
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCu(II)-catalyzed aerobic oxidative coupling of furans with indoles enables expeditious synthesis of indolyl–furans with blue fluorescence†
Shon Gangai‡
a,
Rushil Fernandes‡§
a,
Krishna Mhaskea and
Rishikesh Narayan *ab
*ab
aSchool of Chemical and Materials Sciences, Indian Institute of Technology Goa, GEC Campus, Farmagudi, Goa-403401, India. E-mail: rishikesh.narayan@iitgoa.ac.in
bSchool of Interdisciplinary Life Sciences, Indian Institute of Technology Goa, GEC Campus, Farmagudi, Goa-403401, India
First published on 3rd January 2024
Abstract
With the purpose of incorporating sustainability in chemical processes, there has been a renewed focus on utilizing earth-abundant metal catalysts to expand the repertoire of organic reactions and processes. In this work, we have explored the atom-economic oxidative coupling between two important electron-rich heterocycles – indoles and furans – using commonly available, inexpensive metal catalyst CuCl2·2H2O (<0.25$ per g) to develop an expeditious synthesis of indolyl–furans. Moreover, the reaction proceeded well in the presence of the so-called ‘ultimate oxidant’ – air, without the need for any external ligand or additive. The reaction was found to be scalable and to work even under partially aqueous conditions. This makes the methodology highly economical, practical, operationally simple and sustainable. In addition, the methodology provides direct access to novel indole–furan–thiophene (IFT)-based electron-rich π-conjugated systems, which show green-yellow fluorescence with large Stokes shift and high quantum yields. Mechanistic investigations reveal that the reaction proceeds through chemoselective oxidation of indole by the metal catalyst followed by the nucleophilic attack by furan.
Introduction
Furan represents a versatile class of electron-rich heterocycles with natural origin.1 Furan derivatives are abundant in nature as well as in active pharmaceutical ingredients such as ranitidine (antiulcer), lapatinib (anticancer), furosemide (diuretic) etc. (>12 FDA approved drugs).1 In addition, furan derivatives display a versatile reactivity profile ranging from electrophilic aromatic substitution to oxidative manipulation of the ring to obtain a range of advanced intermediates such as α, β-unsaturated esters, hydroxypyranones, and mono- and oligosaccharides, even leading to the synthesis of polyoxygenated natural products.2 Over the past decades, the importance of furan has attained a new dimension with its ever-increasing use in materials science for a variety of vital applications such as conducting polymers,3a photovoltaics,3b electroluminescent materials, self-healing materials3c etc.3 Short oligomers containing furan are being increasingly researched as possible alternatives to more prevalent thiophene-based π-conjugated compounds due to their favourable materials properties such as solubility in organic solvents, ease of processibility, crystal packing, chromism etc.4 Hence, expanding the repertoire of furan reactivity leading to the synthesis of novel furan derivatives with potential applications in materials science is highly desirable.Indolyl–furanoid, a heterobiaryl scaffold consisting of furan and indole is an important class of compound which shows promising properties for electroluminescent and photovoltaic materials5e besides an impressive bioactivity profile, e.g. as inhibitors against the most-frequently mutated RAS-derived cancers,5c acute myeloid leukemia (AML), matrix metalloproteinase (MMP)5d etc. (Scheme 1a).5 Despite the importance, their syntheses have mostly relied on traditional approaches such as dehydration of furylcarbinols,6a nucleophilic substitution,6b cyclization,6c Stille coupling6d etc. which suffer from multiple obvious disadvantages, most notably the poor atom economy due to the prefunctionalization of the substrates and the presence of redundant substituents in the product.6
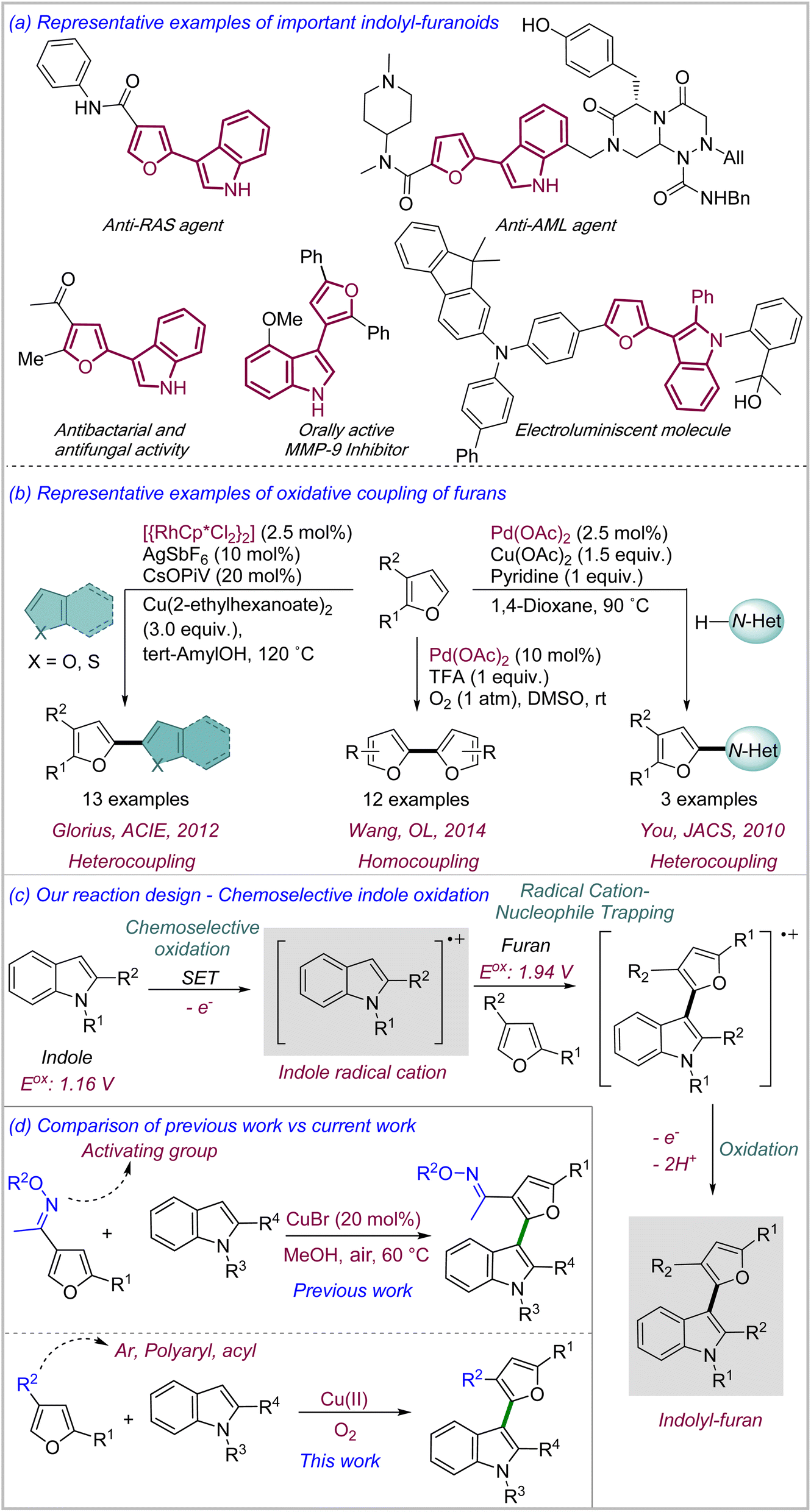 | ||
| Scheme 1 (a) Representative examples of important indolyl–furan derivatives; (b) representative examples of oxidative coupling of furans with other heterocycles for the synthesis of heterobiaryls; (c) our reaction design including the potential synthetic challenges; (d) comparison of previous work and current work. | ||
We sought to develop an oxidative coupling-based methodology for the expeditious synthesis of indole–furan heterobiaryl motif involving direct oxidation of C–H bonds in both furan and indole to form heterobiaryl C–C bond.7 To our surprise, there are only a limited number of oxidative coupling of furans reported in literature, especially for the synthesis of heterobiaryls.8 Moreover, most of these methodologies require Pd(II) or Rh(III) as catalysts with stoichiometric amount of external oxidants including Cu(II) salts. You,8a Glorius,8b and Seayad8c have reported limited examples of direct coupling of furans with N-heterocycles (xanthine, benzimidazole), O/S heterocycles (thiophene, benzofuran) and aryls respectively under relatively harsh conditions involving ligands, additives, high temp etc. (Scheme 1b). Wang has reported the Pd(II)-catalyzed homocoupling of furans.8f Mechanistically, most of these reactions proceed through either base-assisted deprotonation from the α-carbon or electrophilic metalation at the α-carbon of furan as the key steps. In our reaction design of the oxidative coupling of furan with indole, we sought to chemoselectively oxidize indole to its radical cation through single electron transfer (SET) and use the nucleophilicity of furan to trap the electrophilic indole radical cation to generate the key heterocoupling intermediate (Scheme 1c).9 Notably, the two most challenging aspects of the proposed design is: (a) to ensure chemoselective oxidation of indole in the presence of furan to prevent the formation of furan–furan and indole–indole homocoupling products and (b) to prevent the over-oxidation of indolyl–furan products which could also be potential substrates for oxidation through SET. Based on the reactivity demonstrated by Cu-based enzymes10 such as laccase, multicopper oxidase etc., which undertake selective single electron oxidation of electron-rich aromatic substrates in biotransformations while reducing O2 to H2O, we hypothesized that similar catalytic combinations involving earth-abundant metals11 such as Cu, Fe etc. as catalysts along with mild oxidants such as air, oxygen etc. could provide the favorable conditions required for the proposed chemoselective oxidation of indole. As a proof-of-concept, we recently reported our initial findings where Cu(I)/air combination was able to catalyze the direct coupling of a limited set of ‘oxime-ether activated’ furans with indoles (Scheme 1d).12 However, all the cross-coupled products had oxime ether as a redundant substituent which severely limited the scope of the reaction.
