
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIodine-PEG as a unique combination for the metal-free synthesis of flavonoids through iodonium-triiodide ion-pair complexation†
Naveen
Kumar
a,
Navneet
Sharma
b,
Vijay
Kumar
 a,
Vinay
Kumar
a,
Kailash
Jangid
ac,
Bharti
Devi
a,
Ashish Ranjan
Dwivedi
cd,
Kousik
Giri
b,
Rakesh
Kumar
a and
Vinod
Kumar
*a
a,
Vinay
Kumar
a,
Kailash
Jangid
ac,
Bharti
Devi
a,
Ashish Ranjan
Dwivedi
cd,
Kousik
Giri
b,
Rakesh
Kumar
a and
Vinod
Kumar
*a
aLaboratory of Organic and Medicinal Chemistry, Department of Chemistry, Central University of Punjab, Bathinda, Punjab, India-151401. E-mail: vpathania18@gmail.com; vinod.kumar@cup.edu.in; Tel: +911642864214
bDepartment of Computational Sciences, Central University of Punjab, Bathinda, Punjab, India-151401
cDepartment of Pharmaceutical Sciences and Natural Products, Central University of Punjab, Bathinda, Punjab, India-151401
dGitam School of Pharmacy, Hyderabad, Telangana 502329, India
First published on 19th February 2024
Abstract
An efficient metal-free single-step protocol has been developed for the direct synthesis of flavones from 2-hydroxyacetophenone and substituted benzaldehydes. This chemical transformation is exclusively promoted by the iodonium-triiodide ion couple formed through iodine and PEG-400 complexation. The triiodide anion not only helps in the abstraction of a proton from the acetophenone but also promotes the cyclization of intermediate chalcone to the corresponding flavones. The flavones were obtained in very high yields without using any toxic metal catalysts or harsh reaction conditions. The reaction mechanism was established through a series of test reactions and entrapping of reaction intermediates. The developed protocol provides direct access to flavones in high yields under milder reaction conditions with great substrate compatibility, including hydroxylated derivatives.
Introduction
Flavonoids are secondary metabolites widely distributed in many plants, fruits, and vegetables.1 These play a crucial role in the defence mechanism of plants against various pathogens and fungal attacks.2 Polyhydroxy flavonoids impart attractive colours to the flowers and help in the process of pollination by attracting various pollinating agents. Flavonoids are part of the core structural units of many biologically and pharmacologically important moieties.3 Several flavonoids have been reported to possess antioxidant, antimicrobial, anticancer, anti-psychotic, anti-inflammatory, and antiallergic properties.4 Because of the diverse biological properties and numerous pharmacological activities, many flavonoid derivatives have been prepared utilizing various synthetic strategies.Synthesis of flavonoids is carried out through different methods, including Baker–Venkataraman rearrangement,5 Claisen–Schmidt condensation,6 Allan–Robinson reactions,7 coupling reactions, and cyclization through chalcones, etc. Many coupling reactions have been reported involving several metal catalysts8 however, most of these suffers with limitations such as use of toxic metal catalysts, tedious work up and formation of many side products (Scheme 1).
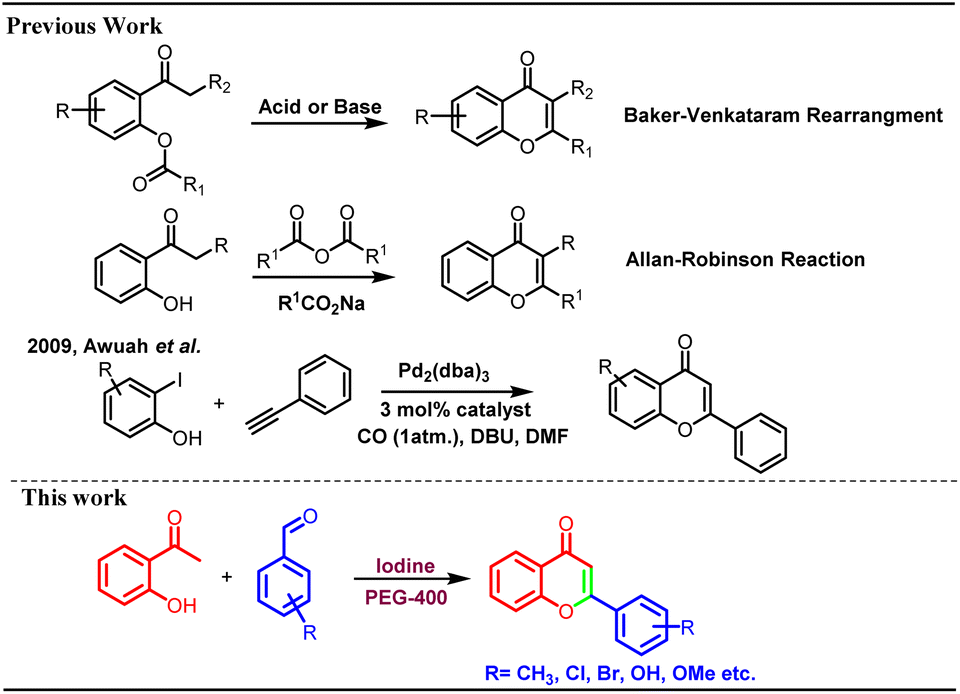 | ||
| Scheme 1 Previous synthetic schemes and present work. | ||
Baker–Venkataraman method has been extensively used for the synthesis of flavonoids9 including total synthesis of dirchromone and related 2-substituted chromones.10 Yatabe et al. synthesized a library of flavones using gold nanoparticles supported on a layered double hydroxide catalyst.11 Kunde et al. described catalytic synthesis of chalcones and flavanones using Zn–Al hydrotalcite adhere ionic liquids however, lower yields were reported.12 In another metal-based protocol, thermally regulated molybdate-based ionic liquids were used for the molecular oxygen generation and one pot oxidative cascade process furnished variety of flavones.13 Similarly, palladium has been used as catalyst for the synthesis of chromones/flavanones through cyclo-carbonylation of ortho-iodophenols with terminal acetylenes in phosphonium salt ionic liquids.14 Awuah et al. reported microwave-assisted one pot synthesis of flavones through Sonogashira-carbonylation-annulation reactions of aryl halides with iodophenols in the presence of palladium catalyst.15 There are many reports for the formation of chalcones in the presence of iodine only.16 Cyclization of 2-hydroxy chalcones can lead to the formation of chromones and other flavonoids under strong basic conditions.17 Chalcones are also cyclized to corresponding flavones using number of oxidizing agents such as DDQ,18 NH4I,19 InBr3/InCl3.20 In addition, molecular I2 (ref. 21) and other iodine-mediated catalytic systems such as I2-DMSO,18,22 I2-SiO2 (ref. 23) and I2–Al2O3 (ref. 24) were explored for the cyclization of chalcones into flavones. Kavala et al. reported iodine catalysed one pot synthesis of flavanones and tetrahydropyrimidines from benzaldehyde, aniline and 2-hydroxyacetophenones via Mannich type reaction however, no flavone formation was reported in this research article.25 Naik et al. reported synthesis of flavones through pyrrolidine and iodine catalysed aldol-Michael-dehydrogenative reaction. In this transformation, pyrrolidine was found to be indispensable and it was observed that no product formation could occur in the absence of pyrrolidine.26 The iodine-PEG combination was utilised in several other valuable transformations.27 We did not come across any report for the single pot synthesis of flavones from 2-hydroxyacetophenone and benzaldehydes exclusively using molecular iodine only under milder conditions. Most of the reported protocols suffer with the disadvantages like use of strong acidic or basic conditions, use of toxic reagents/catalysts, formation of mixture of products such as flavones, flavanones, aurones etc. Thus, there is a scope for the development of efficient protocol for the synthesis of flavones under mild conditions without use of toxic metal catalysts. In the current research work, a single-pot synthesis of flavones is reported from the 2-hydroxy acetophenone and benzaldehydes using molecular iodine in polyethylene glycol. No other supporting reagent or catalyst was used for this transformation. The developed methodology provides direct access to the pharmaceutically important flavonoids/chromones in high yields. The possible mechanism of this chemical transformation was explored, and it was found to be facilitated by iodonium-triiodide anion pair formation through iodine-PEG 400 complexation.
