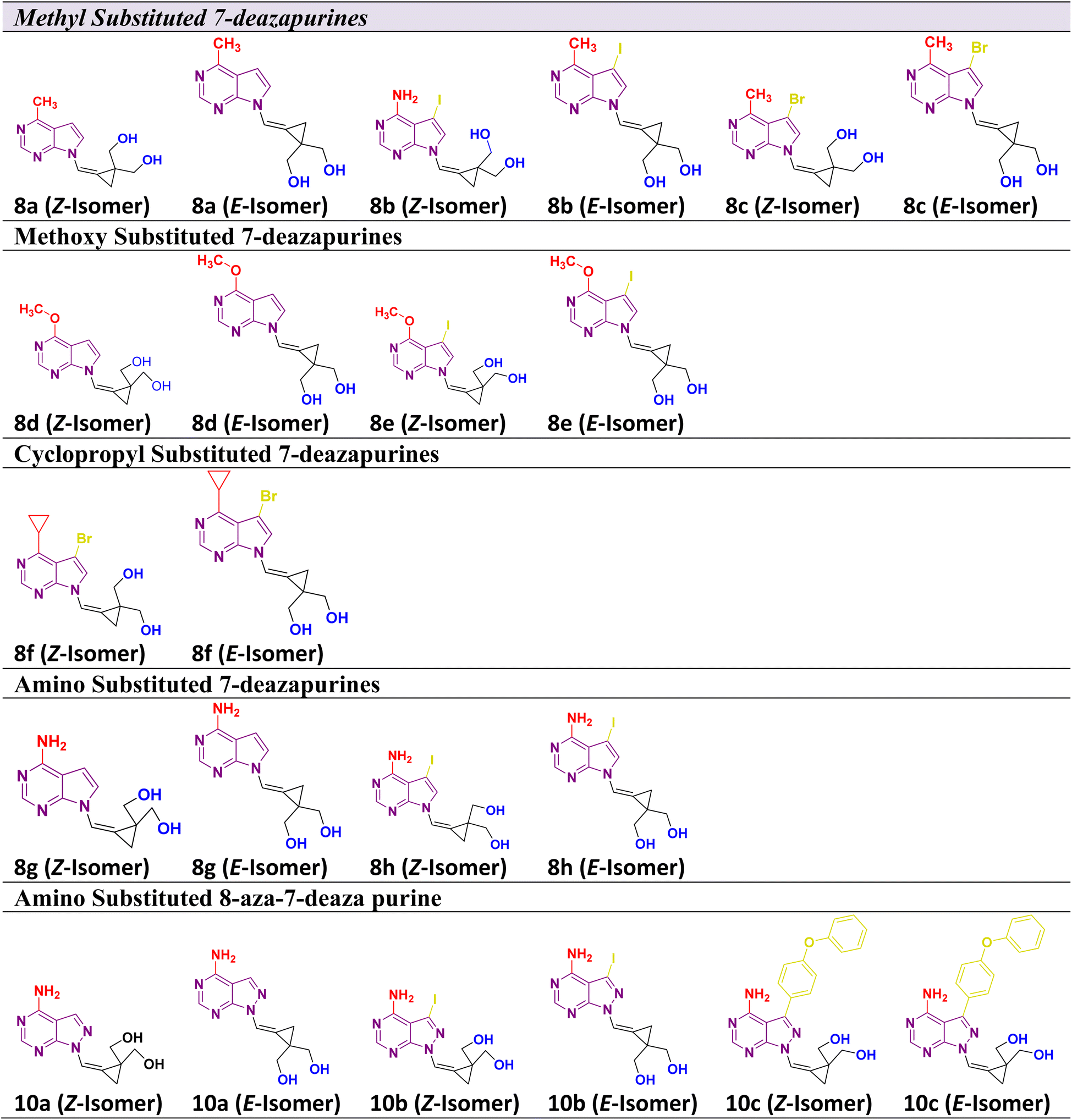
Considering the aforementioned factors, the pyrimidine-based carbocyclic nucleoside derivatives have been achieved with diverse modifications in terms of both structure and functionality (Table 1) (Fig. 3). The evaluation of physicochemical and structural properties through computational methods entails the determination of various essential parameters. These parameters encompass heavy atoms (HA), heavy aromatic atoms (HAA), rotatable bonds (RB), hydrogen bond acceptors (HBA), hydrogen bond donors (HBD), molar refractivity (MR), and total polar surface area (TPSA). The SwissADME tool is employed for this purpose. The results of this analysis have been summarised in Table 1, and the observations are consistent with the established criteria for “drug-likeness” as outlined by the Lipinski, Ghose, Veber, Egan, and Muegge rules.
 |
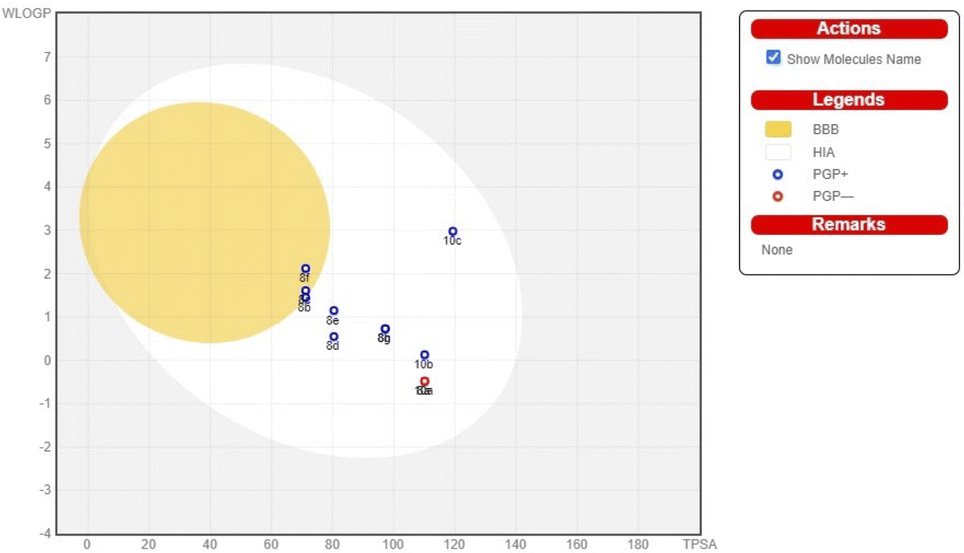 | ||
| Fig. 3 BOILEDEgg of pyrimidine-based carbocyclic nucleoside derivatives. | ||
The pharmacokinetic characteristics of pyrimidine-based carbocyclic nucleoside derivatives, such as gastrointestinal absorption (GIA) and blood–brain–blood barrier (BBB) permeability, have also been projected. The pharmacokinetic behaviours of all pyrimidine-based carbocyclic nucleoside derivatives, including gastrointestinal absorption (GIA) and brain–blood barrier (BBB), have also been predicted. By calculating the lipophilicity (WLOGP versus TPSA), the Brain or IntestinaLEstimateD Permeation method (BOILEDEgg) has been visually shown (Fig. 3).34 It is projected that all molecules fall inside the white ellipse and none fall inside the yellow ellipse and grey region. This indicates that molecules could exhibit superior GIA but poorer BBB properties. All substances whose efflux activity in the Central Nervous System (CNS) was predicted by the P-glycoprotein (PGP).
