DOI:
10.1039/D3RA08962B
(Review Article)
RSC Adv., 2024,
14, 5690-5728
Antineoplastic indole-containing compounds with potential VEGFR inhibitory properties†
Received
31st December 2023
, Accepted 29th January 2024
First published on 14th February 2024
Abstract
Cancer is one of the most significant health challenges worldwide. Various techniques, tools and therapeutics/materials have been developed in the last few decades for the treatment of cancer, together with great interest, funding and efforts from the scientific society. However, all the reported studies and efforts seem insufficient to combat the various types of cancer, especially the advanced ones. The overexpression of tyrosine kinases is associated with cancer proliferation and/or metastasis. VEGF, an important category of tyrosine kinases, and its receptors (VEGFR) are hyper-activated in different cancers. Accordingly, they are known as important factors in the angiogenesis of different tumors and are considered in the development of effective therapeutic approaches for controlling many types of cancer. In this case, targeted therapeutic approaches are preferable to the traditional non-selective approaches to minimize the side effects and drawbacks associated with treatment. Several indole-containing compounds have been identified as effective agents against VEGFR. Herein, we present a summary of the recent indolyl analogs reported within the last decade (2012–2023) with potential antineoplastic and VEGFR inhibitory properties. The most important drugs, natural products, synthesized potent compounds and promising hits/leads are highlighted. Indoles functionalized and conjugated with various heterocycles beside spiroindoles are also considered.
 Dalia R. Aboshouk | Dalia R. Aboshouk, Research Assistant in the National Research Centre, Egypt. Awarded BSc from the Faculty of Science, Alexandria University (Honors Degree, 2013) and MSc in Organic Chemistry from Helwan University (2022). |
 M. Adel Youssef | Prof. M. Adel Youssef, Prof. of Organic Chemistry, Faculty of Science, Helwan University, Egypt. He received his PhD from AL-Azhar University, Egypt in 1976 and became a Professor of Organic Chemistry in 1990. He supervised many MSc and PhD theses, contributing to different branches of chemistry. |
 Mohamed S. Bekheit | Mohamed S. Bekheit, Associate Professor in the National Research Center, Egypt since 2018. He received his PhD from Ain-Shams University, Egypt (2013). His research studies focus on design and synthesis of heterocyclic compounds of potential biological properties, together with interest in organo-phosphorus chemistry with special interest in computational chemistry techniques. He has participated in several national and international research projects. |
 Ahmed R. Hamed | Ahmed Ragab Hamed, Professor of Therapeutic Biochemistry at the National Research Centre, Egypt. He was awarded his PhD in Biomedical Sciences from the University of Nottingham, UK (2005–2009). Subsequently, was granted two Postdoctoral Fellowships in Dundee (Scotland) and UT Health (Houston, Texas). His special interest is in the evaluation of compounds in various cell-based models such as cancer prevention, inflammation inhibition and reversal of cancer multi-drug resistance. He also has experience in cancer cell biology and regulation of several members of the cytoprotective proteome using in vitro cell culture approaches, with experience in investigation the levels of expression of different mammalian proteins using western blotting. |
 Adel S. Girgis | Adel S. Girgis, Emeritus Professor at the National Research Centre (NRC), Egypt. He received his PhD from Cairo University, Egypt in 1997 and was promoted to Full Professor of Organic Chemistry at the National Research Centre, Egypt in 2009. He joined the research group of Prof. Alan R. Katritzky (Center of Heterocyclic Compounds, University of Florida, USA) in 2008 and 2013. He received many national and international research funds. His research studies are focused on the design and synthesis of heterocyclic compounds with potential biological properties, with special interest in computational chemistry techniques. |
1. Introduction
Cancer is one of the most significant health challenges worldwide. It is the second most fatal disease globally after cardiovascular disorders.1 About 19–20 million people are diagnosed with different cancer types every year and many of them lose their life.2 Although many techniques, tools and therapeutics/materials have been developed in the last few decades, none of them are effective in controlling the various types of cancer at different stages, especially the advanced ones.3 To date, this challenge is still a significant task despite the great interest, funding and efforts from the entire scientific society (including research institutes and pharmaceutical companies).4 Recently, progress has been achieved in cancer chemotherapy due to the efforts devoted to developing selective molecular therapeutics. Although traditional non-selective therapeutics are still employed clinically, their drawbacks/side effects and poor survival rates are major problems, limiting their applications. Additionally, the early detection of this disease is challenging, although it is the most appropriate opportunity for curing it.5,6
To date, 90 protein tyrosine kinases have been identified among the known 518 kinases.7 Tyrosine kinases are capable of many diverse cellular functions including growth, proliferation, differentiation and death.8 Tyrosine kinases can catalyze the phosphorylation of tyrosine utilizing the adenosine triphosphate (ATP) molecule, which can be classified into receptor and non-receptor types. The receptor category include trans-membrane, extracellular and intracellular, whereas the non-receptors are intracellular.9 The overexpression of tyrosine kinases is associated with cancer proliferation and/or metastasis, indicating their importance as cancer chemotheraputics.10,11
Angiogenesis is an essential biological process for the formation/extension of new blood capillaries from the vessels present in the vascular system. Thus, it is an essential process for growth, menstruation, embryonic development, wound healing, functional repair and many pathological disorders including cancer. Furthermore, it is one of the main functions for delivering vital supplies including nutrients and oxygen to cells and removal of waste. Angiogenesis is also a critical process for tumor progression and metastasis. Many growth factors (VEGF: vascular endothelial growth factor, EGF: epidermal growth factor, and FGF: fibroblast growth factor) can stimulate angiogenesis.12–15 VEGF can be categorized into different classes (VEGF-A, B, C, and D), which can bind to diverse tyrosine kinase receptors (VEGFR-1, 2, and 3). Notably, VEGF and its receptors are hyper-activated in different cancers, and thus considered an important target for combating this disease. VEGFR-2 is the most well-known factor for angiogenesis of various solid tumors (colon, breast, ovary, lung, skin, renal, head, neck, lymphoma, etc.). Several drugs with inhibitory VEGFR-2 activity have been recognized and clinically approved as anticancer drugs.12,13,16 Anti-angiogenic active agents reduce the vascular permeability and enhance the extravasation of the therapeutic small molecules.17 However, some adverse effects have been reported to be associated with anti-VEGFR therapeutics including dermatologic disorders (skin rash, depigmentation and mucositis), painful hand-foot skin reactions and pruritus.18
Accordingly, multi-targeted inhibitory drugs/candidates have become a recent trend in cancer chemotherapy, attracting significant attention and research interest. Interest in this strategy is attributed to the multifactorial nature of many cancer types.7 Additionally, carcinoma cell initiation and proliferation involve various receptors and signaling pathways. Moreover, multi-target inhibitors can overcome cancer cell resistance, which is an advantage for multi-targeted treatment compared to single-targeted treatment or cocktail of multi-component drugs.7,19
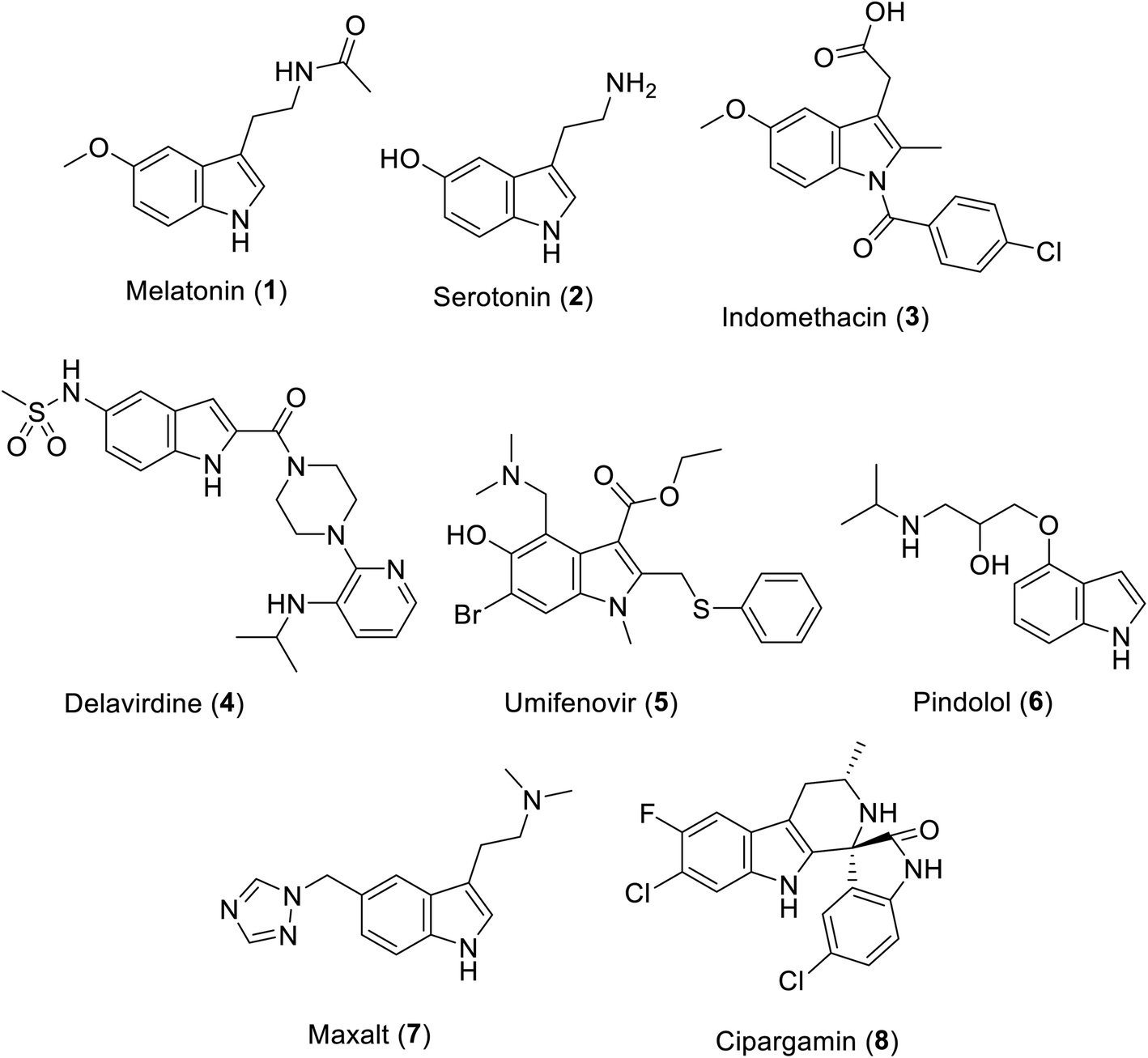
The indolyl scaffold occupies a unique position among the diverse alkaloids due to the wide range of bio-properties of its natural and synthesized analogs. Many natural alkaloids are well known, among which melatonin 1 is a natural hormone biosynthesized in the dark by the pineal gland20 (Fig. 1). Recently, clinical trials supported its positive impact on the prevention or treatment COVID-19 infection upon being administered alone or in combination with other therapeutics.21–24 Serotonin 2 is a neurotransmitter that controls many human functions such as mood, appetite, sleep and social behavior.25
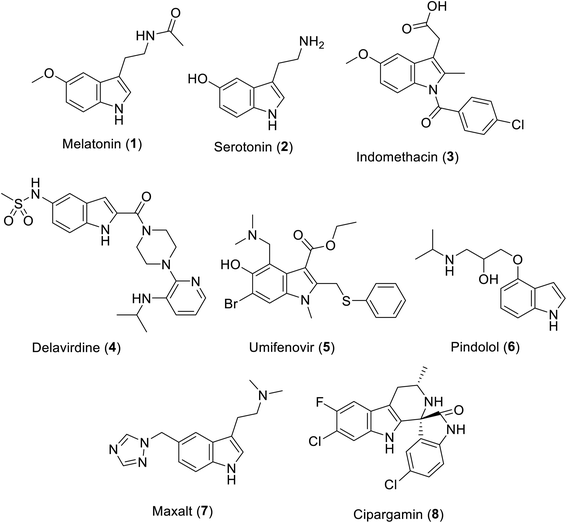 |
| | Fig. 1 Clinically approved indole-containing drugs (1–7) and cipargamin (8). | |
Many indole-containing drugs have been approved and in clinical use for a long time, among which indomethacin 3 is a famous non-steroidal anti-inflammatory drug (approved by the FDA “Food and Drug Administration” since 1965)26 with inhibitory properties against cyclooxygenase (COX), an enzyme controlling the formation of prostaglandin from arachidonic acid.27 Delavirdine 4 (FDA approval in 1997) is an anti-HIV (human immunodeficiency virus) drug.28,29 Umifenovir 5 (approved in Russia and China) is an anti-influenza drug repurposed for the treatment of SARS-CoV-2.30–33 Pindolol 6 (antihypertensive drug, non-cardioselective β-blocker, FDA approval in 1982)34 and Maxalt 7 (antimigraine agent, FDA approval in 1998)35 are also indole analogs. Cipargamin 8 exhibits high efficacy against protein synthesis in Plasmodium falciparum. In addition, it has entered pre-clinical trial investigations as a potential antimalarial drug.36
The current study aims to describe and highlight the indole-containing compounds with potential anti-VEGFR properties. Specifically, the relevant keywords were input in different search engines such as Scopus, ScienceDirect and PubMed. The recent advances in this subject (last decade, 2023–2012) with the greatest diversification revealing promising bio-properties will be discussed.
2. Indole-containing drugs and potent agents
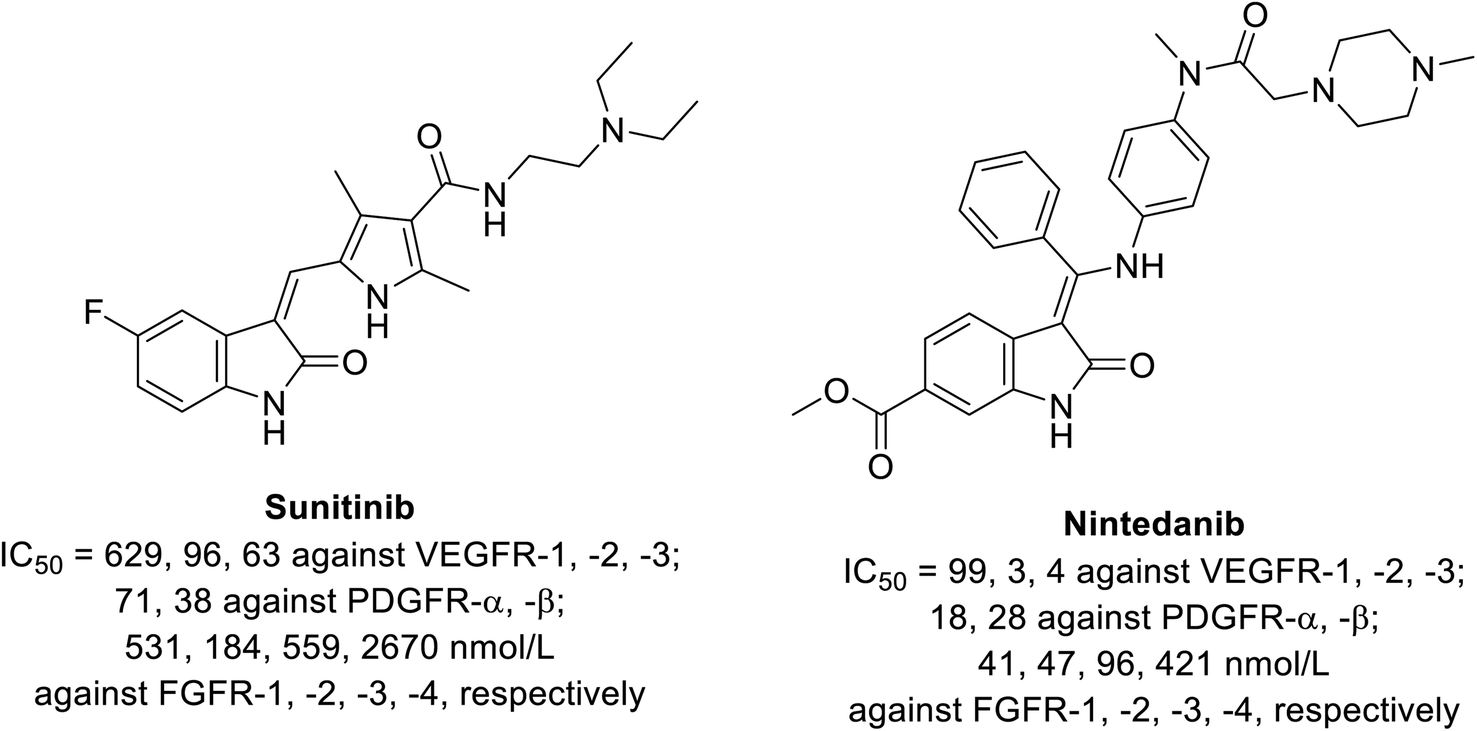
2.1. Sunitinib
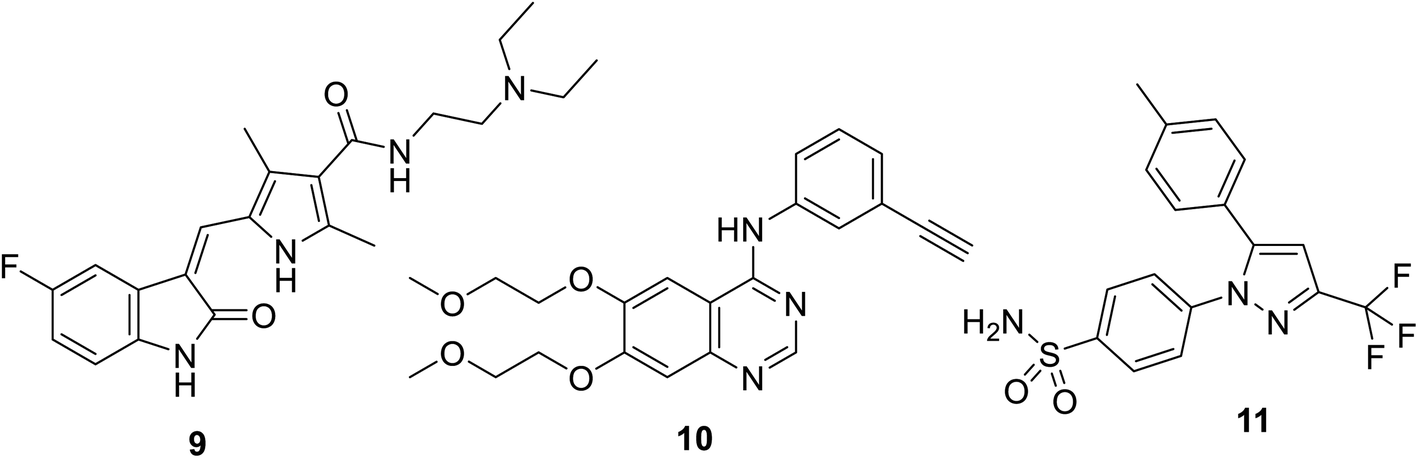
Sunitinib (Sutent) 9 (Fig. 2) is an oral antitumor multi-targeted tyrosine kinase inhibitor (VEGFR-1,-2, -3; PDGFR-α, -β: platelet-derived growth-factor receptor; and c-kit: stem cell factor receptor) against FLT3, which is an FMS-like tyrosine kinase inhibitor with potent anti-angiogenesis properties.16,37–39 It is clinically approved against advanced renal and imatinib-resistant gastrointestinal (FDA approval in 2006) and pancreatic cancers (FDA approval in 2011). It has also been given FDA approval (2017) for adult adjuvant treatment at high risk of renal cancer.40,41 However, due to the side effects/drawbacks (diarrhea, fatigue, hypertension, hematologic toxicities, cardiotoxic effects and hand-foot syndrome)39,42–44 observed during clinical administration, many studies proposed the combination of sunitinib with another chemotherapeutic agent or radiation. This approach can reduce the unintended side effects and enhance the efficacy of the drug.42
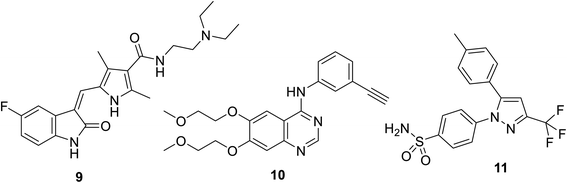 |
| | Fig. 2 Sunitinib (sutent) 9 multi-targeted tyrosine kinase inhibitor, erlotinib 10 (EGFR inhibitor) and celecoxib 11 (COX-inhibitor). | |
Also, sunitinib can be useful in the treatment of melanoma, which was supported by pre-clinical studies revealing the initiation of tumor hypoxia in melanoma xenografts.16 An obvious objective response by solid tumors to sunitinib including metastatic breast, colon, neuroendocrine and non-small cell lung cancers (NSCLC) was reported.39,42,45
A synergistic effect was observed upon the combination of sunitinib with erlotinib 10 (EGFR: epidermal growth factor receptor, inhibitor) for the treatment of NSCLC A549 xenograft mice46 (Fig. 2). Pre-clinical model studies supported that the combination of PRX177561 (CXC4, chemokine receptor type 4 antagonist) with sunitinib enhanced the therapeutic efficacy (reducing the tumor proliferation and extending the disease-free survival) against glioblastoma (brain cancer).47,48 In vivo studies supported that a COX-2 (cyclooxygenase-2) inhibitor (celecoxib 11) can enhance the activity of sunitinib in mice bearing human renal cancer xenografts via the observation of delay in tumor progression.49
2.2. Nintedanib
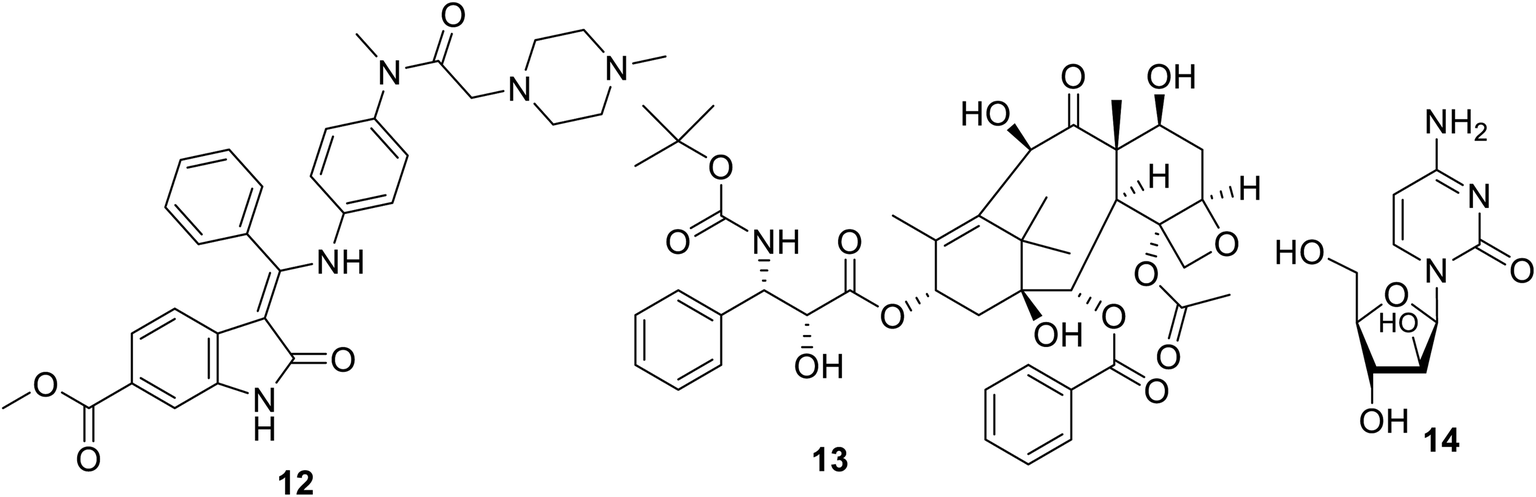
Nintedanib 12 (Ofev and Vargatef) is also an oral multi-targeted tyrosine kinase inhibitor (VEGFR-1, -2, -3; PDGFR-α, -β and FGFR-1, -2, -3, -4: fibroblast growth-factor receptor)50–54 (Fig. 3 and 4). The FDA approved its use for the treatment of idiopathic lung fibrosis (2014), systemic sclerosis-associated interstitial lung disease (2019) and NSCLC (in combination with docetaxel 13).50,52–56 However, the most notable side effects are diarrhea and increase in alanine and aspartate aminotransferase associated with the clinical administration of nintedanib.54
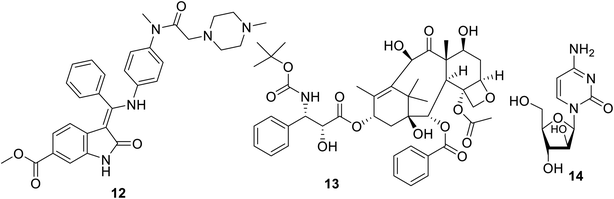 |
| | Fig. 3 Nintedanib 12 (multi-targeted) tyrosine kinase inhibitor, docetaxel 13 and cytarabine 14. | |
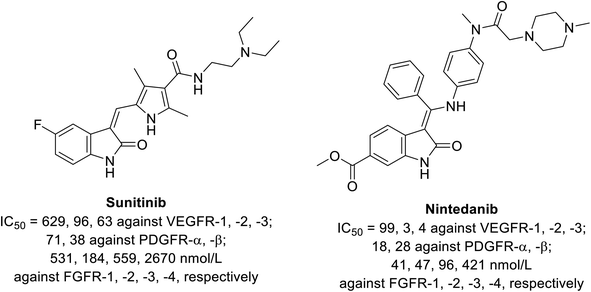 |
| | Fig. 4 Inhibitory properties of sunitinib and nintedanib against tyrosine kinases (VEGFR-1, -2, -3; PDGFR-α, -β and FGFR-1, -2, -3, -4).52 | |
Many studies explained the promising efficacy of nintedanib against different types of cancer. The delay of the proliferation and growth arrest of a mouse prostate (TRAMP: transgenic adenocarcinoma of the mouse prostate) model by nintedanib indicates its promising properties against prostate cancer.57,58 Its clinical trial (phase III) also support its promising properties against ovarian cancer.59,60 A compensatory role was reported for nintedanib towards metastatic colon cancer.61 Meanwhile, clinical trial observation (20 patients, 200 mg twice per day) revealed that there was no considerable effect on salivary gland cancer except in controlling the rate of the disease.62 A phase I clinical study (13 elderly patients) also showed its efficacy towards myeloid leukemic cells, especially when used with cytarabine 14.63
2.3. Anlotinib
Anlotinib 15 (Fig. 5) is an oral multi-targeted tyrosine kinase inhibitor (VEGFR, PDGFR, FGFR and c-kit) approved in China for NSCLC.64–69 Hypertension and gastrointestinal problems are the most significant side effects associated with its administration.65 Finger print loss for about two months was observed during a case study on its treatment of lung cancer.65 It has also been reported that anlotinib is capable of inhibiting lymphangiogenesis and lymphatic metastasis, which is probably due to the suppression of VEGFR-3 phosphorylation.70 Antiproliferation properties were also observed against colon cancer cells (HCT-116 and LOVO).71 Moreover, anlotinib showed promising properties against thyroid and metastatic renal cell cancers.68,72 A phase II clinical study supported the possibility for the therapeutic utilization of anlotinib in combination with oxaliplatin 16 and capecitabine 17 to treat patients with metastatic colon cancer.73
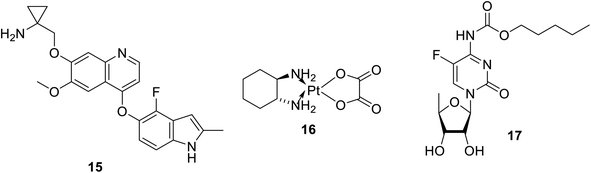 |
| | Fig. 5 Anlotinib 15 (a multi-targeted tyrosine kinase inhibitor), oxaliplatin 16 capecitabine 17. | |
2.4. Surufatinib (HMPL-012, sulfatinib)
Surufatinib (Sulanda) 18 (Fig. 6) is an oral multi-kinase inhibitor with dual function against angiogenesis (VEGFR-1, -2, -3 and FGFR) and tumor immune evasion (CSF-1R: colony stimulating factor-1 receptor). It is approved (in China, 2020) for the treatment of extrapancreatic neuroendocrine tumor (NET).74 However, it has been filed with the FDA for approval for the treatment of advanced NET (2021).75 Hypertension and proteinuria are the most severe adverse effects reported with the administration of Sulanda.76 A phase II clinical study adopting 39 patients (300 mg, 28 day cycles, once daily) supported its possibility for moderate biliary tract cancer.77
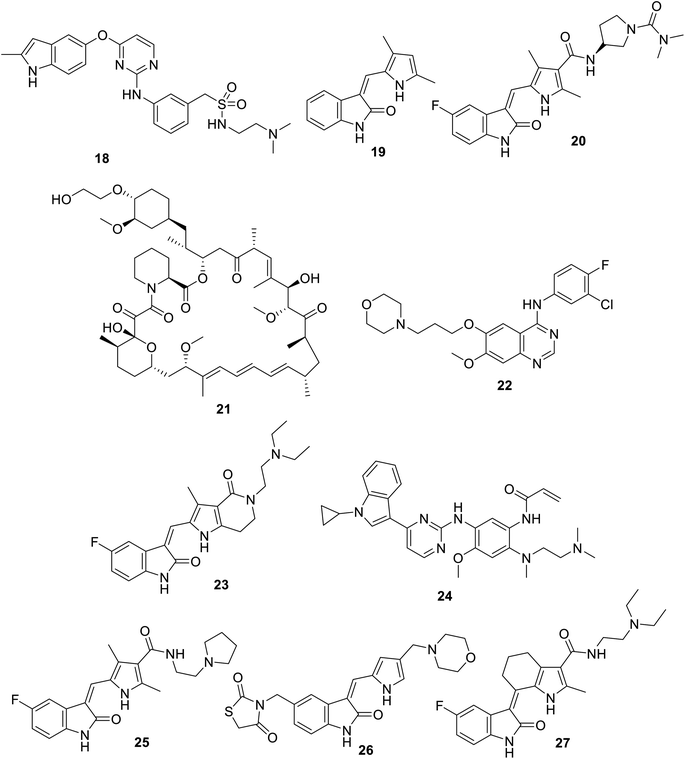 |
| | Fig. 6 Chemical structures of surufatinib 18, SU5416 (semaxanib) 19, vorolanib (CM082) 20, everolimus 21, gefitinib (Iressa) 22, famitinib 23, almonertinib (HS-10296) 24, toceranib (Palladia, Zoetis) 25, S49076 26 and SIM010603 27. | |
2.5. SU5416 (semaxanib)
SU5416 (semaxanib) 19 (Fig. 6) is a human solid tumor antiangiogenic VEGFR inhibitor under investigation.78–80 Its antiproliferation of murine cardiac endothelial cells was reported.80 Also, pathophysiological effects in respiratory disorders were mentioned, which was supported by the lipopolysaccharide-induced acute lung injury in mice via restrain/modulate vascular permeability.79
2.6. Vorolanib
Vorolanib (CM082) 20 (Fig. 6) is an oral VEGFR and PDGFR inhibitor.81 A phase I study (22 patients) exhibited promising results for the combination of vorolanib with everolimus 21 towards renal and neuroendocrine cancers.82 It has also mentioned that vorolanib enhances the antiproliferation and apoptosis properties of gefitinib (Iressa) 22 (EGFR: epidermal growth factor receptor, inhibitor) towards NSCLC cell lines (HCC827 and H3255). This was explained by the strong inhibitory properties of the combined therapeutics on STAT3 phosphorylation compared to that of the mono-therapeutic.83
2.7. Famitinib
Famitinib 23 (Fig. 6) is an oral VEGFR-2 and -3 inhibitor used in trial studies against colon and renal cancers.84,85 A promising synergistic effect was observed against NSCLC upon the combination of famitinib with almonertinib (HS-10296, EGFR inhibitor) 24.85
2.8. Toceranib
Toceranib (Palladia, Zoetis) 25 (Fig. 6) is a multi-targeted tyrosine kinase inhibitor (VEGFR-2, PDGFRs and c-Kit) used as a phosphate salt for the treatment of bone cancer (canine osteosarcoma) in veterinary medicine (dogs). Cell growth inhibition of toceranib phosphate on canine osteosarcoma (Penny and Wall) in vitro was reported.86
2.9. S49076
S49076 26 (Fig. 6) is a VEGF and HIF-1-α (hypoxia-inducible factor 1-alpha) inhibitor.87 The inhibition of VEGF and HIF-1 expression can explain the mode of action of S49076 in ovarian cancer cells.87 The administration of S49076 as a monotherapeutic resulted in the arrest of colon bevacizumab-resistant tumor growth. Moreover, the combination of S49076 and bevacizumab (Avastin, anti-vascular endothelial growth factor antibody) showed total growth inhibition of colon cancer xenograft models.88 A phase I study (103 patients) also revealed its effect on solid tumors (colon, lung, mesothelioma and uveal melanoma) upon oral administration.89
2.10. SIM010603
SIM010603 27 (Fig. 6) is an oral multi-targeted tyrosine kinase inhibitor (VEGFR-2, -3; PDGFR-β and stem cell factor receptor “c-kit”).90 Antiproliferation properties were reported against NCI-H460 (human lung), LLC-SW44 (Lewis lung) and MDA-MB-435 (breast) cancer cells in addition to the inhibition of xenograft tumor growth models and angiogenesis in mice.90 Adverse effects including gastrointestinal, pancreatic and skeletal toxicities, bronchopneumonia and cardiovascular dysfunction were mentioned in the toxicological studies of SIM010603 in rats and dogs (0–20 and 0–10 mg kg−1 per day oral administration, respectively, for 28 followed by 14 recovery days). However, no mortality rates were recorded for dogs receiving 10 mg kg−1.91
3. Natural indole-containing compounds
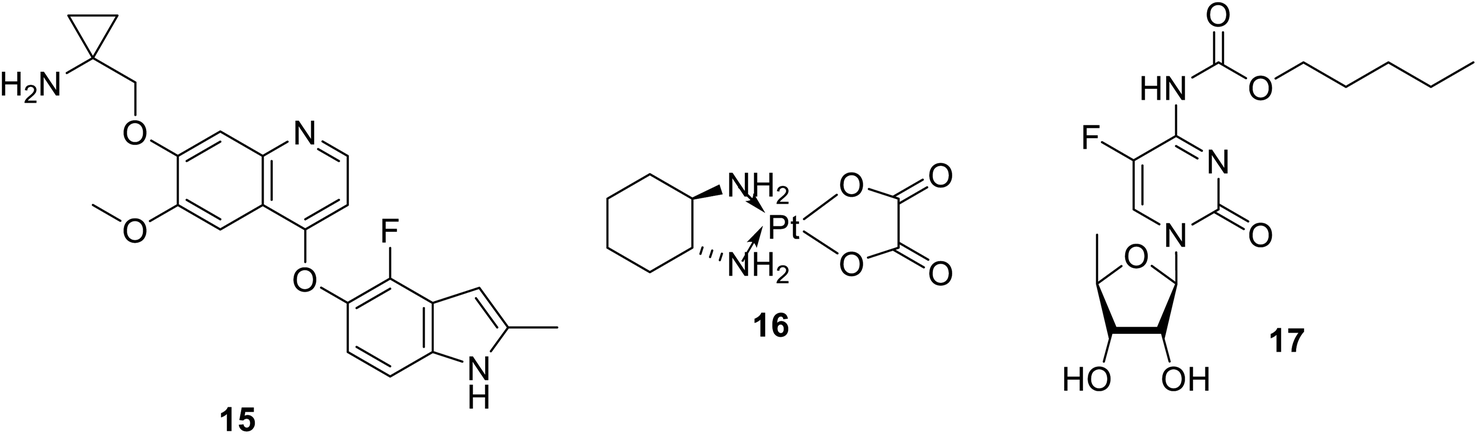
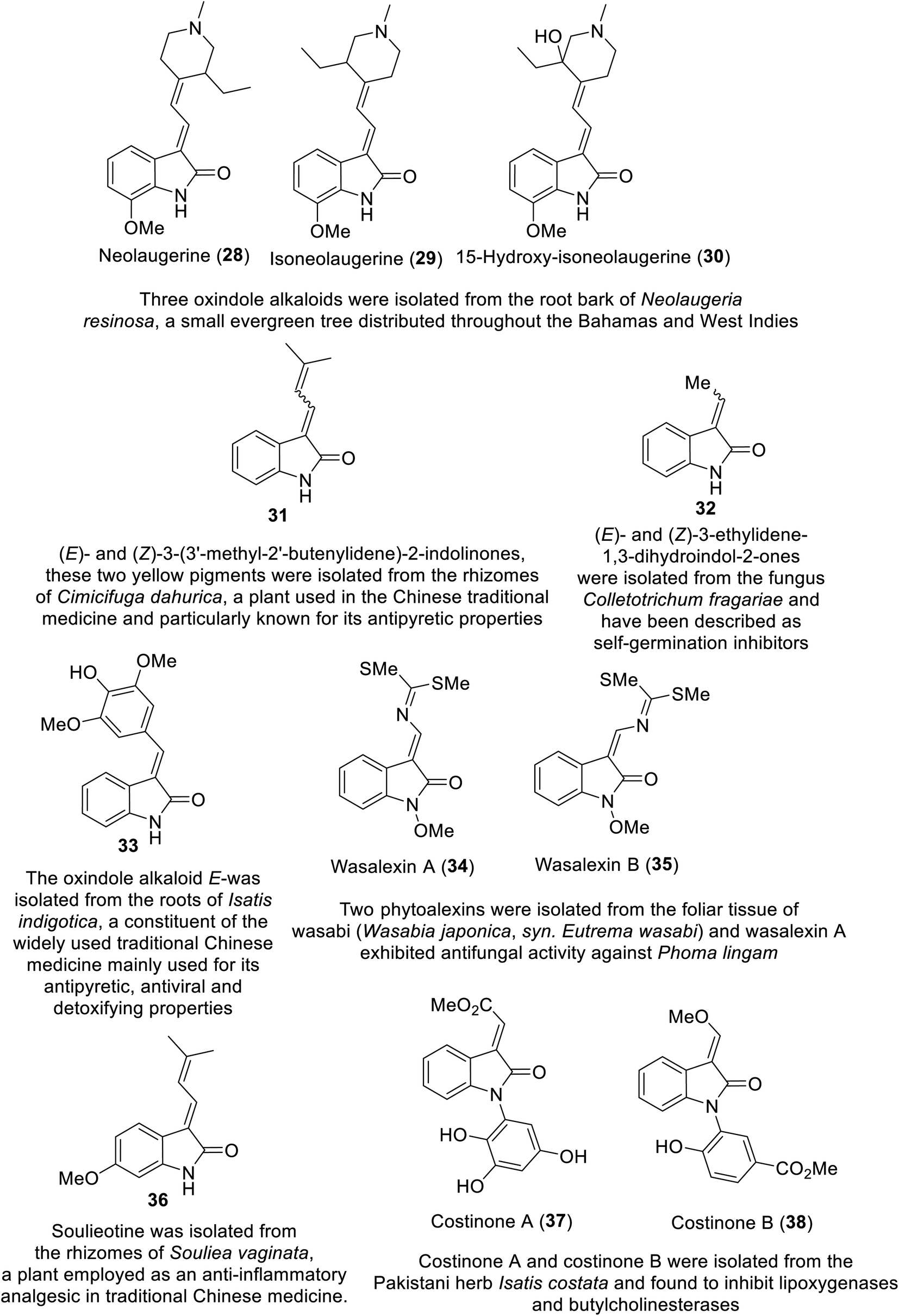
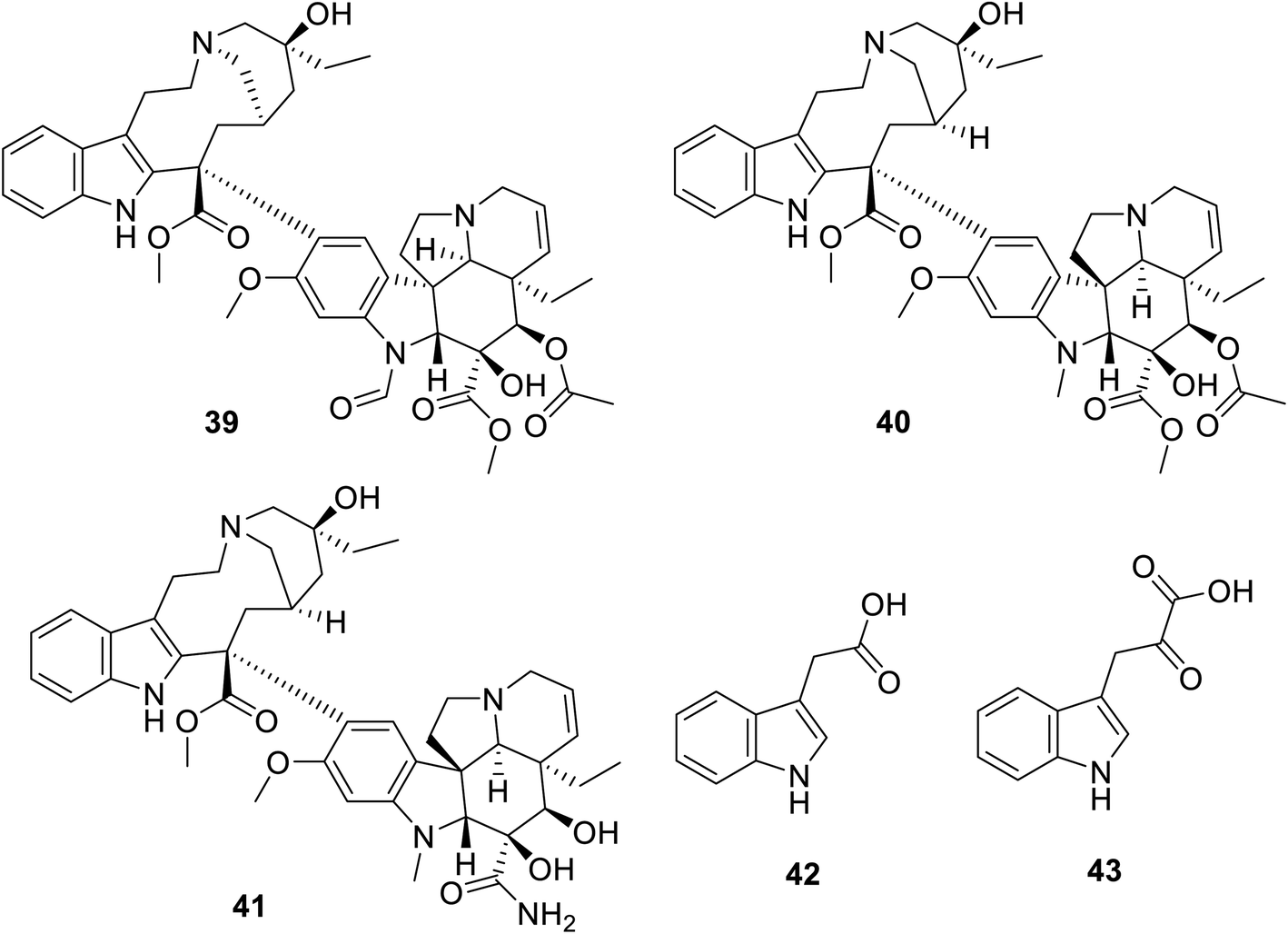
Many natural indolyl derivatives have been identified to possess considerable bio-properties including 28–38, as summarized in Fig. 7.92 Vincristine 39 (Fig. 8) was isolated from Vinca rosea and approved by the FDA (since 1963) for the treatment of Hodgkin's disease, non-Hodgkin's lymphoma and neuroblastoma.93–95 Vinblastine (Velban) 40 is also a Vinca alkaloid approved by the FDA (since 1965) for the treatment of Hodgkin's disease, lymphoma, and testicular and breast cancers.95–97 Vindesine (Eldisine) 41 is a Vinca alkaloid with efficacy against acute lymphocytic leukemia.95,98 3-Indole acetic acid 42, which is present in wine, and 3-indole pyruvic acid 43 (human metabolite) exhibited inhibitory properties against VEGFR-2 with IC50 = 0.9704 and 1.037 mM, respectively99 (Fig. 8).
