
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReusable magnetic mixture of CuFe2O4–Fe2O3 and TiO2 for photocatalytic degradation of pesticides in water†
Dmytro Danilian *a,
Franziska Maria Bundrücka,
Arvo Kikasa,
Tanel Käämbrea,
Hugo Mändara,
Sandro Lehnerb,
Alexander Gogoscd,
Jekaterina Kozlovaa,
Mati Kooka,
Valter Kiiska,
Joosep Linke,
Raivo Sterne,
Angela Ivaskf,
Vambola Kisand*a and
Rainer Pärnaa
*a,
Franziska Maria Bundrücka,
Arvo Kikasa,
Tanel Käämbrea,
Hugo Mändara,
Sandro Lehnerb,
Alexander Gogoscd,
Jekaterina Kozlovaa,
Mati Kooka,
Valter Kiiska,
Joosep Linke,
Raivo Sterne,
Angela Ivaskf,
Vambola Kisand*a and
Rainer Pärnaa
aInstitute of Physics, University of Tartu, W.Ostwaldi 1, Tartu 50411, Estonia. E-mail: vambola.kisand@ut.ee; dmytro.danilian@ut.ee
bLaboratory of Advanced Fibers, Department of Materials Meet Life, Swiss Federal Laboratories for Materials Science and Technology (Empa), Lerchenfeldstrasse 5, St. Gallen 9014, Switzerland
cLaboratory for Particles-Biology Interactions, Department of Materials Meet Life, Swiss Federal Laboratories for Materials Science and Technology (Empa), Lerchenfeldstrasse 5, St. Gallen 9014, Switzerland
dNanoparticle Systems Engineering Laboratory, Institute of Energy and Process Engineering (IEPE), Department of Mechanical and Process Engineering (D-MAVT), ETH Zurich, Sonneggstrasse 3, Zurich 8092, Switzerland
eNational Institute of Chemical Physics & Biophysics, Akadeemia tee 23, 12618, Tallinn, Estonia
fInstitute of Molecular and Cell Biology, University of Tartu, Riia 23, 51010 Tartu, Estonia
First published on 16th April 2024
Abstract
Photocatalysis is a promising treatment method to remove pollutants from water. TiO2-P25 is a commercially available model photocatalyst, which very efficiently degrades organic pollutants under UVA light exposure. However, the collection and the recovery of TiO2-P25 from cleaned water poses significant difficulties, severely limiting its usability. To address this challenge, we have prepared a sintered mixture of TiO2-P25 nanomaterials and magnetic CuFe2O4–Fe2O3 nanocomposites. The mixture material was shown to contain spinel ferrite, hematite and maghemite structures, copper predominantly in Cu2+ and iron predominantly in Fe3+ state. The CuFe2O4–Fe2O3 and TiO2-P25 mixture demonstrated magnetic collectability from processed water and photocatalytic activity, which was evidenced through the successful photodegradation of the herbicide 2,4-D. Our findings suggest that the sintered mixture of CuFe2O4–Fe2O3 and TiO2-P25 holds a promise for improving photocatalytic water treatment, with the potential to overcome current photocatalyst recovery issues.
1 Introduction
Pesticides are chemical compounds that are used in increasing quantities to inhibit the growth or survival of unwanted organisms. Due to their extensive application on soil but also spills and disposal, pesticides may accumulate in different environmental compartments.1 This can lead to contamination of surface water or groundwaters.2 A recent study in Estonia3,4 has demonstrated that both in the water table and in borehole wells, the level of pesticides can exceed the respective regulatory threshold values indicating that pesticide-polluted water, particularly in agricultural and rural areas, may pose a potential health hazard.5In order to overcome the limitations of more conventional water purification procedures at large-scale facilities several new local drinking water treatment technologies (e.g., ozone, ultraviolet, silver ion, ferrate) have been recently developed.6,7 As a promising approach for water treatment, photocatalysis stands out for its innovative potential.8–12 Photocatalysis is a process facilitating light-induced modification of a substance. Therefore, photocatalyst materials can often be useful in transforming harmful pollutants into harmless substances under sunlight. Currently, photocatalytic degradation of harmful organic pollutants can be realistically applied to small to medium size water purification setups, once sufficient efficiency of the process as well as recyclability of the photocatalyst materials are ensured.13–15 An important determinant is also non-toxicity of the reaction end-products, especially when photocatalytic degradation concerns phenolic pollutants.16
During the last three decades, titania (TiO2) has attracted much attention as a robust high-efficiency solar light activated photocatalyst material. However, due to its rather large band gap, TiO2 absorbs only a fraction of sunlight. Its band gap depends on crystal structure and is typically 3.2 eV in the case of anatase and 3.0 eV in the case of rutile, which means it can only use a small part of the solar spectrum. Evonik Aeroxide P25 (formerly Degussa P25) is a flame-made multiphasic TiO2 nanoparticle powder containing anatase and rutile. The past decades have witnessed the wide applications range of P25 as a benchmark material for studying photocatalytic mechanisms, materials, and processes.13 However, the photocatalytic efficiency of TiO2 is restricted by several factors, such as limited absorption in the solar spectrum, short lifetime of photogenerated electrons, and poor collectability after its use.13 Therefore, different methods have been proposed to improve the performance of TiO2. One of such changes involves combination of TiO2 with other oxide semiconductors,17,18 which is expected to contribute both to the increased photocatalytic performance as well as to collectability and therefore, improved reusability.
One viable option to improve the collectability of photocatalytic materials is to supply them with magnetic properties. One known magnetic photocatalyst is ferrimagnetic maghemite (γ-Fe2O3).19 However, maghemite is a material with relatively low stability. The most common form of Fe2O3, hematite (α-Fe2O3) is a stable material with fairly narrow bandgap (2.2 eV), but with very weak magnetic moment due to its canted antiferromagnetic order.20 In addition to poor magnetic properties, hematite has particularly rapid recombination of the photogenerated charge carriers, which critically limits its photocatalytic potential.21,22 A promising strongly magnetic photocatalytic material with high magnetic stability near room temperature (Curie temperature as high as ∼750 K) is CuFe2O4.23 Compared with binary iron oxide-based materials, this material can be seen as potentially more robust against photocorrosion or oxidative changes at ambient conditions as it does not contain ferrous iron (like magnetite) and does not depend on vacancies in structure (like maghemite), both of which tend to lead to fully oxidated stable hematite phase in an oxidative environment. CuFe2O4 material has an even smaller band gap value (∼1.6 eV) than iron oxide materials but again, its photocatalytic efficiency is limited due to charge recombination, inherent for the strong light absorbance, small band gap materials.23 Therefore, currently according to our knowledge, there is no material with sufficient photocatalytic activity and decent magnetic collectability.
In this study, we propose a combination of TiO2 and α-Fe2O3 with CuFe2O4 to design magnetically collectable photocatalyst material.