2.2 Synthesis of the C-4 substituted carbocyclic purine compounds (8a–8h) and (10a–10c)
The 7-deazapurine scaffold can be easily substituted with functional groups at the C-4 position by standard synthetic chemistry. Scheme 1 illustrates a seven-step general synthetic route for the preparation of 8a–8h and 10a–10c. The initial formation of the product was identified by MS analysis with their molecular weight. Their structures were confirmed by 1H-NMR, 13C NMR and FT-IR. Most of the compounds were soluble in polar solvents like DMF and DMSO. To synthesize these compounds, we have performed a de-acetylation reaction between compound 7 and commercially available LiOH·H2O to give the corresponding compounds. The formation of C-4 substituted carbocyclic purines was confirmed by 1H-NMR, 13C NMR and FT-IR. In these compounds, the characteristic methoxy (–methyl) proton shifted to the downfield at δ 4.0, the pyrimidine ring proton was at around δ 8.68 and the pyrrole ring proton at 8.26 ppm (Z-Isomer) and proton at 7.82 ppm (E-isomer). In the 13C NMR spectra, the methoxy (O–Me) appeared around δ 53.5 ppm; the cyclopropyl carbons were found in the region between δ 10.6 to 13.4 ppm. In the 1H-NMR, the aromatic methoxy group protons of compound 8d appeared at δ 4.06–4.04 ppm. In compound 8f, the C-4 substituted cyclopropane CH proton multiplate signal was observed in Z and E isomers at δ 3.12–3.06 and 3.09–3.05 ppm. The 13C NMR of NH2 attached carbon of compounds 8g and 8h was shifted to downfield and appeared at δ 157.2 and the signal for carbon attached to two nitrogen pyrimidine rings was shifted to downfield and appeared at δ 152.4 respectively. The 13C NMR of 8a–8h were recorded in DMSO as an internal standard, and the signal for two hydroxyl methyl attached carbon of cyclopropane ring merged into the DMSO signal. The FT-IR data clearly defined the presence of ν(–C–X) at 600–500 cm−1 in all compounds and ν(–NH for amino) at 3501 cm−1 and ν(–CN for amino) at 1250–1020 cm−1 in compound 8h. Structural confirmation was done by NOESy1D for the 10c (Z-isomer) compound.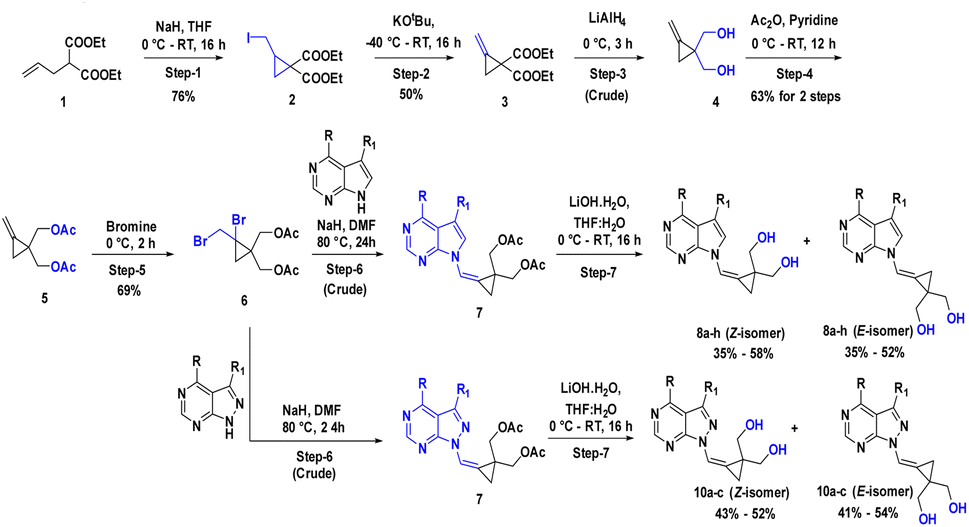 | ||
| Scheme 1 Synthetic scheme for compounds 8a–8h and 10a–10c; Reaction conditions: step-1: NaH, I2, THF, 0 °C RT, 16 h (Y: 76%); step-2: t-BuOK, THF, −40 °C RT, 16 h (Y: 50%); step-3: LiAlH4, THF, 0 °C, 3 h (Y: crude); step-4: Ac2O, pyridine, 0 °C – RT, 12 h (Y: 63% for 2 steps); step-5: Br2, CCl4, 0 °C, 2 h (Y: 69%); step-6: NaH, DMF, 80 °C, 24 h (Y: crude); step-7: LiOH·H2O, THF: H2O, 0 °C RT, 16 h (Y: 35–55% for 2 steps to all compounds). | ||
The alcohol protected (2-bromo-2-(bromomethyl)cyclopropane-1,1-diyl)bis(methylen e) 6 was reacted with a C-4 substituted 7-deaza or 7-halo-7-deazapurines.35,36 N-glycidylation reaction (7a–7h) followed by deacetylation to afford the desired compounds 8a–8h. The key moiety of alcohol protected (2-bromo-2-(bromomethyl)cyclopropane-1,1-diyl)bis(methylene) compound 6 was prepared using the diethyl 2-allylmalonate as a starting material, which on treatment with NaH and Iodination with I2 gave diethyl 2-(iodomethyl)cyclopropane-1,1-dicarboxylate moiety. This compound on double bond formation with KOtBu resulted in diethyl 2-methylenecyclopropane-1,1-dicarboxylate compound 3. Compound 3 was reduced with LiAlH4 reagent resulting in the intermediate 4.37 The compound 4 (ref. 38 and 39) in the presence of acetic anhydride and pyridine resulted in intermediate 5. Bromination with bromine gave the key intermediate 6 (ref. 40 and 41), with this intermediate conducted N-glycidylation reaction with different C-4 substituted 7-deaza or 7-halo-7-deazapurines resulted in their respective products 7a–7h in 65–70% yield. Further, deprotection with LiOH·H2O gave the first set of our target molecules 8a–8h (Table 1). The structures of the final compounds 8a–8h were confirmed by spectral characterization data using 1H-NMR, 13C-NMR, MS, and FT-IR (ESI†). 1H-NMR of compound 8a indicated singlet at δ 8.69 ppm for Pyrimidine ring proton (proton between nitrogen's), the doublet with chemical shift value of δ 8.37–8.36 ppm for pyrrole ring proton (proton adjacent to NH) and 7.68 ppm for alkene proton and the olefin proton was seen in the downfield region of δ 7.68 ppm. The 13C NMR spectra showed C-4 substituted methyl carbon shifted upfield at δ 21.1 and C7 position carbon shifted at δ 101.1 and δ 111.6 for alkene carbon. MS (ESI): m/z = 246.5 (M + H)+. The 13C signal for two hydroxyl methyl attached carbon of cyclopropane ring merged into the DMSO signal. The FT-IR data clearly defined the presence of ν(–C–O for alcohol) and ν(–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C for alkene) at 1029 and 1564 cm−1, respectively.
C for alkene) at 1029 and 1564 cm−1, respectively.