Herein, we report a CuCl2·2H2O-catalyzed general oxidative coupling of a variety of ‘unactivated’ furans with indoles in the presence of air as terminal oxidant. Besides the use of earth-abundant metal as catalyst and air as the green oxidant, the reaction also displays aqueous compatibility, which are the key tenets of sustainable catalysis. Interestingly, we also demonstrate that indole displays divergent reactivity under these conditions depending on their substitution patterns and resultant electronic nature. Finally, a selection of indolyl–furanoid derivatives were found to display blue to green-yellow fluorescence with large Stokes shift generally.
Results and discussion
Optimization of the reaction
We started our investigation with 2,4-diphenylfuran 1a and 1,2-dimethylindole 2a as the probe substrates under aerial conditions using catalytic CuBr in MeOH which showed only minimal reactivity and did not give any significant amount of the cross-coupled product 3a (Table 1, entry 1). This observation prompted us to use more active Cu(CH3CN)4PF6 as the catalyst to induce reactivity. Indeed, the reactivity improved to give 3a in a modest 24% yield with partial conversion (entry 2).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | (x mol%) | Solvent | Oxidant | Yieldb (%) |
| a Conditions: furan (1 equiv.), indole (2 equiv.), catalyst, rt or temperature as mentioned, solvent (as mentioned).b Yields were determined after column chromatography.c Reaction was carried out at 60 °C.d 4 equiv. of H2O2 was used.e 2 equiv. of oxone was used.f 1.2 equiv. of indole was used.g The reaction required 5 equiv. of indole for completion (CA: compressed air balloon; O2: oxygen balloon). | |||||
| 1 | CuBr | 20 | MeOH | Air | <5 |
| 2 | Cu(CH3CN)4PF6 | 20 | MeOH | Air | 24 |
| 3c | CuCl2·2H2O | 30 | MeOH | Air | 22 |
| 4c | CuCl2·2H2O | 30 | EtOH | Air | 36 |
| 5 | CuCl2·2H2O | 30 | EtOH | Air | 47 |
| 6 | CuCl2·2H2O | 30 | MeNO2 | Air | 39 |
| 7 | CuCl2·2H2O | 10 × 3 | EtOH | Air | 63 |
| 8d | CuCl2·2H2O | 20 | EtOH | H2O2 | 38 |
| 9e | CuCl2·2H2O | 20 | EtOH | Oxone | 30 |
| 10 | CuCl2·2H2O | 30 | EtOH | CA | 71 |
| 11 | CuCl2·2H2O | 30 | EtOH/MeCN (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1) :1) |
CA | 81 |
| 12 | CuCl2·2H2O | 30 | EtOH/MeCN (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
CA | 61 |
| 13 | CuCl2·2H2O | 20 | EtOH/MeCN (10:1) |
CA | 60 |
| 14f | CuCl2·2H2O | 30 | EtOH/MeCN (10:1) |
CA | 61 |
| 15 | CuCl | 20 | EtOH | CA | 78 |
| 16 | Anhyd. CuCl2 | 30 | EtOH/MeCN (10:1) |
CA | 54 |
| 17 | FeCl3·6H2O | 20 | Toluene | O2 | 55g |
| 18 | CuCl2·2H2O | 30 | EtOH/MeCN/H2O (10:1:2) |
CA | 72 |
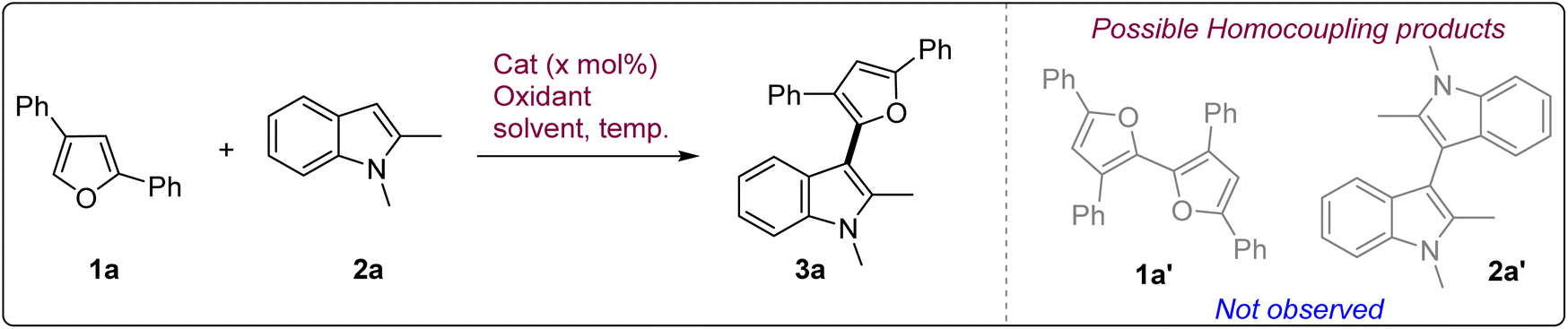
Attempts to improve conversion using Cu(CH3CN)4PF6 in other solvents such as CH3CN, DCE, CHCl3, toluene etc. either under aerial condition or with external oxidants such as oxone, H2O2, DTBP, TBHP etc. either didn't show any improvement in reactivity or proved to be too harsh leading to the oxidative decomposition of indole primarily (Table S1, ESI†). Further exploration of catalysts indicated that CuCl2·2H2O in MeOH upon mild heating at 60 °C was also able to catalyze the reaction partially to give 22% yield (entry 3). Changing the solvent to EtOH improved the conversion and 3a was obtained in encouraging 36% yield (entry 4). Interestingly, reducing the temperature to ambient temperature improved the yield to 47% (entry 5). Attempts to further optimize the reaction using CuCl2·2H2O in different solvents like EtOAc, DMF, DMSO, benzonitrile proved futile and the reaction didn't even initiate in these solvents (Table S1, ESI†). However, nitromethane did show appreciable reactivity with 39% yield (entry 6). During these attempts it was observed that the reaction showed good conversion during the initial phases but slowed down significantly later. Hence, we attempted to add the catalyst batch-wise (3 × 0.1 eq., every 14 h) which led to significantly better 63% yield but still with incomplete conversion (entry 7). We also attempted to use mild external oxidants such as H2O2, oxone etc. to maintain the catalytic nature of the reaction (entries 8 and 9). However, they gave reduced yields of the product on account of multiple side reactions which reinforced the reactive nature of the substrates and hence, the unavoidable need for mild chemoselective oxidative conditions. At this point we presumed that increasing the air pressure over the reaction might increase the concentration of available oxygen which, in turn, might result into improved conversion. Hence, when the reaction was performed using 30 mol% catalyst in EtOH under compressed air conditions, it showed complete conversion in 36 h with 71% yield (entry 10). It was observed that the non-polar furan 1a was not completely soluble in EtOH and hence, when a mixture of EtOH/CH3CN (10:1 ratio) was used, the reaction worked even better to give 3a in an excellent 81% yield (entry 11). Increasing the amount of CH3CN led to slower reaction and decreased yield (entry 12). Reducing the catalyst loading to 20 mol% decreased the yield to 60% (entry 13). Similarly, when 1.2 equiv. of indole was used instead of 2 equiv., 3a was obtained in only 61% yield (entry 14). CuCl was also found to be an effective catalyst with 78% yield but exhibited extremely slow reactivity (entry 15). In comparison to CuCl2·2H2O, anhyd. CuCl2 showed significantly diminished activity to give 3a in a suboptimal yield of 54% (entry 16). FeCl3·6H2O also proved to be a suboptimal catalyst with only 55% yield (entry 17). We also checked the aqueous compatibility of the reaction by performing the reaction in the presence of H2O (20% v/v) (entry 18). To our delight, the reaction showed remarkable aqueous compatibility and 3a could be isolated in 72% yield. Hence, considering the rate as well as efficiency of the reaction we decided to use CuCl2·2H2O (30 mol%) in EtOH/CH3CN mixture under compressed air conditions as the optimal reaction condition for the oxidative coupling of the furans with indoles. Notably, under these conditions, neither of the homocoupling products (1a′ or 2a′) was formed in any significant amount which indicates towards the chemoselective nature of the reaction (Table 1).
