
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced sensitivity in electrochemical detection of ochratoxin A within food samples using ferrocene- and aptamer-tethered gold nanoparticles on disposable electrodes†
Wicem Argoubi a,
Faisal K. Algethami*b and
Noureddine Raouafi*a
a,
Faisal K. Algethami*b and
Noureddine Raouafi*a
aSensors and Biosensors Group, ACE-Lab (LR99ES15), Faculty of Science, University of Tunis El Manar, 2092 Tunis El Manar, Tunisia. E-mail: noureddine.raouafi@fst.utm.tn
bDepartment of Chemistry, College of Science, Imam Mohammad Ibn Saud Islamic University (IMSIU), P.O. Box 90950, Riyadh 11623, Saudi Arabia. E-mail: falgethami@imamu.edu.sa
First published on 7th March 2024
Abstract
Ensuring food security is crucial for public health, and the presence of mycotoxins, produced by fungi in improperly stored processed or unprocessed food, poses a significant threat. This research introduces a novel approach – a disposable aptasensing platform designed for the detection of ochratoxin A (OTA). The platform employs gold-nanostructured screen-printed carbon electrodes functionalized with a ferrocene derivative, serving as an integrated faradaic transducing system, and an anti-OTA aptamer as a bioreceptor site. Detection relies on the ferrocene electrochemical signal changes induced by the aptamer folding in the presence of the target molecule. Remarkably sensitive, the platform detects OTA within the range of 0.5 to 70 ng mL−1 and a detection limit of 11 pg mL−1. This limit is approximately 200 times below the levels stipulated by the European Commission for agricultural commodities. Notably, the sensing device exhibits efficacy in detecting OTA in complex media, such as roasted coffee beans and wine, without the need for sample pretreatment, yielding accurate recoveries. Furthermore, while label-free electrochemical aptasensors have proliferated, this study addresses a gap in understanding the binding mechanisms of some aptasensors. To enhance the experimental findings, a theoretical study was conducted to underscore the specificity of the anti-OTA aptamer as a donor for OTA detection. The molecular docking technique was employed to unveil the key binding region of the aptamer, providing valuable insights into the aptasensor specificity.
Introduction
Diversifying methods beyond antibody-based approaches for highly sensitive detection of analytes of interest is crucial for advancing biochemical analysis.1 Aptamers, stable single-stranded oligonucleotides selected through the SELEX method, are gaining prominence as superior substitutes to antibodies.2 Possessing enhanced affinities toward proteins, small molecules, metallic cations and other macromolecular compounds, aptamers opened new avenues in biosensor development for practical applications, including medical diagnostics and the detection of contaminants in food, beverages or the environment.3–5Mycotoxins, such as ochratoxins A, B and C, aflatoxins B1, B2, G1 and G2, etc., produced by Aspergillus and Penicillium fungi, pose a threat when present in raw and processed food stored under unfavorable conditions.6,7 These toxic metabolites are extensively studied due to the mycotoxicosis they induce in humans and animals upon consuming contaminated food and beverages. Among them, OTA has garnered recent attention and has been identified in various products, including cereal-derived items, dried fruits, spices, beer, and wine.8,9 Regulatory agencies have established maximum toxin levels in foodstuffs to ensure safety. For instance, the European Commission mandates OTA concentrations not exceeding 3 μg kg−1 (7.4 nM) for cereal products, 5 μg kg−1 (12.4 nM) for roasted coffee, and up to 10 μg kg−1 (25 nM) for instant coffee.10 Wine is subject to even stricter limits, requiring levels below 2 μg kg−1 (5 nM) for compliance.11
Various standard techniques, such as thin layer chromatography, HPLC,12 fluorescence spectroscopy, ELISA, etc.,13 are commonly employed to monitor ochratoxin A contamination. Electrochemical methods have gained widespread attention due to their simplicity, cost-effectiveness, ease of operation, high sensitivity, and rapid sensing capabilities.5,14–18 Indeed, they allow the preparation of sensing devices that fit the World Health Organization “ASSURED: Affordable, Sensitive, Selective, User-friendly, Equipment-free and Deliverable to end-users” criteria.19,20 For instance, Gökçe et al.21 developed an aptamer-based electrochemical sensor for OTA detection, demonstrating practical application in samples using pencil graphite electrodes. Rivas et al.22 presented a label-free electrochemical impedimetric aptasensor for OTA quantitation in cocoa beans. Recently, Hou et al. introduced a novel label-free impedimetric aptasensor employing dual-amplification with Nafion-MWCNTs and Au nanopopcorns for highly selective and ultra-sensitive OTA detection.23 Dridi et al. reported a differential conductimetric method for OTA determination using thermolysin embedded in a bovine serum albumin matrix, successfully applied to quantify OTA in spiked olive oil samples.24
This paper details the design of a disposable aptasensing platform for OTA using gold nanoparticles-modified screen-printed carbon electrodes. The platform is functionalized with ferrocene as a transducing system and anti-OTA aptamer as a bioreceptor site. Cyclic voltammetry, electrochemical impedance spectroscopy, and SEM imaging were employed to monitor the aptasensor's preparation steps. The developed aptasensor demonstrates efficacy in detecting OTA, with the biosensor's selectivity and specificity evaluated against interference from competitive mycotoxins using various electrochemical techniques. The aptasensor device was successfully applied to determine OTA levels in roasted coffee beans and wine without requiring sample pretreatment. This work introduces a novel platform for highly sensitive detection, utilizing gold nanoparticles anchored to a carbon surface with minimized agglomeration, which in turn yielded a high current response. This innovative approach incorporates a ferrocene-functionalization lipoic acid ester as a transducing agent, enabling an active surface and eliminating the need for external probes like hexaferrocyanate and ruthenium complexes. Additionally, the study is supported by theoretical research that optimizes and validates the specificity of this detection method, further enhancing its significance.