Coprecipitation method was used to synthesize CuFe2O4–Fe2O3 material with controlled particle size distribution. The synthesized CuFe2O4–Fe2O3 was then mechanically mixed with TiO2-P25, the mix was annealed at 600 °C to allow particles to sinter and form enduring mechanical contact. Finally, the nonmagnetic (titania) particles that had not attached to any magnetic material were excluded by using a strong permanent magnet to lift and collect the particle agglomerates that contained magnetic components. The composition of the obtained material was characterised using multiple analytical methods. The functionality of the material was assessed by measuring the photocatalytic degradation rate of aqueous 2,4-dichlorophenoxyacetic acid (2,4-D) solution as a model for a commonly used pesticide for weed control.24 We demonstrate that the sintered mixture of CuFe2O4–Fe2O3 and TiO2-P25 can be magnetically collected from water solution and reused as a sustainable and effective method for pesticide removal from drinking water.
2 Experimental
2.1 Synthesis of materials and magnetic extraction
The CuFe2O4–Fe2O3 was synthesised using a co-precipitation method. The precursor reagents Cu(NO3)2 × 3H2O (99% purity, trace metals based), the Fe(NO3)3 × 9H2O (99% purity, trace metals based) and NH4OH (98% purity) were all obtained from Sigma-Aldrich. The salts were dissolved in deionised (DI) water (8.7 MΩ cm) to produce solution containing 0.01 M Cu(NO3)2 × 3H2O and 0.02 M Fe(NO3)3 × 9H2O to which a 3.34 M aqueous solution of NH4OH was added dropwise until the pH of the solution reached approximately 11 and the precipitation was complete. The obtained precipitate was washed with DI water five times. Between every wash cycle, the precipitate was left at rest to settle for 15 minutes. Approximately 70% of the total liquid was carefully discarded at each wash cycle. To separate the precipitate from the remaining solution, centrifugation was performed at 2000 rpm for 5 minutes. Obtained precipitates were dried at a temperature of 60 °C for 12 hours. The dried powder was then annealed at 700 °C for 4 hours.To obtain TiO2-P25 with 20 wt% of CuFe2O4–Fe2O3, designated as TCF20-Sc (see ESI Table S1†), the annealed CuFe2O4–Fe2O3 powder was manually mechanically mixed with TiO2 (Evonik Aeroxide P25, formerly Degussa P25) in 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :80 weight ratio using mortar and pestle, for a duration of 30 minutes. The resulting powder mixture was then subjected to additional annealing for 24 hours at 600 °C.
:80 weight ratio using mortar and pestle, for a duration of 30 minutes. The resulting powder mixture was then subjected to additional annealing for 24 hours at 600 °C.
The process of magnetic extraction of the photocatalyst powder was carried out using a NdFeB N45-grade magnet of 20 kg pull force (dimensions 30 mm diameter and 10 mm height). The powder was positioned on a plastic weighing dish approximately 20 mm below a borosilicate glass barrier completely covering the surface area of a plastic weighing dish, then the magnet was placed above the borosilicate barrier. The magnet attracted powder from down to up onto the borosilicate barrier, separating it from weak/non-magnetic particles. Next, the plastic dish with initial material was changed to an empty one, and the magnet was carefully removed from above the barrier, allowing the magnetically separated powder to fall into the new plastic weighing dish. This separation process was repeated five times. As a result, from the initial mass approximately 40–50% was identified as having weak or non-magnetic properties. This portion was designated as “non-magnetic”, referenced as TCF-NM. Adversely, the magnetically extracted fraction of TCF20-S material was designated as TCF-M. The TCF-M material, which has been recycled five times and subsequently characterised, was designated as TCF-M-5c. We note that the 20:80 wt% ratio of P25 and CuFe2O4–Fe2O3 in the ready mixture only applies for TCF20-S. After magnetic separation the respective ratios for TCF-NM and TCF-M were different.
As reference materials, TiO2 (Evonik Aeroxide P25) and the same TiO2-P25 annealed at 600 °C (referred as TiO2-P25 @ 600 °C in text) were used. The latter provides a reference to possible changes in TiO2 component itself when the mixtures were later annealed at similar temperature in order to achieve mechanically robust attachment of the Cu/Fe oxide to the titania particles.
2.2 Material characterisation
178 nm) from rotating anode working at 8.1 kW (45 kV and 180 mA). Powder diffraction database PDF-2 (version 2023) was used for qualitative analysis of phases. Rietveld analysis (program TOPAS 6) was used for a rough estimation of concentrations of the phases.For repeated use, TCF-M material, post 2,4-D degradation, was reclaimed using a magnet, attracting the material to a borosilicate glass barrier. This recovered material was then thoroughly rinsed five times with DI and dried in an oven at 70 °C for 12 hours.
3 Results and discussion
3.1 Morphology of particles and element distribution
Distinct differences in particle morphology and element distribution were observed between TiO2-P25 @ 600 °C, CuFe2O4–Fe2O3 materials and mixtures TCF20-S, TCF-NM, TCF-M, and TCF-M-5c (Fig. 1). STEM results indicate that TiO2-P25 @ 600 °C particles agglomerated and exhibited higher polydispersity compared to the initial TiO2-P25 material.30 The relatively high polydispersity of TiO2-P25 @ 600 °C (particle sizes approx. ≈200 nm down to tens of nm) can be seen from ESI Fig. S1†. Compared with TiO2 materials, CuFe2O4–Fe2O3 showed significantly larger grain sizes (roughly 1 μm to ≈100 nm). However, the mixture materials TCF20-S, TCF-NM and magnetically extracted TCF-M all had similar grain size and morphology with respect to the TiO2. STEM-HAADF (Fig. 1) indicated the presence of elements as expected from materials composition. In TiO2-P25 @ 600 °C the predominant element was Ti while no Cu of Fe were detected. In all other materials, Fe and Cu signals were detected. Interestingly, copper appeared to be homogenously distributed in most of the materials, whereas Fe was observed in nanoscopic clusters with a size of a few nm. After magnetic extraction, both Fe and Cu were only barely detectable in the residual TCF-NM, suggesting successful magnetic removal of Fe rich material from TCF20-S. In contrast, the extracted magnetic fraction (TCF-M) appeared slightly richer in general Fe content as well as in nanoscopic Fe clusters throughout the material, while the distribution of Cu was uniform. The presence of Fe and Cu was also seen in the recycled material TCF-M-5c as seen both, from STEM-EDX maps (see Fig. 1 and ESI S2†) as well as from energy-dispersive X-ray spectra (see ESI Fig. S3†). | ||