Results and discussion
2-Hydroxy acetophenone and benzaldehydes are usually converted into flavones/chromones in a two-step process.28 In the first step, chalcones are synthesized under strong basic, acidic, or other conditions and the second step involves the cyclization of 2-hydroxy chalcones to the corresponding flavones under different catalytic conditions. From the literature search, we have realized that no synthetic methodology is available for the direct conversion of 2-hydroxy acetophenone and benzaldehydes into flavones. Hence, we got interested in the one-pot synthesis of flavones/chromones from the corresponding 2-hydroxy acetophenone and benzaldehydes under milder conditions. Initially, a mixture of 2-hydroxyacetophenone (1a), benzaldehyde (2a), and molecular iodine (0.5 equivalent) was refluxed in ethanol at 80 °C. Notably, the desired product flavone (3a) was obtained in 7% yield along with some other side products and unreacted starting material even after 72 h of refluxing. It was pleasant to see the direct formation of flavone in a single pot. Naik et al. in their report, could not observe the flavone formation under similar reaction conditions. It might be due to the use of low molar concentration (10 mol%) of molecular iodine and shorter reaction time.26 To improve the product yield, we increased the amount of molecular iodine to one equivalent (100 mol%), which slightly improved the yield of 3a (14%) after refluxing for 72 h. Thereafter, we tried different reaction conditions such as elevated temperatures and different solvents like acetonitrile, dimethyl sulfoxide, ethylene glycol, glycerine, toluene, N-methyl pyrrolidone and dimethylformamide (Table 1). However, no product formation could be observed with these solvents, and starting materials remained unreacted in most of the cases even after prolonged heating. Polyethylene glycol (PEG-400) was found to be compatible for this reaction and 3a was obtained in 84% yield at 140 °C within 7 h of optimized reaction conditions (Table 1). It was interesting to see that only PEG as solvent provided the desired product in high yield and all the other solvents were found to be ineffective. The lower molar percentage of molecular iodine with PEG as solvent were also found ineffective (Table 1, entries 12, 13, 14). In addition, replacement of molecular iodine with NaI and CuI as iodine source in PEG yielded no product however KI provided lower yields of the product (Table 1, entries 15, 16, 17). From these test reactions, we concluded that it might be a specific I2-PEG combination assisting this two-step single-pot transformation. Molecular iodine is reported to form a complex with PEG, and it generates an iodonium-triiodide anion pair (PEG-I+·I3−). Thus, some more test reactions were performed to explore the role of triiodide anion in this chemical transformation. A catalyst combination of molecular iodine and KI was used for the generation of triiodide anion and the reaction was performed with different solvents like DMF (Table 1, entry 18), toluene (Table 1, entry 19), and ethanol (Table 1, entry 20). Interestingly, improved yield of the product was obtained with catalyst combination in ethanol as compared to reaction conditions reported in entry 2 (Table 1), and product formation was observed even with DMF and toluene as solvents, clearly demonstrating the involvement of triiodide anion in the facilitation of this reaction.|
|
|||||
|---|---|---|---|---|---|
| Entry | Reagents (equiv.) | Solvents | Temp. (°C) | Time (h) | Yieldb (%) |
| a Reagents and conditions: different reagents were screened as given in Table 1 with varied temp. and equivalencies. b Isolated yield, nd - not detected. | |||||
| 1 | I2 (0.5) | Ethanol | 80 | 72 | 7 |
| 2 | I2 (1.0) | Ethanol | 80 | 72 | 14 |
| 3 | I2 (1.0) | EG | 140 | 72 | nd |
| 4 | I2 (1.0) | Acetonitrile | 80 | 72 | nd |
| 5 | I2 (1.0) | Toluene | 80 | 72 | nd |
| 6 | I2 (1.0) | EtOAc | 80 | 72 | nd |
| 7 | I2 (1.0) | DMF | 140 | 72 | nd |
| 8 | I2 (1.0) | NMP | 140 | 72 | nd |
| 9 | I2 (1.0) | DMSO | 140 | 72 | nd |
| 10 | I2 (1.0) | Xylene | 140 | 72 | nd |
| 11 | I 2 (1.0) | PEG-400 | 140 | 7 | 84 |
| 12 | I2 (0.1) | PEG-400 | 140 | 72 | 13 |
| 13 | I2 (0.3) | PEG-400 | 140 | 72 | 39 |
| 14 | I2 (0.5) | PEG-400 | 140 | 72 | 58 |
| 15 | NaI (1.0) | PEG-400 | 140 | 72 | nd |
| 16 | CuI (1.0) | PEG-400 | 140 | 72 | nd |
| 17 | KI (1.0) | PEG-400 | 140 | 72 | 8 |
| 18 | I2 + KI (1.0) | DMF | 140 | 72 | 28 |
| 19 | I2 + KI (1.0) | Toluene | 80 | 72 | 32 |
| 20 | I2 + KI (1.0) | Ethanol | 80 | 72 | 22 |
| 21 | PhI(OAc)2 | PEG-400 | 140 | 48 | nd |
Thus, the optimized reaction conditions were extended to other substituted benzaldehydes. Different types of benzaldehydes containing electron-releasing and electron-withdrawing groups were used and found compatible with the developed reaction conditions. Chloro, bromo, and cyano substituents were found to be well tolerated under the developed reaction conditions. Most importantly, even mono, and di-hydroxy substituted benzaldehydes provided corresponding flavones in high yield.29 A polyhydroxylated flavonoid derivative, 6,3′,4′-trihydroxyflavone (6,3′4′-HOFL) was synthesized with ease using this protocol. It is reported to possess good anti-oxidant and anti-inflammatory activities in 2D and 3D cell culture models.30 It is worth mentioning that hydroxy benzaldehydes were not reported in the previous studies25,26 and in other reports, reactions were performed by protecting the hydroxy functionality. In addition, the hydroxy-substituted benzaldehydes were reported to display low reactivity due to phenoxide formation and results in the formation of several side products.31 The current protocol was also found to be compatible with 2-thiophenecarboxyaldehyde and nitrobenzaldehyde. However, no product formation was observed with aliphatic aldehydes, probably due to the low boiling points of aliphatic aldehydes. Aliphatic aldehydes have low electrophilicity as compared to the aromatic aldehydes and there is a possibility of self-addition products due to the presence of enolizable protons. Thus, it was concluded that the developed reaction conditions are selective for the aromatic aldehydes (Table 2).