Scope of the reaction
With the optimized reaction conditions, the scope of the reaction was undertaken with variations on the furans at first (Schemes 2). A variety of aryl, heteroaryl as well as polyaromatic substitutions on 5′-(R1) and 3′-positions (R2) of the furan were well tolerated. Mildly activating groups on the 5′-phenyl ring (R1) such as Me–, F–, Cl–, Br– either aided or maintained the reactivity as compared to phenyl. Compound 3b was obtained in 91% yield whereas halide substituted products 3d–3f were obtained in good yields (74–78%) similar to that of 3a. However, relatively stronger electron-donating substituents 2,4-dimethyl gave a significantly decreased yield of 53% in 3c due to the side reactions of furan. On the other hand, strongly electron-withdrawing 2,4-dichloro substituents on phenyl led to inhibited reaction and required heating to furnish the product 3g in a modest 34% yield (54% brsm). Interestingly, the mildness of the reaction conditions allowed the use of 2,2′-thiophene furan as the substrate, wherein the reaction took place through the 5′-position of the furan to selectively give 3h in impressive 70% yield. We also scaled up this reaction at 2.21 mmol (0.5 g) scale to obtain 3h in a good 62% yield which indicates towards the scalability of the reaction. Similarly, the 2,2′-bisfuran substrate also showed appreciable reactivity to give the corresponding product 3i in a modest 28% yield along with the formation of multiple other by-products. Both 3h and 3i contain a conjugated system of three electron-rich heterocycles with the terminal heterocycle containing two unsubstituted positions which could be potentially valuable structural motifs for material science applications.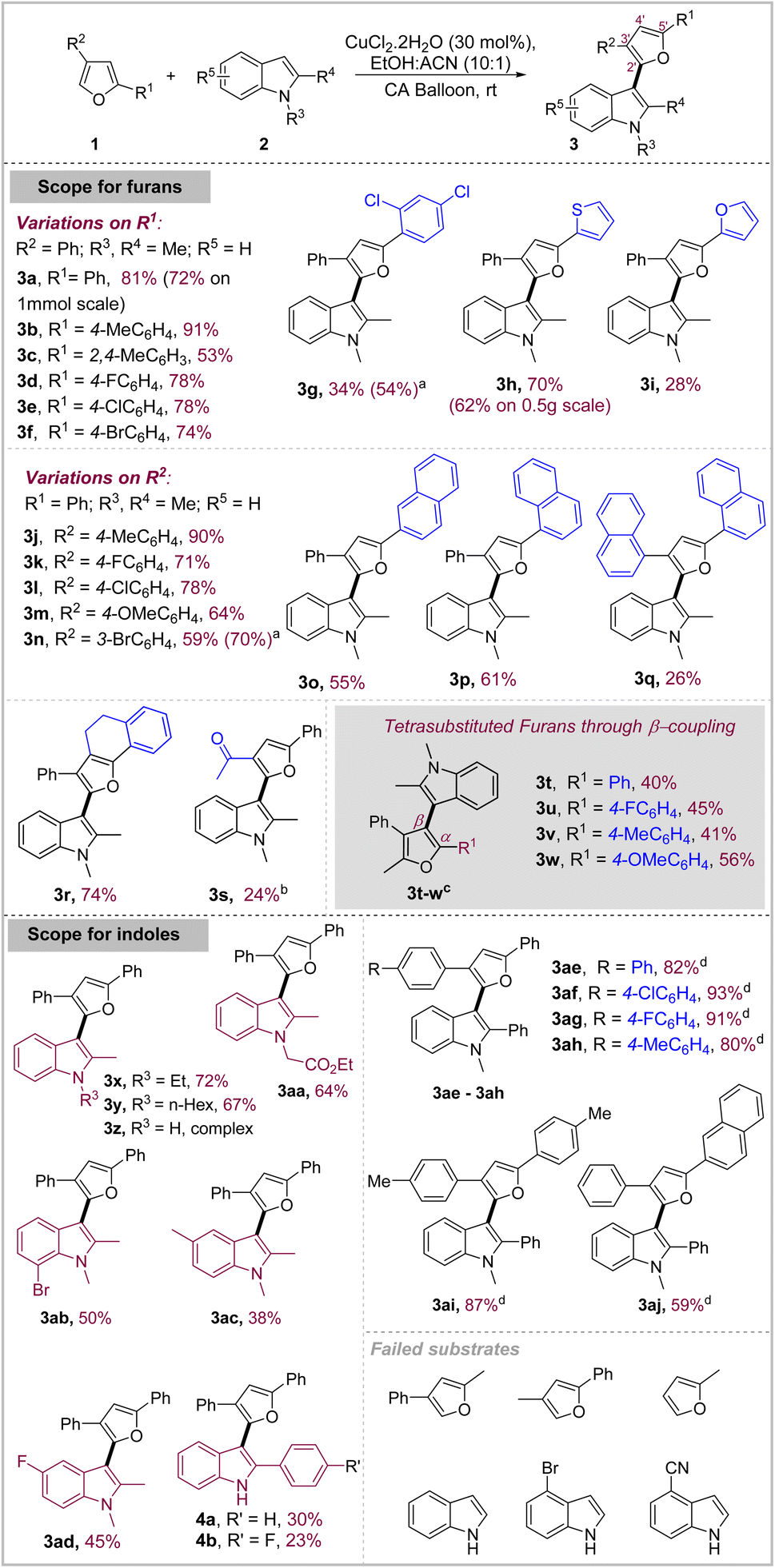 | ||
| Scheme 2 Scope of the reaction under CuCl2·2H2O conditions; abrsm yield; bFeCl3·6H2O (1 eq.) as catalyst; c1–2 eq. of CuCl2·2H2O at 60 °C; d4 eq. of CuCl2·2H2O under ambient conditions (CA: compressed air). | ||
After exploring the reactivity at 5′-position, we turned our attention to 3′-position (R2). Furan substrates with Me–, F– and Cl– substituents on the para position of the 3′-phenyl ring gave the corresponding products 3j–l in 90%, 71% and 78% respectively. However, strongly electron-donating methoxy reacted faster but gave reduced yield of the product 3m (64%), presumably due to partial oxidative degradation of the product in situ. m-Br substituted phenyl gave the product 3n in 59% isolated yield. Since, this oxidative coupling allows synthesis of highly conjugated electron-rich heteropolyaryl motifs, we decided to check if even more conjugated furans with substitutions like α- and β-naphthyl are also viable substrates for this reaction. Towards that, we attempted the suitably substituted furans and found that they reacted with good efficiency to give the corresponding β- and α-naphthyl substituted indolyl–furanoids 3o and 3p in good yields of 55% and 61% respectively. Then, we challenged the reaction with very bulky furan substrate carrying two α-naphthyl groups which also reacted with an acceptable yield of 26% to form 3q despite severe steric crowding on the furan ring. Naturally relevant annulated furan also reacted well to give the corresponding product 3r in a good 74% yield. 3′-acyl substituted furan due to its deactivated nature showed diminished reactivity under optimal conditions but reacted under stoichiometric FeCl3 conditions to give the product 3s in 24% yield. Simple substrates such as furan, 2-methylfuran etc. showed decomposition under these oxidative conditions and failed to give any identifiable product.