| Fig. 1 STEM-HAADF micrographs along with corresponding elemental maps of CuFe2O4–Fe2O3, TiO2-P25 @ 600 °C, TCF20-S, TCF-NM, TCF-M, and TCF-M-5c. | ||
3.2 Crystal phase composition
The crystal phase of the materials was characterized using XRD and Raman spectroscopy. According to XRD, pure commercial TiO2-P25 without thermal treatment consisted predominantly of anatase with a smaller fraction of rutile, as expected (Fig. 2 and Table 1). Annealing the TiO2-P25 at 600 °C for 24 h resulted in partial TiO2 phase transformation from anatase to rutile. The anatase to rutile ratio in the annealed material was close to 1. Indeed, anatase to rutile phase transformation temperature is known to be in the temperature range from 600 to 900 °C.31 | ||
| Fig. 2 XRD patterns of 1 – CuFe2O4–Fe2O3, 2 – TCF-M, 2c5 – TCF-M-5c, 3 – TCF20-S and 4 – TCF-NM, 5 – TiO2-P25 @ 600 °C, 6 – TiO2-P25. The positions of reflection of anatase (A), rutile (R), CuFe2O4 spinel (C) and hematite (H) are shown as vertical bars at the upper part of the figure. | ||
| Material | Crystal phase, wt% | ||||||
|---|---|---|---|---|---|---|---|
| Anatase | Rutile | α-Fe2O3 | CuFe2O4 | α-SiO2 | γ-Fe2O3 | R'wp% | |
| CuFe2O4–Fe2O3 | — | — | 53.5 | 33.9 | 1 | 11.6 | 13.7 |
| TiO2-P25 | 88.9 | 11.1 | — | — | — | — | 10.3 |
| TiO2-P25 @ 600 °C | 51 | 49 | — | — | — | — | 7.7 |
| TCF20-S | 6.3 | 84.5 | 5.7 | 2.6 | 0.3 | 0.6 | 6.3 |
| TCF-NM | 10.8 | 87.2 | 0.9 | 0.5 | 0.2 | 0.4 | 6.1 |
| TCF-M | 6.7 | 69.6 | 12.4 | 8.8 | 1 | 1.5 | 7.2 |
| TCF-M-5c | 6.3 | 88.4 | 5.7 | 3.2 | 0.4 | — | 6.4 |
The XRD pattern of CuFe2O4–Fe2O3 demonstrated the presence of three major phases, hematite (α-Fe2O3), CuFe2O4 spinel and maghemite (γ-Fe2O3), along with minor phase such as α-SiO2. The source of α-SiO2 could be impurities in precursor materials (Table 1). The predominant phase in TCF20-S diffractograms (Fig. 2) was rutile. The other phases in this material were anatase, hematite, CuFe2O4 spinel, and maghemite. After magnetic extraction, the material TCF-M was composed of the same phases as the TCF20-S but with different weights. The TCF-M contained relatively more hematite, but especially CuFe2O4 spinel and maghemite phases. Thus, TCF-M contained magnetic phases in higher concentration compared to TCF20-S. The nonmagnetic leftover material (TCF-NM) displayed primarily rutile and anatase fractions, along with reduced hematite and traces of CuFe2O4 spinel and maghemite (Table 1). Those results strongly suggest that the process of magnetic extraction was efficient in selection of the desired TCF-M materials. XRD results of TCF-M-5c revealed consistent phase composition with TCF-M, but with slightly higher rutile and CuFe2O4, suggesting stability and minimal transformation after five recycling cycles. In addition to XRD, Raman spectroscopy was used to analyse the phase composition of materials (Fig. 3). Raman spectrum confirmed that in TiO2-P25, Raman bands at 144 cm−1 (Eg), 399 cm−1 (B1g), 513 cm−1 (A1g), 519 cm−1 (B1g) and 640 cm−1 (Eg) related to anatase phase, were present.32,33 In case of TiO2-P25 @ 600 °C additional bands at 447 cm−1 (Eg) and 612 cm−1 (A1g) related to rutile phase were detected.34
 | ||
| Fig. 3 The Raman spectra of 1 – CuFe2O4–Fe2O3, 2 – TCF-M, 2c5 – TCF-M-5c, 3 – TCF20-S, and 4 – TCF-NM, 5 – TiO2-P25 @ 600 °C, 6 – TiO2-P25. Dashed lines and capital letters present the positions of TiO2 anatase (A), TiO2 rutile (R), CuFe2O4 (C) or α-Fe2O3 (hematite, H) bands and γ-Fe2O3 (maghemite, M). | ||
Interpretation of Raman spectrum of CuFe2O4–Fe2O3, TCF20-S, TCF-NM, TCF-M and TCF-M-5c (Fig. 3) was not quite straightforward due to overlapping peaks. We assigned the observed Raman bands at 219 cm−1, 283 cm−1, 397 cm−1, 491 cm−1 and 600 cm−1 to hematite35 and CuFe2O4.23 Our interpretation is based on previous findings, where Raman active modes of α-Fe2O3 (tetragonal crystal symmetry) have been detected at 225, 280, 400, 480, 615, and 690 cm−1.36 The Raman active mods of CuFe2O4 spinel have been detected at 215, 278, 481, 586, and 656 cm−1, which are assigned to F2g(1), Eg, F2g(2), F2g(3), and A1g modes, respectively.36 The wide band at 680 cm−1 can be related to maghemite.35 Raman bands of TCF20-S, TCF-NM, TCF-M and TCF-M-5c were mostly related to rutile with minor contribution to anatase. However, the materials demonstrated a wide Raman band at ∼260 cm−1, which arises due to multiphonon process in rutile.37
3.3 Elemental composition
The elemental analysis of the CuFe2O4–Fe2O3 material confirmed the expected composition of the copper-iron oxide system (Table 2). The molar calculations, derived from the ICP-OES data, indicated a ∼6:1 ratio of iron to copper, in line with the presence of (binary) iron oxides as indicated by the XRD analysis (see Section 3.2) and the magnetisation measurements (see Section 3.7). This Fe:Cu ratio of ∼6:1, which roughly corresponds to 1:2 spinel:hematite molar ratio (CuFe2O4 molar mass is 239.1 amu, Fe2O3 molar mass is 159.6 amu), was found to be comparable after introduction of CuFe2O4–Fe2O3 into the TCF20-S mixture. Also, after mixing, Cu and Fe content decreased in neat proportion to each other and in correspondence to the intended composition of 80 wt% TiO2 and 20 wt% spinel-hematite mixture. The relative amount of Cu + Fe in the TCF20-S mixture closely corresponded to the expected value for the mixture of CuFe2O4–Fe2O3 (20% wt.) and TiO2 (80% wt).
| Material | wt% | Atomic% (metals based) | Fe/Cu (at.) | Ti/Cu (at.) | (Cu + Fe) %at. (metals based) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cu | Fe | Ti | Sum (metals) | Cu | Fe | Ti | ||||
| CuFe2O4–Fe2O3 | 11.97 | 62.47 | 0 | 73.43 | 14.5 | 85.5 | 0 | 5.9 | 0 | 100.0 |
| TCF20-S | 2.64 | 14.07 | 49.01 | 65.72 | 3.2 | 19.1 | 77.7 | 6 | 24.6 | 22.3 |
| TCF-NM | 0.42 | 2.45 | 55.82 | 58.69 | 0.55 | 3.6 | 95.8 | 6.6 | 175.8 | 4.2 |
| TCF-M | 6.75 | 33.42 | 30.89 | 71.07 | 7.9 | 44.3 | 47.8 | 5.6 | 6.1 | 52.2 |
| TCF-M-5c | 9.25 | 45.62 | 10.15 | 65.02 | 12.4 | 69.5 | 18.1 | 5.6 | 1.5 | 81.2 |
Upon the magnetic extraction, distinct compositional shifts were noted in the TCF-NM, and TCF-M samples compared with TCF20-S. Specifically, the TCF-NM sample exhibited a marked increase in the Ti/Fe ratio relative to TCF20-S, indicating that the nonmagnetic part remaining after extraction was mainly TiO2. Conversely, the TCF-M material displayed a more balanced Ti and Fe distribution, indicative of successful incorporation of TiO2 within the magnetic fraction. Recycled TCF-M-5c revealed similar atomic Fe/Cu ratio as a TCF-M.
