
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePiers–Rubinsztajn reaction to unlock an 8-step synthesis of 7-hydroxy cannabidiol†
Emanuele Cocco‡
 a,
Debora Iapadre‡a,
Alessandro Brusaa,
Pietro Allegrinib,
Stephen P. Thomasc,
Fabio Pesciaioli*a and
Armando Carlone*ad
a,
Debora Iapadre‡a,
Alessandro Brusaa,
Pietro Allegrinib,
Stephen P. Thomasc,
Fabio Pesciaioli*a and
Armando Carlone*ad
aDepartment of Physical and Chemical Sciences, Università degli Studi dell’Aquila, via Vetoio, 67100, L'Aquila, Italy. E-mail: fabio.pesciaioli@univaq.it; armando.carlone@univaq.it; Web: https://www.carloneresearch.eu/
bIndena SpA, viale Ortles, 12, 20139 Milan, Italy
cEaStCHEM School of Chemistry, The University of Edinburgh, Edinburgh EH9 3FJ, UK
dINSTM, Consorzio Nazionale per la Scienza e Tecnologia dei Materiali, RU L'Aquila, Italy
First published on 16th January 2024
Abstract
A scalable synthesis of 7-hydroxy cannabidiol (7-OH CBD), a primary metabolite of (−)-cannabidiol (CBD), is highly desirable, from an industrial point of view, to enable future clinical trials. A Piers–Rubinsztajn reaction was key to enable a mild deprotection and a concise synthesis of 7-OH CBD from commercially available CBD, in 31% overall yield.
Introduction
7-Hydroxy-cannabidiol (7-OH CBD, 1) is a primary metabolite of cannabidiol (CBD, 2) generated during the first stages of metabolism.1 Since 2018, the European Medicines Agency (EMA) and the U.S. Food and Drug Administration (FDA) have approved the first cannabidiol-based drug in pure form for the treatment of two severe forms of drug-resistant childhood epilepsy, or its intake in combination with commonly used antiepileptic drugs. Cannabidiol (CBD) is one of the most abundant phytocannabinoids obtained from Cannabis sativa and it is well known as a non-psychotic, analgesic, anticonvulsant, and anti-inflammatory compound. The cannabinoids, and their derivatives, have thus gained increasing attention in both academia and industry.2–4 The underlying mechanism by which CBD exerts its therapeutic efficacy has not been fully elucidated,5 though some of the observed effects have tentatively been attributed to metabolites including 7-OH CBD 1. Interest in 7-OH CBD 1 has significantly grown due to a number of effects that were observed in preclinical studies.5–7 Due to the lack of a synthesis of industrial interest, 7-OH CBD 1 has only been used as an analytical standard to date. The development of a robust synthesis is, therefore, critical in enabling any future prospect of targeted clinical trials. Of significant note, and inhibiting many synthetic efforts, is that in the presence of acid CBD, and its metabolites, cyclize to form Δ9-tetrahydrocannabinol (Δ9-THC), and derivatives, it is therefore imperative to plan a feasible synthesis that avoids such acidic reaction conditions.8 The directed oxidation of cannabinoids to obtain hydroxy-derivatives is a reported strategy which involves the use of selenium dioxide (SeO2) as an oxidizing agent.9 Although used for the oxidation of limonene,10 the selectivity of oxidation was low giving a mixture of different regioisomeric hydroxylation products. To the best of our knowledge, the first multistep synthesis of 7-OH CBD 1 was reported by Mechoulam and co-workers starting from commercially available CBD 2.11,12 The authors obtained the target compound in 8 steps, with a key limitation being the demethylation of the protected aryl OH groups requiring the use of methyl magnesium iodide in neat conditions at 210 °C. The forcing nature of this deprotection concisely highlights the challenge that lies in the final deprotection, where acidic conditions are to be avoided.We were interested in developing a synthesis of 7-OH CBD 1 that could be of industrial interest and generate enough material to undertake preclinical studies. Following preliminary discussions and a series of considerations, such as an established industrial process for the preparation of CBD 2 by our industrial partner, (−)-CBD 2 was selected as a key starting material, similarly to Mechoulam's route. At the outset, it was decided that the forcing conditions of the final deprotection had to be avoided, and the route would benefit from being shortened. Therefore, a redesign of the synthesis, along with alternative strategies for the final deprotection, was undertaken. Herein we report a 8-step synthesis to 7-hydroxy cannabidiol 1 starting from (−)-CBD 2 featuring two telescoped transformations, that avoids harsh deprotection conditions (Scheme 1). The application of a Piers–Rubinsztajn reaction, involving B(C6F5)3 (BCF) as a catalyst13 in the presence of pentamethyl disiloxane, allowed us to successfully carry out an acid-free deprotection of cannabidiol derivative under mild conditions.
