
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFreezing-enhanced chlorination of organic pollutants for water treatment†
Duanyang Wua,
Junxue Lia,
Jing Xub and
Wei Cheng *a
*a
aCollege of Resources and Environmental Science, South-Central Minzu University, Wuhan 430074, P.R. China. E-mail: wei.cheng@scuec.edu.cn
bState Key Laboratory of Water Resources and Hydropower Engineering Science, Wuhan University, Wuhan, 430072, PR China
First published on 16th April 2024
Abstract
Freezing has been reported to accelerate chemical reactions and thus affect the fate of pollutants in the environment. However, little research has been conducted on the potential effects of freezing on the chlorination process. This study aimed to explore the freezing-enhanced chlorination process by comparing the oxidation of clofibric acid (CA) by chlorine in ice (at −20 °C) to the same reaction in water (at 25 °C). The degradation of CA, which was negligible in water, was significantly accelerated in ice. This acceleration can be attributed to the freeze concentration effect that occurs during freezing, which excludes solutes such as chlorine, CA and protons from the ice crystals, leading to their accumulated concentration in the liquid brine. The increased concentration of chlorine and protons in the liquid brine leads to higher rates of CA oxidation, supporting the freeze concentration effect as the underlying cause for the accelerated chlorination of CA in ice. Moreover, the chlorine/freezing system was also effective in the degradation of other organic pollutants. This highlights the environmental relevance and significance of freezing-enhanced chlorination in cold regions, particularly for the treatment of organic contaminants.
Introduction
Free chlorine species, such as Cl2, HClO, and ClO−, are commonly employed for disinfection and the effective decomposition of various organic compounds in water treatment processes.1–4 However, the sluggish oxidation kinetics of chlorine impose constraints on its extensive use as a potent chemical oxidant. To overcome this limitation, numerous strategies have been proposed to enhance the effectiveness of chlorination for water remediation. These strategies involve the use of additional oxidants (e.g., H2O2,5 persulfate6 and ozone7), as well as the implementation of various energy inputs (e.g., UV8,9 and solar,10 ultrasound,11 and electrochemical process12,13), and the use of catalysts.14,15 These strategies facilitate the generation of reactive radicals, thereby augmenting the overall efficiency of chlorination. However, it is worth noting that these strategies encounter challenges such as the high cost associated with the utilization of additional oxidants, the substantial increase in energy consumption, and the potential deactivation of catalysts. Hence, there is an ongoing need to develop an energy-efficient approach for activating chlorine.According to the Arrhenius equation, a fundamental principle in chemical kinetics, decreasing the temperature typically slows down reaction rates. This phenomenon has been extensively utilized in practical applications such as long-term food storage and stabilization of volatile or reactive chemicals. Intriguingly, recent investigations have revealed a surprising and counterintuitive finding, wherein certain chemical reactions are notably accelerated under freezing conditions. This phenomenon has been observed across various reaction systems. For example, the reduction of chromate by species such as nitrite,16 arsenite,17 ferrous ions,18 H2O2,19 and phenolic compounds20–22 has demonstrated a remarkable enhancement in reaction rates during the freezing process, surpassing those in the aqueous phase. Additionally, agents like peroxymonosulfate,23,24 IO4−,25 chloride–oxone system,26 H2O2/NO2− (ref. 27) and nitrite28 have exhibited significantly improved efficiency in oxidizing organic pollutants under freezing conditions compared to in aqueous system. These accelerated reactions are not solely confined to homogeneous reactions in aqueous solutions; freezing has also proven to enhance the efficiency of heterogeneous reactions involving soils29 and MnO2 oxides30 and photochemical reactions under irradiation.31–33 The rate accelerations observed during the freezing process can be attributed to various physicochemical mechanisms that are associated with the formation and growth of ice crystals. These mechanisms include:34,35 (i) freeze-concentration, where solutes are expelled from growing ice crystals and consequently become more concentrated in the shrinking liquid brine. The formation of crystals leads to an increase in the concentration of reactants. (ii) Freezing potential, which occurs when specific solutes become incorporated into the ice. This results in the separation and variation in the composition of anions and cations between the different phases, generating an electric potential. (iii) Catalytic effect of the ice surface, whereby the surface of the ice acts as a catalyst, lowering the reaction barrier and facilitating the chemical reaction. (iv) Temperature differences, as the cooling of a solution under laboratory conditions can cause variations in temperature within the sample. This phenomenon can lead to differences in the thermodynamics and kinetics of the reaction. (v) Other factors, such as convection effects, arise when the direction of freezing affects the concentration gradient of the solution. Furthermore, the preferred orientation of reactants in the frozen phase and the disparity in dielectric constants between solutes in water and ice have also been reported as contributing factors.34,35Among these mechanisms, the freeze concentration effect has been widely recognized as the predominant driver for the observed enhancement in reactivity under freezing conditions.16,18,21,25–29,32,33 Several studies have conclusively demonstrated the significant impact of this effect on reaction rates during the freezing process. Notably, freezing induces the transportation of solute molecules away from the slowly-moving ice, resulting in their accumulation and concentration in the unfrozen portion of the solution, known as the liquid brine. This phenomenon, referred to as the freeze concentration effect, effectively amplifies the concentrations of the reactants, thereby creating a favorable environment for chemical reactions to occur. As a consequence, the freeze concentration effect accelerates chemical reaction rates in the liquid brine. Considering that higher concentrations of chlorine favor chlorination efficiency, the freezing process has the potential to enhance chlorination efficiency by enriching the concentrations of free chlorine species. While numerous methods have been employed in conjunction with chlorination to enhance the degradation of organic pollutants,6–8,11–15 the potential of the freezing process to activate free chlorine species remains largely unexplored. Further investigation into this area could provide valuable insights into the utilization of freezing as a means to enhance chlorination efficiency and improve the degradation of organic pollutants in water treatment processes.