These compositional findings align closely with our initial synthetic expectations, confirming the accuracy of our stoichiometric formulations. While slight variations were observed, they remained within the expected range of experimental and measurement precision. The ICP-OES analysis has thus provided a solid validation of the intended elemental makeup of our synthesized materials.
3.4 Composition of materials surface
To obtain information on elemental oxidation states of the very top layer of the materials' surface, XPS measurements were conducted. First of all, as expected, the XPS spectra of TiO2-P25 @ 600° in the Fe 2p and Cu 2p region demonstrated only background noise (i.e. absence Fe and Cu impurities) and therefore is not presented in Fig. 1.XPS spectra in Ti 2p region showed a spin–orbit doublet with peaks at 458.7 eV (2p3/2) and 464.4 eV (2p1/2) characteristic of Ti4+ oxidation state39,40 for all materials containing Ti (Fig. 4a), as expected. No Ti3+ impurity contribution was found in any material, as shown (Fig. 4a). Fe 2p3/2 photoelectron peak (Fig. 4b) was observed at 710.8 eV in case of CuFe2O4–Fe2O3, with a satellite peak at 4.7 eV towards higher binding energies, indicating Fe3+ charge state. Based on Fe 2p binding energies in XPS spectra, it is inferred that the iron ions present in materials synthesised in CuFe2O4–Fe2O3 were in Fe3+ oxidation state.41,42 No Fe2+ contributions were identified in Fe 2p spectra. This agrees with the XRD results, which indicated the presence of only Fe3+ compounds.
 | ||
| Fig. 4 Photoelectron spectra of the Ti 2p region (a), Cu 2p region (b) and the Fe 2p region (c) of 1 – CuFe2O4–Fe2O3, 2 – TCF-M, 2c5 – TCF-M-5c 3 – TCF20-S, 4 – TCF-NM, 5 – TiO2-P25 @ 600 °C. | ||
Fe 2p spectra of mixed materials aligned with expectations (presence of Fe3+), except for TCF-NM. This particular material uniquely demonstrated the absence of Fe 2p peaks, consistent with the anticipated outcome of the magnetic extraction process.
Cu 2p photoelectron spectra for all relevant samples are displayed in Fig. 4c. Cu 2p spectrum of pure CuFe2O4–Fe2O3 had Cu 2p3/2 at 932.5 eV and Cu 2p1/2 at 953 eV. The spectrum also showed a strong satellite line, characteristic of Cu2+,43–45 as expected for Cu contained in ferrite. In the case of TCF20-S, TCF-NM, TCF-M and TCF-M-5c the spectra contained one photoelectron line per spin–orbit component with a weak shoulder at the higher binding energy side, and the (Cu2+) satellite barely observable. Interestingly, XPS spectrum of TCF-M-5c illustrated a further diminished presence of the Cu2+ satellite features, suggesting a reduced intensity of Cu 2p compared to the material before its photocatalytic degradation cycles. This finding suggested the formation of Cu1+ at the surface of the material according to XPS.44,46 However, this finding is contradictory to what XRD and Raman results demonstrated, as these did not indicate any phases related to Cu1+. We therefore consider it plausible that the high Cu1+/Cu2+ ratio we observe in XPS spectra is an effect of beam damage (reduction of Cu under irradiation of the XPS excitation source). Such ‘beam damage’ effect in Cu2+ compounds has been previously reported by others (e.g. in ref. 47 and 48) under varied excitation sources. We find such a scenario more plausible because we observed a slow degradation of the Cu2+ satellite intensity (relative to the main line) even when measuring the Cu 2p XPS of the TCF-M sample. Furthermore, the (relatively faster measurements of) Cu 2p NEXAFS of related samples (vide infra) all showed either pure or strongly dominant Cu2+ charge state.
Comparison of XPS spectra also indicates that the surface region (corresponding to the XPS probe depth of approximately 2 nm at the relevant kinetic energies47) is significantly more copper-rich (see ESI Table S2†) as compared to the bulk composition (viz. from ICP-OES). This suggests that the copper ferrite species is preferably concentrated at the surface of the nanoparticles, which from SEM information were estimated at ∼20–100 nm in size.
3.5 Investigation of Cu and Fe chemical state
To further clarify the chemical state and local environment of Cu and Fe in CuFe2O4–Fe2O3 and TCF20-S, we carried out NEXAFS spectroscopic measurements of selected materials. The fine structure of Cu L23 edge (see ESI Fig. S4†) revealed that copper is mainly in Cu2+ oxidation state in those materials and the fine structure of Fe L23 edge showed that Fe is mainly present as Fe3+. We note that NEXAFS provides a more sensitive tool as concerns the charge state of 3d metal ions than XPS (due to the continuum final state in the latter). NEXAFS measured in total electron yield (TEY) mode also has somewhat larger probe depth and is therefore less sensitive to surface impurities.48 The NEXAFS results indeed do agree well with the analysis results of the XRD data where only Cu2+ compounds were identified.Thus, we can conclude that X-ray irradiation during the lengthy (due to low signal level) XPS measurements may induce reduction of the Cu2+ to Cu1+ (Fig. 4c). This phenomenon has been also previously observed and reported.49
3.6 Optical absorption
The optical absorption spectra of all investigated materials are demonstrated in Fig. 5. Initially measured. In line with previous studies,50 the absorption of TiO2-P25 starts roughly at 400 nm. The absorption spectrum of synthesised black material of CuFe2O4–Fe2O3 covers evenly both the visible and UV range. It is in good accordance with previous investigations.51 TCF20-S material exhibited intensive absorption in the UVA spectrum but also showed absorption in the visible light spectrum. Similar absorption profiles were observed for TCF-NM and TCF-M.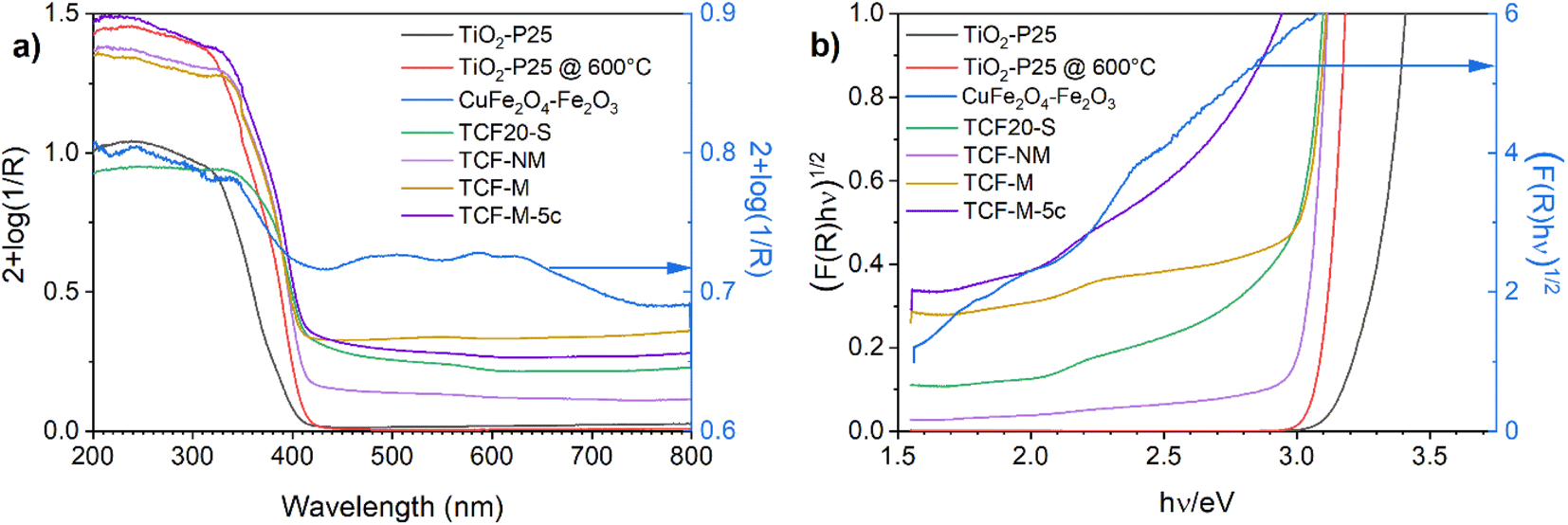 | ||
| Fig. 5 The UV-vis absorption spectra of TiO2-P25, CuFe2O4–Fe2O3, TCF20-S, TCF5-NM, TCF-M and TCF-M-5c materials. (a) Full spectra and (b) spectra plotted as (F(R)(hν))1/2 versus photon energy graphs. Spectra for CuFe2O4–Fe2O3 (blue lines) are zoomed and respective y-axes on the right side are plotted blue. | ||