 | ||
| Scheme 1 Overall synthesis of 7-hydroxy-cannabidiol 1. | ||
Results and discussion
The forward synthesis was started with protecting (−)-CBD 2 on the two phenolic groups with methyl tosylate14 to generate dimethoxy CBD 3 in quantitative yield; a subsequent regioselective epoxidation of the endocyclic double bond on the terpene moiety produced epoxide 4 in high yield. The choice of the protecting group on the phenolic hydroxy groups was restricted to methyl to enable the subsequent epoxide ring-opening reaction,11 as also confirmed in our labs. A Yamamoto rearrangement, enabled a concomitant ring-opening of the epoxide with the removal of the methyl hydrogen, smoothly producing cyclohexanol derivative 5. A strong, bulky base was found to be key for high regio- and chemoselectivity in the epoxide ring-opening reaction.15 LDA proved to be efficient and cost effective for the purpose, providing allylic alcohol 5 in quantitative yield, without any purification needed. An allylic rearrangement of 5, that would directly afford primary alcohol 6, was envisaged; if successful, this would indeed provide an advanced intermediate to 7-OH CBD 1. A conjugated nucleophilic substitution16 on the mesylated-derivative 5a was attempted using hydroxide as a nucleophile in aqueous conditions; to our delight, alcohol 6 was obtained in good yields in a telescoped procedure. It should be noted that, given the multiple steps, complex, biphasic solvent and reagent mixture, the yield of the reaction is scale dependent. Care must be taken to ensure all vessel and mixing parameters are matched. To our delight, the remaining mass balance was unreacted allylic alcohol 5 that could be recovered and subjected to the allylic rearrangement again. To afford 7-OH CBD 1 as the final product, the removal of the methoxy-protecting groups on the phenolic moieties posed a stimulating challenge. Indeed, strong acids or acidic conditions induce a tertiary carbocation formation on the isopropenyl substituent resulting in a cyclization to give the THC skeleton, while strong bases in oxidizing conditions give hydroxy-1,4-benzoquinone derivatives as byproducts. A number of different approaches that would avoid the aforementioned conditions were tested (Scheme 2). Mild Lewis acids were tested, such as FeCl3 and AlCl3 alone or in combination with Et3N or NaI; di-ether 6 was recovered unreacted or the reaction led to decomposition. Attention was then turned to the use of nucleophilic reagents including HPPh2 and tBuOK, which have been exploited in mild deprotection protocols of aryl methyl/benzyl/allylethers,17 but unfortunately only a single phenolic oxygen was deprotected, even under forcing conditions or when subjecting the mono-deprotected product 7 to a second round of the deprotection reaction. Additionally, sodium ethanethiolate has been used to induce a nucleophilic cleavage of the C–O bond, leaving the phenolate as a leaving group. Despite the reported treatment of CBD-ether 3 with 4 equivalents of NaSEt at 150 °C for 5 hours leading to CBD 2,18 in our hands it was unsuccessful yielding only monodeprotection. A similar result was obtained in our trials of NaSEt; the reaction with dimethoxy derivative 6 afforded only mono-deprotected 7, with only trace 7-OH CBD 1 detected by 1H NMR spectroscopy and MS. This issue suggests that in the presence of nucleophiles such as HPPh2 or NaSEt the first demethylation provides a methoxy phenolate that increases the electron density of the aromatic ring, rendering the second deprotection more difficult. While we were facing this challenge, Passarella and co-workers reported the successful deprotection on small scale of dimethoxy CBD 3 to CBD 2 by using a large excess of NaSEt, in two sequential steps, isolating the monodeprotected intermediate.19 Unfortunately, the same protocol did not prove successful in our hands for the deprotection of dimethoxy derivative 6; 7-OH CBD 1 was obtained in very low yield and only detectable by mass spectrometry. Derivatization of phenolate 7 with an easily removable electron-withdrawing group to affect the second deprotection was attempted, along with adjusting the pH, to no avail. | ||
| Scheme 2 Attempted reaction conditions to deprotect methoxy groups on 6. | ||
Eventually, we realized that the Piers–Rubinsztajn reaction20,21 could be the key to a successful deprotection although it had not been reported for the purpose in the context of cannabinoids. We, therefore, investigated whether it could be employed to our aim. The use of tris(pentafluorophenyl) borane (BCF) as a catalyst in the presence of silyl hydrides constitutes a mild and scalable deprotection methodology for aryl ethers.22,23 Lewis basic oxygen species, such as methyl ethers, can attack the activated Si-center of the Lewis complex formed between BCF and a siloxane. A reductive cleavage of both O-methyl groups on the phenolic oxygens brings about the release of methane and the formation of Ar–O–Si bonds in their place, that can be easily cleaved with TBAF in acid-free conditions. More importantly for the case of deprotecting dimethoxy derivative 6, the instant formation of the silyl ether is a key point; capping the negative charge of the phenolate, thus enabling the intermediate monodeprotected species 7 to undergo a second deprotection via nucleophilic attack. Initial tests using CBD-ether 3 as a model substrate were very encouraging; 1H NMR analysis showed the complete formation of CBD 2 as the main product. Unfortunately, subjecting dimethoxy derivative 6 to the same conditions resulted in the unexpected recovery of CBD 2. In hindsight, with the reductive cleavage of methyl groups, the unwanted deoxygenation is also promoted by the reaction system. As demonstrated by Yamamoto, primary and allylic alcohols can be reduced to alkanes using BCF and HSi(Et)3.24 Therefore, a series of protecting groups (see ESI†) were tested on the primary alcohol of an analogue of dimethoxy derivative 6; these had to be stable under the reaction conditions and, ultimately, be removed during the final step of the Piers–Rubinsztajn sequence. TIPS was eventually selected as the protecting group of choice for compound 6. After simple quantitative protection with TIPS, crude 6a is telescoped in the Piers–Rubinsztajn reaction to crude 6b, that is also telescoped to the final deprotection to 1. 7-OH CBD 1 was obtained in a satisfying 54% yield over the three steps (Scheme 3).