Using efficient wastewater treatment technologies is of utmost importance for countries situated in high latitudes or altitudes due to their unique environmental conditions. Conventional wastewater treatment processes, such as the biological process and the electro-Fenton process, face significant challenges in these regions. These limitations are primarily attributed to their reduced effectiveness resulting from factors such as microorganism inactivation, electrode damage, and frozen of electrolytes due to extremely low temperatures that persist for a significant duration of the year. Moreover, implementing conventional wastewater treatment processes in these regions would necessitate a substantial increase in energy consumption to maintain indoor temperatures above the frigid external conditions. Therefore, it is imperative to develop energy-efficient wastewater treatment processes. Recently, the freezing process has been explored to activate IO4− for wastewater treatment, and this IO4−/freezing system has the advantage of operating without external electrical energy, making it well-suited for application in cold regions.25 Drawing an analogy from this, it is plausible to postulate that a chlorine/freezing system may also be effective in the degradation of pollutants. However, the potential of the freezing process for chlorine-induced organic degradation remains largely unexplored and calls for further investigation. Future research in this area holds promise for the development of energy-efficient wastewater treatment processes that are specifically tailored for operation in regions with extreme cold climates.
Thus, in this study, we aimed to investigate the potential of the freeze effect in facilitating chlorination efficiency. To achieve this, we compared the oxidation of organic compounds by chlorine in both water at a temperature of 25 °C and in ice at −20 °C. Clofibric acid (CA), a widely used blood lipid regulator, was selected as the model organic compound because of its persistence and associated environmental concerns.36,37 Varying experimental parameters, including chlorine concentration and pH studied the freezing-induced degradation of CA in the presence of chlorine. Moreover, the underlying mechanisms that drive freezing-enhanced oxidation of CA by chlorine were explored. Additionally, the suitability of the chlorine/freezing system for the degradation of other organic pollutants was also assessed, thereby highlighting the environmental relevance and significance of freezing-accelerated redox conversion of organic pollutants using chlorine in cold regions.