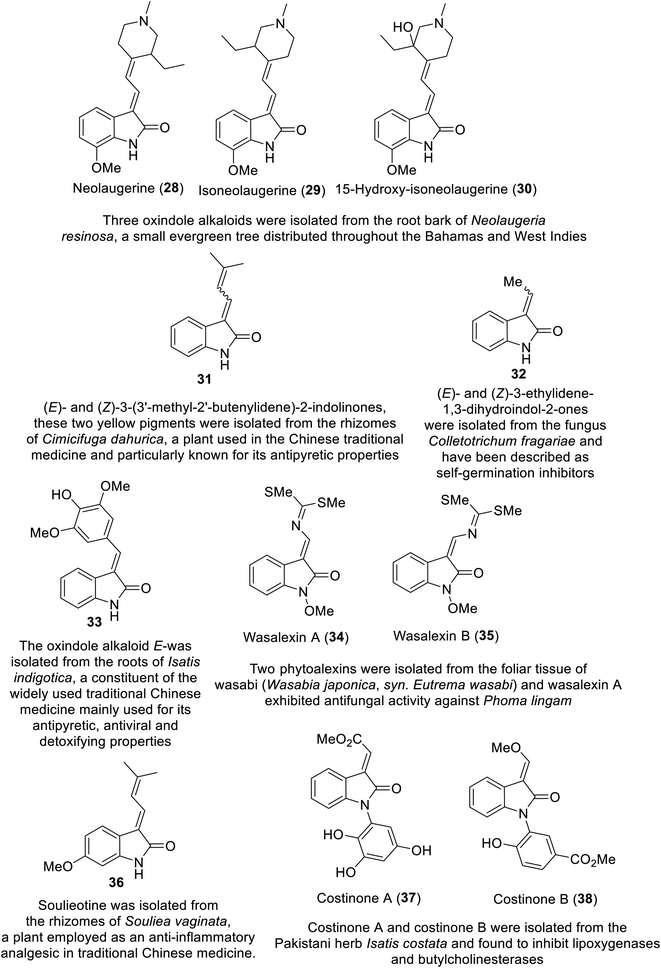 |
| | Fig. 7 Natural indole-containing compounds 28–38 with potential bio-properties. | |
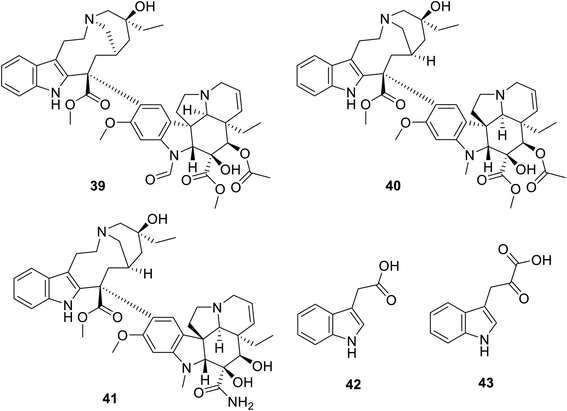 |
| | Fig. 8 Chemical structures of vincristine 39, vinblastine 40, vindesine 41, 3-indole acetic acid 42 and 3-indole pyruvic acid 43. | |
4. Synthesized indole-containing compounds
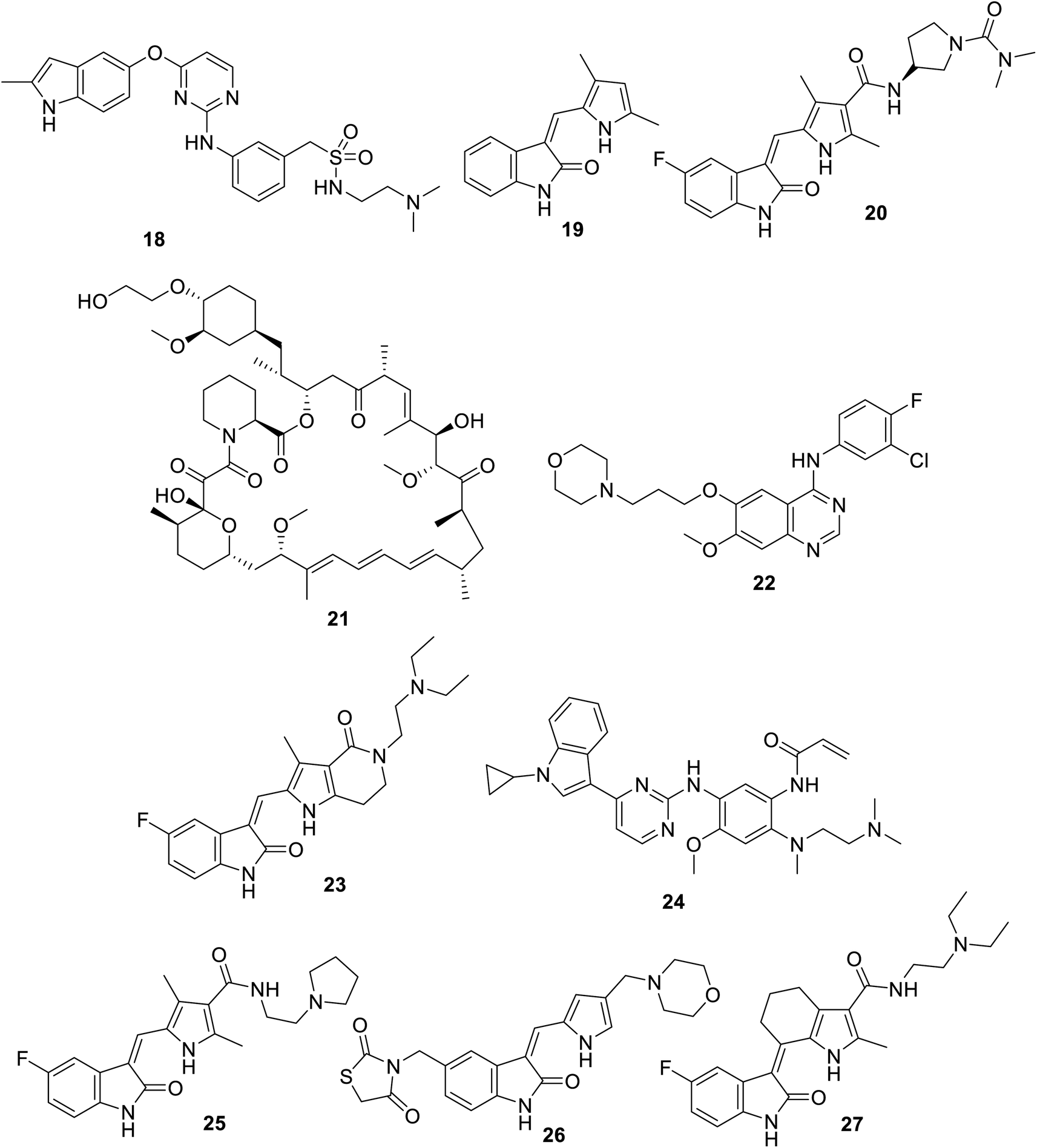
4.1. Indolecarboxamide
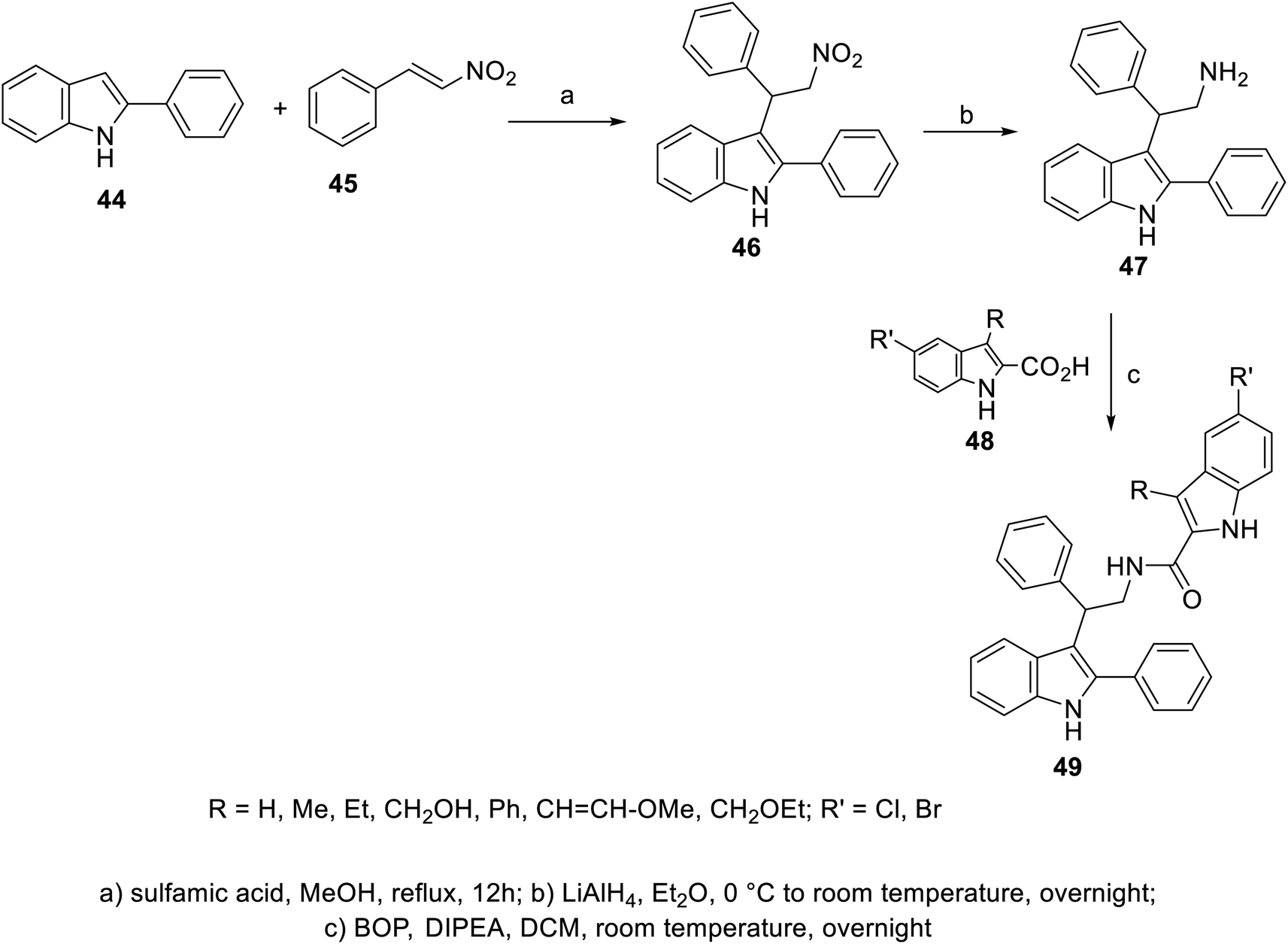
A variety of indole-2-carboxamides 49 was synthesized through the reaction of 2-(1H-indol-3-yl)ethan-1-amine 47 with 2-indolecarboxylic acids 48 in DCM (dichloromethane) containing BOP (benzotriazol-1-yloxytris(dimethylamino)phosphonium hexafluorophosphate) and DIPEA (diisopropyl ethylamine). Compound 47 was obtained through the reduction of nitro analog 46 by LiAlH4 in Et2O (at 0 °C to room temperature) under an inert (nitrogen) atmosphere. The latter 46 was synthesized by the addition reaction of 2-phenylindole 44 to β-nitrostyrene 45 in refluxing MeOH containing sulfamic acid100 (Scheme 1). Greater antiproliferation properties against a variety of human tumor cell lines [MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay, Panc-1 (pancreatic), MCF7 (breast), HT-29 (colon) and A-549 (epithelial)] were revealed (IC50, nM ± SEM “standard error mean”) for some of the prepared 2-indolecarboxamides 49 compared to that of erlotinib. Also, mild VEGFR-2 inhibitory properties were observed by some of the derivatives of 49 relative to that of sorafenib. The most considerable agent was 49e (R = CH2OH, R′ = Cl; IC50 = 44 ± 4, 46 ± 4, 45 ± 4, and 42 ± 4 nM against Panc-1, MCF7, HT-29 and A-549, respectively; IC50 = 1.10 ± 0.08 nM against VEGFR-2) (ESI Fig. S1†). Based on the observed antiproliferation properties, it was concluded that the function/group at the 3-position of the indole-2-carboxamide is the dominant factor, with the bio-properties following the order of H > methoxyvinyl > ethoxymethyl > hydroxymethyl > phenyl.100
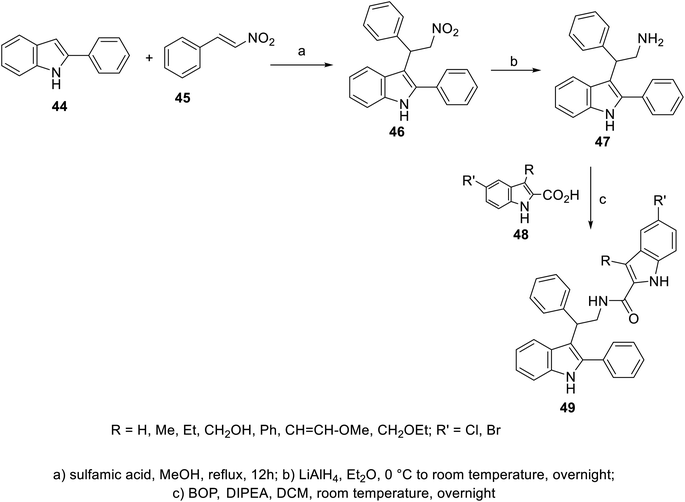 |
| | Scheme 1 Synthesis of indole-2-carboxamides 49. | |
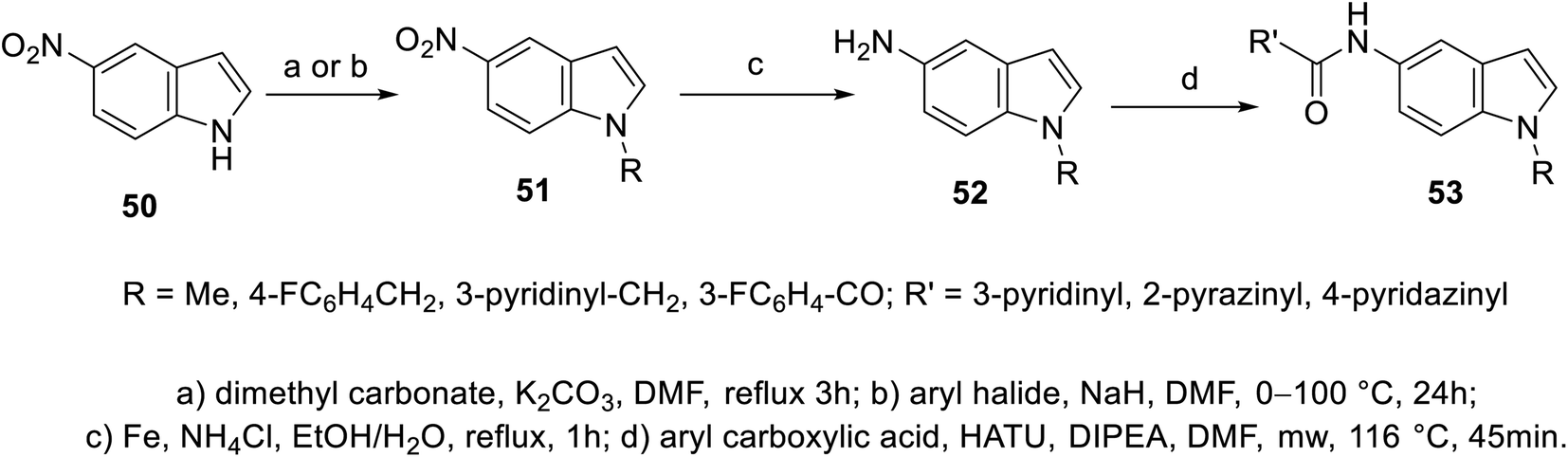
A set of 5-indolecarboxamides 53 was obtained through the reaction of the corresponding amino analogs 52 with the appropriate carboxylic acid in the presence of Hünig's base (N,N-diisopropylethylamine “Hünig's base”, DIPEA) using HATU (hexafluorophosphate azabenzotriazole tetramethyl uronium, coupling agent). 5-Aminoindoles 52 were prepared through the reduction of nitro analogs 51 (Fe, NH4Cl, EtOH/H2O). The latter 51 was obtained through the alkylation of 5-nitroindole 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 101,102 (Scheme 2). Weak VEGFR-2, CDK-1/cyclin B (cyclin-dependent kinase 1), and HER-2 (human epidermal growth factor receptor 2) properties were exhibited by most of the synthesized agents 53 at 10 μM. However, compound 53l [R = CO(3-FC6H4), R′ = 4-pyridazinyl] revealed promising kinase inhibitory activity against CDK-1/cyclin B and HER-2 (activity = 51% and 52%, respectively) at 10 μM101 (ESI Fig. S2†).
101,102 (Scheme 2). Weak VEGFR-2, CDK-1/cyclin B (cyclin-dependent kinase 1), and HER-2 (human epidermal growth factor receptor 2) properties were exhibited by most of the synthesized agents 53 at 10 μM. However, compound 53l [R = CO(3-FC6H4), R′ = 4-pyridazinyl] revealed promising kinase inhibitory activity against CDK-1/cyclin B and HER-2 (activity = 51% and 52%, respectively) at 10 μM101 (ESI Fig. S2†).
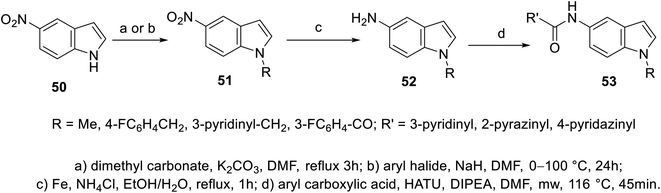 |
| | Scheme 2 Synthesis of 5-indolecarboxamides 53. | |
4.2. Indolyl Schiff bases
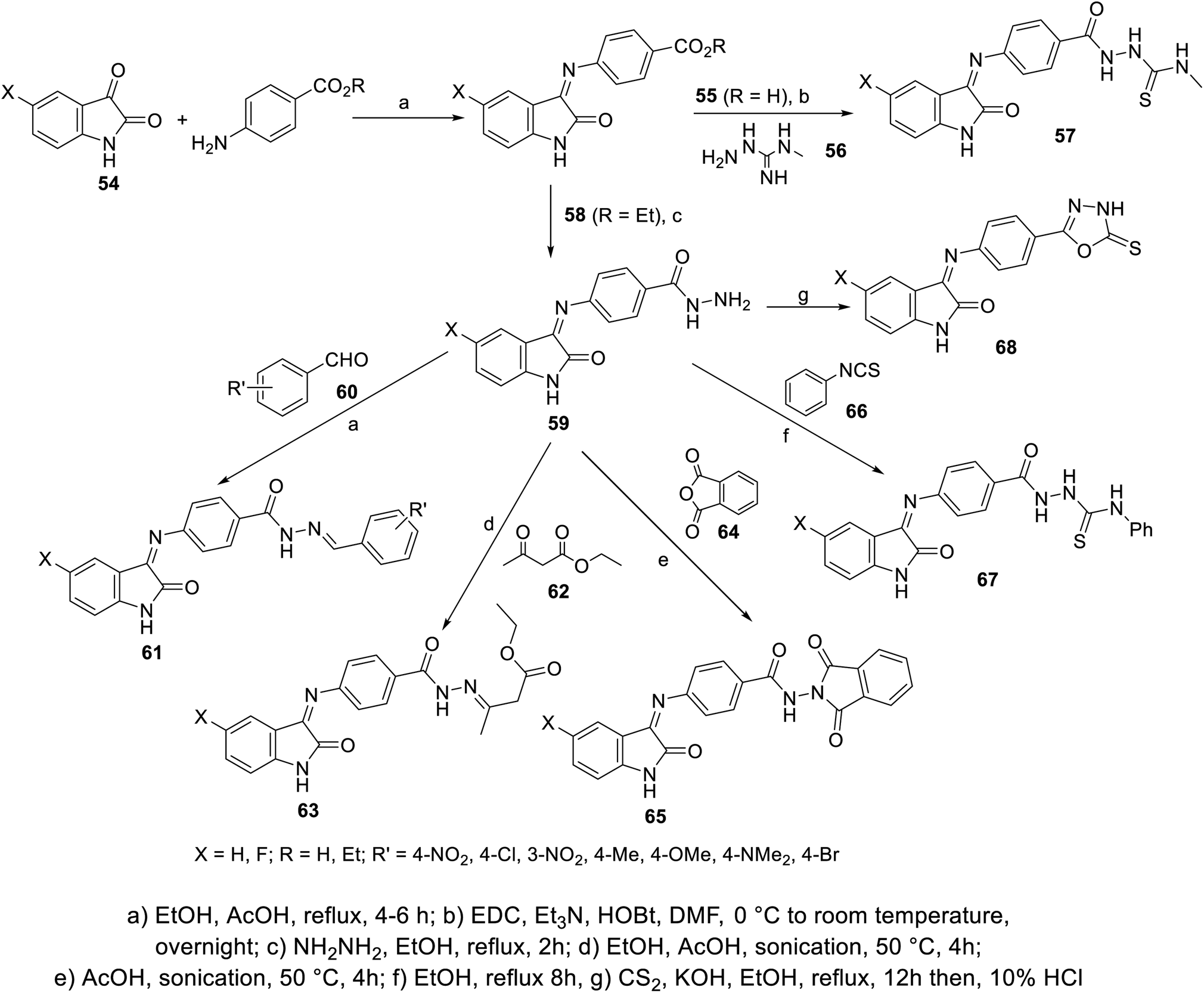
Condensation of isatins 54 with p-aminobenzoic acid (in refluxing EtOH containing a catalytic amount of AcOH) resulted in the formation of Schiff bases 55, which upon reaction with 4-methylthiosemicarbazide 56 afforded the corresponding semicarbazones 57. Similarly, the reaction of isatins 54 with benzocaine (4-aminobenzoic acid ethyl ester) 58, followed by reaction with hydrazine hydrate in refluxing ethanol gave the corresponding hydrazides 59. The latter were subjected to reaction with aromatic aldehydes 60 (in refluxing EtOH containing a catalytic amount of AcOH), ethyl acetoacetate 62 (in EtOH/AcOH, under sonication at 50 °C), phthalic anhydride 64 (in AcOH “glacial” under sonication at 50 °C) or phenyl isothiocyanate 66 (in refluxing EtOH), yielding the corresponding hydrazone 61, 63, dioxoisoindoline 65 and thiourea derivatives 67, respectively. Reaction of 59 with carbon disulfide in refluxing ethanolic KOH followed by acidification with HCl (10%), afforded the corresponding oxadiazolyl derivatives 68 (Scheme 3).
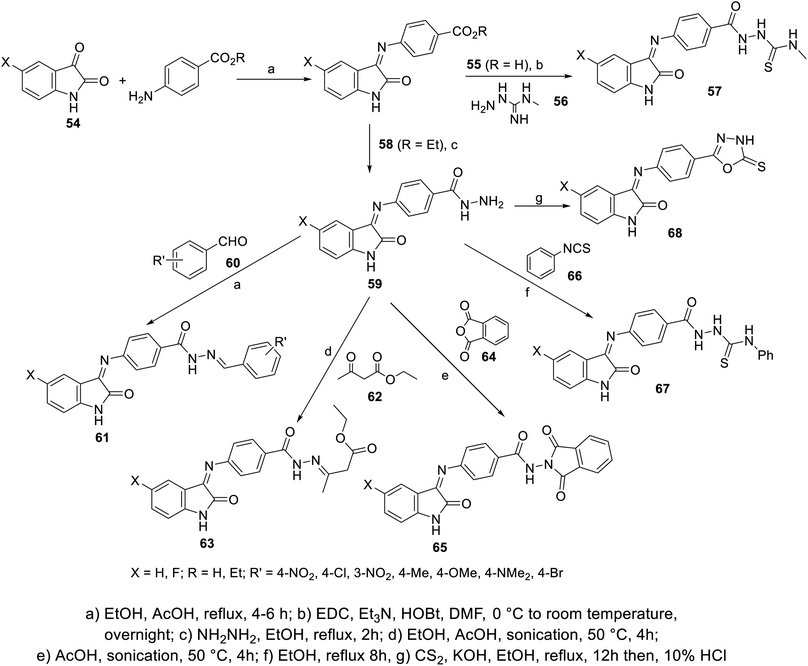 |
| | Scheme 3 Synthesis of indolyl Schiff bases 55, 57–59, 61, 63, 65, 67 and 68. | |
Some of the synthesized agents exhibited promising antiproliferation properties against HepG2 (liver) and MCF7 (breast) cancer cell lines relative to that of sunitinib (MTT assay; IC50, μM ± SD “standard division”) (ESI Fig. S3†). Some of the synthesized fluorinated analogs showed enhanced antitumor properties compared to other prepared derivatives, directing attention on the importance of this substituent in controlling the bio-properties. The VEGFR-2 inhibitory properties determined for the discovered promising agents were consistent with the observed antitumor properties. Compound 67a (X = H) revealed promising antiproliferation and enzymatic inhibitory properties (IC50 = 1.13 ± 0.06, 1.44 ± 0.11 μM against HepG2 and MCF7, respectively; IC50 = 0.078 ± 0.003 μM against VEGFR-2) relative to that of sunitinib (IC50 = 2.23 ± 0.11, 4.77 ± 0.29 μM against HepG2 and MCF7, respectively; IC50 = 0.139 ± 0.007 μM against VEGFR-2)103 (ESI Fig. S3†).
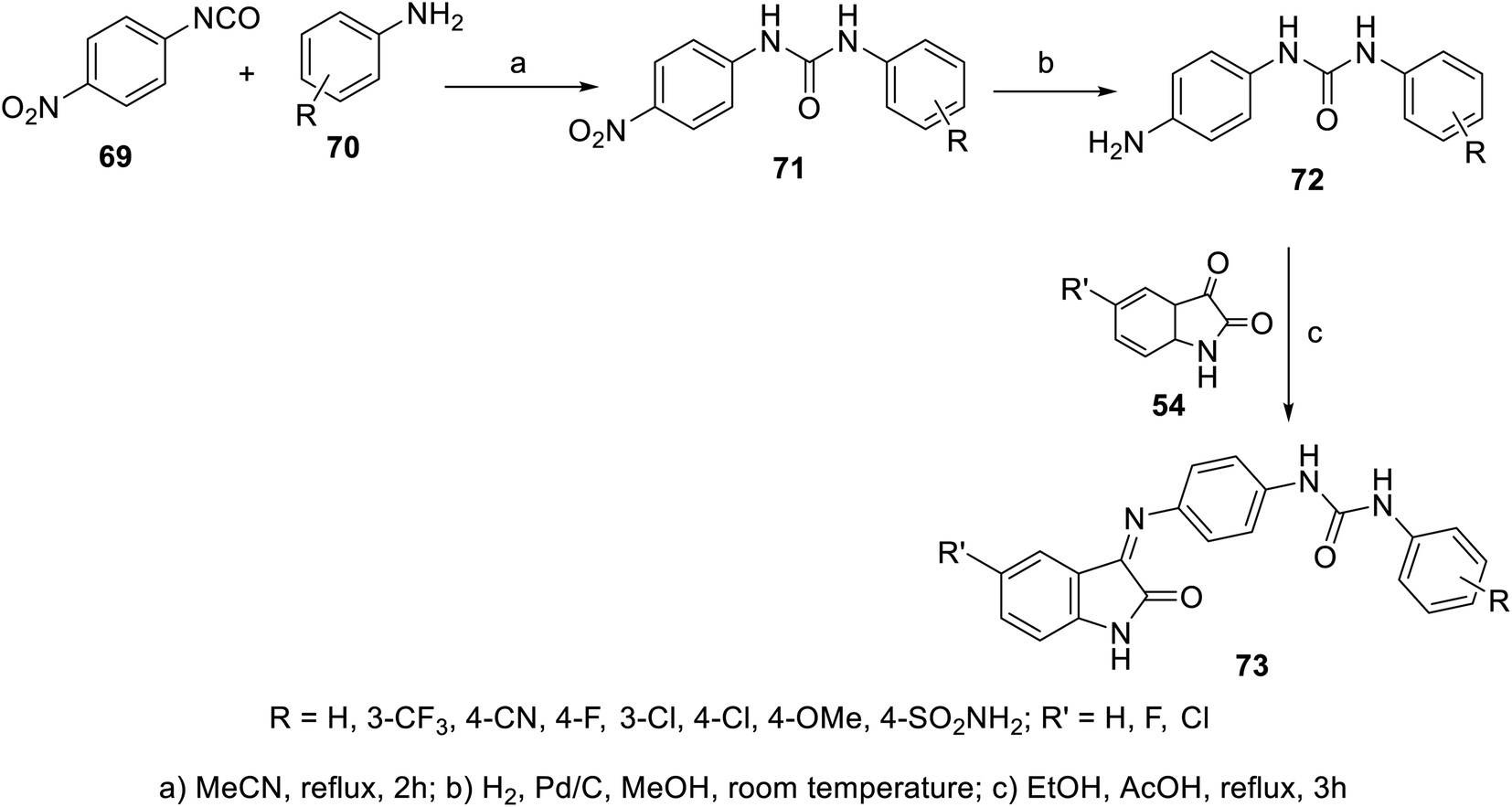
A set of indolyl Schiff bases incorporated in urea 73 was synthesized through the condensation reaction of the appropriate indoles 54 with the corresponding 1-(4-aminophenyl)-3-substituted urea 72. The latter was obtained through reduction (H2, Pd/C, and MeOH) of the corresponding nitro analogs, which were prepared through reaction of 4-nirophenylisocyanate 69 with the appropriate anilines 70 in refluxing acetonitrile104 (Scheme 4). Some of the synthesized Schiff bases revealed promising antiproliferation properties (SRB “sulforhodamine B” technique) against the HepG2 (liver) cancer cell line relative to that of doxorubicin and sorafenib. The VEGFR-2 properties were determined for the discovered promising agents, which showed comparable observations to that of the antiproliferation efficacies. The efficacy observed for most of the synthesized agents followed order of phenyl substitution of 4-Cl > 3-Cl > 3-CF3. The most promising agent discovered was 73x (R = SO2NH2, R′ = Cl; IC50 = 3.15 ± 0.36 and 0.31 ± 0.04 μM for HepG2 cell line and VEGFR-2, respectively), which exhibited comparable activity to that of sorafenib (IC50 = 3.40 ± 0.25, 0.10 ± 0.02 μM for the HepG2 cell line and VEGFR-2, respectively)104 (ESI Fig. S4†).