To synthesize compounds 10a–c, the key building block intermediate, 6(b) was used along with C-4 substituted 8-aza-7-deaza purines42,43 and the same protocol was followed and afford target compounds 10a–c (Table 1) (Scheme 1). In the 1H-NMR spectrum of all the new compounds synthesized from intermediate, 6(b), the characteristic 7-deaza (–CH–) proton signal was observed at δ 8.22 ppm. The two doublets were observed δ 3.65 and 3.52 ppm and one broad singlet was observed for hydroxy at 4.87 ppm. cyclopropyl ring protons ranged at δ 1.40 ppm. Moreover, the broad singlet for pyrimidine ring proton was observed around δ 8.22 ppm. The 13C signal was noticed at δ 116.5 ppm for 7-deaza carbon, and the 13C signal for two hydroxyl methyl attached carbon of cyclopropane ring merged into the DMSO signal, respectively. MS (ESI): m/z = 248.5 (M + H)+. In FT-IR, a strong band for ν(C–O) around 1035 cm−1, ν(–C–O for primary alcohol) 3564 cm−1 was seen δ 1.44 and 4.88 ppm was correlation was observed on 10c (Z-Isomer) by NOESy1D.
2.3 Docking studies
Molecular docking was performed on the synthesized compounds. Further, screening was done using AutoDock Vina for precision (Fig. 4). Among the screened compounds, the compound with the highest docking energy was found to be with 10a (E-Isomer) and 8b (E-Isomer), whereas the compound with the lowest docking energy was 8c (E-Isomer). The compound 10a (E-Isomer) demonstrated a docking energy of −10.3 kcal mol−1. The presence of conventional hydrogen bonding with Gln279, Arg442, Glu277, Asp215, and Arg213 is evident, along with a carbon–hydrogen bond with Asp352. Subsequently, compound 8b (E-Isomer) exhibited the second-highest docking energy of −9.7 kcal mol−1. The presence of conventional hydrogen bonds with Asp69 and Arg442 was observed. Compound 8c (Z-Isomer) had the lowest docking energy, with a value of −7.1 kcal mol−1. Conventional hydrogen bonds with Asp215 and carbon–hydrogen bonds with Asp352 were observed (ESI Table 1†).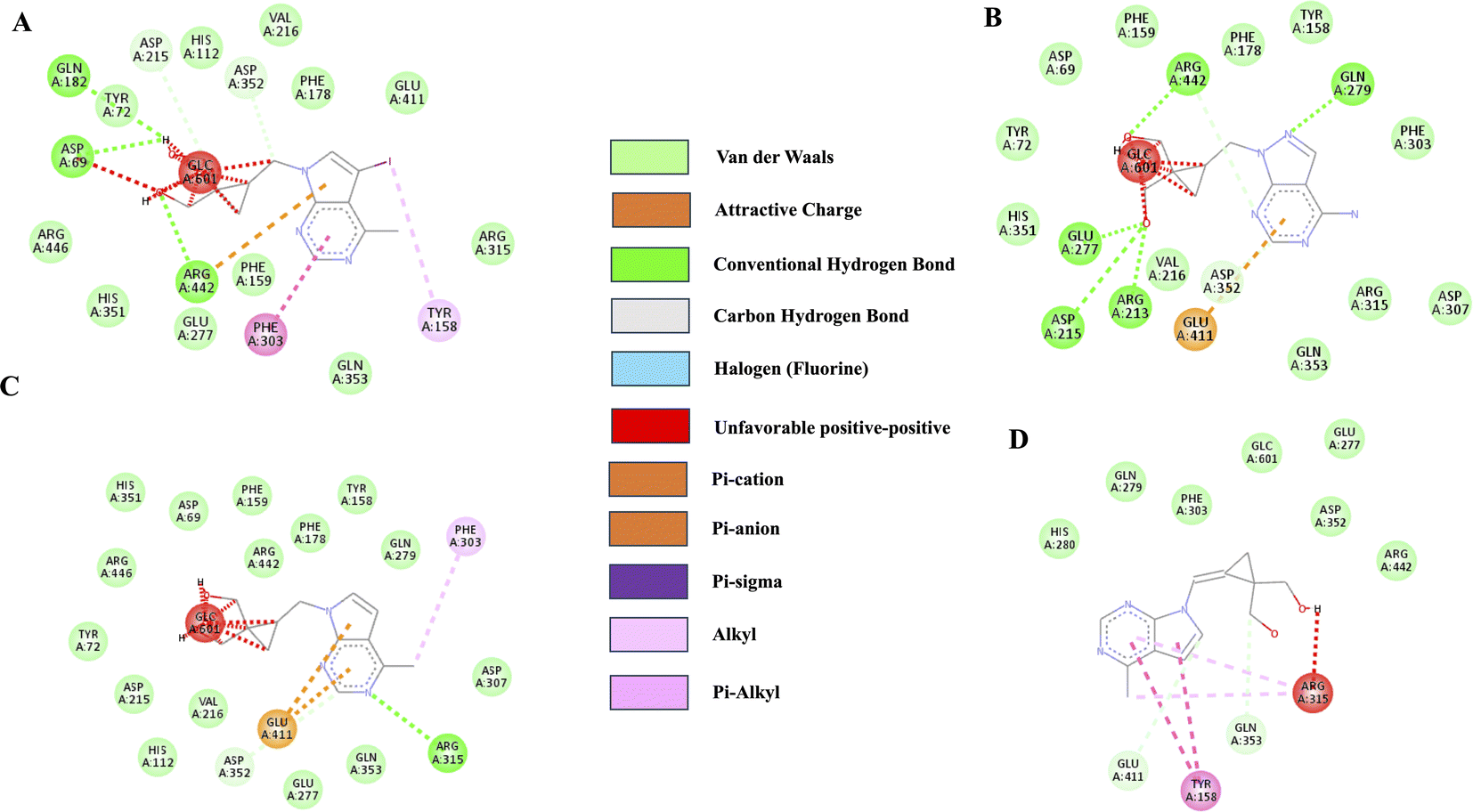 | ||
| Fig. 4 2D Ligand interaction diagrams for docking-compound: (A)8b (E-Isomer), (B) 10a (E-Isomer), (C) 8c (Z-Isomer) and (D) 8a (Z-Isomer) against α-glucosidase (3A4B.PDBID). | ||
2.4 Inhibition of α-glucosidase by C-4 substituted carbocyclic purine compounds
To assess the efficacy of the synthesized derivatives in treating diabetes, an investigation was conducted to determine the inhibitory capability of these compounds against α-glucosidase. The results of this investigation are presented in the table. Acarbose was employed as a positive control, exhibiting an IC50 value of 35.91 nmol. Two compounds, namely 8b (E-Isomer) and 10a (E-Isomer), exhibited favourable IC50 values of 43.292 nmol and 48.638 nmol, respectively. The compounds with relatively low IC50 values were identified as 8f (E-Isomer) and 8c (E-Isomer) with IC50 values of 91.714 nmol and 100.16 nmol, respectively (ESI Table 2†).2.5 Antimicrobial activity
In a recent research, Nguyen et al. conducted a study on the α-glucosidase inhibitory and antimicrobial properties of benzoyl phloroglucinols derived from the fruit of Garcinia schomburgkiana.44 The results of the antimicrobial assessment demonstrated that a newly discovered polyprenylated benzoyl phloroglucinol, referred to as schomburgkianone I (compound 1), as well as a previously identified compound known as guttiferone K (compound 2), exhibited inhibitory effects on Staphylococcus and Enterococcus faecium at the concentration tested. However, no activity against Acinetobacter baumannii was observed.44 In their study, Kaur et al. assessed the antibacterial and anti-biofilm properties of alpha-glucosidase inhibitors derived from the endophytic fungus Alternaria destruens. The efficacy of active fractions AF1 and AF2 derived from Aspergillus destruens was assessed against various bacterial strains including Salmonella enterica, Shigella flexneri, Vibrio cholerae, Escherichia coli, Pseudomonas aeruginosa, Staphylococcus aureus, and Candida albicans. The study demonstrated that AF1 had antibacterial properties against all the pathogens that were examined. In contrast, AF2 only exhibited antibacterial activity against specific pathogens, including Staphylococcus aureus, Vibrio cholerae, Salmonella enterica, and Candida albicans.40
Our findings suggested that among the synthesized compounds, 8bn (E-Isomer) and 10a (E-Isomer), exhibited favourable IC50 values of 43.292 nmol and 48.638 nmol, respectively. The positive control Acarbose has an IC50 value of 35.91 nmol. The IC50 value is associated with drug potency, i.e., the amount of drug necessary to induce the effect, the lower the IC50 value, the more effective the drug.45 Acarbose exhibits a better inhibitory effect against the enzyme α-glucosidase than the produced chemicals. However, in comparison to the derivative 8c (E-Isomer), with an IC50 value of 100.16 nmol, compounds 8b (E-Isomer), and 10a (E-Isomer), are more active, comparatively. Results of docking also showed that the compounds 8b (E-Isomer) and 10a (E-Isomer), showed good docking scores of −9.4 kcal mol−1 and −10.3 kcal mol−1, respectively.