Oxidative coupling through β-position of furan: access to tetrasubstituted furan scaffold
After exploring coupling through α-position of furans, we explored coupling through its β-position, primarily with two objectives (Scheme 2). First, the β-position is less reactive due to its less nucleophilic as well as less acidic nature as compared to α-position, which makes it more difficult to participate in oxidative coupling reactions, either under radical conditions or through metal-directed deprotonative mechanism for oxidative coupling under metal-catalyzed conditions.13 Secondly, densely functionalized furans represent a biologically relevant class of natural products and metabolites owing to their nutritional as well as cardiovascular benefits and anti-inflammatory properties.14The reaction proved to be difficult under catalytic conditions but after some investigation, we found that it was possible to achieve coupling of 1,2-dimethyl indole using stoichiometric amount of CuCl2·2H2O in EtOH under ambient conditions. We explored a limited scope by employing a range of furans to obtain the corresponding β-indolyl furanoids 3t–3w in moderate to good yields (40–56%) (Scheme 2). However, less reactive indoles such as 2-aryl indole and 1-alkyl-2-aryl indole failed to react under this condition.
Interestingly, in contrast to furans, indoles displayed differential reactivity in this oxidative coupling depending on their substitution pattern (Scheme 2). Electron-rich 1,2-dialkyl indoles reacted well in general. 1-Ethyl-2-methyl indole and 1-hexyl-2-methyl indole both reacted well to give corresponding products 3x and 3y in a good 72% and 67% yield respectively. Even the indole carrying synthetically versatile functional group –CH2CO2Et worked well to give 3aa in 64% yield. We then turned our attention to exploring variations on the benzene ring of indole. 5-Me, 5-F and 7-Br substituted indoles reacted to give corresponding products 3ab–3ad in moderate yields (38–50%). Surprisingly, 2-phenyl-1H-indole and 2-(4-fluorophenyl)-indole showed complex reactivity with modest yields (30% and 23%) of the corresponding products 4a and 4b along with the formation of multiple other products which seemed to arise through further reactions of the corresponding products (4a/4b) itself. At least some of the by-products might emanate from the well-known Ullmann-type coupling of unprotected NH-indole under Cu-catalysis in the cross-coupled products. Similarly, the reaction with 1H-indole was found to be complex.
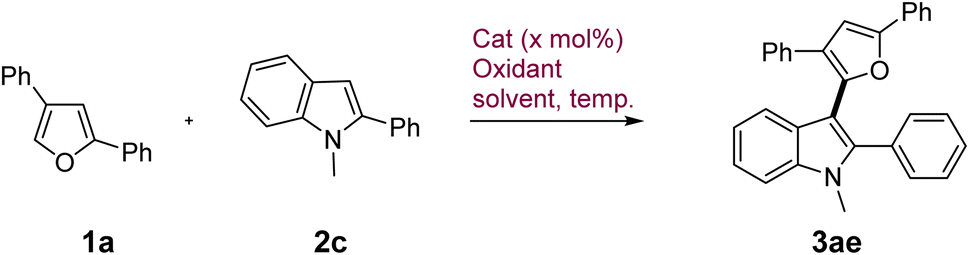
In contrast to these indoles, 1-Me-2-phenyl indole displayed subdued reactivity and gave only limited conversion (∼30%) under catalytic conditions which prompted us to further investigate its reactivity under similar conditions (Table 2, entry 1 and Table S2, ESI†). Attempts to force the conversion with external oxidants such as oxone proved deleterious with even lesser yield of 19% (entry 2) primarily due to the indiscriminate reactivity of furan and indole under this condition. Increasing the CuCl2·2H2O loading to 50 mol% under aerial conditions increased the conversion as well as yield to 34% (entry 3). FeCl3 was able to catalyze the reaction in the presence of O2 as the terminal oxidant under heating but gave modest yield of 38% (entry 4). After investigating various parameters such as oxidants, solvents, temp. etc., it emerged that CuCl2·2H2O provided the mildest and the most chemoselective oxidative conditions for this coupling devoid of any substrate or product decomposition through side reactions. Hence, we attempted the reaction under stoichiometric amount of CuCl2·2H2O wherein 1 equiv. gave 54% yield of the product with 75% conversion which gradually increased to 82% yield with complete conversion in the presence of 4 equiv. of the catalyst (entry 5 and 6). Given the highly inexpensive nature of the catalyst (<0.25$ per g), we decided to use this condition to explore a limited scope of various furans in coupling with 1-Me-2-phenyl indole. Variably substituted phenyl and naphthyl group on furan displayed excellent reactivity to give the products 3ae–3aj in very high yields (59–93%) generally (Scheme 2, bottom).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Loading | Solvent | Oxidant | Yieldb (%) |
| a Conditions: 1a (0.09 mmol, 1 equiv.), 2c (0.36 mmol, 4 equiv.), catalyst, rt or temperature (as mentioned), solvent (2 mL, as mentioned).b Yields were determined after column chromatography.c Reaction mixture was heated at 90 °C.d Significant amount of byproducts were observed in the reaction. | |||||
| 1 | CuCl2·2H2O | 30 mol% | EtOH:ACN |
Air | 20 |
| 2 | CuCl2·2H2O | 30 mol% | MeOH | Oxone | 19 |
| 3 | CuCl2·2H2O | 50 mol% | MeOH | O2 | 34 |
| 4c | FeCl3 | 10 mol% | Toluene | O2 | 38d |
| 5 | CuCl2·2H2O | 1 equiv. | MeOH | Air | 54 |
| 6 | CuCl2·2H2O | 4 equiv. | EtOH | Air | 82 |
Post-synthetic functionalization of the cross-coupled products 3
Thiophene and furan containing short oligomers are versatile building blocks used in material science for various applications. Since, this oxidative coupling provides direct access to novel indole–furan–thiophene (IFT) and indole–furan–furan (IFF) based π-conjugated systems such as 3h and 3i, we focused on devising a modular route which could further extend the length of the conjugated system to tune their photophysical properties if needed (Scheme 3, top). This was achieved through an efficient two-step protocol starting with chemoselective mono-bromination of compound 3h under controlled conditions to obtain α-bromo-thiophene advanced intermediate 5 which could be easily coupled with appropriate boronic acids under aqueous Suzuki coupling conditions to obtain hitherto unknown phenyl- and naphthyl-capped IFT systems 3ak and 3al in excellent yields of 80% and 71% respectively.15 This route represents a general synthetic route to extend the conjugation in IFT systems through Suzuki and other cross-coupling methodologies which could be potentially very versatile.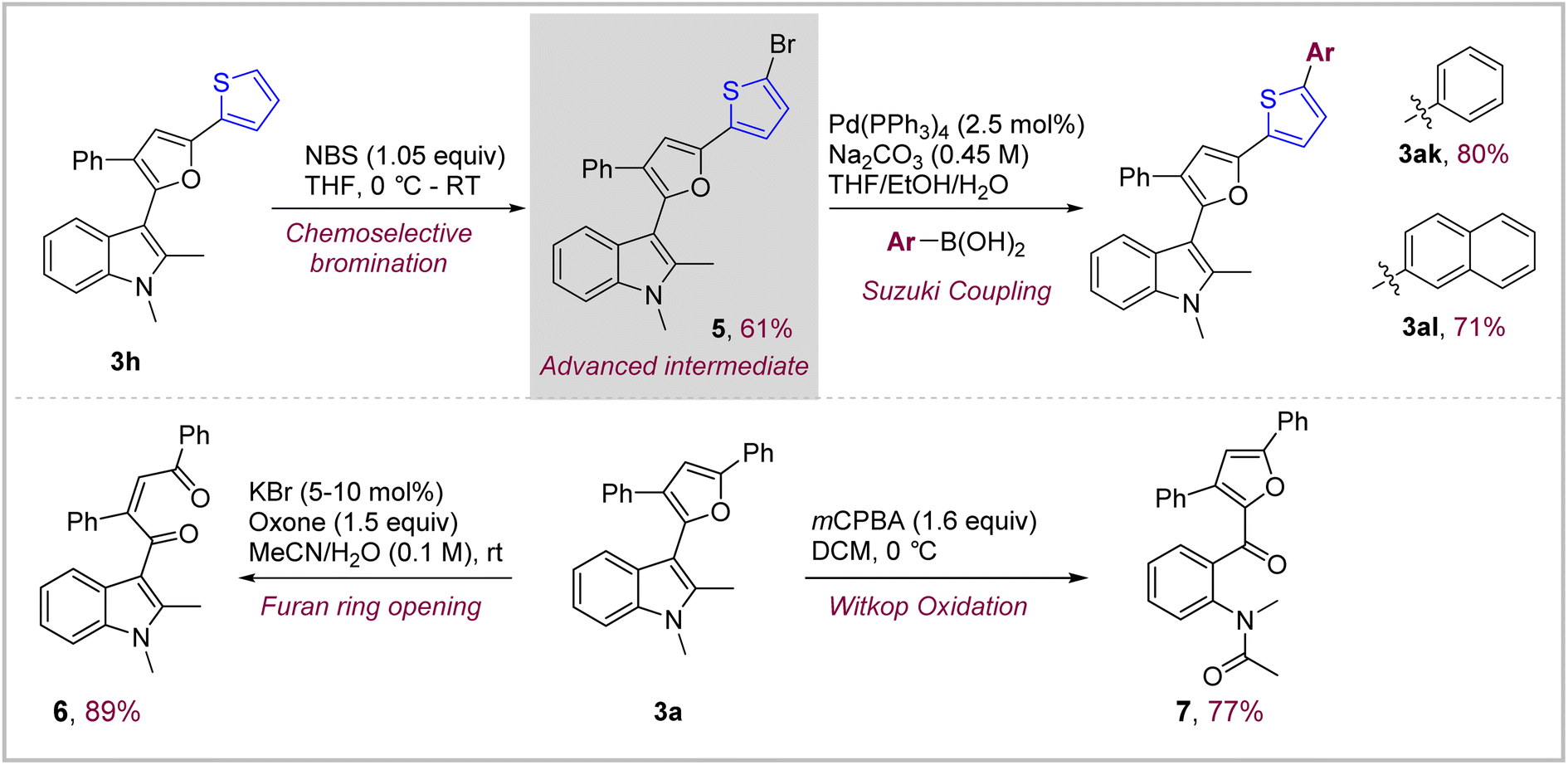 | ||