Materials and methods
Chemicals
All chemicals were of high purity grade and used as received without further purification. Sodium hypochlorite (NaClO) was obtained from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Clofibric acid (CA, >99%), Carbamazepine (CBZ, >99%), 1,4-Dimethoxybenzene (DMOB), Phenol (PE), and Cresol red (CR) were obtained from Aladdin Industrial Corporation (Beijing, China). All other chemicals (e.g., NaOH and HCl) were purchased from Modern Eastern Fine Chemical (Beijing, China). All solutions were prepared in ultrapure water (18.3 MΩ cm) produced using a Milli-Q device (Integral 5, Millipore, U. S.).Experimental procedure
A 50 mL reaction solution was prepared in a beaker, comprising 20 μM of clofibric acid (CA) and 52 μM of sodium hypochlorite (NaClO). The pH of the solution was adjusted to the desired value (usually pH 4) using either HCl or NaOH solution, monitored by a benchtop pH meter (OHAUS, ST3100). Subsequently, 1.25 mL of this solution was transferred into a polypropylene conical tube (volume = 1.5 mL, Thermo Fisher Scientific). These conical tubes, containing the NaClO and organic pollutant, were then placed in a pre-cooled cryogenic ethanol bath at a temperature of −20 °C to initiate freezing. The solution samples were gradually frozen within 10 min (Fig. S1†). The starting point (i.e., t = 0) for the reaction kinetic measurement was defined as the moment the conical tubes containing the aqueous solution were placed in the cryogenic ethanol bath. At predetermined time intervals, a single tube was removed from the cryogenic ethanol bath and transferred to a 25 °C water bath to thaw the frozen solution The thawed solution was immediately analyzed using suitable analytical methods. Control experiments were simultaneously performed in a water bath preset at 25 °C. All reaction times for the experiments were kept within 150 min. In order to further explore the chlorine/freezing system for water treatment, the effects of NaClO concentration and pH on the degradation of CA were investigated. The NaClO concentration was varied within the range of 0–260 μM, while initial pH values were adjusted to a range of 3–10. These experiments followed the same procedure as described above. Additionally, the degradation of other organic pollutants, such as DMOB, CBZ, and PE, was also examined to validate the applicability of the chlorine/freezing system for water treatment purposes. All the experiments were conducted at least twice to confirm the reproducibility, and the presented results are the average values obtained.Chemical analyses
The determination of free chlorine concentration was carried out using a UV/visible spectrophotometer (MAPADA, P3) employing the DPD (N,N-diethyl-p-phenylenedi-amine) method. The DPD reagent reacts with free chlorine, resulting in the formation of color compounds that exhibit significant UV absorbance at 515 nm. CA, DMOB, CBZ and PE were measured using a high-performance liquid chromatograph (HPLC, Ultimate 3000) equipped with a C18 column (250 × 4.6 mm i.d., 5 μm) and a UV diode-array detector (DAD). The eluent consisted of a water/acetonitrile mixture containing 0.1% formic acid, and the flow rate was set at 1 mL min−1 in the isocratic mode. The detailed HPLC method can be found in Table S1.†Results and discussion
Enhanced degradation of clofibric acid (CA) by chlorine in ice
The freezing process expels solutes from ice crystals into the liquid region. Below the eutectic points, temperatures allow for a thin liquid layer to persist among solid ice crystals (at the ice–ice interface) and on their surface (at the ice–air interface).25,38 This confined liquid layer is commonly referred to as liquid brine, ice grain boundary, or micro pocket.25 It is important to note that chemical reactions occur in the liquid region rather than within the solidified ice crystals. In this study, the term “ice” pertains to the binary phase encompassing both ice crystals and liquid brine representing the combination of the solid and liquid compartments, rather than solely referring to the ice-frozen crystals (solid chamber alone). To investigate the impact of the freezing process on chlorination, the degradation of CA by chlorine in ice (i.e., at −20 °C) was compared to that in water (Fig. 1a). The degradation of CA was negligible after 150 min in water at [NaClO] = 52 μM, [CA] = 20 μM, and pH = 4, indicating the recalcitrance of CA towards chlorination. This observation is consistent with previous literature that reported negligible degradation of 5 μM CA by chlorination (50–100 μM) at pH 5.39,40 In contrast, rapid degradation of CA was observed during the freezing process under the same experimental conditions, wherein the concentration of CA in the ice sample gradually decreased over the reaction time, with only ∼3 μM of CA remaining after 20 minutes of reaction. In addition, the degradation of CA in ice was found to be negligible in the absence of chlorine, implying that chlorine plays a crucial role as the sole oxidant in the degradation of CA. These findings highlight that neither freezing nor chlorination alone was sufficient for the effective degradation of CA, indicating the joint requirement of both freezing and chlorine for the degradation of CA. | ||