New α-glucosidase inhibitors are always being explored. In a study, the alpha-glucosidase inhibitor action of novel N-substituted 5-benzylidene-2,4-thiazolidinedione derivatives were performed. All substances demonstrated various degrees of α-glucosidase inhibitory activity as compared to acarbose. When compared to acarbose (IC50 = 67.06 ± 1.24 μM), most compounds displayed extremely considerable inhibitory activity towards α-glucosidase.46 Flavonoids are known to play a function in hypoglycemia via blocking α-glucosidase. A study indicated that the diverse flavonoid compounds exhibited inhibitory activity and good docking score and the presence of Asp 215 was implicated in the hydrogen-bonding interaction between each flavonoid with the enzyme. Also, hydrophobic interactions with Tyr 72, Tyr 158, Phe 159, Phe 178, Asp 352, and Arg 442 were found. These amino acids are recurrent in our synthesized compounds as well.
3 Experimental section
3.1 Chemicals and reagents
All chemicals (reagent grade) used were purchased from Combi-Blocks (USA), Johnson Matthey Co., Ltd (USA) and Enamine Ltd (Ukraine). All the solvents used for the reaction are LR grade. Analytical thin-layer chromatography (TLC) was performed on pre-coated silica gel 60F254 plates, and visualization of TLC was achieved by UV light. Flash column chromatography was undertaken on silica gel (100–200 mesh). 1H NMR was recorded at 400 or 500 MHz, and chemical shifts were quoted in parts per million (ppm) referenced to 0.0 ppm for tetramethyl silane. The following abbreviations were used to describe peak splitting patterns when appropriate: br = broad, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, dd = doublet of doublet. Coupling constants, J, were reported in the hertz unit (Hz). ESI mass spectra were obtained on Agilent and Waters instruments. All the final compounds were purified on GRACE flash chromatography by using C18 reverse-phase columns. The mobile phase was a mixture of water (0.1% formic acid) and acetonitrile. Melting points were recorded on the Buchi M−560 instrument.
3.2 Chemistry
:H2O (7:3 ratio, 10 mL) at 0 °C, LiOH·H2O (1.5 eq.) was added and the reaction mixture was stirred at room temperature for 16 h. The reaction mixture was partitioned between water (30 mL) and DCM (30 mL × 3). The combined organic layer was washed with water (30 mL), and brine (30 mL), dried over anhydrous Na2SO4 and concentrated. The crude was purified by GRACE flash chromatography using a C18 column with water and acetonitrile as an eluent to give the final compound (Z)-(2-((4-methyl-7H-pyrrolo[2,3-d]pyrimidin-7-yl)methylene)cyclopropane-1,1-diyl)dimethanol 8a (Z-Isomer) as an off-white solid (125 mg, 42% Yield for 2 steps) and (E)-(2-((4-methyl-7H-pyrrolo[2,3-d]pyrimidin-7-yl)methylene)cyclopropane-1,1-diyl)dimethanol (8a-E-Isomer) as an off-white solid (105 mg, 40% yield for 2 steps).8a (Z-Isomer): 1H-NMR (400 MHz, DMSO-d6) δ 1.22 (s, 2H, Cyclopropyl CH2, shielding proton), 2.66 (s, 3H), 3.56–3.54 (d, 2H, J = 7.6 Hz, –C(CH2)), 3.66–3.64 (d, 2H, J = 10 Hz, –C(CH2)), 5.08 (brs, 2H, OH), 6.83 (d, 1H, J = 2.8 Hz, Ar–H), 7.68 (brs, 1H), 8.37–8.36 (d, 1H, J = 2.8 Hz, Ar–H), 8.69 (brs, 1H, Ar–H). 13C NMR (400 MHz, DMSO-d6), δ 10.74 (cyclo propyl carbon), 21.14, 30.49, 39.90 (–C attached to hydroxyl methyl) (merged in DMSO moisture),43.85(–C(CH2)2), 62.30 (–C(CH2)2), 101.10 (–C7, pyrrole ring),111.67, 114.76, 117.53, 125.44 (-CC), and 148.45, 151.25, 159.048 (Ar–C). MS (ESI): m/z = 246.5 (M + H)+.
8a (E-Isomer): FT-IR (KBr): ν(–O–H): 3363 cm−1; ν(–C–O): 1029 cm−1; ν(–C–C for aromatic ring): 1564 cm−1; ν(–C–N): 1325 cm−1. 1H-NMR (400 MHz, DMSO-d6) δ 1.54 (s, 2H, cyclopropyl CH2, deshielding proton), 2.66 (s, 3H), 3.54–3.51 (m, 4H, –C(CH2)), 4.78 (brs, 2H, OH), 6.88–6.87 (d, 1H, J = 3.6 Hz, Ar–H), 7.80 (brs, 1H), 7.95–7.94 (d, 1H, J = 3.6 Hz, Ar–H), 8.69 (brs, 1H, Ar–H). 13C NMR (400 MHz, DMSO-d6), δ 13.45 (Cyclo propyl Carbon), 21.17, 28.19, 39.70 (–C attached to hydroxyl methyl) (merged in DMSO moisture), 62.58 (–C(CH2)2), 101.32 (–C7, pyrrole ring),111.30, 115.16, 117.41, 124.47 (–CC), and 148.62, 151.32, 159.23 (Ar–C). MS (ESI): m/z = 246.5 (M + H)+.