| Scheme 3 Post-synthetic functionalization of the cross-coupled products. | ||
Both indole and furan are oxidatively active heterocycles with the possibility to be transformed into a variety of oxygenated products.16 We focused on the chemoselective oxygenative ring opening of both furan and indole in the indolyl–furanoid 3a to obtain highly functionalized indole and furan derivatives (Scheme 3, bottom). This is, of course, challenging given the propensity of both these heterocycles to undergo oxidation simultaneously under similar conditions. Hence, we were delighted to find that it was possible to oxidatively cleave the indole ring selectively in the presence of furan using mCPBA in DCM.17 The indole–furanoid compound 3a containing 1,2-dimethyl groups on indole gave the corresponding acetanilide derivative 7 in 77% yield. On the other hand, we found out that under so called ‘green oxidation’ conditions (cat. KBr, oxone, acetonitrile/water) the furan ring in substrates 3a could be oxidatively cleaved while keeping the indole ring intact to give corresponding indole-substituted 1,4-diketone derivatives 6 in an excellent 89% yield.17b This type of chemoselective oxidation could be highly useful for obtaining highly functionalized indole and furan derivatives as demonstrated here.
Mechanistic experiments and the proposed mechanism
After successfully exploring the scope of the reaction and demonstrating the post-synthetic derivatization potential of the products, we investigated the possible mechanism of the oxidative coupling. Based on the seminal mechanistic work from Stahl and others, we surmised the possibility of the involvement of radical intermediates in the cross-coupling under Cu(II)/O2 conditions.18 In order to probe this, we, at first, checked the feasibility of product formation in the presence of radical traps under the standard conditions (Scheme 4a). Both TEMPO and BHT were found to completely suppress the formation of the product 3a. In fact, 1,2-dimethylindole–BHT adduct 8 could be identified clearly by HRMS ([M + H]+: 364.2655). This is a good indication of the involvement of indole radical species under these conditions. We, then, performed a series of control experiments to ascertain the role of the catalyst, oxidant as well as the fate of the substrates individually under the reaction conditions (Scheme 4b). Product 3a was not formed at all in the absence of the catalyst while both furan and indole were recovered in significant amounts from the reaction, thereby, indicating towards the role of Cu(II) in initiating the reaction. Similarly, when the reaction was performed under inert (N2) atmosphere instead of air, the yield of the reaction decreased significantly to 21% which is reasonable based on the amount of the catalyst (30 mol%) used in the reaction. This indicates towards the role of oxygen in the re-oxidation of Cu(I) to Cu(II), thereby, ensuring the catalytic turnover in the process.18a Further, when both indole and furan were reacted separately under the oxidative conditions, indole 2a showed decomposition as evidenced by multiple spots on the TLC and only 43% could be recovered back. On the other hand, furan 1a remains largely unreacted and >90% was isolated back. These control experiments reveal that most likely indole undergoes preferential oxidation under these conditions which confers chemoselectivity to the reaction.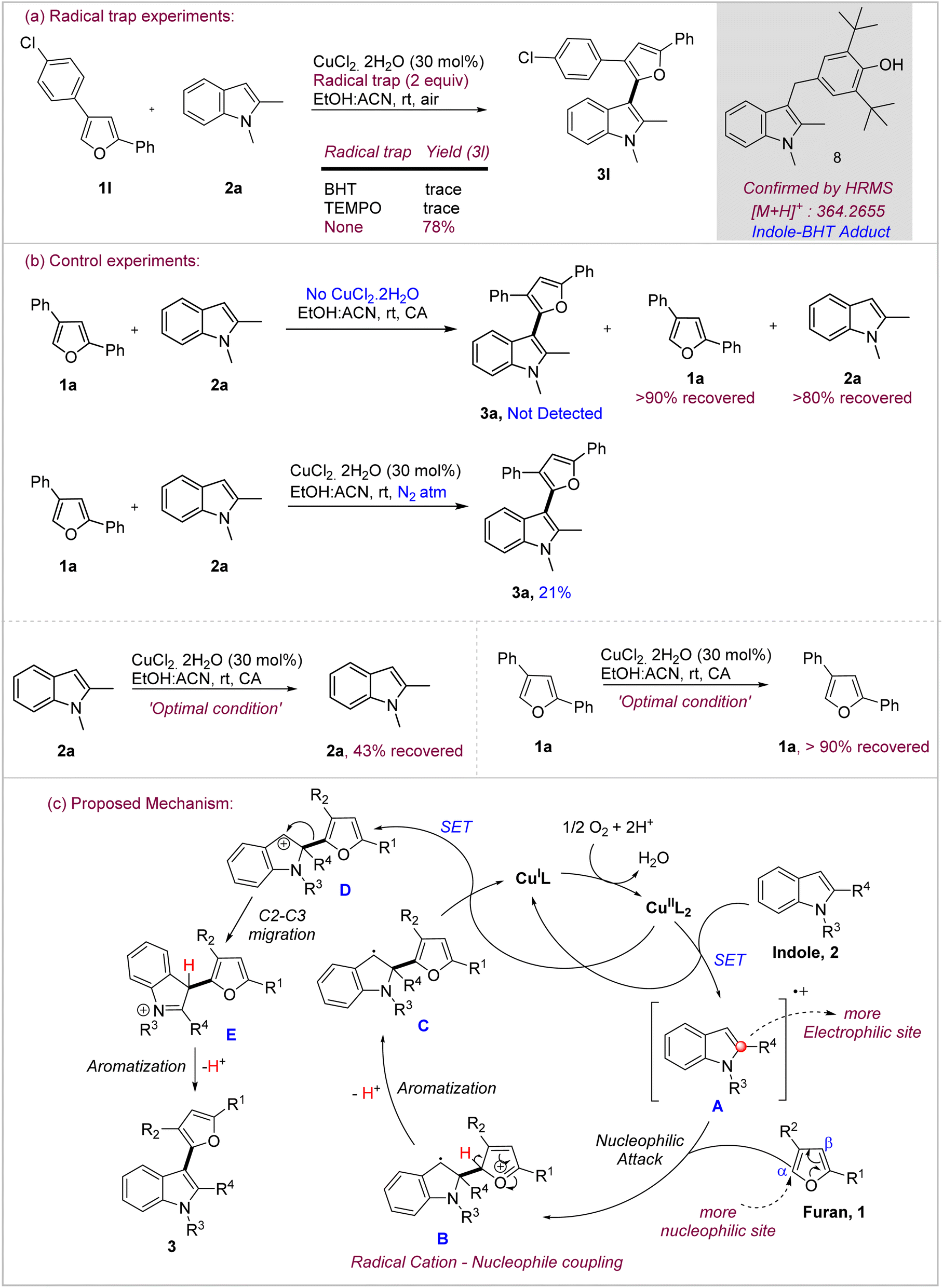 | ||
| Scheme 4 Mechanistic experiments to probe the mechanism of oxidative coupling. (a) Radical trap experiments; (b) control experiments; (c) proposed mechanism. | ||
Considering the evidences obtained through mechanistic experiments as well as literature reports, oxidative coupling seemingly proceeds through radical mechanism involving chemoselective oxidation of indole to its radical cation which acts as the electrophilic species whereas furan owing to its stability under these conditions primarily acts as nucleophile (Scheme 4c). Hence, the reaction is likely initiated by oxidation of indole 2 through single electron transfer to Cu(II) to generate radical cation A. This is followed by the radical cation-nucleophilic coupling as the key bond-forming step in which furan, through its more nucleophilic α-carbon, attacks the radical cation A at its more electrophilic C-2 carbon to form adduct B. Lei, in his recent report19 describing the reactivity of indole radical cations, has concluded that C-2 is the most electrophilic site in the indole radical cation and is the primary site of attack by the nucleophile. Furan in adduct B losses a proton from its α-carbon to regain aromaticity and generate radical C, which undergoes the second single electron transfer to Cu(II) to generate indolinium cation D. At this stage, furan undergoes 1,2-migration20 from C-2 to C-3 of indole to generate cation E which promptly losses a proton from C-3 to regain aromaticity for indole and give the cross-coupled product 3. The migration could be thermodynamically driven due to the possibility for indole aromatization.