| Fig. 1 (a) Degradation of CA in the presence of NaClO, (b) the concentration of NaClO in water and during freezing. Experimental conditions: [NaClO] = 52 μM, [CA] = 20 μM, initial pH 4, water temperature = 25 °C, and freezing temperature = −20 °C. | ||
During the freezing process, the degradation of CA by chlorine was accompanied by a reduction in the chlorine concentration. As shown in Fig. 1b, the concentration of NaClO remained constant in water, while it rapidly decreased during the freezing process. Notably, a significant removal of CA in the frozen NaClO solution was achieved within 20 minutes, with an 84.24% reduction in CA concentration. Extending the reaction time to 150 minutes only resulted in a marginal increase of 6.39% in CA removal. Moreover, the trend in the variation of NaClO concentration during freezing closely mirrored that of the CA concentration under the same experimental conditions (Fig. 1b). The concentration of NaClO during freezing exhibited a rapid decrease within the first 30 min (from 52 μM to 31 μM), followed by a slower change from 30 min to 150 min (from 31 μM to 22 μM), with approximately half of the NaClO remaining in the ice after 150 min. The sluggish oxidation of CA by chlorine in the later stages may be due to the complete solidification of the solutions, as chemical reactions are known to cease when the temperature of ice drops below the eutectic point.41
In order to confirm the limited oxidation of CA by chlorine under complete frozen conditions, an ice sample was subjected to a freeze-thaw process. After the chlorination of CA was halted (specifically, after 20 minutes of reaction), the ice sample was thawed and subsequently refrozen for an additional 20 minutes. This freeze-thaw process was repeated for three cycles within the same batch, with each cycle lasting 20 minutes. It is worth noting that upon refreezing, the degradation of CA resumed (as shown in Fig. S2†). Moreover, the degradation of CA was more pronounced under three cycles of the freeze-thaw process compared to a single cycle lasting 60 minutes. This suggests that while complete freezing temporarily halts CA's oxidative degradation by chlorine, natural freeze-thaw cycles—driven by daily and seasonal temperature fluctuations—can enhance chlorination efficiency. Similar results were also observed for the redox reaction between chromate and nitrite.16
Degradation mechanism of enhanced CA degradation by NaClO during freezing
The freeze concentration effect34,35 is most likely the underlying mechanism responsible for the enhanced reactions observed during freezing. In the freezing/CA/NaClO system, both CA and chlorine as well as protons, are rejected from bulk ice and are accumulated within the liquid brine during freezing. This concentration phenomenon leads to an accumulation of solutes and protons in the liquid brine, subsequently facilitating the acceleration of CA oxidation by chlorine. To investigate the influence of NaClO and proton concentrations on the CA oxidation kinetics, degradation experiments were conducted in aqueous solutions with varying levels of NaClO concentration and/or acidity (pH) (Fig. 2). Compared to the insignificant degradation of CA observed in water with [NaClO] at 52 μM and pH 4, the degradation rate of CA increased to 21.48% within 150 min when the pH was reduced from 4 to 2 (resulting in a 100-fold increase in proton concentration). Furthermore, when the concentration of NaClO was increased by 5 times (52 μM → 260 μM), the degradation rate of CA rose to 47.38%. These results provide clear evidence that the degradation of CA is primary influenced by the concentration of NaClO rather than the concentration of protons. It is vital to note that the degradation of CA during freezing at [NaClO] of 52 μM and pH 4 occurred notably faster compared to that in aqueous solutions with either higher NaClO concentrations or lower pH values. These results indicated that the increase in NaClO concentration induced by freezing is primarily responsible for the degradation of CA.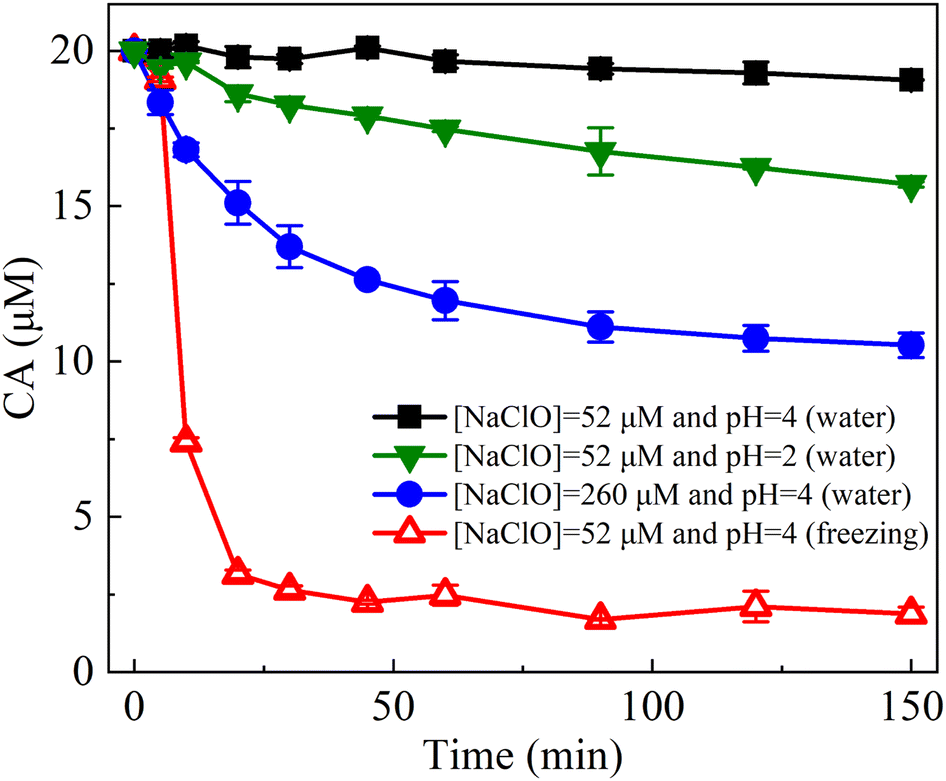 | ||