C), and 148.18, 151.38, 159.77 (Ar–C). MS (ESI): m/z = 372.4 (M + H)+.8b (E-Isomer): Yield: 130 mg (35%). 1H-NMR (400 MHz, DMSO-d6) δ 1.32 (s, 2H, cyclopropyl CH2), 2.88 (s, 3H), 3.53–3.50 (d, J = 12.0 Hz, 2H, (CH2)), 3.68–3.66 (d, J = 8.0 Hz, 2H, (CH2)), 5.09 (brs, 2H, OH), 7.63 (brs, 1H), 8.67 (s, 1H, Ar–H), 8.70 (s, 1H, Ar–H). 13C NMR (400 MHz, DMSO-d6), δ 10.78, 20.44, 30.49, 39.91 (–C attached to hydroxyl methyl) (merged in DMSO moisture), 55.29 (halo substituted carbon, pyrrole ring), 62.22 (–C(CH2)2), 110.05, 115.64, 117.51, 130.79 (–CC), and 148.02, 151.31, 159.58 (Ar–C). MS (ESI): m/z = 372.4 (M + H)+.
C), and 147.71, 151.89, 159.59 (Ar–C). MS (ESI): m/z = 324.5 (M + H)+.8c (Z-Isomer): Yield: 130 mg (36%). FT-IR (KBr): ν(–O–H): 3098 cm−1; ν(–C–O):1032 cm−1; ν(–C–C for aromatic ring): 1562 cm−1; ν(–C–N): 1342 cm−1, ν(–C–Br): 604 cm−1. 1H-NMR (400 MHz, DMSO-d6) δ 1.33 (s, 2H, cyclopropyl CH2), 2.85 (s, 3H), 3.53–3.49 (m, 2H, (CH2)), 3.70–3.66 (m, 2H, (CH2)), 5.09 (brs, 2H, OH), 7.67 (brs, 1H), 8.65 (s, 1H, Ar–H), 8.73 (s, 1H, Ar–H). MS (ESI): m/z = 324.5 (M + H)+.
C), and 150.17, 151.06, 162.30 (Ar–C). MS (ESI): m/z = 262.5 (M + H)+.8d (E-Isomer): Yield: 105 mg (36%). FT-IR (KBr): ν(–O–H): 3327 cm−1; ν(–C–O):1010 cm−1; ν(–C–C for aromatic ring): 1562 cm−1; ν(–C–N): 1310 cm−1. 1H-NMR (400 MHz, DMSO-d6) δ 1.53 (s, 2H, cyclopropyl CH2), 3.54–3.46 (m, 4H, (CH2)), 4.06 (s, 3H), 4.74 (brs, 2H, OH), 6.70–6.69 (d, J = 4.0 Hz, 1H), 7.79 (s, 1H), 7.83–7.82 (d, J = 4.0 Hz, 1H, Ar–H), 8.49 (s, 1H, Ar–H). MS (ESI): m/z = 262.5 (M + H)+. 13C NMR (400 MHz, DMSO-d6), δ 13.41, 28.17, 39.92 (–C attached to hydroxyl methyl) (merged in DMSO moisture), 53.58, 62.58 (–C(CH2)2), 99.88, 104.66, 111.61, 115.23, 122.83 (–CC), and 150.31, 151.14, 162.33 (Ar–C). MS (ESI): m/z = 262.5 (M + H)+.
C), and 148.18, 151.38, 159.77 (Ar–C). MS (ESI): m/z = 388.0 (M + H)+.8e (E-Isomer): Yield: 150 mg (35%). FT-IR (KBr): ν(–O–H): 3327 cm−1; ν(–C–O):1010 cm−1; ν(–C–C for aromatic ring): 1562 cm−1; ν(–C–N): 1310 cm−1, ν(–C–I): 567 cm−1. 1H-NMR (400 MHz, DMSO-d6) δ 1.53 (s, 2H, cyclopropyl CH2), 3.54–3.46 (m, 4H, (CH2)), 4.06 (s, 3H), 4.74 (brs, 2H, OH), 6.70–6.69 (d, J = 4.0 Hz, 1H), 7.79 (s, 1H), 7.83–7.82 (d, J = 4.0 Hz, 1H, Ar–H), 8.49 (s, 1H, Ar–H). MS (ESI): m/z = 262.5 (M + H)+. 13C NMR (400 MHz, DMSO-d6), δ 13.41, 28.17, 39.92 (–C attached to hydroxyl methyl) (merged in DMSO moisture), 55.62 (halo substituted carbon, pyrrole ring), 53.58, 62.58 (–C(CH2)2), 99.88, 104.66, 111.61, 115.23, 122.83 (–CC), and 150.31, 151.14, 162.33 (Ar–C). MS (ESI): m/z = 388.0 (M + H)+.
C), and 147.48, 152.14, 164.37 (Ar–C). MS (ESI): m/z = 350.5 (M + H)+.8f (Z-Isomer): Yield: 170 mg (43%). FT-IR (KBr): ν(–O–H): 3273 cm−1; ν(–C–O): 984 cm−1; ν(–O–C): 1039 cm−1; ν(–C–C for aromatic ring): 1556 cm−1; ν(–C–N): 1338 cm−1, ν(–C–Br): 597 cm−1. 1H-NMR (400 MHz, DMSO-d6) δ 1.26–1.17 (m, 4H, Cyclo propyl CH2), 1.32 (s, 2H, cyclopropyl CH2), 3.09–3.05 (m, 1H, cyclopropyl CH), 3.54–3.49 (m, 2H (CH2)), 3.70–3.66 (m, 2H, (CH2)), 5.09–5.07 (t, J = 8.0 Hz, 2H), 7.66 (brs, 1H), 8.64 (s, 1H, Ar–H), 8.67 (s, 1H, Ar–H). MS (ESI): m/z = 262.5 (M + H)+. 13C NMR (400 MHz, DMSO-d6), δ 12.19, 30.48, 39.92 (–C attached to hydroxyl methyl) (merged in DMSO moisture), 62.23 (–C(CH2)2), 88.85, 111.07, 114.53, 115.63, 125.45 (–CC), and 147.30, 152.05, 164.15 (Ar–C). MS (ESI): m/z = 350.5 (M + H)+.
C), and 148.49, 152.42, 157.21 (Ar–C). MS (ESI): m/z = 247.1 (M + H)+.8g (E-Isomer): Yield: 145 mg (52%). FT-IR (KBr): ν(–O–H): 3273 cm−1; ν(–C–O): 984 cm−1; ν(–O–C): 1039 cm−1; ν(–C–C for aromatic ring): 1556 cm−1; ν(–C–N): 1338 cm−1, ν(–C–Br): 597 cm−1. 1H-NMR (400 MHz, DMSO-d6) δ 1.52 (s, 2H, cyclopropyl CH2), 3.50–3.43 (m, 4H (CH2)2), 4.71 (s, 2H), 6.76 (brs, 2H), 7.64 (brs, 1H), 7.77 (s, 1H, Ar–H), 8.15 (s, 1H, Ar–H). 13C NMR (400 MHz, DMSO-d6), δ 13.45, 28.07, 39.92 (–C attached to hydroxyl methyl) (merged in DMSO moisture), 62.56 (–C(CH2)2), 98.23 (C7 carbon), 102.86, 111.04, 115.00, 125.78 (–CC), and 148.65, 152.48, 157.26 (Ar–C). MS (ESI): m/z = 247.1 (M + H)+.
