
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInducing molecular orientation in solution-processed thin films of fluorene-bithiophene-based copolymer: thermal annealing vs. solvent additive†
Cassia Ferreira Coutinho Pereiraa,
Bruno G. A. L. Borgesa,
Karlison R. A. Sousa bc,
Soheila Holakoeia,
Lucimara S. Romanb,
C. Moyses Araujoef,
Marco Cremonad,
Marlus Koehlerb,
Cleber F. N. Marchiori*e and
Maria Luiza M. Rocco*a
bc,
Soheila Holakoeia,
Lucimara S. Romanb,
C. Moyses Araujoef,
Marco Cremonad,
Marlus Koehlerb,
Cleber F. N. Marchiori*e and
Maria Luiza M. Rocco*a
aInstitute of Chemistry, Federal University of Rio de Janeiro (UFRJ), 21941-909, Rio de Janeiro, RJ, Brazil. E-mail: luiza@iq.ufrj.br
bDepartment of Physics, Federal University of Paraná (UFPR), Centro Politécnico, CP 19081, 81531-900, Curitiba, PR, Brazil
cFundação de Amparo à Pesquisa do Estado do Amazonas – FAPEAM, 69058-030, Manaus, AM, Brazil
dDepartamento de Física, PUC-Rio, 22453-900, Rio de Janeiro, RJ, Brazil
eDepartment of Engineering and Physics, Karlstad University, 65188, Karlstad, Sweden. E-mail: Cleber.Marchiori@kau.se
fMaterials Theory Division, Department of Physics and Astronomy, Uppsala University, 75120, Uppsala, Sweden
First published on 18th March 2024
Abstract
A deep understanding of the factors influencing the morphology of thin films based on conjugated polymers is essential to boost their performance in optoelectronic devices. Herein, we investigated the electronic structure and morphology of thin films of the copolymer poly(9,9-dioctyl-fluorenyl-co-bithiophene) (F8T2) in its pristine form as well as samples processed with the solvent additive 1,8-diiodooctane (DIO) or post-processed through thermal annealing treatment. Measurements were carried out using angle-resolved S K-edge NEXAFS (near-edge X-ray absorption fine structure) in total electron yield (TEY) and fluorescence yield (FY) detection modes. Two main transitions were observed at the S 1s NEXAFS spectra: S 1s → π* and S 1s → σ* (S–C). The observed dichroism pointed to a face-on orientation of the conjugated backbone, which was significantly increased for F8T2 films processed with DIO. Resonant Auger decay spectra were obtained and analyzed using the core-hole clock (CHC) method. An enhancement in the charge transfer process was observed for thermally annealed films, especially for samples processed with DIO, corresponding to an increase in film ordering. Furthermore, the investigated films were characterized using X-ray photoelectron spectroscopy, attesting to the presence of the thiophene unit in the samples and demonstrating that some of its sulfur atoms were positively polarized in the F8T2 films. All these experimental findings were compared with molecular dynamics (MD) simulations of film evaporation with and without DIO. The use of MD, together with mathematical modeling, was able to explain the major effects found in the experiments, including the polarization of sulfur atoms. The simultaneous use of powerful spectroscopic techniques and theoretical methods shed light on key aspects linking film morphology with fabrication procedures.
1. Introduction
Fluorene-based copolymers are highly attractive organic semiconducting materials used for several optoelectronic applications, such as hole transport materials,1–3 field-effect transistors,4,5 light emitting devices6–8 and electrochromic devices,9,10 owing to their well-known synthetic route and chemical and thermal stability. In particular, the poly(9,9-dioctyl-fluorenyl-co-bithiophene) copolymer (F8T2 – chemical structure shown in Fig. 1), a fluorene-based copolymer containing a bithiophene unit as a co-monomer, displays good hole-transporting abilities and appears to be one of the most promising candidates among this class of polymers.11–13 It has been reported that F8T2 can display hole mobilities up to 5 × 10−3 cm2 V−1 s−1. Moreover, F8T2 can organize into crystalline domains upon thermal annealing or even undergo a liquid-crystalline phase transition at 265 °C.12,13 Under such conditions, where the conjugated backbone is oriented, hole mobility can reach values ranging from 0.01 to 0.02 cm2 V−1 s−1.12 | ||
| Fig. 1 Chemical structure representation of (a) the poly(9,9-dioctyl-fluorenyl-co-bithiophene) (F8T2) copolymer, highlighting the fluorene moiety in light blue and the bithiophene unit in dark blue and (b) the 1,8-diiodooctane (DIO) additive. | ||
Hence, many optoelectronic properties of devices based on conjugated copolymers depend on the final morphology reached after thin film fabrication. However, it is extremely difficult to assess how the procedures followed to produce the film can affect its resulting morphology. For instance, the thermal annealing approach is widely employed to improve the charge transport properties of organic semiconductors because it enables the spatial rearrangement of polymer chains, leading to an increase in film ordering, which influences both morphology and charge mobility.12,14–17 The additive 1,8 diiodooctane (DIO) has proven to contribute positively to the morphology of active layers in photovoltaic devices.17–19 The addition of high boiling-point additives, such as DIO, leads to an increase in the solvent evaporation time, enabling molecular reorientation during film formation. This process is used as a strategy to enhance the performance of organic photovoltaic devices (OPVs). However, the charge transfer dynamics of additive-processed films have not been well explored. In this sense, a comparative study on how charge transfer is affected by thermal annealing or solvent additives is timely.
Electron charge transfer (CT) dynamics of excited states in the time scale of femtoseconds (10−15 s) are extremely important for understanding the effect of different (post) processing methods on the electron transport properties in copolymers, such as F8T2. Here, the CT dynamics were derived through the core-hole clock (CHC) method, an elementally sensitive synchrotron-based approach that utilizes the core-hole lifetime of core levels as an internal reference clock, using resonant Auger spectroscopy.20,21
The CHC method works as a two-step model involving core excitation, followed by core-hole non-radiative decay. After the formation of a core-hole, the system reaches a new equilibrium state through radiative or non-radiative processes. In the latter case, once the core vacancy is filled by an electron from a higher occupied level, a second electron is emitted into the continuum (the “Auger electron”). Different decay channels compete in a resonant Auger spectrum, allowing for the application of the CHC method. Resonant excitation leads to the occurrence of two possible processes: recombination of the excited electron and the core-hole, leading to a one-hole (1h) final state (Participator Auger decay) or non-recombination, which leads to a two-hole and one-electron (2h1e) final state (Spectator Auger decay). Additionally, if the excited electron is delocalized from an unoccupied bound state (e.g. the LUMO) to the substrate or the surroundings before the core-hole is filled, we observe a two-hole (2h) final state equivalent to the Normal Auger decay, which typically results from an ionization process.21,22 NEXAFS (near-edge X-ray absorption fine structure) is also a synchrotron-based technique important for correlating the ordering of films with CHC results.
In recent years, our research group has demonstrated the effects of molecular orientation and morphology on the charge transfer dynamics of the thiophene-based polymers12,21,23–25 used in OPVs. In this work, we propose an approach that combines powerful spectroscopic techniques (such as those mentioned above) with molecular dynamics simulation (MD), and mathematical modelling can reveal important links between the final morphology of the film and the procedures followed to produce it. In particular, we present a further study of thin films of F8T2 subjected to thermal annealing or processed with the organic additive DIO (see Fig. 1), with the aim of assessing the effects of those different treatments on molecular organization and electron transfer dynamics.