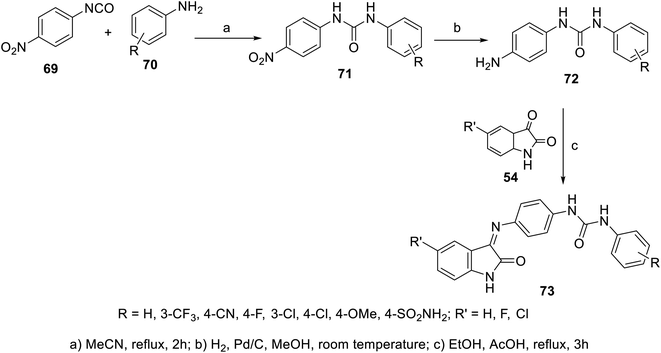 |
| | Scheme 4 Synthesis of indolyl Schiff bases 73. | |
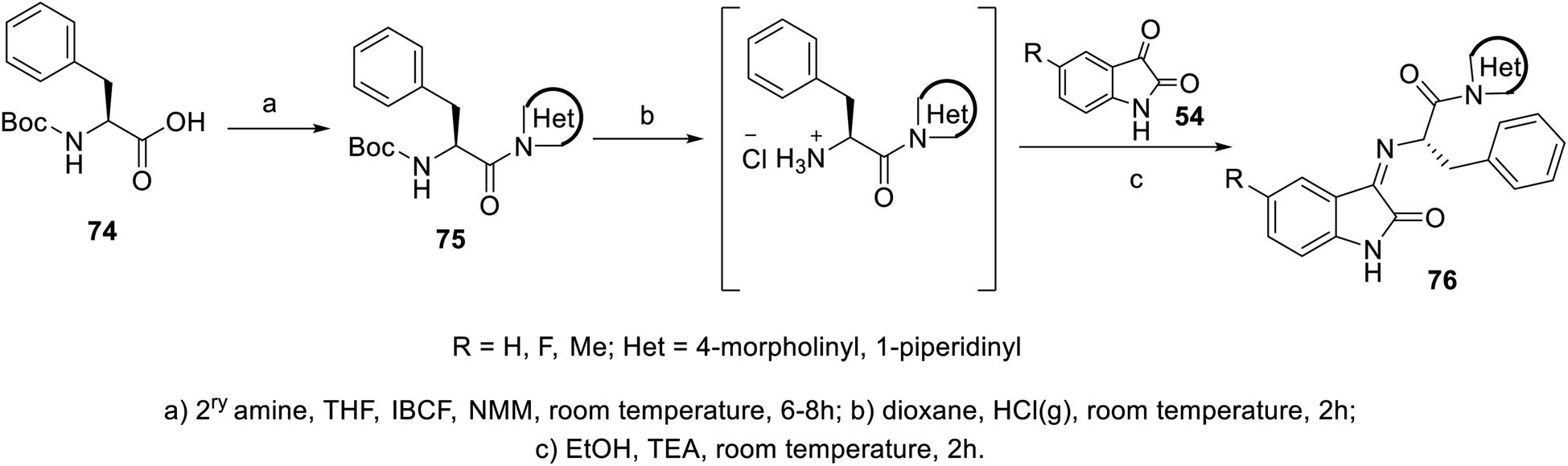
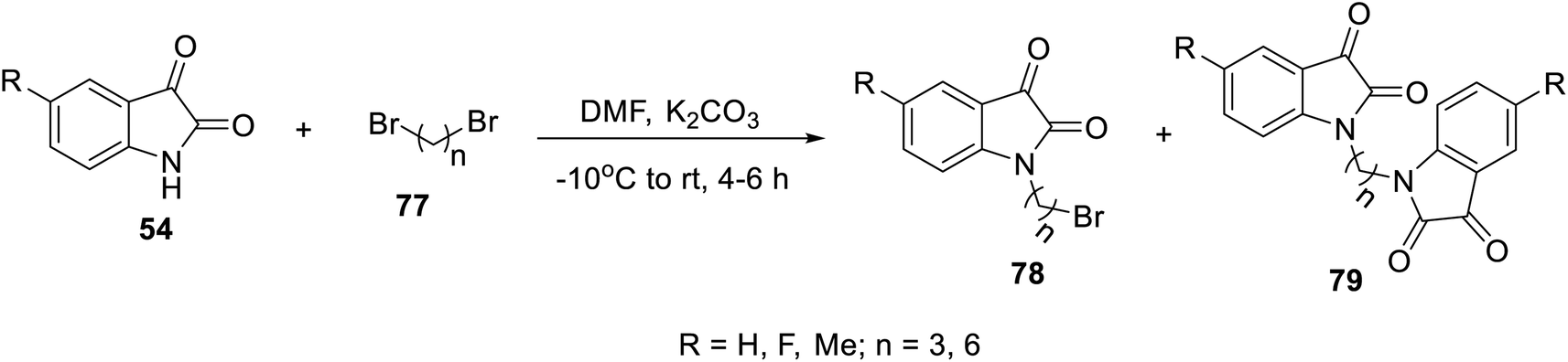
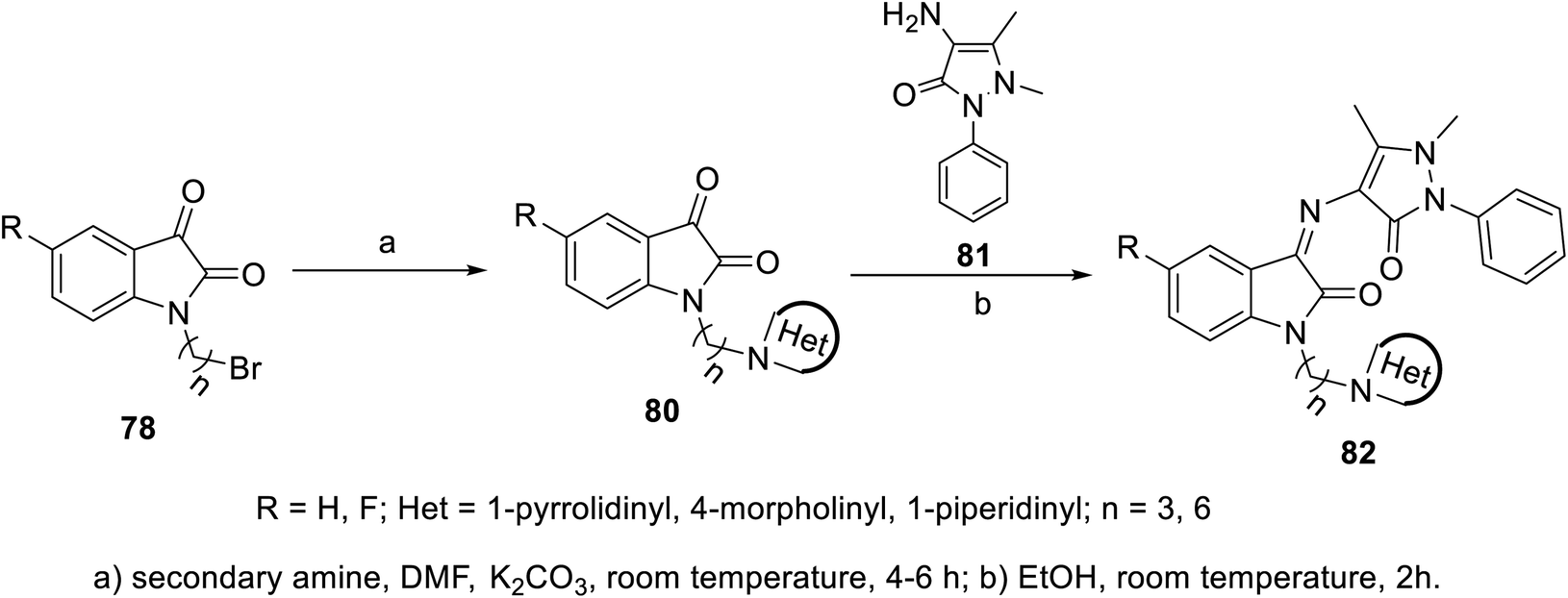
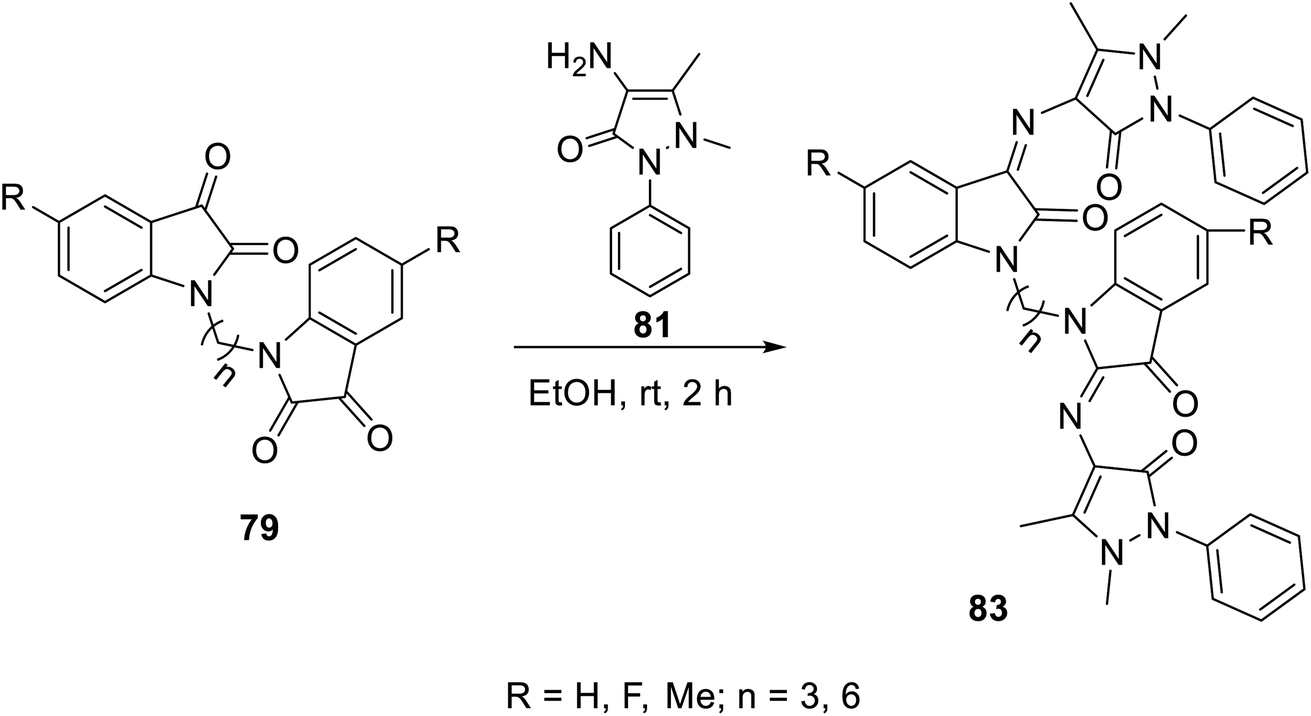
The condensation reaction of isatins 54 with L-phenylalanine secondary amine conjugates (obtained from the reaction of Boc amino acid with secondary amines in THF (tetrahydrofuran) in the presence of IBCF (iso-butyl chloroformate) and NMM (N-methyl morpholine) at room temperature and inert atmosphere followed by removal of the Boc group (HClgas in dioxane)) in EtOH containing triethylamine (TEA) at room temperature gave the corresponding Schiff bases 76 (Scheme 5). Additionally, the reaction of isatins 79/80 with 4-amino antipyrine 81 in ethanolic solution at room temperature afforded the corresponding Schiff bases 83 and 82, respectively. Isatin derivatives 80 were obtained through alkylation with excess dibromoalkane (DMF/K2CO3), which gave monoalkylated derivatives 78 (major products) and bis-isatin derivatives 79 (minor products). The monoalkylated isatins were coupled with secondary amines (DMF/K2CO3 at room temperature) affording the corresponding isatin derivatives 80 (Schemes 6–8).
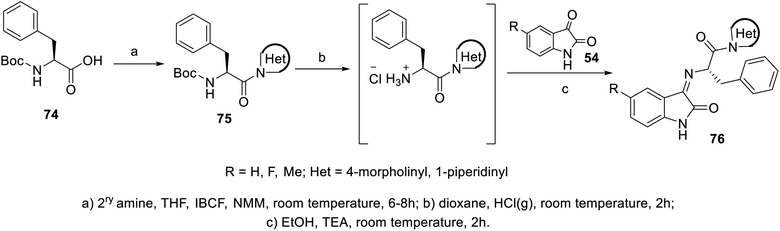 |
| | Scheme 5 Synthesis of indolyl Schiff bases 76. | |
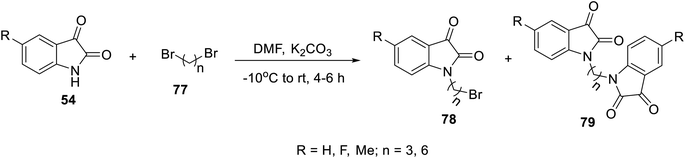 |
| | Scheme 6 Synthesis of mono- 78 and bis-alkylated isatins 79. | |
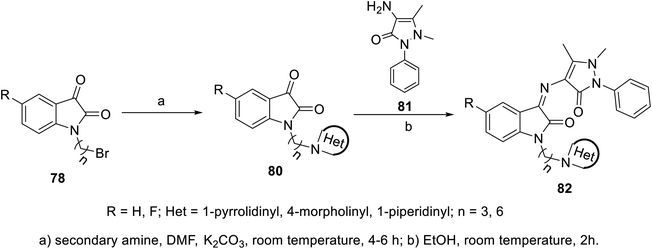 |
| | Scheme 7 Synthesis of indolyl Schiff bases 82. | |
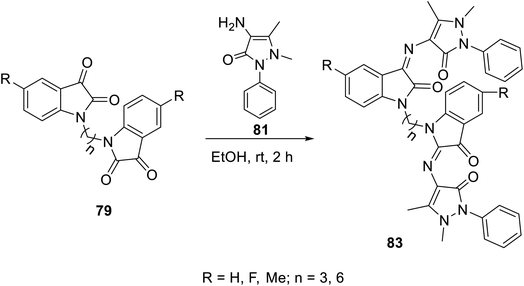 |
| | Scheme 8 Synthesis of indolyl Schiff bases 83. | |
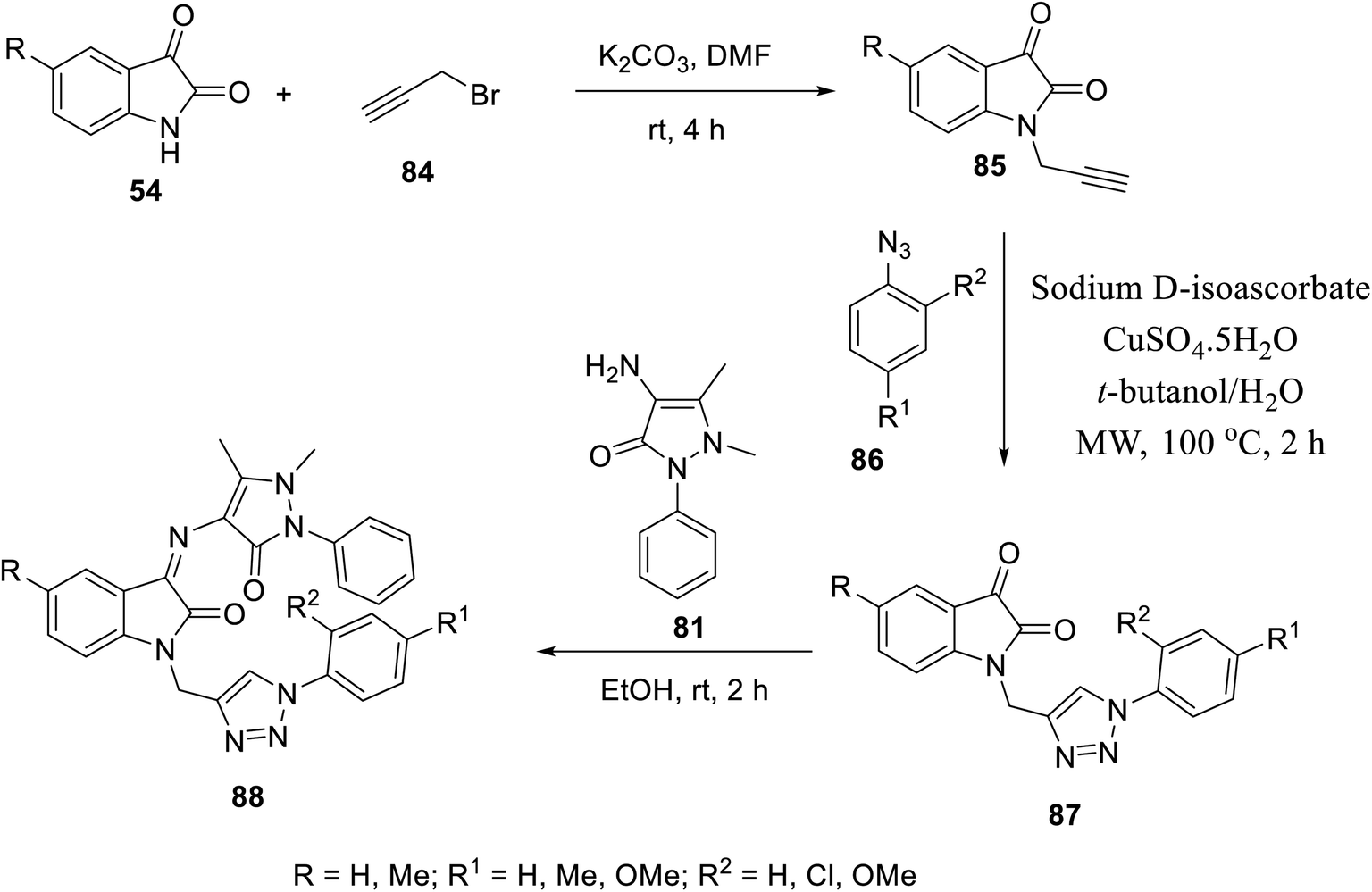
Similarly, Schiff bases 88 were obtained through the condensation of 4-amino antipyrine 81 with isatin triazol conjugates 87 (EtOH, room temperature). Isatin triazol conjugates 87 were synthesized through the click reaction of the appropriate aryl azides 86 with N-propargylated isatins 85 (t-butanol/H2O, CuSO5·5H2O, sodium D-isoascorbate, microwave, 100 °C)15 (Scheme 9).
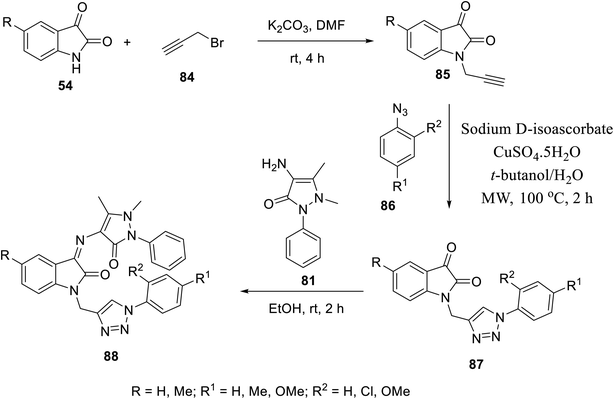 |
| | Scheme 9 Synthesis of indolyl Schiff bases 88. | |
Some of the synthesized Schiff bases exhibited promising antiproliferation properties against MCF7 (breast), HCT116 (colon) and PaCa2 (pancreatic) cancer cell lines (MTT assay) compared to the reference standards (sunitinib and 5-fluorouracil) (ESI Fig. S5†). Compound 88f (R = Me, R1 = H, and R2 = OMe) was the highest potent analog observed against MCF7 (2.1-times potency relative to the standard reference sunitinib). Additionally, some of the compounds prepared (88b, 88d and 88f) exhibited higher efficacies than that of the standard drug 5-flurouracil (approved drug for colon cancer105). The safe profile of all the tested analogs (IC50 = >50.00 μM) against the non-cancer RPE1 cell line is good support, especially for the high potent analogs towards more detailed studies for assigning promising hits. The CAM assay (chick chorioallantoic membrane) of fertilized chicken eggs in addition to VEGFR-2 inhibitory properties (% inhibition ± SD utilizing the IC50 values observed against MCF7 cell line of the tested agents relative to that of sunitinib) (ESI Fig. S5†) support their capability towards antiangiogenesis.15
4.3. Indolyl hydrazones
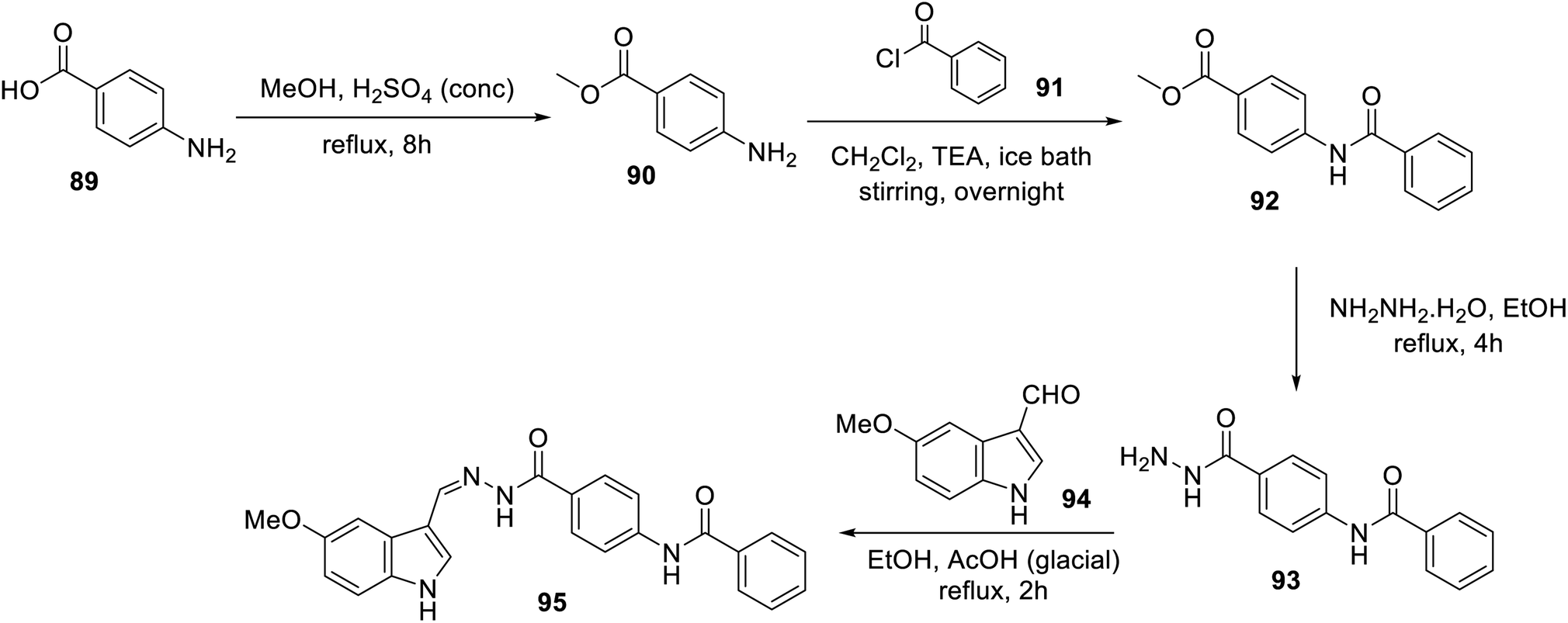
An ethanolic solution of 3-indolecarbaldehyde derivative 94 and hydrazide analog 93 in the presence of a catalytic amount of acetic acid under reflux afforded hydrazone 95 (Scheme 10). Hydrazone 95 exhibited considerably higher antiproliferation properties (MTT assay) against the MCF-7 and HCT116 cell lines (IC50 = 12.93 ± 0.54 and 11.52 ± 0.70 μM, respectively) compared to that of sorafenib (IC50 = 4.32 ± 0.33 and 7.28 ± 0.53 μM, respectively) with a safety profile against the W138 (non-cancer) cell line. Furthermore, the VEGFR-2 inhibitory properties of 95 (IC50 = 25 ± 1.29 nM) were comparable to that of the standard reference sorafenib (IC50 = 35 ± 1.34 nM). This study was supported by diverse computational studies (molecular docking “PDB: 2OH4, MOE2014 software”; MD “molecular dynamics, CHARMM-GUI web server, GROMACS 2021 engine”; DFT “density functional theory, Gaussian 09” and ADMET “absorption, distribution, metabolism, excretion and toxicity; Discovery Studio 4.0”).106
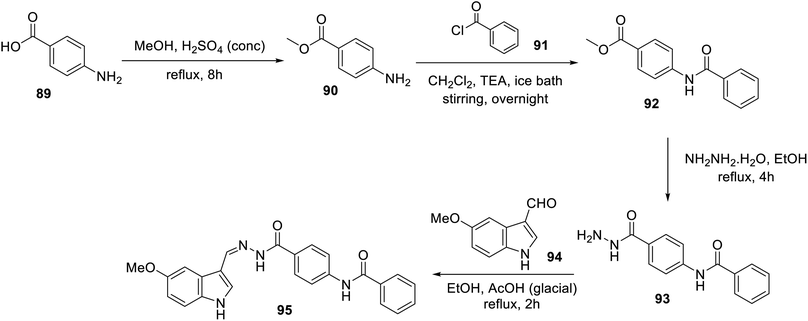 |
| | Scheme 10 Synthesis of hydrazone derivative 95. | |
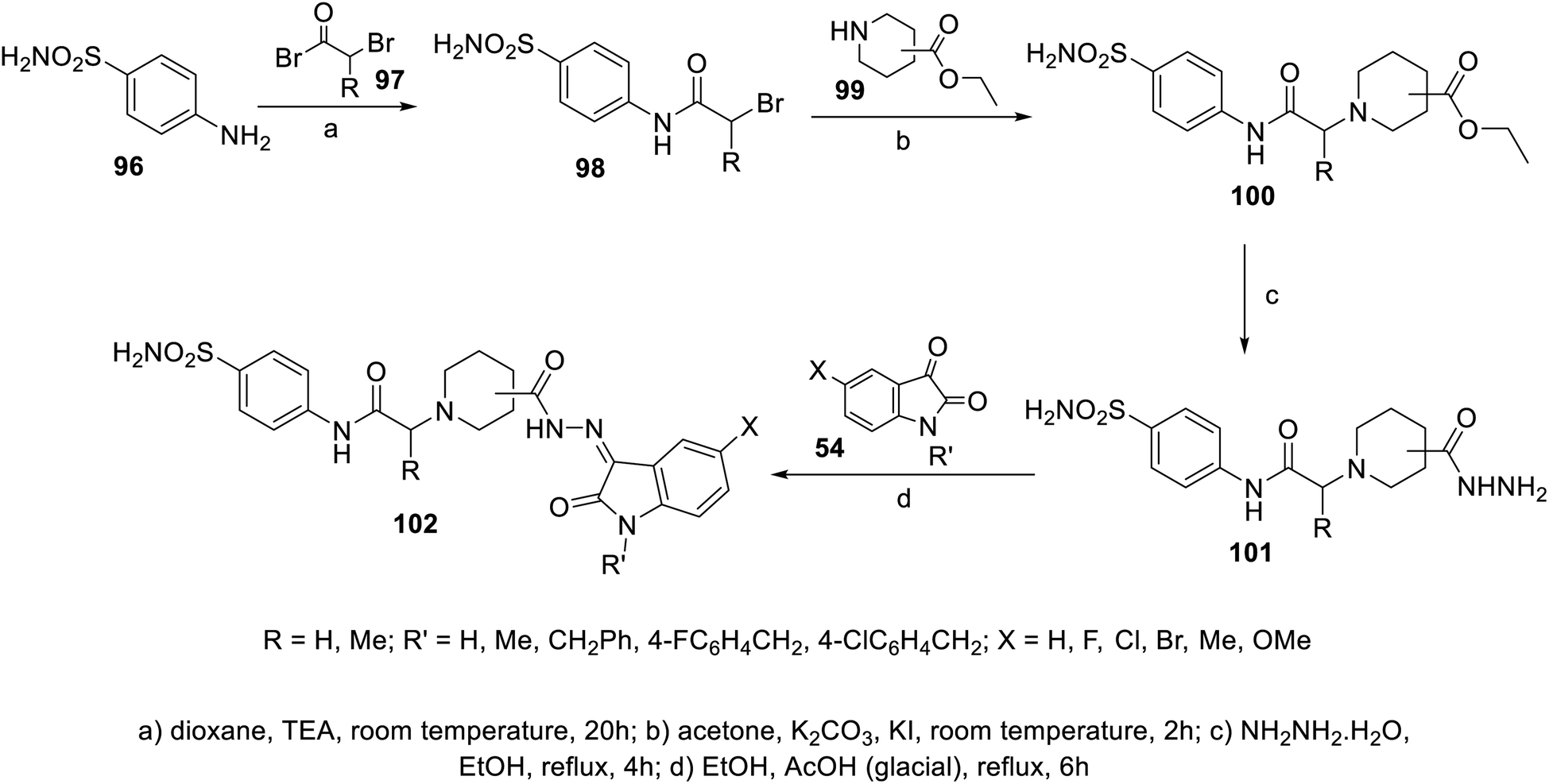
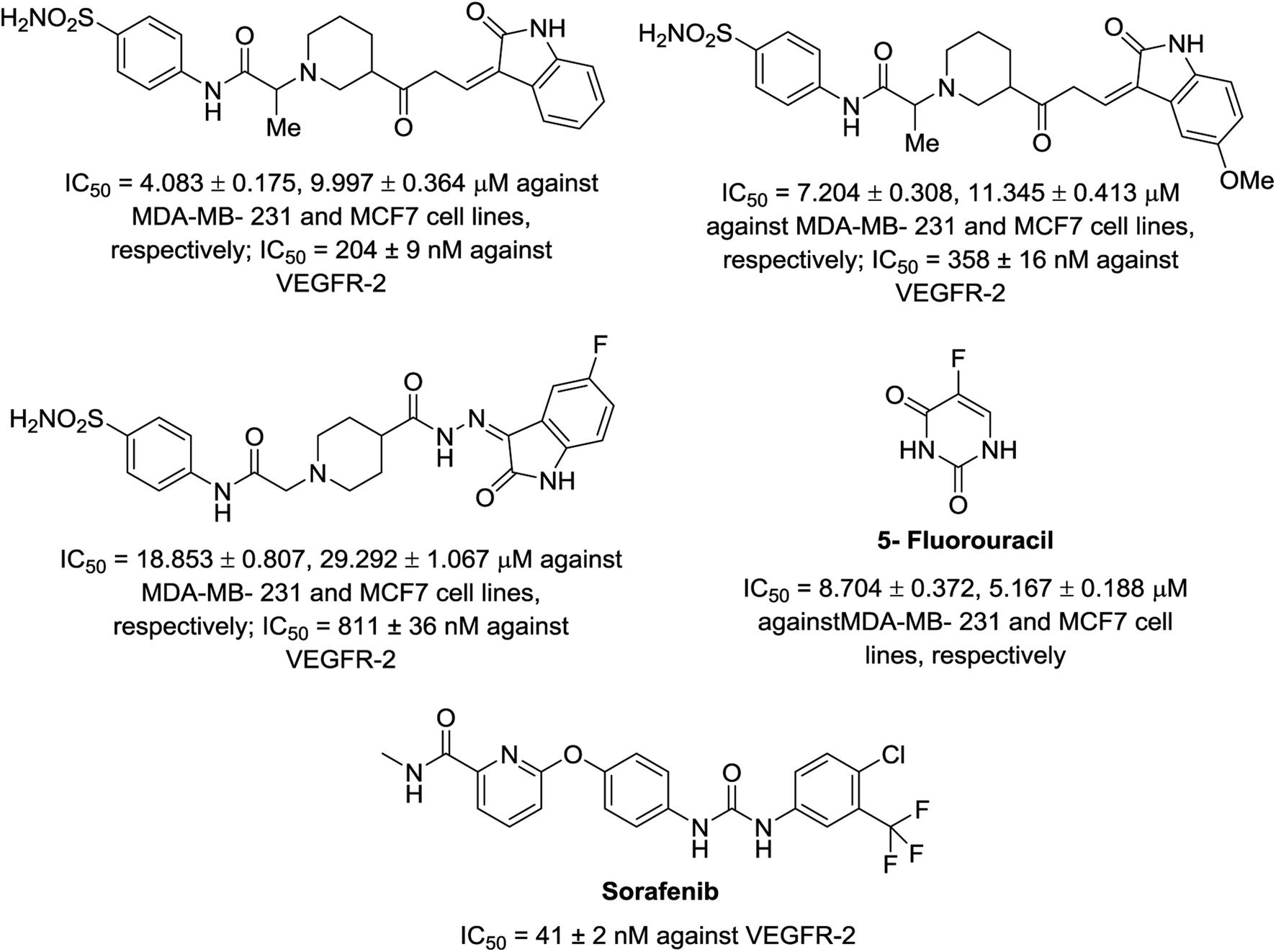
A series of indolyl hydrazones incorporating benzenesulfonamides 102 was synthesized through the condensation of the appropriate isatins 54 with hydrazides 101 in refluxing ethanol containing a catalytic amount of acetic acid. Hydrazides 101 were obtained through the reaction of hydrazine hydrate with the corresponding ethyl esters 100 in refluxing ethanol (Scheme 11). Some of the synthesized hydrazones 102 revealed considerable antiproliferation activities against MDA-MB-231 and MCF7 (breast cancer cell lines) compared to that of 5-fluorouracil, and VEGFR-2 inhibitory properties compared to that of sorafenib (Fig. 9). Inhibitory properties against carbonic anhydrase (CA) hCAI, II, IX, XII were also observed by hydrazones 102 compared to the standard reference acetazolamide.107
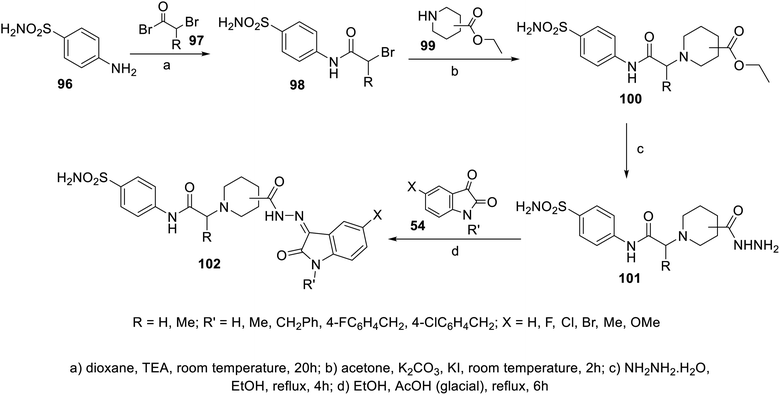 |
| | Scheme 11 Synthesis of hydrazones 102. | |
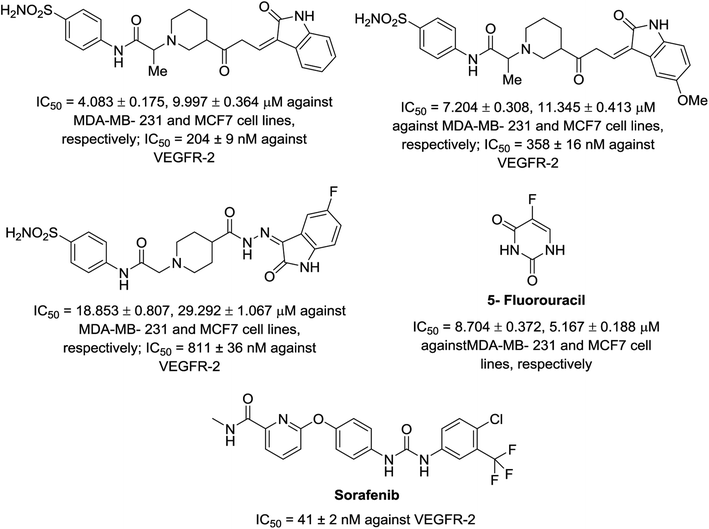 |
| | Fig. 9 Antiproliferation (in μM ± SD) and inhibitory properties against VEGFR-2 (in μM ± SD) of the tested hydrazones 102 and standard references (5-fluorouracil and sorafenib), respectively. | |
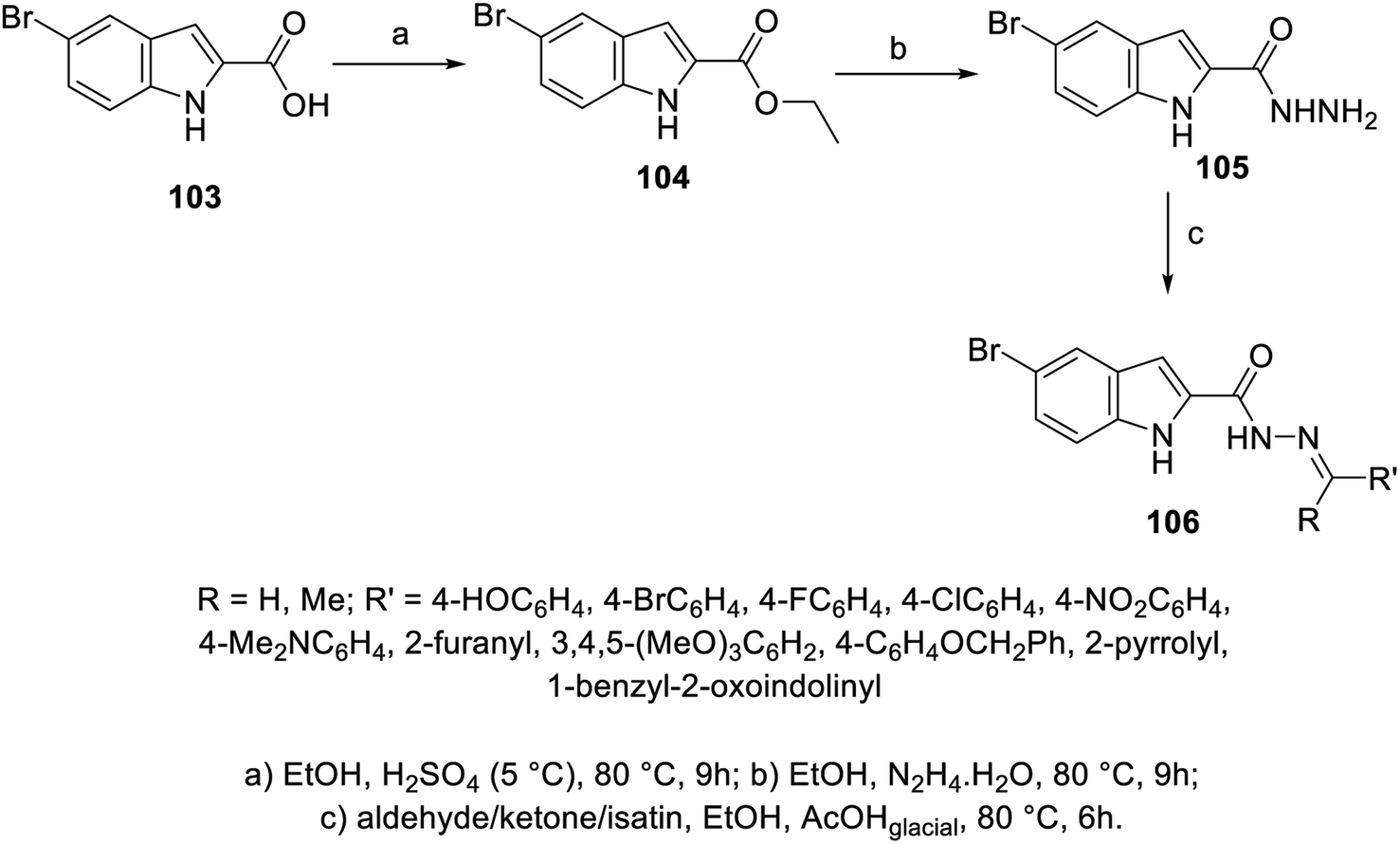
A set of hydrazones 106a–k was prepared through the condensation reaction of 5-bromo-2-indolecarbohydrazide 105 (obtained from the reaction of hydrazine hydrate with ethyl ester of indole derivative 104 in refluxing EtOH) with the appropriate aldehyde, ketone or isatin derivative in refluxing EtOH containing a few drops of glacial AcOH (catalytic amount) (Scheme 12). Mild antiproliferation properties were exhibited by the synthesized hydrazones 106a–k against HepG2 (liver), HeLa (cervical) and PC3 (prostate) cancer cell lines (MTT assay) with modest VEGFR-2 inhibitory activity compared to the standard reference (sorafenib; IC50 = 6.2 ± 1.1, 11.7 ± 1.3, 19.0 ± 1.2, and 15.3 ± 1.8 μM against HepG2, HeLa, PC3 and WI-38 respectively; EC50 = 57.1 ± 3.0 nM ± SEM against VEGFR-2). Furthermore, the most promising agent 106e (R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) 4-Me2NC6H4, R′H; IC50 = 14.3 ± 2.0, 22.2 ± 2.3, 36.2 ± 3.1, and 25.9 ± 2.1 μM against HepG2, HeLa, PC3 and WI-38, respectively) was screened against WI-38 (normal lung fibroblasts; EC50 = 102.6 ± 3.1 nM ± SEM against VEGFR-2) to confirm its safety index108 (ESI Fig. S6†).
4-Me2NC6H4, R′H; IC50 = 14.3 ± 2.0, 22.2 ± 2.3, 36.2 ± 3.1, and 25.9 ± 2.1 μM against HepG2, HeLa, PC3 and WI-38, respectively) was screened against WI-38 (normal lung fibroblasts; EC50 = 102.6 ± 3.1 nM ± SEM against VEGFR-2) to confirm its safety index108 (ESI Fig. S6†).