C, aromatic): 1565 cm−1; ν(–C–O): 984 cm−1; ν(–O–C): 1021 cm−1; ν(–C–N): 1270 cm−1, ν(–C–I): 647 cm−1. 1H-NMR (400 MHz, DMSO-d6) δ 1.43 (s, 2H, cyclopropyl CH2), 3.49–3.47 (d, J = 2H, –C(CH2)), 3.68–3.65 (d, J = 12 HZ, 2H, –C(CH2)), 4.70 (s 2H, OH), 6.72 (br, 1H, amine proton), 7.51 (s, 1H), 8.01 (br, 1H, Ar–H), 8.26 (s, 1H, Ar–H). 13C NMR (400 MHz, DMSO-d6), δ 10.66, 30.46, 39.91 (–C attached to hydroxyl methyl) (merged in DMSO moisture), 62.48 (–C(CH2)2), 92.27 (C7 carbon), 103.36, 112.20, 117.34 (–CC), and 151.85, 156.77, 157.81 (Ar–C). MS (ESI): m/z = 374.5 (M + H)+.8h (E-Isomer): Yield: 160 mg (38%). 1H-NMR (400 MHz, DMSO-d6) δ 1.51 (s, 2H, cyclopropyl CH2), 3.50–3.44 (m, 4H (CH2)2), 4.70 (s, 2H), 7.65–7.64 (t, J = 4 Hz, 1H, Ar–H), 8.25 (s, 1H, Ar–H). 13C NMR (400 MHz, DMSO-d6), δ 14.56, 28.34, 39.92 (–C attached to hydroxyl methyl) (merged in DMSO moisture), 62.61 (–C(CH2)2), 92.44 (C7 carbon), 103.19, 111.90, 117.58 (–CC), and 152.46, 156.43, 157.66 (Ar–C). MS (ESI): m/z = 374.5 (M + H)+.
C), and 151.88, 156.88, 158.24 (Ar–C). MS (ESI): m/z = 248.5 (M + H)+.10a (Z-Isomer): Yield: 145 mg (52%). FT-IR (KBr): ν(–O–H): 3183 cm−1; ν(–C–H): 2865 cm−1; ν(–N–H bending): 1665 cm−1; ν(–CC, aromatic): 1565 cm−1; ν(–C–O): 984 cm−1; ν(–C–O): 1032 cm−1; ν(–C–N): 1291 cm−1. 1H-NMR (400 MHz, DMSO-d6) δ 1.40 (s, 2H, cyclopropyl CH2), 3.52–3.48 (m, 2H (CH2)2), 3.65–3.61 (m, 2H (CH2)2), 4.87 (s, 2H), 7.58 (s, 1H, Ar–H), 8.03 (br, 1H), 8.24 (s, 2H, Ar–H). 13C NMR (400 MHz, DMSO-d6), δ 11.06, 30.56, 39.92 (–C attached to hydroxyl methyl) (merged in DMSO moisture), 62.66 (–C(CH2)2), 100.21, 112.69, 116.50 (C7 carbon), 134.14 (–CC), and 151.88, 156.88, 158.24 (Ar–C). MS (ESI): m/z = 248.5 (M + H)+.
C), and 151.85, 156.77, 157.81 (Ar–C). FT-IR (KBr): ν(–O–H): 3232 cm−1; ν(–C–H): 2871 cm−1; ν(–N–H bending): 1633 cm−1; ν(–CC, aromatic): 1564 cm−1; ν(–C–O): 1021 cm−1; ν(–C–N): 1270 cm−1; ν(–C–I): 647 cm−1. MS (ESI): m/z = 374.5 (M + H)+.10b (E-Isomer): Yield: 190 mg (45%). 1H-NMR (400 MHz, DMSO-d6) δ 1.51 (s, 2H, cyclopropyl CH2), 3.52–3.44 (m, 4H (CH2)2), 4.70 (s, 2H), 7.65–7.64 (t, J = 4.0 Hz, 1H, Ar–H), 8.25 (s, 2H, Ar–H). 13C NMR (400 MHz, DMSO-d6), δ 14.56, 28.34, 39.92 (–C attached to hydroxyl methyl) (merged in DMSO moisture), 62.61 (–C(CH2)2), 92.44 (C7 carbon), 103.19, 111.90, 117.58 (–CC), and 152.46, 156.43, 157.66 (Ar–C). MS (ESI): m/z = 374.5 (M + H)+.
C), and 145.14 (C7 carbon), 152.99, 155.98, 156.65, 157.72, 158.33 (Ar–C). FT-IR (KBr): ν(–C–O–C): 1243 cm−1; ν(–O–H): 3060 cm−1; ν(–C–H): 2039 cm−1; ν(–N–H bending): 1649 cm−1; ν(–CC, aromatic): 1565 cm−1; ν(–C–OH): 1039 cm−1; ν(–C–N): 1243 cm−1. MS (ESI): m/z = 416.7 (M + H)+.10c (E-Isomer): Yield: 190 mg (41%). 1H-NMR (400 MHz, DMSO-d6) δ 1.52 (s, 2H, cyclopropyl CH2), 3.51–3.47 (m, 4H (CH2)2), 4.72 (s, 2H), 7.21–7.13 (m, 5H), 7.46–7.44 (t, J = 8.0 Hz, 2H, Ar–H), 7.71–7.69 (d, J = 8.0 Hz, 2H), 7.79 (s, 1H), 8.30 (s, 2H, Ar–H). 13C NMR (400 MHz, DMSO-d6), δ 14.68, 28.31, 39.92 (–C attached to hydroxyl methyl) (merged in DMSO moisture), 62.74 (–C(CH2)2), 97.53, 112.20, 117.03, 119.03, 123.84, 127.51, 130.13 (–CC), and 144.85 (C7 carbon), 153.56, 156.17, 156.24, 157.38, 158.11 (Ar–C). MS (ESI): m/z = 416.7 (M + H)+.