As examples of the outcomes from this study, we highlight that there is an increase in film ordering when DIO is used as an additive. This result is supported by the total electron and fluorescence yields of the NEXAFS spectra and charge transfer times from the Auger decay spectra. In addition, MD simulations of solvent evaporation/film formation suggest that an improved order is achieved owing to the higher concentration of syn conformers induced by the presence of DIO.
Finally, our work also demonstrates that even unexpected experimental findings, the shift in sulfur polarization (detected by XPS), can be successfully explained by subtle variations in the film morphology induced by the presence of DIO. The outcomes of this study strengthen the usefulness of the strategy proposed herein to detect indistinct effects relating film morphology to the procedures followed during deposition.
2. Experimental section
The F8T2 films were spin-coated at 900 rpm for 80 seconds onto FTO/PEDOT:PPS substrates from a chlorobenzene solution (4.0 mg mL−1). Two different films were produced from this solution. One of them was not subjected to any post-processing treatment, such as cast, and the other was subjected to thermal annealing at 100 °C for 15 minutes in an inert atmosphere. Another processing condition was also investigated, viz. the addition of DIO (1% v/v) to the solution with the same concentration as the previous one and stirred overnight. The film was deposited using the same parameters as the other two films.Near-edge X-ray absorption fine structure (NEXAFS) and resonant Auger decay measurements were performed using a soft X-ray spectroscopy (SXS) beamline at the Brazilian Synchrotron Light Source (LNLS). The beamline has an energy resolution of 0.38 eV at the sulfur K-edge using a Si(111) double-crystal monochromator. Energy calibration was performed through a well-known value for the LIII transition (2p3/2 → 4d) of metallic molybdenum. The NEXAFS spectra were measured at both total electron yield (TEY) and fluorescence yield (FY) modes, with X-ray incidence angles of 20°, 30°, 55° and 85°, to evaluate polarization dependence.
Resonant Auger spectra (RAS) were measured at the same beamline, with a pass energy of 20 eV. A combination of Gaussian (G) and Lorentzian (L) functions was used in the fitting of the spectra with SpecLabs' software CasaXPS® (version 2.3.22PR1.0), while the background correction was performed using an adjustable function, following the procedure reported before.23
X-ray Photoelectron Spectroscopy (XPS) measurements were performed with a Thermo Fisher Scientific spectrometer ESCALAB 250Xi using monochromatic Al Kα source. The spectra were measured at a pressure of 10−9 mbar, and the electron energy analyzer operated with pass energies of 100 eV and 25 eV for survey and high-resolution spectra, respectively. Energy calibration was performed using the well-known value of Au 4f7/2 (84.0 eV).
3. Theoretical methods
Helpful insights into the polymer chain organization at the molecular level were obtained by classical molecular dynamics (MD) simulations of the F8T2 film formation from solution when it is processed with and without the additive DIO. These simulations mimic film growth and final morphology by developing an “in silico” solvent evaporation with and without the presence of DIO. We basically followed the same procedures described in previous works26–28 (details on the methods applied to develop these simulations, such as the force field used (OPLS) can be found in the ESI section†). For comparison, two systems were prepared: (i) one with the F8T2 oligomers formed by 16 repeated units (8.84 kDa, approximately 240 angstrom long) randomly mixed with chlorobenzene (CB) molecules (the main solvent); (ii) the second system with a small volume fraction of DIO molecules (0.47%) added to the “solution” of F8T2+CB. Initially, all the F8T2 oligomers were assumed to have an anti conformation (as depicted in Fig. 1), which corresponds to a dihedral angle between the thiophene rings that minimizes the total energy. Both simulated systems have the same dimensions (30.0 × 30.0 × 60.0) nm3.The simulation of solvent evaporation was carried out through steps adapted from the protocol provided by Alessandri et al.29 In this protocol, many sequential simulations are performed so that 2% of CB molecules are removed from the system in each step. The simulation stops when all solvent molecules are removed from the system without further equilibration. The entire MD was performed in the NVT ensemble with a temperature environment and a pressure of 1.0 atm. Fig. S1† shows some instant frames of the evaporation process, including the final stationary morphology. The resulting F8T2 film has 100 oligomers. More details on the molecular dynamics procedures followed here can be found in the ESI section.†
X-ray absorption (XAS) calculation was performed for a pre-optimized oligomeric model chain containing 2 repeating units. These calculations were performed using the Time-Dependent Density Functional Theory at M06/6-311G(d,p) as implemented in the ORCA software.30
4. Results and discussion
XPS spectra were obtained to confirm the chemical composition of all F8T2 samples and the quality of the films. A survey spectrum for each sample was also measured, with all the expected elements for each film identified (see Fig. S1 in the ESI file†). The survey spectra also detected the presence of small quantities of silicon from the glass substrate, which may indicate the formation of a thinner film or an agglomerate (inhomogeneous). The C 1s core level spectra for the F8T2 pristine (processed without DIO and thermal annealing) annealed and DIO-processed films (see Fig. S2 in the ESI†) are very similar, all displaying the same four expected components at about the same binding energies. The most intense peaks at around 284.6 eV correspond to sp2 hybridized carbon atoms and are attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C groups, while the peaks at about 285.1 eV correspond to carbon atoms that belong to C–C and C–H groups and relate to atoms in α and β positions in the thiophene rings.31–33