| Fig. 2 Effect of NaClO concentration increase and/or pH decrease on the degradation of CA by NaClO in water. Experimental conditions: [NaClO] = 52 μM or 260 μM, [CA] = 20 μM, initial pH 4 or 2, water temperature = 25 °C, and freezing temperature = −20 °C. | ||
The estimation of pH in frozen solutions is typically performed by measuring the UV-vis absorption spectra of cresol red (CR) as a pH indicator after freezing. However, in this study, CR exhibited a pronounced susceptibility to oxidation in the presence of chlorine, rendering it unsuitable for accurate pH estimation. As illustrated in Fig. S3–S5,† the addition of chlorine caused rapid oxidation of CR, resulting in the disappearance of the characteristic peak at 434 nm (corresponding to the monoprotonated CR). Additionally, no new peaks emerged at 518 nm (diprotonated CR) and 573 nm (deprotonated CR). Hence, the determination of pH using CR as a pH indicator in the frozen state is not feasible within this specific system. Previous studies have reported that freezing processes typically lead to a decrease in pH by 2 to 4 units.25,42 Therefore, it is plausible to suggest that the pH values of frozen NaClO solutions may reach potentially as low as 0–2. As a result, the concentrated protons in the liquid brine could also contribute to CA degradation. Overall, the simultaneous increase in NaClO concentration and decrease in pH induced by freezing synergistically facilitate the oxidation of CA by chlorine. The enhanced degradation of CA can be attributed to the increased concentration of solutes and protons resulting from the freeze concentration effect.
Freezing-induced degradation of CA by NaClO under various conditions
To further verify the concentration effect of chlorine and protons, the degradation of CA during freezing was examined under varying pH and NaClO concentrations. Fig. 3 shows the pH-dependent degradation kinetics of CA by NaClO from pH 3 to pH 10 during the freezing process. Notably, the degradation of CA decreased as the pH increased within the range of 4–10, the removal of CA decreased from 90.63% to 0 as the pH increased from 4 to 10. About 30% CA could be degraded under neutral pH conditions, while degradation became negligible above pH 8 (Fig. 3a). Furthermore, the degradation of CA by chlorine during freezing exhibited first-order kinetics under different pH conditions. The rates of CA degradation, as represented by the first-order rate constants, followed the same sequence as the CA removal efficiency (Fig. 3b). | ||
| Fig. 3 (a) Effect of pH on the NaClO-mediated degradation of CA during freezing, (b) comparison of the first-order rate constants of CA under different pH. Experimental conditions: [NaClO] = 52 μM, [CA] = 20 μM, pH 3–10, and freezing temperature = −20 °C. | ||
This pH-dependent degradation of CA is influenced by the chlorine speciation as a function of pH. Previous studies have shown that the reactivity of chlorine species decreases in the order of Cl2 > HOCl > OCl−,43,44 and the dominant chlorine species shifts from Cl2 to HOCl and then to OCl− with increasing pH.43,45 Consequently, lower pH conditions favor the oxidation of CA by chlorine. However, in contrast to the general degradation trend observed above pH 4, the efficiency of CA degradation at pH 3 was slower than that at pH 4. This disparity can potentially be attributed to factors such as CA solubility and the chlorine speciation. It has been estimated that the concentration of protons in the liquid brine increases significantly, reaching 2 to 4 orders of magnitude higher than that in an aqueous solution,19,42,46 Assuming a 4-order magnitude decrease in pH due to freezing, the pH levels decreases from 4 and 3 down to 1 and −1 following the freezing process. Under this condition, the concentration of gaseous Cl2 is expected to be higher at pH 3 than that at pH 4 during freezing, while the solubility of CA at pH 3 is anticipated to be lower than that at pH 4, which resulted in a diffusion limitation for the reaction between CA and chlorine. Overall, a higher concentration of solid CA at pH 3 and limited interaction between gaseous Cl2 and solid form of CA can result in the lower degradation efficiency of CA at pH 3 compared to that at pH 4.
Fig. 4 shows the degradation of CA during freezing in the presence of different NaClO concentrations at pH 4. Obviously, the degradation efficiency of CA increased with the rising of NaClO concentration.