3.3 α-Glucosidase inhibitory assay
The α-glucosidase inhibitory activity was assessed using the method described by Pistia Brueggeman and Hollingsworth, 2001,47 with minor adjustments. Plant extracts, in volumes of 50 μL, were incubated with different concentrations ranging from 12.5 to 400 μg mL−1. The incubation was carried out with 10 μL of α-glucosidase (maltase) enzyme solution (1 U mL−1) for 20 minutes at a temperature of 37 °C. Additionally, 125 μL of 0.1 M phosphate buffer (pH 6.8) was added. Following 20 minutes, the reaction was initiated by adding 20 μL of 1 M pNPG (substrate), and the resulting mixture was allowed to incubate for 30 minutes. The reaction was halted by introducing 0.1 N of Na2CO3 (50 μL), and the ultimate absorbance was quantified at 405 nm. Acarbose was employed as a positive control, with doses ranging from 12.5 to 400 μg mL−1. Enzyme activity was calculated as:| (ODblank − ODsample)/ODblank × 100 |
A single unit of the enzyme can be precisely defined as the quantity of α-glucosidase enzyme necessary to produce one micromole of the product (p-nitrophenol) from the substrate (p-nitrophenyl-α-D-glucopyranoside) within one minute. The IC50, which is the concentration needed to inhibit 50% of the enzyme activity, was determined by fitting a regression equation to a plot of concentration (ranging from 12.5–400 μg mL−1) on the x-axis and % inhibition on the y-axis for various extracts and fractions.48
3.4 Antimicrobial activity
The agar well diffusion method was employed to determine the antibacterial activity.49,50 The antibacterial efficacy of the synthesized compounds was assessed against four bacterial strains that included two Gram-positive bacteria, Staphylococcus epidermis (MTCC 2044), Bacillus cereus (MTCC2128) (Gram-positive) and two Gram-negative bacteria, Escherichia coli (MTCC2412), Klebsiella pneumonia (MTCC 2451). Different test compounds were introduced to wells perforated on an agar surface, with each well containing a concentration of 1 mg mL−1. The bacterial strains were cultured in sterilized Mueller Hinton Broth (MHB) and incubated for 18 hours at 37 °C. The radius of the inhibition zone (RIZ) surrounding each well was measured in millimetres to assess the antibacterial activity. The experiments were conducted in duplicate. Aspergillus niger was utilized to assess the antifungal activity of the compounds. The fungal cultures were plated using the selective media known as Potato Dextrose Agar. The plates were subjected to incubation at a temperature of 37 °C for a duration ranging from 72 to 96 hours. Following this, the plates were checked for the zone of inhibition.3.5 Swiss ADME
4 Conclusion
Individuals with diabetes are susceptible to numerous extra diseases due to a compromised immune system. Carbocyclic nucleosides possessing antibacterial and anti-inflammatory characteristics may offer significant advantages in the prevention of recurring infections and the facilitation of wound healing in patients with diabetes. Our research aimed to evaluate the α-glucosidase inhibitory activity of synthetic substances and their viability as therapeutic agents for the treatment of diabetes. Staphylococcus (a Gram-positive bacteria) and Klebsiella (a Gram-negative bacteria) were used as test organisms for antimicrobial activity, and the results showed that none of the chemicals examined showed any antimicrobial action. Compound 10b (Z-Isomer) was shown to inhibit the growth of the Gram-positive bacterium Bacillus cereus. At 100 μl, the compound showed a peak inhibitory effect of 2.2 ± 0.25 mm. Inhibitory concentrations of 10a (Z-Isomer) against the Gram-negative bacteria Escherichia coli were determined to be 75 μl (0.9 ± 0.05 mm) and 100 μl (1.2 ± 0.15 mm), respectively. There was no evidence of anti-fungal efficacy. Although the antibacterial properties of carbocyclic nucleosides show potential, additional investigation is required to clarify their modes of operation and assess their effectiveness and safety in diabetic individuals. The IC50 values of 8b (E-Isomer) and 10a (E-Isomer) were 43.292 nmol and 48.638 nmol, respectively, indicating that these two compounds are promising. Docking energies of 10.3 kcal mol−1 for compound 10a (E-Isomer) and −9.4 kcal mol−1 for compound 8b (E-Isomer) stood out among the produced compounds. Additional investigation is required to comprehensively comprehend the processes by which carbocyclic nucleosides function, the range of their effectiveness, and their clinical usefulness in the treatment of diabetes.Ethical statement
This article does not contain any studies with animals performed by any of the authors.Consent for publication
We authorize you to publish the article without any conflict.Data availability
All data generated or analysed during this study are included in this published article and its ESI† files.Author contributions
Mrs Kalyani Mallidi: investigation, formal analysis, methodology, and writing – original draft. Prof. Rambabu Gundla: validation, project administration and supervision. Dr Parameshwar Makam: conceptualisation, software, and writing – original draft. Dr Naresh Kumar Katari: data curation, and writing – review & editing. Prof. Sreekantha Babu Jonnalagadda: visualization, resources, and funding acquisition.Conflicts of interest
The authors declare that they have no competing interests/competing interests.Acknowledgements
The authors wish to thank the management of GITAM for the support and facilities.Notes and references
- L. The, Lancet, 2017, 389, 2163–2262 CrossRef PubMed.
- O. A. Ojo, H. S. Ibrahim, D. E. Rotimi, A. D. Ogunlakin and A. B. Ojo, Med. Nov. Technol. Devices, 2023, 19, 100247 CrossRef.
- C. J. Bailey and C. Day, Br. Med. Bull., 2018, 126(1), 123–137 CrossRef CAS PubMed.
- L. A. DiMeglio, C. Evans-Molina and R. A. Oram, Lancet, 2018, 391, 2449–2462 CrossRef PubMed.
- A. A. S. Akil, E. Yassin, A. Al-Maraghi, E. Aliyev, K. Al-Malki and K. A. Fakhro, J. Transl. Med., 2021, 19(1), 137 CrossRef CAS PubMed.
- K. J. Rotenberg, C. Bharathi and H. Davies, Eat. Behav., 2017, 26, 167–170 CrossRef.
- C. Langenberg and L. A. Lotta, Lancet, 2018, 391, 2463–2474 CrossRef CAS.
- I. Kyrou, C. Tsigos, C. Mavrogianni, G. Cardon, V. Van Stappen, J. Latomme, J. Kivelä, K. Wikström, K. Tsochev, A. Nanasi, C. Semanova, R. Mateo-Gallego, I. Lamiquiz-Moneo, G. Dafoulas, P. Timpel, P. E. H. Schwarz, V. Iotova, T. Tankova, K. Makrilakis, Y. Manios, J. Lindström, P. Schwarz, L. Annemans, I. Garamendi, M. Kontogianni, O. Androutsos, K. Tsoutsoulopoulou, C. Katsarou, E. Karaglani, I. Qira, E. Skoufas, K. Maragkopoulou, A. Tsiafitsa, I. Sotiropoulou, M. Tsolakos, E. Argyri, M. Nikolaou, E.-A. Vampouli, C. Filippou, K. Gatsiou, E. Dimitriadis, T. Laatikainen, P. Valve, E. Levälahti, E. Virtanen, R. Willems, I. Panchyrz, M. Holland, S. Liatis, C.-P. Lambrinou, A. Giannopoulou, L. Tsirigoti, E. Fappa, C. Anastasiou, K. Zachari, L. Rabemananjara, M. S. de Sabata, W. Ko, L. Moreno, F. Civeira, G. Bueno, P. De Miguel-Etayo, E. M. Gonzalez-Gil, M. I. Mesana, G. Vicente-Rodriguez, G. Rodriguez, L. Baila-Rueda, A. Cenarro, E. Jarauta, N. Usheva, N. Chakarova, S. Galcheva, R. Dimova, Y. Bocheva, Z. Radkova, V. Marinova, Y. Bazdarska, T. Stefanova, I. Rurik, T. Ungvari, Z. Jancsó, A. Nánási, L. Kolozsvári, C. Semánova, R. Martinez, M. Tong, K. Joutsenniemi and K. Wendel-Mitoraj, BMC Endocr. Disord., 2020, 20(1), 134 CrossRef PubMed.
- S. D. Ray, A. Hussain, A. Niha, M. Krmic, A. Jalshgrari, D. Genis and J. Reji, Anti Diabetic Agents, in Encyclopedia of Toxicology, ed. P. Wexler, Academic Press, Oxford, 4th edn, 2024, pp. 573–589 Search PubMed.
- C. M. Khoo, Diabetes Mellitus Treatment, ed. S.R. Quah, Academic Press, Oxford, 2017, pp. 288–293 Search PubMed.
- A. Berbudi, N. Rahmadika, A. I. Tjahjadi and R. Ruslami, Curr. Diabetes Rev., 2020, 16(5), 442–449 Search PubMed.
- G. Daryabor, M. R. Atashzar, D. Kabelitz, S. Meri and K. Kalantar, Front. Immunol., 2020, 11 Search PubMed.