Absorption edges of TCF20-S, TCF-NM, TCF-M and TCF-M-5c are shifted towards longer wavelengths compared with TiO2-P25 (see Fig. 5a). The weak absorption at 560 nm is close to the band gap edge in hematite, maghemite and CuFe2O4.40
In order to estimate bandgaps from the diffuse reflectance data, the materials were analysed by using the Kubelka–Munk (K–M) theory from a plot (F(R)(hν))1/2 vs. hν (eV) where F(R) = (1 − R)2 (2R)−1 is K–M function and hν is the photon energy in eV, as an intercept of the energy axis with a tangential straight line of the linear part of the graphs (Fig. 5b).52 The absorption threshold values estimated from the Fig. 5b for the TiO2-P25 (indirect optical bandgap) was 3.32 eV. TiO2-P25 @ 600 °C material has an absorption threshold of 3.12 eV, which is due to the higher content of rutile in this material compared with TiO2-P25. The CuFe2O4–Fe2O3 had several absorption thresholds, since it was composed of hematite, maghemite and CuFe2O4 spinel (Fig. 5b, blue line). The bandgap of hematite was 2.1 eV, which qualitatively agrees with an earlier theoretical estimate53,54 TCF20-S had an absorption threshold of 2.97 eV, TCF-NM of 3.03 eV and TCF-M of 3.0 eV. The TCF20-S, TCF-M and TCF-M-5c had an additional absorption threshold at 2.1 eV, which was related to hematite. This result provides proof that materials absorb more visible light compared to pure TiO2-P25.
3.7 Magnetic properties
The magnetic properties of the synthesised photocatalysts were analysed to probe the content of different magnetic phases. Further, it is also important to understand their behaviour under different magnetic fields, which is crucial to ensure robust magnetic collectability if proposed for real application in water treatment systems. These properties measured include bulk saturation magnetisation values, coercivity, and the critical temperatures (TC).The two materials with the strongest magnetic response were investigated (CuFe2O4–Fe2O3, TCF-M and TCF-M-5c). Table S3 (see ESI)† presents the magnetic properties of the prepared materials, with data including saturation magnetisation at 2 T, coercivity, and critical temperatures, as well as reference values from the literature for magnetic crystalline phases identified earlier by XRD.
For CuFe2O4–Fe2O3 material, the saturation magnetisation was recorded as 13.5 emu g−1, and the coercivity was measured at 212 Oe. This material exhibited three distinct magnetic ordering temperatures at 859 K, 763 K, and 248 K, TC1, TC2 and TC3 respectively (Fig. 6a).
 | ||
| Fig. 6 The temperature dependence of the magnetisation (a) and the magnetization curves at room temperature (300 K) (b) of CuFe2O4–Fe2O3, TCF-M and TCF-M-5c materials. Initial zero field cooled (ZFC) and field cooled (FC) temperature dependencies reveal three magnetic transitions. The extrema in the temperature derivative curve dM dT−1 of the latter (Inset) are labelled by the estimates of the magnetic ordering temperatures in the CuFe2O4–Fe2O3 material. | ||
In the case of TCF-M material, the saturation magnetisation at 2 T reduced to 6.12 emu g−1. However, the coercivity increased to 428 Oe. The magnetic ordering temperatures for this material were observed at 852 K, 732 K, and 265 K. The analysis of TCF-M-5c demonstrated a significant alteration, notably the absence of the ∼760 K transition, suggesting a phase composition change. This alteration, confirmed by the sole presence of the 844 K transition in both zero field cooled (ZFC) and field cooled (FC) magnetisation as well as derivative of FC line, aligns with XRD results indicating stability and subtle phase adjustments after multiple usage and recycling cycles.
Ordering at TC2 at around 731–763 K could be assigned to that of spinel ferrite CuFe2O4 with a known bulk Curie temp of 728 K.57 The highest ordering temperature TC1 at 840–859 K would match well with TC = 858 K of magnetite Fe3O4 (ref. 57) or, more plausibly (given the annealing parameters), maghemite.55–59 We could not identify magnetite independently from the copper spinel from the XRD data with which it has almost overlapping reflexes (while for the same reason its presence cannot be ruled out). Although XRD confirms ferrimagnetic maghemite in CuFe2O4–Fe2O3 (11.6%) and in TCF-M (1.5% maghemite), its distinction from magnetite is more conclusively identified by magnetic analysis, specifically through the dM dT−1 measurements, given their overlapping XRD reflexes. Minute changes (on the order of 10 K) in this critical temperature together with the rather broad temperature range of the observed phase transition rather suggest the presence of both these phases with minor differences in their relative weights due to sample processing causing the apparent shift of the center of gravity of the transition. TC2 seemed to shift to slightly lower temperatures during repeated high temperature measurements and hysteresis at room temperature (Fig. 6b) changed slightly after the high temperature measurement procedure, further suggesting that the TC2 corresponds to ordering temperature of the metastable maghemite. Small decrease of magnetisation while cooling below ∼250 K at TC3 is assigned to the so-called Morin transition of hematite, below which hematite is perfectly antiferromagnetic and above a slightly canted antiferromagnet and thus possesses a weak magnetic moment.57
The measured saturation magnetization value can be compared to the weighted sum Msample of the component magnetisations using the simple formula Msample = w1 × M1 + w2 × M2 + … + wn × Mn,57 where wi (i = 1…n) are the weight fractions of all the phases derived from the XRD data and listed in Table 1 and Mi are the corresponding bulk magnetisation values from literature listed in Table S3.†57 Based on phase composition, the measured magnetisation 13.5 emu g−1 at room temperature is comparable but slightly less than the calculated 17.5 emu g−1 value expected for the CuFe2O4–Fe2O3. In the case of TCF-M the recorded value of 6.12 emu g−1 is higher than the expected value of 3.39 emu g−1. This analysis reaffirms the XRD findings, suggesting the magnetic characterization of TCF-M-5c reflects the noted phase stability and the observed decrease in Cu concentration, supporting the inference of slight compositional shifts post-recycling.