| a Reagents and conditions: acetophenones, benzaldehydes, I2, PEG-400, 140 °C, 4–7 h. |
|---|
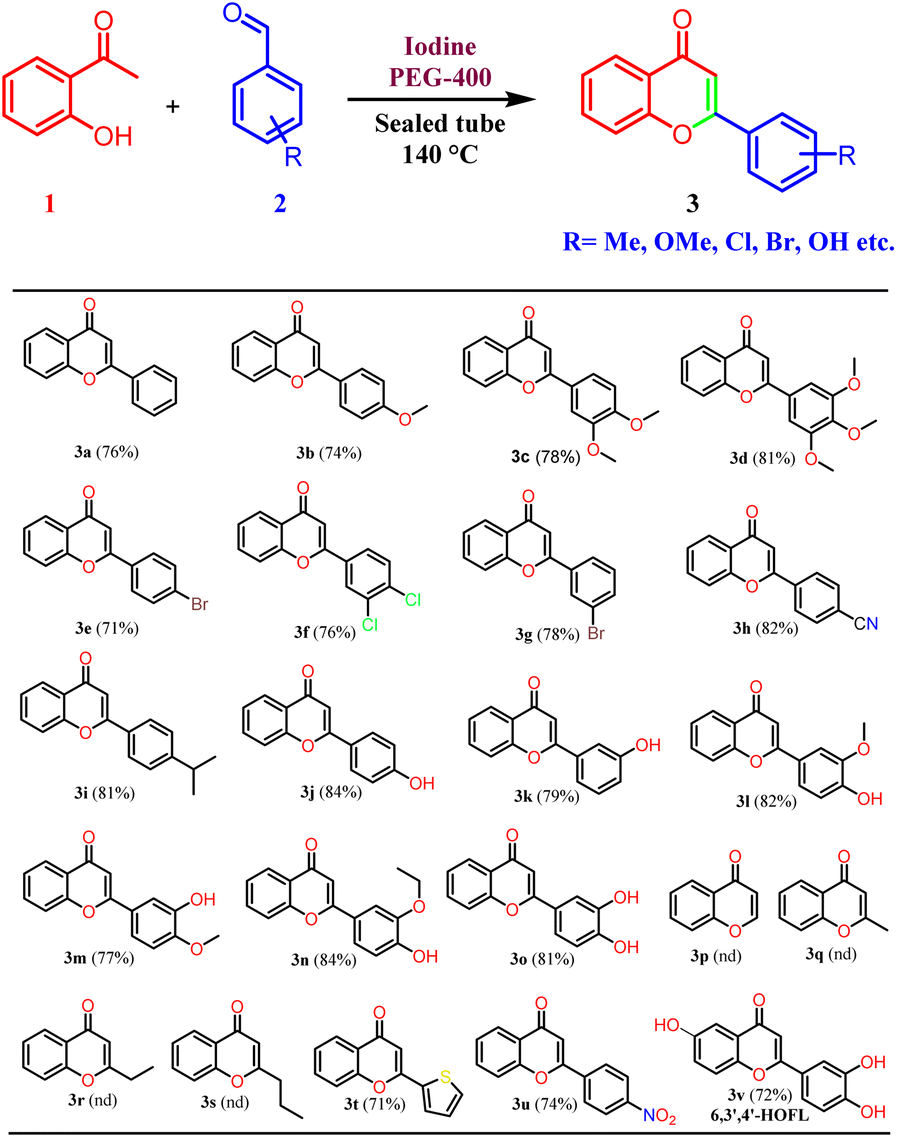
|
Plausible reaction mechanism
The current two-step single-pot chemical transformation is believed to proceed through triiodide anion formation. There are reports indicating that PEG-400 and higher can make a complex with neutral molecular iodine by trapping iodonium ion in a crown ethers-like cage and generating an iodide anion (I−), which subsequently changes to triiodide anion (I3−) by reacting with another molecule of iodine (Fig. 1).32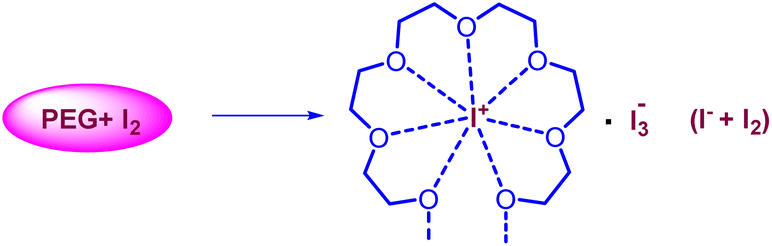 | ||
| Fig. 1 Formation of I3− anion from iodide and molecular iodine. | ||
Disproportionation of two molecules of molecular iodine also leads to the formation of triiodide anion.33 It has been found that the negative charge of the triiodide anion is highly localized on the terminal iodine atom, resulting in the formation of two kinds of iodine–iodine bonds based on the bond lengths. The shorter I–I bond (approx. 2.8708 Å) dissociates in the excited state, whereas the longer one (approx. 3.0048 Å) dissociates in the ground state.34 The disproportionation in the bond length of these I–I bonds explains the electron acceptor as well as the electron-donating nature of the triiodide anion.35 Further, Rogachev and group also explained the potential acceptor and donor nature of I3− anion through the localized highest occupied molecular orbital (HOMO) and low-lying lowest unoccupied molecular orbital (LUMO) of the anion.36 We have confirmed the formation of triiodide anion in the reaction mixture through UV absorption bands near 290 nm and 360 nm (Fig. 2A).37 As evident from Fig. 2, no peaks were observed for the formation of triiodide anion when molecular iodine was used with THF, NMP, ethanol, ethylene glycol, ethylacetate, DMSO, DMF, AcCN, xylene, toluene, and only a band for molecular iodine was observed at 500 nm. While strong bands were observed at 290 nm and 360 nm with PEG-400. In Fig. 2B, the addition of KI along with molecular iodine in other solvents (ethylene glycol, DMF, ethanol) promoted triiodide anion formation, and bands were observed at 290 nm and 360 nm. It was also observed that the band for molecular iodine completely disappeared after 15 min of heating (Fig. 2B), and the whole of the iodine converted into triiodide anion.