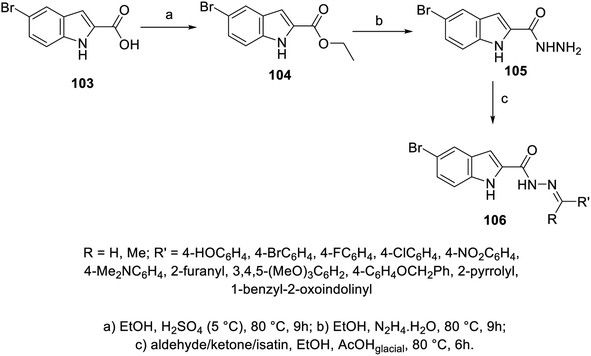 |
| | Scheme 12 Synthesis of hydrazones 106. | |
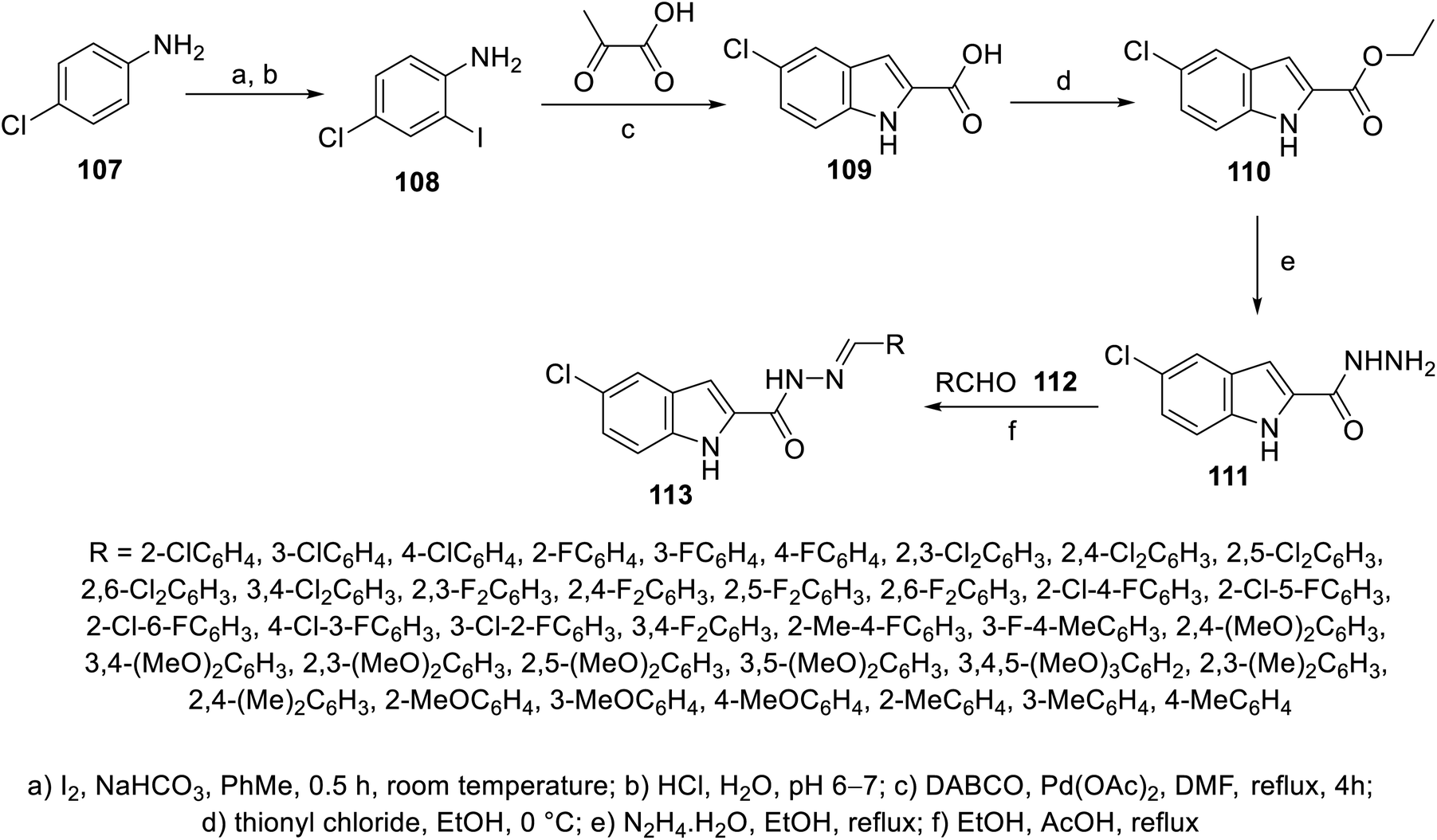
Similarly, 5-chloroindolyl hydrazones 113 were obtained through the reaction of the corresponding hydrazide 111 and aromatic aldehydes 112 in refluxing ethanol containing AcOH as a catalyst (Scheme 13). Considerable antiproliferation properties against HCT116 and SW489 (colon) cancer in addition to MRC-5 (non-cancer human) cell lines were exhibited by some of the synthesized hydrazones compared to the standard references (cisplatin “GI50 = 7.67 ± 3.4, 4.43 ± 2.1, and 3.82 ± 1.9 μM against HCT116, SW480 and MRC-5, respectively”, sorafenib “GI50 = 4.17 ± 2.5, 2.02 ± 1.2, and 30.81 ± 10.6 μM against HCT116, SW480 and MRC-5, respectively” and sunitinib “GI50 = 15.84 ± 1.7, 1.09 ± 0.9, > 100 μM against HCT116, SW480 and MRC-5, respectively”) (ESI Fig. S7†). Compound 113x (R = 2,4-(MeO)2C6H3; GI50 = 8.10 ± 1.8, 7.90 ± 1.3, and >100 μM against HCT116, SW480 and MRC-5, respectively) was the most promising agent synthesized. Its anti-angiogenesis effect was supported by CAM proliferation and HUVEC (human umbilical vein endothelial cell) migration.109
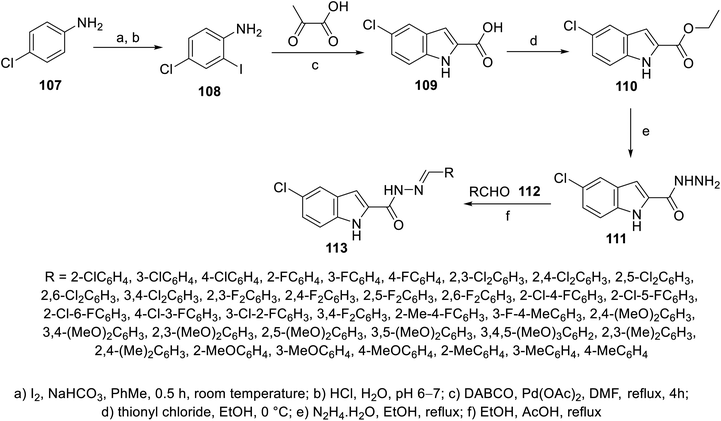 |
| | Scheme 13 Synthesis of indolyl hydrazones 113. | |
4.4. 2-Oxoindolin-3-ylidenes
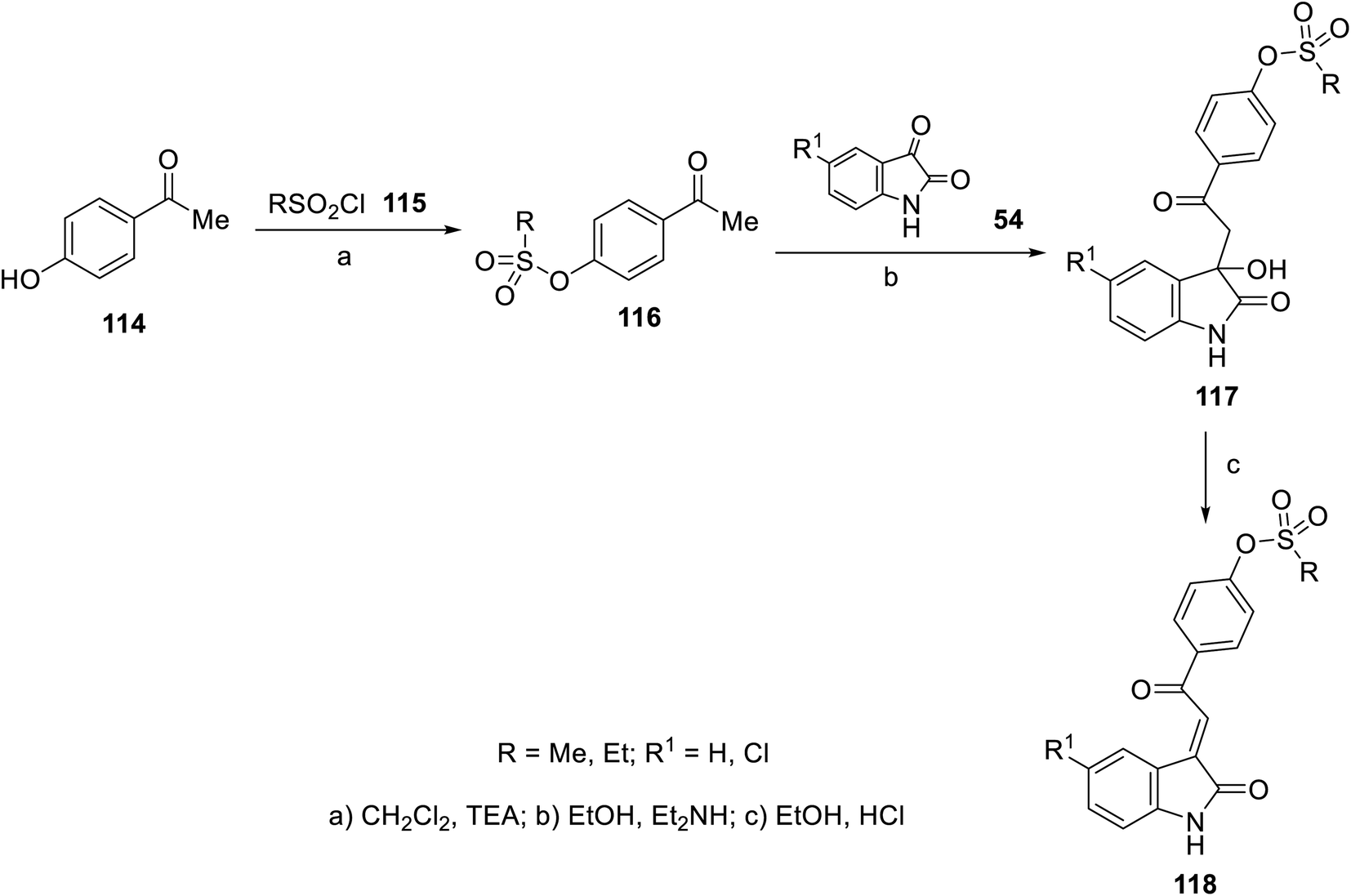
The FDA approval for the use of sunitinib and nintedanib as potent multi-targeted tyrosine kinase inhibitors against many cancer types40,41,55,56 has inspired many researchers to utilize the 2-oxoindolin-3-ylidene scaffold for optimizing novel antitumor active agents. A variety of 2-oxoindolin-3-ylidenes bearing alkanesulfonate 118 was synthesized in a two-step reaction in excellent yields. The reaction of isatins 54 in EtOH containing a quantitative amount of Et2NH gave the corresponding 3-hydroxy-2-oxoindole derivatives 117. Acidic dehydration (EtOH and HCl) of 117 gave the corresponding 11892 (Scheme 14).
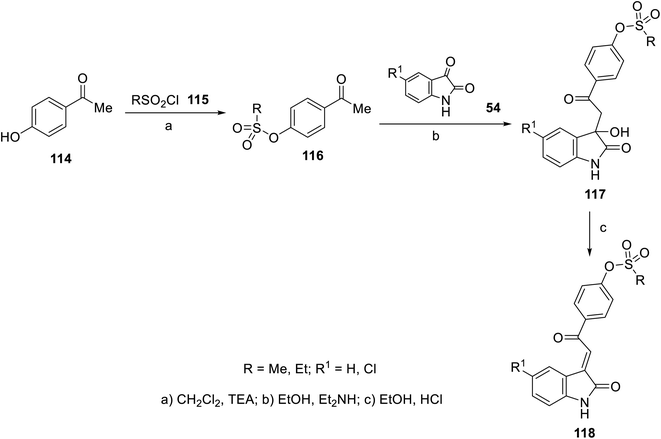 |
| | Scheme 14 Synthesis of 2-oxoindolin-3-ylidenes 118. | |
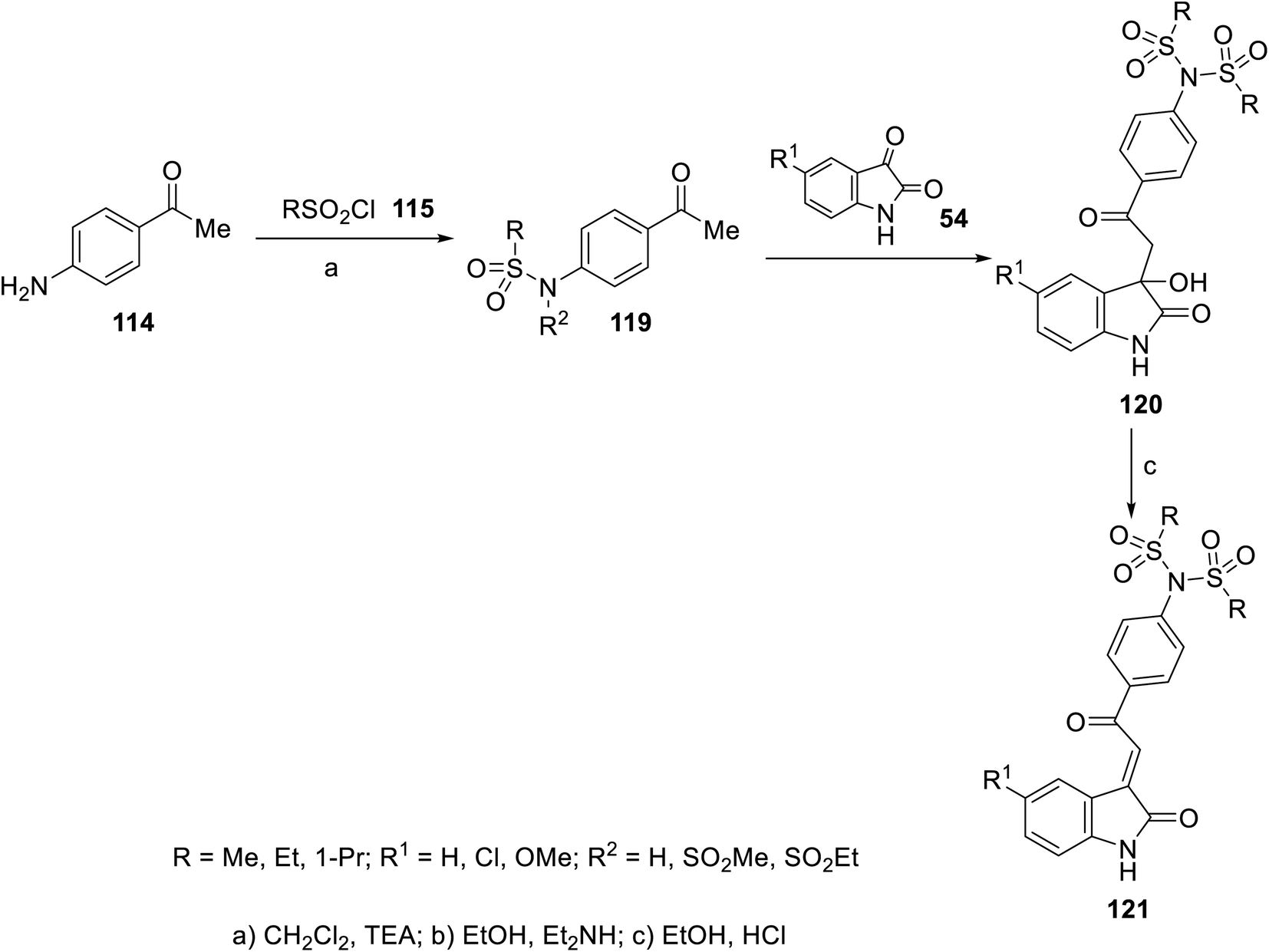
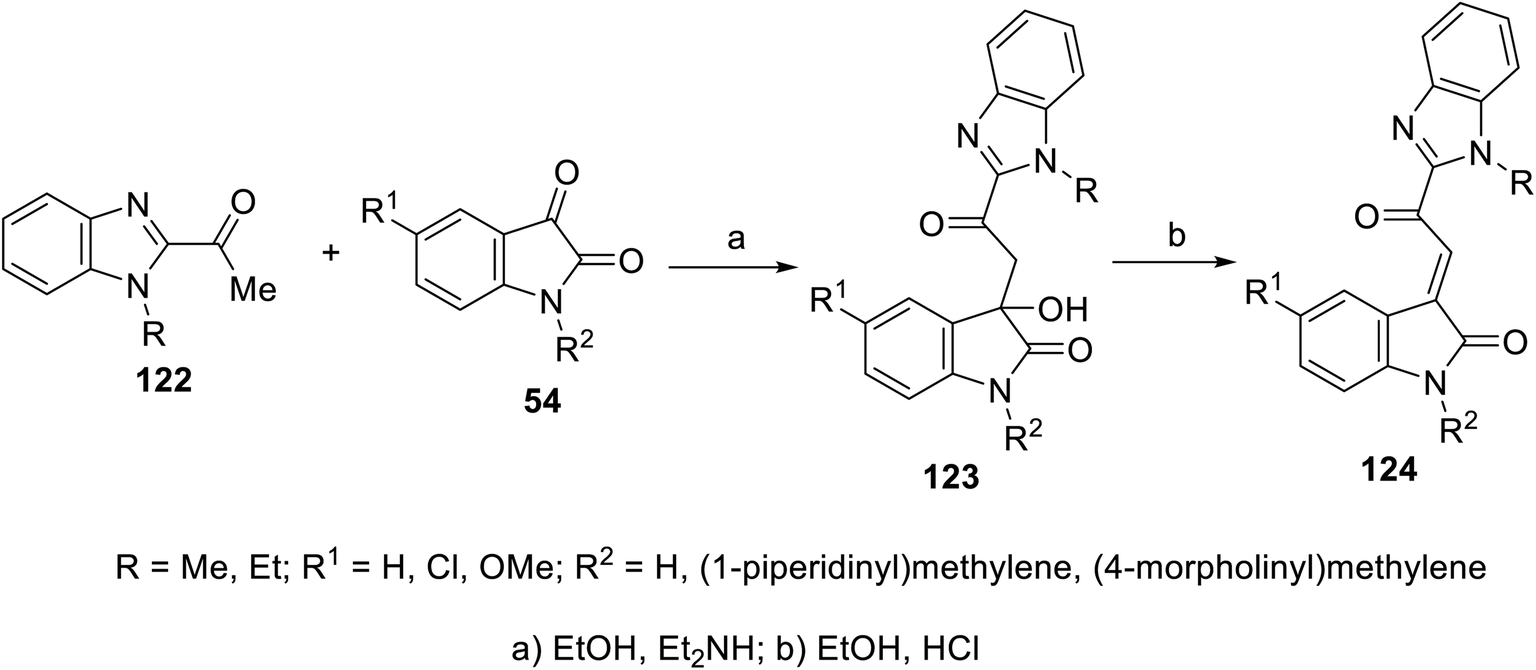
2-Oxoindolin-3-ylidenes incorporating alkanesulfonamide 121 were formed starting from the corresponding alkanesulfonamide derivatives 119 utilizing a similar reaction sequence. It was mentioned that the reaction of isatins 54 with 119 bearing a mono-propanesulfonamide function gave the corresponding 120. This is presumably via the elimination of propylsulfonate from 119 under the applied basic condition92 (Scheme 15). Similarly, 2-oxoindolin-3-ylidenes connected to benzimidazolyl heterocycle 124 were prepared following the same reaction sequence utilizing 2-acetylbenimidazoles 12292 (Scheme 16).
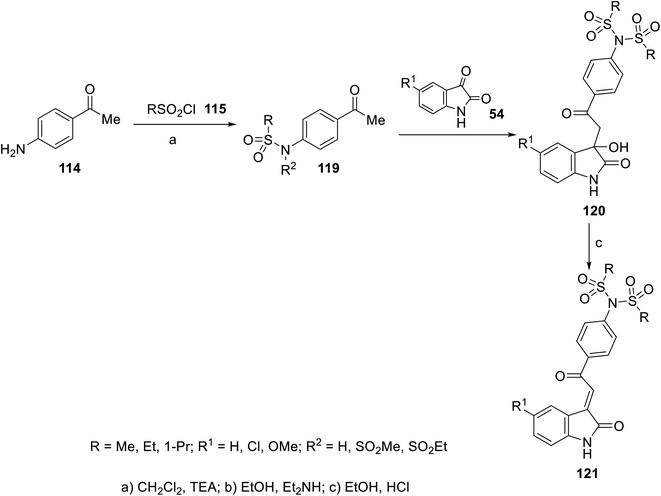 |
| | Scheme 15 Synthesis of 2-oxoindolin-3-ylidenes 121. | |
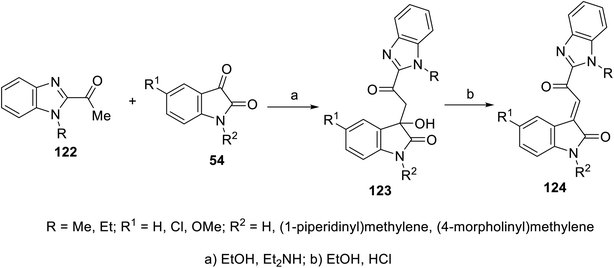 |
| | Scheme 16 Synthesis of 2-oxoindolin-3-ylidenes 124. | |
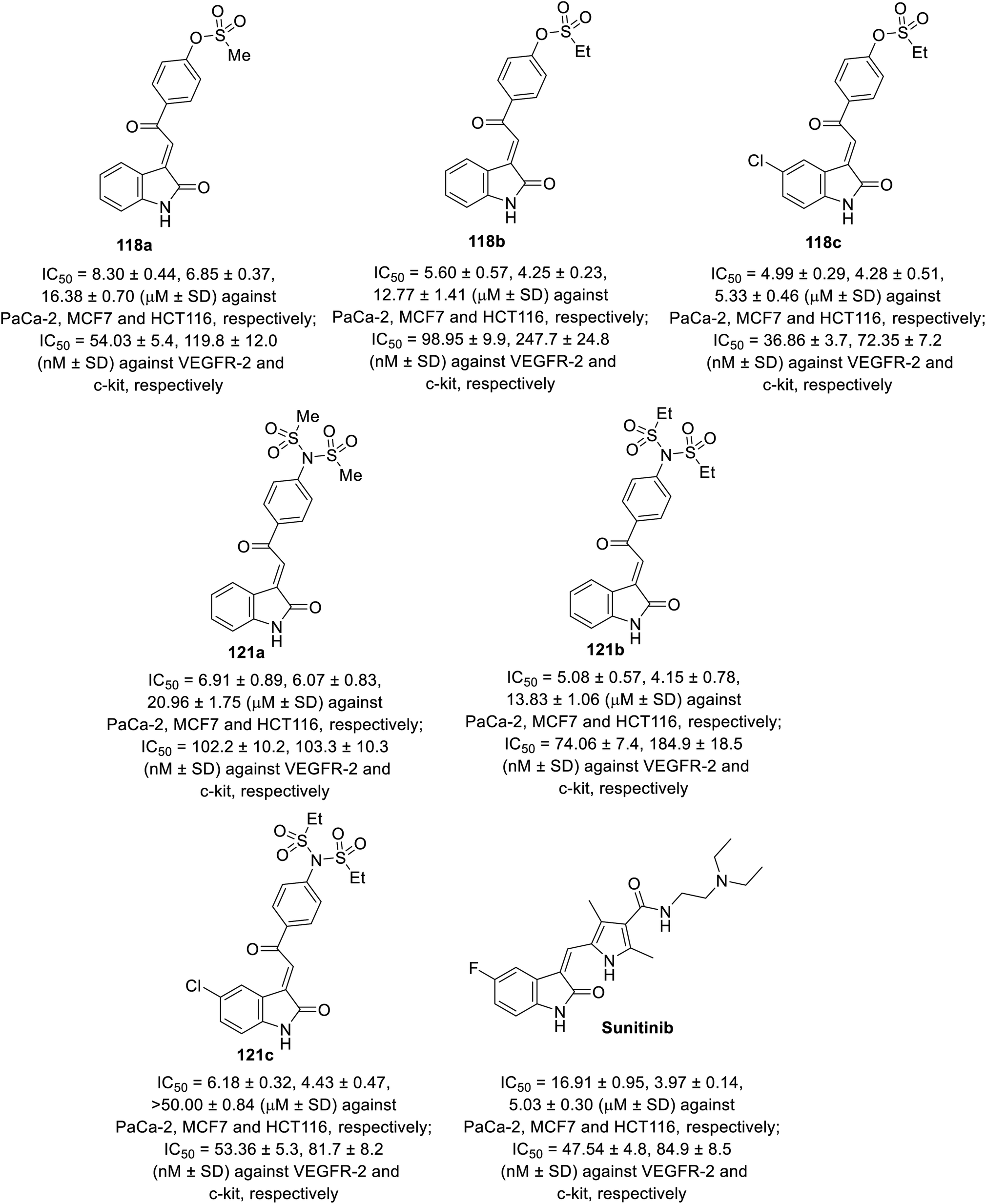
Some of the synthesized agents 118 and 121 exhibited antiproliferation properties (MTT assay) against PaCa-2 (pancreatic), MCF7 (breast) and HCT116 (colon) cancer cell lines with potency comparable to that of sunitinib. The safety index of the tested agents was established through screening against the RPE1 (retinal pigment epithelium) normal cell line. The inhibitory properties of the tested agents against VEGFR-2 and c-kit were comparable to that of the antiproliferation results (Fig. 10). Their anti-angiogenesis properties were also supported by the CAM assay.92
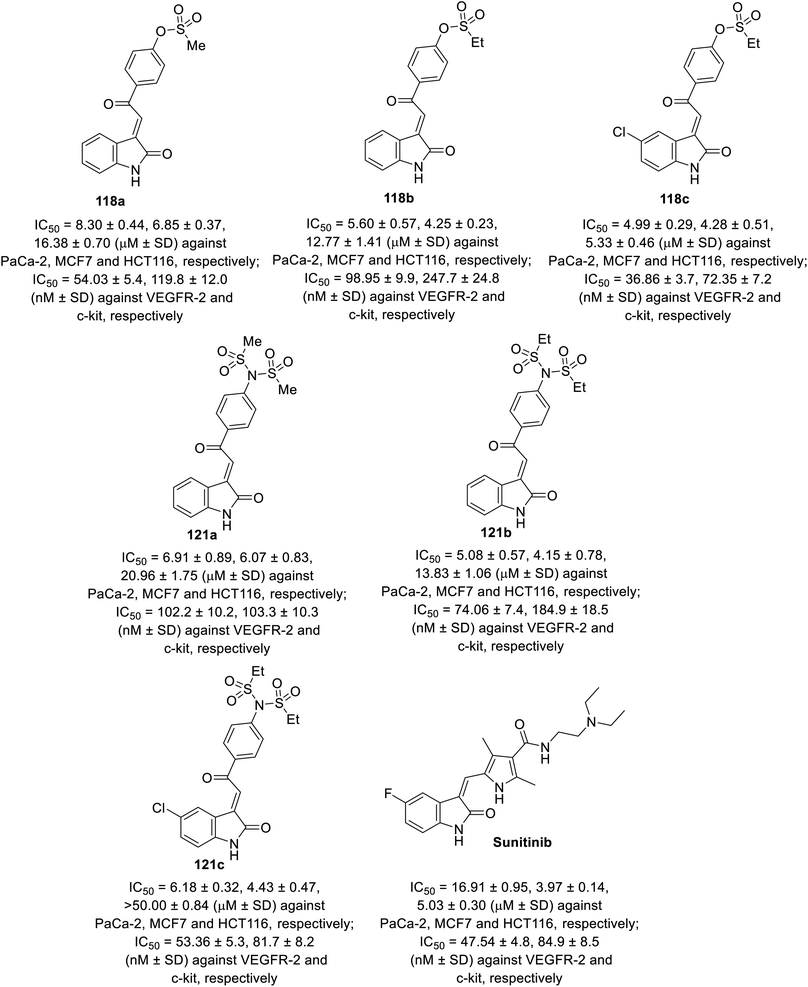 |
| | Fig. 10 Antiproliferation and tyrosine kinase (VEGFR-2, c-kir) inhibitory properties of 118a–c, 121a–c and sunitinib. | |
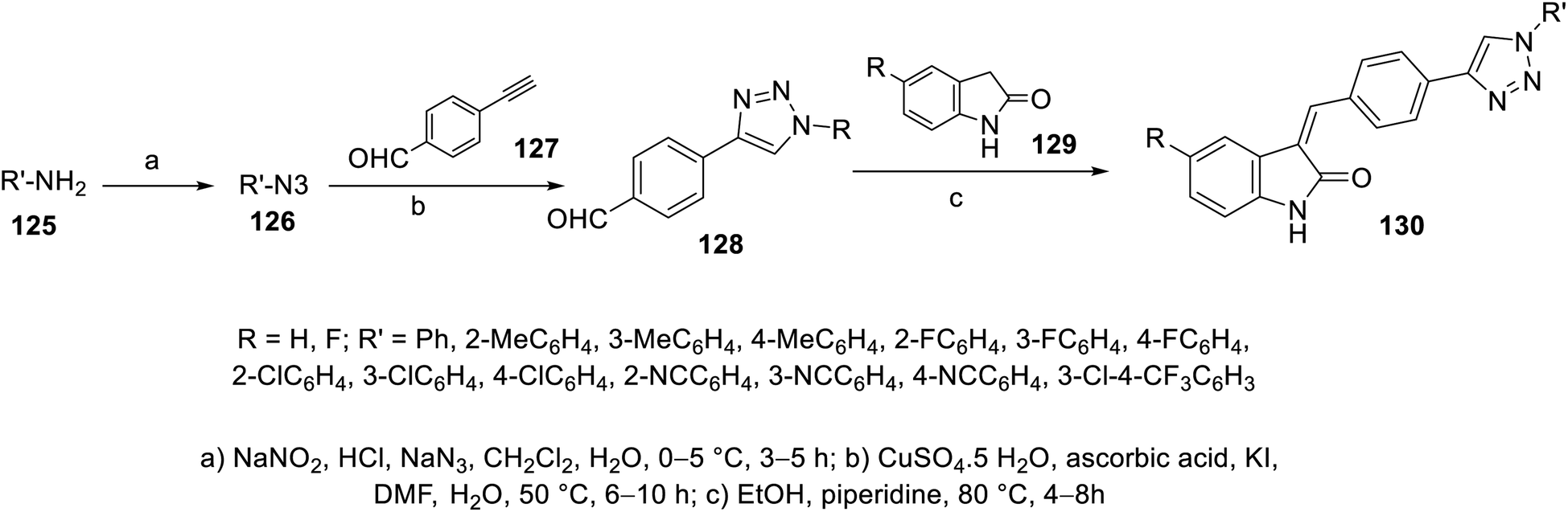
Condensation of 2-indolinones 129 with aryl aldehydes substituted with 1,2,3-triazolyl heterocycle 128 in refluxing ethanol containing a catalytic amount of piperidine gave the corresponding 2-oxoindolin-3-ylidenes 130. The aryl aldehydes bearing 1,2,3-triazolyl heterocycle 128 were obtained through the click reaction of 4-ethynylbenzaldehyde 127 and azidobenzene 126 in DMF containing aqueous CuSO4·5H2O, ascorbic acid and a catalytic amount of KI110 (Scheme 17). Based on the VEGFR-2 inhibitory properties of the tested 130, it was observed that the substituent of the phenyl ring attached to the triazolyl nitrogen possesses a significant effect on the observed bio-properties. The potency against VEGFR-2 followed the order of 4-CH3 > 3-CH3 > 2-CH3 > H. The antiproliferation properties (CCK-8 assay) were studied against HT-29 (colon), MKN-45 (gastric) and HUVEC (umbilical vein endothelial) cancer cells. Compound 130d (R = H, R′ = 4-MeCH4; IC50 = 1.61 ± 0.45, 1.92 ± 0.37, and 7.94 ± 0.36 μM against HT-29, MKN-45 and HUVEC, respectively; IC50 = 26.38 ± 1.09 nM against VEGFR-2) was the most promising agent synthesized with antiproliferation and anti-VEGFR-2 properties comparable to that of sunitinib (IC50 = 10.34 ± 0.96, 9.25 ± 0.77, and 6.37 ± 0.59 μM against HT-29, MKN-45 and HUVEC, respectively; IC50 = 83.20 ± 1.36 nM against VEGFR-2) (ESI Fig. S8†). Also, its angiogenesis properties were supported by a zebrafish labeling model assay.110
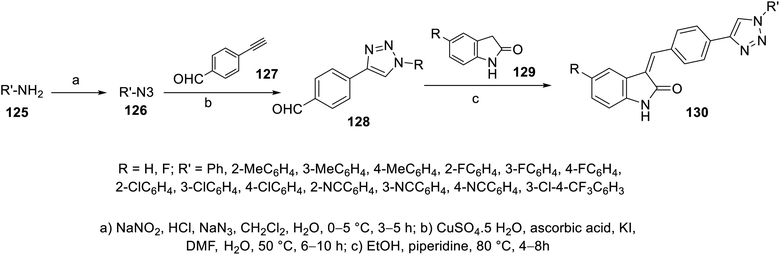 |
| | Scheme 17 Synthesis of 2-oxoindolin-3-ylidenes 130. | |
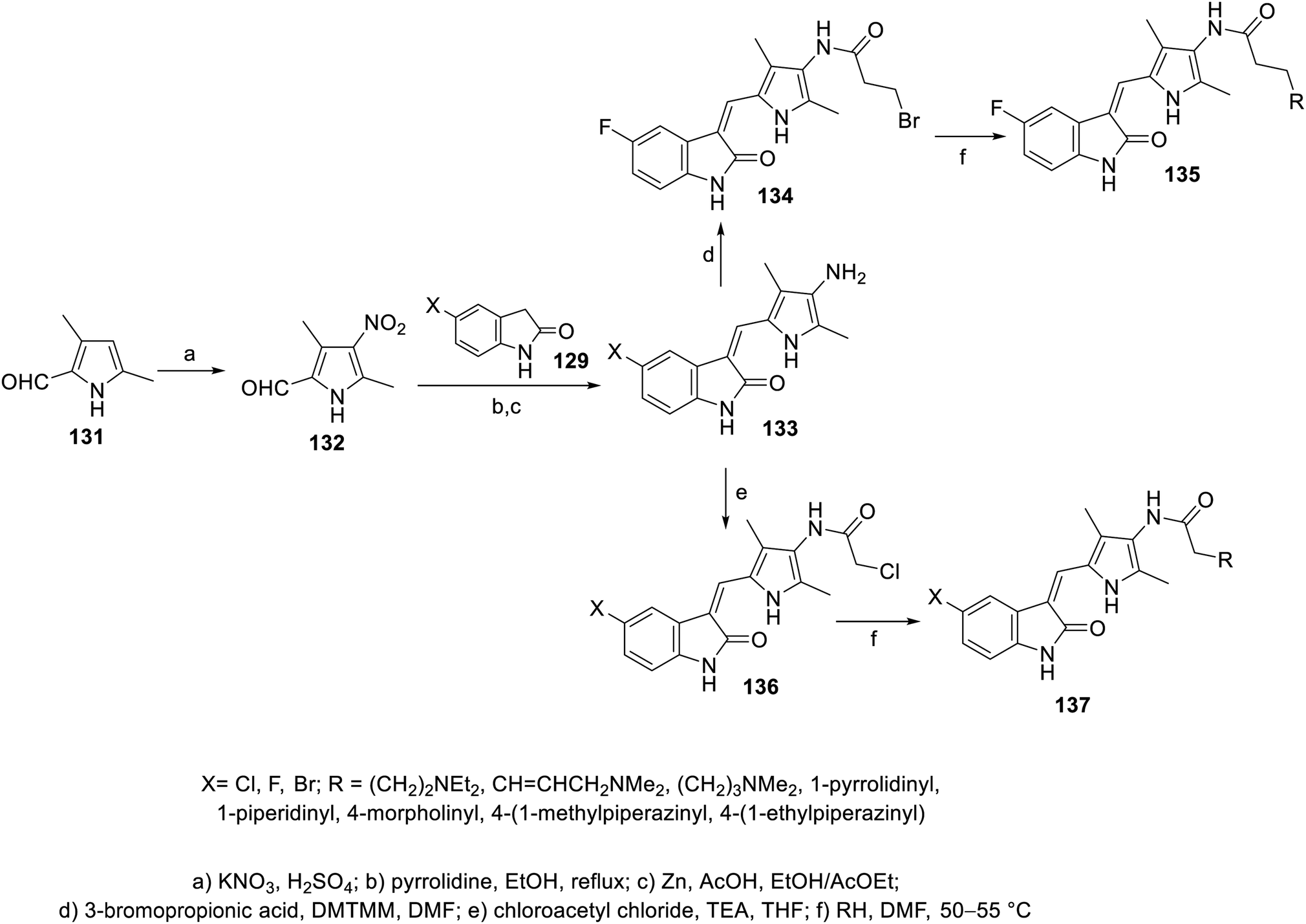
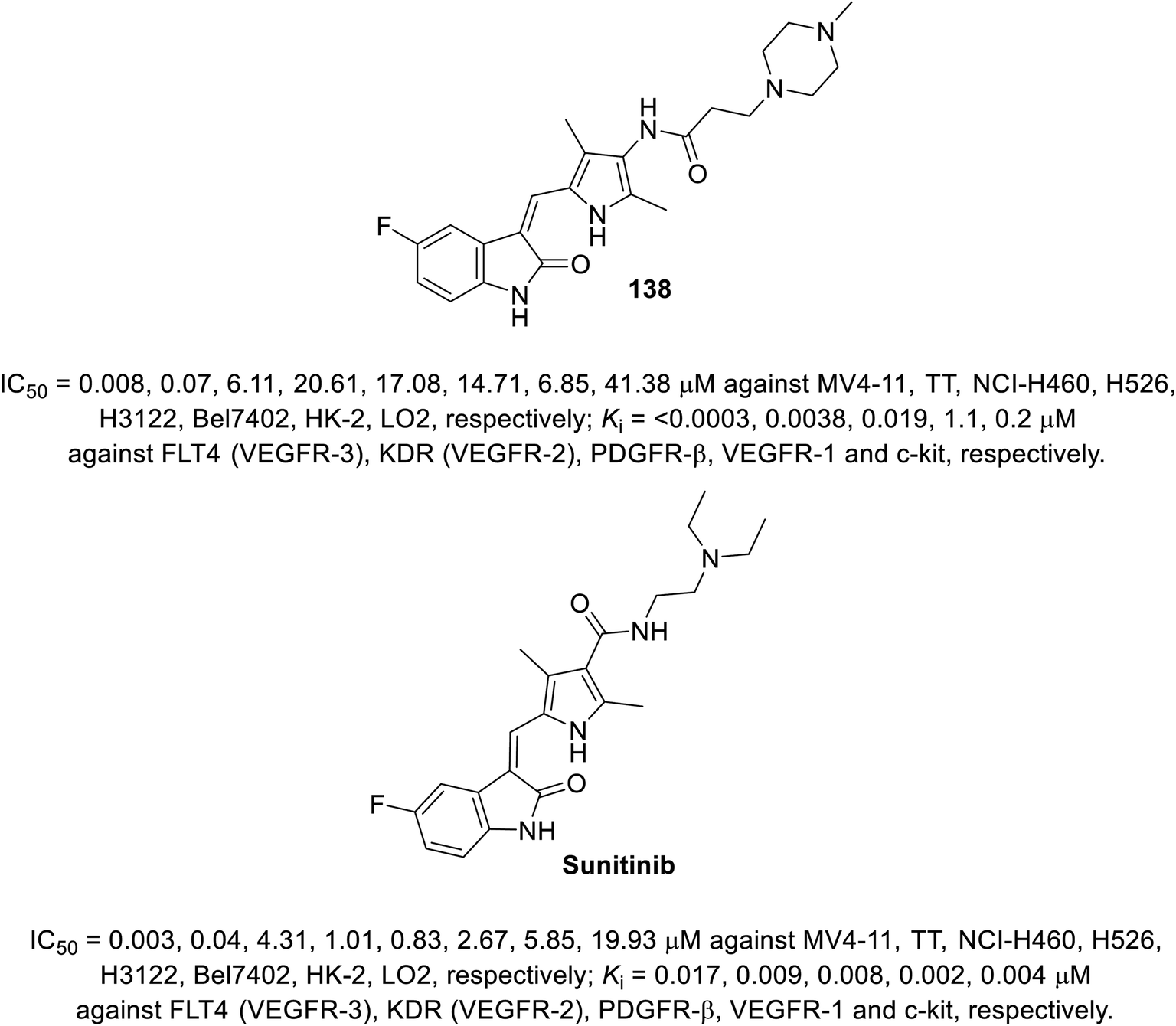
A series of 2-oxoindolin-3-ylidenes connected to pyrrole heterocycle 135 and 137 with high structural resemblance to sunitinib was synthesized via the condensation of 2-indolinones 129 with 2-pyrrolecarbaldehyde 132, followed by the reduction of the nitro group (Zn/AcOH). Subsequently, the reaction of 133 with chloroacetyl chloride (TEA, THF) or 3-bromopropionic acid (DMTMM: 4-(4,6-dimethoxytriazine)chlorinated-4-methylmorpholine, DMF) with various amines finally furnished 135 and 137 (Scheme 18). Based on their physicochemical properties and rat aortic ring assay, a few of the synthesized analogs was selected for in vitro testing (MTT assay) considering HT-29 (colon) and NCI-H460 (non-small cell lung) cancer cell lines, and compound 138 was selected for more sophisticated cell line and targeted receptor assays (Fig. 11) Compound 138 was nominated as a promising drug candidate based on its observed bio-properties compared to that of sunitinib.39
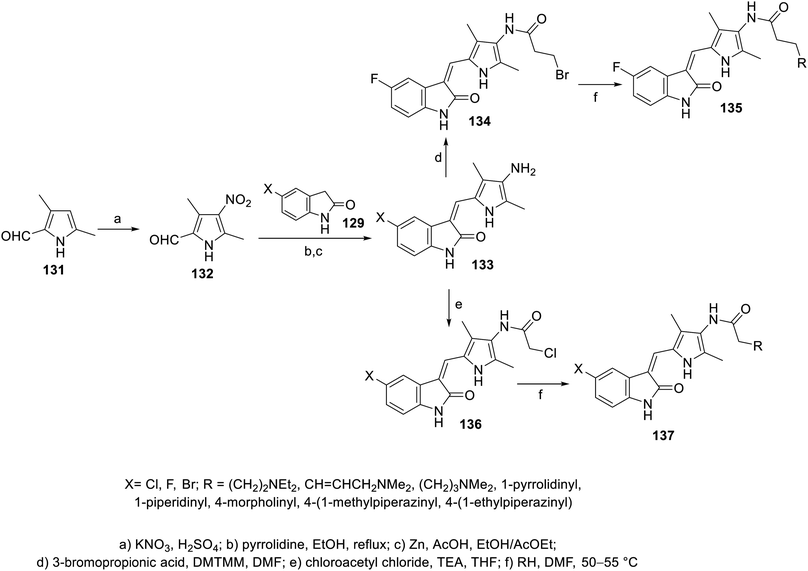 |
| | Scheme 18 Synthesis of 2-oxoindolin-3-ylidenes 135 and 137. | |
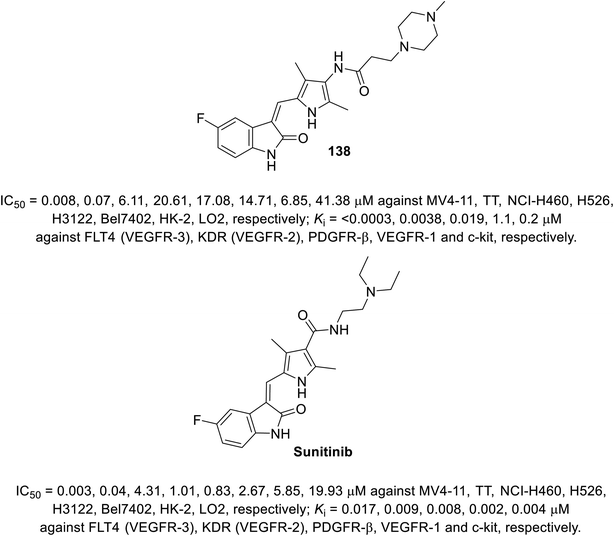 |
| | Fig. 11 Antiproliferation and receptor inhibitory properties of 138 and sunitinib. | |
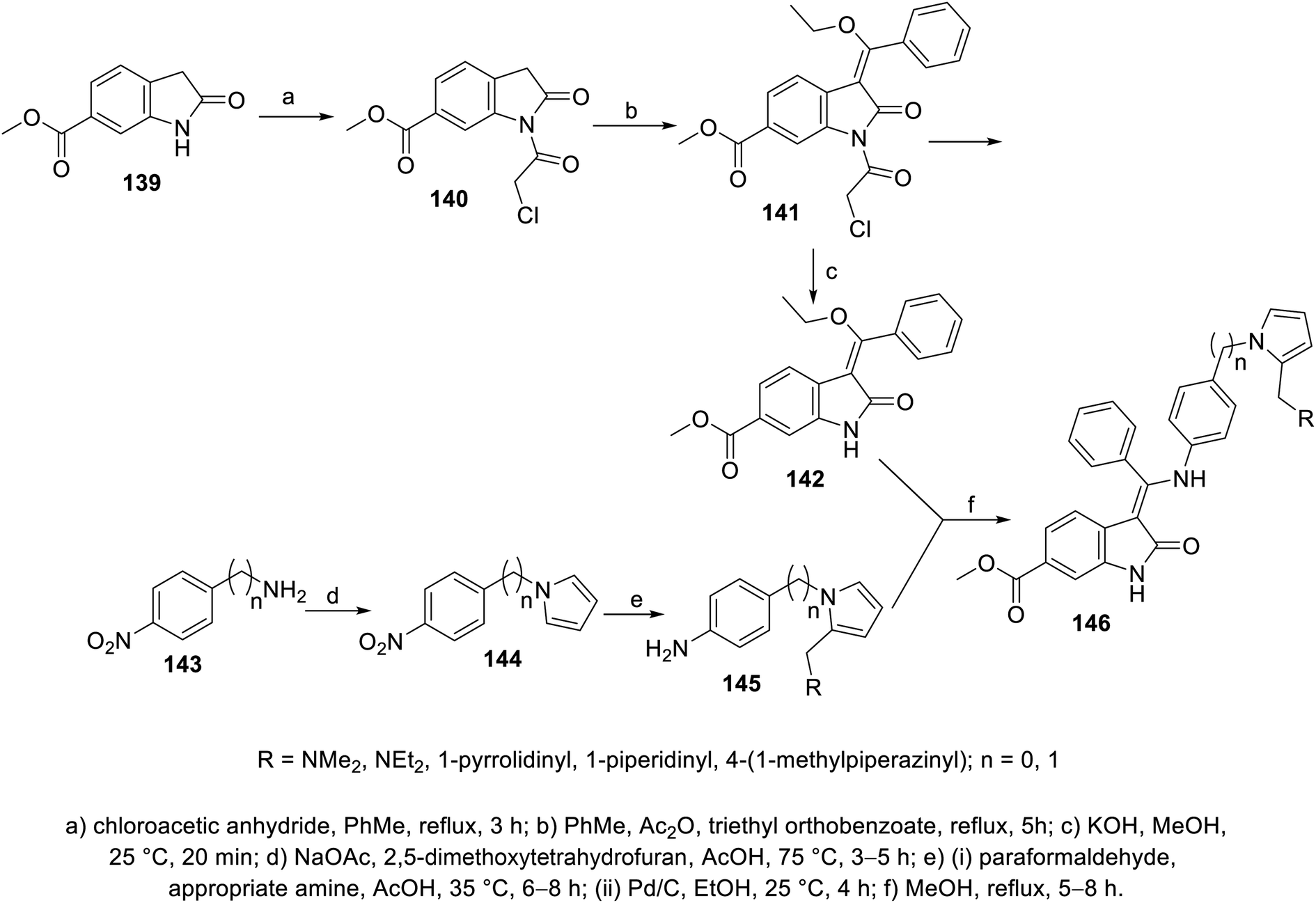
2-Oxoindolin-3-ylidene 141 was obtained through the condensation of 2-indolinone 140 with triethyl orthobenzoate in boiling toluene (110 °C) in the presence of acetic anhydride. The removal of the chloroacetyl group (KOH, MeOH, room temperature), followed by reaction with 4-(1H-pyrrol-1-yl)aniline derivatives 145 gave 2-oxoindolin-3-ylidene 146 (Scheme 19). Some of the synthesized agents 146a (RNMe2) and 146e (R = 1-methyl-4-piperazinyl) exhibited considerable VEGFR-2 and PDGFR-β inhibitory properties compared to that of nintedanib. The most promising agents observed were screened for antiproliferation properties (MTT assay) against HT-29 (colon), SK-OV-3 (ovarian) and HeLa (cervical) cancer cells, exhibiting promising activities (146a; IC50 = 51.7, 14.3 nM against VEGFR-2 and PDGFR-β, respectively; IC50 = 0.98 ± 0.11, 5.22 ± 0.36, 53.25 ± 1.20 μM against HT-29, SK-OV-3 and HeLa cells, respectively and 146e; IC50 = 38.0, 83.17 nM against VEGFR-2 and PDGFR-β, respectively; IC50 = 3.12 ± 0.27, 25.87 ± 1.32, 30.42 ± 1.98 μM against HT-29, SK-OV-3 and HeLa cells, respectively) compared to that of nintedanib (IC50 = 3.3, 3.7 nM against VEGFR-2 and PDGFR-β, respectively; IC50 = 4.90 ± 0.65, 28.76 ± 2.13, 51.65 ± 2.68 μM against HT-29, SK-OV-3 and HeLa cells, respectively)111 (ESI Fig. S9†).