We conclude that magnetisation measurements provide a confirmation of the phase composition determined by XRD and even further removes some remaining ambiguity. Saturated magnetic moment at room temperature matches the value calculated by the weighted average of magnetisation of all phases reasonably well, at least in the order of magnitude level. All features of temperature dependence of magnetisation can be well explained by the Curie temperature of CuFe2O4, γ-Fe2O3/Fe3O4 and by Morin transition temperature of α-Fe2O3. The Néel temperature of α-Fe2O3 remained above the temperatures used during this study so was not experimentally confirmed.
3.8 Photocatalytic activity
Photocatalytic activity of pure TiO2-P25, TiO2-P25 @ 600 °C, CuFe2O4–Fe2O3, TCF20-S, TCF-NM and TCF-M measured as degradation of model herbicide pollutant 2,4-D is demonstrated in Fig. 7. The performance of pure 2,4-D provided a baseline for comparison, indicating the potential decay of this mode pollutant under UVA light. According to our results, 2,4-D was rather stable under UVA and only slight (10%) degradation was observed after prolonged (18 h) irradiation. It is important to note that in our experimental set-up photocatalytic activity was measured without any stirring and with the studied materials placed on the surface of the 2,4-D solution. Such a set-up may result in lower photocatalytic activity than one would achieve with stirring. However, degradation of 2,4-D was well detectable with all the tested materials (see ESI Fig. S5†). | ||
| Fig. 7 Photocatalytic degradation of 2,4-D in aqueous solution over time under UVA irradiation by various photocatalytic materials: TCF20-S, TCF-M, TCF-NM and TiO2 600 °C. In addition, the degradation of a pure 2,4-D aqueous solution is shown. | ||
The photodegradation process of 2,4-D is relatively well described and involves transformation of 2,4-D into chlorinated intermediates and then into compounds like 1,2,4-benzenetriol and chlorohydroquinone, which undergo ring-opening and hydrolysis reactions catalysed by the TiO2 surface, leading to complete mineralisation into carbon dioxide, water, and gaseous products60–62
Photodegradation of 2,4-D by pure TiO2-P25 was not assessed due to opacity of the respective suspension but TiO2-P25 @ 600 °C demonstrated the highest degradation rate across all materials, achieving 20% loss of 2,4 D within 1 h, 40% loss of 2,4 D within 3 h and nearly complete degradation of 2,4-D (around 94.6%) after 18 h (Fig. 7). Within the first 1.5 hours of UVA irradiation, both TCF20-S and TCF-M materials exhibited comparable degradation rates. After this time frame, magnetically extracted TCF-M material showed a higher rate of degradation, achieving 26% of 2,4-D degradation by 3 hours, whereas the TCF20-S reached a slightly lower level of degradation by the same time. TCF-NM material demonstrated relatively good results, degrading 16% of 2,4 D within 1 h and 30% of 2,4 D degradation within 3 h and nearly complete degradation of 2,4-D (around 93%) after 18 hours.
The superior photocatalytic activity of pure TiO2-P25 @ 600 °C when compared with TCF20-S, TCF-M, and TCF-NM materials can be attributed to its relatively higher anatase content while in the case of the latter TiO2 has mostly rutile crystal structure (see Table 1). Therefore, from photocatalytic activity results it is clear that the photocatalytic component in the synthesized materials is TiO2 and CuFe2O4–Fe2O3 works as a magnetic addition. TCF-M exhibits lower photocatalytic activity as it contains 20% less photocatalytic material than pure TiO2-P25 @ 600 °C.
Despite the higher degradation rate of pure TiO2-P25 @ 600 °C and TCF-NM compared with TCF-M, their very fine size (less than 100 nm, even in size of tens of nm) and non-magnetic nature presents challenges for low-cost recovery and recycling. Therefore, despite its relatively lower photocatalytic activity, the magnetic properties of TCF-M can be used to easily collect this material from the environment and repeatedly used as photocatalyst material. In Fig. 8 the photocatalytic activity of TCF-M after five cycles (use and magnetic extraction) is demonstrated. Only a slight decrease in the photocatalytic degradation rate through subsequent cycles was observed while the photocatalytic efficiency of this material remains commendably high. This stability of photocatalytic and 2,4-D degrading activity of TCF-M material after repeated use and collection from water strongly recommends the potential of this material as a reliable and practical choice for water purification applications, including recyclability.
 | ||
| Fig. 8 Time-dependent photocatalytic degradation of 2,4-D in aqueous solution using TCF-M, under UVA irradiation, after five cycles of use. For comparison, the degradation trend of a pure 2,4-D aqueous solution is also depicted. | ||
4 Conclusions
In the present study we propose a sintered mixture of TiO2-P25 and CuFe2O4–Fe2O3 to obtain a magnetically collectable photocatalyst. CuFe2O4–Fe2O3 material was synthesized using co-precipitation method. The obtained material was then mechanically mixed with TiO2-P25 and annealed at 600 °C to form a photocatalytic material (TCF20-S). TCF20-S material was further separated to magnetically collectable part (TCF-M) and non-magnetic residue (TCF-NM) and properties and performance of those fractions were compared. Additionally, we extended our analysis to TCF-M material that had been re-used over 5 cycles (TCF-M-5c), to assess its durability and sustained photocatalytic activity.The synthesized materials had fine grain sizes ranging from 20 to several hundreds nanometers. Elemental distribution of TCF-M demonstrated heterogeneously dispersed nanoscopic Fe and uniformly distributed Cu particles. Elemental composition measured with ICP-OES aligned closely with our initial synthetic expectations and confirmed that the TCF-M sample had a balanced Ti/Fe atomic ratio indicating the effective incorporation of TiO2 into the magnetic fraction. The XRD patterns of TCF20-S, TCF-NM, TCF-M and TCF-M-5c showed the presence of rutile, anatase, hematite (α-Fe2O3), CuFe2O4 spinel and maghemite (γ-Fe2O3). According to XPS and NEXAFS, titanium was in Ti4+, copper was predominantly in Cu2+ and iron was mainly in Fe3+ state in all materials. All materials involving TiO2, exhibited intense absorption in the UVA spectrum, but in case of TCF20-S, TCF-NM, TCF-M and TCF-M-5c also absorption in the visible light spectrum was observed. The latter was due to the presence of Fe2O3 and CuFe2O4 in material, which have absorption in the visible range. The saturation magnetic moment at room temperature is consistent with the calculated magnetisation of all crystal phases present and temperature dependence of magnetisation reveals magnetic transition temperatures that are expected for given phase composition. Improved magnetic properties make studied mixtures well suitable for magnetic collection in water treatment processes.
Photocatalytic activity of materials was evaluated as degradation of the model herbicide pollutant 2,4-D under UVA. Out of all the tested materials pure TiO2-P25 @ 600 °C exhibited the highest photocatalytic activity, which was due to its predominant anatase content in the crystal phase. TCF-NM, TCF20-S and TCF-M materials showed lower photocatalytic activity than TiO2-P25 @ 600 likely due to the relatively higher presence of rutile phase combined with general lower TiO2 content. However, while TiO2-P25 @ 600 °C can not be recycled from water after photocatalytic reaction due to its fine gram size, TCF-M shows a potency for easy removal due to its magnetic properties. The latter was proven by 5 cycles of use, collection and reuse of the material. Therefore, we see a remarkable potential for this material in water purification.