The principle of the method is depicted in Fig. 1.
 | ||
| Fig. 1 Schematic sketch of the aptasensor preparation and its use for OTA sensing using differential pulse voltammetry. | ||
Materials and methods
Apparatuses
All electrochemical measurements, including cyclic voltammetry (CV), electrochemical impedance spectroscopy (EIS), and differential pulse voltammetry (DPV), were conducted using a Metrohm-Autolab PGSTAT M204 electrochemical workstation (Utrecht, Netherlands). The acquired data were processed using NOVA v2.13 software. Screen-printed carbon electrodes (SPCEs) on PET sheets, featuring a 3 mm disk as the carbon working electrode, a printed Ag/AgCl reference electrode, and a carbon counter electrode, were employed for the electrochemical experiments. The electrodes were fabricated using a DEK-248 screen printer from DEK International (Weymouth, UK).For imaging purposes, SEM micrographs and TEM analyses were carried out using a Thermo Fisher Scientific Quanta 650 FEG E-SEM scanning electron microscope (MA, US).
Chemicals and biochemicals
Lipoic acid (99%), ferrocenylmethanol (97%), diméthylaminopyridine (99%), HAuCl4·3H2O (99%), dicyclohexylcarbimide (99%), sodium citrate (99%), mercaptoethanol (99%), phosphate-buffered saline (PBS) tablets, and all solvents were purchased from Sigma-Aldrich (Germany) and used as received without further purification. The 3′-thiolated DNA aptamer (5′-GAT CGG GTG TGG GTG GGG TAA AGG GAG CAT CGG ACA AAA AAA AAA AAA AAA AAA-C6H12-SH-3′),25 ochratoxin A (OTA), ochratoxin B (OTB), picrotoxin (PIC) were purchased from Isogen Life Science (Spain). The phosphate-buffered saline solution (PBS, pH = 7.4) was prepared by dissolving PBS tablets in deionized water according to the manufacturer recommendations. Tris–EDTA (TE) buffer formed 10 mM TRIS and 1 mM EDTA at pH = 7.5 was used to prepare the aptamer solution. Deionized water, produced using MilliQ system (>18.2 MΩ cm) purchased from Millipore Inc., was used for the preparation of all the solutions.Preparation of the platform
The synthesis of ferrocenyl lipoic acid ester (FcL) followed established literature procedures.26–31 Gold nanoparticles with a size of 20 nm were prepared using the Turkevich method.32 Electrodes underwent an initial cleansing and activation process through 5 voltammetric cycles ranging from 0.0 to +1.5 V in a 0.5 M H2SO4 acidic solution, followed by thorough rinsing with deionized water.Subsequently, 50 μL of AuNPs was applied to the electrode surface, initiating a two-step nanostructuring process. The first step involved cycling from +0.8 to +1.5 V at a sweep rate of 50 mV s−1 to oxidize Au(0) to Au(III). The second step consisted of cycling from +0.8 to 0.0 V at the same sweep rate to reduce Au(III) back to Au(0). This chemical treatment facilitated the adsorption of gold nanoparticles onto the carbon surface.
In a separate solution, 50 μL of a 0.1 M ethanolic solution of FcL was diluted in 450 μL of deionized water to prepare a 10 mM (or 1 mM in the discussion part) FcL solution. The nanostructured electrode was immersed in this solution for several hours to form a dense FcL self-assembled monolayer on the surface of the gold nanoparticles.
The aptamer, dissolved in TE buffer at a concentration of 10 μM, was cast onto the surface of the gold-modified electrode and incubated in a closed container at 4 °C for 16 hours to prevent solvent evaporation. Subsequently, the modified electrode underwent rinsing with deionized water and was immersed for 20 minutes in a 1 mM mercaptoethanol solution. This step served to remove physically adsorbed aptamers and block the bare electrode surface by binding to the gold surface.