Photophysical study
π-Conjugated electron-rich systems represent valuable building blocks for the organic materials.21 Various furan and thiophene containing conjugated molecules have been extensively studied for their materials properties. Hence, we decided to study the photophysical properties of these novel indole–furan–thiophene (IFT) analogs (3h, 3i, 3ak, 3al) along with other representative indolyl–furans (3a, 3ae, 3s, 3t) from our collection (Scheme 5).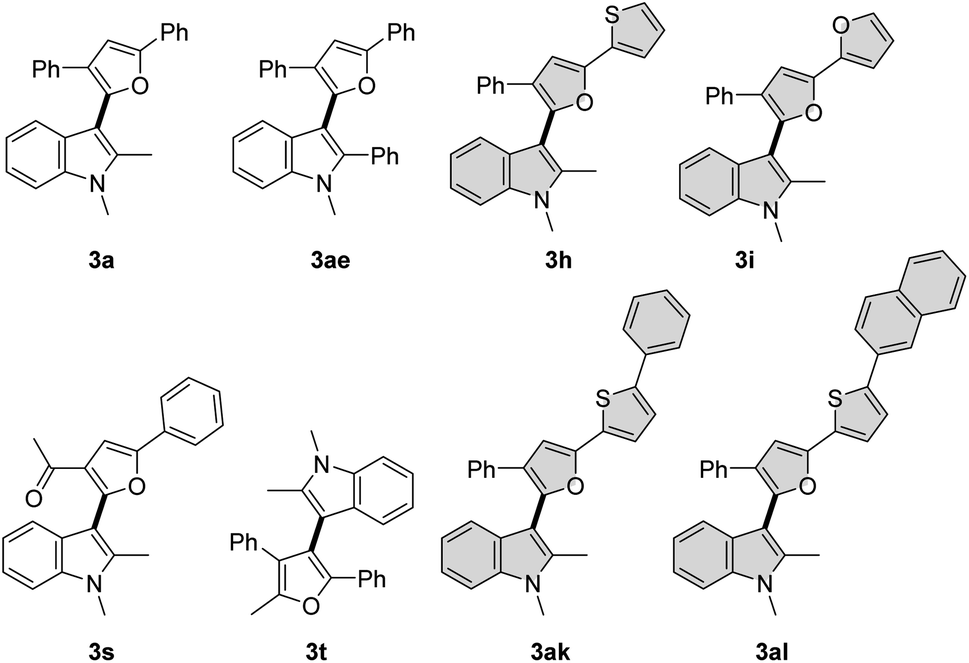 | ||
| Scheme 5 Examples of cross-coupled products screened for photophysical studies. | ||
Absorption and emission spectra for the compounds were recorded in polar aprotic acetonitrile (ACN) and polar protic methanol (MeOH) as the solvents (Fig. 1, Table 3 and Fig. S1, S2†). All the indole–furan heterobiaryl derivatives were found to absorb in the ultraviolet (UV) region with absorption maxima (λmaxabs) generally in the range of 285–320 nm (Table 3). However, the long-conjugated phenyl and naphthyl IFT derivatives 3ak and 3al show absorption maxima at significantly longer wavelengths around 360 nm and 375 nm respectively. As for the emission, the compounds were found to emit in the near blue region (410–440 nm). On the other hand, compounds 3ak and 3al emit in the blue-green region around 496 nm and 520 nm respectively. Interestingly, all the compounds show large Stokes shift of >120 nm with compounds 3ae and 3s showing significantly bigger shifts of 165 nm and 187 nm respectively. It is important to note that large Stokes shift (>80 nm) is a desirable property of biological fluorophores.22 Long conjugated compounds 3ak and 3al also displayed high quantum yields in both the solvents, which coupled with their excellent absorption–emission profile makes them valuable building blocks for further exploration and applications. Expedient access to these potentially useful compounds further validates the utility of our oxidative methodology.
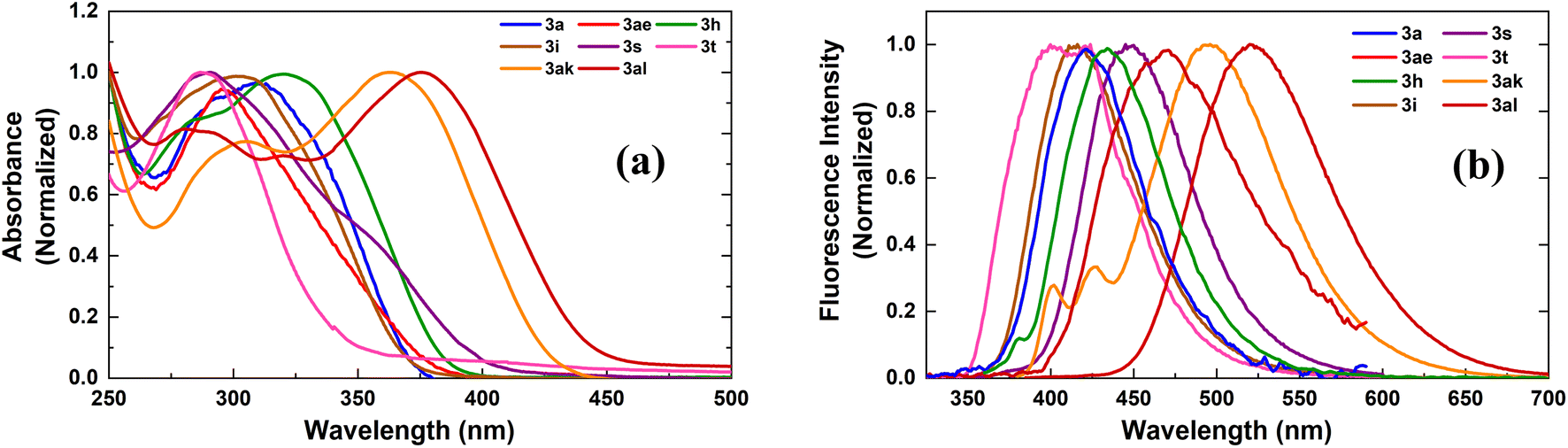 | ||
| Fig. 1 Normalized (a) absorption and (b) emission spectra of compounds in MeOH. Emission spectra were collected by exciting the samples at individual λmaxabs. | ||
| Entry | Sample | Solvent | λmaxabs (nm) | λemi (nm) | Stokes shift (nm) | ϕf |
|---|---|---|---|---|---|---|
| 1 | 3a | ACN | 300 | 420 | 120 | 0.16 |
| MeOH | 0.12 | |||||
| 2 | 3ae | ACN | 295 | 460 | 165 | 0.18 |
| MeOH | 0.16 | |||||
| 3 | 3t | ACN | 287 | 423 | 136 | 0.04 |
| MeOH | 287 | 408 | 121 | 0.04 | ||
| 4 | 3s | ACN | 290 | 423 | 133 | 0.08 |
| MeOH | 288 | 475 | 187 | 0.01 | ||
| 5 | 3i | ACN | 303 | 416 | 113 | 0.15 |
| MeOH | 304 | 411 | 107 | 0.12 | ||
| 6 | 3h | ACN | 320 | 435 | 115 | 0.42 |
| MeOH | 318 | 428 | 110 | 0.10 | ||
| 7 | 3ak | ACN | 363 | 496 | 133 | 0.47 |
| MeOH | 360 | 482 | 122 | 0.57 | ||
| 8 | 3al | ACN | 375 | 520 | 145 | 0.35 |
| MeOH | 375 | 503 | 128 | 0.22 |
Conclusion
In conclusion, we have demonstrated a sustainable methodology for the oxidative coupling of indoles with furans under aerobic base-metal catalyzed conditions, which mimic the SET reactivity of copper-based enzymes in nature. The developed methodology incorporates multiple aspects of sustainable and green catalysis such as the use of earth abundant metals, oxidative nature of the coupling, use of air as the terminal oxidant, less hazardous solvents, and aqueous compatibility. Besides the efficiency and sustainability of the catalytic conditions, we have also demonstrated a wide scope of this reaction with respect to both the substrates. Among the various post-synthetic transformations of the cross-coupled products presented, the most remarkable is the hitherto unknown oxygenative ring-opening of indole as well as furan core in a chemoselective manner in indole–furanoids. To demonstrate the applicative importance of the methodology, we show that some selected indolyl–furans and their derivatives possess interesting photophysical properties with large Stokes shift values with potential for development as biological fluorophores. In essence, this methodology seeks to react ubiquitous, biologically relevant heterocycles such as indole and furan as building blocks using sustainable catalysis to create a useful scaffold.Abbreviations
| CA | Compressed air |
| TBHP | Tetrabutyl hydrogen peroxide |
| DTBP | Di-tertbutyl peroxide |
| DMF | Dimethyl formamide |
| DMSO | Dimethyl sulfoxide |
| DCE | Dichloroethane |
| HFIP | 1,1,1,3,3,3-Hexafluoroisopropanol |
| CAN | Acetonitrile |
| brsm | Based on recovered starting material |
| HRMS | High resolution mass spectrometry |
| NMR | Nuclear magnetic resonance |
| SET | Single electron transfer |
| IFT | Indole–furan–thiophene |
Author contributions