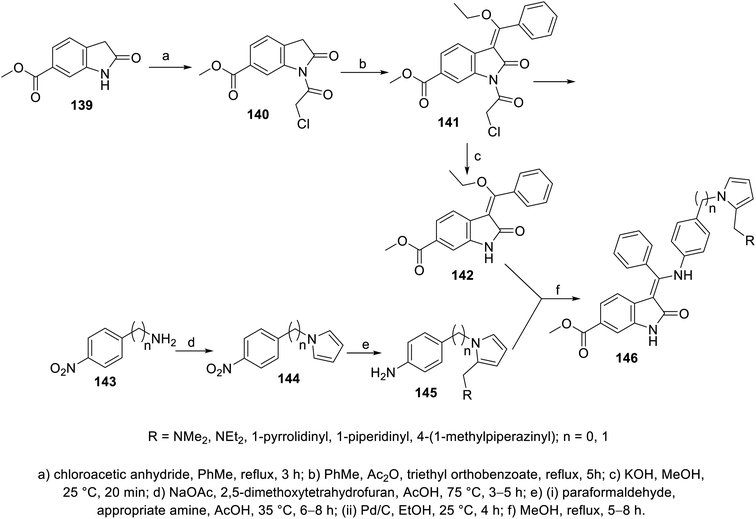 |
| | Scheme 19 Synthesis of 2-oxoindolin-3-ylidenes 146. | |
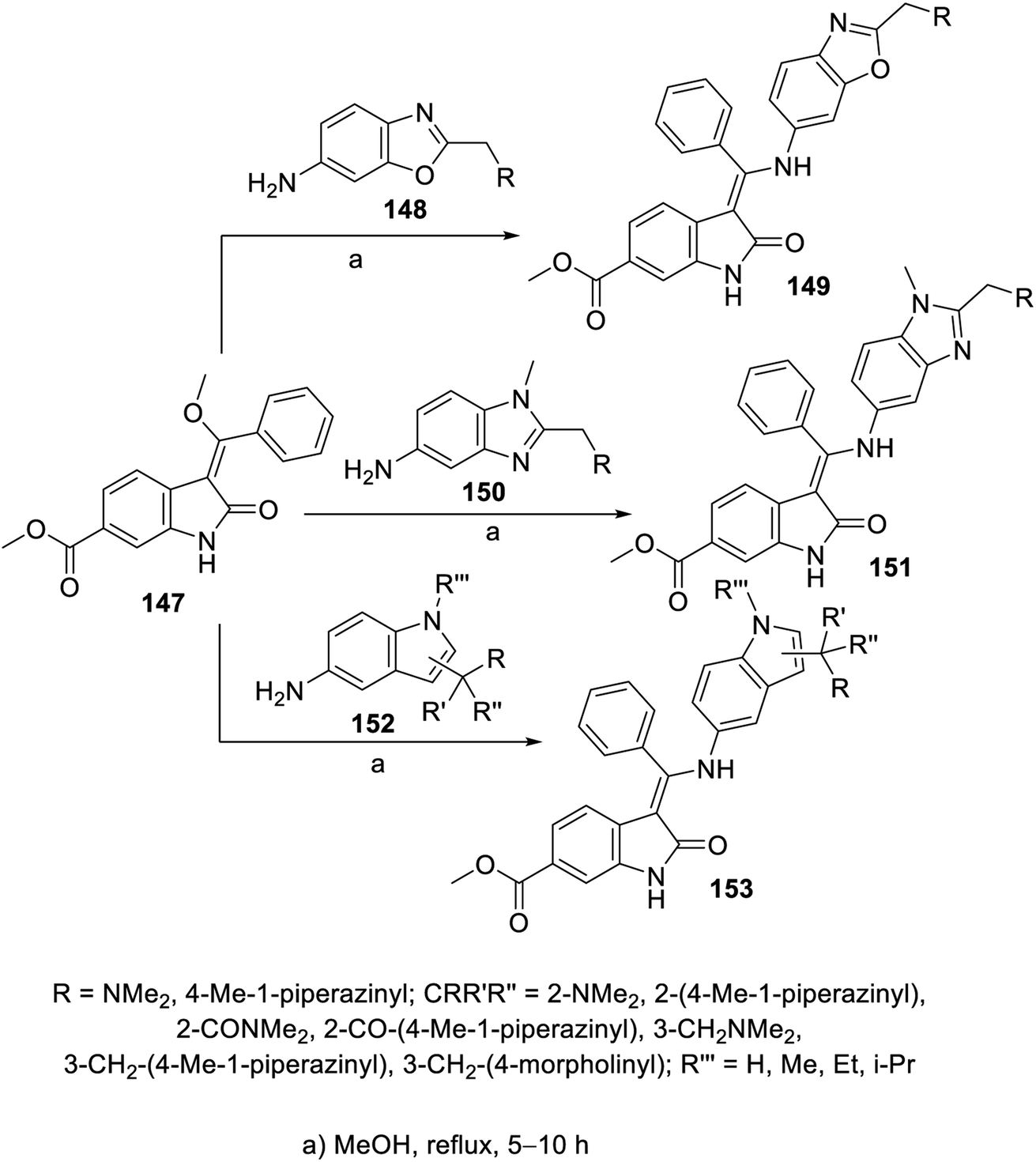
Similarly, the reaction of 2-oxoindolin-3-ylidene 147 with various amine-containing heterocycles (benzoxazole 148, benzimidazole 150 and indole 152) in refluxing methanol yielded the corresponding 2-oxoindolin-3-ylidenes 149, 151 and 153, respectively (Scheme 20). Some of the synthesized agents revealed promising antiproliferation activity (MTT assay) against the A549 (NSCLC), MCF7 (breast) and HT-29 (colon) cancer cell lines with enzymatic inhibitory properties against VEGFR-2 and PDGFR-β (ESI Fig. S10†). It was noticed that the prepared benzoxazolyl analogs were not tolerated due to either their weak or complete inactivity towards the tested enzymes. Meanwhile, the synthesized compounds with indolyl heterocycle 153 exhibited considerable antiproliferation properties against the MCF7 and/or HT-29 cell lines. Compound 153c (CR′R′′R′′′ = 2-[4-methylpiperazin-1-yl)carbonyl] was the most effective agent synthesized with promising inhibitory properties against VEGFR-2, -3 and PDGFR-α, PDGFR-β (IC50 = 69.1, 18.2, 4.4 and 22.0 nM, respectively) compared to nintedanib (IC50 = 8.5, 3.2, 2.3 and 3.5 nM, respectively). Furthermore, the safe proliferation behavior of 153c against the HEK293T (human embryonic kidney) normal cell line supports its use as a promising candidate.112
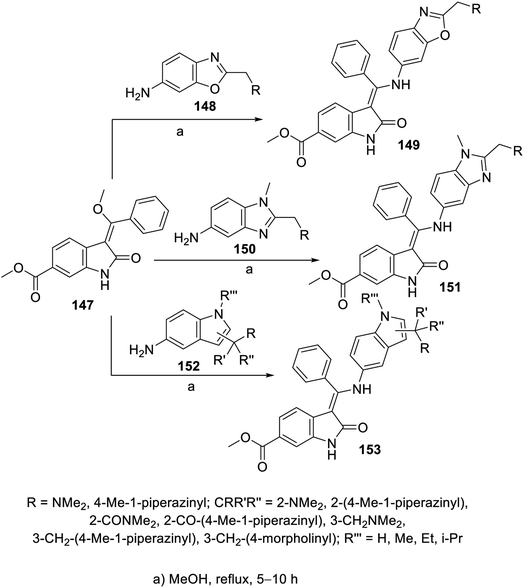 |
| | Scheme 20 Synthesis of 2-oxoindolin-3-ylidenes 149, 151 and 153. | |
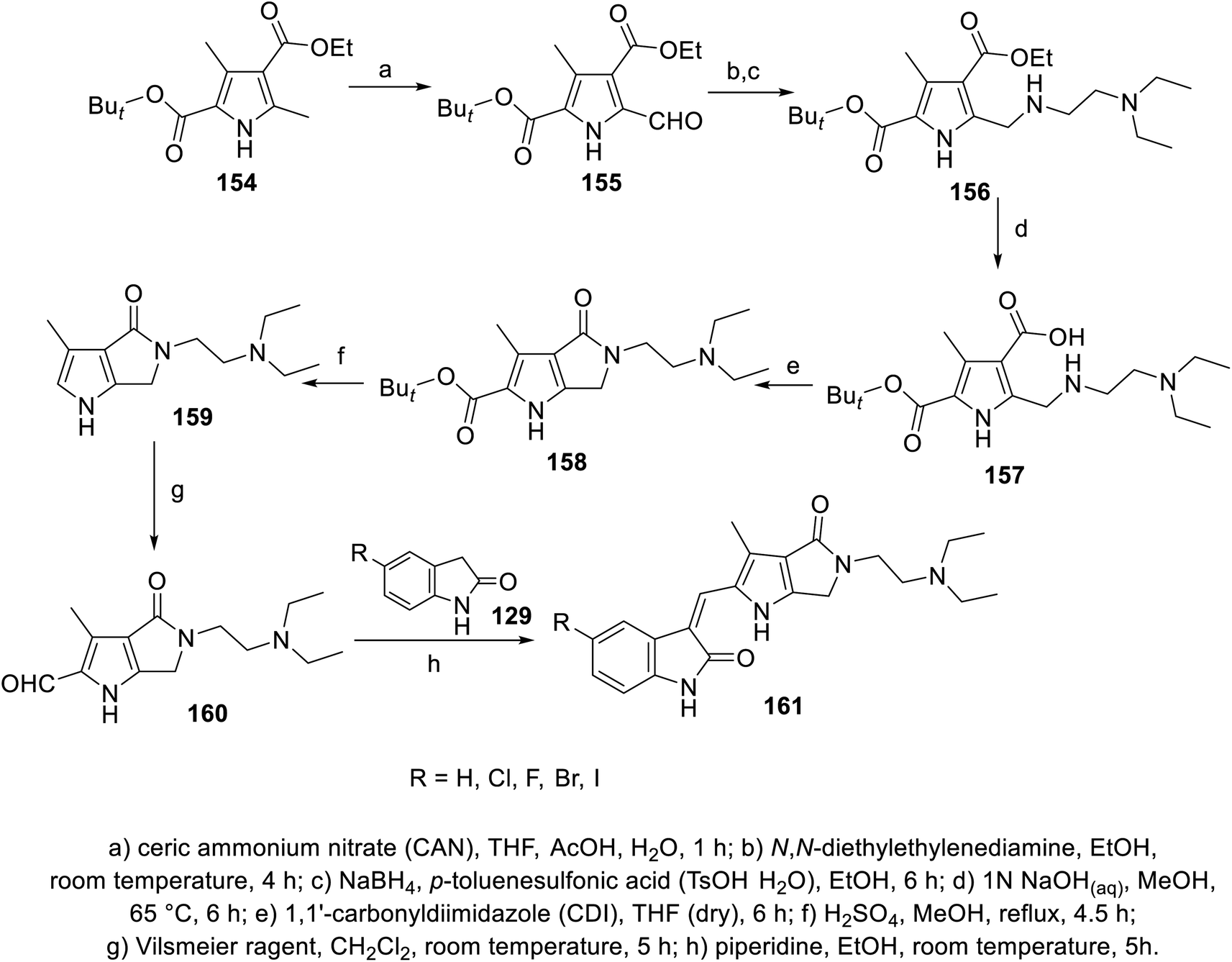
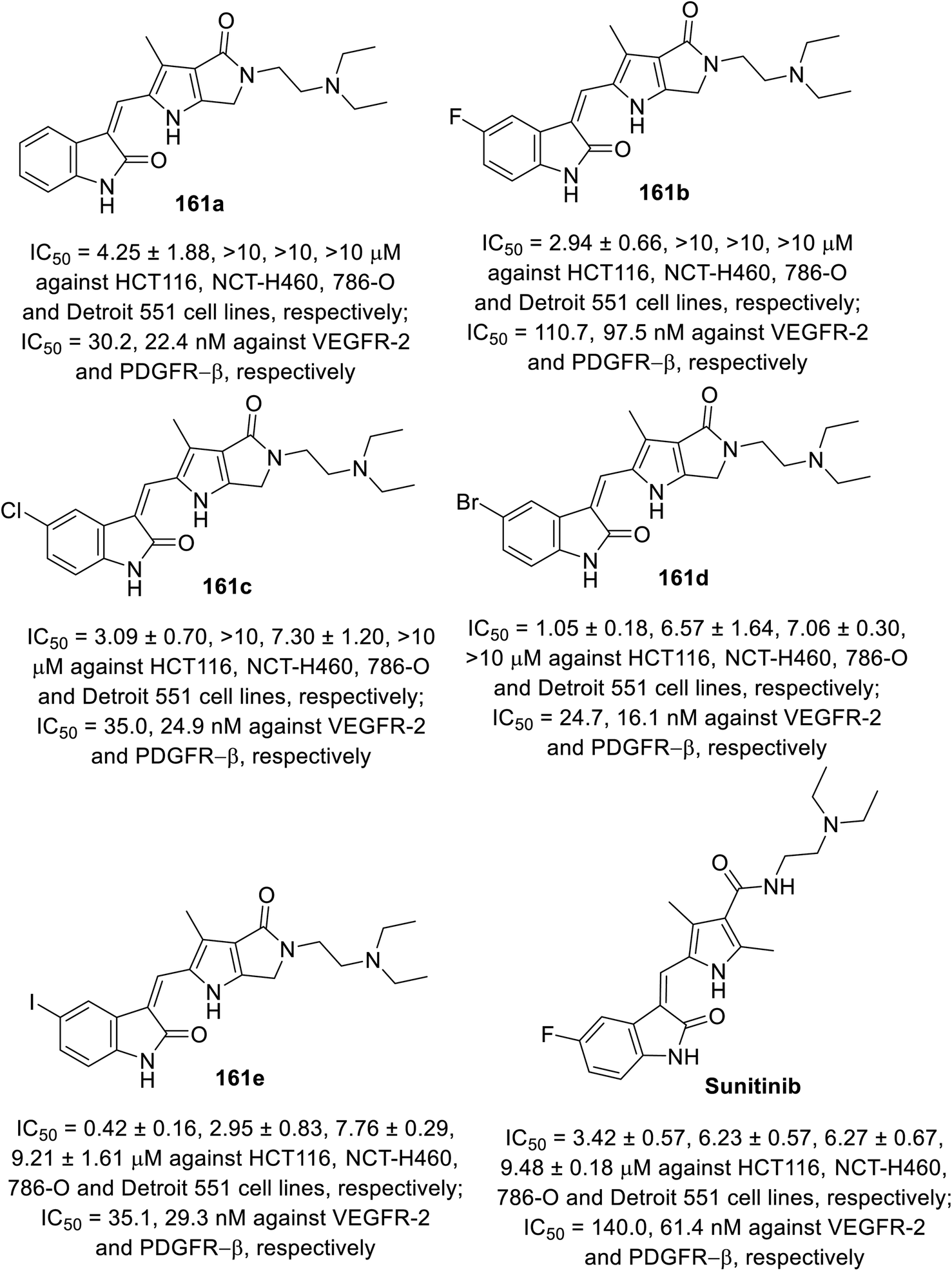
2-Oxoindolin-3-ylidenes linked to pyrrolo[3,4-b]pyrrol-2-yl 161 were synthesized through the condensation of 2-indolinones 129 with pyrrolo[3,4-b]pyrrole-2-carbaldehyde 160 (Scheme 21). The synthesized agents were screened for their antiproliferation properties (MTT assay) against HCT116 (colon), NCT-H460 (NSCLC), and 786-O (renal) cancer in addition to Detroit 551 (fibroblast) normal cell line. Iodoindolinone analog 161e (R = I) exhibited promising properties against HCT116 and NCT-H460 with potency higher than that of sunitinib (IC50 = 0.42 ± 0.16, 2.95 ± 0.83; 3.42 ± 0.57, 6.23 ± 0.57 μM for 161e and sunitinib, respectively). Bromo- 161d and iodoindolinones 161e showed considerable inhibitory properties against VEGFR-2 and PDGFR-β compared to that of sunitinib (IC50 = 24.7, 16.1; 35.1, 29.3; and 140.0, 61.4 nM against VEGFR-2 and PDGFR-β for 161d and 161e and sunitinib, respectively)113 (Fig. 12).
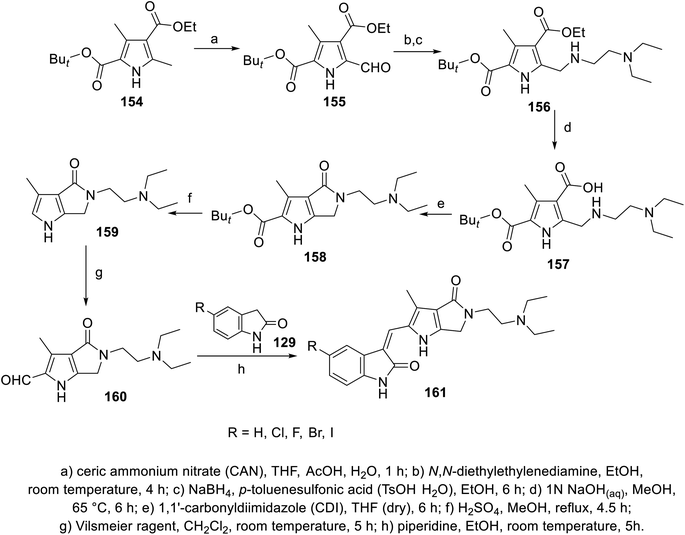 |
| | Scheme 21 Synthesis of 2-oxoindolin-3-ylidenes 161. | |
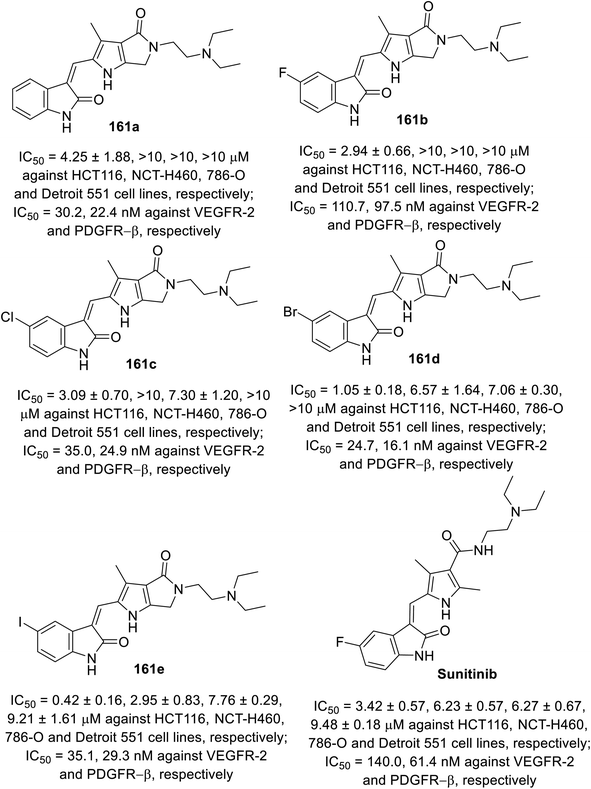 |
| | Fig. 12 Antiproliferation and enzymatic inhibitory properties of 161a–e and sunitinib. | |
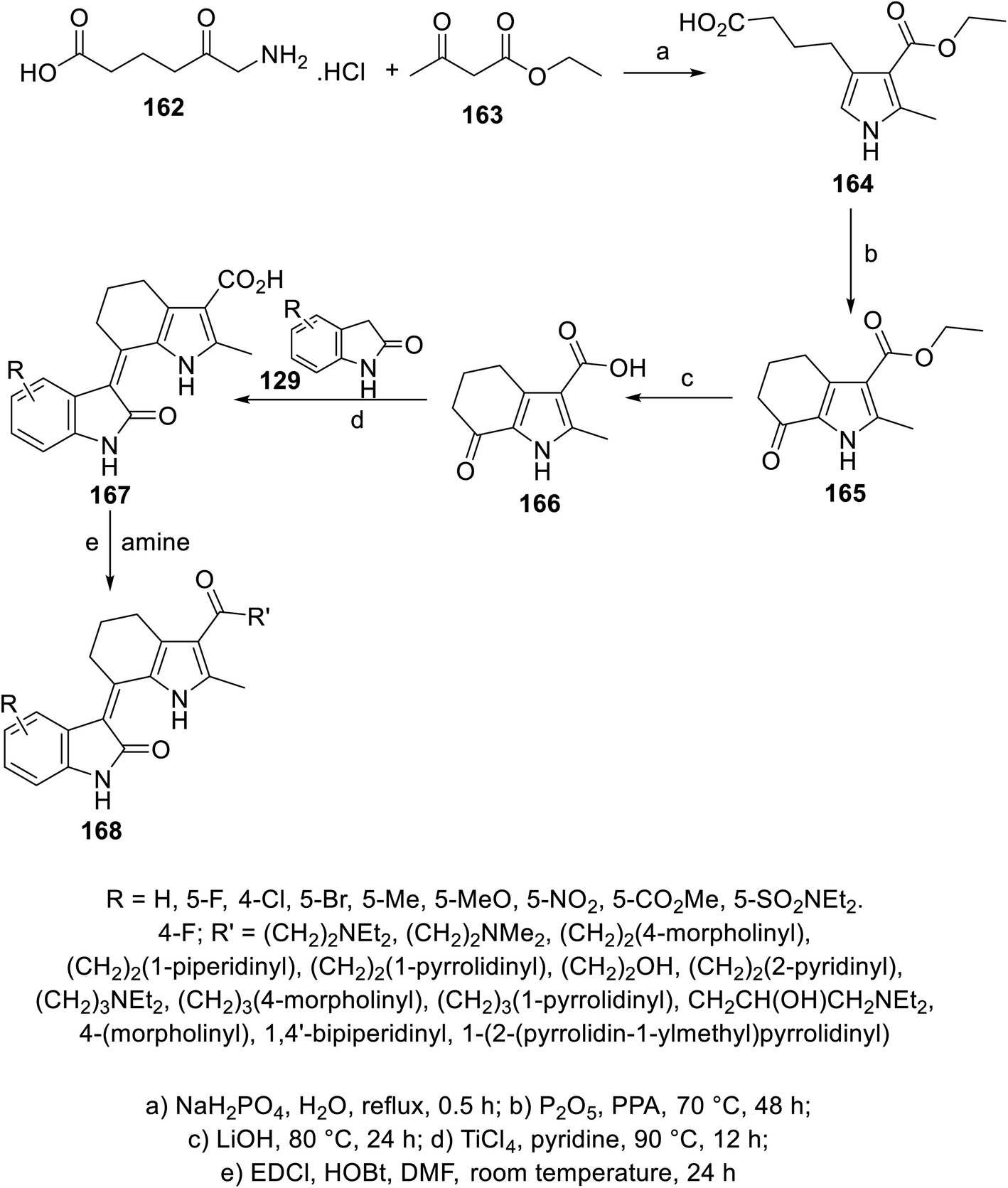
A series of 2-oxoindolin-3-ylidenes connected to pyrrolo[b]cyclohexyl heterocycle 168 was prepared via the condensation (pyridine containing TiCl4 “catalyst”) of the ketonic function of 1H-indole-3-carboxylate 166 with 2-indolinones 129, followed by reaction with either primary or secondary amines (in DMF containing EDCI: 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide and HOBt: hydroxybenzotriazole at room temperature) (Scheme 22). Some of the synthesized agents 168 showed promising inhibitory properties against different/multi-targeted tyrosine kinases (VEGFR-2, PDGFR-β and c-kit) compared to that of sunitinib. Furthermore, some of the promising agents were tested for their antiproliferation properties (MTT assay) against BXPC-3 (pancreatic), T24 (bladder), BGC (gastric), HEPG2 (liver) and HT29 (colon) cancer cell lines. Among them, compound 168c (R = 5-Cl, R′ = NH(CH2)2NEt2) was one of the most promising candidates discovered with antiproliferation (IC50 = 1.95, 1.83, 2.03, 3.14, and 6.48 μM against BXPC-3, T24, BGC, HEPG2 and HT29, respectively) and enzymatic inhibitory properties (IC50 = 2.6, 12.1, and 2.7 nM against VEGFR-2, PDGFR-β and c-kit, respectively) comparable to that of sunitinib (IC50 = 3.63, 2.44, 4.78, 5.61 and 1.47 μM against BXPC-3, T24, BGC, HEPG2 and HT29; IC50 = 4.0, 10.6, and 8.9 nM against VEGFR-2, PDGFR-β and c-kit, respectively)114 (ESI Fig. S11†).
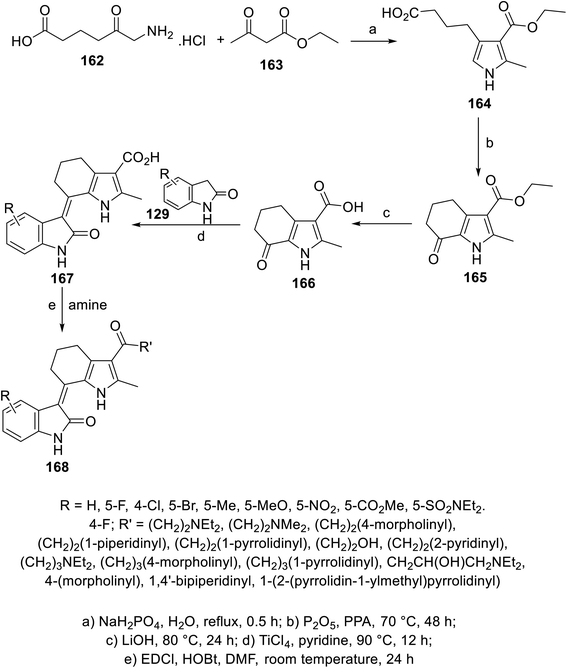 |
| | Scheme 22 Synthesis of 2-oxoindolin-3-ylidenes 168. | |
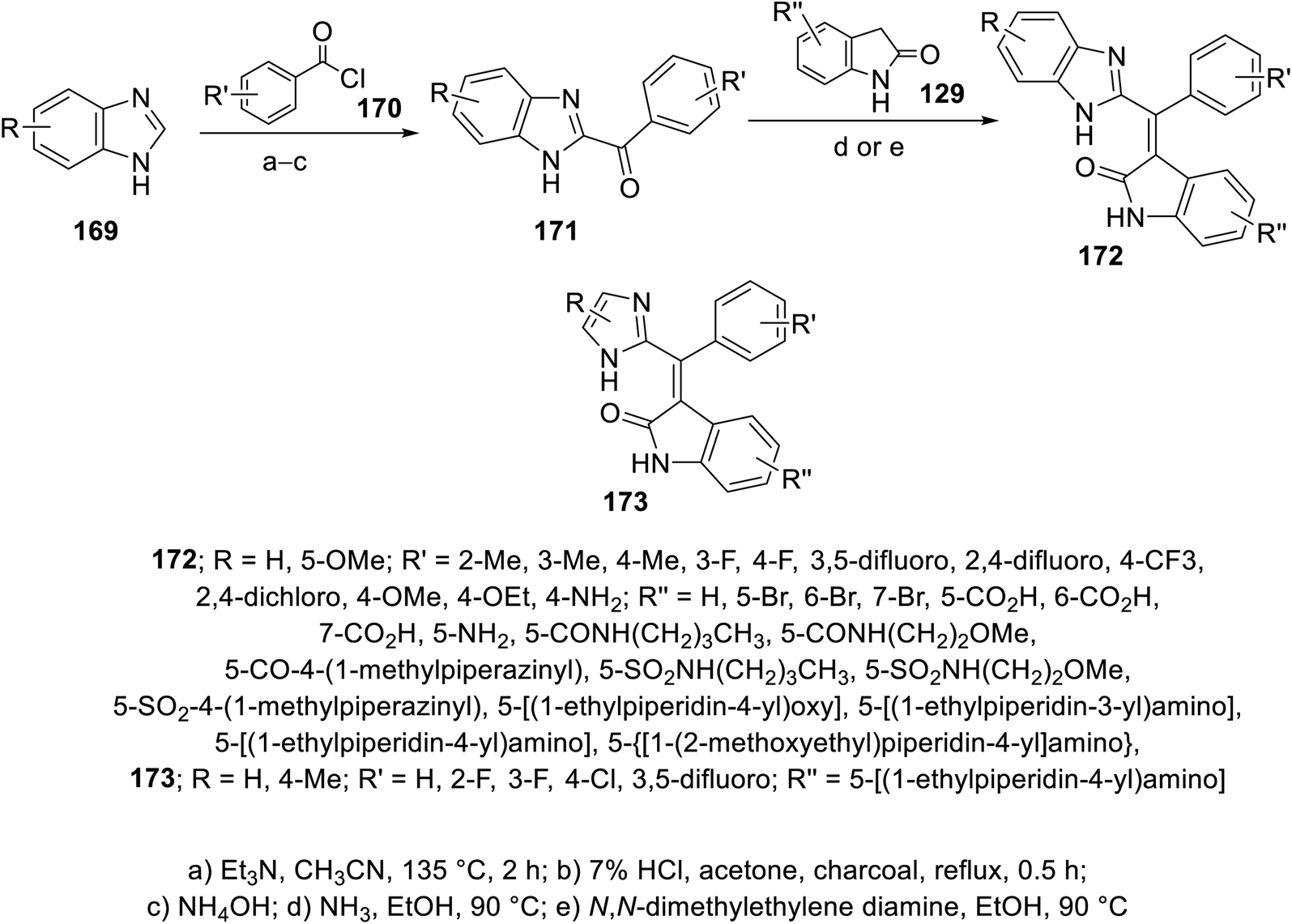
The reaction of benzimidazoles 169 with benzoyl chlorides 170 at 135 °C in the presence of TEA led to the formation of N- and C-diacylated benzimidazoles. The N-acyl function was removed upon refluxing with 7% aqueous HCl. Condensation of the formed benzimidazolyl ketones 171 with 2-indolinones 129 in a sealed tube (90 °C) in EtOH/NH3 gave the targeted 2-oxoindolin-3-ylidenes 172. Similarly imidazolyl connected to 2-oxoindolin-3-ylidenes 173 were obtained (Scheme 23).115 Some of the synthesized 2-oxoindolin-3-ylidenes 172/173 revealed promising enzymatic inhibitory properties against VEGFR-1 (Flt-1), VEGFR-2 (KDR), FGFR-1 and PDGFR-α compared to sunitinib and SU6668 (standard references). Compounds 172p (R = R′ = H, R′′ = 5-CO2H), 172t (R = H, R′ = 4-Me, R′′ = 5-CO2H) and 172v (R = 5-OMe, R′ = H, R′′ = 5-CO2H) exhibited considerable potencies against VEGFR-2 compared to the standard references used (IC50 = 4, 5, and 3 nM, respectively). Additionally, compounds 172af {R = 5-OMe, R′ = H, R′′ = 5-NH[4-(N-ethylpiperidinyl)]} and 173c {R = H, R′ = 4-Cl, R′′ = 5-NH[4-(N-ethylpiperidinyl)]} (IC50 = 10, 4 nM, respectively) showed promising enzymatic inhibitory, ADME properties and cellular potencies115 (ESI Fig. S12†).