Electrochemical measurements
Voltametric measurements were conducted in triplicate by applying 50 μL of a phosphate-buffered saline solution containing the analyte onto the working electrode. CV and DPV curves were recorded at a sweep rate of 50 mV s−1 (pulse amplitude: 5 mV and pulse time: 100 ms) within the potential range of −0.1 to +0.4 V. Measurements were taken both before and after sequential additions of 2 μL solutions to achieve OTA concentrations of 0.5, 5, 10, 30, 50, and 70 ng mL−1 in PBS.For the selectivity tests, 10 μL of a 100 ng mL−1 solution of OTB and PIC were applied to the biosensor's surface. The DPV current was measured and compared to that obtained for 10 μL of a 30 ng mL−1 OTA solution. Specificity was assessed through a comparative analysis of currents obtained for 10 μL solutions containing 30 ng mL−1 of OTA and a mixture of OTB (30 ng mL−1), picrotoxin (100 ng mL−1), and OTA (30 ng mL−1). Chemical structures of OTA, OTB and PIC are given in Fig. S7 from the ESI.†
In silico study
The molecular docking between aptamer and OTA was conducted using AutoDock Vina.37 Subsequently, Discovery Studio Visualizer was used to predict and visualize the interactions between aptamer and OTA.38
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles. Detailed MD simulations using the complex structures were conducted with the CHARMM36.40 The complex is solvated in a cubic box with keeping 10 Å between the complex and the edge of the solvated box. Sodium and chloride ions were added to neutralize the charge of the system. In all simulations the condition was set at the room temperature (310 K) and the atmospheric pressure (1 bar) to closely mimic the general experiment conditions. Subsequently, the fully temperature and pressure equilibrated system was used as the initial configuration for the MD production dynamic analysis. All simulations were conducted using a 2 fs time step.
000 cycles. Detailed MD simulations using the complex structures were conducted with the CHARMM36.40 The complex is solvated in a cubic box with keeping 10 Å between the complex and the edge of the solvated box. Sodium and chloride ions were added to neutralize the charge of the system. In all simulations the condition was set at the room temperature (310 K) and the atmospheric pressure (1 bar) to closely mimic the general experiment conditions. Subsequently, the fully temperature and pressure equilibrated system was used as the initial configuration for the MD production dynamic analysis. All simulations were conducted using a 2 fs time step.Results and discussion
Preparation of the modified electrodes
Following 20 cycles, particulates began forming dense agglomerates (Fig. 2B). In comparison to other methods, such as passive and electrochemical gold salt reduction for depositing gold nanoparticles,45 this technique has several advantages. These include size homogeneity, high dispersibility into the carbon graphite surface, and stability in the electrochemical response (Table S1 and Fig. S2†). Indeed, drop-casted gold nanoparticles have been observed to being washed out during the stepwise preparation and cleansing of the bioelectrodes.46 Additionally, the deposited particles from the salt reduction have been shown to form large agglomerates, as demonstrated by Argoubi et al.28