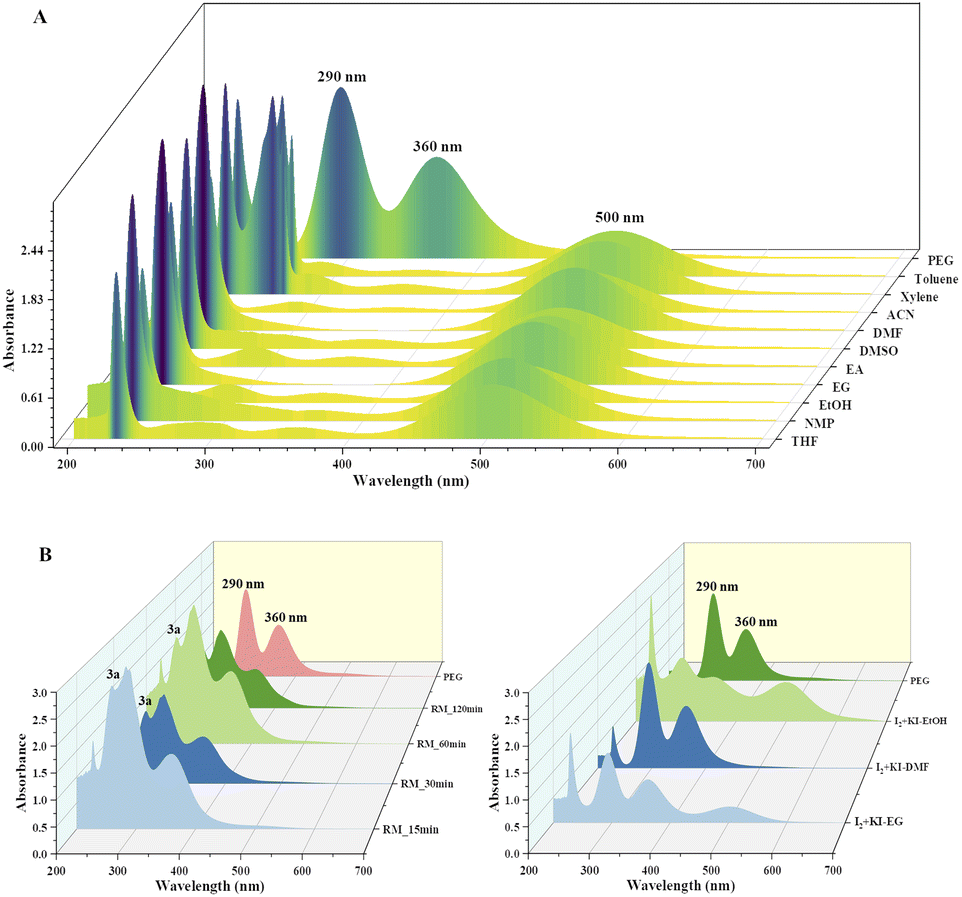 | ||
| Fig. 2 UV-visible spectrum of (A) various iodine and solvent mixtures depicting the formation of I3− anion, peaks at 290 nm and 360 nm for the I3− and peak at 500 nm for I2; (B) formation of I3− anion by the reaction of iodine and KI in solvents and in reaction mixture (PEG) at different time intervals. | ||
It was hypothesized that due to disproportionation, the I3− anion abstracts the proton through the longer iodine–iodide bond.38 In addition, iodine plays an active role in the polarization of carbonyl and olefinic bond during the cyclization of intermediate chalcones to the corresponding chromones. Deshidi and group demonstrated the polarisation of oxygen (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond) molecule by molecular iodine through halogen bond formation for the synthesis of α-ketoamides39 while Liang et al. showed the keto–enol tautomerization of cyclohexanone through interaction with iodonium ion.40 It is reported that iodine also plays a central role in α,β-dehydrogenation reactions.41 Thus, it was proposed that the reaction is initiated and assisted by the formation of PEG-iodonium-triiodide ion pair. Acetophenones and benzaldehydes condense to form chalcones as intermediates, which, through oxidative cyclization, convert to corresponding chromones. To get the mechanistic insights, we performed some controlled reactions. In a first test reaction, a mixture of 2-hydroxy acetophenone, benzaldehyde, molecular iodine, and PEG-400 was refluxed at 140 °C for one hour, and the reaction was stopped and quenched to trap and identify the intermediates. The reaction mixture was subjected to EIMS analysis, and four intermediates were identified as shown in Fig. 3. The quenched reaction mixture was worked up and purified through column chromatography. The two reaction intermediates (chalcone, B, and flavanone, C) were isolated, purified, and characterized through 1H and 13C NMR (see ESI† for details). Given below are some of the mass spectra of the identified intermediates.
O bond) molecule by molecular iodine through halogen bond formation for the synthesis of α-ketoamides39 while Liang et al. showed the keto–enol tautomerization of cyclohexanone through interaction with iodonium ion.40 It is reported that iodine also plays a central role in α,β-dehydrogenation reactions.41 Thus, it was proposed that the reaction is initiated and assisted by the formation of PEG-iodonium-triiodide ion pair. Acetophenones and benzaldehydes condense to form chalcones as intermediates, which, through oxidative cyclization, convert to corresponding chromones. To get the mechanistic insights, we performed some controlled reactions. In a first test reaction, a mixture of 2-hydroxy acetophenone, benzaldehyde, molecular iodine, and PEG-400 was refluxed at 140 °C for one hour, and the reaction was stopped and quenched to trap and identify the intermediates. The reaction mixture was subjected to EIMS analysis, and four intermediates were identified as shown in Fig. 3. The quenched reaction mixture was worked up and purified through column chromatography. The two reaction intermediates (chalcone, B, and flavanone, C) were isolated, purified, and characterized through 1H and 13C NMR (see ESI† for details). Given below are some of the mass spectra of the identified intermediates.
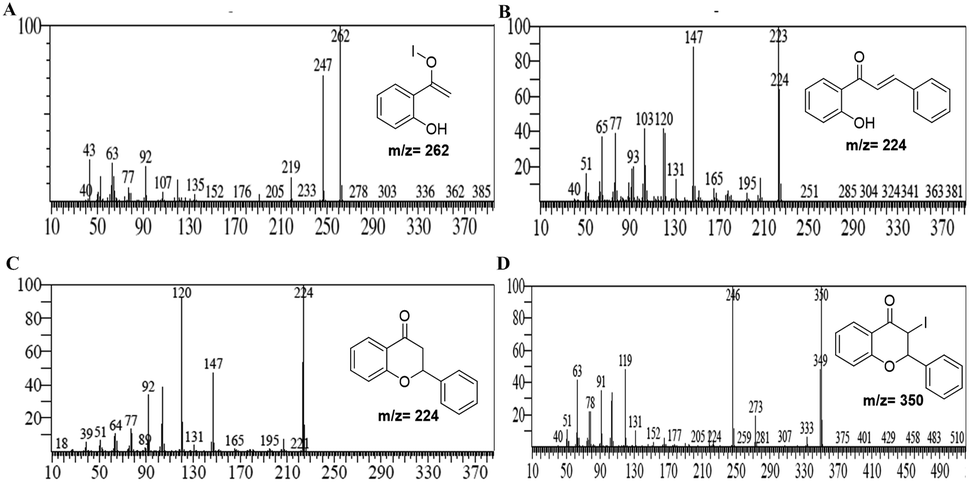 | ||
| Fig. 3 Mass spectra of the identified intermediates; (A) formation of 1-(2-hydroxyphenyl)vinylhypoiodite intermediate; (B) formation of chalcone intermediate; (C) formation of chromanone intermediate; (D) formation of 3-iodo-chromanone intermediate. | ||
In the 2nd test reaction, chalcone was synthesized through Claisen–Schmidt condensation of 2-hydroxy acetophenone and benzaldehyde. Thereafter, the chalcone was treated with molecular iodine in PEG-400 at 140 °C. After completion of the reaction, the flavone was successfully obtained thus further confirming that the reaction might be proceeding through the formation of chalcone-intermediate. Based on the results obtained from all the test reactions and the UV-visible spectra of the reaction and solvent mixtures in iodine, we proposed a reaction mechanism as given below (Fig. 4).