C groups, while the peaks at about 285.1 eV correspond to carbon atoms that belong to C–C and C–H groups and relate to atoms in α and β positions in the thiophene rings.31–33
The smaller peaks at around 285.9 eV and 287.1 eV are assigned, respectively, to C atoms bonded to the S atom (C–S), confirming the presence of the thiophene unit, and C–O groups, indicating the occurrence of a small oxidation process at the surface.33,34 A detailed description of the binding energies, full width at half maximum (FWHM) and atomic percentage is presented in Tables S1 and S2 in the ESI file.†
The S 2p core-level XPS spectra for the three different processing conditions, viz. pristine, annealed and processed with the additive DIO, are shown in Fig. 2. An intriguing feature was observed when comparing the pristine film with the annealed or DIO-processed ones. The spectrum for the pristine sample (Fig. 2a) is resolved with two main doublets, with the BE for each S 2p3/2 peak lying at about 163.8 eV and 164.5 eV. The first one, at a lower binding energy, can be attributed to neutral thiophene units (S–C),34,35 while the second peak, shifted by approximately +0.7 eV, indicates that some of the sulfur atoms in the thiophene units are positively polarized or partially charged.32,36
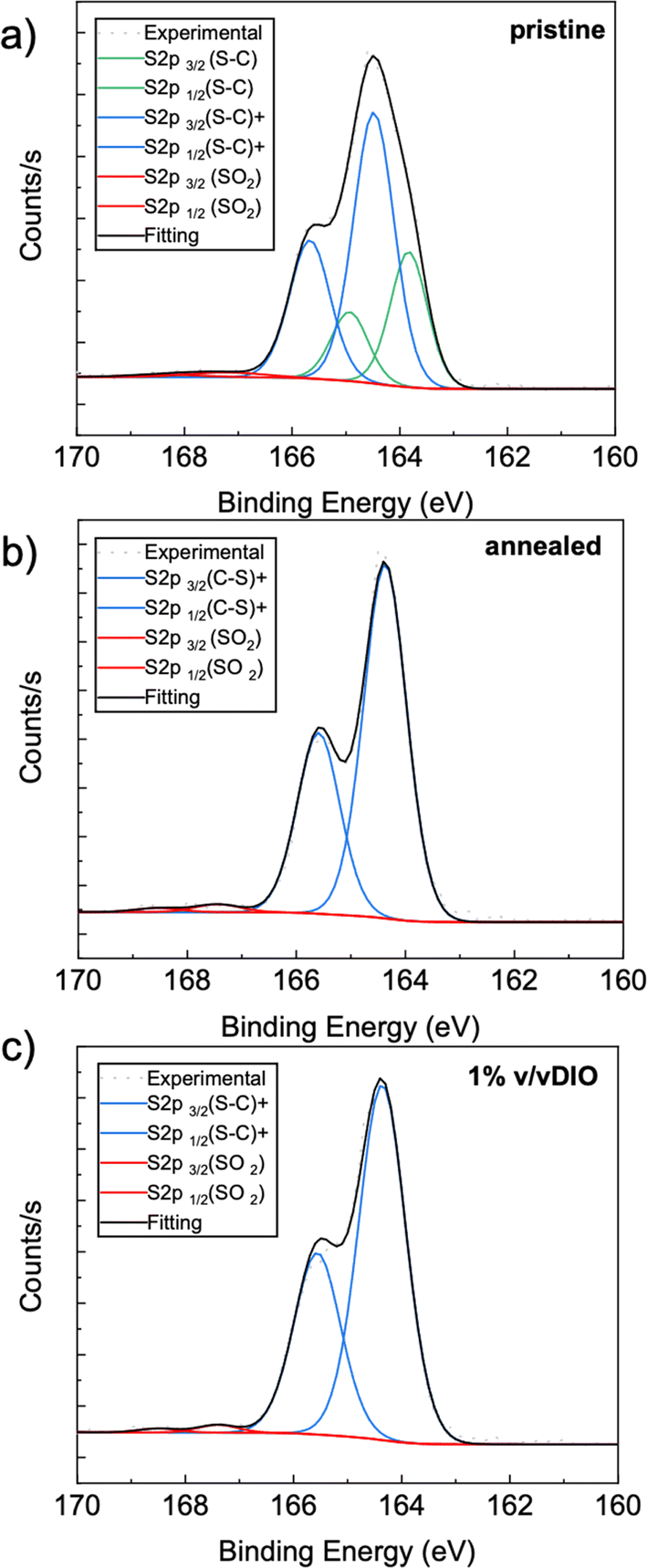 | ||
| Fig. 2 S 2p core-level spectra of F8T2. (a) pristine, (b) thermal annealed at 100 °C and (c) processed from a solution containing 1% v/v of DIO. | ||
Kang et al. observed two spin–orbit split doublets in the S 2p core-level spectra of PBT-perchlorate complexes, with the BE for each of the S 2p3/2 peaks lying about 1.0 eV apart from each other.36 They proposed that the second peak at a higher binding energy is due to the existence of some sulfur atoms in a more positive environment due to charge extraction from some of the thiophene units as a result of increased oxidation. However, after thermal annealing or when the film is processed from a solution containing the DIO as an additive, only one doublet at 164.4 eV is observed, as depicted in Fig. 2b and c. The observed BE values for sulfur in the thiophene rings may suggest that both thermal annealing and the additive promote some changes in the molecular organization, resulting in inter/intrachain interactions that stabilize the state in which the S atoms have a more positive partial charge. To the best of our knowledge, this feature has not been well discussed in the literature. Consideration of such an intriguing outcome is provided in the discussion section. It is worth mentioning that the S 2p spectra for all samples display an extra doublet at a higher BE (the S 2p3/2 peak at about 167.4 eV) because of very small surface oxidation, potentially indicating the presence of sulfone groups (SO2).37,38
To gain a better understanding of the effects of processing methods on film organization at the molecular level, we conducted angular-resolved X-ray absorption spectroscopy studies. Before delving into angular dependence, let us first understand the features of the sulfur K-edge spectrum. Fig. 3 shows the total electron yield TEY NEXAFS spectrum obtained for the F8T2 film. The spectrum displays a sharp peak, centered at 2472.6 eV, corresponding to electronic transitions from its sulfur 1s electron to unoccupied molecular orbitals,39 followed by broad bands at higher photon energies.
 | ||
| Fig. 3 Experimental (red) and calculated (blue) sulfur K-edge NEXAFS spectrum of F8T2. The red vertical bars represent the oscillator strength of each calculated electronic transition. The natural transition orbitals (isovalue = 0.01) for the most intense transitions labeled as 1 and 2 are shown on the right-hand side. The calculated results were obtained from TD-DFT calculation at M06/6-311G (d,p) theory level. | ||
Through the minimum values in the second derivative, depicted in Fig. S3,† we observe that the main peak is a result of the overlapping of two transitions, identified as 1 and 2. The calculated natural transition orbitals (NTOs), shown on the right-hand side of Fig. 3, confirm that those electronic transitions correspond to S 1s → π* and S 1s → σ*(S–C) transitions, respectively, at 2471.7 eV and 2472.6 eV, a well-known assignment for gas-phase thiophene40 and other thiophene-based polymers.21,25,41
Broader bands at higher photon energies are attributed to excitations containing the Rydberg character and σ*(C–C) shape resonances appearing above the sulfur 1s ionization potential (2475.5 eV).12 Similar structures were previously reported in the absorption spectra of condensed thiophene42 and thiophene-based polymers.12,23,43
Fig. 4 shows the TEY NEXAFS spectra following sulfur K-edge photoexcitation for the F8T2 films obtained as a function of X-ray incident angles (AR-NEXAFS, depicted in Fig. 4a–c). The corresponding difference spectra are presented in Fig. S4 in the ESI file† for photon energies around the main peak.
 | ||
| Fig. 4 Angular dependence of sulfur K-edge TEY and FY NEXAFS spectra for F8T2 films under the following conditions: (a and d) pristine, (b and e) thermal annealed at 100 °C, and (c and f) processed with DIO. | ||
For the pristine F8T2 film, we note a clear dichroism when analyzing the angular dependence of the F8T2 TEY absorption spectrum (Fig. 4a), which indicates the existence of a preferential molecular orientation in the film. The intensity of the second transition, associated with an unoccupied σ* orbital, increases with an increase in the incidence angle, reaching its maximum intensity at normal incidence, while the first transition, associated with the π* orbital, exhibits the opposite behavior. Because the transition dipole moment for the π* resonance is perpendicular to the molecular plane of the thiophene unit, this result reveals that the thiophene units in the polymer backbone present an orientation parallel to the substrate (face-on orientation).
Fig. 4b and c show the spectra obtained for F8T2 films annealed and DIO processed, respectively. No significant change in the angular dependence behavior of these spectra was observed in comparison to the pristine film. However, we observed a gradual increase in the dichroism displayed in these spectra, reaching a maximum for the F8T2 film processed with the additive DIO (Fig. 4c).