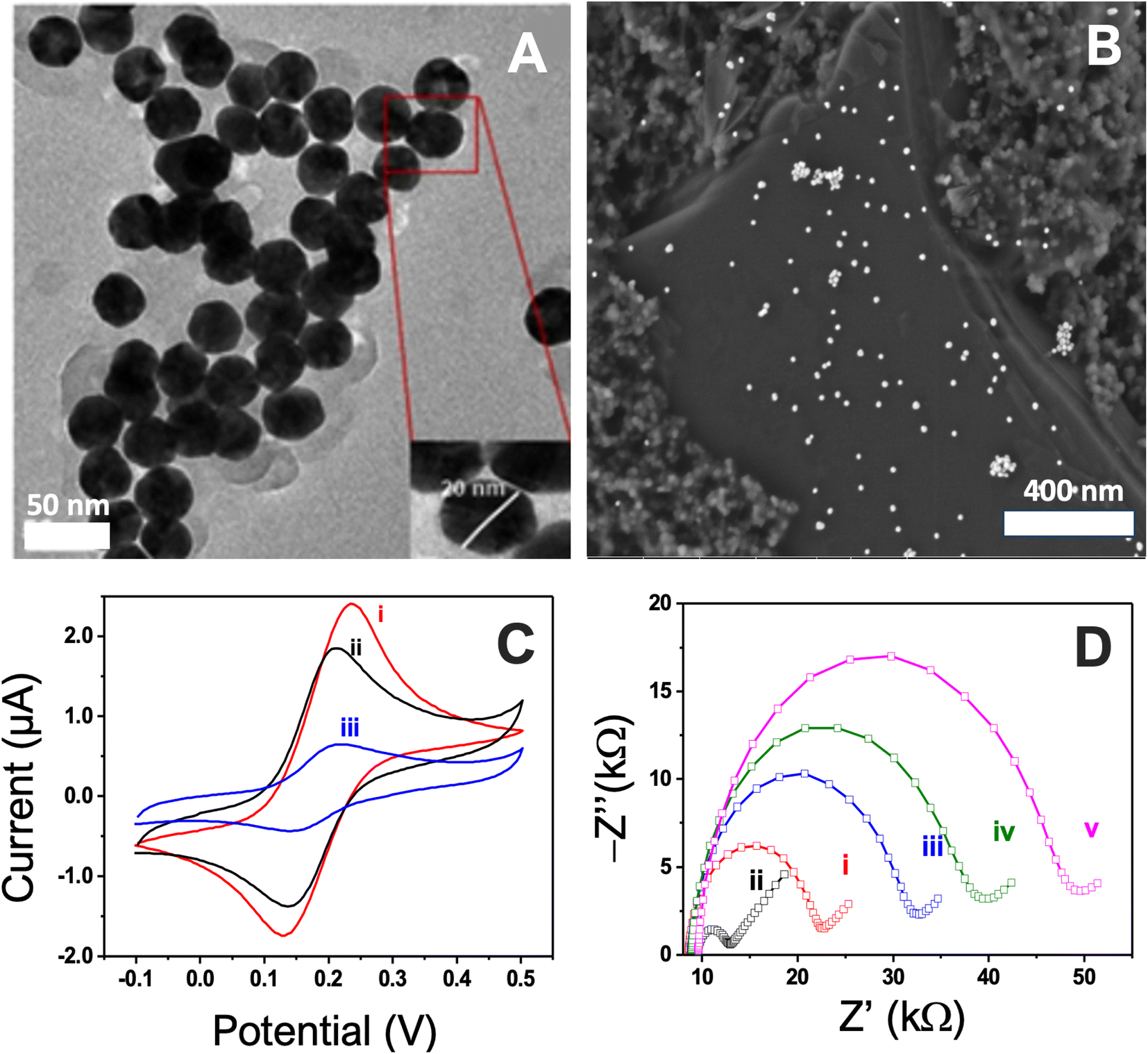 | ||
| Fig. 2 TEM (A), SEM (B) carbon surface obtained after nanostructuration using 20 cyclic sweeps, (C) CV curves of (i) functionalization of AuNPs by ferrocene derivative, (ii) conjugation with the aptamer, (iii) coverage with mercaptoethanol and (D) stepwise characterization of the surface modification using EIS, (i) bare SPCE, (ii) AuNPs nanostructured electrode, (iii) functionalization of AuNPs by ferrocene derivative, (iv) conjugation with the aptamer, (v) coverage with mercaptoethanol. | ||
Optimization of the working parameters
 | ||
| Fig. 3 (A) Optimization of (A) pH of the working buffer solution and (B) the incubation of FcL, (C) calibration curve for OTA aptasensing in the range of 0.5 to 70 ng mL−1. (D) Selectivity tests in presence of 100 ng mL−1 of picrotoxin (PIC), 100 ng mL−1 of ochratoxin B (OTA), a mixture of 100 ng mL−1 of PIC and 100 ng mL−1 of OTB compared to 30 ng mL−1 of OTA. MIX bar is related to the electrode challenged with 30 ng mL−1 of OTA, 100 ng mL−1 of OTB and 100 ng mL−1 of PIC. | ||
Aptasensing of ochratoxin A
The DPV results revealed a proportional decrease in current upon successive additions of OTA aliquots containing known concentrations of the analyte to the device. This phenomenon can be attributed to the blocking effect arising from the formation of a biocomplex between the Aptamer and OTA, as illustrated in the DPV curves presented in Fig. S3.†The linear regression equation was fitted to ΔI (nA) = 0.10736 × [OTA] (ng mL−1) + 2.1087 (r = 0.993) (Fig. 3C). The estimated limits of detection and quantification were determined as 11 and 33 pg mL−1, respectively. These values were calculated based on the concentration of toxin corresponding to the 3.3 and 10 times “s/m” ratio, where “s” represents the standard deviation of the blank signal (three replicates), and “m” denotes the slope of the related calibration curve. The linear range spans from 0.5 to 70 ng mL−1, with a limit of detection of 11 pg mL−1. Importantly, this range encompasses the authorized levels of OTA in various commodities such as cereal-processed goods and beverages like wine and grape juice. Furthermore, the limit of detection is approximately 180 times below the lowest permissible limit, underscoring the utility of this method for accurately determining OTA levels in widely consumed products.
Selectivity of OTA aptasensor
In assessing the selectivity of OTA detection for practical implementation, we compared the current variations induced by other mycotoxins, namely ochratoxin B and picrotoxin (Fig. S7†). As depicted in Fig. 3D (DPV curves are given in Fig. S5†), the changes in current intensity induced by these two mycotoxins were minimal, even at concentrations three times higher than that of the target mycotoxin. For the specificity test, we introduced a mixture of OTB and picrotoxin followed by a mixture of OTA, OTB, and picrotoxin to the aptasensor. Interestingly, only the latter mixture induced a significant current drop, indicating a high specificity of the sensing platform for OTA. Despite the structural similarity between OTA and OTB, the latter did not interfere with the aptasensor response. This ability to distinguish between them is crucial for the selectivity of the aptasensing platform.Comparison with literature