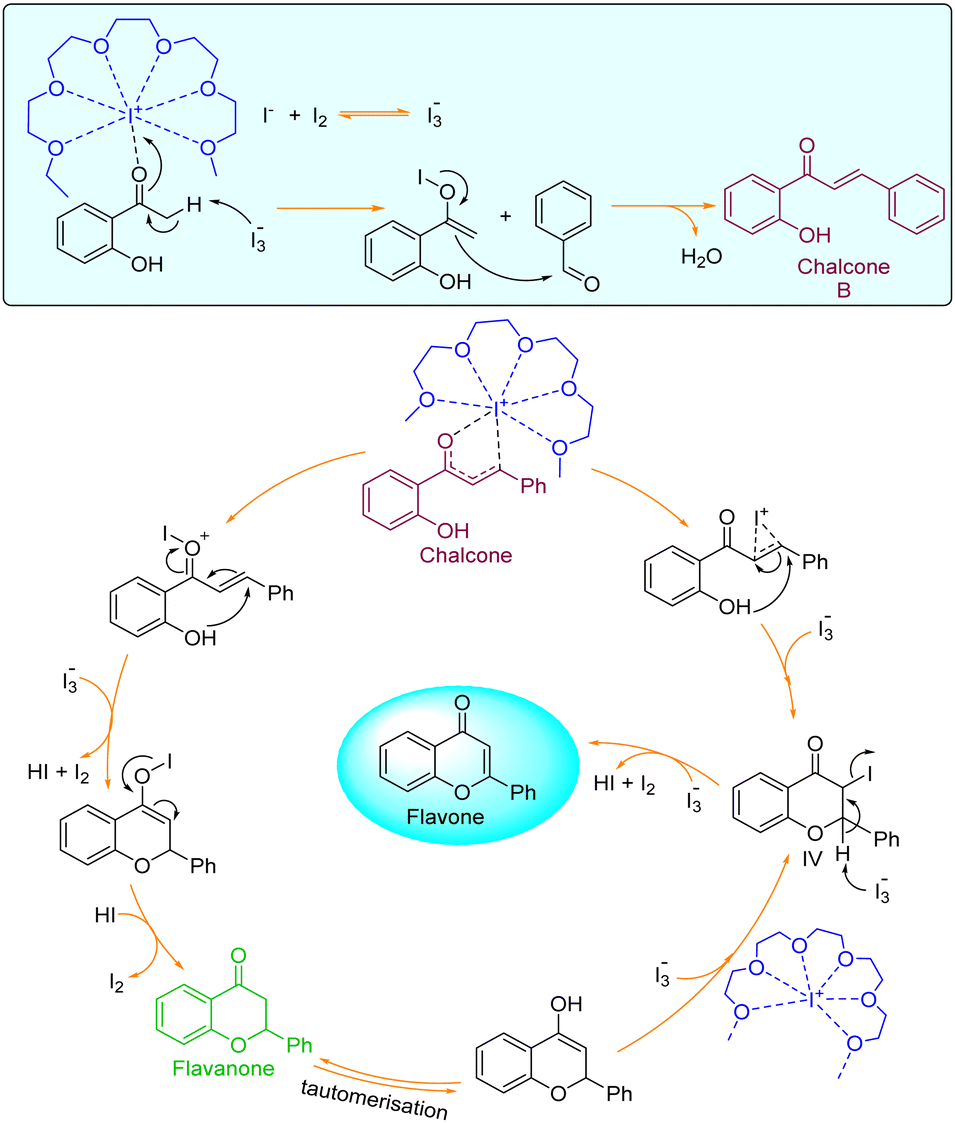 | ||
| Fig. 4 Plausible mechanism for the formation of flavones. | ||
DFT studies
To access the feasibility of the reaction progression and to support the reaction mechanism, DFT calculations were performed using ORCA 5.0.3 software with PBE0 functional. Herein, it shows that the 1st step of the reaction proceeds with the formation of stable intermediate B (chalcone, ΔGS = −33.52 kcal mol−1) through an exergonic aldol condensation reaction. It is also evidenced that the formation of major product flavone (C) may proceed through two pathways. The first is through oxidative cyclization of chalcone to flavone (ΔGS = −49.18 kcal mol−1) directly via transition state TS_1 (ΔGS = 13.15 kcal mol−1). Secondly, it may change to another stable intermediate flavanone (D, ΔGS = −12.10 kcal mol−1), which subsequently converts to flavone through a barrierless transformation. This transformation of chalcone to either flavanone or flavone is exergonic in nature in both cases, but the energy barrier of the transformation through TS_1 to flavone is 10.36 kcal mol−1 more as compared to TS_2 to flavanone. This showed that the major product, flavone (C), is 37.08 kcal mol−1 lower in energy than flavanone, which is untimely converted to flavone by dehydrohalogenation. Transformation of both, the chalcone and flavanone to flavone proceeds through a common intermediate (IV, ΔGS = −35.83 kcal mol−1) with the loss of HI. The DFT analyses concur the formation of the final product through two stable intermediates (chalcone and flavanone), with the overall release of energy (Fig. 5).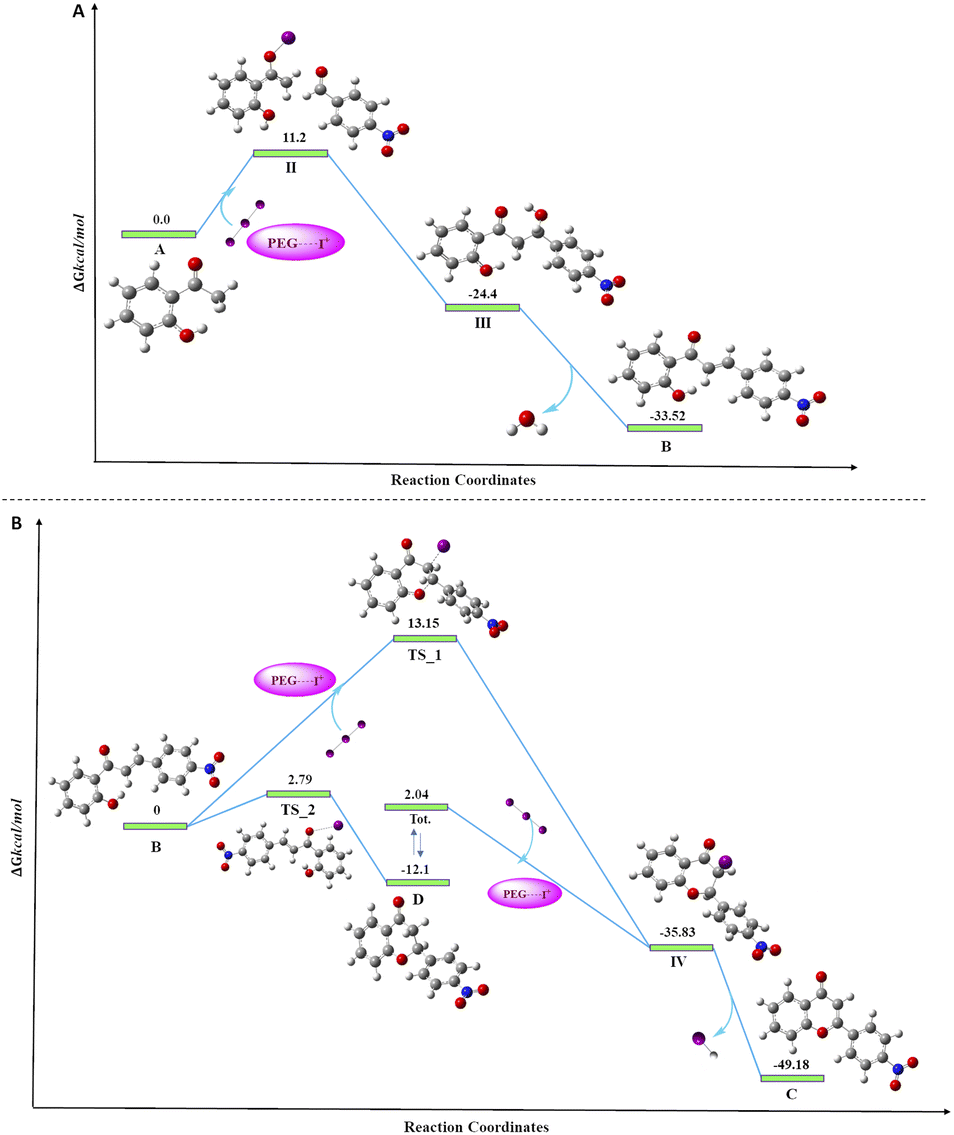 | ||
| Fig. 5 Free energy diagram for the progress of the reaction, (A) the formation of chalcone; (B) the formation of flavone from chalcone. | ||
In a similar test reaction, when 2-hydroxy acetophenone was reacted with p-methoxy benzaldehyde under the same reaction conditions, and it was quenched after one hour, two new side products (Scheme 2, VII, and VIII) were obtained along with the expected chalcone and flavone. Compounds VII and VIII were purified through column chromatography and characterized through 1H and 13C NMR (ESI†). These side products were formed through double addition of para-methoxy-benzaldehyde and both side products were found to be isomers. In another test reaction with para hydroxybenzaldehyde, flavone was obtained as a major product while chalcone was obtained as a minor product when the reaction was stopped within an hour. Similarly, with para nitro-benzaldehyde flavanone and chalcone were obtained as intermediate products after one hour of the reaction time. The spectroscopic data of all these intermediates (chalcone and flavanone) from unsubstituted benzaldehyde, p-OH, and p-NO2 substituted benzaldehydes are reported in the ESI.†
