
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel phenylthiazoles with a tert-butyl moiety: promising antimicrobial activity against multidrug-resistant pathogens with enhanced ADME properties†
Mohamed Hagras *a,
Abdelrahman A. Abuelkhira,
Nader S. Abutalebbc,
Ahmed M. Helala,
Iten M. Fawzyd,
Maghawry Hegazye,
Mohamed N. Seleembf and
Abdelrahman S. Mayhoub*ag
*a,
Abdelrahman A. Abuelkhira,
Nader S. Abutalebbc,
Ahmed M. Helala,
Iten M. Fawzyd,
Maghawry Hegazye,
Mohamed N. Seleembf and
Abdelrahman S. Mayhoub*ag
aDepartment of Pharmaceutical Organic Chemistry, College of Pharmacy (Boys), Al-Azhar University, Cairo 11884, Egypt. E-mail: m.hagrs@azhar.edu.eg; amayhoub@azhar.edu.eg
bDepartment of Biomedical Sciences and Pathobiology, Virginia-Maryland College of Veterinary Medicine, Virginia Polytechnic Institute and State University, Blacksburg, Virginia 24061, USA
cDepartment of Microbiology and Immunology, Faculty of Pharmacy, Zagazig University, Zagazig 44519, Egypt
dDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Future University in Egypt, 11835, Cairo, Egypt
eBiochemistry and Molecular Biology Department, Faculty of Pharmacy (Boys), Al-Azhar University, Cairo 11884, Egypt
fCenter for One Health Research, Virginia Polytechnic Institute and State University, Blacksburg, Virginia 24061, USA
gUniversity of Science and Technology, Nanoscience Program, Zewail City of Science and Technology, October Gardens, 6th of October, Giza 12578, Egypt
First published on 3rd January 2024
Abstract
The structure–activity relationship of a new tert-butylphenylthiazole series, with a pyrimidine linker, was investigated. We wished to expand knowledge of this novel class of antibiotics by generating 21 new derivatives bearing ≥2 heteroatoms in their side chains. Their activity was examined against isolates of methicillin-resistant Staphylococcus aureus (MRSA), Clostridium difficile, Escherichia coli, Neisseria gonorrhoeae, and Candida albicans. Two compounds with 1,2-diaminocyclohexane as a nitrogenous side chain showed promising activity against the highly infectious MRSA USA300 strain, with a minimum inhibitory concentration (MIC) of 4 μg mL−1. One of these two compounds demonstrated potent activity against C. difficile, with a MIC of 4 μg mL−1. Moderate activities against a C. difficile strain with a MIC of 8 μg mL−1 were noted. Some new compounds possessed antifungal activity against a wild fluconazole-resistant C. albicans strain, with MIC values of 4–16 μg mL−1. ADME and metabolism-simulation studies were performed for the most promising compound and compared with lead compounds. Our results revealed that one compound possessed greater penetration of bacterial membranes and metabolic resistance, which aided a longer duration of action against MRSA.
1. Introduction
Antimicrobial resistance (AMR) is among the most relevant health problems of this century.1,2 Methicillin-resistant Staphylococcus aureus (MRSA) is one of the main reasons for persistent infections in humans.3 MRSA causes severe morbidity and mortality worldwide so, in 2017 the World Health Organization considered it to be a high-priority multidrug-resistant pathogen.4 MRSA is responsible for serious infections resistant to most antibiotics on the market, such as skin and soft-tissue infections, bacteremia, infective endocarditis, osteomyelitis, and pneumonia. Moreover, MRSA is often responsible for infections due to indwelling catheters, prosthetic devices, and implants.5–7 A significant gene (mecA) confers to MRSA the ability to grow undisturbed in the presence of penicillin-like antibiotics. mecA is found in all MRSA strains and encodes penicillin binding protein 2a (PBP2a).8 Clostridium difficile has been recognized worldwide as the leading cause of nosocomial diarrhea, which is associated with substantial morbidity and mortality.9,10 Recent reports have suggested an increase in the incidence and severity of C. difficile infection.11 Based on statistical analyses from the US Centers of Disease Control and Prevention, each year, more than 250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 people need hospital care and at least 14000 people die from C. difficile infection in the USA.12
000 people need hospital care and at least 14000 people die from C. difficile infection in the USA.12
AMR has reached alarming levels globally.13,14 Each year, ∼750000 deaths due to AMR are reported, and the toll is likely to grow to 10 million by 2050.13 Treatments for bacterial infections, including β-lactam antibiotics,15 macrolides,16 fluoroquinolones.16–19 glycopeptides (chiefly vancomycin)20 and oxazolidinones (primarily linezolid), are increasingly becoming inadequate.21 Therefore, there is an urgent need to synthesize novel antibiotics to overcome the main mechanisms for bacterial resistance.22
Previously, phenylthiazoles were reported to be new scaffolds with wide antimicrobial activity against multidrug-resistant strains of S. aureus, including MRSA and VRSA.23 Deep-dive investigations on lead compound 1a revealed its efficacy against bacteria arose from its ability to block the construction of their cell walls.24 Prior to this finding, a thorough analysis of the structure–activity relationship (SAR) of 1a showed that it possessed a central thiazole ring attached to two unique features: a cationic aminoguanidine moiety (highlighted in red in Fig. 1) at position C5 and a lipophilic n-butyl moiety (highlighted in blue) at C2. These two key features were vital for the potent activity against MRSA of 1a, so removal of any one of them led to a complete loss of activity.23 The bacterial target was not determined at the time of discovery of lead compound 1a, so a structure-based drug-design approach could not be applied for further optimization of the structure. Instead, a lead compound-based drug-design approach had been adopted and two important structural elements were identified: a guanidine moiety (colored red in Fig. 1) and a lipophilic tail (colored blue).23
 | ||
| Fig. 1 Developmental progress of phenylthiazole antibiotics and a new approach to improve metabolic stability and activity. | ||
The initial series of phenylthiazole compounds (represented by compound 1a in Fig. 1) suffered from a short half-life (t1/2),23 which limited systemic pharmacological application. A comprehensive metabolic study referred to the butyl benzylic carbon as a metabolic “soft spot”,25 which replaced an oxygen atom to provide derivatives 1b with a longer duration of action.25 On the other hand, modifications related to the cationic head (where the readily hydrolysable C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N was incorporated in a heterocyclic linker system) led to the potent and metabolically stable analogue 1c (Fig. 1).26
N was incorporated in a heterocyclic linker system) led to the potent and metabolically stable analogue 1c (Fig. 1).26
This structural modification of the cationic head resulted in several phenylthiazoles bearing, at thiazole position-5, a pyrimidine ring connected with different amines, guanidine, or a guanidine-like moiety.26 A biological study of this generation indicated that cationic side chains with more than one heteroatom (N or O) had greater antibacterial action, and drew attention to the role of the distance and configuration of this moiety upon activity (Fig. 1).26
The present study investigated a dual tactic to maximize pharmacodynamic and pharmacokinetic profiles by blocking the metabolic soft spot (benzylic C–H oxidation) and preventing hydrolysis of the Schiff bond. The first tactic was achieved via replacement of the n-butyl of lead compound 1a with a tertiary isostere, whereas the second tactic was achieved by incorporating it in the rigid system (pyrimidine linker).26,27 In addition, the SAR at the nitrogenous head was studied carefully. A new set of derivatives was designed to cover all possibilities of the distance between the heteroatoms and/or different spatial configurations of the cationic head (Fig. 1).