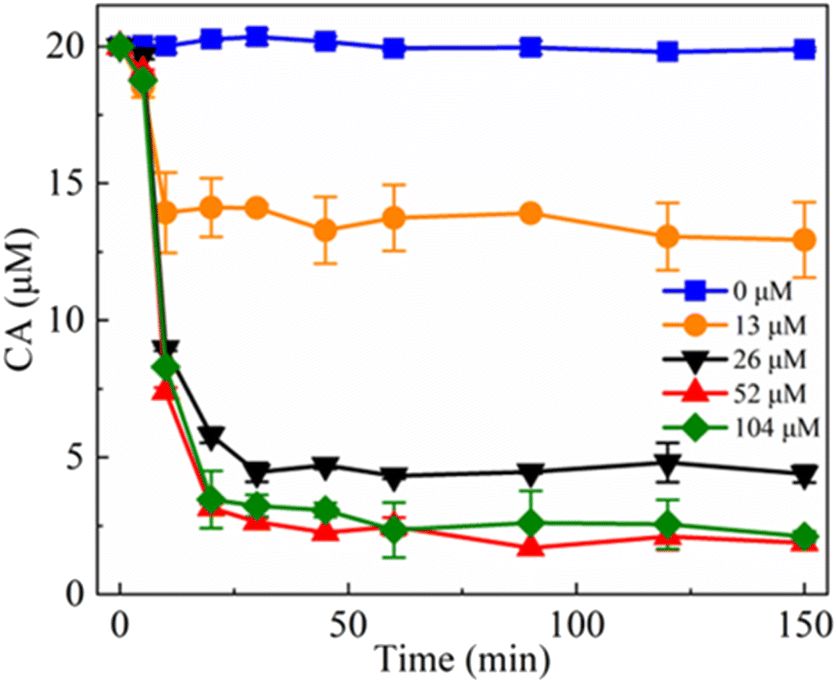 | ||
| Fig. 4 Effect of NaClO dosage on the NaClO-mediated degradation of CA during freezing, experimental conditions: [NaClO] = 0–104 μM, [CA] = 20 μM, pH 4.0, and freezing temperature = −20 °C. | ||
As the chlorine concentration increased from 0 μM to 52 μM, the removal of CA increased from 0 to 90.63% over a period of 150 minutes. This result further supports the role of freeze concentration effect as the primary mechanism for enhanced chlorination of CA during freezing. However, it is notable that the degradation efficiency of CA did not exhibit further improvement when the NaClO concentration was raised from 52 μM to 104 μM. This phenomenon can be attributed to a marginal amount of CA being incorporated into the frozen ice crystals, thereby becoming isolated from the chlorine. Similar results were also obtained at pH 3, where the degradation of CA displayed a similar increasing trend with the elevation of NaClO concentration (Fig. S6†).
Applicability of the chlorine/freezing system for other organic pollutants
In order to assess the practical viability of the chlorine/freezing system for water treatment, the degradation kinetics of different organic pollutants, such as PE, CBZ, and DMOB, were measured and compared in both ice and aqueous solutions (see Fig. 5a). While the degradation rate of organic pollutants in the chlorine/freezing system varied depending on the specific pollutant type, freezing clearly enhanced the degradation of all pollutants tested. The degradation of organic pollutants followed first-order kinetics, and the first-order rate constants in freezing conditions were significantly higher compared to those observed in water (Fig. 5b). These compelling findings substantiate the potential utility of the chlorine/freezing system for the effective degradation of diverse organic pollutants, thus establishing it as a promising method for water treatment applications. | ||
| Fig. 5 (a) The degradation of various organic pollutants (CA, PE, DMOB and CBZ) on the NaClO-mediated process in water and during freezing, (b) comparison of the first-order rate constants in water and during freezing. Experimental conditions: [NaClO] = 52 μM, [pollutants] = 20 μM, pH 4, and freezing temperature = −20 °C. | ||
Furthermore, the chlorine/freezing system presents the notable advantage of not requiring any subsequent physical post-treatments. This renders it a highly advantageous method for water treatment, particularly in regions characterized by cold climates such as permafrost areas, polar regions, high latitudes, and midlatitudes during the winter season. Overall, the chlorine/freezing system stands out as an environmentally friendly and energy-efficient wastewater treatment approach for cold regions.