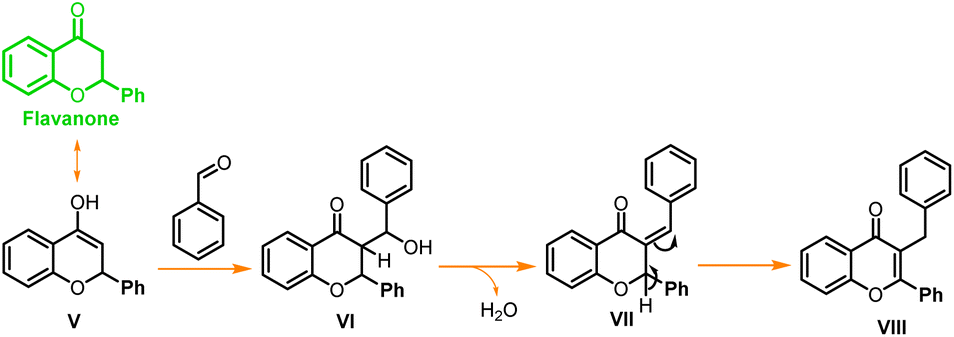 | ||
| Scheme 2 Plausible mechanism for the formation of side products from flavanone. | ||
Conclusion
Flavones are an important class of compounds, and limited protocols are reported for their synthesis. Most of the synthetic methodologies for the flavones make use of toxic metal catalysts. In the current research article, a single-step synthesis of flavones was achieved from inexpensive and abundantly available acetophenones and benzaldehydes. Molecular iodine was used as a mild reagent in PEG-400 as solvent. The developed conditions were found to be compatible with range of substituted benzaldehydes including hydroxy-substituted derivatives. A reasonable mechanism has been proposed for this two-step single-pot chemical transformation through a series of test reactions. The iodine-PEG-400 combination was anticipated to play a crucial role in this reaction. UV-visible spectra showed the formation of the PEG iodonium-triiodide anion complex. The reaction proceeds through the formation of stable intermediates chalcone and flavanone. The DFT studies also supported the proposed mechanism. Thus, the developed protocol provides an efficient route for the direct excess of flavones with great tolerability for different substrates and can be explored for the synthesis of new library of medicinally important chromone derivatives.Experimental section
General information
All the chemicals were purchased from Sigma Aldrich and were used as received. The compounds synthesized by the general procedure described below and were characterised by mass spectrometry and 1H and 13C NMR spectra recorded on 600 MHz JEOL NMR spectrometer in either CDCl3 or DMSO-d6 with TMS as an internal reference at Central University of Punjab, Bathinda (1H NMR: TMS at 0.00 ppm, CDCl3 at 7.26 ppm, DMSO-d6 at 2.5 ppm; 13C NMR: CDCl3 at 77.16 ppm, DMSO-d6 at 39.52 ppm). The spectroscopic data of all the synthesized compounds are consistent with the reported datas.42General synthetic procedure for (3a–3v)
A mixture of 2-hydroxyacetophenone (0.5 g, 3.67 mmol), respective benzaldehyde (1 equiv.), and iodine (0.47 g, 3.67 mmol) were taken in a sealed tube in PEG-400 as solvent (2 ml) and heated at 140 °C for 4 to 7 hours. The completion of the reaction was checked by thin-layer chromatography. On completion of the reaction, it was cooled to room temperature, iodine was quenched by 10% sodium thiosulphate solution and extracted with ethyl acetate. As PEG-400 is soluble in water, the traces from the ethyl acetate layer were removed by several water washes. The crude product was purified on silica gel by column chromatography using pet. ether/ethyl acetate (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 6:1) as eluent to give the desired product (70–84% yield).
:1 to 6:1) as eluent to give the desired product (70–84% yield).