While more sensitive electrochemical approaches for ochratoxin A detection, such as those involving methylene blue (MB) intercalation,49 a fluorescent aptamer-based biosensor utilizing a cascade strand displacement reaction,50 polythionine and IrO2 nanoparticles22 or DNAzyme/aptamer and enzymatic reading51 have been recently reported, our aptasensing platform offers notable analytical advantages. The current approach attains an exceptionally low limit of detection without the need for amplification or the intercalation of redox species into the DNA structure or using carbonaceous nanomaterials. This achievement is made possible by leveraging a novel nanostructured faradaic platform. Consequently, our approach is simpler and more cost-effective, providing a wide range of response and one of the lowest reported detection limits for differential pulse voltammetry with ferrocene labeling in OTA detection (Table 1).| Platform | Detection technique | Linear range (ng mL−1) | LoD (ng mL−1) | Ref. |
|---|---|---|---|---|
| SPCE, oxidation of amines using HDMA and MB tagged anti-OTA | DPV | 0.01–5.0 | 0.01 | 49 |
| OTA@AP@MBs/cDNA cascade strand displacement reaction | FL | 1–2000 | 0.63 | 50 |
| Single-walled carbon nanohorns | FL | 8.0–200.0 | 6.80 | 52 |
| Aptamer/AuNPs | Colorimetry | 0.5–6.0 | 0.4 | 53 |
| Aptamer/Au@Fe3O4NPs | Colorimetry | 0.5–100 | 0.03 | 54 |
| Aptamer/DNAzyme/Fe3O4NPs/glucometer | Amperometry | 0.001–250 | 0.11 | 51 |
| Aptamer/thionine-IrO2NPs/SPCE | EIS | 0.005–40 | 0.005 | 22 |
| Aptamer/diazonium/SPCE | EIS | 0.15–2.5 | 0.15 | 55 |
| Aptamer/FcL/AuNPs/SPCE | DPV | 0.5–70 | 0.011 | This work |
Determination of OTA levels in real samples
To assess the practical reliability of the proposed sensing system, the aptasensor was employed to determine the recoveries of five different OTA concentrations using standard addition methods in ground coffee beans and French wine samples9 (DPV curves are given in Fig. S6†). The results, as presented in Table 2, reveal recoveries within the range of 97.4–97.2% and 107.2–108.0% respectively for the two samples. These findings affirm that the proposed sensing system is suitable for the analysis of OTA in agricultural commodities. The aptasensor's robust performance in the quantitative determination of mycotoxins in food samples, without the need for sample pretreatment or concentration, holds significant promise for enhancing quality control in food safety. The system provides a simple, rapid, and sensitive testing solution for monitoring mycotoxins in agricultural products.| Samples | Detected (D) | Added (A) | Detected after addition (R) | % Recovery |
|---|---|---|---|---|
| 100 × (R − D)/A | ||||
| Wine | 1.10 | 5.00 | 6.46 | 107.20 ± 3.60 |
| 10.00 | 11.90 | 108.00 ± 3.20 | ||
| Coffee | 11.40 | 5 | 16.27 | 97.40 ± 2.40 |
| 10 | 21.12 | 97.20 ± 2.85 |
In silico study
In silico simulations, by molecular docking and molecular dynamics, can provide several useful information on the binding site, the interaction energies and study degradation kinetics,56–60 especially if coupled to experimental techniques.57,61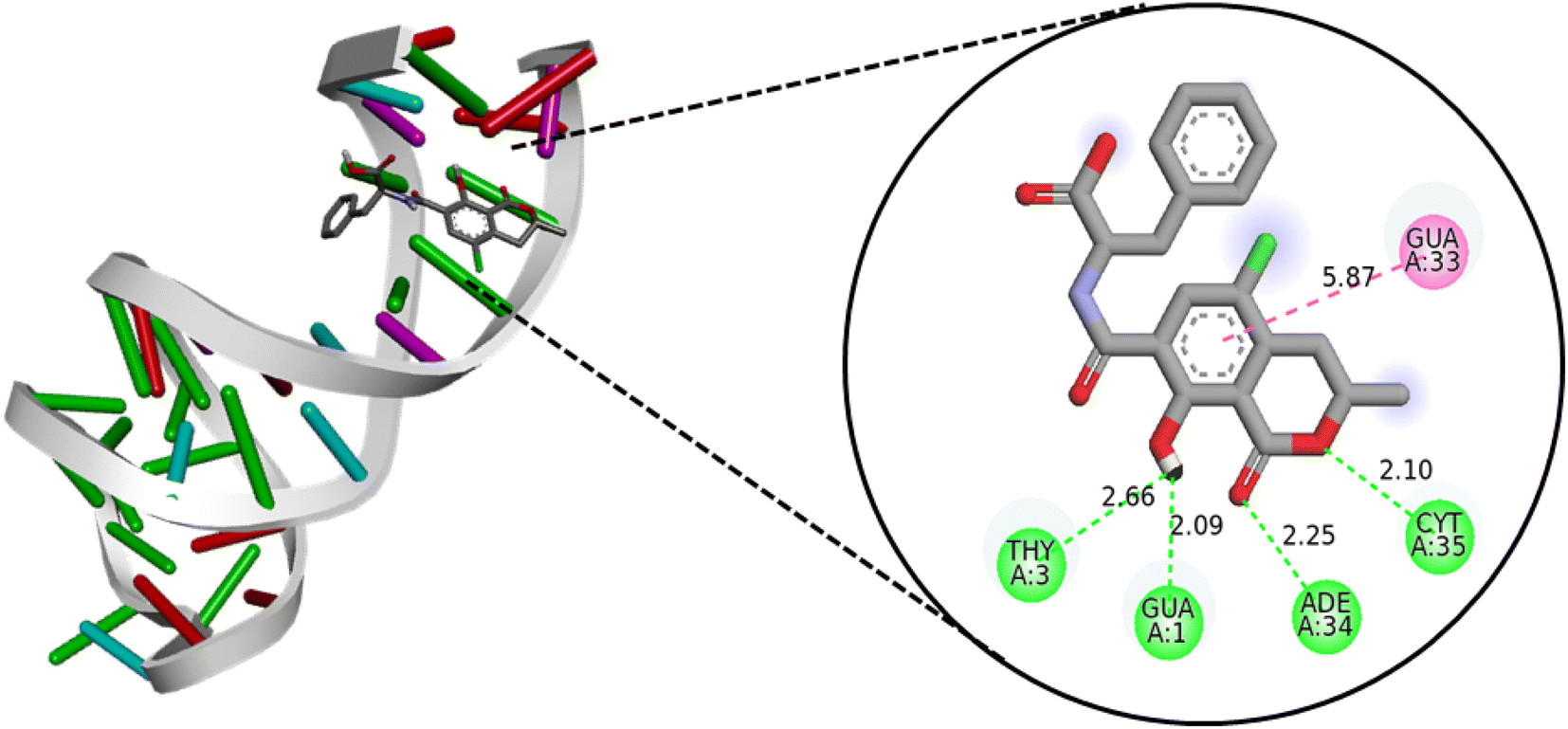 | ||
| Fig. 4 Interaction of ochratoxin A with the aptamer obtained by docking using Vina software visualized with Discovery Studio. | ||
 | ||