Conclusions
The present study provides compelling evidence of the significant acceleration of chlorine-mediated oxidation of CA during the freezing process, a reaction that is typically negligible in aqueous solutions at ambient temperatures. The accelerated CA oxidation observed in chlorine/freezing system can be primarily ascribed to the accumulation of CA, chlorine, and protons at the ice grain boundaries, creating a favorable condition for the redox reaction between CA and chlorine namely low pH and high solute concentration. Moreover, the chlorine/freezing system has also effectively degraded other organic pollutants, suggesting its potential feasibility for water treatment in cold environments. This innovative chlorine/freezing system offers an advantage of operating without the need for external electrical energy in cold regions. This is particularly significant as it aligns with global efforts to combat climate change and achieve carbon neutrality, making it a valuable contribution for countries located in low-temperature regions.Author contributions
DW and WC designed the study. DW and JL carried out the experiments and wrote the manuscript. JX and WC: wrote, reviewed and edited the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundations of China (NSFC22006165) and the Fundamental Research Funds for the Central Universities (CZQ21012).Notes and references
- S. Gao, Z. Zhao, Y. Xu, J. Tian, H. Qi, W. Lin and F. Cui, J. Hazard. Mater., 2014, 274, 258–269 CrossRef CAS PubMed.
- D. L. Sedlak and U. von Gunten, Science, 2011, 331, 42–43 CrossRef CAS PubMed.
- J. L. Acero, F. J. Benitez, F. J. Real, G. Roldan and E. Rodriguez, Chem. Eng. J., 2013, 219, 43–50 CrossRef CAS.
- W. L. Wang, Q. Y. Wu, Y. Du, N. Huang and H. Y. Hu, Water Res., 2018, 129, 115–122 CrossRef CAS PubMed.
- X. Zhao, J. Jiang, S. Y. Pang, C. T. Guan, J. Li, Z. Wang, J. Ma and C. W. Luo, Chemosphere, 2019, 221, 270–277 CrossRef CAS PubMed.
- M. Y. Xu, J. Deng, A. H. Cai, C. Ye, X. Y. Ma, Q. S. Li, S. Q. Zhou and X. Y. Li, Chem. Eng. J., 2021, 413, 127533 CrossRef CAS.
- N. Huang, W.-L. Wang, Z.-B. Xu, M.-Y. Lee, Q.-Y. Wu and H.-Y. Hu, Chem. Eng. J., 2020, 382, 122856 CrossRef CAS.
- R. Yin, L. Ling and C. Shang, Water Res., 2018, 142, 452–458 CrossRef CAS PubMed.
- J. Fang, Y. Fu and C. Shang, Environ. Sci. Technol., 2014, 48, 1859–1868 CrossRef CAS PubMed.
- S. S. Cheng, X. R. Zhang, W. H. Song, Y. H. Pan, D. Lambropoulou, Y. Zhong, Y. Du, J. X. Nie and X. Yang, Sci. Total Environ., 2019, 682, 629–638 CrossRef CAS PubMed.
- Y. R. Huang, S. K. Ding, L. Li, Q. Y. Liao, W. H. Chu and H. Z. Li, Water Res., 2021, 201, 117334 CrossRef CAS PubMed.
- I. Sánchez-Montes, G. O. S. Santos, A. J. dos Santos, C. H. M. Fernandes, R. S. Souto, P. Chelme-Ayala, M. G. El-Din and M. R. V. Lanza, Sci. Total Environ., 2023, 878, 163047 CrossRef PubMed.
- S. Liang, L. Zhu, J. Hua, W. Duan, P. T. Yang, S. L. Wang, C. Wei, C. Liu and C. Feng, Environ. Sci. Technol., 2020, 54, 6406–6414 CrossRef CAS PubMed.
- Z. H. Cheng, L. Ling, Z. H. Wu, J. Y. Fang, P. Westerhoff and C. Shang, Environ. Sci. Technol., 2020, 54, 11584–11593 CrossRef CAS PubMed.
- H. Zhang, Z. Li, X. Zhou, X. Lu, H. Gu and J. Ma, Sci. Total Environ., 2022, 853, 158345 CrossRef CAS PubMed.
- K. Kim, H. Y. Chung, J. Ju and J. Kim, Sci. Total Environ., 2017, 590–591, 107–113 CrossRef CAS PubMed.
- K. Kim and W. Choi, Environ. Sci. Technol., 2011, 45, 2202–2208 CrossRef CAS PubMed.
- Q. A. Nguyen, B. Kim, H. Y. Chung, A. Q. K. Nguyen, J. Kim and K. Kim, Ecotoxicol. Environ. Saf., 2021, 208, 111735 CrossRef CAS PubMed.
- K. Kim, J. Kim, A. D. Bokare, W. Choi, H. I. Yoon and J. Kim, Environ. Sci. Technol., 2015, 49, 10937–10944 CrossRef CAS PubMed.
- J. Ju, J. Kim, L. Vetrakova, J. Seo, D. Heger, C. Lee, H. I. Yoon, K. Kim and J. Kim, J. Hazard. Mater., 2017, 329, 330–338 CrossRef CAS PubMed.