2-(3-Hydroxyphenyl)-4H-chromen-4-one (3k)
Off white solid, 79% yield. M.pt.: 206–208 °C. 1H NMR (600 MHz, DMSO) δ 6.94 (s, 1H), 7.03 (dd, J = 8.4, 2.4 Hz, 1H), 7.39 (t, J = 7.8 Hz, 1H), 7.46 (t, J = 1.8 Hz, 1H), 7.53 (dd, J = 15.6, 7.2 Hz, 2H), 7.78 (d, J = 8.4, 1H), 7.83–7.86 (m, 1H), 8.07 (dd, J = 7.8, 1.2 Hz, 1H), 9.91 (s, 1H); 13C NMR (150 MHz, DMSO) δ 106.8, 112.7, 117.1, 118.4, 118.7, 123.2, 124.7, 125.4, 130.1, 132.3, 134.2, 155.5, 157.8, 162.6, 176.9. MS (EI, m/z): 238.
2-(3,4-Dihydroxyphenyl)-4H-chromen-4-one (3o)
A brown solid, 81% yield. M.pt.: 242–244 °C. 1H NMR (600 MHz, DMSO) δ 6.77 (s, 1H), 6.93 (d, J = 9.0 Hz, 1H), 7.47 (dt, J = 6.6, 1.8 Hz, 2H), 7.5 (d, J = 6.6 Hz, 1H), 7.73 (d, J = 7.8 Hz, 1H), 7.80–7.83 (m, 1H), 8.05 (dd, J = 7.8, 1.2 Hz, 1H), 9.44 (s, 1H), 9.86 (s, 1H); 13C NMR (150 MHz, DMSO) δ 104.7, 113.3, 115.9, 118.1, 118.7, 121.8, 123.2, 124.6, 125.2, 133.9, 145.6, 149.3, 155.4, 136.1, 176.6. MS (EI, m/z): 254.
Spectroscopic data of some isolated intermediates/side products
DFT calculations
All the DFT calculations were performed using the ORCA 5.0.3 software.43 Density functional theory was employed with PBE0 functional,44 dispersion corrections based on tight binding partial charges (D4)45 along with Ahlrichs and co-workers balanced polarised double zeta basis set (def2-svp).46 RIJCOSX47 approximation was used throughout to speed up the calculations. Transition states were calculated by implementing Nudged-Elastic-Band method of Asgeirsson et al.48 All minima on the potential energy surface were verified by calculating the vibrational frequencies using the same level of theory. Furthermore, for refinement in results, a higher valence polarised triple zeta basis set (def2-tzvp) was employed with PBE0 functional to calculate the single point-energy of the whole system. The solute–solvent interaction is described by the conductor-like polarizable continuum model (CPCM).49 The dielectric constant & refractive index of PEG-400 is taken as 11.6 and 54, respectively.Conflicts of interest
There are no conflicts to declare.Acknowledgements
VK is thankful to the Council of Scientific and Industrial Research New Delhi for the financial grant No. 02/(0354)/19/EMRII. NK, NS, and Vijay Kumar are thankful to the University Grant Commission for providing Senior Research Fellowship. Vinay Kumar is thankful to CSIR for SRF. KJ and ARD are thankful to ICMR (45/29/2022-/BIO/BMS) and DST-SERB, respectively, for their fellowship grants.Notes and references
- N. Shen, T. Wang, Q. Gan, S. Liu, L. Wang and B. Jin, Food Chem., 2022, 383, 132531 CrossRef CAS PubMed.
- D. Treutter, Environ. Chem. Lett., 2006, 4, 147–157 CrossRef CAS.
- S. K Sharma, S. Kumar, K. Chand, A. Kathuria, A. Gupta and R. Jain, Curr. Med. Chem., 2011, 18, 3825–3852 CrossRef.
- (a) J. Reis, A. Gaspar, N. Milhazes and F. Borges, J. Med. Chem., 2017, 60, 7941–7957 CrossRef CAS PubMed; (b) M. Irfan, S. ul Hassan and U. Saleem, Pharm. Chem. J., 2020, 54, 241–257 CrossRef; (c) L. K. Tsou, M. Lara-Tejero, J. RoseFigura, Z. J. Zhang, Y.-C. Wang, J. S. Yount, M. Lefebre, P. D. Dossa, J. Kato and F. Guan, J. Am. Chem. Soc., 2016, 138, 2209–2218 CrossRef CAS; (d) M. Singh, M. Kaur and O. Silakari, Eur. J. Med. Chem., 2014, 84, 206–239 CrossRef CAS.
- D. Ameen and T. J. Snape, Synth., 2015, 47, 141–158 CAS.
- (a) D. A. Williams, C. Smith and Y. Zhang, Tetrahedron Lett., 2013, 54, 4292–4295 CrossRef CAS; (b) S. Patel and U. Shah, Synth., 2017, 10, 403–406 CAS.
- J. J. Li, in Name Reactions, Springer, 2014, pp. 8–9 Search PubMed.
- (a) N. Anitha, K. V. Reddy and Y. J. Rao, Heterocycl. Commun., 2014, 20, 129–132 CrossRef CAS; (b) W. Mansour, M. Fettouhi and B. El Ali, ACS Omega, 2020, 5, 32515–32529 CrossRef CAS PubMed.
- (a) B. P. Reddy and G. D. Krupadanam, J. Heterocycl. Chem., 1996, 33, 1561–1565 CrossRef CAS; (b) A. Fougerousse, E. Gonzalez and R. Brouillard, J. Org. Chem., 2000, 65, 583–586 CrossRef CAS PubMed; (c) N. Thasana and S. Ruchirawat, Tetrahedron Lett., 2002, 43, 4515–4517 CrossRef CAS.
- A. St-Gelais, J. Alsarraf, J. Legault, C. Gauthier and A. Pichette, Org. Lett., 2018, 20, 7424–7428 CrossRef CAS.
- T. Yatabe, X. Jin, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2015, 54, 13302–13306 CrossRef CAS.
- L. B. Kunde, S. M. Gade, V. S. Kalyani and S. P. Gupte, Catal. Commun., 2009, 10, 1881–1888 CrossRef CAS.
- Z. Song, W. Huang, Y. Zhou, Z.-Q. Tian, Z.-M. Li and D.-J. Tao, Green Chem., 2020, 22, 103–109 RSC.
- (a) X. Zhao, J. Zhou, S. Lin, X. Jin and R. Liu, Org. Lett., 2017, 19, 976–979 CrossRef CAS PubMed; (b) Q. Yang and H. Alper, J. Org. Chem., 2010, 75, 948–950 CrossRef CAS PubMed.
- E. Awuah and A. Capretta, Org. Lett., 2009, 11, 3210–3213 CrossRef CAS.
- (a) K. V. Sashidhara, J. N. Rosaiah and A. Kumar, Synth. Commun., 2009, 39, 2288–2296 CrossRef CAS; (b) D. Kakati and J. C. Sarma, Chem. Cent. J., 2011, 5, 1–5 CrossRef PubMed; (c) H. Wang and J. Zeng, Can. J. Chem., 2009, 87, 1209–1212 CrossRef CAS.
- H. Chawla and S. Sharma, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1987, 26, 1075–1077 Search PubMed.
- P. Venkatesan and T. Maruthavanan, Bull. Chem. Soc. Ethiop., 2011, 25, 419–425 CrossRef CAS.