| Fig. 5 Time-dependent RMSD (A) and RoG (B) plots for the associated and the free aptamer for the last 20 ns of the simulation time. Per-residue contribution plot for the aptamer in mechanics energy (C). | ||
Furthermore, the RoG results, as depicted in Fig. 5B, align with the RMSD analysis, indicating that the associated aptamer is more stable than the free one, and the system has achieved convergence. To better understand this stability, an analysis of hydrogen bond interactions between the ligand and the receptor was conducted. The results indicate that OTA primarily interacts with six nucleotides from the aptamer – GUA1, ADE2, THY3, GUA25, ADE26, and ADE34 – with a frequency of occupancy exceeding 50% throughout the simulation time (Table S3†). These findings are consistent with the docking results, confirming the stability of the interaction pose between the aptamer and OTA.
Further analysis reveals that CYT28, ADE29, and THY30 nucleotides from the aptamer play significant roles in the mechanical energy (ΔEelec and ΔEVdW), as illustrated in Fig. 5C. The calculated free energy for the solvated complex formed by the association of the aptamer and OTA is −9.22 kcal mol−1, with a substantial contribution from ΔEelec and ΔEVdW. To comprehend the effect of different energy contribution terms, the results are detailed in Table S4.†
These results enable the prediction of interaction poses and identify residues involved in non-bonded interactions with OTA. The affinity and stability of the formed complex can be reliably predicted using these findings. The free binding energy further confirms the system's convergence, supported by reasonable values of RMSD and RoG. The stability observed is attributed to the formation of hydrogen bonds between the ligand and six nucleotides from the aptamer, along with three electrostatic interactions with three different nucleotides from the aptamer.
Conclusion
This study presents a straightforward design for an aptasensing device targeting ochratoxin A, employing gold nanoparticles, a ferrocene derivative, and an anti-OTA aptamer. The platform capitalizes on the alterations in the ferrocene signal induced by the folding of the aptamer in the presence of OTA, which avoid using hexacyanoferrate or ruthenium complexes for signal transduction. The device exhibits remarkable sensitivity, detecting OTA within the range of 0.5 to 70 ng mL−1, with a limit of detection of 11 pg mL−1. This detection limit falls below the threshold levels established by the EC for agricultural commodities. The aptasensing device was successfully applied to quantify OTA in complex untreated media, specifically coffee beans, and French wine, achieving good recoveries (97.20 ± 2.85 and 108.00 ± 3.20) without the need for sample pretreatment. Additionally, an in silico evaluation, encompassing structural similarity, molecular docking, and molecular dynamics, was conducted to unveil the key binding region of the OTA aptamer through molecular docking technology.Data availability
Data will be made available on request.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at Imam Mohammad Ibn Saud Islamic University (IMSIU) for funding and supporting this work through Research Partnership Program no RP-21-09-69.References
- G. Atik, N. M. Kilic, N. Horzum, D. Odaci and S. Timur, ACS Appl. Mater. Interfaces, 2023, 15, 24109–24119 CrossRef CAS PubMed
.