The manuscript was written by R. N. with help from S. G. and K. M. The reactions were planned by R. N., R. F., S. G., K. M. and carried out by S. G., R. F., K. M. Photophysical studies were carried out by S. G. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support provided by SERB (CRG/2022/006652) and the infrastructural support by IIT Goa. K. M. and S. G. would like to thank IIT Goa for the fellowship. The support provided by Dipti Naik in the synthesis of certain compounds and Arkaprava Chowdhury (IIT Bombay) in the steady state photophysical measurements of two molecules is appreciated.References
- (a) J. B. Sperry and D. L. Wright, Curr. Opin. Drug Discovery Dev., 2011, 8, 723–740 Search PubMed; (b) R. A. Craig and B. M. Stoltz, Chem. Rev., 2017, 117, 7878–7909 CrossRef CAS.
- (a) F. Yoshimara, M. Sasaki, I. Hattori, K. Komatsu, M. Sakai, K. Tanino and M. Miyashita, Chem.–Eur. J., 2009, 15, 6626–6644 CrossRef PubMed; (b) H. K. Lee, K. F. Chan, C. W. Hui, H. K. Yim, X. W. Wu and H. N. C. Wong, Pure Appl. Chem., 2005, 77, 139–143 CrossRef CAS; (c) X. Yu and G. O'Doherty, De Novo Synthesis in Carbohydrate Chemistry: From Furans to Monosaccharides and Oligosaccharides, in Chemical Glycobiology, ed. X. Chen, R. Halcomb and P. G. Wang, American Chemical Society, Washington DC, 2008, pp. 3–28 Search PubMed; (d) T. Montagnon, M. Tofi and G. Vassilikogiannakis, Acc. Chem. Res., 2008, 41, 1001–1011 CrossRef CAS PubMed; (e) B. H. Lipshutz, Chem. Rev., 1986, 86, 795–819 CrossRef CAS; (f) H. D. Hao and D. Trauner, J. Am. Chem. Soc., 2017, 139, 4117–4122 CrossRef CAS.
- (a) P. A. Peart and J. D. Tovar, Macromolecules, 2009, 42, 4449–4455 CrossRef CAS; (b) T. Umeyama, T. Takamatsu, N. Tezuka, Y. Matano, Y. Araki, T. Wada, O. Yoshikawa, T. Sagawa, S. Yoshikawa and H. Imahori, J. Phys. Chem. C, 2009, 113, 10798–10806 CrossRef CAS; (c) E. B. Murphy and F. Wudl, Prog. Polym. Sci., 2010, 35, 223–251 CrossRef CAS; (d) L. Fritze, M. Fest, A. Helbig, T. Bischof, I. Krummenacher, H. Braunschweig, M. Finze and H. Helten, Macromolecules, 2021, 54, 7653–7665 CrossRef CAS; (e) O. Gidron and M. Bendikov, Angew. Chem., Int. Ed., 2014, 53, 2546–2555 CrossRef CAS; (f) J. C. Bijleveld, B. P. Karsten, S. G. J. Mathijssen, M. M. Wienk, D. M. de Leeuw and R. A. J. Janssen, J. Mater. Chem., 2011, 21, 1600–1606 RSC.
- (a) H. Cao and P. A. Rupar, Chem.–Eur. J., 2017, 23, 14670–14678 CrossRef CAS PubMed; (b) Y. Miyata, T. Nishinaga and K. Komatsu, J. Org. Chem., 2005, 70, 1147–1153 CrossRef CAS PubMed; (c) O. Gidron, A. Dadvand, Y. Sheynin, M. Bendikov and D. F. Perepichka, Chem. Commun., 2011, 47, 1976–1978 RSC.
- (a) S. C. Kuo, F. Y. Lee, T. M. Huang, C. M. Teng, O. Lee, C. Y. Wu, C. S. Hwang and C. Y. Hung, US Pat., Appl. Publ., US20060106041A120060518, 2006 Search PubMed; (b) A. El-Mekabaty and H. M. El-Shora, Chem. Heterocycl. Compd., 2018, 54, 618–624 CrossRef CAS; (c) X. Deng, H. Wang, T. Zeng, T. Zhang and T. Jiang, WO2018210314, 2018; (d) P. Jaisankar, S. Swarnakar, S. Chatterjee, S. Verma, M. Mandal and S. R. Chaudhuri, US Pat., US20180230135, 2018 Search PubMed; (e) A. C. Morera, P. S. Bigorra, M. V. Ruiz and G. G. M. Adelaida, PCT Int. Appl., WO2016042194, 2016; (f) P. Diana, A. Carbone, P. Barraja, G. Kelter, H. H. Fiebig and G. Cirrincione, Bioorg. Med. Chem., 2010, 18, 4524–4529 CrossRef CAS PubMed; (g) S. Y. Hyun, S. U. Jung and D. W. Kim, Repub. Korean Kongkae Taeho Kongbo, KR2015133097A20151127, 2015.
- (a) J. Xu, Y. Luo, H. Xu, Z. Chen, M. Miao and H. Ren, J. Org. Chem., 2017, 82, 3561–3570 CrossRef CAS PubMed; (b) C. Liu, L. Zhou, W. Huang, M. Wang and Y. Gu, Adv. Synth. Catal., 2016, 358, 900–918 CrossRef CAS; (c) M. M. Campbell, N. Cosford, L. Zongli and M. Sainsbury, Tetrahedron, 1987, 43, 1117–1122 CrossRef CAS; (d) M. Dowlut, D. Mallik and M. G. Organ, Chem.–Eur. J., 2010, 16, 4279–4283 CrossRef CAS PubMed; (e) D. K. Li, J. Y. Tan, W. Deng and Z. Y. Xu, Tetrahedron, 2021, 99, 132407 CrossRef CAS; (f) S. Chatterjee, P. Bhattacharjee, G. L. Butterfoss, A. Achari and P. Jaisankar, RSC Adv., 2019, 9, 22384–22388 RSC.
- Oxidative cross-coupling reactions. (a) C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780–1824 CrossRef CAS; (b) C. Liu, D. Liu and A. Lei, Acc. Chem. Res., 2014, 47, 3459–3470 CrossRef CAS PubMed; (c) N. Gulzar, B. Schweizer-Chaput and M. Klussmann, Catal. Sci. Technol., 2014, 4, 2778–2796 RSC; (d) A. Purtsas, M. Rosenkranz, E. Dmitrieva, O. Kataeva and H. -J. Knölker, Chem.–Eur. J., 2022, 28, e202104292 CrossRef CAS PubMed; (e) R. F. Frische, G. Theumer, O. Kataeva and H.-J. Knölker, Angew. Chem., Int. Ed., 2017, 56, 549–553 CrossRef PubMed; (f) A. Purtsas, O. Kataeva and H.-J. Knölker, Chem.–Eur. J., 2020, 26, 2499–2508 CrossRef CAS PubMed.