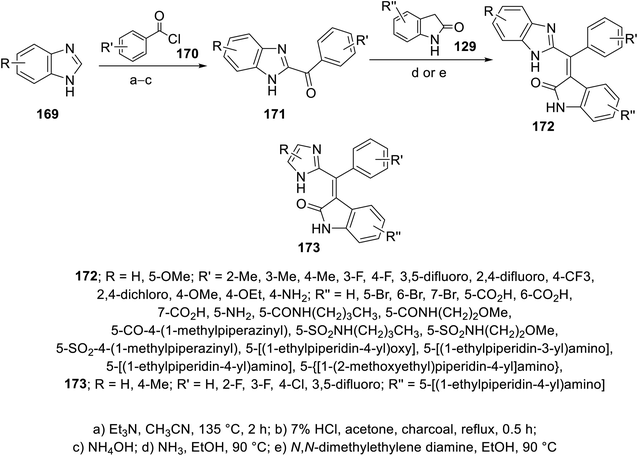 |
| | Scheme 23 Synthesis of 2-oxoindolin-3-ylidenes 172 and 173. | |
5. Indole heterocycle conjugates
Bio-conjugation is one of the most powerful and attractive rational drug design strategies used for the development of new drug candidates by connecting two or more therapeutic pharmacophoric functions/fragments/agents.116,117 Many conjugates have been reported to possess diverse potential bio-properties, among which vasodilation is useful for smooth muscle relaxation,118,119 anti-inflammatory, analgesic,120–123 antibacterial,124–126 antimycobacterial,127 antimalarial,128 antiviral including anti-SARS-CoV-2,129–133 HCV “hepatitis C viruses” and chikungunyan,134 anti-parasitic135 and antitumor.136,137
5.1. Indole triazole conjugates
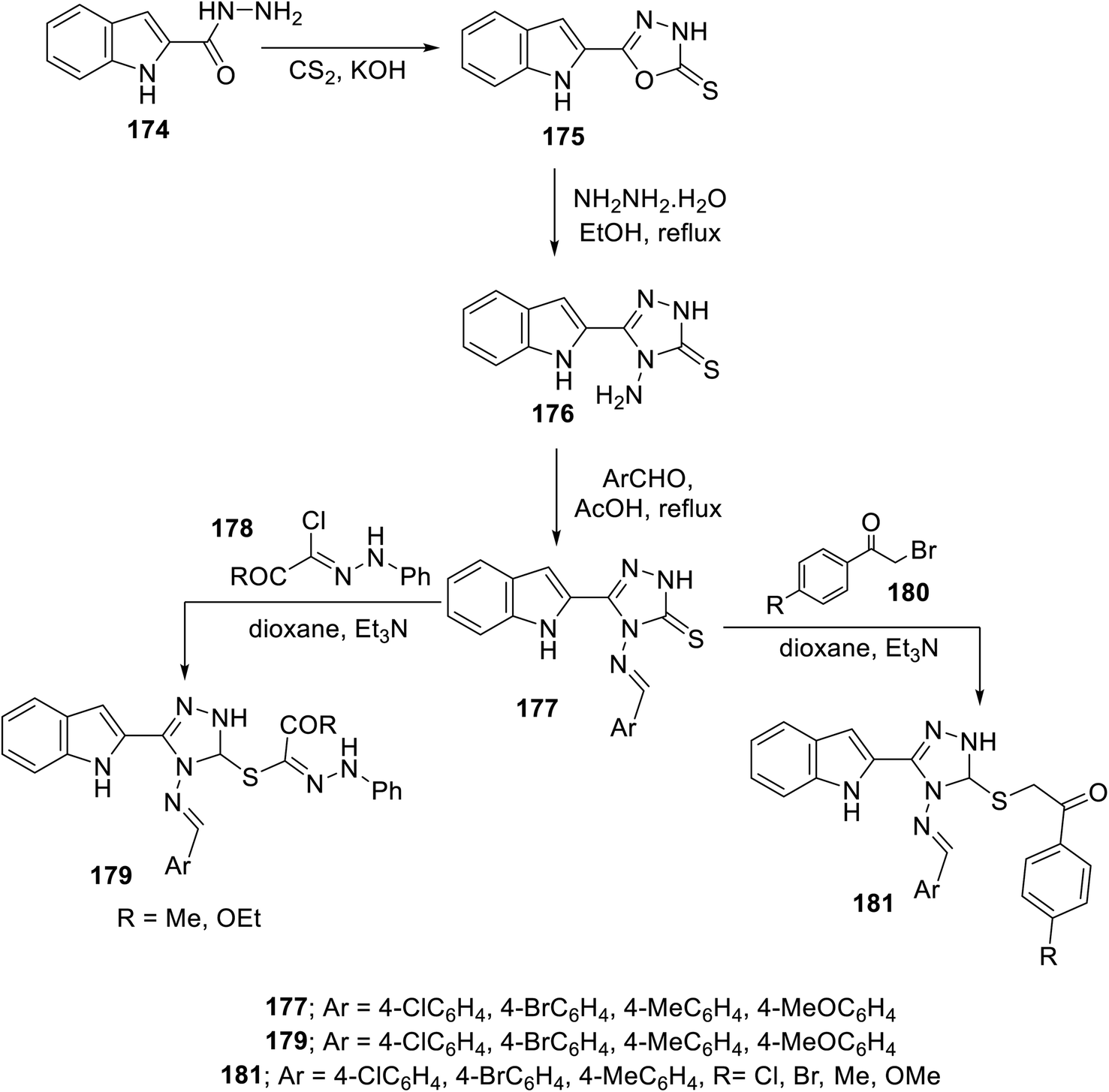
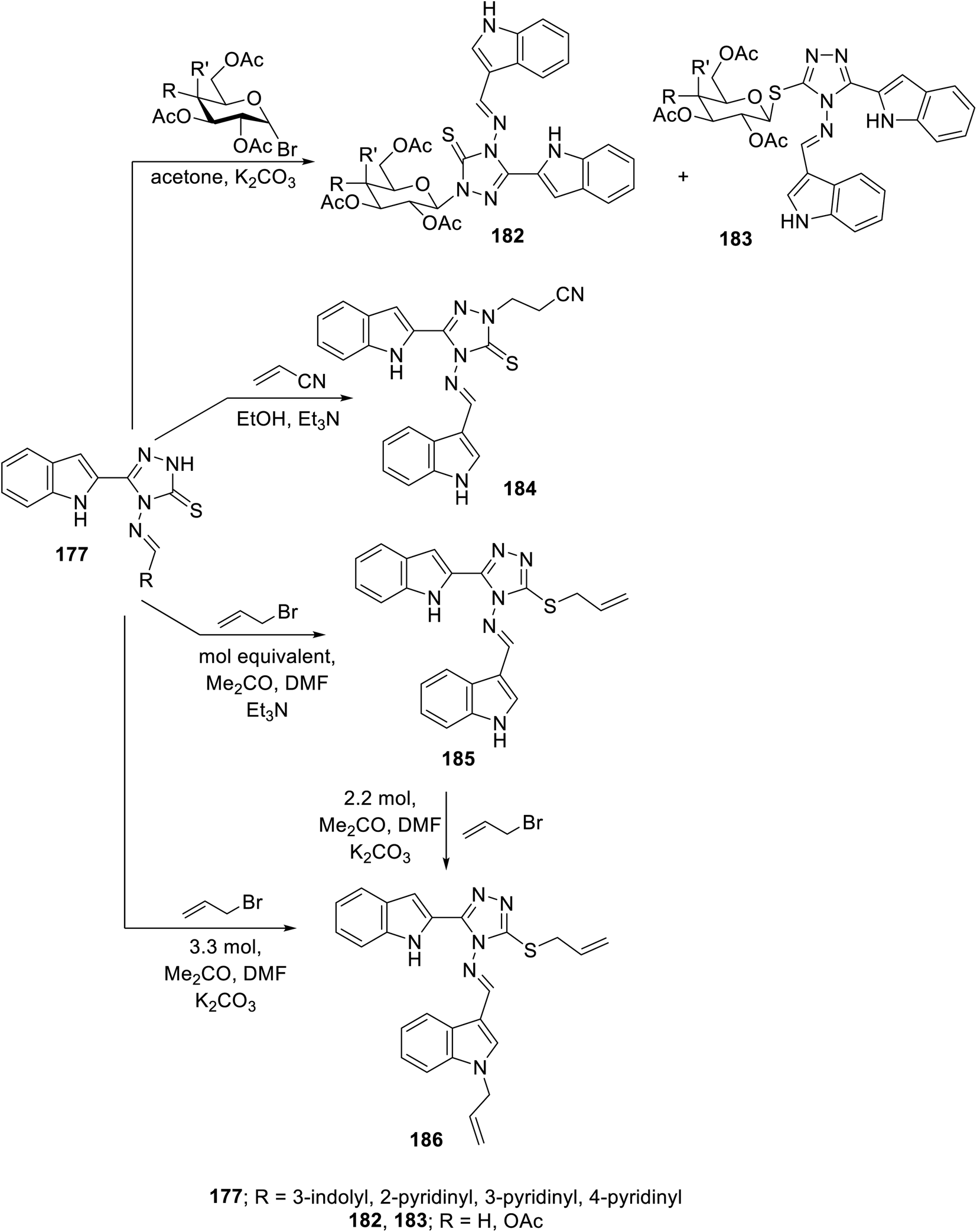
The condensation reaction (in refluxing acetic acid) of the appropriate aromatic aldehyde with indole triazole conjugate bearing an amino function 176 (obtained through reaction of 2-indolecarbohydrazide 174 with CS2/KOH followed by hydrazonolysis (NH2NH2·H2O in refluxing EtOH)) afforded the corresponding Schiff base 177. The reaction of 177 with hydrazonoyl chlorides 178 or phenacyl bromides 180 (in dioxane, Et3N) afforded the corresponding conjugates 179 and 181138 (Scheme 24). Some of the synthesized agents were observed to exhibit considerable VEGFR-2 inhibitory and antiproliferation properties (MTT assay, against human renal cancer cell lines CAKI-1 and A498). The most promising agents discovered were 179c (Ar = 4-H3CC6H4; IC50 = 0.075 ± 0.002 μM against VEGFR-2; IC50 = 3.23 ± 0.15 and 2.05 ± 0.09 μM against CAKI-1 and A498 cell lines, respectively) and 181g (Ar = 4-MeOC6H4, R = Cl; IC50 = 0.071 ± 0.002 μM against VEGFR-2; IC50 = 0.89 ± 0.04 and 2.2 ± 0.1 μM against CAKI-1 and A498 cell lines, respectively) compared to sunitinib (IC50 = 0.075 ± 0.002 μM against VEGFR-2; IC50 = 4.93 ± 0.16, 1.25 ± 0.04 μM against CAKI-1 and A498 cell lines, respectively) with a safety profile upon testing against the RPTEC/TERT1 non-cancer cell line138 (ESI Fig. S13†).
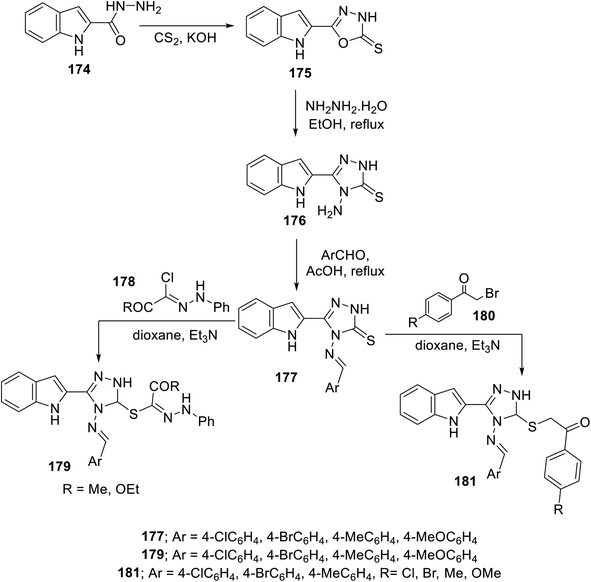 |
| | Scheme 24 Synthesis of indole triazole conjugates 179 and 181. | |
Glycosylation of Schiff base 177 with either acetobromoglucose or acetobromogalactose (K2CO3, acetone at room temperature) gave a mixture of N-glycosylated 182 and S-analog 183. However, the reaction of 177 with acrylonitrile (Michael acceptor) in refluxing ethanol containing Et3N afforded the corresponding N-substituted Michael adduct 184. Meanwhile, the reaction of 177 with allyl bromide (equimolar values) in acetone/DMF (1:1 v/v) containing Et3N at room temperature as a basic catalyst gave the S-allylated analog 185. Meanwhile, utilizing K2CO3 in the same reaction (using 3.3 molar value equivalent of allyl bromide relative to 177), the allylation of both the indolyl nitrogen and S-function of triazolyl heterocycle 186 was achieved. The same product was obtained upon reacting S-allylated analog 185 under the same conditions (2.2 molar equivalent value) (Scheme 25). The antiproliferation properties of the synthesized conjugates 182–186 were studied against the MCF7 (breast) and HepG2 (liver) cancer cell lines (MTT assay) utilizing 5-flurouracil and sorafenib as reference standards. Also, the VEGFR-2 inhibitory properties were studied for the most effective antiproliferative agent observed, supporting that compound 185 is a promising conjugate (IC50 = 1.18 ± 0.15 and 7.09 ± 0.67 μM against MCF7 and HepG2 cell lines; IC50 = 19.8 ± 1.58 nM against VEGFR-2, respectively) compared to sorafenib (IC50 = 2.13 ± 0.24 and 3.24 ± 0.23 μM against MCF7 and HepG2 cell lines; IC50 = 30.0 nM against VEGFR-2, respectively). The safe profile of compound 185 was supported through antiproliferation studies against the MDA-MB-231 non-cancer cell line (triple-negative breast cancer) and normal breast cells (MCF-10A) (IC50 = 10.49, 24.76 μM, against MDA-MB-231 and MCF-10A, respectively)139 (ESI Fig. S14†).
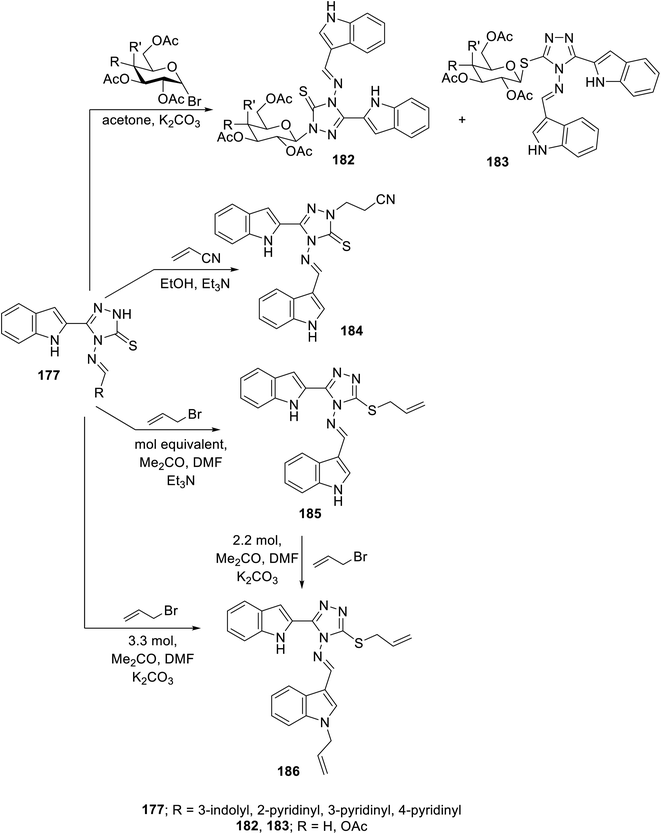 |
| | Scheme 25 Synthesis of indole triazole conjugates 182–186. | |
5.2. Indole benzimidazole conjugates
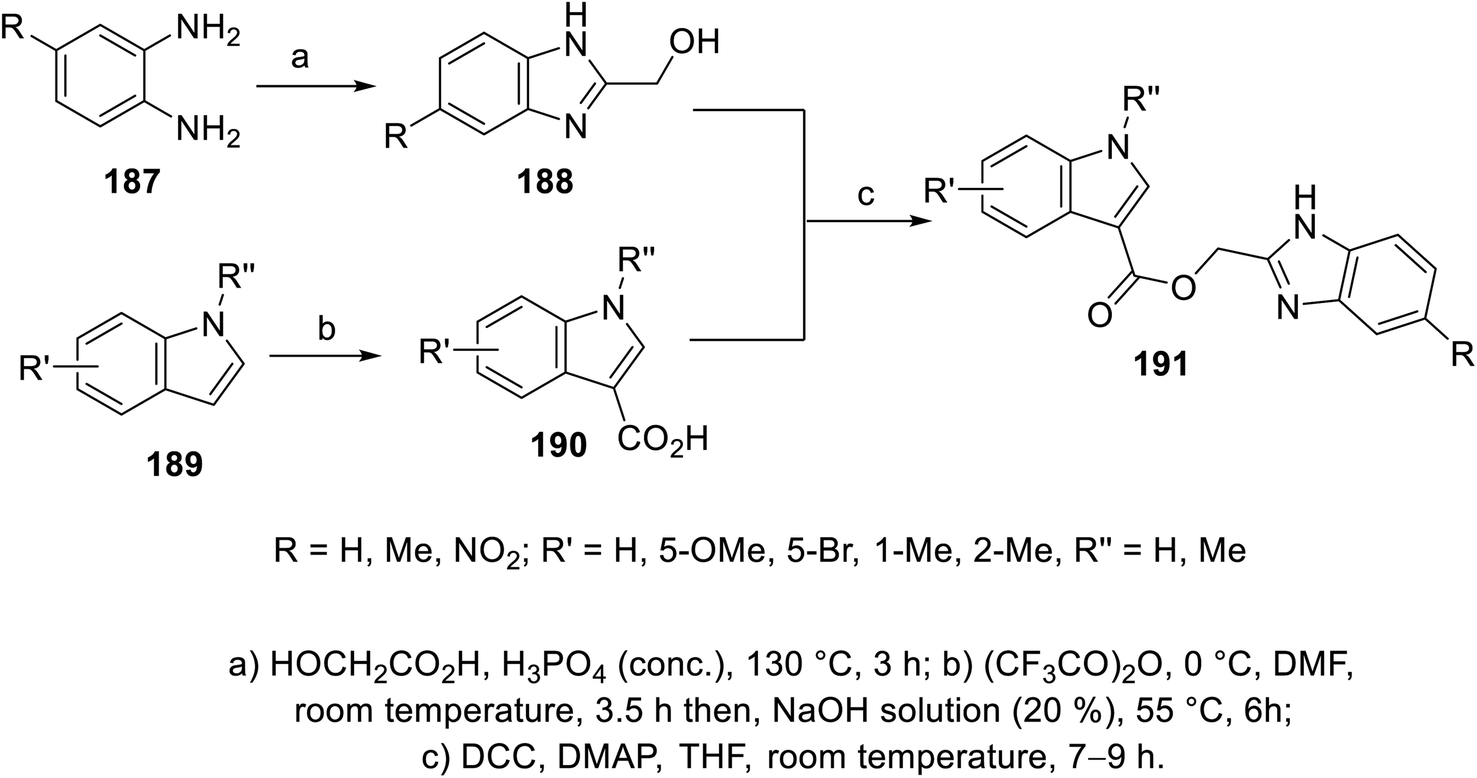
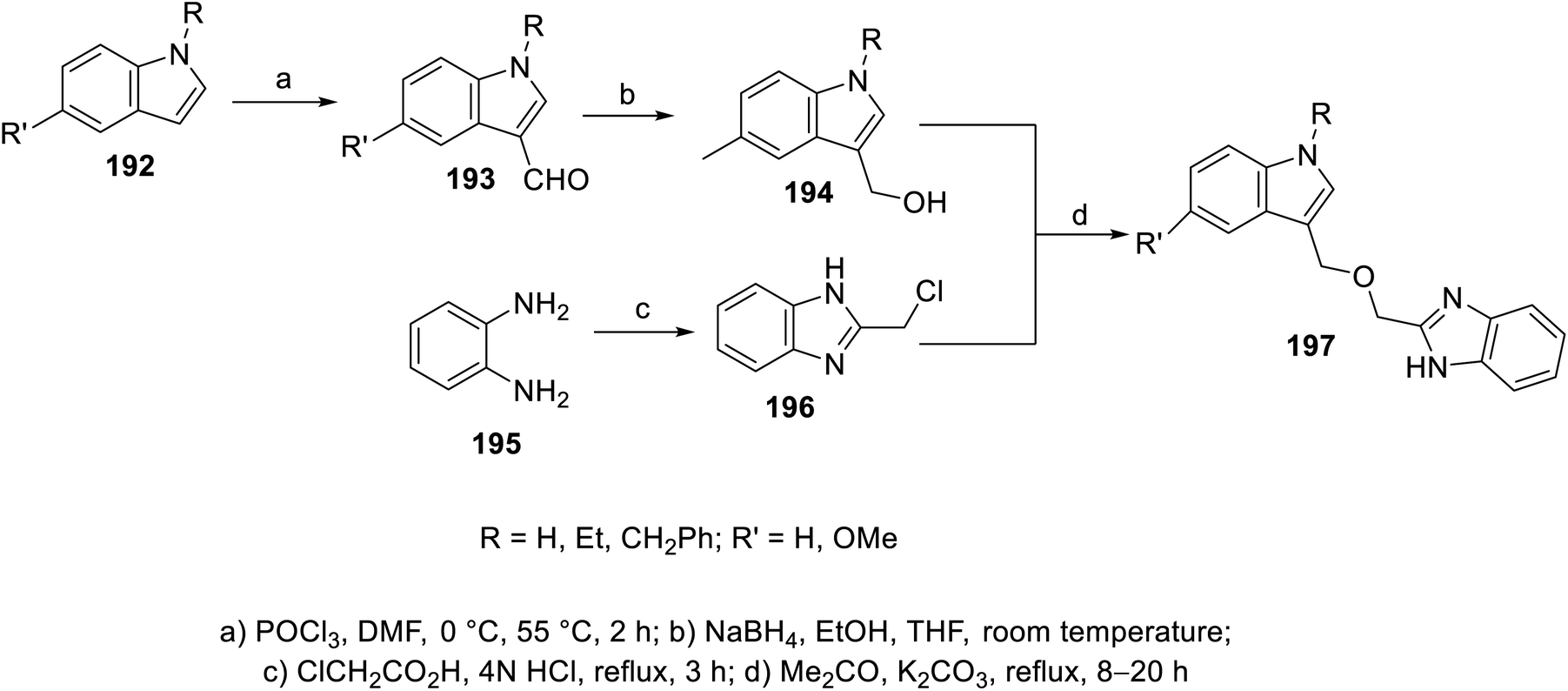
The esterification reaction of 3-indolecarboxylic acid 190 (obtained through the reaction of indole analogs 189 with (CF3CO)2O in DMF followed by treatment with 20% NaOH solution at 55 °C) with 2-hydroxymethyl benzimidazole 188 (synthesized through the reaction of the appropriate o-phenylenediamine 187 with hydroxyacetic acid in conc. H3PO4 at 130 °C) in THF containing DMAP (dimethylaminopyridine) and (DCC) (dicyclohexylcarbodiimide) at room temperature afforded indole benzimidazole conjugates 191 (Scheme 26). Additionally, indole benzimidazole conjugates 197 were obtained via the reaction of 2-chloromethyl-1H-benzimidazoles 196 (prepared by refluxing the appropriate o-phenylenediamine 195 with chloroacetic acid in 4 N HCl) with the appropriate (1H-indol-3-yl)methanol 194 (obtained through the formylation of the appropriate indolyl analogs “POCl3, DMF, 55 °C” followed by NaBH4 reduction) in refluxing acetone containing anhydrous K2CO3 (Scheme 27). Some of the synthesized conjugates exhibited considerable VEGFR-2 inhibitory properties (ESI Fig. S15†). Among them, the most promising was 191j (R = R′ = H, R′′ = Me; % inhibition of VEGFR-2 at 10 μM = 6.0) relative to sunitinib/SU11248 (% inhibition of VEGFR-2 at 10 μM = 98.1).140
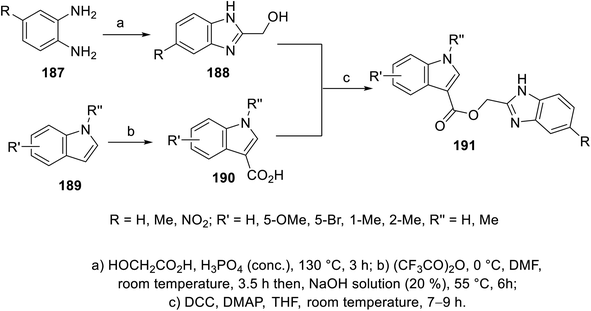 |
| | Scheme 26 Synthesis of indole benzimidazole conjugates 191. | |
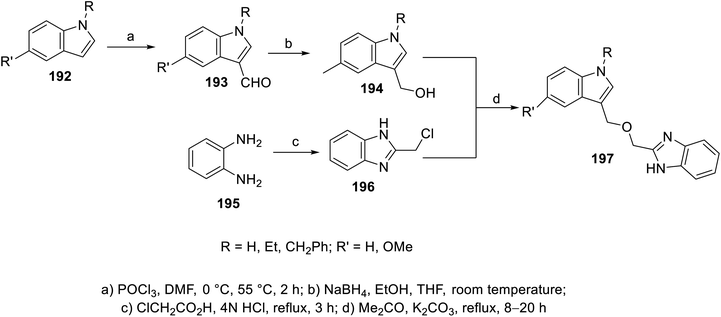 |
| | Scheme 27 Synthesis of indole benzimidazole conjugates 197. | |
5.3. Indole benzothiazole conjugates
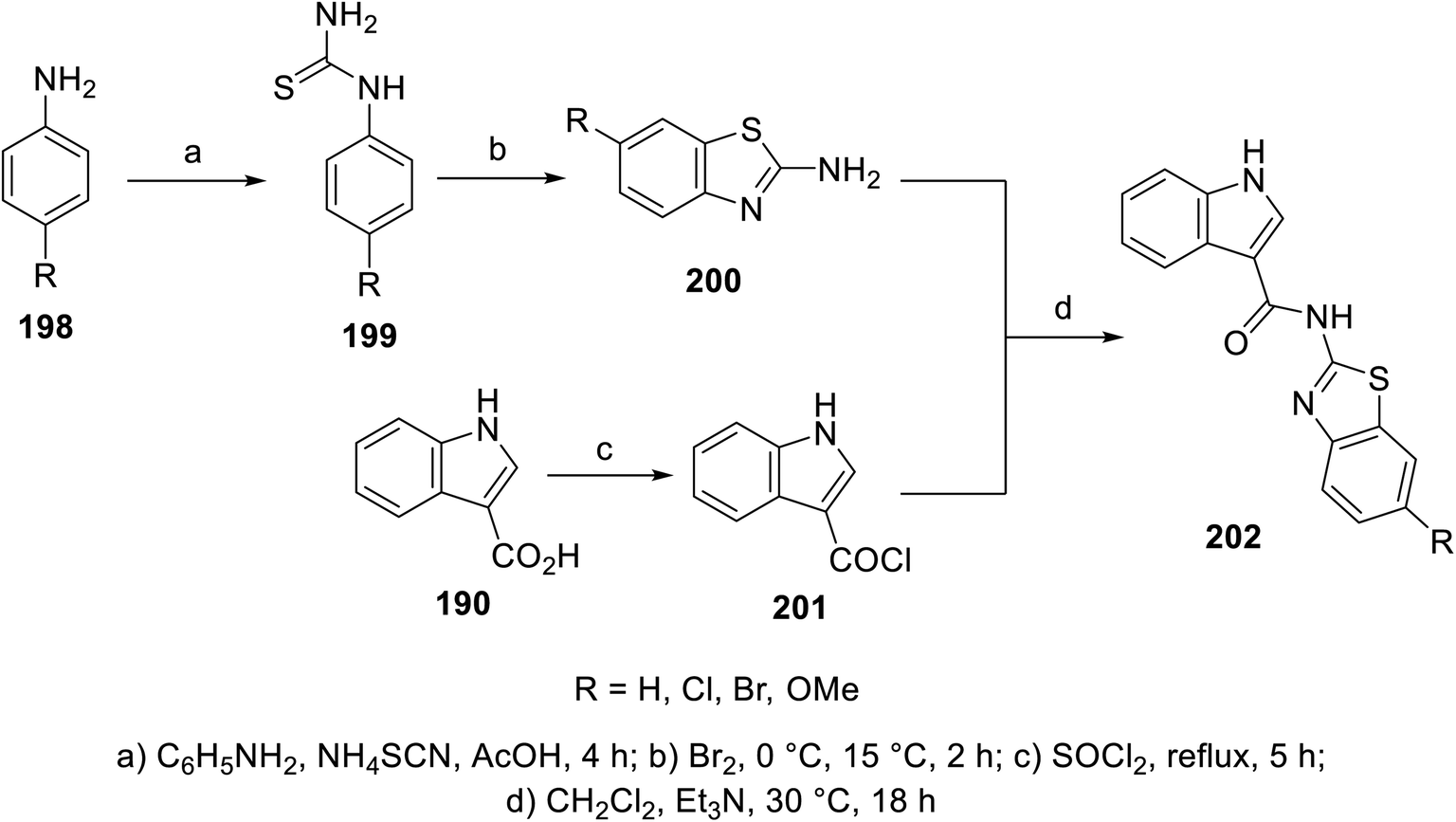
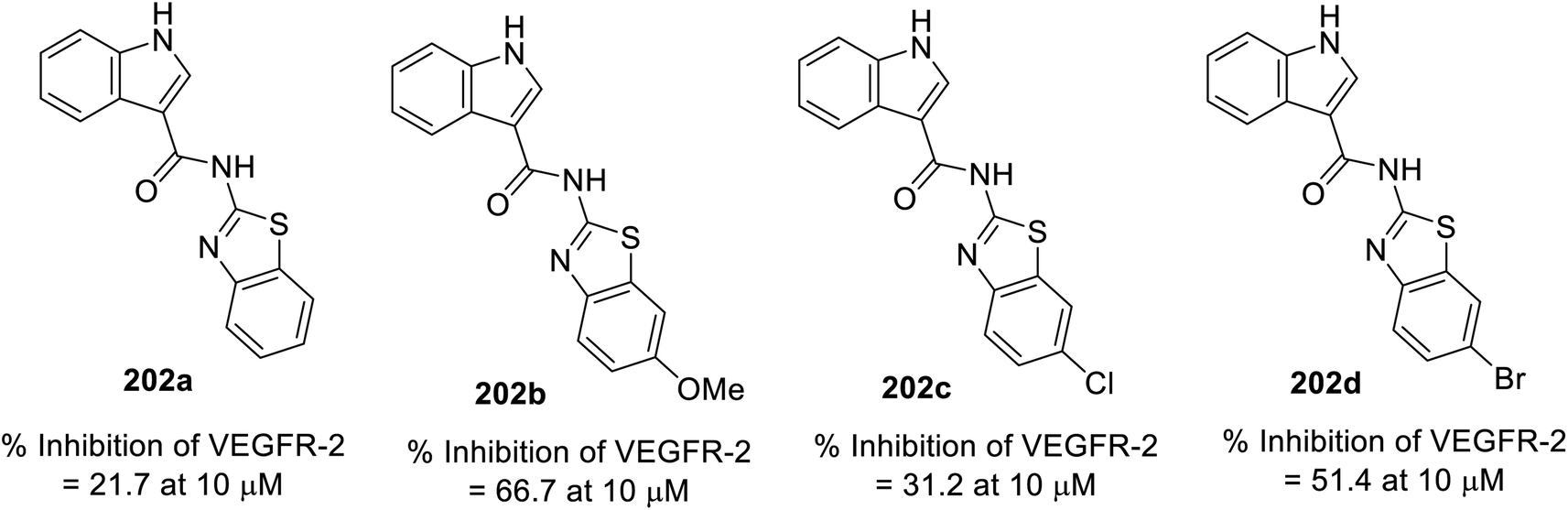
A variety of indole benzothiazole conjugates 202 was obtained through the reaction of 3-indole carbonyl chloride 201 (obtained by refluxing 3-indolecarboxylic acid 190 with thionyl chloride) with 2-aminobenzothiazoles 200 (in CH2Cl2 containing Et3N at room temperature) (Scheme 28). The synthesized conjugates exhibited considerable inhibitory properties (Fig. 13) compared to that of sunitinib (% inhibition of VEGFR-2 at 10 μM = 98.1).140
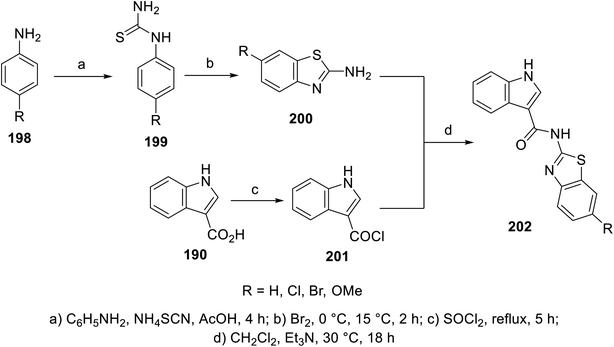 |
| | Scheme 28 Synthesis of indole benzothiazole conjugates 202. | |
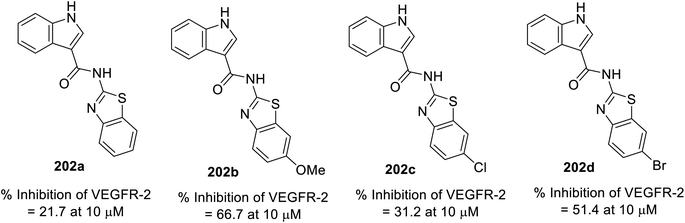 |
| | Fig. 13 % Inhibitory properties of VEGFR-2 by indole benzothiazole conjugates 202 at 10 μM. | |
5.4. Indole–pyrimidine conjugates
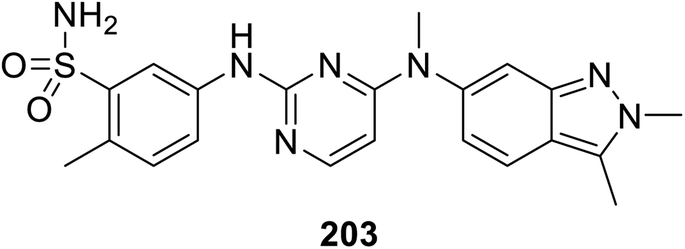
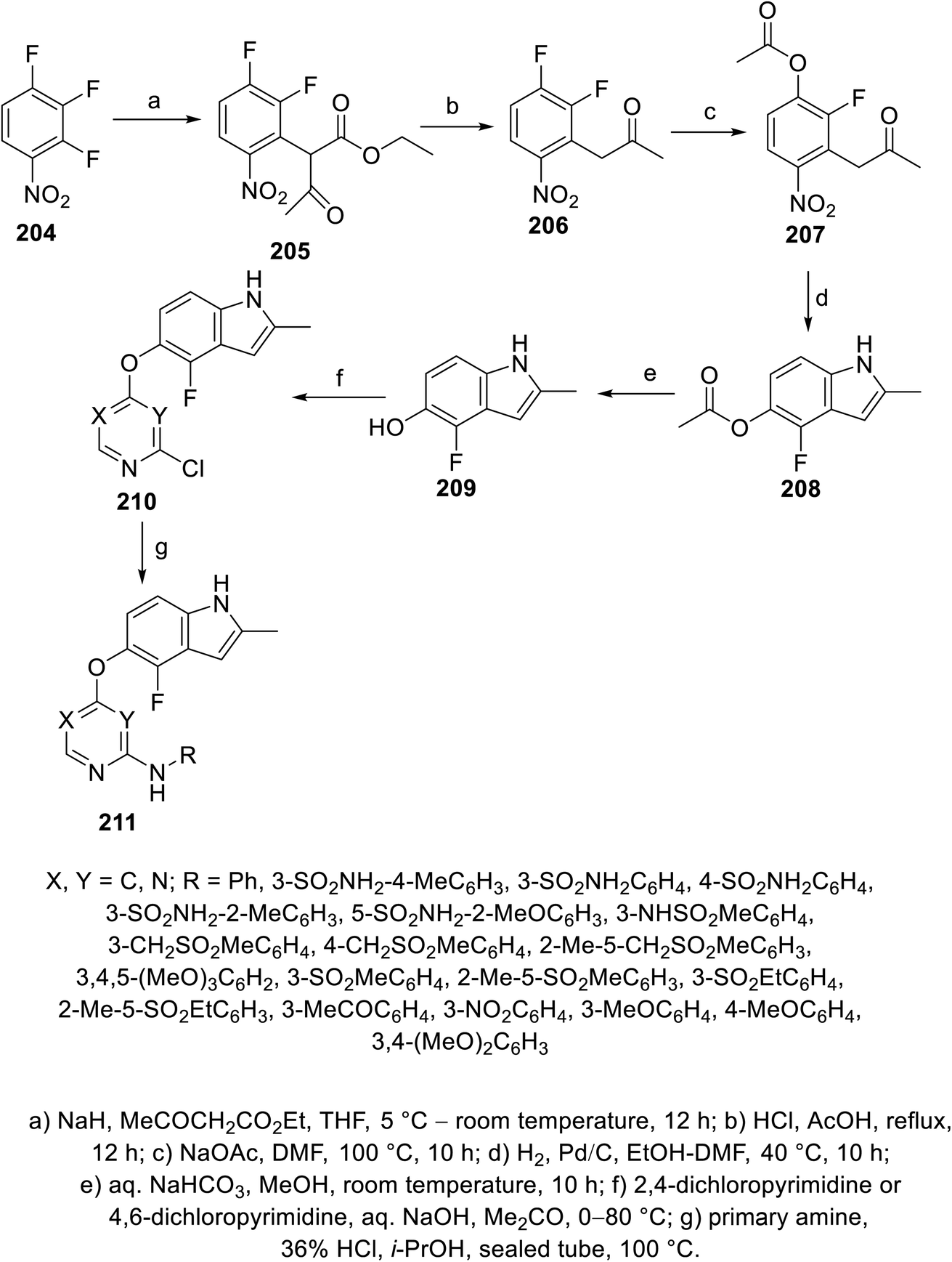
Pazopanib 203 (Fig. 14) is an FDA approved VEGFR-2 inhibitor for the treatment of advanced renal cell cancer (2009) and soft tissue sarcoma.141,142 In this molecule, the pyrimidinyl pharmacophoric heterocycle is attached to an indazole heterocycle. This inspired the design, construction and VEGFR-2 inhibitory properties investigation of indole pyrimidine conjugates linked through an ether linkage.143 A set of indole pyrimidine conjugates 211 was synthesized through the coupling of 5-hydroxyindole derivative 209 with 2,4-dicholoropyrimidine or 4,6-dicholoropyrimidine in Me2CO containing aq. NaOH, followed by reaction with various primary amines (HCl “36%”, i-PrOH, sealed tube, 100 °C) (Scheme 29). Some of the synthesized conjugates 211 were observed to exhibit considerable VEGFR-2 inhibitory properties. Among them, the most promising was 211k [R = 3-(MeSO2C6H4)CH2] compared to sunitinib (IC50 = 0.0038 ± 0.0033, 0.0022 ± 0.0005 μM, respectively) (ESI Fig. S16†). Additionally, compound 211k exhibited inhibitory properties against VEGFR-1, VEGFR-3, PDGFR-α and PDGFR-β (IC50 = 40.4, 515.8, 24.1 and 33.6 nM, respectively) and can be nominated as multi-targeted tyrosine kinase inhibitor.143
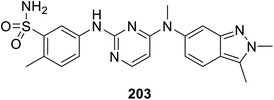 |
| | Fig. 14 Pazopanib VEGFR-2 inhibitor FDA approved for treatment of advanced renal cell cancer and soft tissue sarcoma. | |
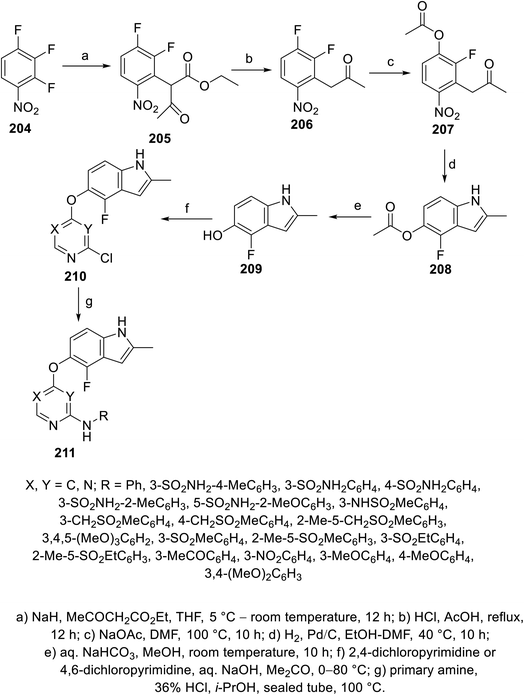 |
| | Scheme 29 Synthesis of indole-pyrimidine conjugates 211. | |
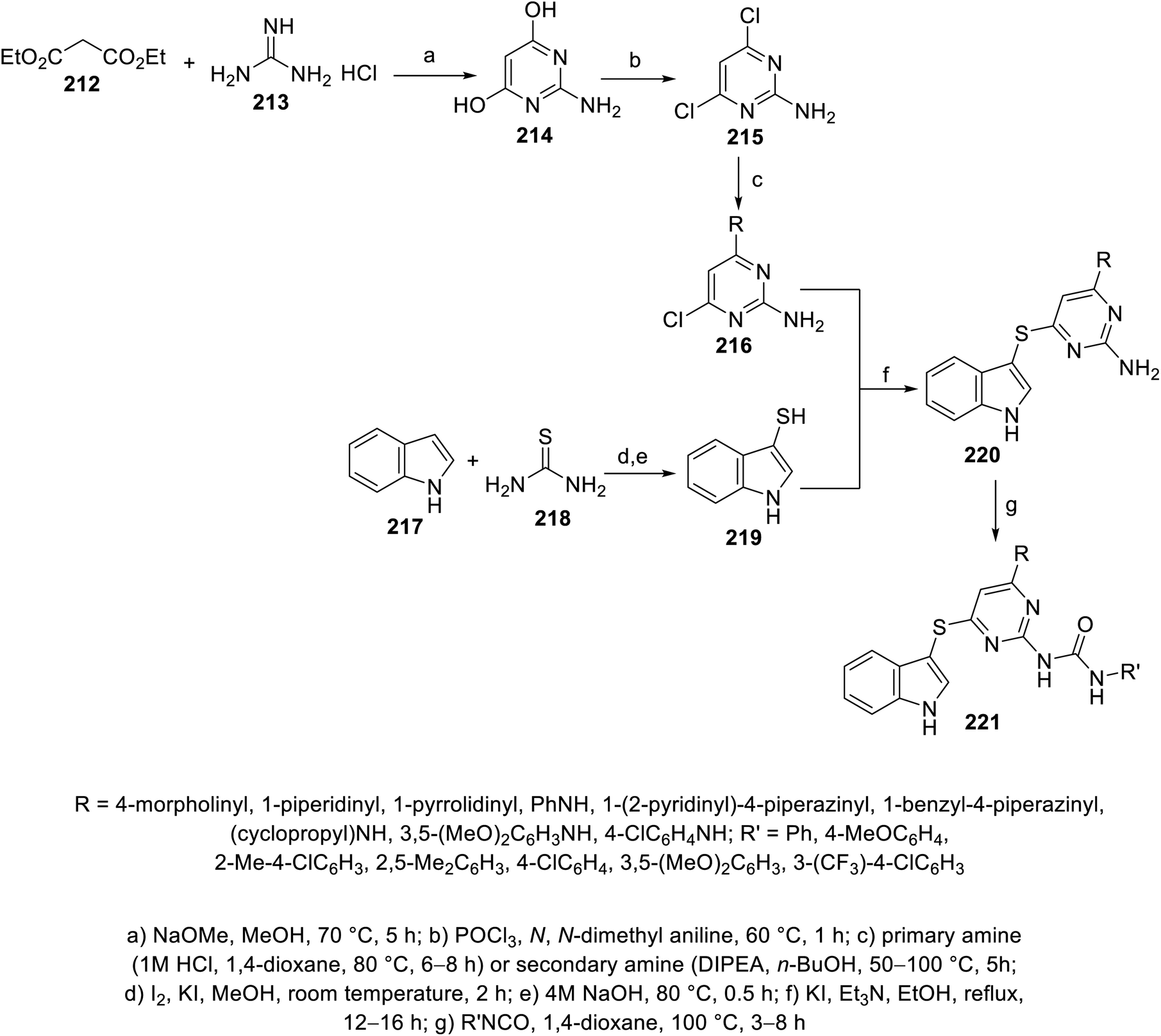
Indole pyrimidine conjugates 221 connected through a thioether linkage were also reported. The reaction of indole-3-thiol 219 with 6-chloro-2,4-diaminopyrimidines 216 (obtained from condensation of diethylmalonate 212 and guanidine hydrochloride 213 under basic condition, followed by chlorination with POCl3, and then reaction with either primary or secondary amine) in refluxing ethanol containing Et3N and KI gave 220, which was reacted with isocyanate in refluxing 1,4-dioxane, finally giving 221144 (Scheme 30). Some of the synthesized agents exhibited promising antiproliferation properties (MTT assay) against A549 (lung), PC-3 (prostate), MDAMB-231 (breast) and HepG2 (liver) human cancer cell lines with promising VEFGR-2 properties compared to that of sorafenib. The most promising was 221k (R = 1-pyrrolidinyl, R′ = 4-ClC6H4; IC50 = 6.41 ± 0.81, 10.42 ± 0.78, 5.85 ± 0.71, and 7.87 ± 1.18 μM against A549, PC-3, MDAMB-231 and HepG2, respectively with % inhibition = 0.33% ± 0.04% of VEGFR-2 at 10 μM) compared to that of Sorafenib (IC50 = 7.43 ± 0.81, 9.77 ± 1.12, 11.84 ± 1.25, 5.78 ± 0.41 μM against A549, PC-3, MDAMB-231 and HepG2, respectively with % inhibition = 1.21% ± 0.02% of VEGFR-2 at 10 μM)144 (ESI Fig. S17†).