- S. Liu, F. Zhao, K. Xu, M. Cao, M. Sohail, B. Li and X. Zhang, Anal. Chim. Acta, 2023, 342044 Search PubMed
- K. Y. Goud, K. K. Reddy, M. Satyanarayana, S. Kummari and K. V. Gobi, Microchim. Acta, 2019, 187, 29 CrossRef PubMed
- F. K. Algethami, A. Rabti, M. Mastouri, S. Ben Aoun, B. Y. Abdulkhair and N. Raouafi, Talanta, 2023, 258, 124445 CrossRef CAS
- F. Algethami, A. Rabti, M. Mastouri, B. Abdulkhair, S. Ben Aoun and N. Raouafi, RSC Adv., 2023, 13, 21336–21344 RSC
- R. A. El-Sayed, A. B. Jebur, W. Kang and F. M. El-Demerdash, J. Future Foods, 2022, 2, 91–102 CrossRef
- S. Ben Aissa, R. K. Mishra, N. Raouafi and J. L. Marty, in Nanosensors, CRC Press, 2023, p. 205 Search PubMed
- L. Wang, Q. Wang, S. Wang, R. Cai, Y. Yuan, T. Yue and Z. Wang, Curr. Res. Food Sci., 2022, 5, 1539–1549 CrossRef CAS PubMed
- N. Mejri-Omrani, A. Miodek, B. Zribi, M. Marrakchi, M. Hamdi, J.-L. Marty and H. Korri-Youssoufi, Anal. Chim. Acta, 2016, 920, 37–46 CrossRef CAS PubMed
- I. M. Hwang, J. Y. Jeong, B. Park, J. Y. Choi, N. Khan, N. Jamila, B. R. Yoon and J. S. Kim, Food Addit. Contam., Part A: Chem., Anal., Control, Exposure Risk Assess., 2023, 40, 1275–1284 CrossRef CAS
- W. Chtioui, S. Heleno, Q. Migheli and P. Rodrigues, Int. J. Food Microbiol., 2023, 407, 110425 CrossRef CAS PubMed
- F. Kardani, A. Z. Jelyani, M. Rashedinia, S. Shariati, M. Hashemi, S. M. A. Noori and M. Mahdavinia, Food Chemistry Advances, 2023, 3, 100490 CrossRef
- J. Zhang, D. Xu, Y. Zhang, Z. Luo, Y. Zhao, X. Zheng, H. Yang and Y. Zhou, Spectrochim. Acta, Part A, 2024, 304, 123312 CrossRef CAS PubMed
- M. Zouari, S. Campuzano, J. Pingarrón and N. Raouafi, Biosens. Bioelectron., 2017, 91, 40–45 CrossRef CAS PubMed
- F. K. Algethami, A. Rabti, M. Mastouri, S. Ben Aoun, L. S. Alqarni, M. R. Elamin and N. Raouafi, Microchim. Acta, 2023, 190, 434 CrossRef CAS PubMed
- S. Baachaoui, M. Mastouri, M. Meftah, B. Yaacoubi-Loueslati and N. Raouafi, Biosensors, 2023, 13, 240 CrossRef CAS
- S. Baachaoui, W. Mabrouk, K. Charradi, B. Slimi, A. M. Ramadan, R. M. I. Elsamra, A. Alhussein, S. M. A. S. Keshk and N. Raouafi, R. Soc. Open Sci., 2023, 10, 230294 CrossRef CAS PubMed
- B. Ouedraogo, S. Baachaoui, A. Tall, I. Tapsoba and N. Raouafi, Microchim. Acta, 2023, 190, 316 CrossRef CAS PubMed
- S. Smith, J. G. Korvink, D. Mager and K. Land, RSC Adv., 2018, 8, 34012–34034 RSC
- J. A. Otoo and T. S. Schlappi, Biosensors, 2022, 12, 124 CrossRef CAS PubMed
- G. Gökçe, S. B. Aissa, K. Nemčeková, G. Catanante, N. Raouafi and J.-L. Marty, Food Control, 2020, 115, 107271 CrossRef
- L. Rivas, C. C. Mayorga-Martinez, D. Quesada-González, A. Zamora-Gálvez, A. de la Escosura-Muñiz and A. Merkoçi, Anal. Chem., 2015, 87, 5167–5172 CrossRef CAS PubMed
- Y. Hou, N. Long, Q. Xu, Y. Li, P. Song, M. Yang, J. Wang, L. Zhou, P. Sheng and W. Kong, Food Chem., 2023, 403, 134375 CrossRef CAS PubMed
- F. Dridi, M. Marrakchi, M. Gargouri, J. Saulnier, N. Jaffrezic-Renault and F. Lagarde, Anal. Methods, 2015, 7, 8954–8960 RSC
- L. Barthelmebs, J. Jonca, A. Hayat, B. Prieto-Simon and J.-L. Marty, Food Control, 2011, 22, 737–743 CrossRef CAS
- A. Mars, C. Parolo, N. Raouafi, K. Boujlel and A. Merkoçi, J. Mater. Chem. B, 2013, 1, 2951–2955 RSC
- A. Mars, C. Parolo, A. de la Escosura-Muñiz, N. Raouafi and A. Merkoçi, Electroanalysis, 2016, 28, 1795–1802 CrossRef CAS
- W. Argoubi, M. Saadaoui, S. Ben Aoun and N. Raouafi, Beilstein J. Nanotechnol., 2015, 6, 1840–1852 CrossRef CAS PubMed
- M. Li, L.-Q. Jiang, L. Lin, Y.-F. Li, D.-L. Yu, L.-L. Cui and X.-Q. He, J. Solid State Electrochem., 2014, 18, 2743–2753 CrossRef CAS
- M. Zouari, S. Campuzano, J. Pingarrón and N. Raouafi, Electrochim. Acta, 2018, 262, 39–47 CrossRef CAS
- E. Vargas, E. Povedano, V. Montiel, R. Torrente-Rodriguez, M. Zouari, J. Montoya, N. Raouafi, S. Campuzano and J. Pingarron, Sensors, 2018, 18, 863 CrossRef PubMed
- J. Kimling, M. Maier, B. Okenve, V. Kotaidis, H. Ballot and A. Plech, J. Phys. Chem. B, 2006, 110, 15700–15707 CrossRef CAS PubMed
- M. Antczak, M. Popenda, T. Zok, J. Sarzynska, T. Ratajczak, K. Tomczyk, R. W. Adamiak and M. Szachniuk, Acta Biochim. Pol., 2016, 63(4), 737–744 CAS
- S. Li, W. K. Olson and X.-J. Lu, Nucleic Acids Res., 2019, 47(W1), W26–W34 CrossRef CAS PubMed
- X.-J. Lu and W. K. Olson, Nat. Protoc., 2008, 3, 1213–1227 CrossRef CAS PubMed
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30(16), 2785–2791 CrossRef CAS PubMed
- J. Eberhardt, D. Santos-Martins, A. F. Tillack and S. Forli, J. Chem. Inf. Model., 2021, 61, 3891–3898 CrossRef CAS
- BIOVIA Discovery Studio Visualizer, https://discover.3ds.com/discovery-studio-visualizer-download Search PubMed.