- P. S. Kulkarni, D. D. Kondhare, R. Varala and P. K. Zubaidha, J. Serb. Chem. Soc., 2013, 78, 909–916 CrossRef CAS.
- N. Ahmed, H. Ali and J. E. van Lier, Tetrahedron Lett., 2005, 46, 253–256 CrossRef CAS.
- K. V. Sashidhara, M. Kumar and A. Kumar, Tetrahedron Lett., 2012, 53, 2355–2359 CrossRef CAS.
- M. Masih, T. Banerjee, B. Banerjee and A. Pal, Int. J. Pharm. Pharm. Sci., 2011, 3, 51–54 Search PubMed.
- K. Rajesh Babu, K. Vijaya Kumar, M. Vijaya and V. Madhavarao, Int. J. Pharm. Technol, 2012, 4, 3943–3950 CAS.
- S. R. Sarda, W. N. Jadhav and R. P. Pawar, Int. J. Chem. Tech. Res., 2009, 1, 539–543 CAS.
- V. Kavala, C. Lin, C.-W. Kuo, H. Fang and C.-F. Yao, Tetrahedron, 2012, 68, 1321–1329 CrossRef CAS.
- M. M. Naik, S. G. Tilve and V. P. Kamat, Tetrahedron Lett., 2014, 55, 3340–3343 CrossRef CAS.
- (a) W. Lui, G. Lu, B. Xiao and C. Xie, RSC Adv., 2018, 8(54), 30860–30867 RSC; (b) D. Guttman and T. Higuchi, J. Am. Pharm. Assoc., 1955, 44, 668–678 CrossRef CAS PubMed; (c) A. K. Pathan, P. Patil, A. Shinde and S. Zangade, Curr. Green Chem., 2021, 8(2), 166–173 CrossRef CAS.
- E. S. VH, S. Matsjeh, T. D. Wahyuningsih, M. Mustofa and T. Redjeki, Indones. J. Chem., 2012, 12, 146–151 CrossRef.
- M. Mellado, A. Madrid, Ú. Martínez, J. Mella, C. O. Salas and M. Cuellar, Chem. Pap., 2018, 72, 703–709 CrossRef CAS.
- X. Wang, Y. Cao, S. Chen, X. Yang, J. Bian and D. Huang, Antioxid, 2023, 12, 204 CrossRef CAS.
- (a) G. Sipos and F. Sirokman, Nature, 1964, 202, 489 CrossRef CAS; (b) P. Kulkarni, P. Swami and P. Zubaidha, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2013, 43, 617–620 CrossRef CAS.
- S. Hemalatha, B. Chandani and D. Balasubramanian, Spectrosc. Lett., 1979, 12, 535–541 CrossRef CAS.
- S. Aronson, P. Epstein, D. B. Aronson and G. Wieder, J. Phys. Chem., 1982, 86, 1035–1037 CrossRef CAS.
- J. Heo, J. G. Kim, E. H. Choi, H. Ki, D.-S. Ahn, J. Kim, S. Lee and H. Ihee, Nat. Commun., 2022, 13, 522 CrossRef CAS PubMed.
- E. Bartashevich, V. Batalov, I. Yushina, A. Stash and Y. Chen, Acta Crystallogr., Sect. C: Struct. Chem., 2016, 72, 341–345 CrossRef CAS PubMed.
- A. Y. Rogachev and R. Hoffmann, Inorg. Chem., 2013, 52, 7161–7171 CrossRef CAS.
- A. Nimmrich, M. R. Panman, O. Berntsson, E. Biasin, S. Niebling, J. Petersson, M. Hoernke, A. Björling, E. Gustavsson and T. B. van Driel, J. Am. Chem. Soc., 2023, 145, 15754–15765 CrossRef CAS PubMed.
- (a) P. A. Sathe, K. S. Vadagaonkar, M. V. Vhatkar, L. Melone and A. C. Chaskar, ChemistrySelect, 2018, 3, 277–283 CrossRef CAS; (b) Y.-P. Zhu, M. Lian, F.-C. Jia, M.-C. Liu, J.-J. Yuan, Q.-H. Gao and A.-X. Wu, Chem. Commun., 2012, 48, 9086–9088 RSC; (c) Y. Cao, L. Liu, T. Huang and T. Chen, New J. Chem., 2020, 44, 8697–8701 RSC.
- R. Deshidi, M. Kumar, S. Devari and B. A. Shah, Chem. Commun., 2014, 50, 9533–9535 RSC.
- Y.-F. Liang, X. Li, X. Wang, M. Zou, C. Tang, Y. Liang, S. Song and N. Jiao, J. Am. Chem. Soc., 2016, 138, 12271–12277 CrossRef CAS PubMed.
- (a) Y. Cao, L. Lui, T. Huang and T. Chen, New J. Chem., 2020, 44, 8697–8701 RSC; (b) Q. Gao, M. Wu, K. Zhang, N. Yang, M. Lui, J. Li, L. Fang, S. Bai and Y. Xu, Org. Lett., 2020, 22, 5645–5649 CrossRef CAS PubMed; (c) D. Li, Asian J. Org. Chem., 2023, 12, e202300012 CrossRef CAS.
- (a) M.-L. Ma, M. Li, J.-J. Gou, T. Y. Ruan, H.-S. Jin, L.-H. Zhang, L. C. Wu, X. Y. Li, Y. H. Hu and K. Wen, Biomed. Chem., 2014, 22(21), 6117–6123 CAS; (b) J. Y. An, H.-H. Lee, J. S. Shin, H. S. Yoo, J. S. Park, S. H. Son, S. W. Kim, J. Yu, J. Lee and K. T. Lee, Bioorg. Med. Chem. Lett., 2017, 27, 2613–2616 CrossRef CAS; (c) V. N. Badavath, S. Ciftci-Yabanoglu, S. Bhakat, A. K. Timiri, B. N. Sinha, G. Ucar, M. E. Soliman and V. Jayaprakash, Bio. Chem., 2015, 58, 72–80 CrossRef CAS; (d) Z. Du, H. Ng, K. Zhang, H. Zeng and J. Wang, Org. Biochem., 2011, 9, 6930–6933 CAS; (e) X. Zhao, R. Fang, F. Wang, X. Kong and Y. Li, JACS Au, 2023, 3, 185–194 CrossRef CAS PubMed.
- (a) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2(1), 73–78 CAS; (b) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8(1), e1327 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- (a) E. Caldeweyher, C. Bannwarth and S. Grimme, J. Chem. Phys., 2017, 147, 034112 CrossRef; (b) E. Caldeweyher, S. Ehlert, A. Hansen, H. Neugebauer, S. Spicher, C. Bannwarth and S. Grimme, Chem. Phys., 2019, 150, 154122 Search PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356(1–3), 98–109 CrossRef CAS.
- V. Ásgeirsson, B. O. Birgisson, R. Bjornsson, U. Becker, F. Neese, C. Riplinger and H. Jónsson, J. Chem. Theory Comput., 2021, 17, 4929–4945 CrossRef PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Quantum mechanical continuum solvation models, Chem. Rev., 2005, 105(8), 2999–3094 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra08810c |
| This journal is © The Royal Society of Chemistry 2024 |