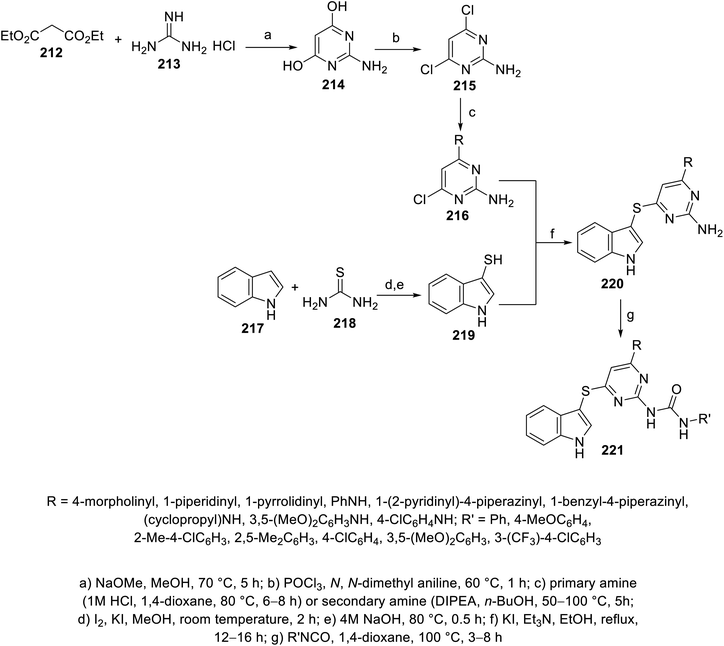 |
| | Scheme 30 Synthesis of indole-pyrimidine conjugates 221. | |
5.5. Indole chromene conjugates
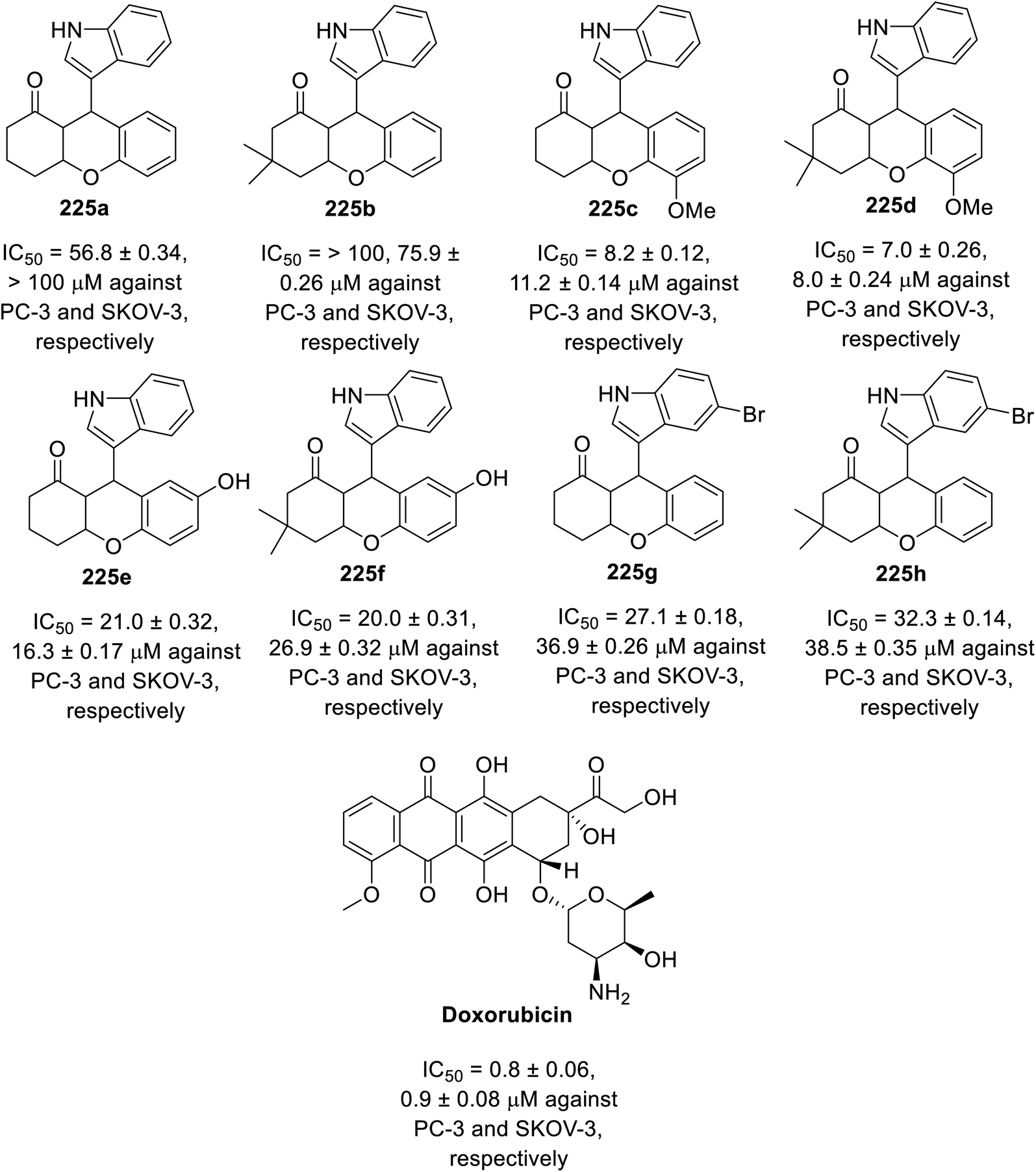
A multi-component eco-friendly synthetic procedure was employed for the preparation of indole chromene conjugates 225 via the one-pot reaction of salicaldehydes 222, with the appropriate 1,3-cyclohaxanones 223 and indole derivative 224 in refluxing water containing DBU (1,8-diazabicyclo[5.4.0]undec-7-ene). The reaction was assumed to take place via the base-catalyzed Michael addition of indole to the Knoevenagel adduct formed from the condensation of the salicaldehyde with 1,3-cyclohexanone assisted by the basic catalysis of DBU145 (Scheme 31). The synthesized conjugates 225 exhibited mild antitumor properties (MTT assay) against the PC-3 (prostate) and SKOV-3 (ovarian) cancer cell lines compared to that of doxorubicin (Fig. 15). Their VEGFR-2 inhibitory properties were reported based on the molecular modeling technique (PDB ID: 4ASD, AutoDock Tools 4.2).145 However, theoretical (molecular modeling) studies are insufficient supporting evidence for assigning the mode of action, and thus experimental data is required.
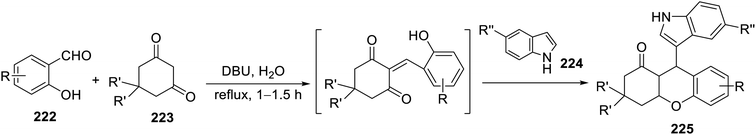 |
| | Scheme 31 Synthesis of indole chromene conjugates 225. | |
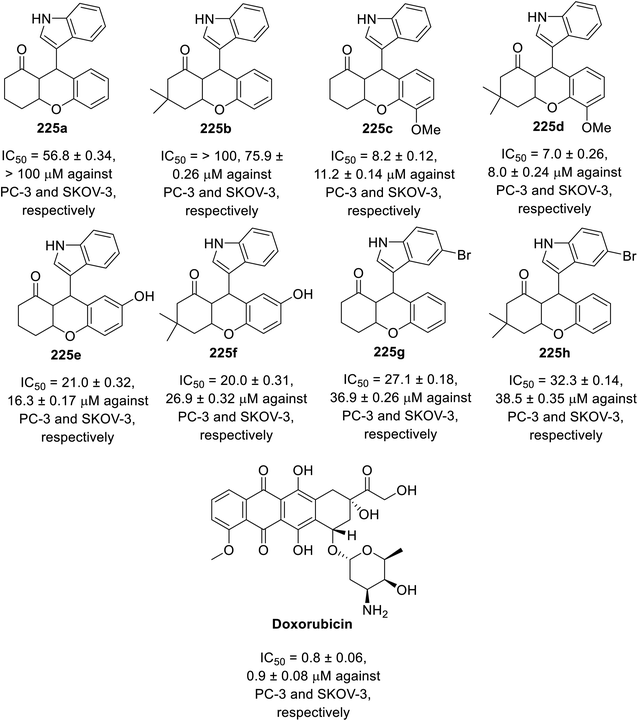 |
| | Fig. 15 Antiproliferation properties of indole chromene conjugates 225 and doxorubicin (standard reference). | |
6. Spiroindoles
Spiroindoles occupy a unique place in heterocyclic chemistry due to the versatile bio-properties of their natural and synthetic analogs146 (exemplified by antibacterial, antifungal,147–151 antimycobacterial,152–154 antiviral,19,155 anticancer,156–160 antimalarial,161,162 anti-inflammatory163 and antihyperglycemic164,165). [3 + 2]-Dipolar cycloaddition of azomethine ylides derived from isatin derivatives with α-amino acids to the exocyclic olefinic linkage derived from alicycles or heterocycles is an accessible synthetic approach successfully used for the construction of various spiroindoles with regio- and stereoselectivity.146,166–168 Many antitumor active agents against various human tumor cell lines were optimized via the above-mentioned synthetic approach; however, the VEGFR mode of action was only assigned for a few analogs.169,170
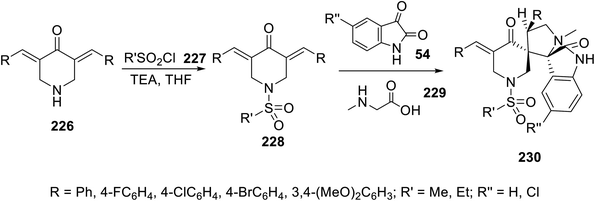
Spiroindoles 230 were regioselectively synthesized through the one-pot three-component reaction of 3,5-diylidene-4-piperidones 228 and azomethine, which were formed by the condensation of isatins 54 and sarcosine 229 in refluxing ethanol (Scheme 32). The stereochemical structure of 230 was established by single crystal X-ray studies. Promising antiproliferation properties were exhibited by some of the synthesized spiroindoles 230 against the MCF7 (breast), HCT116 (colon), A431 (skin squamous) and PaCa2 (pancreatic) cancer cell lines (MTT assay) compared to the standard references (sunitinib and 5-fluorouracil). Safe behaviors against the non-cancer RPE1 cell line were revealed by the synthesized agents. Considerable multi-targeted inhibitory properties (western blotting technique) were exhibited by the synthesized agents against VEGFR-2 and EGFR. The SAR (structure–activity relationship) study indicated that the chloro-substituted indolyl heterocycle can optimize efficient agents against the tested cancer cell lines. Compound 231n (R = 4-BrC6H4, R′ = Me, R′′ = H) was the most promising agent observed (IC50 = 3.597 ± 0.19, 3.236 ± 0.27, 2.434 ± 0.18, 12.500 ± 0.67, and 14.894 ± 1.61 μM against the MCF7, HCT116, A431, PaCa-2 and RPE1 cell lines, respectively; % inhibition = 61.3% and 65.6% against VEGFR-2 and EGFR at the IC50 value observed against MCF7, respectively) compared to sunitinib (IC50 = 3.97 ± 0.32, 9.67 ± 0.22, and 16.91 ± 0.95 μM against MCF7, HCT116, and PaCa-2 cell lines, respectively; % inhibition = 74.7% and 81.4% against VEGFR-2 and EGFR at IC50 value observed against MCF7, respectively)19 (ESI Fig. S18†).
 |
| | Scheme 32 Synthesis of spiroindoles 230. | |
7. Conclusion
Tyrosine kinases are capable of many diverse cellular functions including growth, proliferation, differentiation and death. VEGFR is one of the targeted therapeutic approaches that is preferable to the classical non-selective therapies to minimize the associated side effects or drawbacks. VEGF is an important category of tyrosine kinases, which can stimulate angiogenesis. VEGFR-2 is the most well-known factor in the angiogenesis of different solid tumors (colon, breast, ovary, lung, skin, renal, head, neck, lymphoma, etc.). Indolyl therapeutics have been approved against some serious types of cancer. Additionally, research efforts identified natural and synthesized antitumor indole-containing compounds with promising anti-VEGFR properties. Computational/theoretical studies can assist in designing and identifying novel hits/leads of anti-VEGFR agents; however, without experimental supporting their enzymatic properties, the predictions cannot be considered for further investigations or utilization.
Abbreviations
| ADMET | Absorption, distribution, metabolism, excretion and toxicity |
| AMD | Age-related macular degeneration |
| ATP | Adenosine triphosphate |
| BOP | Benzotriazol-1-yloxytris(dimethylamino)phosphonium hexafluorophosphate |
| CA | Carbonic anhydrase |
| CAM | Chick chorioallantoic membrane |
| CDK-1 | Cyclin-dependent kinase 1 |
| c-kit | Stem cell factor receptor |
| COX | Cyclooxygenase |
| COX-2 | Cyclooxygenase-2 |
| CSF-1R | Colony stimulating factor-1 receptor |
| DBU | 1,8-Diazabicyclo[5.4.0]undec-7-ene |
| DCM | Dichloromethane |
| DFT | Density function theory |
| DIPEA | N,N-Diisopropylethylamine |
| DMTMM | 4-(4,6-Dimethoxytriazine)chlorinated 4-methylmorpholine |
| EDCI | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| EGF | Epidermal growth factor |
| EGFR | Epidermal growth factor receptor |
| FDA | Food and drug administration |
| FGF | Fibroblast growth factor |
| FGFR | Fibroblast growth-factor receptor |
| HATU | Hexafluorophosphate azabenzotriazole tetramethyl uronium |
| HCV | Hepatitis C viruses |
| HER-2 | Human epidermal growth factor receptor 2 |
| HIF-1-α | Hypoxia-inducible factor-1-alph |
| HIV | Human immunodeficiency virus |
| HOBt | Hydroxybenzotriazole |
| HUVEC | Human umbilical vein endothelial cell |
| IBCF | Iso-butyl chloroformate |
| MD | Molecular dynamic |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| NET | Neuroendocrine tumor |
| NSCLC | Non-small cell lung cancer |
| NT | Not tested |
| PDGFR | Platelet-derived growth-factor receptor |
| SAR | Structure–activity relationship |
| SD | Standard division |
| SEM | Standard error mean |
| SRB | Sulforhodamine B |
| TEA | Triethylamine |
| THF | Tetrahydrofuran |
| TRAMP | Transgenic adenocarcinoma of the mouse prostate |
| VEGF | Vascular endothelial growth factor |
| VEGFR | Vascular endothelial growth factor receptor |
Author contributions
Conceptualization, A. S. G.; methodology, A. S. G. and D. R. A.; data analyses, A. S. G.; writing – original draft preparation, A. S. G. and D. R. A.; review and editing, A. S. G., D. R. A., M. S. B. and A. R. H.; supervision, A. S. G. and M. A. Y. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
There is no conflict to declare.
Acknowledgements
This work was supported financially by National Research Centre, Egypt, project ID: 13060103.
References
- G.-F. Zha, H.-L. Qin, B. G. M. Youssif, M. W. Amjad, M. A. G. Raja, A. H. Abdelazeem and S. N. A. Bukhari, Discovery of potential anticancer multi-targeted ligustrazine based cyclohexanone and oxime analogs overcoming the cancer multidrug resistance, Eur. J. Med. Chem., 2017, 135, 34–48, DOI:10.1016/j.ejmech.2017.04.025.
- B. S. Chhikara and K. Parang, Global cancer statistics 2022: the trends projection analysis, Chem. Biol. Lett., 2023, 10, 451 Search PubMed.
- R. L. Siegel, K. D. Miller, N. S. Wagle and A. Jemal, Cancer statistics, 2023, Ca-Cancer J. Clin., 2023, 73, 17–48, DOI:10.3322/caac.21763.
- J. Ferlay, I. Soerjomataram, R. Dikshit, S. Eser, C. Mathers, M. Rebelo, D. M. Parkin, D. Forman and F. Bray, Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012, Int. J. Cancer, 2015, 136, 359–386, DOI:10.1002/ijc.29210.
- L. K. Penny and H. M. Wallace, The challenges for cancer chemoprevention, Chem. Soc. Rev., 2015, 44, 8836–8847, 10.1039/c5cs00705d.
- K. T. Jha, A. Shome, Chahat and P. A. Chawla, Recent advances in nitrogen-containing heterocyclic compounds as receptor tyrosine kinase inhibitors for the treatment of cancer: biological activity and structural activity relationship, Bioorg. Chem., 2023, 138, 106680, DOI:10.1016/j.bioorg.2023.106680.
- O. M. Soltan, M. E. Shoman, S. A. Abdel-Aziz, A. Narumi, H. Konno and M. Abdel-Aziz, Molecular hybrids: a five-year survey on structures of multiple targeted hybrids of protein kinase inhibitors for cancer therapy, Eur. J. Med. Chem., 2021, 225, 113768, DOI:10.1016/j.ejmech.2021.113768.
- D. R. Robinson, Y.-M. Wu and S.-F. Lin, The protein tyrosine kinase family of the human genome, Oncogene, 2000, 19, 5548–5557, DOI:10.1038/sj.onc.1203957.
- D. Mondal, S. Abdul Amin, M. Moinul, K. Das, T. Jha and S. Gayen, How the structural properties of the indole derivatives are important in kinase targeted drug design?: a case study on tyrosine kinase inhibitors, Bioorg. Med. Chem., 2022, 53, 116534, DOI:10.1016/j.bmc.2021.116534.
- A. M. Srour, S. S. Panda, A. Mostafa, W. Fayad, M. A. El-Manawaty, A. A. F. Soliman, Y. Moatasim, A. El Taweel, M. F. Abdelhameed, M. S. Bekheit, M. A. Ali and A. S. Girgis, Synthesis of aspirin-curcumin mimic conjugates of potential antitumor and anti-SARS-CoV-2 properties, Bioorg. Chem., 2021, 117, 105466, DOI:10.1016/j.bioorg.2021.105466.
- X. Liang, Q. Yang, P. Wu, C. He, L. Yin, F. Xu, Z. Yin, G. Yue, Y. Zou, L. Li, X. Song, C. Lv, W. Zhang and B. Jing, The synthesis review of the approved Tyrosine kinase inhibitors for anticancer therapy in 2015–2020, Bioorg. Chem., 2021, 113, 105011, DOI:10.1016/j.bioorg.2021.105011.
- A. M. Abuzenadah, F. Al-Sayes, S. S. M. Alam, M. Hoque, S. Karim, I. M. R. Hussain and S. Tabrez, Elucidating antiangiogenic potential of Rauwolfia serpentina: VEGFR-2 targeting-based molecular docking study, Evidence-Based Complementary Altern. Med., 2022, 6224666, DOI:10.1155/2022/6224666.
- T. A. Farghaly, W. A. Al-Hasani and H. G. Abdulwahab, An updated patent review of VEGFR-2 inhibitors (2017-present), Expert Opin. Ther. Pat., 2021, 31, 989–1007, DOI:10.1080/13543776.2021.1935872.
- S. Török and B. Döme, Possibilities for inhibiting tumor-induced angiogenesis: results with multi-target tyrosine kinase inhibitors, Magy. Onkol., 2012, 56, 3–15 Search PubMed.
- I. A. Seliem, S. S. Panda, A. S. Girgis, Q. L. Tran, M. F. Said, M. S. Bekheit, A. Abdelnaser, S. Nasr, W. Fayad, A. A. F. Soliman, R. Sakhuja, T. S. Ibrahim, Z. K. M. Abdel-Samii and A. M. M. Al-Mahmoudy, Development of isatin-based Schiff bases targeting VEGFR-2 inhibition: synthesis, characterization, antiproliferative properties, and QSAR studies, ChemMedChem, 2022, 17, e202200164, DOI:10.1002/cmdc.202200164.
- M. Sabbah, A. Najem, M. Krayem, A. Awada, F. Journe and G. E. Ghanem, RTK inhibitors in melanoma: from bench to bedside, Cancers, 2021, 13, 1685, DOI:10.3390/cancers13071685.
- A. Weiss, D. Bonvin, R. H. Berndsen, E. Scherrer, T. J. Wong, P. J. Dyson, A. W. Griffioen and P. Nowak-Sliwinska, Angiostatic treatment prior to chemo- or photodynamic therapy improves anti-tumor efficacy, Sci. Rep., 2015, 5, 8990, DOI:10.1038/srep08990.
- P. R. Massey, J. S. Okman, J. Wilkerson and E. W. Cowen, Tyrosine kinase inhibitors directed against the vascular endothelial growth factor receptor (VEGFR) have distinct cutaneous toxicity profiles: a meta-analysis and review of the literature, Support. Care Cancer, 2015, 23, 1827–1835, DOI:10.1007/s00520-014-2520-9.
- N. G. Fawazy, S. S. Panda, A. Mostafa, B. M. Kariuki, M. S. Bekheit, Y. Moatasim, O. Kutkat, W. Fayad, M. A. El-Manawaty, A. A. F. Soliman, R. A. El-Shiekh, A. M. Srour, R. F. Barghash and A. S. Girgis, Development of spiro-3-indolin-2-one containing compounds of antiproliferative and anti-SARS-CoV-2 properties, Sci. Rep., 2022, 12, 13880, DOI:10.1038/s41598-022-17883-9.
- R. Begum, A. N. M. Mamun-Or-Rashid, T. T. Lucy, K. Pramanik, B. K. Sil, N. Mukerjee, P. Tagde, M. Yagi and Y. Yonei, Potential therapeutic approach of melatonin against omicron and some other variants of SARS-CoV-2, Molecules, 2022, 27, 6934, DOI:10.3390/molecules27206934.
- A. S. Girgis, S. S. Panda, B. M. Kariuki, M. S. Bekheit, R. F. Barghash and D. R. Aboshouk, Indole-Based Compounds as Potential Drug Candidates for SARS-CoV-2, Molecules, 2023, 28, 6603, DOI:10.3390/molecules28186603.
- R. J. Reiter, R. Sharma, F. Simko, A. Dominguez-Rodriguez, J. Tesarik, R. L. Neel, A. T. Slominski, K. Kleszczynski, V. M. Martin-Gimenez, W. Manucha and D. P. Cardinali, Melatonin: highlighting its use as a potential treatment for SARS-CoV-2 infection, Cell. Mol. Life Sci., 2022, 79, 143, DOI:10.1007/s00018-021-04102-3.
- G. García, M. Rodriguez-Rubio, A. R. Mariblanca, L. M. de Soto, L. D. García, J. M. Villatoro, J. Q. Parada, E. S. Meseguer, M. J. Rosales, J. González, J. R. Arribas, A. J. Carcas, P. de la Oliva and A. M. Borobia, A randomized multicenter clinical trial to evaluate the efficacy of melatonin in the prophylaxis of SARS-CoV-2 infection in high-risk contacts (MeCOVID Trial): a structured summary of a study protocol for a randomised controlled trial, Trials, 2020, 21, 466, DOI:10.1186/s13063-020-04436-6.
- Z. T. Hasan, M. Q. Y. M. A. Al Atrakji and A. K. Mehuaiden, The effect of Melatonin on thrombosis, sepsis and mortality rate in COVID-19 patients, Int. J. Infect. Dis., 2022, 114, 79–84, DOI:10.1016/j.ijid.2021.10.012.
- https://go.drugbank.com/drugs/DB08839, (accessed on Dec. 11, 2023).
- https://www.drugs.com/history/tivorbex.html, (accessed on Dec. 11, 2023).
- R. M. Bokhtia, S. S. Panda, A. S. Girgis, N. Samir, M. F. Said, A. Abdelnaser, S. Nasr, M. S. Bekheit, A. S. Dawood, H. Sharma, M. Wade, S. K. Sharma and A. M. Ghanim, New NSAID conjugates as potent and selective COX-2 inhibitors: synthesis, molecular modeling and biological investigation, Molecules, 2023, 28, 1945, DOI:10.3390/molecules28041945.
- https://www.drugs.com/dosage/delavirdine.html, (accessed on Dec. 11, 2023).
- G. Li, Y. Wang and E. De Clercq, Approved HIV reverse transcriptase inhibitors in the past decade, Acta Pharm. Sin. B, 2022, 12, 1567–1590, DOI:10.1016/j.apsb.2021.11.009.
- https://go.drugbank.com/drugs/DB13609, (accessed on Dec. 11, 2023).
- https://www.drugs.com/international/umifenovir.html, (accessed on Dec. 11, 2023).
- A. Shuster, D. Pechalrieu, C. B. Jackson, D. Abegg, H. Choe and A. Adibekian, Clinical antiviral drug arbidol inhibits infection by SARS-CoV-2 and variants through direct binding to the spike protein, ACS Chem. Biol., 2021, 16, 2845–2851, DOI:10.1021/acschembio.1c00756.
- X. Wang, R. Cao, H. Zhang, J. Liu, M. Xu, H. Hu, Y. Li, L. Zhao, W. Li, X. Sun, X. Yang, Z. Shi, F. Deng, Z. Hu, W. Zhong and M. Wang, The anti-influenza virus drug, arbidol is an efficient inhibitor of SARS-CoV-2 in vitro, Cell Discovery, 2020, 6, 28, DOI:10.1038/s41421-020-0169-8.
- https://www.drugs.com/mtm/pindolol.html, (accessed on Dec. 11, 2023).
- https://www.drugs.com/maxalt.html, (accessed on Dec. 11, 2023).
- B. Yu, D.-Q. Yu and H.-M. Liu, Spirooxindoles: promising scaffolds for anticancer agents, Eur. J. Med. Chem., 2015, 97, 673–698, DOI:10.1016/j.ejmech.2014.06.056.
- J. Jin, Y. Xie, J.-S. Zhang, J.-Q. Wang, S.-J. Dai, W.-f. He, S.-Y. Li, C. R. Ashby Jr., Z.-S. Chen and Q. He, Sunitinib resistance in renal cell carcinoma: from molecular mechanisms to predictive biomarkers, Drug Resistance Updates, 2023, 67, 100929, DOI:10.1016/j.drup.2023.100929.
- F. M. Guida, M. Santoni, A. Conti, L. Burattini, A. Savini, T. Zeppola, M. Caricato, S. Cascinu, G. Tonini and D. Santini, Alternative dosing schedules for sunitinib as a treatment of patients with metastatic renal cell carcinoma, Crit. Rev. Oncol. Hematol., 2014, 92, 208–217, DOI:10.1016/j.critrevonc.2014.07.006.
- L. Zhang, Q. Zheng, Y. Yang, H. Zhou, X. Gong, S. Zhao and C. Fan, Synthesis and in vivo SAR study of indolin-2-one-based multi-targeted inhibitors as potential anticancer agents, Eur. J. Med. Chem., 2014, 82, 139–151, DOI:10.1016/j.ejmech.2014.05.051.
- https://go.drugbank.com/drugs/DB01268, (accessed on Dec. 11, 2023).
- https://www.drugs.com/history/sutent.html, (accessed on Dec. 11, 2023).
- X. Zheng, Y. Zhang, L. Zhang, W. Xu, W. Ma, R. Sun and H. Zeng, Synergistic inhibition of sunitinib and ethaselen against human colorectal cancer cells proliferation, Biomed. Pharmacother., 2016, 83, 212–220, DOI:10.1016/j.biopha.2016.06.040.
- L. A. Leon and L. M. Antón-Aparicio, Treatment of sunitinib-induced hypertension in solid tumors by nitric oxid donors, Redox Biol., 2015, 5, 413–423, DOI:10.1016/j.redox.2015.09.036.
- A. M. J. Thijs, C. M. L. van Herpen, V. Verweij, J. Pertijs, P. H. H. van den Broek, W. T. A. van der Graaf and G. A. Rongen, Impaired endothelium-dependent vasodilation does not initiate the development of sunitinib-associated hypertension, J. Hypertens., 2015, 33, 2075–2082, DOI:10.1097/HJH.0000000000000662.
- L. Q. Chow and S. G. Eckhardt, Sunitinib: from rational design to clinical efficacy, J. Clin. Oncol., 2007, 25, 884–896, DOI:10.1200/JCO.2006.06.3602.
- J.-Y. Li, Y.-P. Ren, Y. Yuan, S.-M. JI, S.-P. Zhou, L.-J. Wang, Z.-Z. Mou, L. Li, W. Lu and T.-Y. Zhou, Preclinical PK/PD model for combined administration of erlotinib and sunitinib in the treatment of A549 human NSCLC xenograft mice, Acta Pharmacol. Sin., 2016, 37, 930–940, DOI:10.1038/aps.2016.55.
- G. L. Gravina, A. Mancini, F. Marampon, A. Colapietro, S. D. Monache, R. Sferra, F. Vitale, P. J. Richardson, L. Patient, S. Burbidge and C. Festuccia, The brain-penetrating CXCR4 antagonist, PRX177561, increases the antitumor effects of bevacizumab and sunitinib in preclinical models of human glioblastoma, J. Hematol. Oncol., 2017, 10, 5, DOI:10.1186/s13045-016-0377-8.
- G. L. Gravina, A. Mancini, A. Colapietro, F. Vitale, A. Vetuschi, S. Pompili, G. Rossi, F. Marampon, P. J. Richardson, L. Patient, L. Patient, S. Burbidge and C. Festuccia, The novel CXCR4 antagonist, PRX177561, reduces tumor cell proliferation and accelerates cancer stem cell differentiation in glioblastoma preclinical models, Tumour Biol., 2017, 39 DOI:10.1177/1010428317695528.
- X. Wang, L. Zhang, A. O'Neill, B. Bahamon, D. C. Alsop, J. W. Mier, S. N. Goldberg, S. Signoretti, M. B. Atkins, C. G. Wood and R. S. Bhatt, Cox-2 inhibition enhances the activity of sunitinib in human renal cell carcinoma xenografts, Br. J. Cancer, 2013, 108, 319–326, DOI:10.1038/bjc.2012.591.
- A. Jamadar, S. M. Suma, S. Mathew, T. A. Fields, D. P. Wallace, J. P. Calvet and R. Rao, The tyrosine-kinase inhibitor Nintedanib ameliorates autosomal-dominant polycystic kidney disease, Cell Death Dis., 2021, 12, 947, DOI:10.1038/s41419-021-04248-9.
- C. Landi, L. Bergantini, P. Cameli, M. d'Alessandro, A. Carleo, E. Shaba, P. Rottoli, L. Bini and E. Bargagli, Idiopathic pulmonary fibrosis serum proteomic analysis before and after nintedanib therapy, Sci. Rep., 2020, 10, 9378, DOI:10.1038/s41598-020-66296-z.
- F. Hilberg, U. Tontsch-Grunt, A. Baum, A. T. Le, R. C. Doebele, S. Lieb, D. Gianni, T. Voss, P. Garin-Chesa, C. Haslinger and N. Kraut, Triple angiokinase inhibitor Nintedanib directly inhibits tumor cell growth and induces tumor shrinkage via blocking oncogenic receptor tyrosine kinases, J. Pharmacol. Exp. Ther., 2018, 364, 494–503, DOI:10.1124/jpet.117.244129.
- M. C. Riesco-Martinez, A. S. Torre and R. García-Carbonero, Safety and efficacy of nintedanib for the treatment of metastatic colorectal cancer, Expert Opin. Invest. Drugs, 2017, 26, 1295–1305, DOI:10.1080/13543784.2017.1385762.
- R. Kurzrock and D. J. Stewart, Exploring the benefit/risk associated with antiangiogenic agents for the treatment of non–small cell lung cancer patients, Clin. Cancer Res., 2017, 23, 1137–1148, DOI:10.1158/1078-0432.CCR-16-1968.
- https://go.drugbank.com/drugs/DB09079, (accessed on Dec. 11, 2023).
- https://www.drugs.com/history/ofev.html, (accessed on Dec. 11, 2023).
- R. F. da Silva, T. P. Banzato, L. F. Alves, J. E. Carvalho, R. Agarwal and V. H. A. Cagnon, Antiangiogenic therapy with Nintedanib affects hypoxia, angiogenesis and apoptosis in the ventral prostate of TRAMP animals, Cell Tissue Res., 2020, 379, 407–420, DOI:10.1007/s00441-019-03091-x.
- R. F. da Silva, E. Nogueira-Pangrazi, L. A. Kido, F. Montico, S. Arana, D. Kumar, K. Raina, R. Agarwal and V. H. A. Cagnon, Nintedanib antiangiogenic inhibitor effectiveness in delaying adenocarcinoma progression in Transgenic Adenocarcinoma of the Mouse Prostate (TRAMP), J. Biomed. Sci., 2017, 24, 31, DOI:10.1186/s12929-017-0334-z.
- S. Khalique and S. Banerjee, Nintedanib in ovarian cancer, Expert Opin. Invest. Drugs, 2017, 26, 1073–1081, DOI:10.1080/13543784.2017.1353599.
- G. J. Roth, R. Binder, F. Colbatzky, C. Dallinger, R. Schlenker-Herceg, F. Hilberg, S.-L. Wollin and R. Kaiser, Nintedanib: from discovery to the clinic, J. Med. Chem., 2015, 58, 1053–1063, DOI:10.1021/jm501562a.