- T. U. Han, J. Kim and K. Kim, Environ. Res., 2021, 197, 111059 CrossRef CAS PubMed.
- T. U. Han, J. Kim and K. Kim, J. Ind. Eng. Chem., 2021, 100, 310–316 CrossRef CAS.
- N. T. H. Le, J. Ju, B. Kim, M. S. Kim, C. Lee, S. Kim, W. Choi, K. Kim and J. Kim, Chem. Eng. J., 2020, 388, 124226 CrossRef CAS.
- Y. Y. Ahn, J. Kim and K. Kim, Sci. Total Environ., 2021, 785, 147369 CrossRef CAS PubMed.
- Y. Choi, H. I. Yoon, C. Lee, L. Vetrakova, D. Heger, K. Kim and J. Kim, Environ. Sci. Technol., 2018, 52, 5378–5385 CrossRef CAS PubMed.
- K. Kim, N. T. H. Le, A. Q. K. Nguyen, Y.-Y. Ahn, B. Kim, G. Shin, W. Choi and J. Kim, Chem. Eng. J., 2022, 428, 131134 CrossRef CAS.
- Y. Y. Ahn, J. Kim and K. Kim, Environ. Sci. Technol., 2022, 56, 2323–2333 CrossRef CAS PubMed.
- F. Sun, Y. Xiao, D. Wu, W. Zhu and Y. Zhou, Chem. Eng. J., 2017, 327, 1128–1134 CrossRef CAS.
- J. Du, K. Kim, S. Son, D. Pan, S. Kim and W. Choi, Environ. Sci. Technol., 2023, 57, 5317–5326 CrossRef CAS PubMed.
- O. Monfort and K. Hanna, Environ. Chem. Lett., 2019, 17, 1391–1396 CrossRef CAS.
- S. Xue, C. Wang, Z. Zhang, Y. Song and Q. Liu, Chemosphere, 2016, 144, 816–826 CrossRef CAS PubMed.
- A. M. Grannas, A. R. Bausch and K. M. Mahanna, J. Phys. Chem. A, 2007, 111, 11043–11049 CrossRef CAS PubMed.
- D. W. Min and W. Choi, Environ. Sci. Technol., 2017, 51, 8368–8375 CrossRef CAS PubMed.
- R. O'Concubhair and J. R. Sodeau, Acc. Chem. Res., 2013, 46, 2716–2724 CrossRef PubMed.
- N. Takenaka and H. Bandow, J. Phys. Chem. A, 2007, 111, 8780–8786 CrossRef CAS PubMed.
- A. Barra Caracciolo, E. Topp and P. Grenni, J. Pharm. Biomed. Res., 2015, 106, 25–36 CrossRef CAS PubMed.
- I. Sirés, C. Arias, P. L. Cabot, F. Centellas, J. A. Garrido, R. M. Rodríguez and E. Brillas, Chemosphere, 2007, 66, 1660–1669 CrossRef PubMed.
- S. C. Park, E. S. Moon and H. Kang, Phys. Chem. Chem. Phys., 2010, 12, 12000–12011 RSC.
- X. J. Kong, Z. H. Wu, Z. R. Ren, K. H. Guo, S. D. Hou, Z. C. Hua, X. C. Li and J. Y. Fang, Water Res., 2018, 137, 242–250 CrossRef CAS PubMed.
- L. Wang, J. Fang, X. Zhang, X. Xu, X. Kong, Z. Wu, Z. Hua, Z. Ren and K. Guo, Chemosphere, 2019, 226, 123–131 CrossRef CAS PubMed.
- N. Takenaka, M. Tanaka, K. Okitsu and H. Bandow, J. Phys. Chem. A, 2006, 110, 10628–10632 CrossRef CAS PubMed.
- D. Heger, J. Klánová and P. Klán, J. Phys. Chem. B, 2006, 110, 1277–1287 CrossRef CAS PubMed.
- M. Deborde and U. von Gunten, Water Res., 2008, 42, 13–51 CrossRef CAS PubMed.
- S. S. Lau, K. P. Reber and A. L. Roberts, Environ. Sci. Technol., 2019, 53, 11133–11141 CrossRef CAS PubMed.
- C. K. Remucal and D. Manley, J. Environ. Sci. Water Resour., 2016, 2, 565–579 CAS.
- C. Robinson, C. S. Boxe, M. I. Guzmán, A. J. Colussi and M. R. Hoffmann, J. Phys. Chem. B, 2006, 110, 7613–7616 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra00081a |
| This journal is © The Royal Society of Chemistry 2024 |