An increased dichroism corresponds to a higher degree of ordering of the polymeric film. These results indicate that additive DIO leads to a more ordered film among all the samples analyzed, which can have an impact on charge transport properties. In a previous study,44 some of the co-authors investigated the effect of thermal annealing and the DIO on the electrical properties and its impact on the device performance in OPV using F8T2 as a light absorbing material. It was observed that an increase in hole mobility was responsible for the DIO-processed films, which is even higher than the increase promoted by thermal annealing. This result is in line with the molecular orientation observed in the AR-NEXAFS measurements.
The preferential molecular orientation is not only a surface effect, but it is also present in the bulk environment. The fluorescence yield (FY) NEXAFS spectra (shown in Fig. 4d–f) exhibit the same behavior observed for TEY, which is a surface-sensitive measurement. The same sharp peak centered at 2472.6 eV was observed for the bulk of the F8T2 films made up of two transitions: S 1s → π* and S 1s → σ*(S–C) at 2471.7 eV and 2472.6 eV, respectively. This result reveals that the molecular order is present throughout the bulk.
To assess the electron delocalization times, sulfur KL2,3L2,3 Auger decay spectra (RAS) were obtained at four photon energies labeled A–D (see Fig. S3 in the ESI file†), where A = 2471.7 eV, B = 2472.6 eV, C = 2474.3 eV, and D = 2475.2 eV. The spectra are shown in Fig. S5 in the ESI file.† Structures in red, blue, green and purple correspond to a normal Auger peak, π* and σ*(S–C) spectator peaks and a second spectator channel, respectively. The spectrum with a photon energy of 2472.6 eV (labeled B) corresponds to the resonance maximum (1s–σ*). The RAS spectra measured at 2480.4 eV photon energy (labeled E in Fig. S3†), above the S 1s ionization potential, were used to identify the non-resonant contribution for each sample.
The Auger decay spectra show a structure, in red, of photon energy around 2112 eV, which is attributed to a 1D2 normal Auger component. Two other main features can be observed in the spectra obtained for the photon energy labeled B in Fig. S5 and S6† for all samples, which are attributed to spectator Auger peaks. The first one, in blue, is located at a kinetic energy of around 2114 eV and corresponds to 1s–π* transitions, while the second one, in green, is located at a kinetic energy of around 2115 eV and corresponds to 1s–σ* (S–C) transitions. By comparing the spectra obtained for photon energies labeled A (2471.7 eV) and B (2472.6 eV) regarding the position and intensity of the peaks, we can clearly observe that the two spectator peaks disperse in energy with the increase in the photon energy and that their intensities switch between spectra, as expected for spectator channels.
Analysis of the RAS spectra obtained for all F8T2 samples confirms that the main resonance observed in the NEXAFS spectrum is, in fact, composed of two transitions, observed as two narrow and well-separated peaks by resonant Auger spectroscopy. This result reinforces the importance of resonant Auger spectroscopy in experimentally identifying states that are not well resolved by NEXAFS, especially when a theoretical assessment is unavailable.
Charge transfer times (τCT) were calculated from the ratio between the intensities of the spectator signals and the normal Auger signal using the equation τCT = (Ispectator/Inormal Auger) × τCH, where τCH = 1.27 fs represents the S-1s core-hole lifetime.45 The accessible range of τCT is from one tenth to around ten times the τCH, in this case, from approximately 0.13 fs to 12.7 fs. The results obtained for pristine, annealed and DIO-processed films excited at photon energies labeled A–D are presented in Table 1. The significantly lower non-resonant contribution at the 1s–π* photon energy for the F8T2 film in comparison to the total signal limits the accuracy of some τCT values that were calculated.22
| Label | Photon energy (eV) | τCT (fs) | ||
|---|---|---|---|---|
| Pristine | Annealed | DIO | ||
| A (1s–π*) | 2471.7 | 16.9 | 12.6 | 10.1 |
| B (1s–σ*) | 2472.6 | 12.9 | 11.4 | 8.4 |
| C | 2474.3 | 4.0 | 3.7 | 3.2 |
| D | 2475.2 | 0.4 | 0.2 | 0.2 |
The τCT values calculated from the RAS spectra at resonance maximum (1s–σ*) are in the same order as those previously obtained for other thiophene-based polymers.21,24,25 A clear improvement in the charge transfer process can be observed in annealed and DIO-processed films when compared to the pristine F8T2 film at resonance maximum photon energy (2472.6 eV). The most significant improvement was observed for the films processed with the additive DIO, which showed the lowest τCT values for all photon energies. Such improvement can be attributed to an increase in film ordering, as supported by the results obtained from the NEXAFS spectra, which showed higher dichroism for the DIO-processed sample, followed by improvement in the chain packing, resulting in an increased electronic coupling.
Given the encouraging benefits resulting from the use of DIO, we decided to in depth investigate the effects of the additive on film formation. We then performed an atomic-scale molecular dynamics simulation of film formation from a model solution with and without the presence of DIO. The simulations start with 100% of the BT dihedral angles set to the anti conformation (as drawn in Fig. 1), which is the geometry derived from energy minimization calculations using the DFT method (see ESI† for more details). In the very first moments of MD evaporation (t = 1 ns, red curves in Fig. 5a), a fraction of the oligomers already rotates to the symmetrical syn conformation. However, there is a significant variation in the population of this conformer with the addition of DIO. For the CB only system, 0.7% of the conformers switch to the syn conformation compared to 18.8% in the system with DIO. 27.0 ns after the beginning of the “evaporation” process, the fraction of syn conformations increases to 16.0% and 39.34% in the systems without and with additives, respectively (see Fig. 5). After complete evaporation of the solvent, the dihedral population stabilizes and does not show significant variations over time. In the final steps of the process, the oligomers are already close enough to each other so that steric hindrances block further rotations of the thiophenes (even in the presence of DIO).
 | ||
| Fig. 5 (a) Dihedral population of syn and anti conformations between the thiophenes of the F8T2 polymer without (left) and with (right) DIO additives during CB solvent evaporation. (b) Normalized end-to-end length distribution of the F8T2 oligomers in the final configuration of MD evaporation with and without DIO. | ||
This further supports the hypothesis proposed in ref. 44 that the additive induces an anti to syn conversion of the BT units along the F8T2 backbone. One possible reason for this transition is the DIO-induced variation in the torsional energy barrier between the thiophene rings of the BT moiety. The proximity of the additive creates electrostatic shielding of the partial charges in the sulfur atoms, which decreases the activation energy necessary to achieve the syn conformation. The positive charge in the iodine is higher than that in the S atoms, so the electrostatic repulsion produced by the proximity of one I atom to one S atom of the thiophene ring facilitates the rotation of this ring toward the syn conformation. This hypothesis was investigated by tracking the radial pair distribution g(rij) (and also by directly calculating the average distances rij between the I and S atoms) during the MD evaporation process. These parameters are plotted in Fig. S8.† The radial pair distribution basically quantifies the relative density of iodine atoms around the S atoms at a given time along the evaporation. It is clear that during the final steps of the MD process, the iodine atoms of the DIO form a first solvation layer near the sulfur atoms of the F8T2 with an average ratio of ≃4.15 Å. This finding is confirmed by the time evolution of rij that decreases fast from 10.5 Å for the initial instant of the evaporation to saturate at ≃4.15 Å for later times (see Fig. S7 in the ESI file†).