- J. C. Phillips, D. J. Hardy, J. D. C. Maia, J. E. Stone, J. V. Ribeiro, R. C. Bernardi, R. Buch, G. Fiorin, J. Hénin, W. Jiang, R. McGreevy, M. C. R. Melo, B. K. Radak, R. D. Skeel, A. Singharoy, Y. Wang, B. Roux, A. Aksimentiev, Z. Luthey-Schulten, L. V. Kalé, K. Schulten, C. Chipot and E. Tajkhorshid, J. Chem. Phys., 2020, 153(4), 044130 CrossRef CAS
- J. Huang and A. D. MacKerell, J. Comput. Chem., 2013, 34(25), 2135–2145 CrossRef CAS PubMed
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14(1), 33–38 CrossRef CAS
- M. Scheurer, P. Rodenkirch, M. Siggel, R. C. Bernardi, K. Schulten, E. Tajkhorshid and T. Rudack, Biophys. J., 2018, 114(3), 577–583 CrossRef CAS PubMed
- B. R. I. Miller, T. D. Jr. McGee, J. M. Swails, N. Homeyer, H. Gohlke and A. E. Roitberg, J. Chem. Theory Comput., 2012, 8, 3314–3321 CrossRef CAS PubMed
- H. Liu and T. Hou, Bioinformatics, 2016, 32(14), 2216–2218 CrossRef CAS PubMed
- D. Antuña-Jiménez, M. B. González-García, D. Hernández-Santos and P. Fanjul-Bolado, Biosensors, 2020, 10, 9 CrossRef PubMed
- J. T. Steven, V. B. Golovko, B. Johannessen and A. T. Marshall, Electrochim. Acta, 2016, 187, 593–604 CrossRef CAS
- R. Sahli, C. Fave, N. Raouafi, K. Boujlel, B. Schöllhorn and B. Limoges, Langmuir, 2013, 29, 5360–5368 CrossRef CAS PubMed
- M. Hamami, N. Raouafi and H. Korri-Youssoufi, Appl. Sci., 2021, 11, 1382 CrossRef CAS
- G. Catanante, R. K. Mishra, A. Hayat and J.-L. Marty, Talanta, 2016, 153, 138–144 CrossRef CAS PubMed
- B. Han, C. Fang, L. Sha, M. Jalalah, M. S. Al-Assiri, F. A. Harraz and Y. Cao, Food Chem., 2021, 338, 127827 CrossRef CAS PubMed
- S. Zhang, Y. Luan, M. Xiong, J. Zhang, R. Lake and Y. Lu, ACS Appl. Mater. Interfaces, 2021, 13, 9472–9481 CrossRef CAS PubMed
- L. Lv, C. Cui, C. Liang, W. Quan, S. Wang and Z. Guo, Food Control, 2016, 60, 296–301 CrossRef CAS
- C. Yang, Y. Wang, J.-L. Marty and X. Yang, Biosens. Bioelectron., 2011, 26, 2724–2727 CrossRef CAS PubMed
- C. Wang, J. Qian, K. Wang, X. Yang, Q. Liu, N. Hao, C. Wang, X. Dong and X. Huang, Biosens. Bioelectron., 2016, 77, 1183–1191 CrossRef CAS PubMed
- R. K. Mishra, A. Hayat, G. Catanante, C. Ocaña and J.-L. Marty, Anal. Chim. Acta, 2015, 889, 106–112 CrossRef CAS PubMed
- S. J. Lee, J. Cho, B.-H. Lee, D. Hwang and J.-W. Park, Biomedicines, 2023, 11, 356 CrossRef CAS PubMed
- S. Ben Aissa, M. Mastouri, G. Catanante, N. Raouafi and J. L. Marty, Antibiotics, 2020, 9, 860 CrossRef PubMed
- M. Mastouri, S. Baachaoui, A. Mosbah and N. Raouafi, RSC Adv., 2022, 12, 13003–13013 RSC
- J. Santos, T. Castro, A. Venâncio and C. Silva, Heliyon, 2023, 9, e19921 CrossRef CAS PubMed
- Y. Xie, L. A. Eriksson and R. Zhang, Nucleic Acids Res., 2020, 48, 6471–6480 CrossRef CAS PubMed
- F. A. Azri, J. Selamat, R. Sukor, N. A. Yusof, N. H. A. Raston, S. Eissa, M. Zourob and R. Chinnappan, Anal. Bioanal. Chem., 2021, 413, 3861–3872 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra08567h |
| This journal is © The Royal Society of Chemistry 2024 |