Author contributions
Dmytro Danilian: investigation, validation, formal analysis, visualization, data curation, methodology, conceptualization, writing – original draft. Franziska Maria Bundrück: investigation, formal analysis. Arvo Kikas: investigation, formal analysis. Tanel Käämbre: investigation, formal analysis, writing – review & editing. Hugo Mändar: investigation, formal analysis, resources, visualization, writing – review & editing. Sandro Lehner: investigation. Alexander Gogos: investigation, formal analysis, resources, visualization, writing – review & editing. Jekaterina Kozlova: investigation, formal analysis, visualization. Mati Kook investigation, formal analysis. Valter Kiisk: formal analysis, resources. Joosep Link: investigation, formal analysis, visualization. Raivo Stern: investigation, formal analysis, resources, funding acquisition. Angela Ivask: writing – review & editing, funding acquisition, supervision. Vambola Kisand: writing – review & editing, funding acquisition, resources, supervision. Rainer Pärna: conceptualization, methodology, formal analysis, writing – review & editing, resources, funding acquisition, supervision, project administration.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
Dmytro Danilian, Vambola Kisand and Angela Ivask's work was supported by Estonian Research Council grant PRG1496. Rainer Pärna's work was supported by Estonian Research Council grants MOBTP145, TT20 and PRG629. Work in Tartu was partly supported also by Estonian Centre of Excellence in Research project “Advanced materials and high-technology devices for sustainable energetics, sensorics and nanoelectronics” TK141, “Center of excellence in sustainable green hydrogen and energy technologies” (TK210), and University of Tartu Development Fund (grant PLTFYARENG53). Magnetic characterisation in Tallinn was supported by the number of Estonian Research Council and EU grants (PRG4, PRG1702, TK134, and IUT23-9). We acknowledge MAX IV Laboratory for time on Beamline FinEstBeaMS under Proposal 20220275. Research conducted at MAX IV, a Swedish national user facility, is supported by the Swedish Research council under contract 2018-07152, the Swedish Governmental Agency for Innovation Systems under contract 2018-04969, and Formas under contract 2019-02496. The FinEstBeAMS beamline operation costs were partially supported within the MAX-TEENUS project (grant no. 2014-2020.4.01.20-0278) by the ERDF funding in Estonia awarded to University of Tartu. The research was partly conducted using the NAMUR+ core facility funded by project TT13 “Center of nanomaterials technologies and research (NAMUR+)”. We also like to thank Andreas Voegelin and Ralf Kägi (EAWAG, Switzerland) for access to their HF facilities. Finally, we would like to thank the Scientific Center for Optical and Electron Microscopy (ScopeM) of ETH Zurich for access to their microscopes.References
- M. Syafrudin, R. A. Kristanti, A. Yuniarto, T. Hadibarata, J. Rhee, W. A. Al-onazi, T. S. Algarni, A. H. Almarri and A. M. Al-Mohaimeed, Int. J. Environ. Res. Public Health, 2021, 18, 468 CrossRef CAS PubMed.
- R. Z. Marsala, E. Capri, E. Russo, M. Bisagni, R. Colla, L. Lucini, A. Gallo and N. A. Suciu, Sci. Total Environ., 2020, 736, 139730 CrossRef PubMed.
- Ü. Leisk and R. Rebane, Taimekaitsevahendite Jääkide Sisalduse Ja Dünaamika Uuring Pinna- Ja Põhjavees, Estonian Environmental Research Centre, 2018 Search PubMed.
- V. Kõrgmaa, E. Usin, Ü. Leisk, M. Laht, V. Värk, S. Otsmaa, K. Pachel, J. Jaaku, M. Kriipsalu, K. Pehme, I. Tamm, L. Albreht, M. Lukk, L. Liepkalns, A. Marandi, J. Pärn, V. Raidla and K. Vooro, Hajaasustuspiirkondade Joogivee Kvaliteedi Ja -süsteemide Uuring, Eesti Keskkonnauuringute Keskus OÜ, 2020 Search PubMed.
- F. H. M. Tang, M. Lenzen, A. McBratney and F. Maggi, Nat. Geosci., 2021, 14, 206–210 CrossRef CAS.
- A. F. Gilca, C. Teodosiu, S. Fiore and C. P. Musteret, Chemosphere, 2020, 259, 127476 CrossRef CAS.
- V. K. Gupta and I. Ali, Environmental Water, 2013, pp. 1–27 Search PubMed.
- G. Ren, H. Han, Y. Wang, S. Liu, J. Zhao, X. Meng and Z. Li, Nanomaterials, 2021, 11, 1804 CrossRef CAS PubMed.
- H. He, Z. Luo and C. Yu, J. Alloys Compd., 2020, 816, 152652 CrossRef CAS.
- H. He, Z. Luo and C. Yu, Colloids Surf., A, 2021, 613, 126099 CrossRef CAS.
- H. He, Z. Luo, Z.-Y. Tang and C. Yu, Appl. Surf. Sci., 2019, 490, 460–468 CrossRef CAS.
- F. Yi, J. Ma, C. Lin, H. Zhang, Y. Qian, H. Jin and K. Zhang, Chem. Eng. J., 2022, 427, 132028 CrossRef CAS.
- S. Anandan, Y. Ikuma and K. Niwa, Solid State Phenom., 2010, 162, 239–260 CAS.
- F. Yi, J. Ma, C. Lin, L. Wang, H. Zhang, Y. Qian and K. Zhang, J. Alloys Compd., 2020, 821, 153557 CrossRef CAS.
- H. He, J. Jiang, Z. Luo, D. Li, M. Shi, H. Sun, J. Chen, C. Chen, B. Deng and C. Yu, Colloids Surf., A, 2023, 667, 131357 CrossRef CAS.
- Z. Xu, Y. Ren, X. Deng, M. Xu, W. Chai, X. Qian and Z. Bian, Adv. Energy Sustainability Res., 2022, 3(11), 2200105 CrossRef CAS.
- V. Kisand, U. Joost, V. Reedo, R. Pärna, T. Tätte, J. Shulga, A. Saar, L. Matisen, A. Kikas and I. Kink, Appl. Surf. Sci., 2010, 256, 4538–4542 CrossRef CAS.
- Q. Guo, C. Zhou, Z. Ma and X. Yang, Adv. Mater., 2019, 31, e1901997 CrossRef PubMed.
- M. Mishra and D.-M. Chun, Appl. Catal., A, 2015, 498, 126–141 CrossRef CAS.
- R. M. Cornell and U. Schwertmann, The Iron Oxides, 2003 Search PubMed.
- M. R. A. Kumar, B. Abebe, H. P. Nagaswarupa, H. C. A. Murthy, C. R. Ravikumar and F. K. Sabir, Sci. Rep., 2020, 10, 1249 CrossRef CAS PubMed.
- W. Bootluck, T. Chittrakarn, K. Techato and W. Khongnakorn, J. Environ. Chem. Eng., 2021, 9, 105660 CrossRef CAS.
- R. S. Yadav, J. Havlica, J. Masilko, L. Kalina, J. Wasserbauer, M. Hajdúchová, V. Enev, I. Kuřitka and Z. Kožáková, J. Supercond. Novel Magn., 2016, 29, 759–769 CrossRef CAS.