The anti → syn transition induces a planarization of the bithiophene moiety, which tends to spiralize the polymer backbone. The spiral configuration of the chain minimizes intrachain interactions created by the proximity between the S atoms in the BT. Consequently, the polymeric chains of syn conformers are expected to have shorter end-to-end distances (R) compared to chains of anti conformers. This effect can be observed in Fig. 5b, which depicts the oligomer's population distribution as a function of the end-to-end distance. This distance was plotted in units of the total length (L), which corresponded to a completely stretched oligomer. One can see that the average end-to-end distance of the polymer in the presence of DIO peaks is around 0.41L. However, the population distribution of the film evaporated without DIO peaks at approximately 0.63L. These results further support the conclusion that the use of DIO significantly increases the concentration of oligomers with the syn conformation.
Fig. 6 shows the radial distribution g(rij) that quantifies the average interaction distance that separates pairs of moieties, computed for films evaporated with and without additives. These distances were evaluated considering the center of each group so that BT–BT corresponded to the interaction between bithiophene pairs, BT-FL between the fluorene and bithiophene pairs, and FL–FL between fluorene pairs. To interpret these results, it is important to point out that g(rij) is a relative quantity that compares the local density of group-to-group distances to the average density of distance pairs along the whole film. Hence, g(rij) = 1.0 means that the concentration of pairs separated by the distance rij is statistically equal to the average concentration for any other distance considered within the simulation box.
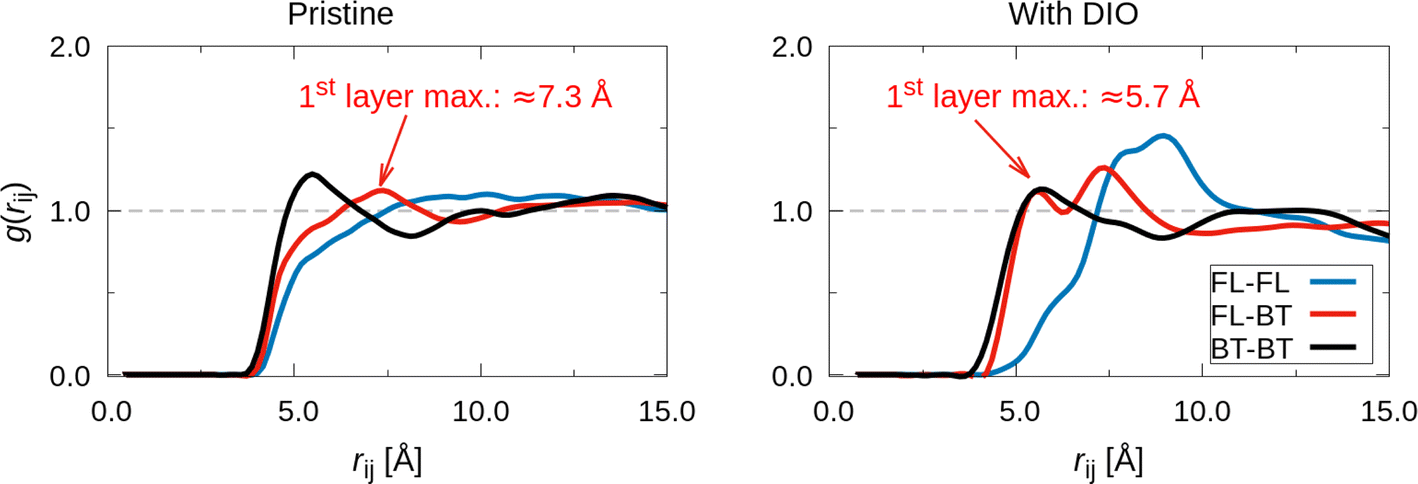 | ||
| Fig. 6 Radial distribution g(rij) of pairs between fluorene (FL) and bithiophene (BT) functional groups of different polymers. The distance was considered between the geometric centers of each functional group. | ||
The radial distribution (see Fig. 6) indicates that the BT group preferentially interacts with another BT in the film produced without DIO because there is a well distinct peak in the BT–BT g(rij) at approximately 5.7 Å. However, g(rij) has a maximum of around 7.3 Å for the BT-FL pairs, which is considerably longer compared to the average distance between bithiophene groups. The increase in the interaction distance for the BT-FL might be related to the exclusion volume effect created by the alkyl side chains attached to the fluorene moiety. The majority of anti conformations of the BT moiety in films prepared without additives favors a linear configuration of the oligomer chain with an alternated orientation of the alkyl chains along both sizes of the main backbone. This enhances the exclusion volume around the whole space surrounding each oligomer.
However, the g(rij) profile changes significantly when DIO is included in the evaporation process. Although the BT–BT interaction remains strong, its concentration decreases in favor of a higher density of BT-FL interactions. In addition, these BT-FL interactions tend to be stronger because the average distance between these groups decreases from 7.3 Å to 5.7 Å. One possible explanation for this effect might be linked to the preferential configuration of the backbone when a considerable number of BT groups are in the syn conformation. As mentioned above, the chain tends to assume a spiral configuration with the alkyl chains of the fluorene preferentially oriented in the direction of the spiraling axis.44 This orientation of the alkyl chains pointing in a preferential direction tends to reduce the exclusion volume effect so that a closer approximation of FL groups belonging to a nearby oligomer is possible from the side with a lower concentration of alkyl chains. By still considering the films prepared with DIO, there is also a second peak of the BT-FL g(rij) at 7.3 Å. This layer corresponds to the first peak in the film without DIO. However, this maximum is much better defined in the presence of the additive. A similar effect is found for the distribution of FL–FL pairs. The associated g(rij) in the CB only system does not show a distinct layer but seems to be spread over the simulation box with a distribution close to the average density for rij ≥ 7.0 Å. However, there is a well-defined maximum of g(rij) at 8 Å in the film evaporated with DIO. The presence of sharp peaks in the radial distributions for all the pairs considered suggests that the film with DIO has a higher degree of short-range order compared to the film produced without DIO additives.
5. Discussion
Our experimental and theoretical results show that the use of the additive DIO to solution process thin films of F8T2 induces changes in the polymeric chain organization, similar to thermal annealing. The order induced by the additive enhanced the preferential face-on orientation, as observed by the AR-NEXAFS measurements. Moreover, the induced order promotes a faster charge transfer process owing to the improved π–π stacking.The MD results help us to gain insight into the experimental observations from the detailed spectroscopic study reported above. In particular, the MD simulation confirms the higher ordering of the polymeric film after the use of DIO, which is in accordance with the angular dependence of the sulfur K-edge NEXAFS spectra. The evaporation with DIO tends to increase the concentration of syn conformers and decrease the dihedral angles between the thiophene rings of the BT group (see Fig. S7†). This planarization effect helps to induce a more ordered packing morphology of the chains. One important effect found in the MD simulations is shown in the radial distribution shown in Fig. 6. This indicates that DIO induces a shorter average separation between the BT and fluorene groups after the solvent evaporation simulation. Indeed, g(rij) for the BT-FL separation peaks at 5.7 Å in the DIO-processed film is very close to the distance between these two moieties in a single F8T2 chain. This behavior occurs because chains in the syn conformer tend to spiralize, approaching different points on the backbone. Because the long octyl side chains are attached to the FL group, the increased concentration of syn branches favors the interaction between the aliphatic chains and the BT moiety of the same (or adjacent) backbones. This trend is confirmed by the radial distribution g(rij) between the side chains and the bithiophene group (see Fig. S8†). This function has a peak for rij < 5.0 Å, which strongly suggests that the atoms belonging to the alkyl side chains can approach the BT ring (the highest peak of this radial distribution is around rij ≈ 6.0 Å). In fact, we were able to identify some structures in the DIO film with aliphatic chains closely interacting with one thiophene ring of the BT moiety (Fig. S9† exemplifies some of these structures).