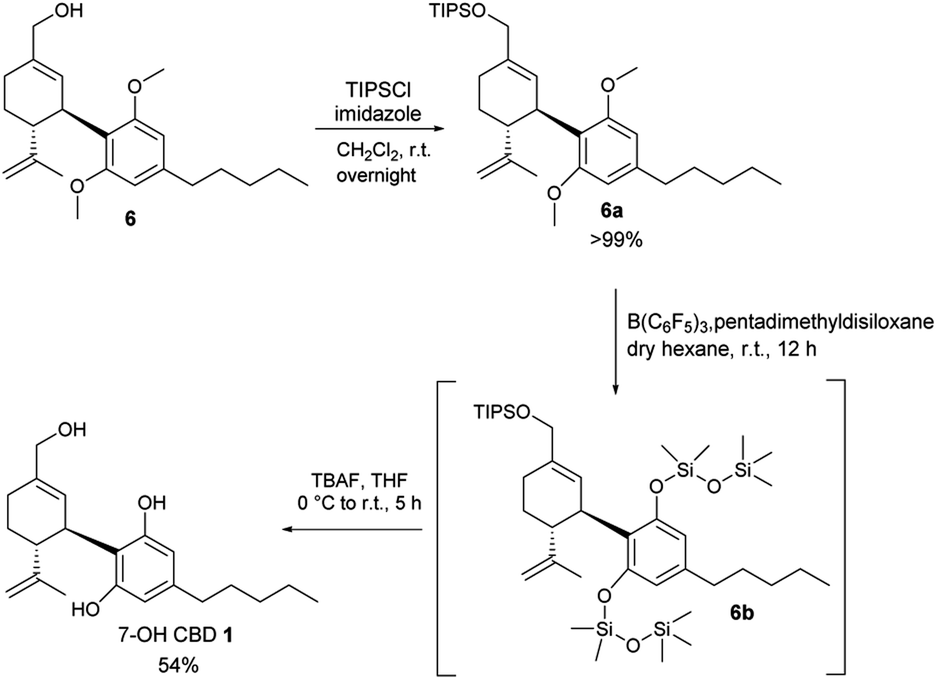 | ||
| Scheme 3 Deprotection of 6 in mild conditions employing Piers–Rubinsztajn reaction. | ||
Conclusions
In conclusion, we report the development of an 8-step synthesis, with two telescoped transformations, of 7-hydroxy-cannabidiol 1 in 31% overall yield. The synthesis features an allylic rearrangement and a Piers–Rubinsztajn reaction as key steps; in particular, the development of the final removal of the O-methyl groups was critical to efficiently effect the deprotection reaction under more mild and safer conditions. Interestingly, this type of deprotection of cannabidiol derivatives is reported for the first time, to the best of our knowledge, and we believe that such a mild deprotection will be highly attractive for cannabidiol chemistry. Further work is underway in our labs to access similar derivatives and undertake research for clinical studies on the pharmacological activity of 7-hydroxy-cannabidiol.Author contributions
E. C. and D. I. conceptualization, data curation, formal analysis, investigation, methodology. A. B. data curation, formal analysis, investigation, methodology. P. A. supervision. S. P. T. funding acquisition and supervision. F. P. and A. C. conceptualization, project administration, funding acquisition and supervision. D. I. and A. C. writing – original draft. All writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Indena SpA is gratefully acknowledged for in-kind donation. Prof. Filippo Romiti is acknowledged for fruitful discussions. We also thank Prof. Alex Adronov for insights. E. C., S. T., and A. C. thank the Royal Society of Chemistry for a Research Enablement Grant (E22-1692522330). D. I. acknowledges the Italian National PhD programme in Catalysis. F. P. thanks Ministero dell'Università e della Ricerca (PON-AIM1842894, CUP E18D19000560001) for funding this research. A. B. and A. C. acknowledge funding by the European Union – NextGenerationEU under the Italian Ministry of University and Research (MUR) National Innovation Ecosystem grant ECS00000041 – VITALITY– CUP E13C22001060006.Notes and references
- H. Li, Y. Liu, D. Tian, L. Tian, X. Ju, L. Qi, Y. Wang and C. Liang, Eur. J. Med. Chem., 2020, 192, 112163 CrossRef CAS PubMed.