- A. Rossi, T. P. Latiano, P. Parente, C. Chiarazzo, F. Limosani, G. Di Maggio and E. Maiello, The potential role of Nintedanid in treating colorectal cancer, Expert Opin, Pharmacother., 2017, 18, 1153–1162, DOI:10.1080/14656566.2017.1346086.
- Y. Kim, S. J. Lee, J. Y. Lee, S.-H. Lee, J.-M. Sun, K. Park, H. J. An, J. Y. Cho, E. J. Kang, H.-Y. Lee, J. Kim, B. Keam, H. R. Kim, K. E. Lee, M. Y. Choi, K. H. Lee and M.-J. Ahn, Clinical trial of Nintedanib in patients with recurrent or metastatic salivary gland cancer of the head and neck: a multicenter phase 2 study (Korean Cancer Study Group HN14-01), Cancer, 2017, 123, 1958–1964, DOI:10.1002/cncr.30537.
- C. Schliemann, J. Gerss, S. Wiebe, J.-H. Mikesch, N. Knoblauch, T. Sauer, L. Angenendt, T. Kewitz, M. Urban, T. Butterfass-Bahloul, S. Edemir, K. Vehring, C. Müller-Tidow, W. E. Berdel and U. Krug, A phase I dose escalation study of the triple angiokinase inhibitor Nintedanib combined with low-dose cytarabine in elderly patients with acute myeloid leukemia, PLoS One, 2016, 11, e0164499, DOI:10.1371/journal.pone.0164499.
- https://go.drugbank.com/drugs/DB11885, (accessed on Dec. 11, 2023).
- L. Xie, Y. Feng and J. Sun, Fingerprint loss during combination therapy using osimertinib and anlotinib: a case report, J. Clin. Pharm. Ther., 2022, 47, 248–250, DOI:10.1111/jcpt.13480.
- J. Liang, Z. Jin, J. Kuang, H. Feng, Q. Zhao, Z. Yang, L. Zhan, B. Shen, J. Yan, W. Cai, X. Cheng and W. Qiu, The role of anlotinib-mediated EGFR blockade in a positive feedback loop of CXCL11-EGF-EGFR signalling in anaplastic thyroid cancer angiogenesis, Br. J. Cancer, 2021, 125, 390–401, DOI:10.1038/s41416-021-01340-x.
- S. Li, Anlotinib: a novel targeted drug for bone and soft tissue sarcoma, Front. Oncol., 2021, 11, 664853, DOI:10.3389/fonc.2021.664853.
- G. Shen, F. Zheng, D. Ren, F. Du, Q. Dong, Z. Wang, F. Zhao, R. Ahmad and J. Zhao, Anlotinib: a novel multi-targeting tyrosine kinase inhibitor in clinical development, J. Hematol. Oncol., 2018, 11, 120, DOI:10.1186/s13045-018-0664-7.
- Y. Y. Syed, Anlotinib: first global approval, Drugs, 2018, 78, 1057–1062, DOI:10.1007/s40265-018-0939-x.
- T. Qin, Z. Liu, J. Wang, J. Xia, S. Liu, Y. Jia, H. Liu and K. Li, Anlotinib suppresses lymphangiogenesis and lymphatic metastasis in lung adenocarcinoma through a process potentially involving VEGFR-3 signaling, Cancer Biol. Med., 2020, 17, 753–767, DOI:10.20892/j.issn.2095-3941.2020.0024.
- Z.-X. Jia, Z. Zhang, Z. Li, A. Li, Y.-N. Xie, H.-J. Wu, Z.-B. Yang, H.-M. Zhang and X.-M. Zhang, Anlotinib inhibits the progress of colorectal cancer cells by antagonizing VEGFR/JAK2/STAT3 axis, Eur. Rev. Med. Pharmacol. Sci., 2021, 25, 2331–2343, DOI:10.26355/eurrev_202103_25272.
- D. Li, Y. Chi, X. Chen, M. Ge, Y. Zhang, Z. Guo, J. Wang, J. Chen, J. Zhang, Y. Cheng, Z. Li, H. Liu, J. Qin, J. Zhu, R. Cheng, Z. Xu, X. Zheng, P. Tang and M. Gao, Anlotinib in locally advanced or metastatic medullary thyroid carcinoma: a randomized, double-blind phase IIB trial, Clin. Cancer Res., 2021, 27, 3567–3575, DOI:10.1158/1078-0432.CCR-20-2950.
- Y. Liu, Q. Xiao, J. He, H. Hu, J. Du, Y. Zhu, J. Chen, Z. Liu, J. Wang, L. Sun, D. Xu, J. Li, X. Liao, J. Wang, Y. Cai, C. Cai, Z. Jin, L. Wang, Y. Yuan and K. Ding, Phase II study of anlotinib in combination with oxaliplatin and capecitabine for patients with RAS/BRAF wild-type metastatic colorectal adenocarcinoma as the first-line therapy, BMC Med., 2022, 20, 155, DOI:10.1186/s12916-022-02357-6.
- Y. Y. Syed, Surufatinib: first approval, Drugs, 2021, 81, 727–732, DOI:10.1007/s40265-021-01489-y.
- https://www.drugs.com/history/surufatinib.html, (accessed on Dec. 11, 2023).
- X. Lu, S. Yan, K. A. Koral and Z. Chen, Surufatinib for the treatment of advanced extrapancreatic neuroendocrine tumors, Expert Rev. Anticancer Ther., 2021, 21, 917–926, DOI:10.1080/14737140.2021.1944110.
- J. Xu, Y. Bai, H. Sun, C. Bai, R. Jia, Y. Li, W. Zhang, L. Liu, C. Huang, M. Guan, J. Zhou and W. Su, A single-arm, multicenter, open-label phase 2 trial of Surufatinib in patients with unresectable or metastatic biliary tract cancer, Cancer, 2021, 127, 3975–3984, DOI:10.1002/cncr.33803.
- https://go.drugbank.com/articles/A24188, (accessed on Dec. 11, 2023).
- X. Huang, J. Zhu, Y. Jiang, C. Xu, Q. Lv, D. Yu, K. Shi, Z. Ruan and Y. Wang, SU5416 attenuated lipopolysaccharide-induced acute lung injury in mice by modulating properties of vascular endothelial cells, Drug Des., Dev. Ther., 2019, 13, 1763–1772, DOI:10.2147/DDDT.S188858.
- G. L. Peloquin, L. Johnston, M. Damarla, R. L. Damico, P. M. Hassoun and T. M. Kolb, SU5416 does not attenuate early RV angiogenesis in the murine chronic hypoxia PH model, Respir. Res., 2019, 20, 123, DOI:10.1186/s12931-019-1079-x.
- https://go.drugbank.com/drugs/DB15247, (accessed on Dec. 11, 2023).
- K. S. Pedersen, P. M. Grierson, J. Picus, A. C. Lockhart, B. J. Roth, J. Liu, A. Morton, E. Chan, J. Huffman, C. Liang, A. Wang-Gillam and B. Tan, Vorolanib (X-82), an oral anti-VEGFR/PDGFR/CSF1R tyrosine kinase inhibitor, with everolimus in solid tumors: results of a phase I study, Invest. New Drugs, 2021, 39, 1298–1305, DOI:10.1007/s10637-021-01093-7.
- K. Zhang, L. Wang, A. Wei, X. Jia and X. Liu, CM082, a novel angiogenesis inhibitor, enhances the antitumor activity of gefitinib on epidermal growth factor receptor mutant non-small cell lung cancer in vitro and in vivo, Thorac. Cancer, 2020, 11, 1566–1577, DOI:10.1111/1759-7714.13430.
- https://go.drugbank.com/drugs/DB11741, (accessed on Dec. 11, 2023).
- M. Zhang, H. Quan, L. Fu, Y. Li, H. Fu and L. Lou, Third-generation EGFR inhibitor HS-10296 in combination with famitinib, a multi-targeted tyrosine kinase inhibitor, exerts synergistic antitumor effects through enhanced inhibition of downstream signaling in EGFR-mutant non-small cell lung cancer cells, Thorac. Cancer, 2021, 12, 1210–1218, DOI:10.1111/1759-7714.13902.
- R. Sánchez-Céspedes, P. Accornero, S. Miretti, E. Martignani, F. Gattino, L. Maniscalco, C. Gola, S. Iussich, M. Martano, E. Morello, P. Buracco, L. Aresu and R. De Maria, In vitro and in vivo effects of toceranib phosphate on canine osteosarcoma cell lines and xenograft orthotopic models, Vet. Comp. Oncol., 2020, 18, 117–127, DOI:10.1111/vco.12562.
- https://go.drugbank.com/drugs/DB16821, (accessed on Dec. 11, 2023).
- M. F. Burbridge, C. J. Bossard, C. Saunier, I. Fejes, A. Bruno, S. Léonce, G. Ferry, G. Da Violante, F. Bouzom, V. Cattan, A. Jacquet-Bescond, P. M. Comoglio, B. P. Lockhart, J. A. Boutin, A. Cordi, J.-C. Ortuno, A. Pierré, J. A. Hickman, F. H. Cruzalegui and S. Depil, S49076 is a novel kinase inhibitor of MET, AXL, and FGFR with strong preclinical activity alone and in association with bevacizumab, Mol. Cancer Ther., 2013, 12, 1749–1762, DOI:10.1158/1535-7163.MCT-13-0075.
- J. Rodon, S. Postel-Vinay, A. Hollebecque, P. Nuciforo, A. Azaro, V. Cattan, L. Marfai, I. Sudey, K. Brendel, A. Delmas, S. Malasse and J.-C. Soria, First-in-human phase I study of oral S49076, a unique MET/AXL/FGFR inhibitor, in advanced solid tumours, Eur. J. Cancer, 2017, 81, 142–150, DOI:10.1016/j.ejca.2017.05.007.
- D. Wang, F. Tang, S. Wang, Z. Jiang and L. Zhang, Preclinical anti-angiogenesis and anti-tumor activity of SIM010603, an oral, multi-targets receptor tyrosine kinases inhibitor, Cancer Chemother. Pharmacol., 2012, 69, 173–183, DOI:10.1007/s00280-011-1681-1.
- Y. Mao, Z. Xia, X. Zhang, Y. Zong, L. Zhu, B. Yuan and G. Lu, Evaluation of subchronic toxicity of SIM010603, a potent inhibitor of receptor tyrosine kinase, after 28-day repeated oral administration in SD rats and beagle dogs, Food Chem. Toxicol., 2012, 50, 1256–1270, DOI:10.1016/j.fct.2012.02.007.
- A. S. Girgis, S. S. Panda, A. M. Srour, A. Abdelnaser, S. Nasr, Y. Moatasim, O. Kutkat, A. El Taweel, A. Kandeil, A. Mostafa, M. A. Ali, N. G. Fawzy, M. S. Bekheit, E. M. Shalaby, L. Gigli, W. Fayad and A. A. F. Soliman, 3-Alkenyl-2-oxindoles: synthesis, antiproliferative and antiviral properties against SARS-CoV-2, Bioorg. Chem., 2021, 114, 105131, DOI:10.1016/j.bioorg.2021.105131.
- https://www.drugs.com/mtm/vincristine.html, (accessed on Dec. 11, 2023).
- https://go.drugbank.com/drugs/DB00541, (accessed on Dec. 11, 2023).
- D. Mondal, S. Abdul Amin, M. Moinul, K. Das, T. Jha and S. Gayen, How the structural properties of the indole derivatives are important in kinase targeted drug design?: a case study on tyrosine kinase inhibitors, Bioorg. Med. Chem., 2022, 53, 116534, DOI:10.1016/j.bmc.2021.116534.
- https://www.drugs.com/mtm/vinblastine.html, (accessed on Dec. 11, 2023).
- https://go.drugbank.com/drugs/DB00570, (accessed on Dec. 11, 2023).
- https://go.drugbank.com/drugs/DB00309, (accessed on Dec. 11, 2023).
- M. Gallardo-Fernández, A. B. Cerezo, R. Hornedo-Ortega, A. M. Troncoso and M. C. Garcia-Parrilla, Anti-VEGF effect of bioactive indolic compounds and hydroxytyrosol metabolites, Foods, 2022, 11, 526, DOI:10.3390/foods11040526.
- L. H. Al-Wahaibi, A. F. Mohammed, M. H. Abdelrahman, L. Trembleau and B. G. M. Youssif, Design, synthesis, and biological evaluation of indole-2-carboxamides as potential multi-target antiproliferative agents, Pharmaceuticals, 2023, 16, 1039, DOI:10.3390/ph16071039.
- A. Elkamhawy, U. M. Ammar, S. Paik, M. H. Abdellattif, M. H. Elsherbeny, K. Lee and E. J. Roh, Scaffold repurposing of in-house small molecule candidates leads to discovery of first-in-class CDK-1/HER-2 dual inhibitors: in vitro and in silico screening, Molecules, 2021, 26, 5324, DOI:10.3390/molecules26175324.
- A. Elkamhawy, S. Paik, H. J. Kim, J.-H. Park, A. M. Londhe, K. Lee, A. N. Pae, K. D. Park and E. J. Roh, Discovery of N-(1-(3-fluorobenzoyl)-1H-indol-5-yl)pyrazine-2-carboxamide: a novel, selective, and competitive indole-based lead inhibitor for human monoamine oxidase B, J. Enzyme Inhib. Med. Chem., 2020, 35, 1568–1580, DOI:10.1080/14756366.2020.1800666.
- M. A. Abdelgawad, A. M. Hayallah, S. N. A. Bukhari, A. Musa, M. Elmowafy, H. M. Abdel-Rahman and M. K. Abd El-Gaber, Design, synthesis, molecular modeling, and anticancer evaluation of new VEGFR-2 inhibitors based on the indolin-2-One scaffold, Pharmaceuticals, 2022, 15, 1416, DOI:10.3390/ph15111416.
- W. M. Eldehna, M. Fares, H. S. Ibrahim, M. H. Aly, S. Zada, M. M. Ali, S. M. Abou-Seri, H. A. Abdel-Aziz and D. A. Abou El Ella, Indoline ureas as potential anti-hepatocellular carcinoma agents targeting VEGFR-2: synthesis, in vitro biological evaluation and molecular docking, Eur. J. Med. Chem., 2015, 100, 89–97, DOI:10.1016/j.ejmech.2015.05.040.
- https://www.cancer.gov/about-cancer/treatment/drugs/fluorouracil, (accessed on Dec. 11, 2023).
- E. B. Elkaeed, R. G. Yousef, H. Elkady, I. M. M. Gobaara, A. A. Alsfouk, D. Z. Husein, I. M. Ibrahim, A. M. Metwaly and I. H. Eissa, The assessment of anticancer and VEGFR-2 inhibitory activities of a new 1H-indole derivative: in silico and in vitro approaches, Processes, 2022, 10, 1391, DOI:10.3390/pr10071391.
- S. Saied, M. Shaldam, M. M. Elbadawi, S. Giovannuzzi, A. Nocentini, H. Almahli, R. Salem, T. M. Ibrahim, C. T. Supuran and W. M. Eldehna, Discovery of indolinone-bearing benzenesulfonamides as new dual carbonic anhydrase and VEGFR-2 inhibitors possessing anticancer and pro-apoptotic properties, Eur. J. Med. Chem., 2023, 259, 115707, DOI:10.1016/j.ejmech.2023.115707.
- O. M. Hassan, A. A. R. Mahmood, A. H. Hamzah and L. H. Tahtamouni, Design, synthesis, and molecular docking studies of 5-bromoindole-2-carboxylic acid hydrazone derivatives: in vitro anticancer and VEGFR-2 inhibitory effects, ChemistrySelect, 2022, 7, e202203726, DOI:10.1002/slct.202203726.
- J. Zhang, T. Liu, M. Chen, F. Liu, X. Liu, J. Zhang, J. Lin and Y. Jin, Synthesis and biological evaluation of indole-2-carbohydrazide derivatives as anticancer agents with anti-angiogenic and antiproliferative activities, ChemMedChem, 2018, 13, 1181–1192, DOI:10.1002/cmdc.201800033.
- D.-P. Wang, K.-L. Liu, X.-Y. Li, G.-Q. Lu, W.-H. Xue, X.-H. Qian, K. Mohamed and F.-H. Meng, Design, synthesis, and in vitro and in vivo anti-angiogenesis study of a novel vascular endothelial growth factor receptor-2 (VEGFR-2) inhibitor based on 1,2,3-triazole scaffold, Eur. J. Med. Chem., 2021, 211, 113083, DOI:10.1016/j.ejmech.2020.113083.
- M. Qin, S. Yan, L. Wang, H. Zhang, Y. Tian, Y. Zhao and P. Gong, Novel 6-methoxycarbonyl indolinones bearing a pyrrole Mannich base moiety as angiokinase inhibitors, Bioorg. Med. Chem., 2017, 25, 1778–1786, DOI:10.1016/j.bmc.2017.01.039.
- M. Qin, Y. Tian, X. Sun, S. Yu, J. Xia, P. Gong, H. Zhang and Y. Zhao, Novel methyl indolinone-6-carboxylates containing an indole moiety as angiokinase inhibitors, Eur. J. Med. Chem., 2017, 139, 492–502, DOI:10.1016/j.ejmech.2017.08.031.
- T.-H. Yang, C.-I. Lee, W.-H. Huang and A.-R. Lee, Synthesis and evaluation of novel 2-pyrrolidone-fused (2-oxoindolin-3-ylidene)methylpyrrole derivatives as potential multi-target tyrosine kinase receptor inhibitors, Molecules, 2017, 22, 913, DOI:10.3390/molecules22060913.
- L. Ding, F. Tang, W. Huang, Q. Jin, H. Shen and P. Wei, Design, synthesis, and biological evaluation of novel 3-pyrrolo[b]cyclohexylene-2-dihydroindolinone derivatives as potent receptor tyrosine kinase inhibitors, Bioorg. Med. Chem. Lett., 2013, 23, 5630–5633, DOI:10.1016/j.bmcl.2013.08.037.
- M. H. Kim, A. L. Tsuhako, E. W. Co, D. T. Aftab, F. Bentzien, J. Chen, W. Cheng, S. Engst, L. Goon, R. R. Klein, D. T. Le, M. Mac, J. J. Parks, F. Qian, M. Rodriquez, T. J. Stout, J. H. Till, K.-A. Won, X. Wu, F. M. Yakes, P. Yu, W. Zhang, Y. Zhao, P. Lamb, J. M. Nuss and W. Xu, The design, synthesis, and biological evaluation of potent receptor tyrosine kinase inhibitors, Bioorg. Med. Chem. Lett., 2012, 22, 4979–4985, DOI:10.1016/j.bmcl.2012.06.029.
- S. Patel, N. Challagundla, R. A. Rajput and S. Mishra, Design, synthesis, characterization and anticancer activity evaluation of deoxycholic acid-chalcone conjugates, Bioorg. Chem., 2022, 127, 106036, DOI:10.1016/j.bioorg.2022.106036.
- R. Dudchak, M. Podolak, S. Holota, O. Szewczyk-Roszczenko, P. Roszczenko, A. Bielawska, R. Lesyk and K. Bielawski, Click chemistry in the synthesis of antibody-drug conjugates, Bioorg. Chem., 2024, 143, 106982, DOI:10.1016/j.bioorg.2023.106982.
- Z. M. Nofal, A. M. Srour, W. I. El-Eraky, D. O. Saleh and A. S. Girgis, Rational design, synthesis and QSAR study of vasorelaxant active 3-pyridinecarbonitriles incorporating 1H-benzimidazol-2-yl function, Eur. J. Med. Chem., 2013, 63, 14–21, DOI:10.1016/j.ejmech.2013.01.042.
- A. S. Girgis, A. Kalmouch and M. Ellithey, Synthesis of novel vasodilatory active nicotinate esters with amino acid function, Bioorg. Med. Chem., 2006, 14, 8488–8494, DOI:10.1016/j.bmc.2006.08.041.
- A. M. Ghanim, A. S. Girgis, B. M. Kariuki, N. Samir, M. F. Said, A. Abdelnaser, S. Nasr, M. S. Bekheit, M. F. Abdelhameed, A. J. Almalki, T. S. Ibrahim and S. S. Panda, Design and synthesis of ibuprofen-quinoline conjugates as potential anti-inflammatory and analgesic drug candidates, Bioorg. Chem., 2022, 119, 105557, DOI:10.1016/j.bioorg.2021.105557.
- S. S. Panda, A. S. Girgis, S. J. Thomas, J. E. Capito, R. F. George, A. Salman, M. A. El-Manawaty and A. Samir, Synthesis, pharmacological profile and 2D-QSAR studies of curcumin-amino acid conjugates as potential drug candidates, Eur. J. Med. Chem., 2020, 196, 112293, DOI:10.1016/j.ejmech.2020.112293.
- S. S. Panda, A. S. Girgis, H. H. Honkanadavar, R. F. George and A. M. Srour, Synthesis of new ibuprofen hybrid conjugates as potential anti-inflammatory and analgesic agents, Future Med. Chem., 2020, 12, 1369–1386, DOI:10.4155/fmc-2020-0109.
- A. D. Tiwari, S. S. Panda, A. S. Girgis, S. Sahu, R. F. George, A. M. Srour, B. La Starza, A. M. Asiri, C. D. Hall and A. R. Katritzky, Microwave assisted synthesis and QSAR study of novel NSAID acetaminophen conjugates with amino acid linkers, Org. Biomol. Chem., 2014, 12, 7238–7249, 10.1039/c4ob01281j.
- I. A. Seliem, S. S. Panda, A. S. Girgis, Y. I. Nagy, R. F. George, W. Fayad, N. G. Fawzy, T. S. Ibrahim, A. M. M. Al-Mahmoudy, R. Sakhuja and Z. K. M. Abdel-samii, Design, synthesis, antimicrobial and DNA gyrase inhibitory properties of fluoroquinolone-dichloroacetic acid hybrids, Chem. Biol. Drug Des., 2020, 95, 248–259, DOI:10.1111/cbdd.13638.
- H. M. Faidallah, A. S. Girgis, A. D. Tiwari, H. H. Honkanadavar, S. J. Thomas, A. Samir, A. Kalmouch, K. A. Alamry, K. A. Khan, T. S. Ibrahim, A. M. M. EL-Mahmoudy, A. M. Asiri and S. S. Pand, Synthesis, antibacterial properties and 2D-QSAR studies of quinolone-triazole conjugates, Eur. J. Med. Chem., 2018, 143, 1524–1534, DOI:10.1016/j.ejmech.2017.10.042.
- S. S. Panda, O. S. Detistov, A. S. Girgis, P. P. Mohapatra, A. Samir and A. R. Katritzky, Synthesis and molecular modeling of antimicrobial active fluoroquinolone–pyrazine conjugates with amino acid linkers, Bioorg. Med. Chem. Lett., 2016, 26, 2198–2205, DOI:10.1016/j.bmcl.2016.03.062.
- S. S. Panda, A. S. Girgis, B. B. Mishra, M. Elagawany, V. Devarapalli, W. F. Littlefield, A. Samir, W. Fayad, N. G. Fawzy, A. M. Srour and R. M. Bokhtia, Synthesis, computational studies, antimycobacterial and antibacterial properties of pyrazinoic acid-isoniazid hybrid conjugates, RSC Adv., 2019, 9, 20450–20462, 10.1039/c9ra03380g.
- H. M. Faidallah, S. S. Panda, J. C. Serrano, A. S. Girgis, K. A. Khan, K. A. Alamry, T. Therathanakorn, M. J. Meyers, F. M. Sverdrup, C. S. Eikhoff, S. G. Getchell and A. R. Katritzky, Synthesis, antimalarial properties and 2D-QSAR studies of novel triazole-quinine conjugates, Bioorg. Med. Chem., 2016, 24, 3527–3539, DOI:10.1016/j.bmc.2016.05.060.
- Q. Zhou, Y. Luo, Y. Zhu, Q. Chen, J. Qiu, F. Cong, Y. Li and X. Zhang, Nonsteroidal anti-inflammatory drugs (NSAIDs) and nucleotide analog GS-441524 conjugates with potent in vivo efficacy against coronaviruses, Eur. J. Med. Chem., 2023, 249, 115113, DOI:10.1016/j.ejmech.2023.115113.
- N. S. El-Sayed, A. S. Jureka, M. R. Edwards, S. Lohan, C. G. Williams, P. T. Keiser, R. A. Davey, J. Totonchy, R. K. Tiwari, C. F. Basler and K. Parang, Synthesis and antiviral activity of fatty acyl conjugates of remdesivir against severe acute respiratory syndrome coronavirus 2 and Ebola virus, Eur. J. Med. Chem., 2021, 226, 113862, DOI:10.1016/j.ejmech.2021.113862.
- I. A. Seliem, S. S. Panda, A. S. Girgis, Y. Moatasim, A. Kandeil, A. Mostafa, M. A. Ali, E. S. Nossier, F. Rasslan, A. M. Srour, R. Sakhuja, T. S. Ibrahim, Z. K. M. Abdel-samii and A. M. M. Al-Mahmoudy, New quinoline-triazole conjugates: synthesis, and antiviral properties against SARS-CoV-2, Bioorg. Chem., 2021, 114, 105117, DOI:10.1016/j.bioorg.2021.105117.
- I. A. Seliem, A. S. Girgis, Y. Moatasim, A. Kandeil, A. Mostafa, M. A. Ali, M. S. Bekheit and S. S. Panda, New pyrazine conjugates: synthesis, computational studies, and antiviral properties against SARS-CoV-2, ChemMedChem, 2021, 16, 3418–3427, DOI:10.1002/cmdc.202100476.
- K. A. Wyman, A. S. Girgis, P. S. Surapaneni, J. M. Moore, N. M. Abo Shama, S. H. Mahmoud, A. Mostafa, R. F. Barghash, Z. Juan, R. D. Dobaria, A. J. Almalki, T. S. Ibrahim and S. S. Panda, Synthesis of potential antiviral agents for SARS-CoV-2 using molecular hybridization approach, Molecules, 2022, 27, 5923, DOI:10.3390/molecules27185923.
- J. R. Hwu, M. Kapoor, N. K. Gupta, S.-C. Tsay, W.-C. Huang, K.-T. Tan, Y.-C. Hu, P. Lyssen and J. Neyts, Synthesis and antiviral activities of quinazolinamine–coumarin conjugates toward chikungunya and hepatitis C viruses, Eur. J. Med. Chem., 2022, 232, 114164, DOI:10.1016/j.ejmech.2022.114164.
- N. Mishriky, F. M. Asaad, Y. A. Ibrahim and A. S. Girgis, New 2-pyrazolines of anticipated molluscicidal activity, Pharmazie, 1996, 51, 544–548 CAS.
- M. Chen, L. Cai, Y. Xiang, L. Zhong and J. Shi, Advances in non-radioactive PSMA-targeted small molecule-drug conjugates in the treatment of prostate cancer, Bioorg. Chem., 2023, 141, 106889, DOI:10.1016/j.bioorg.2023.106889.
- M. A. Youssef, S. S. Panda, D. R. Aboshouk, M. F. Said, A. El Taweel, M. GabAllah, W. Fayad, A. A. F. Soliman, A. Mostafa, N. G. Fawzy and A. S. Girgis, Novel curcumin mimics: design, synthesis, biological properties and computational studies of piperidone–piperazine conjugates, ChemistrySelect, 2022, 7, e202201406, DOI:10.1002/slct.202201406.
- S. A. Al-Hussain, T. A. Farghaly, M. E. A. Zaki, H. G. Abdulwahab, N. T. Al-Qurashi and Z. A. Muhammad, Discovery of novel indolyl-1,2,4-triazole hybrids as potent vascular endothelial growth factor receptor-2 (VEGFR-2) inhibitors with potential anti-renal cancer activity, Bioorg. Chem., 2020, 105, 104330, DOI:10.1016/j.bioorg.2020.104330.
- M. S. Nafie and A. T. A. Boraei, Exploration of novel VEGFR2 tyrosine kinase inhibitors via design and synthesis of new alkylated indolyl-triazole Schiff bases for targeting breast cancer, Bioorg. Chem., 2022, 122, 105708, DOI:10.1016/j.bioorg.2022.105708.
- C. Zhang, D. Xu, J. Wang and C. Kang, Efficient synthesis and biological activity of novel indole derivatives as VEGFR-2 tyrosine kinase inhibitors, Russ. J. Gen. Chem., 2017, 87, 3006–3016, DOI:10.1134/S1070363217120465.
- https://www.drugs.com/history/votrient.html, (accessed on Dec. 11, 2023).
- https://go.drugbank.com/drugs/DB06589, (accessed on Dec. 11, 2023).
- G.-R. Gao, M.-Y. Li, L.-J. Tong, L.-X. Wei, J. Ding, H. Xie and W.-H. Duan, Design, synthesis and biological evaluation of O-linked indoles as VEGFR-2 kinase inhibitors (I), Chin. Chem. Lett., 2015, 26, 1165–1168, DOI:10.1016/j.cclet.2015.07.016.
- S. Sana, V. G. Reddy, S. Bhandari, T. S. Reddy, R. Tokala, A. P. Sakla, S. K. Bhargava and N. Shankaraiah, Exploration of carbamide derived pyrimidine-thioindole conjugates as potential VEGFR-2 inhibitors with anti-angiogenesis effect, Eur. J. Med. Chem., 2020, 200, 112457, DOI:10.1016/j.ejmech.2020.112457.
- K. Ravi, B. N. Dhoddi and J. Pochampally, DBU-mediated efficient synthesis of indole-conjugated chromene derivatives and their anticancer evaluation and molecular modeling study, Russ. J. Org. Chem., 2023, 59, 907–914, DOI:10.1134/S1070428023050214.
- S. S. Panda, A. S. Girgis, M. N. Aziz and M. S. Bekheit, Spirooxindole: a versatile niologically active heterocyclic scaffold, Molecules, 2023, 28, 618, DOI:10.3390/molecules2802061.
- A. Pandey, A. Pandey, R. Dubey, R. Kant and J. Pandey, Synthesis and computational studies of potent antimicrobial and anticancer indolone scaffolds with spiro cyclopropyl moiety as a novel design element, J. Indian Chem. Soc., 2022, 99, 100539, DOI:10.1016/j.jics.2022.100539.
- M. A. Salem, A. Ragab, A. A. Askar, A. El-Khalafawy and A. H. Makhlouf, One-pot synthesis and molecular docking of some new spiropyranindol-2-one derivatives as immunomodulatory agents and in vitro antimicrobial potential with DNA gyrase inhibitor, Eur. J. Med. Chem., 2020, 188, 111977, DOI:10.1016/j.ejmech.2019.111977.
- A. Jarrahpour, Z. Jowkar, Z. Haghighijoo, R. Heiran, J. A. Rad, V. Sinou, F. Rouvier, C. Latour, J. M. Brunel and N. Özdemir, Synthesis, in-vitro biological evaluation, and molecular docking study of novel spiro-β-lactam-isatin hybrids, Med. Chem. Res., 2022, 31, 1026–1034, DOI:10.1007/s00044-022-02898-8.
- M. Bolous, N. Arumugam, A. I. Almansour, R. S. Kumar, K. Maruoka, V. C. Antharam and S. Thangamani, Broad-spectrum antifungal activity of spirooxindolo-pyrrolidine tethered indole/imidazole hybrid heterocycles against fungal pathogens, Bioorg. Med. Chem. Lett., 2019, 29, 2059–2063, DOI:10.1016/j.bmcl.2019.07.022.
- M. Moghaddam-Manesh, E. Sheikhhosseini, D. Ghazanfari and M. Akhgar, Synthesis of novel 2-oxospiro[indoline-3,4′-[1,3]dithiine]-5′-carbonitrile derivatives by new spiro[indoline-3,4′-[1,3]dithiine]@Cu(NO3)2 supported on Fe3O4@gly@CE MNPs as efficient catalyst and evaluation of biological activity, Bioorg. Chem., 2020, 98, 103751, DOI:10.1016/j.bioorg.2020.103751.
- V. V. Vintonyak, K. Warburg, B. Over, K. Hübel, D. Rauh and H. Waldmann, Identification and further development of thiazolidinones spiro-fused to indolin-2-ones as potent and selective inhibitors of Mycobacterium tuberculosis protein tyrosine phosphatase B, Tetrahedron, 2011, 67, 6713–6729, DOI:10.1016/j.tet.2011.04.026.
- M. A. Borad, M. N. Bhoi, S. K. Rathwa, M. S. Vasava, H. D. Patel, C. N. Patel, H. A. Pandya, E. A. Pithawala and J. J. Georrge, Microwave-assisted ZrSiO2 catalysed synthesis, characterization and computational study of novel spiro[indole-thiazolidines] derivatives as anti-tubercular agents, Interdiscip. Sci.: Comput. Life Sci., 2018, 10, 411–418, DOI:10.1007/s12539-016-0195-2.
- M. A. Borad, D. J. Jethava, M. N. Bhoi, C. N. Patel, H. A. Pandya and H. D. Patel, Novel isoniazid-spirooxindole derivatives: design, synthesis, biological evaluation, in silico ADMET prediction and computational studies, J. Mol. Struct., 2020, 1222, 128881, DOI:10.1016/j.molstruc.2020.128881.
- J. Xu, X. Xie, N. Ye, J. Zou, H. Chen, M. A. White, P.-Y. Shi and J. Zhou, Design, synthesis, and biological evaluation of substituted 4,6-dihydrospiro[[1,2,3]triazolo[4,5-b]pyridine-7,3′-indoline]-2′,5(3H)-dione analogues as potent NS4B inhibitors for the treatment of dengue virus infection, J. Med. Chem., 2019, 62, 7941–7960, DOI:10.1021/acs.jmedchem.9b00698.
- A. Gollner, H. Weinstabl, J. E. Fuchs, D. Rudolph, G. Garavel, K. S. Hofbauer, J. Karolyi-Oezguer, G. Gmaschitz, W. H. Hela, N. Kerres, E. Grondal, P. Werni, J. Ramharter, J. Broeker and D. B. McConnell, Targeted synthesis of complex spiro [3H-indole-3,2’-pyrrolidin]-2(1H)-ones by intramolecular cyclization of azomethine ylides: highly potent MDM2–p53 inhibitors, ChemMedChem, 2019, 14, 88–93, DOI:10.1002/cmdc.201800617.
- M. Espadinha, E. A. Lopes, V. Marques, J. D. Amaral, D. J. V. A. dos Santos, M. Mori, S. Daniele, R. Piccarducci, E. Zappelli, C. Martini, C. M. P. Rodrigues and M. M. M. Santos, Discovery of MDM2-p53 and MDM4-p53 protein-protein interactions small molecule dual inhibitors, Eur. J. Med. Chem., 2022, 241, 114637, DOI:10.1016/j.ejmech.2022.114637.
- K. Murali, H. A. Sparkes and K. J. R. Prasad, Regio- and stereoselective synthesis of dispirooxindolepyrrolocarbazole hybrids via 1,3-dipolar cycloaddition reactions: cytotoxic activity and SAR studies, Eur. J. Med. Chem., 2018, 143, 292–305, DOI:10.1016/j.ejmech.2017.11.039.
- R. F. George, N. S. M. Ismail, J. Stawinski and A. S. Girgis, Design, synthesis and QSAR studies of dispiroindole derivatives as new antiproliferative agents, Eur. J. Med. Chem., 2013, 68, 339–351, DOI:10.1016/j.ejmech.2013.07.035.
- A. S. Girgis, A. F. Mabied, J. Stawinski, L. Hegazy, R. F. George, H. Farag, E. M. Shalaby and I. S. Ahmed Farag, Synthesis and DFT studies of an antitumor active spiro-oxindole, New J. Chem., 2015, 39, 8017, 10.1039/c5nj01109d.
- E. A. Lopes, R. Mestre, D. Fontinha, J. Legac, J. V. Pei, M. Sanches-Vaz, M. Mori, A. M. Lehane, P. J. Rosenthal, M. Prudêncio and M. M. M. Santos, Discovery of spirooxadiazoline oxindoles with dual-stage antimalarial activity, Eur. J. Med. Chem., 2022, 236, 114324, DOI:10.1016/j.ejmech.2022.114324.
- B. Mathebula, K. R. Butsi, R. L. van Zyl, N. C. J. van Vuuren, H. C. Hoppe, J. P. Michael, C. B. de Koning and A. L. Rousseau, Preparation and antiplasmodial activity of 3',4'-dihydro-1'H-spiro(indoline-3,2'-quinolin)-2-ones, Chem. Biol. Drug Des., 2019, 94, 1849–1858, DOI:10.1111/cbdd.13598.
- R. S. Kumar, P. Antonisamy, A. I. Almansour, N. Arumugam, G. Periyasami, M. Altaf, H.-R. Kim and K.-B. Kwon, Functionalized spirooxindole-indolizine hybrids: stereoselective green synthesis and evaluation of anti-inflammatory effect involving TNF-α and nitrite inhibition, Eur. J. Med. Chem., 2018, 152, 417–423, DOI:10.1016/j.ejmech.2018.04.060.
- M. B. Hammouda, S. Boudriga, K. Hamden, M. Askri, M. Knorr, C. Strohmann, L. Brieger, A. Krupp, E. Anouar, M. Snoussi, K. Aouadi and A. Kadri, New spiropyrrolothiazole derivatives bearing an oxazolone moiety as potential antidiabetic agent: design, synthesis, crystal structure, Hirshfeld surface analysis, ADME and molecular docking studies, J. Mol. Struct., 2022, 1254, 132398, DOI:10.1016/j.molstruc.2022.132398.
- A. Kaur, B. Singh, B. Vyas and O. Silakari, Synthesis and biological activity of 4-aryl-3-benzoyl-5-phenylspiro[pyrrolidine-2.3'-indolin]-2'-one derivatives as novel potent inhibitors of advanced glycation end product, Eur. J. Med. Chem., 2014, 79, 282–289, DOI:10.1016/j.ejmech.2014.04.022.
- S. S. Panda, M. N. Aziz, J. Stawinski and A. S. Girgis, Azomethine ylides–Versatile synthons for pyrrolidinyl-heterocyclic compounds, Molecules, 2023, 28, 668, DOI:10.3390/molecules28020668.
- A. S. Girgis, S. S. Panda, I. S. Ahmed Farag, A. M. El-Shabiny, A. M. Moustafa, N. S. M. Ismail, G. G. Pillai, C. S. Panda, C. D. Hall and A. R. Katritzky, Synthesis, and QSAR analysis of anti-oncological active spiro-alkaloids, Org. Biomol. Chem., 2015, 13, 1741–1753, 10.1039/c4ob02149e.
- A. S. Girgis, S. S. Panda, A. M. Srour, H. Farag, N. S. M. Ismail, M. Elgendy, A. K. Abdel-Aziz and A. R. Katritzky, Rational design, synthesis and molecular modeling studies of novel anti-oncological alkaloids against melanoma, Org. Biomol. Chem., 2015, 13, 6619–6633, 10.1039/c5ob00410a.
- A. S. Girgis, S. S. Panda, E. M. Shalaby, A. F. Mabied, P. J. Steel, C. D. Hall and A. R. Katritzky, Regioselective synthesis and theoretical studies of an anti-neoplastic fluoro-substituted dispirooxindole, RSC Adv., 2015, 5, 14780–14787, 10.1039/c4ra13433h.
- A. S. Girgis, S. S. Panda, M. N. Aziz, P. J. Steel, C. D. Hall and A. R. Katritzky, Rational design, synthesis, and 2D-QSAR study of anti-oncological alkaloids against hepatoma and cervical carcinoma, RSC Adv., 2015, 5, 28554–28569, 10.1039/c4ra16663a.
|
| This journal is © The Royal Society of Chemistry 2024 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
M. Adel Youssef
b,
Mohamed S. Bekheit
a,
M. Adel Youssef
b,
Mohamed S. Bekheit