- Furan-based oxidative coupling: (a) P. Xi, F. Yang, S. Qin, D. Zhao, J. Lan, G. Gao, C. Hu and J. You, J. Am. Chem. Soc., 2010, 132, 1822–1824 CrossRef CAS PubMed; (b) N. Kuhl, M. N. Hopkinson and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 8230–8234 CrossRef CAS; (c) N. A. B. Juwaini, J. K. Peng Ng and J. Seayad, ACS Catal., 2012, 2, 1787–1791 CrossRef CAS; (d) J. Tsuji and H. Nagashima, Tetrahedron Lett., 1984, 40, 2699–2702 CrossRef CAS; (e) B. Liu, Y. Huang, J. Lan, F. Song and J. You, Chem. Sci., 2013, 4, 2163–2167 RSC; (f) N. N. Li, Y. L. Zhang, S. Mao, Y. R. Gao, D. D. Guo and Y. Q. Wang, Org. Lett., 2014, 16, 2732–2735 CrossRef CAS PubMed.
- (a) C. F. Gürtler, S. Blechert and E. Steckhan, Angew. Chem., Int. Ed., 1995, 34, 1900–1901 CrossRef; (b) S. E. Walden and R. A. Wheeler, J. Chem. Soc., Perkin Trans. 2, 1996, 2, 2663–2672 RSC; (c) U. Haberl, E. Steckhan, S. Blechert and O. Wiest, Chem.–Eur. J., 1999, 5, 2859–2865 CrossRef CAS.
- (a) G. Janusz, A. Pawlik, U. Świderska-Burek, J. Polak, J. Sulej, A. Jarosz-Wilkołazka and A. Paszczyński, Int. J. Mol. Sci., 2020, 21, 966 CrossRef CAS; (b) E. I. Solomon, U. M. Sundaram and T. E. Machonkin, Chem. Rev., 1996, 96, 2563–2606 CrossRef CAS; (c) F. Peter Guengerich, ACS Catal., 2018, 8, 10964–10976 CrossRef.
- (a) R. M. Bullock, J. G. Chen, L. Gagliardi, P. J. Chirik, O. K. Farha, C. H. Hendon, C. W. Jones, J. A. Keith, J. Klosin, S. D. Minteer, R. H. Morris, A. T. Radosevich, T. B. Rauchfuss, N. A. Strotman, A. Vojvodic, T. R. Ward, J. Y. Yang and Y. Surendranath, Science, 2020, 369, eabc3183 CrossRef CAS; (b) B. Su, Z. C. Cao and Z. J. Shi, Acc. Chem. Res., 2015, 48, 886–896 CrossRef CAS PubMed.
- R. Fernandes, K. Mhaske, R. Balhara, G. Jindal and R. Narayan, Chem.–Asian J., 2022, 17, e202101369 CrossRef CAS PubMed.
- (a) H. W. Gschwend and H. R. Rodriguez, Org. React., 2005, 26, 1–360 Search PubMed; (b) E. J. Bures and B. A. Keay, Tetrahedron Lett., 1988, 29, 1247 CrossRef CAS; (c) A. L. Gottumukkala and H. Doucet, Adv. Synth. Catal., 2008, 350, 2183–2188 CrossRef CAS.
- (a) L. Xu, A. J. Sinclair, M. Faiza, D. Li, X. Han, H. Yin and Y. Wang, Prog. Lipid Res., 2017, 68, 119–137 CrossRef CAS; (b) A. P. Riley, C. E. Groer, D. Young, A. W. Ewald, B. M. Kivell and T. E. Prisinzano, J. Med. Chem., 2014, 57, 10464–40475 CrossRef CAS.
- (a) Y. Chen, P. Shen and T. Cao, Nat. Commun., 2021, 12, 6165 CrossRef CAS; (b) P. Aillard, D. Dova, V. Magne, P. Retailleau, S. Cauteruccio, E. Licandro, A. Voituriez and A. Marinetti, Chem. Commun., 2016, 52, 10984–10987 RSC.
- For the oxidation reactions of furan: (a) A. S. Makarov, M. G. Uchuskin and I. V. Trushkov, Synthesis, 2018, 50, 3059–3086 CrossRef CAS; (b) J. Xu, L. Liang, H. Zheng, Y. R. Chi and R. Tong, Nat. Commun., 2019, 4754 CrossRef.
- (a) M. Nandakumar, R. Sivasakthikumaran and A. K. Mohanakrishnan, Eur. J. Org Chem., 2012, 19, 3647–3657 CrossRef; (b) J. Xu, L. Liang, H. Zheng, Y. R. Chi and R. Tong, Nat. Commun., 2019, 10, 4754 CrossRef.
- (a) S. E. Allen, R. R. Walvoord, R. Padilla-Salinas and M. C. Kozlowski, Chem. Rev., 2013, 113, 6234–6458 CrossRef CAS PubMed; (b) A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062–11087 CrossRef CAS; (c) J. Zhang, S. Torabi Kohlbouni and B. Borhan, Org. Lett., 2019, 21, 14–17 CrossRef CAS PubMed; (d) S. D. McCann and S. S. Stahl, Acc. Chem. Res., 2015, 48, 1756–1766 CrossRef CAS; (e) J. E. Nutting, K. Mao and S. S. Stahl, J. Am. Chem. Soc., 2021, 143, 10565–10570 CrossRef CAS ; for a review about the state of mechanism of oxidative couplings:; (f) I. Funes-Ardoiz and F. Maseras, ACS Catal., 2018, 8, 1161–1172 CrossRef CAS.
- X. Liu, D. Yang, Z. Liu, Y. Wang, Y. Liu, S. Wang, P. Wang, H. Cong, Y. H. Chen, L. Lu, X. Qi, H. Yi and A. Lei, J. Am. Chem. Soc., 2023, 145, 3175–3186 CrossRef CAS PubMed.
- K. Sun, S. Liu, P. M. Bec and T. G. Driver, Angew. Chem., Int. Ed., 2011, 50, 1702–1706 CrossRef CAS PubMed.
- (a) U. Scherf and E. J. W. List, Adv. Mater., 2002, 14, 477 CrossRef CAS; (b) J. L. Bredas, J. P. Calbert, D. A. da Silva Filho and J. Cornil, Proc. Natl. Acad. Sci. U.S.A., 2002, 99, 5804–5809 CrossRef CAS; (c) S. C. Rasmussen, S. J. Evenson and C. B. McCausland, Chem. Commun., 2015, 51, 4528–4543 RSC; (d) B. Zheng and L. Huo, Small Methods, 2021, 5, 2100493 CrossRef CAS PubMed; (e) M. A. Perkins, L. M. Cline and G. S. Tschumper, J. Phys. Chem. A, 2021, 125, 6228–6237 CrossRef CAS PubMed; (f) J. S. de Melo, F. Elisei and R. S. Becker, J. Chem. Phys., 2002, 117, 4428–4435 CrossRef; (g) R. Fernandes, K. Mhaske and R. Narayan, Tetrahedron, 2022, 103, 132553 CrossRef CAS.
- (a) L. D. Lavis and R. T. Raines, ACS Chem. Biol., 2014, 9, 855–866 CrossRef CAS PubMed; (b) X. Wu, X. Sun, Z. Guo, J. Tang, Y. Shen, T. D. James, H. Tian and W. Zhu, J. Am. Chem. Soc., 2014, 136, 3579–3588 CrossRef CAS; (c) J. F. Araneda, W. E. Piers, B. Heyne, M. Parvez and R. McDonald, Angew. Chem., Int. Ed., 2011, 50, 12214–12217 CrossRef CAS; (d) D. M. Shcherbakova, M. A. Hink, L. Joosen, T. W. J. Gadella and V. V. Verkhusha, J. Am. Chem. Soc., 2012, 134, 7913–7923 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Optimization tables, reaction conditions, general methods, spectral and characterization details copy of 1H, 13C, 19F NMR spectra etc. See DOI: https://doi.org/10.1039/d3ra08226a |
| ‡ Both the authors contributed equally. |
| § Present address: Syngenta Biosciences Pvt. Ltd., Goa, India. |
| This journal is © The Royal Society of Chemistry 2024 |