2. Results and discussion
2.1. Chemistry
The methylsulfonyl derivative 7 was readily obtained, as reported previously,28 by allowing enaminone 4 to react with thiourea followed by methylation with dimethyl sulfate and oxidation of the corresponding thioether 6 using m-chloroperbenzoic acid (MCPBA) (Scheme 1). In an attempt to use an alternative shorter pathway, enaminone 4 was allowed to react with S-methylisothiourea, Unfortunately, the required product (compound 6) could not be separated from the reaction mixture with the desirable purity, and most of the yield was lost during successive chromatographic separations. The inserted S-methyl group of compound 6 was represented by one extra singlet signal in the aliphatic region of the 1H NMR spectrum at 2.77 ppm. This signal was shifted downfield to 3.47 ppm upon oxidation of thioether 6 to the methylsulfonyl analogue 7. Finally, the methylsulphonyl intermediate 7 was utilized to complete a series of optimized tert-butylphenylthiazoles. Hence, the methylsulphonyl moiety was reacted with 21 nucleophiles to afford the corresponding final products 8–28, respectively (Scheme 1). These nucleophiles included primary and secondary amines. The structures of this set of novel compounds were confirmed by spectral and elemental analyses (see Experimental section). | ||
| Scheme 1 Reagents and conditions: (a) absolute EtOH, 3-chloropentane-2,4-dione, heat at reflux, 12 h; (b) DMF–DMA heat at 80 °C, 8 h; (c) thiourea, KOH, EtOH, heat at reflux, 8 h; (d) dimethyl sulfate, KOH, H2O, stirring at 23 °C, 2 h; (e) MCPBA, dry DCM, stirring at 23 °C, 16 h; (f) appropriate amine, dry DMF, heat at 80 °C for 0.5–8 h. | ||
2.2. Biological results and discussion
Studying the antimicrobial activity of the newly synthesized compounds with a two-carbon unit distance between the two heteroatoms (compounds 8–14) provided considerable SAR information. In brief, ethylenediamine-containing compound 8 did not have any microbial activity. Addition of two alkyl groups at the second nitrogen (compound 10) resulted in an increase in antimicrobial activity against C. difficile. Replacement of the free amino group with a hydroxyl group provided (compound 9) with one-fold enhanced antibacterial activity against C. difficile and weak activity against MRSA because the minimum inhibitory concentration (MIC) value of 9 was 32 μg mL−1 vs. MRSA and 16 μg mL−1 vs. C. difficile. The second set of structural optimizations included an increase in the distance to three carbon units to give compounds 12–16. Propylenediamine derivative 12 maintained the antibacterial activity of compound 9 in addition to moderate antifungal activity with an MIC of 8 μg mL−1. The hydroxyl analogue (compound 13) was more potent than compound 12 with one-fold activity (MIC = 16 μg mL−1) vs. MRSA and four-fold activity against C. difficile (MIC = 4 μg mL−1). All other derivatives with a second hydroxyl group at the adjacent carbon with different chirality (stereoisomers 14, 15, 16 and 17) maintained activity against Gram-positive bacteria. The only exceptions among the hydroxylated derivatives were the diol positional isomers 14 and 17, which reduced the activity against C. difficile with MIC values of 16 and 32 μg mL−1, respectively. This result suggested that compounds 14 and 17 might possess selective activity against Gram-positive bacteria or specifically C. difficile. This observation might add an additional clinical value because compounds 14 and 17 are not expected to disturb the normal human microbiota (Table 1).
|
|||||||
|---|---|---|---|---|---|---|---|
| Cpd | Side chain | MRSA NRS384 (MRSA USA300) | E. coli JW55031 (TolC mutant) | E. coli BW25113 (wild-type strain) | Clostridium difficile ATCC BAA 1870 | Candida albicans ATCC 64124 | Neisseria gonorrhoeae 194 |
| 1a | NA | 4.8 | NT | NT | NT | NT | NT |
| 8 |  |
>64 | >64 | >64 | >64 | 64 | >64 |
| 9 |  |
32 | >64 | >64 | 16 | >64 | >64 |
| 10 |  |
>64 | >64 | >64 | 32 | 64 | >64 |
| 11 |  |
>64 | >64 | >64 | >64 | >64 | >64 |
| 12 |  |
32 | 32 | >64 | 16 | 8 | >64 |
| 13 |  |
16 | >64 | >64 | 4 | >64 | >64 |
| 14 |  |
>64 | >64 | >64 | 16 | >64 | >64 |
| 15 | 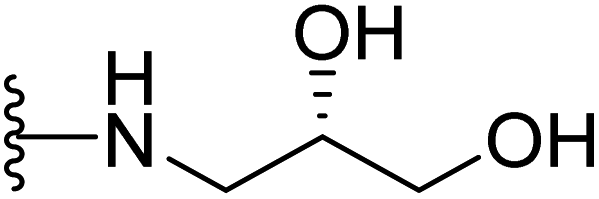 |
32 | >64 | >64 | 16 | >64 | >64 |
| 16 |  |
32 | >64 | >64 | 8 | >64 | >64 |
| 17 | 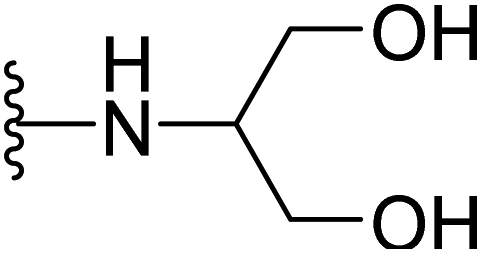 |
>64 | >64 | >64 | 32 | >64 | >64 |
| 18 |  |
16 | 16 | >64 | 32 | 16 | >64 |
| 19 |  |
4 | >64 | >64 | 8 | >64 | >64 |
| 20 |  |
4 | 8 | >64 | 4 | 4 | >64 |
| 21 |  |
16 | 16 | >64 | 8 | 16 | >64 |
| 22 |  |
16 | 16 | >64 | >64 | >64 | >64 |
| 23 |  |
>64 | >64 | >64 | 32 | >64 | >64 |
| 24 |  |
>64 | >64 | >64 | 64 | >64 | >64 |
| 25 |  |
32 | >64 | >64 | 32 | >64 | >64 |
| 26 |  |
16 | >64 | >64 | 16 | >64 | >64 |
| 27 |  |
16 | >64 | >64 | 16 | >64 | >64 |
| 28 |  |
16 | >64 | >64 | 8 | >64 | >64 |
| Linezolid | NA | 1 | 8 | >64 | NT | NT | |
| Vancomycin | NA | 1 | NT | NT | NT | NT | |
| Gentamicin | NA | NT | ≤0.5 | ≤0.5 | NT | NT | |
| Cefixime | NA | NT | NT | NT | NT | 1 | |
| Amphotericin B | NA | NT | NT | NT | 1 | NT | |
| Fluconazole | NA | NT | NT | NT | >64 | NT | |
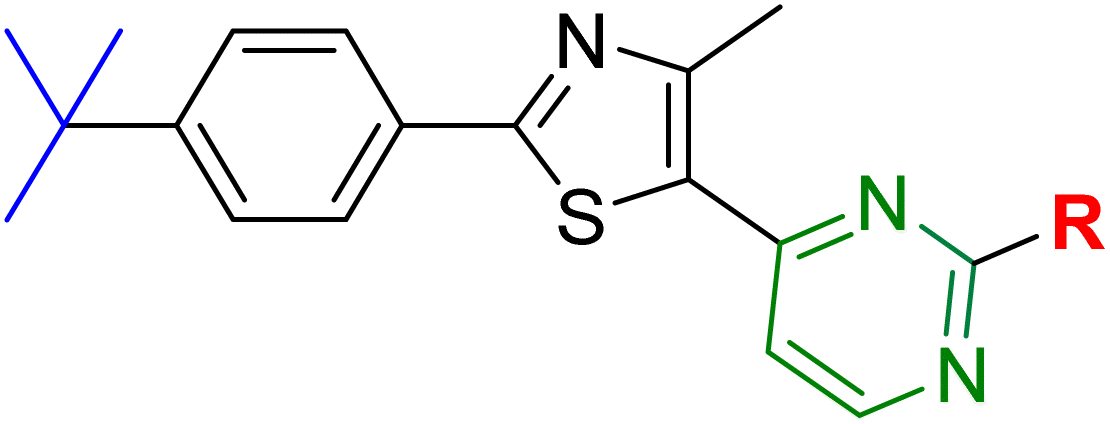
Encouraged by the previous result, the next set of structures 18–28 included addition of a second polar atom (N or O) at a two-carbon-unit distance with different possibilities for spatial configurations. For the diamino-cyclohexane derivatives 18, 19 and 20, the trans-1,2-diamino isomer (compound 20) showed good anti-MRSA activity with an MIC value of 4 μg mL−1 (near to that of the lead structure 1a) in addition to potent activity against C. difficile and Candida species and moderate activity against Gram-negative bacteria. Changing the stereo configuration of the two amino group to cis (compound 19) maintained the anti-MRSA activity with an MIC value of 4 μg mL−1 but decreased activity against tested Escherichia coli, C. difficile, and Candida albicans (Table 1). In addition, compound 18 (1,4-diamino isomer) maintained moderate potency against the E. coli JW55031 strain, MRSA USA300, and C. albicans.
On the other hand, pyrrolidine-containing derivatives 22–28 showed mixed effects against microbes. The amino pyrrolidine 22 had moderate activity against MRSA and E. coli JW55031 and showed a MIC value of 16 μg mL−1, respectively. Hydroxymethylpyrrolidines 24 were completely void of antimicrobial activity. Carboximidine and dimethylamino pyrrolidine derivatives 25–28 showed moderate activity against MRSA and C. difficile strains.
In brief, our SAR study revealed the impact of structural modifications on the antimicrobial activity of newly synthesized compounds. Ethylenediamine (compound 8) lacked activity, but addition of alkyl groups (compound 10) or hydroxylation (compound 9) enhanced activity against C. difficile. Increasing the carbon distance (compounds 12–16) maintained activity, and introduction of a second polar atom (compounds 18–28) enabled further elucidation of the SAR. Pyrrolidine-containing derivatives (compounds 22–28) exhibited mixed effects. These findings provided valuable insights into the potential of these compounds as antimicrobial agents.
 | ||
| Fig. 2 Analyses of the toxicity of compound 20 (tested in triplicates at 8, 16, 32 and 64 μg mL−1) against human breast cancer (MCF-7) cells using the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay. Results are presented as percent viable cells relative to DMSO (negative control) to determine a baseline measure for the cytotoxic impact of each compound. Absorbance values represent an average of three samples analyzed for each compound. Error bars represent standard deviation values. Data were analyzed via two-way ANOVA with post hoc Dunnett's test for multiple comparisons. * denotes a significant difference (P < 0.05) between values obtained for compounds and DMSO. | ||
2.3. Absorption, distribution, metabolism, and excretion (ADME) studies
p calculations. Adopting the SWISS-ADME method, the three compounds were subjected to calculations of ADME molecular properties and count of cytochrome P450 (CYP) inhibition using five strains. The results are shown in Table 2, Fig. 3 and 4. The results were also validated using drug-likeness calculations to predict Mollogp for hydrophobicity comparison, pKa, and model score of drug-likeness (Fig. 5).
| Software | SWISS-ADME | DSa | SWISS-ADME | DS | MolSoft | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Parameters vs. compounds | Solubility | LogS |
GI absorption | CYP 1A2 inhibitor | CYP 2C19 inhibitor | CYP 2C9 inhibitor | CYP 2D6 inhibitor | CYP 3A4 inhibitor | CYP2D6 applicability MDp value | PPB availability | Hepatotoxic availability | Logp o/w |
Alogp |
Mollogp |
pKa (most basic/acidic groups) | Model score |
| a DS: Discovery Studio 4.1 software.b Mod.: moderate. | ||||||||||||||||
| Lead 1a | Mod | 1.22 × 10−2 mg mL | High | Yes | Yes | Yes | No | Yes | 1.15996 × 10−5 | 16.07 | 13.51 | 3.08 | 3.699 | 4.48 | 1.02 | 0.68 |
| Lead 1c | Mod | 9.40 × 10−3 mg mL | Low | Yes | Yes | Yes | No | Yes | 5.20627 × 10−6 | 15.45 | 11.71 | 3.25 | 4.339 | 4.44 | 0.82 | 1.47 |
| Comp. 20 | Modb | 4.92 × 10−4 mg mL | High | No | Yes | No | No | Yes | 1.63303 × 10−5 | 13.07 | 10.82 | 4.20 | 4.325 | 5.34 | 0.81 | 0.68 |
 | ||
| Fig. 3 Radar map of the ADME calculations for lead compound 1a, lead compound 1c, and compound 20. | ||
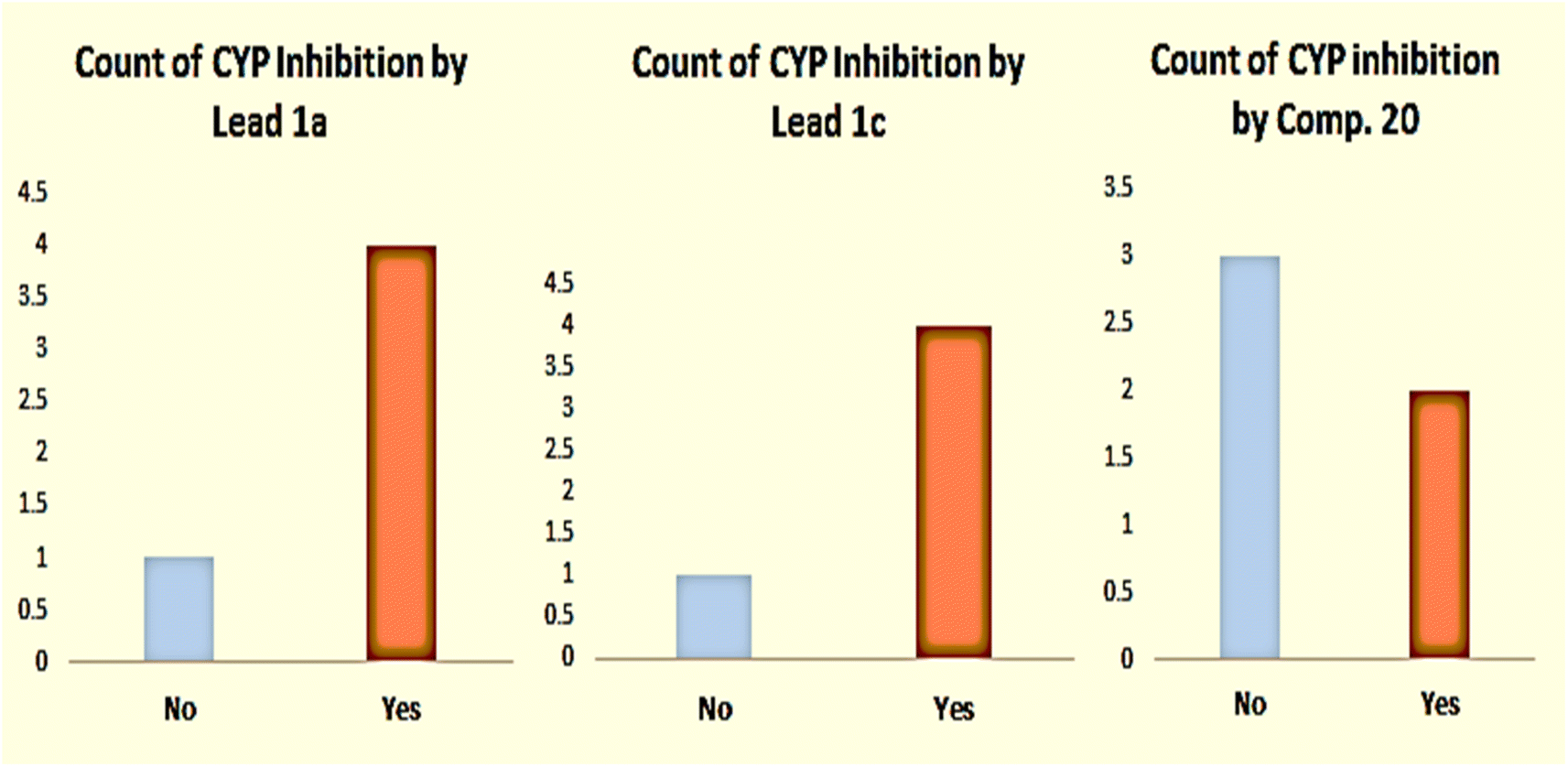 | ||
| Fig. 4 Count of CYP inhibition of five strains by lead compound 1a, lead compound 1c, and compound 20. | ||
 | ||
| Fig. 5 Drug-likeness model obtained from calculations of molecular properties of lead compound 1a, lead compound 1c, and compound 20. | ||
The results shown above displayed clear evidence of low interaction of compound 20 with metabolic enzymes compared with that of the other two lead compounds, which suggested metabolic stability. Moreover, the hydrophobicity of compound 20 expressed in different forms of logp was higher than those of the lead compounds, which provided proof of efficient permeability and penetration of bacterial membranes, suggesting that it was a more bioavailable drug.
 | ||
| Fig. 6 Pharmacokinetic analysis of lead compound 1a and compound 20. | ||
 | ||
| Fig. 7 Half-life of lead compound 1a and compound 20. | ||
Owing to the tertiary butyl group rather than an n-alkyl chain, the hydrophobicity of compound 20 was higher and penetration of bacterial membranes was greater. Also, the bulky steric effect of the group hindered the metabolic process, so a longer duration of biological activity was observed.32,33
3. Conclusions
Dual modification on lead compound 1a (via a t-butyl moiety and pyrimidine linker) provided a new series of phenylthiazoles characterized by broad-spectrum antimicrobial activity. Among the tested nitrogenous moieties, compounds 19 and 20, with a two-carbon distance between the two hetero atoms through a six-membered ring, possessed the most potent activity against the highly infectious MRSA USA300 strain with an MIC value of 4 μg mL−1. Compounds 13 and 20 exhibited potent activity against C. difficile. Four other derivatives (16, 19, 21, and 28) exhibited moderate activity against C. difficile with MIC values of 8 μg mL−1, respectively. Compounds 20, 21, and 22 exhibited some activity against TolC-mutant E. coli, and they did not show activity against wild-type E. coli nor N. gonorrhoeae, suggesting that these compounds might be removed from Gram-negative bacteria by efflux pumps. Also, compounds 12, 18, 20, and 21 exhibited activity against a C. albicans strain with MIC values of 4–16 μg mL−1.4. Experimental
4.1. Chemistry
General procedure. An appropriate amine (0.4 mmol) was added to a solution of compound 7 (0.1 g, 0.25 mmol) in dry DMF (5 mL). The reaction mixture was heated at 80 °C for 4–8 h, and then poured over iced water (50 mL). The formed solid was filtered and washed with 50% ethanol and recrystallized from absolute ethanol or extracted by ethyl acetate. Then, it was dried over MgSO4 and evaporated under reduced pressure to give the products. The physical properties and spectral analysis of isolated products are shown below.
N-{4-[2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl]pyrimidin-2-yl}ethane-1,2-diamine (8). Following the general procedure and using ethylenediamine (21 μL, 0.4 mmol), compound 8 was obtained as a yellowish-brown solid (90 mg, 91%) mp = 137 °C; 1H NMR (DMSO-d6) δ: 8.32 (d, J = 5.2 Hz, 1H), 7.86 (d, J = 7.6 Hz, 2H), 7.48 (d, J = 7.6 Hz, 2H), 7.11 (brs, 1H), 6.85 (d, J = 5.2 Hz, 1H), 3.48–3.45 (m, 2H), 2.72 (s, 3H), 2.58 (t, J = 12 Hz, 2H), 1.64 (brs, 1H), 1.30 (s, 9H); 13C NMR (DMSO-d6) δ: 166.6, 162.1, 159.0, 158.3, 154.1, 153.1, 131.6, 130.5, 126.5, 126.4, 106.5, 56.5, 44.4, 35.1, 31.3, 18.7; MS (m/z) 367; anal. calc. for: (C20H25N5S, Mwt = 367): C, 65.36; H, 6.86; N, 19.06; found: C, 65.46; H, 6.94; N, 19.11%.
2-{(4-[2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl)pyrimidin-2-yl]amino}ethan-1-ol (9). Following the general procedure and using 2-aminoethan-1-ol (21 μL, 0.4 mmol), compound 9 was obtained as yellowish-white solid (90 mg, 92%) mp = 123 °C; 1H NMR (DMSO-d6) δ: 8.35 (d, J = 5.2 Hz, 1H), 7.89 (d, J = 7.6 Hz, 2H), 7.53 (d, J = 7.6 Hz, 2H), 7.11 (brs, 1H), 6.89 (d, J = 5.2 Hz, 1H), 4.68 (brs, 2H), 3.65 (d, J = 8 Hz, 2H), 3.54 (d, J = 8 Hz, 2H), 2.70 (s, 3H), 1.30 (s, 9H); 13C NMR (DMSO-d6) δ: 166.6, 162.1, 159.7, 157.9, 154.2, 153.8, 131.6, 130.5, 126.5, 126.4, 106.5, 60.1, 43.4, 35.1, 31.3, 18.7; MS (m/z) 368; anal. calc. for: (C20H24N4OS, Mwt = 368): C, 65.19; H, 6.57; N, 15.20; found: C, 65.27; H, 6.65; N, 15.26%.
N-{4-[2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl]pyrimidin-2-yl}-N,N-dimethylethane-1,2-diamine (10). Following the general procedure and using N,N-dimethylenediamine (32 μL, 0.4 mmol), compound 10 was obtained as yellow solid (90 mg, 91%) mp = 117 °C; 1H NMR (DMSO-d6) δ: 8.33 (d, J = 5.2 Hz, 1H), 7.87 (d, J = 7.6 Hz, 2H), 7.53 (d, J = 7.6 Hz, 2H), 7.04 (brs, 1H), 6.88 (d, J = 5.2 Hz, 1H), 3.42–3.38 (m, 2H), 2.70 (s, 3H), 2.41 (t, J = 8 Hz, 2H), 2.18 (s, 6H), 1.28 (s, 9H); 13C NMR (DMSO-d6) δ: 166.6, 162.4, 160.7, 159.4, 154.0, 153.4, 131.6, 130.5, 126.5, 126.4, 106.5, 58.4, 45.7, 44.7, 35.1, 31.3, 18.6; MS (m/z) 395; anal. calc. for: (C22H29N5S, Mwt = 395): C, 66.80; H, 7.39; N, 17.70; found: C, 66.89; H, 7.45; N, 17.77%.
2-{[4-(2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl)pyrimidin-2-yl]amino}acetimidamide (11). Following the general procedure and using 2-aminoacetimidamide dihydrobromide (90 mg, 0.4 mmol) and anhydrous potassium carbonate (0.1 g, 0.7 mmol), compound 11 was obtained as a brown solid (70 mg, 74%) mp = 189 °C; 1H NMR (DMSO-d6) δ: 8.73 (brs, 2H), 8.32 (d, J = 5.2 Hz, 1H), 7.83 (d, J = 7.6 Hz, 2H), 7.53 (d, J = 7.6 Hz, 2H), 6.90 (d, J = 5.2 Hz, 1H), 6.68 (brs, 2H), 3.19 (s, 2H), 2.68 (s, 3H), 1.29 (s, 9H); 13C NMR (DMSO-d6) δ: 166.6, 163.7, 159.6, 159.0, 158.3, 153.9, 153.5, 131.6, 130.5, 126.5, 126.4, 106.9, 45.1, 39.3, 35.1, 31.3, 18.6; MS (m/z) 380; anal. calc. for: (C20H24N6S, Mwt = 380): C, 63.13; H, 6.36; N, 22.09; found: C, C, 63.21; H, 6.43; N, 22.19%.
N-{4-[2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl]pyrimidin-2-yl}propane-1,3-diamine (12). Following the general procedure and using propane-1,3-diamine (27 μL, 0.4 mmol), compound 12 was obtained as a buff solid (91 mg, 89%) mp = 165 °C; 1H NMR (DMSO-d6) δ: 8.36 (d, J = 5.2 Hz, 1H), 7.89 (d, J = 7.6 Hz, 2H), 7.53 (d, J = 7.6 Hz, 2H), 7.33 (brs, 1H), 6.87 (d, J = 5.2 Hz, 1H), 3.45–3.41 (m, 2H), 2.70 (s, 3H), 2.63 (t, J = 12 Hz, 2H), 1.66 (brs, 2H), 1.30 (s, 9H), 1.15–1.13 (m, 2H); 13C NMR (DMSO-d6) δ: 166.3, 159.0, 158.2, 157.2, 154.5, 153.1, 131.6, 130.5, 126.7, 126.4, 106.5, 46.1, 45.1, 34.3, 32.3, 31.2, 18.7; MS (m/z) 381; anal. calc. for: (C21H27N5S, Mwt = 381): C, 66.11; H, 7.13; N, 18.36; found: C, 66.17; H, 7.19; N, 18.43%.
3-{[4-(2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl)pyrimidin-2-yl]amino}propan-1-ol (13). Following the general procedure and using 3-aminopropan-1-ol (27 μL, 0.4 mmol), compound 13 was obtained as a yellowish-brown solid (93 mg, 83%) mp = 110 °C; 1H NMR (DMSO-d6) δ: 8.33 (d, J = 5.2 Hz, 1H), 7.88 (d, J = 7.6 Hz, 2H), 7.53 (d, J = 7.6 Hz, 2H), 7.19 (brs, 1H), 6.87 (d, J = 5.2 Hz, 1H), 4.45 (brs, 1H), 3.50–3.35 (m, 4H), 2.69 (s, 3H), 1.73–1.67 (m, 2H), 1.29 (s, 9H); 13C NMR (DMSO-d6) δ: 166.6, 162.5, 159.4, 157.6, 154.0, 153.4, 131.9, 130.5, 126.5, 126.4, 106.5, 59.2, 38.5, 35.1, 32.5, 31.3, 18.6; MS (m/z) 382; anal. calc. for: (C21H26N4OS, Mwt = 382): C, 65.94; H, 6.85; N, 14.65; found: C, 66.01; H, 6.93; N, 14.74%.
3-{[4-(2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl)pyrimidin-2-yl]amino}propane-1,2-diol (14). Following the general procedure and using 3-aminopropane-1,2-diol (33 μL, 0.4 mmol), compound 14 was obtained as a yellowish-white solid (72 mg, 63%) mp = 106 °C; 1H NMR (DMSO-d6) δ: 8.32 (d, J = 5.2 Hz, 1H), 7.87 (d, J = 7.6 Hz, 2H), 7.51 (d, J = 7.6 Hz, 2H), 6.99 (brs, 1H), 6.86 (d, J = 5.2 Hz, 1H), 4.81 (brs, 1H), 4.59 (brs, 1H), 3.73–3.61 (m, 1H), 3.47–3.42 (m, 2H), 3.27–3.21 (m, 2H), 2.69 (s, 3H), 1.28 (s, 9H); 13C NMR (DMSO-d6) δ: 166.7, 162.5, 159.4, 158.5, 154.0, 153.5, 131.7, 130.5, 126.4, 126.0, 106.8, 70.6, 64.5, 44.7, 35.0, 31.3, 18.6; MS (m/z) 398; anal. calc. for: (C21H26N4O2S, Mwt = 398): C, 63.29; H, 6.58; N, 14.06; found: C, 63.35; H, 6.66; N, 14.11%.
(R)-3-{(4-[2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl)pyrimidin-2-yl]amino}propane-1,2-diol (15). Following the general procedure and using (R)-3-aminopropane-1,2-diol (33 mg, 0.4 mmol), compound 15 was obtained as a brown solid (60 mg, 57%) mp = 166 °C; 1H NMR (DMSO-d6) δ: 8.34 (d, J = 5.2 Hz, 1H), 7.86 (d, J = 7.6 Hz, 2H), 7.52 (d, J = 7.6 Hz, 2H), 6.99 (brs, 1H), 6.87 (d, J = 5.2 Hz, 1H), 4.77 (brs, 1H), 4.54 (brs, 1H), 3.73–3.60 (m, 1H), 3.47–3.41 (m, 2H), 3.27–3.21 (m, 2H), 2.69 (s, 3H), 1.29 (s, 9H); 13C NMR (DMSO-d6) δ: 166.6, 162.5, 159.3, 158.7, 154.0, 153.5, 131.6, 130.5, 126.8, 126.4, 106.8, 70.6, 64.5, 44.8, 35.1, 30.5, 18.6; MS (m/z) 398; anal. calc. for: (C21H26N4O2S, Mwt = 398): C, 63.29; H, 6.58; N, 14.06; found: C, 63.37; H, 6.68; N, 14.10%.
(S)-3-{(4-[2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl)pyrimidin-2-yl]amino}propane-1,2-diol (16). Following the general procedure and using (S)-3-aminopropane-1,2-diol (33 mg, 0.4 mmol), compound 16 was obtained as a yellow solid (73 mg, 75%) mp = 159 °C; 1H NMR (DMSO-d6) δ: 8.33 (d, J = 5.2 Hz, 1H), 7.85 (d, J = 7.6 Hz, 2H), 7.51 (d, J = 7.6 Hz, 2H), 6.98 (brs, 1H), 6.87 (d, J = 5.2 Hz, 1H), 4.78 (brs, 1H), 4.57 (brs, 1H), 3.74–3.61 (m, 1H), 3.53–3.46 (m, 2H), 3.26–3.22 (m, 2H), 2.69 (s, 3H), 1.28 (s, 9H); 13C NMR (DMSO-d6) δ: 166.7, 162.1, 159.4, 158.2, 154.0, 153.5, 131.7, 130.5, 126.4, 126.0, 106.8, 70.6, 64.5, 44.7, 37.1, 31.3, 18.6; MS (m/z) 398; anal. calc. for: (C21H26N4O2S, Mwt = 398): C, 63.29; H, 6.58; N, 14.06; found: C, 63.38; H, 6.68; N, 14.11%.
2-{[4-(2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl)pyrimidin-2-yl]amino}propane-1,3-diol (17). Following the general procedure and using 2-aminopropane-1,3-diol (33 mg, 0.4 mmol), compound 17 was obtained as a yellow solid (80 mg, 81%) mp = 154 °C; 1H NMR (DMSO-d6) δ: 8.34 (d, J = 5.2 Hz, 1H), 7.88 (d, J = 7.6 Hz, 2H), 7.52 (d, J = 7.6 Hz, 2H), 6.89 (d, J = 5.2 Hz, 1H), 6.66 (brs, 1H), 4.63 (brs, 2H), 3.97–3.93 (m, 1H), 3.61–3.59 (m, 4H), 2.69 (s, 3H), 1.29 (s, 9H); 13C NMR (DMSO-d6) δ: 166.6, 162.3, 159.2, 157.9, 154.0, 153.5, 131.7, 130.5, 126.0, 125.7, 106.9, 60.6, 55.0, 35.1, 30.8, 18.7; MS (m/z) 398; anal. calc. for: (C21H26N4O2S, Mwt = 398): C, 63.29; H, 6.58; N, 14.06; found: C, 63.37; H, 6.67; N, 14.12%.
N-{4-[2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl]pyrimidin-2-yl}cyclohexane-trans-1,4-diamine (18). Following the general procedure and using trans-1,4-diaminocyclohexane (42 mg, 0.4 mmol), compound 18 was obtained as a brown solid (100 mg, 92%) mp = 193 °C; 1H NMR (DMSO-d6) δ: 8.31 (d, J = 5.2 Hz, 1H), 7.87 (d, J = 7.6 Hz, 2H), 7.56 (d, J = 7.6 Hz, 2H), 7.10 (brs, 1H), 6.91 (d, J = 5.2 Hz, 1H), 3.67–3.64 (m, 2H), 2.69 (s, 3H), 2.0–1.79 (m, 4H), 1.60 (brs, 2H), 1.29 (s, 9H) 1.20 (m, 4H); 13C NMR (DMSO-d6) δ: 166.3, 161.8, 159.7, 157.6, 154.0, 153.8, 131.9, 130.5, 126.5, 126.4, 106.5, 54.8, 50.1, 36.9, 35.1, 34.6, 31.3, 18.5; MS (m/z) 421; anal. calc. for: (C24H31N5S, Mwt = 421): C, 68.37; H, 7.41; N, 16.61; found: C, 68.43; H, 7.47; N, 16.67.
N-{4-[2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl]pyrimidin-2-yl}cyclohexane-cis-1,2-diamine (19). Following the general procedure and using (±)-cis-1,2-diaminocyclohexane (42 μL, 0.4 mmol), compound 19 was obtained as a brown solid (71 mg, 62%) mp = 116 °C; 1H NMR (DMSO-d6) δ: 8.33 (d, J = 5.2 Hz, 1H), 7.82 (d, J = 7.6 Hz, 2H), 7.46 (d, J = 7.6 Hz, 2H), 6.82 (d, J = 5.2 Hz, 1H), 5.60 (brs, 1H), 3.99–3.93 (m, 2H), 2.67 (s, 3H), 1.95–1.38 (m, 8H), 1.26 (s, 9H), 1.60 (brs, 2H); 13C NMR (DMSO-d6) δ: 166.6, 159.5, 159.1, 157.8, 153.8, 153.4, 131.6, 130.5, 126.4, 126.0, 105.9, 62.4, 54.8, 37.2, 35.0, 31.2, 26.4, 22.5, 19.7, 18.7; MS (m/z) 421; anal. calc. for: (C24H31N5S, Mwt = 421): C, 68.37; H, 7.41; N, 16.61; found: C, 68.45; H, 7.45; N, 16.66.
N-{4-[2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl]pyrimidin-2-yl}cyclohexane-trans-1,2-diamine (20). Following the general procedure and using (±)-trans-1,2-diaminocyclohexane (42 μL, 0.4 mmol), compound 20 was obtained as a brown solid (100 mg, 89%) mp = 111 °C; 1H NMR (DMSO-d6) δ: 8.31 (d, J = 5.2 Hz, 1H), 7.86 (d, J = 7.6 Hz, 2H), 7.53 (d, J = 7.6 Hz, 2H), 7.11 (brs, 1H), 6.88 (d, J = 5.2 Hz, 1H), 3.48–3.43 (m, 2H), 2.68 (s, 3H), 2.05–1.86 (m, 3H), 1.64 (brs, 2H), 1.30 (s, 9H), 1.22–1.05 (m, 5H); 13C NMR (DMSO-d6) δ: 166.6, 162.1, 159.0, 157.2, 154.0, 153.5, 131.9, 130.5, 126.5, 126.4, 106.5, 61.4, 54.2, 37.1, 35.1, 31.3, 25.3, 22.5, 19.7, 18.6; MS (m/z) 421; anal. calc. for: (C24H31N5S, Mwt = 421): C, 68.37; H, 7.41; N, 16.61; found: C, 68.44; H, 7.46; N, 16.64.
4-{2-[4-(tert-Butyl)phenyl]-4-methylthiazol-5-yl}-N-(pyrrolidin-3-yl)pyrimidin-2-amine (21). Following the general procedure and using 3-amino-pyrrolidine dihydrochloride (60 mg, 0.4 mmol) and anhydrous potassium carbonate (0.1 g, 0.7 mmol), compound 21 was obtained as yellowish white solid (100 mg, 94%) mp = 115 °C; 1H NMR (DMSO-d6) δ: 8.41 (d, J = 5.2 Hz, 1H), 8.0 (brs, 1H), 7.89 (d, J = 7.6 Hz, 2H), 7.52 (d, J = 7.6 Hz, 2H), 6.90 (d, J = 5.2 Hz, 1H), 3.66–3.48 (m, 3H), 2.71 (s, 3H), 2.09–2.08 (m, 2H), 1.88 (brs, 1H), 1.73–1.70 (m, 2H), 1.30 (s, 9H); 13C NMR (DMSO-d6) δ: 166.6, 160.1, 159.2, 158.0, 154.0, 153.4, 131.6, 130.5, 126.5, 126.4, 106.1, 54.5, 51.0, 45.2, 35.1, 33.6, 31.3, 18.6; MS (m/z) 393; anal. calc. for: (C22H27N5S, Mwt = 393): C, 67.14; H, 6.92; N, 17.80; found: C, 67.22; H, 6.99; N, 17.87%.
4-{2-[4-(tert-Butyl)phenyl]-4-methylthiazol-5-yl}-N-(piperidin-2-ylmethyl)pyrimidin-2-amine (22). Following the general procedure and using 2-(aminomethyl)piperidine (42 μL, 0.4 mmol), compound 22 was obtained as a yellow solid (99 mg, 90%) mp = 146 °C; 1H NMR (DMSO-d6) δ: 8.33 (d, J = 5.2 Hz, 1H), 7.88 (d, J = 7.6 Hz, 2H), 7.53 (d, J = 7.6 Hz, 2H), 7.21 (brs, 1H), 6.88 (d, J = 5.2 Hz, 1H), 4.01–3.98 (m, 1H), 3.18–3.14 (m, 2H), 2.95–2.87 (m, 2H), 2.70 (s, 3H), 1.75–1.44 (m, 6H), 1.26 (s, 9H) 1.04 (brs, 1H); 13C NMR (DMSO-d6) δ: 166.6, 162.6, 159.7, 159.0, 154.0, 153.4, 131.6, 130.5, 126.7, 126.4, 106.7, 70.1, 60.7, 56.1, 46.5, 35.1, 31.3, 26.2, 24.5, 18.6; MS (m/z) 421; anal. calc. for: (C24H31N5S, Mwt = 421): C, 68.37; H, 7.41; N, 16.61; found: C, 68.44; H, 7.48; N, 16.66.
(R)-{1-[4-(2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl)pyrimidin-2-yl]pyrrolidin-2-yl}methanol (23). Following the general procedure and using (R)-(+)-2-pyrrolidinemethanol (37 μL, 0.4 mmol), compound 23 was obtained as a yellowish-green solid (90 mg, 85%) mp = 136 °C; 1H NMR (DMSO-d6) δ: 8.39 (d, J = 5.2 Hz, 1H), 7.89 (d, J = 7.6 Hz, 2H), 7.53 (d, J = 7.6 Hz, 2H), 6.92 (d, J = 5.2 Hz, 1H), 4.85 (brs, 1H), 4.18–4.08 (m, 2H), 3.67–3.60 (m, 1H), 3.46–3.34 (m, 2H), 2.71 (s, 3H), 2.05–1.86 (m, 4H), 1.29 (s, 9H); 13C NMR (DMSO-d6) δ: 166.6, 160.2, 159.4, 158.3, 154.0, 153.8, 131.9, 130.5, 126.5, 126.4, 106.3, 61.8, 59.0, 47.7, 35.1, 31.3, 28.1, 22.9, 18.6; MS (m/z) 408; anal. calc. for: (C23H28N4OS, Mwt = 408): C, 67.62; H, 6.91; N, 13.71; found: C, C, 67.69; H, 6.97; N, 13.75%.
(S)-{1-[4-(2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl]pyrimidin-2-yl}pyrrolidin-2-yl)methanol (24). Following the general procedure and using (S)-(−)-2-pyrrolidinemethanol (37 μL, 0.4 mmol), compound 24 was obtained as a brown solid (100 mg, 96%) mp = 125 °C; 1H NMR (DMSO-d6) δ: 8.39 (d, J = 5.2 Hz, 1H), 7.88 (d, J = 7.6 Hz, 2H), 7.52 (d, J = 7.6 Hz, 2H), 6.89 (d, J = 5.2 Hz, 1H), 4.75 (brs, 1H), 4.18–4.12 (m, 2H), 3.67–3.63 (m, 1H), 3.52–3.46 (m, 2H), 2.69 (s, 3H), 2.03–1.89 (m, 4H), 1.29 (s, 9H); 13C NMR (DMSO-d6) δ: 166.6, 160.2, 160.1, 159.0, 154.0, 153.4, 131.6, 130.5, 126.5, 126.4, 106.3, 61.1, 59.4, 47.4, 35.1, 31.3, 28.0, 23.2, 18.7; MS (m/z) 408; anal. calc. for: (C23H28N4OS, Mwt = 408): C, 67.62; H, 6.91; N, 13.71; found: C, C, 67.70; H, 6.99; N, 13.77%.
(R)-1-{4-[2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl]pyrimidin-2-yl}-N,N-dimethylpyrrolidin-3-amine (25). Following the general procedure and using (R)-(+)-3-(dimethylamino)pyrrolidine dihydrochloride (71 mg, 0.4 mmol) and anhydrous potassium carbonate (0.1 g, 0.7 mmol), compound 25 was obtained as a yellow solid (99 mg, 91%) mp = 127 °C; 1H NMR (DMSO-d6) δ: 8.38 (d, J = 5.2 Hz, 1H), 7.89 (d, J = 7.6 Hz, 2H), 7.55 (d, J = 7.6 Hz, 2H), 6.93 (d, J = 5.2 Hz, 1H), 3.82–3.69 (m, 4H), 3.14–3.10 (m, 1H), 2.72 (s, 3H), 2.19 (s, 6H), 1.82–1.78 (m, 2H), 1.32 (s, 9H); 13C NMR (DMSO-d6) δ: 166.6, 159.9, 159.2, 158.0, 154.0, 153.7, 131.5, 130.5, 126.5, 126.4, 106.3, 64.9, 50.3, 45.9, 44.2, 35.1, 31.3, 28.8, 18.7; MS (m/z) 421; anal. calc. for: (C24H31N5S, Mwt = 421): C, 68.37; H, 7.41; N, 16.61; found: C, 68.42; H, 7.48; N, 16.65%.
(S)-1-{4-[2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl]pyrimidin-2-yl}-N,N-dimethylpyrrolidin-3-amine (26). Following the general procedure and using (S)-(−)-3-(dimethylamino)pyrrolidine (42 μL, 0.4 mmol), compound 26 was obtained as a yellow solid (81 mg, 73%) mp = 136 °C; 1H NMR (DMSO-d6) δ: 8.35 (d, J = 5.2 Hz, 1H), 7.84 (d, J = 7.6 Hz, 2H), 7.51 (d, J = 7.6 Hz, 2H), 6.88 (d, J = 5.2 Hz, 1H), 3.74–3.66 (m, 4H), 3.19–3.17 (m, 1H), 2.69 (s, 3H), 2.17 (s, 6H), 1.77–1.70 (m, 2H), 1.28 (s, 9H); 13C NMR (DMSO-d6) δ: 166.6, 159.8, 159.1, 158.0, 153.9, 153.7, 131.4, 130.5, 126.7, 126.4, 106.2, 56.1, 50.9, 45.9, 44.2, 35.0, 31.3, 29.7, 18.7; MS (m/z) 421; anal. calc. for: (C24H31N5S, Mwt = 421): C, 68.37; H, 7.41; N, 16.61; found: C, 68.42; H, 7.46; N, 16.67%.
(S)-1-{4-[2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl]pyrimidin-2-yl}pyrrolidine-2-carboxamide (27). Following the general procedure and using L-prolinamide (42 mg, 0.4 mmol), compound 28 was obtained as a yellowish-white solid (82 mg, 74%) mp = 110 °C; 1H NMR (DMSO-d6) δ: 8.34 (d, J = 5.2 Hz, 1H), 7.86 (d, J = 7.6 Hz, 2H), 7.52 (d, J = 7.6 Hz, 2H), 7.30 (brs, 2H), 6.89 (d, J = 5.2 Hz, 1H), 4.42–4.40 (m, 1H), 3.67–3.50 (m, 2H), 2.71 (s, 3H), 2.22–2.18 (m, 1H), 1.95–1.90 (m, 3H), 1.29 (s, 9H); 13C NMR (DMSO-d6) δ: 175.3, 166.6, 162.4, 159.7, 158.6, 154.5, 153.8, 131.2, 130.5, 126.0, 125.6, 106.9, 60.7, 46.8, 35.1, 31.2, 30.8, 23.9, 18.6; MS (m/z) 421; anal. calc. for: (C23H27N5OS, Mwt = 421): C, 65.53; H, 6.46; N, 16.61; found: C, 65.60; H, 6.51; N, 16.65%.
(R)-1-{4-[2-(4-(tert-Butyl)phenyl)-4-methylthiazol-5-yl]pyrimidin-2-yl}pyrrolidine-2-carboxamide (28). Following the general procedure and using D-prolinamide (42 mg, 0.4 mmol), compound 28 was obtained as a yellowish-white solid (100 mg, 90%) mp = 115 °C; 1H NMR (DMSO-d6) δ: 8.40 (d, J = 5.2 Hz, 1H), 7.88 (d, J = 7.6 Hz, 2H), 7.51 (d, J = 7.6 Hz, 2H), 7.38 (brs, 2H), 6.92 (d, J = 5.2 Hz, 1H), 4.46–4.43 (m, 1H), 3.60–3.56 (m, 2H), 2.68 (s, 3H), 2.20–2.19 (m, 1H), 1.95–1.90 (m, 3H), 1.29 (s, 9H); 13C NMR (DMSO-d6) δ: 175.0, 166.6, 160.1, 159.0, 158.6, 154.0, 153.8, 131.9, 130.5, 126.5, 126.4, 106.8, 60.6, 47.6, 35.1, 31.3, 31.1, 23.6, 18.6; MS (m/z) 421; anal. calc. for: (C23H27N5OS, Mwt = 421): C, 65.53; H, 6.46; N, 16.61; found: C, 65.62; H, 6.53; N, 16.69%.
4.2. Microbiological assay
4.3. Studies on metabolism simulation
To validate the metabolic resistance and bioavailability of new compound 20 compared with lead compounds 1a and 1c, certain molecular parameters were calculated, and metabolism simulation was performed. First, calculations using SWISS-ADME, MolSoft, and Discovery Studio 4.1 were conducted for data comparison and validation from discrete sources. Compound 19 was similar in all properties to compound 20 because they were structural isomers, so compound 20 was designated for studies. The structures of compounds were drawn using ChemDraw v.2014 and saved in a readable mol extension. Online free platforms such as SWISSADME and MolSoft were selected to ascertain expected molecular properties and expected interaction with prominent metabolic enzymes via structural upload and RUN. Validation through calculations of ADME descriptors using Discovery Studio after compound preparation and application of CHArMm forcefield was done. Metabolism simulation was developed using PK-Sim software after data insertion of pKa, molecular properties, and assuming drug-dose administration as a single intravenous infusion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This paper is based on work supported by Science, Technology and Innovation Funding Authority under grant number 43229 for young researchers.References
- D. Schillaci, V. Spanò, B. Parrino, A. Carbone, A. Montalbano, P. Barraja, P. Diana, G. Cirrincione and S. Cascioferro, J. Med. Chem., 2017, 60, 8268–8297 CrossRef CAS PubMed.
- B. Parrino, D. Carbone, G. Cirrincione, P. Diana and S. Cascioferro, Futur. Med. Chem., 2019, 12, 326 Search PubMed.
- B. Aslam, W. Wang, M. I. Arshad, M. Khurshid, S. Muzammil, M. H. Rasool, M. A. Nisar, R. F. Alvi, M. A. Aslam and M. U. Qamar, Infect. Drug Resist., 2018, 11, 1645 CrossRef CAS PubMed.
- S. S. Kadri, Crit. Care Med., 2020, 7176261 Search PubMed.
- A. Hassoun, P. K. Linden and B. Friedman, Crit. Care, 2017, 21, 211 CrossRef PubMed.
- A. S. Lee, H. de Lencastre, J. Garau, J. Kluytmans, S. Malhotra-Kumar, A. Peschel and S. Harbarth, Nat. Rev. Dis. Primers, 2018, 4, 1–23 Search PubMed.
- R. M. Hassan, M. G. Elanany, M. M. Mostafa, R. H. A. Yousef and S. T. Salem, J. Microbiol., Immunol. Infect., 2023, 56(4), 802–814 CrossRef CAS PubMed.
- J. Fishovitz, J. A. Hermoso, M. Chang and S. Mobashery, IUBMB Life, 2014, 66, 572–577 CrossRef CAS PubMed.
- P. Wiegand, D. Nathwani, M. Wilcox, J. Stephens, A. Shelbaya and S. Haider, J. Hosp. Infect., 2012, 81, 1–14 CrossRef CAS.
- P. A. Myer, A. Mannalithara, G. Singh, G. Singh, P. J. Pasricha and U. Ladabaum, Am. J. Gastroenterol., 2013, 108, 1496–1507 CrossRef PubMed.
- L. C. McDonald, M. Owings and D. B. Jernigan, Emerg. Infect. Dis., 2006, 12, 3291455 Search PubMed.
- M. L. Manning, J. Pfeiffer and E. L. Larson, Am. J. Infect. Control, 2016, 44, 1454–1457 CrossRef PubMed.
- H. Grundmann, M. Aires-de-Sousa, J. Boyce and E. Tiemersma, Lancet, 2006, 368, 874–885 CrossRef PubMed.
- L. Ravindar, S. Bukhari, K. Rakesh, H. Manukumar, H. Vivek, N. Mallesha, Z.-Z. Xie and H.-L. Qin, Bioorg. Chem., 2018, 81, 107–118 CrossRef CAS PubMed.
- H. F. Chambers, N. Engl. J. Med., 2005, 352, 1485–1487 CrossRef CAS PubMed.
- G. J. Moran, A. Krishnadasan, R. J. Gorwitz, G. E. Fosheim, L. K. McDougal, R. B. Carey and D. A. Talan, N. Engl. J. Med., 2006, 355, 666–674 CrossRef CAS PubMed.
- B. W. Frazee, J. Lynn, E. D. Charlebois, L. Lambert, D. Lowery and F. Perdreau-Remington, Ann. Emerg. Med., 2005, 45, 311–320 CrossRef PubMed.
- S. K. Fridkin, J. C. Hageman, M. Morrison, L. T. Sanza, K. Como-Sabetti, J. A. Jernigan, K. Harriman, L. H. Harrison, R. Lynfield and M. M. Farley, N. Engl. J. Med., 2005, 352, 1436–1444 CrossRef CAS.
- G. J. Moran, R. N. Amii, F. M. Abrahamian and D. A. Talan, Emerging Infect. Dis., 2005, 11, 928 CrossRef.
- K. Hiramatsu, Lancet Infect. Dis., 2001, 1, 147–155 CrossRef CAS.
- P. Wilson, J. Andrews, R. Charlesworth, R. Walesby, M. Singer, D. Farrell and M. Robbins, J. Antimicrob. Chemother., 2003, 51, 186–188 CrossRef CAS PubMed.
- S. Lin, J.-J. Koh, T. T. Aung, F. Lim, J. Li, H. Zou, L. Wang, R. Lakshminarayanan, C. Verma and Y. Wang, J. Med. Chem., 2017, 60, 1362–1378 CrossRef CAS PubMed.
- H. Mohammad, A. S. Mayhoub, A. Ghafoor, M. Soofi, R. A. Alajlouni, M. Cushman and M. N. Seleem, J. Med. Chem., 2014, 57, 1609–1615 CrossRef CAS PubMed.
- I. Eid, M. M. Elsebaei, H. Mohammad, M. Hagras, C. E. Peters, Y. A. Hegazy, B. Cooper, J. Pogliano, K. Pogliano and H. S. Abulkhair, Eur. J. Med. Chem., 2017, 139, 665–673 CrossRef CAS PubMed.
- E. Yahia, H. Mohammad, T. M. Abdelghany, E. Fayed, M. N. Seleem and A. S. Mayhoub, Eur. J. Med. Chem., 2017, 126, 604–613 CrossRef CAS PubMed.
- M. A. Seleem, A. M. Disouky, H. Mohammad, T. M. Abdelghany, A. S. Mancy, S. A. Bayoumi, A. Elshafeey, A. El-Morsy, M. N. Seleem and A. S. Mayhoub, J. Med. Chem., 2016, 59, 4900–4912 CrossRef CAS PubMed.
- Y. Cheng, F. Zhang, T. A. Rano, Z. Lu, W. A. Schleif, L. Gabryelski, D. B. Olsen, M. Stahlhut, C. A. Rutkowski and J. H. Lin, Bioorg. Med. Chem. Lett., 2002, 12, 2419–2422 CrossRef CAS PubMed.
- A. Kotb, N. S. Abutaleb, M. A. Seleem, M. Hagras, H. Mohammad, A. Bayoumi, A. Ghiaty, M. N. Seleem and A. S. Mayhoub, Eur. J. Med. Chem., 2018, 151, 110–120 CrossRef CAS PubMed.
- Clinical and Laboratory Standards Institute, M7-A9, Wayne, PA, 9th edn, 2012 Search PubMed.
- Clinical and Laboratory Standards Institute, M29, Wayne, PA, 2007 Search PubMed.
- Clinical and Laboratory Standards Institute, Approved Standard M27-A3, Wayne, PA, 3rd edn, 2008 Search PubMed.
- S. B. Olasupo, A. Uzairu, G. A. Shallangwa and S. Uba, Future J. Pharm. Sci., 2021, 7, 1–10 CrossRef.
- D. Teutonico, M. Block, L. Kuepfer, J. Solodenko, T. Eissing and K. Coboeken, Oral Drug Delivery for Modified Release Formulations, 2022, pp. 375–389 Search PubMed.
- P. Wayne, CLSI Document M27-A2, 2002 Search PubMed.
- T. A. Geers and A. M. Donabedian, Antimicrob. Agents Chemother., 1989, 33, 233–234 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra07619a |
| This journal is © The Royal Society of Chemistry 2024 |