- J. Chávez-Reyes, C. E. Escárcega-González, E. Chavira-Suárez, A. León-Buitimea, P. Vázquez-León, J. R. Morones-Ramírez, C. M. Villalón, A. Quintanar-Stephano and B. A. Marichal-Cancino, Public Health Front., 2021, 9, 559595 CrossRef.
- J. Casqueiro and C. Alves, Indian J. Endocrinol. Metab., 2012, 1(1), 2230–8210 Search PubMed.
- S. Erener, Mol. Metab., 2020, 39, 101044 CrossRef CAS PubMed.
- B. J. Eckhardt and R. M. Gulick, Infect. Dis., 2017, 1293–1308 Search PubMed.
- P. D. Kennewell, Compr. Med. Chem. II, 2007, 1, 97–249 Search PubMed.
- S. Rachakonda and L. Cartee, Curr. Med. Chem., 2004, 11(6), 775–793 CrossRef CAS PubMed.
- D. Xia, F. Wang and M. J. Parmely, Biochem. Pharmacol., 2000, 60(5), 717–727 CrossRef CAS PubMed.
- S. M. Daluge, S. S. Good, M. B. Faletto, W. H. Miller, M. H. St Clair, L. R. Boone, M. Tisdale, N. R. Parry, J. E. Reardon, R. E. Dornsife, D. R. Averett and T. A. Krenitsky, Antimicrob. Agents Chemother., 1997, 41(5), 1082–1093 CrossRef CAS PubMed.
- J. Huuskonen, D. J. Livingstone and D. T. Manallack, SAR QSAR Environ. Res., 2008, 19(3–4), 191–212 CrossRef CAS PubMed.
- M. Yazdanian, S. L. Glynn, J. L. Wright and A. Hawi, Pharm. Res., 1998, 15(9), 1490–1494 CrossRef CAS PubMed.
- P. Artursson, A.-L. Ungell and J.-E. Löfroth, Pharm. Res., 1993, 10(8), 1123–1129 CrossRef CAS PubMed.
- M. Sharifi and T. Ghafourian, AAPS J., 2014, 16(1), 65–78 CrossRef CAS PubMed.
- M. V. Varma, Y. Lai and A. F. El-Kattan, Adv. Drug Delivery Rev., 2017, 116, 92–99 CrossRef CAS PubMed.
- S. Struck, U. Schmidt, B. Gruening, I. S. Jaeger, J. Hossbach and R. Preissner, Toxicity versus Potency: Elucidation of Toxicity Properties Discriminating between Toxins, Drugs, and Natural Compounds, Imperial College Press, 2008, pp. 231–242 Search PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23(1–3), 3–25 CrossRef CAS.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46(1–3), 3–26 CrossRef CAS PubMed.
- I. Kola and J. Landis, Nat. Rev. Drug Discovery, 2004, 3(8), 711–716 CrossRef CAS PubMed.
- D. F. Veber, S. R. Johnson, H.-Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45(12), 2615–2623 CrossRef CAS.
- P. D. Leeson and A. M. Davis, J. Med. Chem., 2004, 47(25), 6338–6348 CrossRef CAS.
- M. P. Gleeson, A. Hersey, D. Montanari and J. Overington, Nat. Rev. Drug Discovery, 2011, 10(3), 197–208 CrossRef CAS PubMed.
- S. Khojasteh, H. Wong and C. C. A. Hop, Drug Metabolism and Pharmacokinetics Quick Guide, Springer, New York, 2011, pp. 165–181 Search PubMed.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7(1), 42717 CrossRef PubMed.
- M. Hocek, P. Nauš, R. Pohl, I. Votruba, P. A. Furman, P. M. Tharnish and M. J. Otto, J. Med. Chem., 2005, 48(18), 5869–5873 CrossRef CAS PubMed.
- L. A. Agrofoglio, I. Gillaizeau and Y. Saito, Chem. Rev., 2003, 103(5), 1875–1916 CrossRef CAS.
- Y.-L. Qiu, M. B. Ksebati, R. G. Ptak, B. Y. Fan, J. M. Breitenbach, J.-S. Lin, Y.-C. Cheng, E. R. Kern, J. C. Drach and J. Zemlicka, J. Med. Chem., 1998, 41(1), 10–23 CrossRef CAS.
- R. J. Rybak, J. Zemlicka, Y. L. Qiu, C. B. Hartline and E. R. Kern, Antiviral Res., 1999, 43(3), 175–188 CrossRef CAS.
- R. J. Rybak, C. B. Hartline, Y. L. Qiu, J. Zemlicka, E. Harden, G. Marshall, J. P. Sommadossi and E. R. Kern, Antimicrob. Agents Chemother., 2000, 44(6), 1506–1511 CrossRef CAS PubMed.
- Z. Yan, E. R. Kern, E. Gullen, Y. C. Cheng, J. C. Drach and J. Zemlicka, J. Med. Chem., 2005, 48(1), 91–99 CrossRef CAS PubMed.
- D. Ramesh, B. G. Vijayakumar and T. Kannan, ChemMedChem, 2021, 16(9), 1403–1419 CrossRef CAS PubMed.
- M. Kasula, M. Toyama, R. Samunuri, F. Rozy, M. Yadav, C. Bal, A. K. Jha, M. Baba and A. Sharon, Bioorg. Med. Chem. Lett., 2016, 26(16), 3945–3949 CrossRef CAS PubMed.
- R. Samunuri, A. K. Jha and C. Bal, Nucleosides, Nucleotides Nucleic Acids, 2019, 38(6), 391–399 CrossRef CAS PubMed.
- H. T. Nguyen, T. T. Nguyen, T. H. Duong, N. M. Tran, C. H. Nguyen, T. H. Nguyen and J. Sichaem, Molecules, 2022, 27(8), 2574 CrossRef CAS PubMed.
- C. T. Meyer, D. J. Wooten, B. B. Paudel, J. Bauer, K. N. Hardeman, D. Westover, C. M. Lovly, L. A. Harris, D. R. Tyson and V. Quaranta, Cell Syst., 2019, 8(2), 97–108 CrossRef CAS PubMed.
- V. M. Patil, K. N. Tilekar, N. M. Upadhyay and C. S. Ramaa, ChemistrySelect, 2022, 7(1), e202103848 CrossRef CAS.
- G. Pistia-Brueggeman and R. I. Hollingsworth, Tetrahedron, 2001, 57(42), 8773–8778 CrossRef CAS.
- A. Bhatia, B. Singh, R. Arora and S. Arora, BMC Complementary Altern. Med., 2019, 19(1), 74 CrossRef.
- S. Magaldi, S. Mata-Essayag and C. Hartung de Capriles, Int. J. Infect. Dis., 2004, 8, 39–45 CrossRef CAS.
- C. Valgas, S. M. De Souza, E. F. Smânia and A. Smânia Jr, Braz. J. Microbiol., 2007, 38, 369–380 CrossRef.
- B. Bakchi, A. D. Krishna, E. Sreecharan, V. B. J. Ganesh, M. Niharika, S. Maharshi, S. B. Puttagunta, D. K. Sigalapalli, R. R. Bhandare and A. B. Shaik, J. Mol. Struct., 2022, 1259, 132712 CrossRef CAS.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25(13), 1605–1612 CrossRef CAS.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31(2), 455–461 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra00304g |
| This journal is © The Royal Society of Chemistry 2024 |