- S. P. Kamble, S. P. Deosarkar, S. B. Sawant, J. A. Moulijn and V. G. Pangarkar, Ind. Eng. Chem. Res., 2004, 43, 8178–8187 CrossRef CAS.
- N. Fairley, V. Fernandez, M. Richard-Plouet, C. Guillot-Deudon, J. Walton, E. Smith, D. Flahaut, M. Greiner, M. Biesinger, S. Tougaard, D. Morgan and J. Baltrusaitis, Appl. Surf. Sci. Adv., 2021, 5, 100112 CrossRef.
- R. Pärna, R. Sankari, E. Kukk, E. Nõmmiste, M. Valden, M. Lastusaari, K. Kooser, K. Kokko, M. Hirsimäki, S. Urpelainen, P. Turunen, A. Kivimäki, V. Pankratov, L. Reisberg, F. Hennies, H. Tarawneh, R. Nyholm and M. Huttula, Nucl. Instrum. Methods Phys. Res., Sect. A, 2017, 859, 83–89 CrossRef.
- W. Wang, A. Kivimäki, K. Chernenko, R. Pärna, T. Käämbre, E. Kukk, K. Kokko, M. Valden, M. Hirsimäki, M. Kirm and M. Huttula, J. Phys.: Conf. Ser., 2022, 2380, 012048 CrossRef.
- M. Kumar, M. N. Islam, F. L. Terry, M. J. Freeman, A. Chan, M. Neelakandan and T. Manzur, Appl. Opt., 2012, 51, 2794 CrossRef CAS PubMed.
- QD Application Note 1500-021 Rev. B0, 2020 Search PubMed.
- M. M. Viana, V. F. Soares and N. D. S. Mohallem, Ceram. Int., 2010, 36, 2047–2053 CrossRef CAS.
- D. A. H. Hanaor and C. C. Sorrell, J. Mater. Sci., 2011, 46, 855–874 CrossRef CAS.
- T. Ohsaka, F. Izumi and Y. Fujiki, J. Raman Spectrosc., 1978, 7, 321–324 CrossRef.
- R. Liang, A. Hu, W. Li and Y. N. Zhou, J. Nanopart. Res., 2013, 15, 1990 CrossRef.
- Lj. D. Arsov, C. Kormann and W. Plieth, J. Raman Spectrosc., 1991, 22, 573–575 CrossRef CAS.
- S. P. Schwaminger, P. Fraga-García, F. Selbach, F. G. Hein, E. C. Fuß, R. Surya, H.-C. Roth, S. A. Blank-Shim, F. E. Wagner, S. Heissler and S. Berensmeier, Adsorption, 2017, 23, 281–292 CrossRef CAS.
- L. S. Mdletshe, P. R. Makgwane and S. S. Ray, Nanomaterials, 2019, 9, 1140 CrossRef CAS PubMed.
- S. Challagulla, K. Tarafder, R. Ganesan and S. Roy, Sci. Rep., 2017, 7, 8783 CrossRef PubMed.
- B. H. Toby, Powder Diffr., 2006, 21, 67–70 CrossRef CAS.
- E. McCafferty and J. P. Wightman, Surf. Interface Anal., 1998, 26, 549–564 CrossRef CAS.
- J. J. Yeh and I. Lindau, At. Data Nucl. Data Tables, 1985, 32, 1–155 CrossRef CAS.
- T. Yamashita and P. Hayes, Appl. Surf. Sci., 2008, 254, 2441–2449 CrossRef CAS.
- Q. Qin, Y. Liu, X. Li, T. Sun and Y. Xu, RSC Adv., 2018, 8, 1071–1077 RSC.
- H. He, J. Xiao, Z. Liu, B. Yang, D. Wang, X. Peng, L. Zeng, Z. Li, L. Lei, M. Qiu and Y. Hou, Chem. Eng. J., 2023, 453, 139751 CrossRef CAS.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-Ray Photoelectron Spectroscopy, Perkim-Elmer Corporation, Eden Prairie, 1992 Search PubMed.
- G. van der Laan, C. Westra, C. Haas and G. A. Sawatzky, Phys. Rev. B: Condens. Matter Mater. Phys., 1981, 23, 4369–4380 CrossRef CAS.
- H. He, L. Zeng, X. Peng, Z. Liu, D. Wang, B. Yang, Z. Li, L. Lei, S. Wang and Y. Hou, Chem. Eng. J., 2023, 451, 138628 CrossRef CAS.
- L. Trotochaud, A. R. Head, S. Pletincx, O. Karslıoǧlu, Y. Yu, A. Waldner, L. Kyhl, T. Hauffman, H. Terryn, B. Eichhorn and H. Bluhm, J. Phys. Chem. B, 2018, 122, 1000–1008 CrossRef CAS PubMed.
- Z. Li, J. Lyu and M. Ge, J. Mater. Sci., 2018, 53, 15081–15095 CrossRef CAS.
- S. Tanuma, C. J. Powell and D. R. Penn, Surf. Interface Anal., 1994, 21, 165–176 CrossRef CAS.
- J. Stöhr, NEXAFS Spectroscopy, Springer, Berlin, Heidelberg, 2017 Search PubMed.
- C. C. Chusuei, M. A. Brookshier and D. W. Goodman, Langmuir, 1999, 15, 2806–2808 CrossRef CAS.
- X. Wang, S. O. Pehkonen, J. Rämö, M. Väänänen, J. G. Highfield and K. Laasonen, Catal. Sci. Technol., 2011, 2, 784–793 RSC.
- A. Meidanchi and H. Ansari, J. Cluster Sci., 2021, 32, 657–663 CrossRef CAS.
- P. Makuła, M. Pacia and W. Macyk, J. Phys. Chem. Lett., 2018, 9, 6814–6817 CrossRef PubMed.
- B. D. Cullity and C. D. Graham, Introduction to Magnetic Materials, IEEE Press, 2008 Search PubMed.
- C. Xia, Y. Jia, M. Tao and Q. Zhang, Phys. Lett. A, 2013, 377, 1943–1947 CrossRef CAS.
- S. Piccinin, Phys. Chem. Chem. Phys., 2019, 21, 2957–2967 RSC.
- A. U. Gehring, H. Fischer, M. Louvel, K. Kunze and P. G. Weidler, Geophys. J. Int., 2009, 179, 1361–1371 CrossRef CAS.
- M. I. Dar and S. A. Shivashankar, RSC Adv., 2013, 4, 4105–4113 RSC.
- F. Amiri, M. Dehghani, Z. Amiri, S. Yousefinejad and A. Azhdarpoor, Water Sci. Technol., 2021, 83, 3110–3122 CrossRef CAS PubMed.
- S. S, K. L. Nagashree, T. Maiyalagan and G. Keerthiga, Appl. Surf. Sci., 2018, 449, 371–379 CrossRef.
- M. Abdennouri, A. Elhalil, M. Farnane, H. Tounsadi, F. Z. Mahjoubi, R. Elmoubarki, M. Sadiq, L. Khamar, A. Galadi, M. Baâlala, M. Bensitel, Y. E. Hafiane, A. Smith and N. Barka, J. Saudi Chem. Soc., 2015, 19, 485–493 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra00094c |
| This journal is © The Royal Society of Chemistry 2024 |