Aiming at explaining how this subtle morphological variation can influence the binding energy of the 2p electron in sulfur, we build a simplified electrostatic interaction model. Given the complexity of the system, which involves many particle and force fields acting on them, a simple model that represents the essential features of the real situation can be a valuable tool for accessing the physical phenomena behind this BE shift. A detailed description of such a model is presented in the ESI file.† It essentially assumes an effective nuclear charge (Zeff) to account for the electrostatic shielding effect produced by the negatively charged electrons in the inner shells of the atom. According to this H atom-like model, a charge variation Δq ≈ 0.0148 e is enough to promote a shift in BE of 0.7 eV. This partial charge can come from a polarization generated by changes in ΔF ≈ 0.035 V Å−1 in the electric field surrounding the bithiophene units.
The question arises now as to whether morphological variations induced by the additive (as indicated by the MD evaporation) can produce variations in the local electric field surrounding the sulfur atom in the range ≥0.040 V Å−1. We addressed this question by considering the simplified two-dimensional scheme depicted in Fig. 7a. In this scheme, the relevant atoms in the system are represented by point charges following a geometrical distribution scaled from typical bond lengths derived from DFT calculations of F8T2 oligomers (see Table S5† for details). The net partial charges in the atoms are also estimated from DFT calculations and are typical of this kind of molecular system. The electric potential at every point is numerically calculated by adding the potential created by each charge so that the electric field is determined by taking the gradient of this potential along a determined direction. Using this simple tool, ΔF can be easily evaluated in the xy plane by considering rFL–S (the coordinate measures the distance of the S point (see Fig. 7a) to the origin of the coordinate system) and ![[r with combining macron]](https://www.rsc.org/images/entities/i_char_0072_0304.gif) AL–S (the coordinate references the average distance of S to the charges in the nearest alkyl lateral chain). These coordinates are associated with the radial distributions in Fig. 6 and S8c,† respectively.
AL–S (the coordinate references the average distance of S to the charges in the nearest alkyl lateral chain). These coordinates are associated with the radial distributions in Fig. 6 and S8c,† respectively.
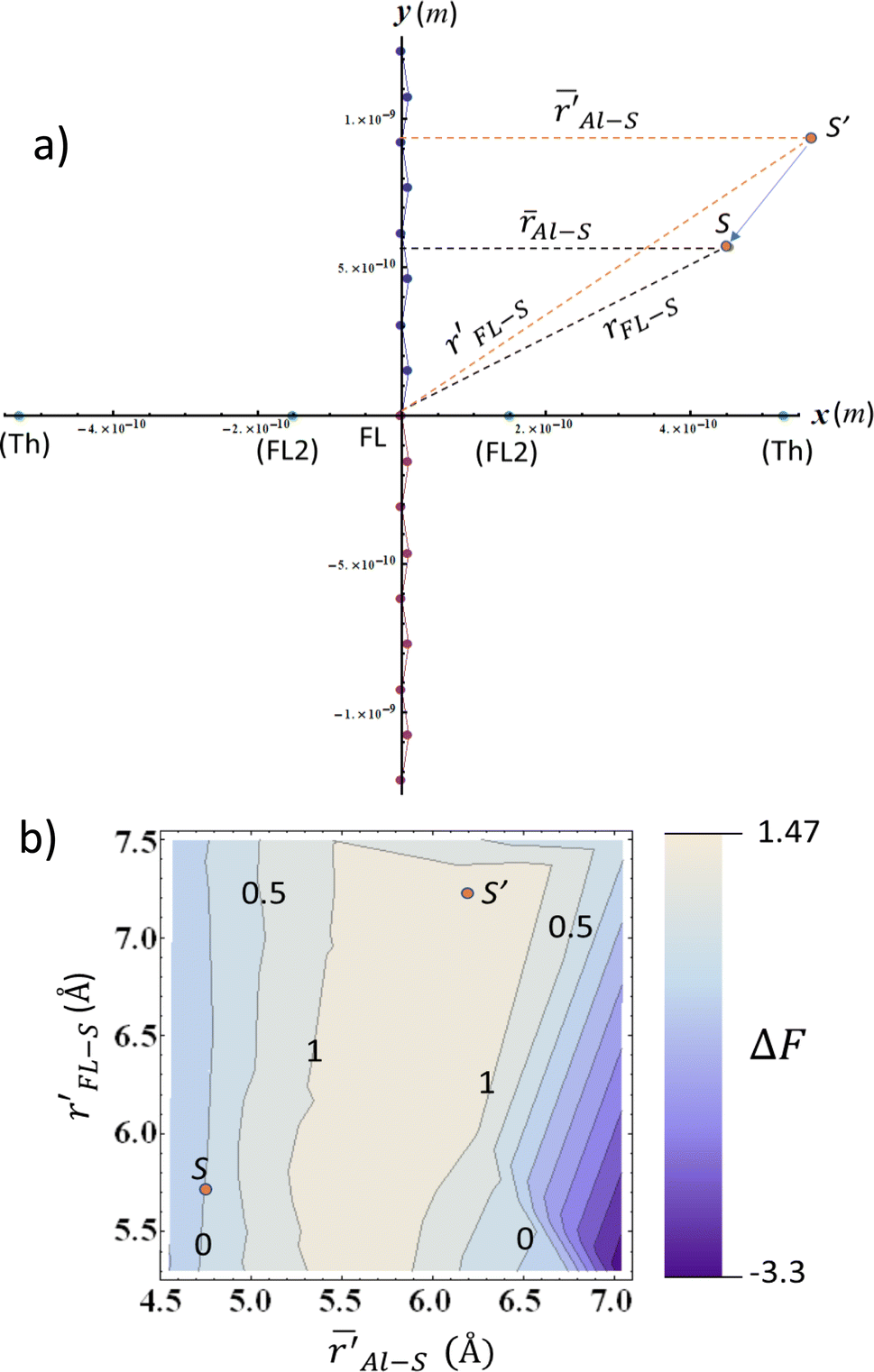 | ||
Fig. 7 (a) Simplified model of the electrostatic environment filled by the S atom (represented by points S and S′) in the vicinities of an F8T2 alkyl lateral chain. The coordinates rFL–S (r′FL–S) and  are the distances of S (S′) to the origin and the average distance to the nearest octyl side chain, respectively. The atomic charges are represented by point charges with values obtained from DFT calculations. The charge coordinates are also in accordance with the bond lengths and angles extracted from the DFT. The FL point in the origin is associated with the net charge of the central pentagonal ring of the FL group, whereas FL2 represents the net charge on the remaining three carbon atoms of the hexagonal ring. Finally, the point Th represents the net charge of the closest thiophene ring belonging to the BT group. See Table S22† for details of charge coordinates and magnitudes. (b) Normalized ΔF along the are the distances of S (S′) to the origin and the average distance to the nearest octyl side chain, respectively. The atomic charges are represented by point charges with values obtained from DFT calculations. The charge coordinates are also in accordance with the bond lengths and angles extracted from the DFT. The FL point in the origin is associated with the net charge of the central pentagonal ring of the FL group, whereas FL2 represents the net charge on the remaining three carbon atoms of the hexagonal ring. Finally, the point Th represents the net charge of the closest thiophene ring belonging to the BT group. See Table S22† for details of charge coordinates and magnitudes. (b) Normalized ΔF along the 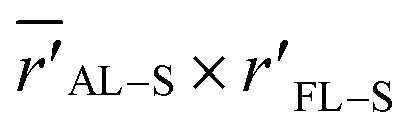 space relative to the coordinates of the point S in the bottom left corner. The variation in the electric field was calculated following the system described in (a). The coordinate of S(S′) corresponds approximately to the peaks of the radial distributions for MD evaporation with (without) DIO. The contour lines indicate the points with constant ΔF so that ΔF = 0.040 V Å−1 is the line with contour 1. space relative to the coordinates of the point S in the bottom left corner. The variation in the electric field was calculated following the system described in (a). The coordinate of S(S′) corresponds approximately to the peaks of the radial distributions for MD evaporation with (without) DIO. The contour lines indicate the points with constant ΔF so that ΔF = 0.040 V Å−1 is the line with contour 1. | ||
As depicted in Fig. 7a, we then map ΔF in the points of the 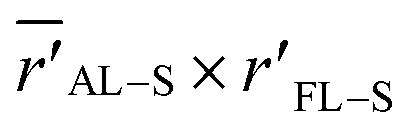 space that produces variations in the local electric field in the range ≥0.040 V Å−1. These calculations were performed by considering a reference point S with coordinates AL–S and rFL–S that corresponds to the peaks of the DIO radial distributions of Fig. 6 (for the BT-FL interaction) and Fig. S8c.† This point is highlighted near the bottom left corner of Fig. 7b. In this figure, the contour lines represent the points with a constant ΔF (normalized by the critical value 0.040 V Å−1). Any starting point S′ within the gray area limited by contour 1 in Fig. 7b feels a variation of F high enough to induce a 0.7 eV (or higher) shift in the BE when displaced toward point S. As illustrated in Fig. 7b, we also highlight the point S′ associated with the peaks of the respective radial distributions without DIO. One can observe that this point is located within the critical gray area, very close to the ΔF = 0.040 V Å−1 border. Although derived using simple modeling arguments, these results strongly suggest that the BE shifts are indeed due to an average decrease in the distance between the BT group and the aliphatic chains in the FL moiety promoted by processing the materials using the additive.
space that produces variations in the local electric field in the range ≥0.040 V Å−1. These calculations were performed by considering a reference point S with coordinates AL–S and rFL–S that corresponds to the peaks of the DIO radial distributions of Fig. 6 (for the BT-FL interaction) and Fig. S8c.† This point is highlighted near the bottom left corner of Fig. 7b. In this figure, the contour lines represent the points with a constant ΔF (normalized by the critical value 0.040 V Å−1). Any starting point S′ within the gray area limited by contour 1 in Fig. 7b feels a variation of F high enough to induce a 0.7 eV (or higher) shift in the BE when displaced toward point S. As illustrated in Fig. 7b, we also highlight the point S′ associated with the peaks of the respective radial distributions without DIO. One can observe that this point is located within the critical gray area, very close to the ΔF = 0.040 V Å−1 border. Although derived using simple modeling arguments, these results strongly suggest that the BE shifts are indeed due to an average decrease in the distance between the BT group and the aliphatic chains in the FL moiety promoted by processing the materials using the additive.
6. Conclusions
We employed a methodology that combines powerful spectroscopic techniques with molecular dynamics simulation (MD) and mathematical modelling to assess important correlations between the final morphology of the film and the procedures followed to produce it, viz. the effects of thermal annealing versus the additive processing in thin films of F8T2.XPS analysis was efficient in determining the chemical composition of the surface of the films, confirming the presence of the thiophene unit in the samples. Additionally, the core-hole clock method was employed to investigate the charge transfer dynamics in the F8T2 thin films. The effect of thermal annealing and additive processing was monitored through charge transfer times (τCT), with significant enhancement in the charge transfer process being observed, especially for the DIO-processed samples. This is attributed to a higher ordering of the polymeric film in accordance with the results obtained from analyzing the angular dependence of the sulfur K-edge NEXAFS spectra, which showed the strongest dichroism for the F8T2 film processed with DIO.
Spectroscopic techniques and theoretical simulations indicate that the ordering of F8T2 films rises as a result of the presence of DIO molecules in the solution. Essentially, there is a conformational transition induced by the additive that increases the concentration of syn conformers relative to the film processed without DIO. It was found that those conformers tended to have lower dihedral angles between the thiophene rings of the BT group. The planarization of the chain favors a more ordered morphology, which can improve the electronic properties of the film that are related to charge transport.
Finally, XPS measurements revealed a shift in sulfur polarization with the DIO processing and thermal annealing treatment. Here, we demonstrate that this shift is due to variations in the electrostatic environment that change the electric field in the vicinities of the S atom. This effect is induced by subtle modifications in the film morphology induced by the additive or annealing. The DIO promotes a stronger interaction of the S atoms in the BT group with the FL group and the lateral octyl chains. These results reinforce the usefulness of the strategy proposed here to detect indistinct effects relating film morphology to the procedures followed during deposition.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by the LNLS – National Synchrotron Light Laboratory, Brazil. The authors acknowledge the financial support from CNPq (308930/2019-3), CAPES, CT-INFRA FINEP and FAPERJ (E-26/202.623/2019). CM and CMA thank the Swedish National Infrastructure for Computing (SNIC) at the PDC Center for High-Performance Computing and National Supercomputer Center at Linköping University (NSC) and CMA thanks the STandUP for energy collaboration for financial support.References
- C.-r. Hao, X. Zong, Y. Cheng, M. Zhao, M. Luo, Y. Zhanga and S. Xue, Sustainable Energy Fuels, 2021, 5, 5548–5556 RSC.
- T. Wu, D. Wang, X. Jiang, X. Ge, F. Guo, T. Ye, S. Gao and Y. Zhang, Adv. Mater. Interfaces, 2022, 9, 2200923 CrossRef CAS.
- X. Wu, Y. Wu, S. Peng, L. Xiao, Z. Xiao, W. Zhang, G. Ren, Y. Min and Y. Liu, RRL Sol., 2023, 7(17), 2300333 CrossRef.
- Y. M. Kim, E. Lim, I.-N. Kang, B.-J. Jung, J. Lee, B. W. Koo, L.-M. Do and H.-K. Shim, Macromolecules, 2006, 39(12), 4081–4085 CrossRef CAS.
- E. Lim, Y. M. Kim, J.-I. Lee, B.-J. Jung, N. S. Cho, J. Lee, L.-M. Do and H.-K. Shim, J. Polym. Sci., Part A: Polym. Chem., 2006, 44(16), 4709–4721 CrossRef CAS.
- L. Xie, et al., Adv. Funct. Mater., 2023, 33, 2303947 CrossRef.
- G. Huseynova, J.-H. Lee, Y. H. Kim and J. Lee, Org. Electron., 2020, 86, 105900 CrossRef CAS.
- W. Huang, et al, Adv. Funct. Mater., 2022, 32(5), 2106564 CrossRef.
- B. B. Carbas, Polymer, 2022, 254, 125040 CrossRef.
- L. Scalon, A. L. Neto, L. O. Araujo, S. Zaioncz, J. B. Floriano, A. G. Macedo, C. M. Araujo, C. F. N. Marchiori and P. C. Rodrigues, ACS Appl. Polym. Mater., 2021, 3(8), 4223–4233 CrossRef CAS.
- J.-H. Huang, C.-Y. Yang, Z.-Y. Ho, D. Kekuda, M.-C. Wu, F.-C. Chien, P. Chen, C.-W. Chu and K.-C. Ho, Org. Electron., 2009, 10(1), 27–33 CrossRef CAS.
- Y. Garcia-Basabe, N. A. D. Yamamoto, L. S. Roman and M. L. M. Rocco, Phys. Chem. Chem. Phys., 2015, 17(17), 11244–11251 RSC.
- O. Kettner, et al., Synth. Met., 2016, 220(1), 162–173 CrossRef CAS.
- H. Wang, Y. Zheng, L. Zhang and J. Yu, Sol. Energy Mater. Sol. Cells, 2014, 128, 215–220 CrossRef CAS.
- J.-H. Huang, C.-Y. Yang, Z.-Y. Ho, D. Kekuda, M.-C. Wu, F.-C. Chien, P. Chen, C.-W. Chu and K.-C. Ho, Org. Electron., 2009, 10(1), 27–33 CrossRef CAS.
- Z. Yi, W. Ni, Q. Zhang, M. Li, B. Kan, X. Wana and Y. Chen, J. Mater. Chem. C, 2014, 2(35), 7247–7255 RSC.
- L. Chen, M. Wu, G. Shao, J. Hu, G. He, T. Bu, J.-P. Yi and J. Xia, R. Soc. Open Sci., 2018, 5(5), 172041 CrossRef PubMed.
- Y. Liang, Z. Xu, J. Xia, S.-T. Tsai, Y. Wu, G. Li, C. Ray and L. Yu, Adv. Mater., 2010, 22(20), E135–E138 CrossRef CAS PubMed.
- X. Liu, Y. Cai, X. Huang, R. Zhang and X. Sun, J. Mater. Chem. C, 2013, 5(12), 3188–3194 RSC.
- L. Cao, M. Yang, L. Yuan, N. Nerngchamnong, Y.-P. Feng, A. T. S. Wee, D.-C. Qi and C. A. Nijhuis, J. Phys.: Condens.Matter, 2016, 28(9), 094006 CrossRef PubMed.
- C. Arantes, B. G. A. L. Borges, B. Beck, G. Araujo, L. S. Roman and M. L. M. Rocco, J. Phys. Chem. C, 2013, 117(16), 8208–8213 CrossRef CAS.
- D. Menzel, Chem. Soc. Rev., 2008, 37(10), 2212–2223 RSC.
- Y. Garcia-Basabe, B. G. A. L. Borges, D. C. Silva, A. G. Macedo, L. Micaroni, L. S. Roman and M. L. M. Rocco, Org. Electron., 2013, 14(11), 2980–2986 CrossRef CAS.
- Y. Garcia-Basabe, C. F. N. Marchiori, B. G. A. L. Borges, N. A. D. Yamamoto, A. G. Macedo, M. Koehler, L. S. Roman and M. L. M. Rocco, J. Appl. Phys., 2014, 115(13), 134901 CrossRef.
- B. G. A. L. Borges, L. S. Roman and M. L. M. Rocco, Top. Catal., 2019, 62(12), 1004–1010 CrossRef CAS.
- K. R. d. A. Sousa, L. Benatto, L. Wouk, L. S. Roman and M. Koehler, Phys. Chem. Chem. Phys., 2020, 22(17), 9693–9702 RSC.
- L. Benatto, K. R. d. A. Sousa and M. Koehler, J. Phys. Chem. C, 2020, 124(25), 13580–13591 CrossRef CAS.
- L. Benatto, C. A. M. Moraes, G. Candiotto, K. R. d. A. Sousa, J. P. A. Souza, L. S. Roman and M. Koehler, J. Mater. Chem. A, 2021, 9(48), 27568–27585 RSC.
- R. Alessandri, J. J. Uusitalo, A. H. de Vries, R. W. Havenith and S. J. Marrink, J. Am. Chem. Soc., 2017, 139(10), 3697–3705 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2(1), 73–78 CAS.
- T. Y. Leung, W. F. Man, P. K. Lim, W. C. Chan, F. Gaspari and S. Zukotynski, J. Non-Cryst. Solids, 1999, 254(1–3), 156–160 CrossRef CAS.
- M. Bouabdallaoui, Z. Aouzal, S. Ben Jadi, A. El Jaouhari, M. Bazzaoui, G. Lévi, J. Aubard and E. A. Bazzaoui, J. Solid State Electrochem., 2017, 21(12), 3519–3532 CrossRef CAS.
- A. Lachkar, A. Selmani and E. Sacher, Synth. Met., 1995, 72(1), 73–80 CrossRef CAS.
- M. Vásquez, G. J. Cruz, M. G. Olayo, T. Timoshina, J. Morales and R. Olayo, Polymer, 2006, 37(23), 7864–7870 CrossRef.
- J. Riga, P. Snauwaert, A. De Pryck, R. Lazzaroni, J. P. Boutique, J. J. Verbist, J. L. Brédas, J. M. André and C. Taliani, Synth. Met., 1987, 21(1–3), 223–228 CrossRef CAS.
- E. T. Kang, K. G. Neoh and K. L. Tan, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44(19), 10461–10469 CrossRef CAS PubMed.
- G. Beamson and D. Briggs. High Resolution XPS of Organic Polymers, John Wiley & Sons, Chichester (GB), 1992 Search PubMed.
- G. Zotti, S. Zecchin, G. Schiavon and L. Groenendaal, Chem. Matter, 2000, 12(10), 2996–3005 CrossRef CAS.
- J. R. Santa Rita, C. Arantes, G. Araújo, L. S. Roman, L. Micaroni and M. L. M. Rocco, J. Electron Spectrosc. Relat. Phenom., 2011, 184(3–6), 265–269 CrossRef.
- A. P. Hitchcok, J. A. Horsley and J. Stöhr, J. Chem. Phys., 1986, 85(9), 4835–4848 CrossRef.
- Y. Garcia-Basabe, G. G. Parra, M. B. Barioni, C. D. Mendoza, F. C. Vincentin and D. G. Larrudé, Phys. Chem. Chem. Phys., 2019, 21(42), 23521–23532 RSC.
- M. L. M. Rocco, T. Sekiguchi and Y. Baba, J. Vac. Sci. Technol., A, 2006, 24(6), 2117–2121 CrossRef CAS.
- M. L. M. Rocco, D. E. Weibel, L. S. Roman and L. Micaroni, Surf. Sci., 2004, 560(1–3), 45–52 CrossRef CAS.
- O. D. Lourenço, L. Benatto, C. F. N. Marchiori, H. C. Avila, N. A. D. Yamamoto, C. K. Oliveira, M. G. E. da Luz, M. Cremona, M. Koehler and L. S. Roman, J. Phys. Chem., 2017, 121(29), 16035–16044 Search PubMed.
- J. L. Campbell and T. Papp, At. Data Nucl. Data Tables, 2001, 77(1), 1–56 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra08066h |
| This journal is © The Royal Society of Chemistry 2024 |