- A. Maiocchi, J. Barbieri, V. Fasano and D. Passarella, ChemistrySelect, 2022, 7, e202202400 CrossRef CAS.
- J. A. A. Grimm, H. Zhou, R. Properzi, M. Leutzsch, G. Bistoni, J. Nienhaus and B. List, Nature, 2023, 615, 634–639 CrossRef CAS PubMed.
- G. Appendino and P. Allegrini, Chim. Ind., 2018, 6, 12–16 Search PubMed.
- J. L. Beers, D. Fu and K. D. Jackson, Drug Metab. Dispos., 2021, 49, 882–891 CrossRef CAS PubMed.
- C. Stott, N. Jones, B. Whalley, G. Stephens and C. Williams, US Pat., US 2017/0209390A1, 2017 Search PubMed.
- Y. Li, Q. Wu, X. Li, L. S. Von Tungeln, F. A. Beland, D. Petibone, L. Guo, P. Cournoyer, S. Choudhuri and S. Chen, Food Chem. Toxicol., 2022, 159, 112722 CrossRef CAS PubMed.
- R. K. Razdan, in The Total Synthesis of Natural Products, Wiley, 2007, vol. 4, pp. 185–262 Search PubMed.
- A. Breuer, C. G. Haj, M. V. Fogaça, F. V. Gomes, N. R. Silva, J. F. Pedrazzi, E. A. D. Bel, J. C. Hallak, J. A. Crippa, A. W. Zuardi, R. Mechoulam and F. S. Guimarães, PLoS One, 2016, 11, e0158779 CrossRef PubMed.
- S. Yoshitsugu, Bull. Chem. Soc. Jpn., 1969, 42, 3348–3349 CrossRef.
- S. Tchilibon and R. Mechoulam, Org. Lett., 2000, 2, 3301–3303 CrossRef CAS PubMed.
- L. O. Hanus, S. Tchilibon, D. E. Ponde, A. Breuer, E. Frideb and R. Mechoulam, Org. Biomol. Chem., 2005, 3, 1116–1123 RSC.
- V. Nori, F. Pesciaioli, A. Sinibaldi, G. Giorgianni and A. Carlone, Catalysts, 2022, 12, 5 CrossRef CAS.
- M. A. Arai, Y. Yamaguchi and M. Ishibashi, Org. Biomol. Chem., 2017, 15, 5025–5032 RSC.
- A. Yasuda, S. Tanaka, K. Oshima, H. Yamamoto and H. Nozaki, J. Am. Chem. Soc., 1974, 96, 6513–6514 CrossRef CAS.
- K. McAulay and J. S. Clark, Chem.–Eur. J., 2017, 23, 9761–9765 CrossRef CAS PubMed.
- W. Pan, C. Li, H. Zhu, F. Li, T. Li and W. Zhao, Org. Biomol. Chem., 2021, 19, 7633–7640 RSC.
- K. Abdur-Rashid, W. Jia and K. Abdur-Rashid, WO 2020/232545A1, 2020.
- P. Marzullo, A. Maiocchi, G. Paladino, U. Ciriello, L. Lo Presti and D. Passarella, Eur. J. Org. Chem., 2022, 22, e202200392 CrossRef.
- S. Rubinsztajn and J. A. Cella, Macromolecules, 2005, 38, 1061–1063 CrossRef CAS.
- J. M. Blackwell, E. R. Sonmor, T. Scoccitti and W. E. Piers, Org. Lett., 2000, 2, 3921–3923 CrossRef CAS PubMed.
- A. A. Torrens, A. L. Ly, D. Fong and A. Adronov, Eur. J. Org. Chem., 2022, e202200570 CrossRef CAS.
- T. A. Bender, J. A. Dabrowski and M. R. Gagne, ACS Catal., 2016, 6, 8399–8403 CrossRef CAS.
- V. Gevorgyan, M. Rubin, S. Benson, J. X. Liu and Y. Yamamoto, J. Org. Chem., 2000, 65, 6179–6186 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra08392f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |