
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation and characterization of sulphur and zinc oxide Co-doped graphitic carbon nitride for photo-assisted removal of Safranin-O dye†
Azmat Ali Khana,
Abbas Khan *a,
Sumayya Khana,
Nasrullah Shaha,
Ajmal Khanb,
Faheem Nawazc,
Asaad Khalidd,
Afnan Jane and
Ahmed Al-Harrasi*b
*a,
Sumayya Khana,
Nasrullah Shaha,
Ajmal Khanb,
Faheem Nawazc,
Asaad Khalidd,
Afnan Jane and
Ahmed Al-Harrasi*b
aDepartment of Chemistry, Abdul Wali Khan University, Mardan 23200, KP, Pakistan. E-mail: asaster45@gmail.com; abbas80@awkum.edu.pk; muhammadtalhay42@gmail.com; nasrullah@awkum.edu.pk; abbas053@gmail.com; Fax: +92-937-542188; Tel: +92-3408467885
bNatural and Medical Sciences Research Center, University of Nizwa, PO Box 33, 616 Birkat Al Mauz, Nizwa, Oman. E-mail: ajmalkhan@unizwa.edu.om; aharrasi@unizwa.edu.om
cDepartment of Environmental Science, Faculty of Life Sciences & Informatics, Balochistan University of Information Technology, Engineering and Management Sciences (BUITEMS), Quetta, Pakistan. E-mail: faheem.nawaz@buitms.edu.pk
dSubstance Abuse and Toxicology Research Center, Jazan University, PO Box: 114, Jazan 45142, Saudi Arabia. E-mail: akahmed@jazanu.edu.sa
eDepartment of Biochemistry, Faculty of Medicine, Umm Al-Qura University, Makkah, Kingdom of Saudi Arabia. E-mail: Amjan@uqu.edu.sa
First published on 15th March 2024
Abstract
Recently, there has been significant interest in photocatalytic reactions involving graphitic carbon nitride (g-C3N4) due to its sp2-hybridized carbon and nitrogen content and it is an ideal candidate for blending with other materials to enhance performance. Here, we have synthesized and analyzed both doped and undoped g-C3N4 nanoparticles. Specifically, we co-doped sulfur (S) into g-C3N4, integrated it with ZnO particles, and investigated the photocatalytic potential of these nanocomposites to remove Safranin-O dye. The initial step involved the preparation of pure g-C3N4 through calcination of urea. Subsequently, S-g-C3N4 was synthesized by calcining a mixture of urea and thiourea with a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. Finally, the ZnO–S-g-C3N4 composite was synthesized using the liquid exfoliation technique, with distilled water serving as the exfoliating solvent. These samples were characterized by advanced techniques, including UV-Vis spectroscopy, Fourier-transform infrared spectroscopy (FTIR), X-ray Diffraction (XRD), energy dispersive X-ray (EDX) and scanning electron microscopy (SEM), to assess their crystallinity, morphology, optical properties, and phase purity. Subsequently, these nanocomposites were employed in catalytic and photocatalytic processes to remove the Safranin-O dye (SO). The results highlighted the formation of Z-scheme junction responsible for ZnO–S-g-C3N4's significant performance improvement. The comparison of results demonstrated that S-g-C3N4 and ZnO–S-g-C3N4 composites revealed an effective removal of Safranin-O dye in the presence of UV-light as compared to pure g-C3N4, as it was attributed to the phenomenon of improved separation of photogenerated charge carriers as a result of heterojunction formation between S-g-C3N4 and ZnO interfaces. In addition to improving photocatalytic performance, this study presents a facile route for producing ZnO–S-g-C3N4 composite with superior adsorption capabilities and selectivity.
:1 ratio. Finally, the ZnO–S-g-C3N4 composite was synthesized using the liquid exfoliation technique, with distilled water serving as the exfoliating solvent. These samples were characterized by advanced techniques, including UV-Vis spectroscopy, Fourier-transform infrared spectroscopy (FTIR), X-ray Diffraction (XRD), energy dispersive X-ray (EDX) and scanning electron microscopy (SEM), to assess their crystallinity, morphology, optical properties, and phase purity. Subsequently, these nanocomposites were employed in catalytic and photocatalytic processes to remove the Safranin-O dye (SO). The results highlighted the formation of Z-scheme junction responsible for ZnO–S-g-C3N4's significant performance improvement. The comparison of results demonstrated that S-g-C3N4 and ZnO–S-g-C3N4 composites revealed an effective removal of Safranin-O dye in the presence of UV-light as compared to pure g-C3N4, as it was attributed to the phenomenon of improved separation of photogenerated charge carriers as a result of heterojunction formation between S-g-C3N4 and ZnO interfaces. In addition to improving photocatalytic performance, this study presents a facile route for producing ZnO–S-g-C3N4 composite with superior adsorption capabilities and selectivity.
1. Introduction
The polymeric semiconductor graphitic carbon nitride (g-C3N4) has an appealing tri-s-triazine ring electrical structure and a medium band gap. It is highly durable against chemical and heat impacts. When the conduction and valence band levels are exactly in balance, photogenerated electron–hole pairs can be separated.1,2 Due to the polymeric nature and weak van der Waals interaction between adjoining layers, large interlayer spacing like those in graphite is observed, aiding in intercalation with superior physicochemical features.3 For years, the work of M. Humayun, W. Pi, Y. Yuan et al.,4 has sparked considerable interest in photocatalysis utilizing semiconductors. Recently, metal oxide semiconductors with a wide band gap have been developed, such as TiO2,5,6 ZnO,7 and NiO,8,9 have been utilized to destroy organic contaminants through photodegradation. Due to its low cost, chemical stability, and non-toxic makeup, polymeric photocatalyst g-C3N4 has gained recognition on a global scale.Numerous modification techniques have been developed over the years to increase the applicability of g-C3N4.10 Heteroatom doping is an effective way to alter g-C3N4's electronic configuration and increase its density of surface charges, leading to notable changes in its properties and applications.11 Numerous investigations have revealed that, compared to pristine g-C3N4, doped g-C3N4 demonstrates a more desired energy band structure, higher solar light usage, increased photocarrier separation efficiency, and improved photoactivity.12 The features of the g-C3N4 matrix, such as dye adsorption and light absorption, can be enhanced by doping it with other inorganic, organic metals or compounds.13 Among various elements, sulphur (S) doping on g-C3N4 alters the reaction kinetics for transferring photogenerated charge carriers to lower the band gap energy, which increases solar-energy absorption.14 The g-C3N4 behaves like a conductor as a result of S-doping. The non-uniform distribution of S-over g-C3N4 collects excited electrons, whereas undoped portions act as a photo anode. The entire setting encourages charge separation and higher photocatalytic activity. S-doping causes significant changes in the electrical structure of g-C3N4, narrowing the band gap and converting the material into a conductor, which are particularly noticeable.15 The length between the S–C bond (1.75 A) is longer than that of the original C–N bond (1.35 A) in terms of sulfur doping chemistry.
Additionally, due to the S and N bonds' differing electronegativity, the band gap of the S-g-C3N4 sheet is 0.20 eV, which is substantially less than the pristine g-C3N4 sheet's band gap of 2.70 eV. This could enable more efficient charge transfer.16 Due to its resemblance to graphene, g-C3N4 has received much attention in recent studies.17 It has abundant uses in gas sensors, solar cells, and energy conversion.18 Polycondensation of common monomers is a typical synthetic method for producing g-C3N4.19 It possesses fascinating properties such as excellent thermal stability and a high in-plane nitrogen concentration.20 The composition and physical characteristics of carbon nitrides can be changed by adding heteroatoms, which also engineer the molecular orbital shape and position necessary to improve the material's reactivity and selectivity. By charge transfer complexation or interaction with semiconductor materials, this then aids in creating composites with a high electron density system inside the catalyst.21
Water splitting has been extensively researched using g-C3N422 and the removal of organic pollutants in the presence of visible light irradiation23 due to its low band gap energy range of 2.7–2.9 eV.24 Nevertheless, it cannot be used in photocatalysis because of its low precise surface area, quick recombination of photogenerated e−–h+ couples, poor adsorption, and poor dispersion.23 Numerous methods have been researched to modify g-C3N4 in order to increase its photooxidation capacities to get rid of these drawbacks, including customizing the morphologies,25 doping with metal and non-metal ions,26 as well as enhancing pore development to increase surface area and adsorption sites.27 The synthesis of mesoporous g-C3N4 is also an effective approach for improving the catalytic activity of g-C3N4.28,29 Additionally, the photocatalytic activity can be enhanced by combining g-C3N4 with other semiconductors, preventing charge carrier recombination caused by visible light. Several g-C3N4 based composites, such as TiO2/g-C3N4,30 ZnO/g-C3N4,7 Bi2WO6/g-C3N4,31 and Ag/g-C3N4,32 have been reported to have good photocatalytic activity.
In this context, the synergetic effects of graphitic carbon nitride (g-C3N4) doped with sulphur and zinc oxide offer a promising opportunity. Incorporating sulphur and zinc oxide into g-C3N4 forms a novel nanocomposite with potential applications in electroflocculation for wastewater treatment. Although, electroflocculation has shown promise as an alternative approach for pollutant removal, it faces limitations that hinder its widespread adoption. One major limitation of conventional electroflocculation methods is the high energy requirement, which leads to increased operational costs and limits its feasibility for large-scale wastewater treatment.33 Additionally, the removal efficiency of electroflocculation can be limited, particularly for contaminants that are difficult to flocculate and sediment. These challenges emphasize the need for innovative materials and approaches to enhance the efficiency and effectiveness of electroflocculation for advanced wastewater treatment.
Our current study delved into the fascinating realm of co-doping of sulphur and ZnO with g-C3N4. By incorporating sulphur and zinc oxide into the structure of g-C3N4, we aimed to enhance its performance and expand its range of applications. The doping effects improved photocatalytic activity, enhanced charge carrier mobility, and extended absorption spectrum. This research opens up exciting possibilities for developing advanced materials with improved properties, offering promising prospects for various fields such as renewable energy, environmental remediation, and heterogeneous catalysis.
2. Experimental work
2.1. Chemicals used
The chemicals used in the preparation of g-C3N4, S-g-C3N4, ZnO and ZnO–S-g-C3N4 are Urea, thiourea, ethanol, zinc acetate di-hydrate, KOH, ethylene glycol, Safranin-O dye, and distilled water. All these chemicals were of sound purity and used without further treatments.2.2. Synthesis of g-C3N4
12 grams of urea dissolved in a 1:1 ratio in a mixture of ethanol and water. The solution was then heated to 100 °C while being continuously stirred. The white precipitates were obtained after the solvent evaporated completely. Aluminum foil was then placed over it before being moved to a crucible, preferably one made of alumina. These precipitates were then calcined at 550 °C in the muffle furnace for three hours. After that, it was permitted to a comfortable temperature. The pale yellow substance was turned into a powder for further testing. The following is the chemical process.
2.3. Synthesis of sulphur doped g-C3N4
Sulphur doped g-C3N4 was synthesized using urea and thiourea as a precursor. Various mixtures of urea and thiourea were heated at 550 °C for 3 hours with different weight ratios (X:Y), X (urea) and Y (thiourea); such as 3:1, 4:1, and 5:1. Among these mixtures, the S-g-C3N4 materials exhibited higher photocatalytic capacity. Notably, the sample with a 3:1 composition demonstrated the most active photocatalytic performance. This superiority can be attributed to the higher concentration of doped sulfur in the g-C3N4 matrix, which reduced the band gap. Theoretical calculations support these findings, showing that the band gap of sulphur doped g-C3N4 material is decreased compared to that of pure g-C3N4. Scheme 1 provides a summary of the procedure.
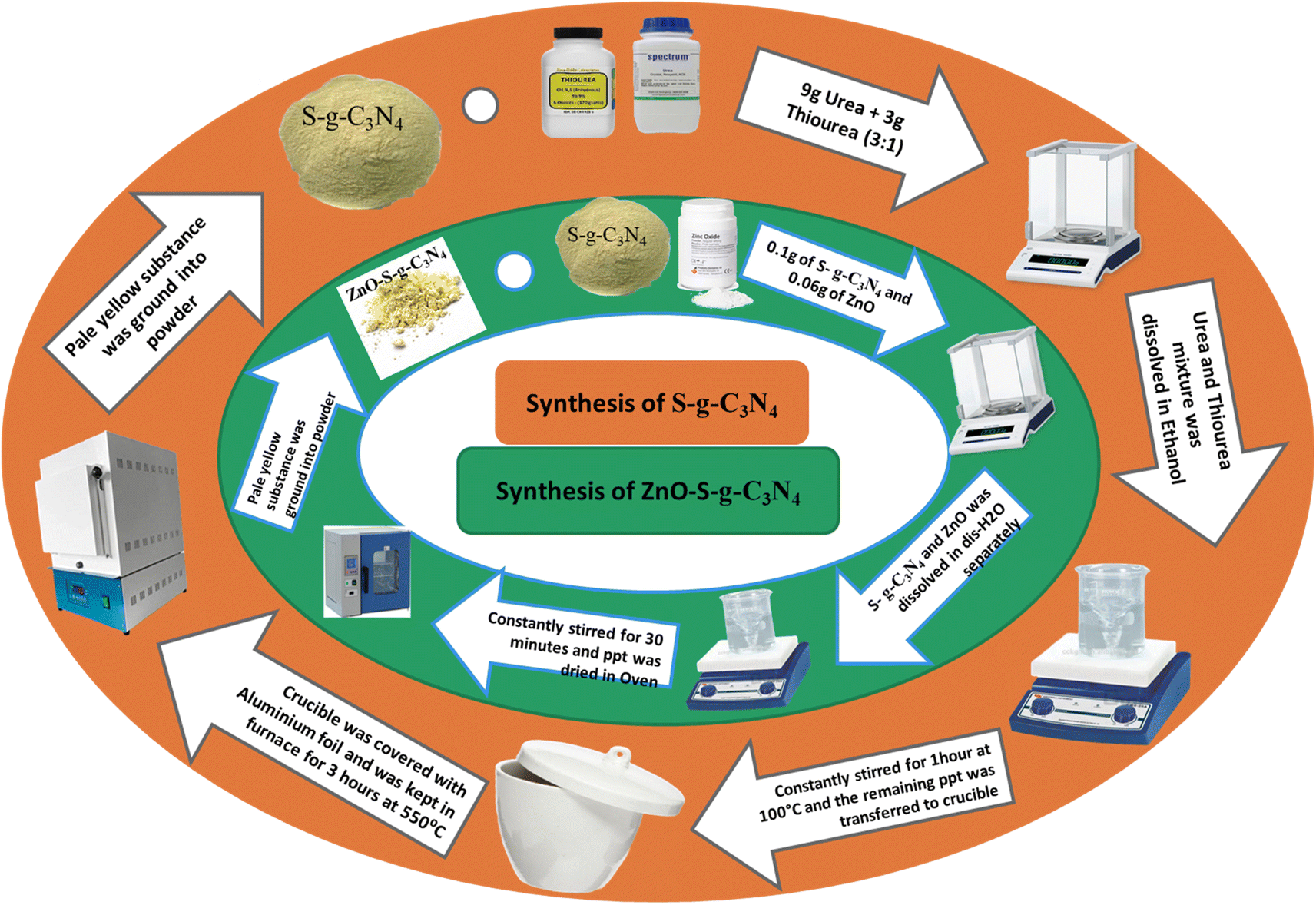 | ||
| Scheme 1 Schematic representation for the synthesis of S-g-C3N4 and ZnO–S-g-C3N4. | ||
2.4. Synthesis of ZnO nanoparticles
ZnO nanoparticles were prepared using the co-precipitation technique with ethylene glycol as solvent. To fabricate the ZnO nanoparticles, 40 mL of ethylene glycol and 4 grams of zinc acetate dihydrate were added and stirred continuously until a completely transparent solution was achieved. Subsequently, a potassium hydroxide KOH solution was added dropwise to the mixture, maintaining constant stirring. The system's pH was monitored using pH paper, and potassium hydroxide was added until a pH of 9.5 was gradually obtained. After reaching the desired pH, the solvent was completely evaporated, while the suspension was kept in motion by continuous stirring at 210 °C. The resulting dry solid was collected and washed repeatedly with deionized water. These particles were then separated through centrifugation at 4000 rpm for 20 minutes. Following separation, the ZnO nanoparticles were grinded into fine powder using an agate mortar and subsequently calcined for 4 hours at 600 °C.2.5. Synthesis of ZnO–S-g-C3N4
The ZnO–S-g-C3N4 nanocomposite was synthesized by dissolving 0.1 g of S-g-C3N4 and 0.06 g of ZnO nanoparticles in separate 30 mL portions of distilled water. Subsequently, the two solutions were mixed while being constantly stirred. After combining the solutions, the suspension was subjected to constant stirring for 30 minutes in a darkened environment with no light to achieve adsorption equilibrium. The resulting material was then dried in an oven. Scheme 1 summarizes the procedure.2.6. Photocatalytic degradation
The degradation/removal of Safranin-O dye (SO) from wastewater was accomplished by using the prepared nanomaterials in catalysis and photocatalysis, with the help of a 15 W UV-light bulb as light source. Additionally, it was covered to stop light from straying and to focus it on the reaction. Different experiments were conducted by adding specific amounts of catalysts (0.1 g) to a 10 ppm solution of SO and stirring constantly. The equilibrium of dye molecules that are adsorbed and desorbed on the surface of nanocomposites was typically verified by swirling the dye solution in the dark for 20 minutes before subjecting it to UV light irradiation. A predetermined volume of dye solutions was collected and centrifuged at 4000 rpm for 5–10 minutes at regular intervals. Double beam UV-Visible spectrophotometer (Cary 100 Bio) was used and the dye's absorbance at maximum absorption wavelength was measured to evaluate its concentration during photocatalytic degradation. However, the percentage of removal of the SO dye was assessed as follows:
 | (1) |
3. Results and discussion
3.1. UV-Visible absorption spectroscopy
The UV-Visible absorption studies were carried out by dispersing the nanocomposites in ethanol and then analyzed the solution using a UV-Visible Spectrometer Lamda-25 (PerkinElmer). Fig. 1 shows the UV-Visible spectra acquired in the 200–800 nm wavelength range. The energy bandgaps of nanocomposites were determined using the following Tauc plot equation (eqn (2)):| (αhv)γ = A(hν − Eg) | (2) |
 | ||
| Fig. 1 UV-Visible results of ZnO, g-C3N4, S-g-C3N4 and ZnO–S-g-C3N4 nanocomposite. | ||
 | ||
| Fig. 2 Bandgap energy results of g-C3N4, ZnO, S-g-C3N4 and ZnO–S-g-C3N4 nanocomposite. | ||
The absorption characteristics of the synthesized materials were identified using UV-Visible spectroscopy. The UV-Visible spectrum of pure g-C3N4 exhibits an absorption pattern of typically semiconductors, spanning the range from 200 to 450 nm. This spectrum results from charge transfer processes, specifically the transition from the populated valence band of nitrogen atoms (2p orbitals) to the conduction band of carbon atoms (2p orbitals) in carbon nitride.35 Fig. 1 shows the UV-Vis absorption spectra of the prepared samples, including ZnO, g-C3N4, S-g-C3N4, and ZnO–S-g-C3N4. Notably, a prominent absorption band appears in the range of 300–350 nm. This band is associated with electronic transitions in aromatic 1,3,5-triazine compounds, which aligns with the absorption characteristics of this type of transition. These results corroborate the presence of structures based on s-triazine rings, supporting the conclusions drawn from the EDX and FTIR analyses. In Fig. 1, the UV-Visible spectroscopy data for ZnO nanoparticles reveal two absorption peaks at wavelengths of 267 nm and 380 nm. These dual peaks are attributed to the elongated shape of the nanoparticles.36 The peak at 380 nm corresponds to the intrinsic band gap absorption of ZnO, arising from electron transitions between the valence band and the conduction band (O2p → Zn3d). The broad nature of this peak indicates that the particles are on a nanoscale and possess a narrow particle size distribution.37
Tauc plots were utilized to determine the consistent energy bandgap of the provided materials using UV-Visible absorption spectra, as shown in Fig. 2. The bandgap energy of ZnO was determined to be 2.4 eV. This accurately characterizes ZnO as a semiconductor material with excellent photoelectric properties in the ultraviolet range but limited reactivity to visible light.38,39 The absorption edge of g-C3N4 is 300–350 nm, which corresponds to the direct bandgap of 2.3 eV. Likewise, the bandgap energy of S-g-C3N4 (Eg = 2.44 eV) is given more instinctively in Fig. 2.40 It is attributed to the bandgap alignment resulting from the coupling of semiconductor materials. This indicates that the composite photocatalyst exhibits enhanced response to visible light, which significantly enhances its performance. The bandgap energy of ZnO–S-g-C3N4 is approximately 2.13 eV (Fig. 2). It was observed that a slight increase in the bandgap occurred upon doping sulfur into graphitic carbon nitride. This could be attributed to the smaller atomic radius of sulfur atoms compared to carbon and nitrogen atoms, which may introduce energy levels into the bandgap. These new levels act as traps for electrons, making it more difficult for them to move through the material. As a result, the bandgap increases.40. The increased bandgap makes the material more suitable for photocatalytic applications, as it can absorb high energy photons. Similarly, doping sulfur can also improve the charge separation and transfer efficiency, thereby enhancing the photocatalytic activity of the resulting ZnO–S-g-C3N4 composite. The observed variations in the overall behavior of ZnO–S-g-C3N4 are promising from an application point of views.
3.2. XRD analysis
The X-ray diffraction (XRD) technique is a powerful tool for examining a compound's structure, crystallite size, phase information, and the presence of contaminants within the matrix.41 In our study, XRD was instrumental in understanding how doping influenced changes in phase morphology and crystal size in the generated nanomaterials.As illustrated in Fig. 3, the XRD patterns reveal the crystalline nature of the prepared (a) g-C3N4, (b) ZnO, (c) S-g-C3N4, and (d) ZnO–S-g-C3N4 nanocomposites. The XRD pattern of g-C3N4 exhibits diffraction peaks at approximately 27.2°, corresponding to the (002) plane of hexagonal g-C3N4 geometry.42 All the synthesized samples exhibit two distinct XRD peaks, (100) and (002) planes, with 2θ angles of approximately 13.1° and 27.2°, respectively, indicative of the typical g-C3N4 structure. The smaller intense peak observed at 13.1° is associated with the (100) plane, likely related to the in-plane structural packing motif of g-C3N4, such as the distance between nitride pores. Upon the addition of ZnO, the intensity of this smaller peak decreased, suggesting a significant interaction between the g-C3N4 host and the newly introduced ZnO. This interaction influenced the nitride pore structure and altered the hole-to-hole distance.7 The peak at a higher angle (002) is attributed to the periodic facial stacking of motifs found in conjugated aromatic systems, highlighting the distinctive graphitic structure of g-C3N4. In the case of ZnO–S-g-C3N4 (Fig. 3(b)), distinct diffraction peaks at 2θ values of 31.23, 33.89, 35.63, 46.88, 55.88, 67.09, 62.28, and 67.36 are observed. These peaks correspond to the hexagonal-structured wurtzite ZnO, confirming that the heterostructures are composed of both g-C3N4 and ZnO phases, respectively.43
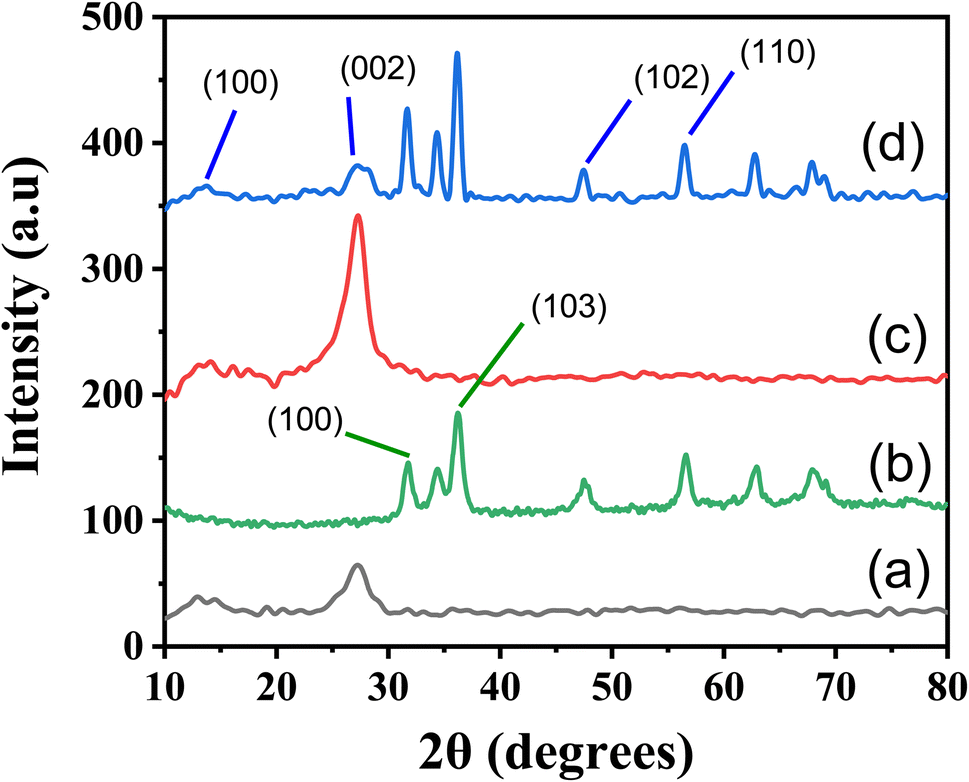 | ||
| Fig. 3 XRD spectrum of g-C3N4 (a), ZnO (b), S-g-C3N4 (c) and ZnO–S-g-C3N4 (d) nanocomposite. | ||
Additionally, the average crystallite size (Dp) calculated, shown in Table S1† from the diffraction peaks using Scherrer's equation.
3.3. FTIR analysis
In order to study chemical bonding and functional groups in molecules, FT-IR analysis is a crucial technique. The FTIR analysis of the produced nanocomposites employed an FT-IR spectrometer from PerkinElmer's series 100 with a resolution of 5 cm−1 and a wavenumber range of 4000–400 cm−1. FT-IR results are displayed in Fig. 4(a) and (b). Fig. 4(a) shows that a peak around 1641 cm−1 corresponds to the stretching vibration band of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N. While the stretching vibrations for rings of C–N bonds are clearly apparent at 1533 and 1700 cm−1.44 Likewise, the peak at 810 cm−1 indicates the out-of-plane bending mode of heptazine rings.45–47 Another peak in the range of 2354 cm−1 may be due to the presence of adsorbed CO2 from the atmosphere/air.16 The peak of sulphur was not seen in the spectra of S-g-C3N4 due to its insignificant existence Fig. 4(a). The stretching vibration of the Zn–O bond is attributed to the small peak at 467 cm−1 in Fig. 4(a) of the ZnO–S-g-C3N4 FT-IR spectrum. The peak near 3743 cm−1 is mainly caused by the amine groups and adsorbed water molecules on the surface of g-C3N4.48 Moreover, the band around 3143 cm−1 is also ascribed may be due to stretching modes of the OH of adsorbed H2O.49 In general, the presence of the identical peaks in the ZnO–S-g-C3N4 sample corroborated the compositional assembly, however the peaks intensities decreased slightly.
N. While the stretching vibrations for rings of C–N bonds are clearly apparent at 1533 and 1700 cm−1.44 Likewise, the peak at 810 cm−1 indicates the out-of-plane bending mode of heptazine rings.45–47 Another peak in the range of 2354 cm−1 may be due to the presence of adsorbed CO2 from the atmosphere/air.16 The peak of sulphur was not seen in the spectra of S-g-C3N4 due to its insignificant existence Fig. 4(a). The stretching vibration of the Zn–O bond is attributed to the small peak at 467 cm−1 in Fig. 4(a) of the ZnO–S-g-C3N4 FT-IR spectrum. The peak near 3743 cm−1 is mainly caused by the amine groups and adsorbed water molecules on the surface of g-C3N4.48 Moreover, the band around 3143 cm−1 is also ascribed may be due to stretching modes of the OH of adsorbed H2O.49 In general, the presence of the identical peaks in the ZnO–S-g-C3N4 sample corroborated the compositional assembly, however the peaks intensities decreased slightly.
 | ||
| Fig. 4 FTIR spectrum of (a): g-C3N4, S-g-C3N4, ZnO–S-g-C3N4 and (b) ZnO nanocomposite. | ||
Fig. 4(b) displays the FTIR spectra of ZnO in the 4000–400 cm−1 range. The formation of ZnO nanoparticles is evident from the appearance of peaks at 3324 cm−1 and 1632 cm−1, corresponding to the stretching and bending vibrations of O–H groups in water molecules. Additionally, a peak at approximately 564 cm−1 indicates the presence of a metal oxide bond (Zn–O).50 Furthermore, the peak observed at 2120 cm−1 can be attributed to the presence of CO2 molecules in the surrounding air.51
3.4. SEM and TEM analysis
SEM images shown in Fig. 5 represent the texture of g-C3N4, S-g-C3N4, and ZnO–S-g-C3N4. SEM micrographs of g-C3N4, S-g-C3N4, and ZnO–S-g-C3N4 are also presented for comparison. It is clear from Fig. 5(a) and (b) that the g-C3N4 samples showed aggregated, smooth, slate-like particles made up of lamellar structures. The layered structure of sample S-g-C3N4 was verified by microscopic observation. In gas-templating synthesis, layered microstructure is produced by the dense assembly of many irregular nanosheets.42,52 Interestingly, the pristine g-C3N4 photocatalyst has a non-porous microstructure with tightly connected nanolayers Fig. 5(a) and (b), whereas the S-doped g-C3N4 has a porous microstructure Fig. 5(c) and (d), which is expected to be caused by the emission of ammonia gas during the thermal polymerization of urea and thiourea mixture. The porous, layered structure of the S-doped g-C3N4 photocatalyst has already been hypothesized to be favorable for the development of reactive sites on the catalyst's surface, improving the charge carriers separation and transfer,53 which is directly related to raising the photocatalyst performance. | ||
| Fig. 5 SEM images of g-C3N4 (a and b), S-g-C3N4 (c and d) and ZnO–S-g-C3N4 (e and f) nanocomposites. | ||
Also, it was shown that ZnO–S-g-C3N4 grew into a quench interconnected elongated fiber network after adding ZnO to S-g-C3N4 Fig. 5(e) and (f). A flimsy framework of ZnO–S-g-C3N4 with a higher surface area and porosity was formed when [ZnCO3]2 and [Zn(OH)2]3 interacted with a mixture of urea and thiourea, according to SEM analysis. This occurred most likely as a result of the influence of the gasses generated during the thermal polymerization process.7 Such heterojunction morphology is praised for increasing the material's surface area, active sites, and corresponding photocatalytic activity.54,55
The TEM examination of g-C3N4 reveals that the material has a nanosheet structure, which would be advantageous for the sensing mechanism. The higher surface area could be due to the layered and flaky organization of the g-C3N4.56 Porous and flocculent structures in the nanomaterials facilitate the departure of charge carriers and thus suppressing recombination (Fig. 6).
 | ||
| Fig. 6 TEM images of g-C3N4 (a), S-g-C3N4 (b) and ZnO–S-g-C3N4 (c) nanocomposites. | ||
3.5. Energy dispersive X-ray spectroscopy (EDX)
Energy dispersive X-ray spectroscopy (EDX) microanalysis was used to check the elemental composition of all the samples. Energy-dispersive X-ray spectroscopy patterns of the g-C3N4, S-g-C3N4, ZnO–S-g-C3N4, and all the hybrid microgels is depicted in Fig. 7, respectively. The EDX result demonstrates the corresponding peaks of the elements. The EDX spectrum of g-C3N4 Fig. 7(a), revealed the existence of carbon (C) and nitrogen (N). According to EDX results of S-g-C3N4 (Fig. 7b) the main elements were C (carbon), N (nitrogen) and S (sulphur). The percentage composition of all the components found in the ZnO–S-g-C3N4 (Fig. 7c) nanocomposite was in the order C > N > O > K > Zn > S. This reveals that C and N have higher content ratios, whereas Zn and S have the lowest concentrations. The identified elements found in the EDX spectrums, confirm the fruitful synthesis of nanomaterials.57 The mass percent and atomic percent compositions of all the elements are provided in Table S2.† | ||
| Fig. 7 The elemental composition images of g-C3N4 (a), S-g-C3N4 (b) and ZnO–S-g-C3N4 (c) nanocomposites from EDX. | ||
3.6. Photocatalytic activity
The effectiveness of photocatalysis of the produced samples was evaluated by monitoring the Safranin-O degradation under UV light. Using a light source, the photocatalytic reaction was successfully carried out. 0.1 g of photocatalysts were added to 100 mL of SO solution with 10 ppm of SO. In order to achieve equilibrium between adsorption and desorption, the suspension from the preceding illustration was stirred at room temperature for 30 minutes without light.58 The liquid was then agitated vigorously while being exposed to ionizing radiation. The solution was periodically extracted to a volume of 4 mL and centrifuged to ensure the solid catalyst was removed. An ultraviolet-visible spectrometer was used to determine the catalyst's dye adsorption capacity and dye degradation efficiency. The amount of already removed dye can be calculated using the formula shown in eqn (3).| Efficiency of degradation (%) = (Co − Ct/Co) × 100 | (3) |
To execute the kinetic studies of the dye degradation process, the specified first-order equation (eqn (4)) was employed for considering the data.59
| ln(Ct/Co) = −kapp t | (4) |
Fig. S1 and S2† depict the decrease in the absorbance at various time intervals of SO dye in the presence of UV light and dark conditions by synthesized g-C3N4 nanoparticles. Under normal conditions, 10 ppm SO dye solutions were prepared and 0.1 g catalyst dosage was applied; the reaction took up to 360 minutes under UV light. In dark conditions, g-C3N4 removed less dye than in light conditions. The irradiation suspensions were collected at predefined time intervals. The progression of the degradation was demonstrated by the decreasing SO absorbance over time as displayed in Fig. S1 and S2(a).† As evident from Fig. S1 and S2(b),† the percentage of degradation efficiency increased with time and reached a maximum of 360 and 480 minutes. These results also demonstrate that 20% of the dye was eliminated in the dark in 480 minutes, but in the presence of light, the catalyst removed 69% of SO in 360 minutes. These results indicate that g-C3N4 exhibits higher efficiency in the removal of SO dye in the presence of light compared to its removal in the absence of light. The degradation process of SO dye can be tracked by monitoring changes in the absorbance ratio (Ct/C0) over time, as illustrated in Fig. S1 and S2(c).† These figures reveal the degradation of SO dye with g-C3N4 as a catalyst, where quantitative and qualitative spectral fluctuations occur over time.
Furthermore, Fig. S1 and S2(d)† depict the plot of the pseudo-first-order degradation of SO dye. The data suggest that the reaction follows a pseudo-first-order equation, as indicated by the high values of R2 (R2 = 0.94660 in the presence of light and R2 = 0.97152 in the absence of light).
The values of the rate constant (K1), as these numbers for the first-order kinetics, further corroborate this pattern. The values 0.00249, and 0.00041 min−1 were used for eliminating SO dye under light and dark conditions, respectively (see Table S3†).
Fig. S3(a) and S4(a)† show the degradation of SO with the S-g-C3N4 catalyst in light and dark, as well as the qualitative and quantitative changes seen with the double beam UV-Visible spectrophotometer. Additionally, the results showed that the SO intensity reduces when the irradiation period is lengthened. This is because of the gradual removal (breakdown) of SO molecules with the passage of time. Furthermore, it is found that the degradation efficiency rises with time and touched a maximum at 270 and 480 minutes, as displayed in Fig. S3(b) and S4(b).† These findings also show that 36.22% of SO was removed in 480 minutes in the absence of light, while in light, the catalyst removed 88.54% of SO in 270 minutes. These results show that the S-g-C3N4 has virtuous efficiency in the degradation of SO dye both in the absence and presence of light, but it works more effectively in the presence of light than in totally dark conditions. Furthermore, the plots in Fig. S3(c) and S4(c)† demonstrate the degradation progress in terms of the absorbance ratio (Ct/C0) versus time. These figures illustrate the degradation of SO dye using the S-g-C3N4 catalyst, showing both quantitative and qualitative spectral changes over time. Fig. S3(d) and S4(d)† also present the pseudo-first-order relation for the degradation process, confirming that it follows pseudo-first-order kinetics. The high values of R2 (R2 = 0.90922 in the presence of light and R2 = 0.84046 in the absence of light) support this pattern. Moreover, the rate constant (K1) values, indicative of the first-order kinetics, were estimated to be 0.00690 in the presence of light and 0.00066 min−1 in the absence of light (see Table S3†).
Furthermore, absorbance versus wavelength plots of SO dye in light and in the dark for various time periods are shown, using ZnO–S-g-C3N4 nanocomposite as a catalyst are illustrated in Fig. 8(a) and 9(a). The graphs clearly specify that the intensity of the absorption peaks of SO lowers from the maximum wavelengths with increasing irradiation period. This is because of effective removal of SO molecules with time. However, after adding ZnO–S-g-C3N4; SO demonstrated a sharp peak due to the π–π* transition and a shoulder peak because of dimeric and polymeric π stacked forms of SO in water.60 Fig. 8(b) and 9(b) clearly show that the removal efficiency increases with time, as represented by the plot, at 150 and 480 minutes, respectively, the value reaches its maximum. And that 87% dye elimination happened at 150 minutes in light and 58.64% in dark at 480 minutes, respectively. The plot of absorbance ratio (Ct/C0) versus time is utilized for the removal process, as shown in Fig. 8(c) and 9(c). Fig. 8(d) and 9(d) show the plots of pseudo first-order relations for the SO removal with ZnO–S-g-C3N4 catalyst. As confirmed, the degradation data follow a pseudo-first-order equation as shown by the R2 = 0.89186 under light conditions. The first-order rate constant (K1) value is 0.00989. Similarly, under dark conditions, the R2 = 0.75568, and the first-order rate constant (K1) is equal to 0.00144 per minute.
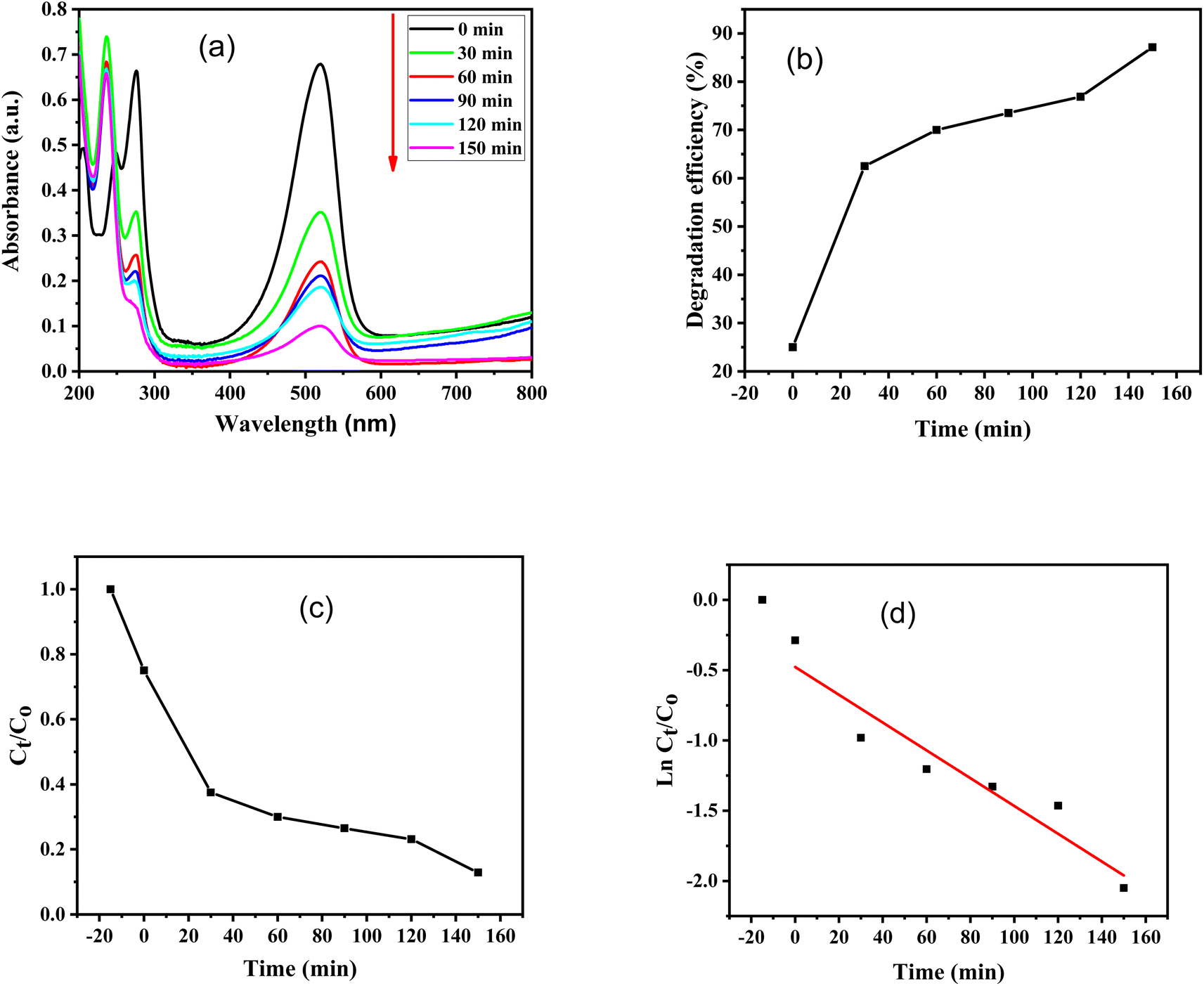 | ||
| Fig. 8 SO degradation with the aid of 0.1 g ZnO–S-g-C3N4 in the presence of light; (a) change in UV-Visible absorbance spectra at different time intervals, (b) degradation efficiency, (c) absorbance ratio (Ct/C0) versus time, (d) plot of pseudo-first-order kinetics of degradation. | ||
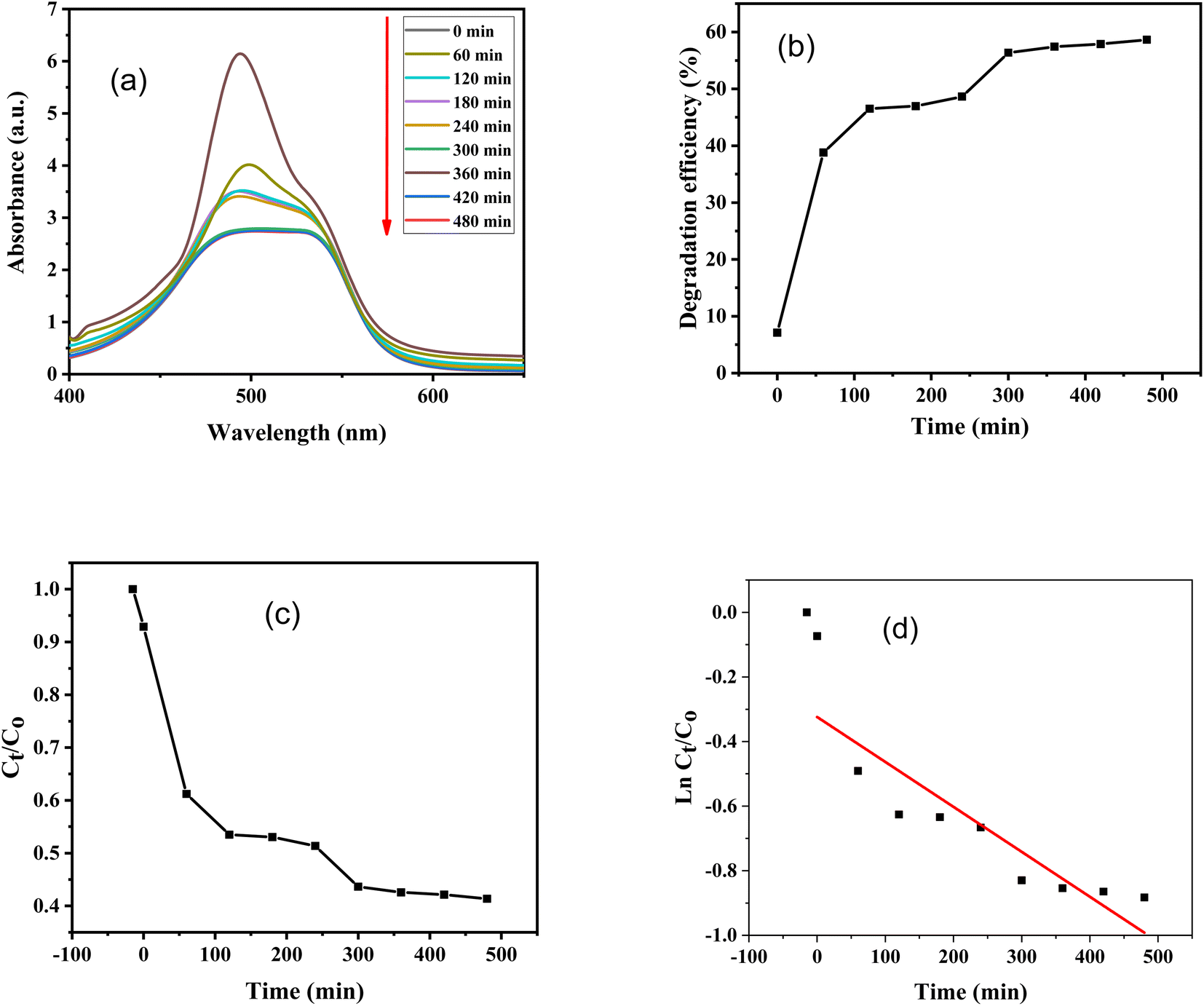 | ||
| Fig. 9 SO degradation with the aid of 0.1 g ZnO–S-g-C3N4 in the absence of light (dark condition); (a) change in UV-Visible absorbance spectra at different time intervals, (b) degradation efficiency, (c) absorbance ratio (Ct/C0) versus time, (d) plot of pseudo-first-order kinetics of degradation. | ||
A brief comparison of the dye removal performance of the synthesized nanocomposites with a few previously published nanocomposite materials and dyes is shown in Table S3.† The ability of different nanocomposites to remove pollutants might differ depending on the precise pollutant being targeted or the exact nanomaterial being employed. This variability can be attributable to a number of variables, such as variations in light, pH, the concentration of catalysts and pollutants, and differences in chemical structures and reactivity toward pollutants. The dyes compatibility with the nanoparticles may also impact the efficiency of removal. Several surface phenomena occur when a catalyst such as ZnO–S-g-C3N4 is used to remove SO dye. The surface of the ZnO–S-g-C3N4 contains active sites where SO molecules may link due to electrostatic interactions, hydrogen bonding, and van der Waals forces. This adsorption process aids in the concentration of dye molecules on the catalyst surface. The photocatalytic action of ZnO is activated after SO molecules are deposited onto the surface of catalyst. When ZnO absorbs photons from light, electron–hole pairs are formed. These photo-induced charge carriers can participate in redox processes, which degrade the SO dye molecules into simpler, less toxic compounds. The S-g-C3N4 can improve the chemical reaction among the dye molecules and the surface of the catalyst. The surface and SO molecules may establish covalent bonds as a result, assisting in their breakdown. So, the adsorption of SO molecules on the surface of the ZnO–S-g-C3N4 catalyst is followed by photocatalytic and possibly chemisorptive activities that helps in the breakdown or elimination of the dye from the polluted water.
It can be concluded that the degradation potential of ZnO–S-g-C3N4 has enhanced as compared to g-C3N4, S-g-C3N4. This could be due to the lower band gap energy of ZnO–S-g-C3N4 nanocomposite, and leads to an enhanced response to light absorption. A decline in photoactivity noticed for g-C3N4 and S-g-C3N4 as compared to ZnO–S-g-C3N4, perhaps because the g-C3N4 and possesses extra defects that act as the target for electron–hole recombination, slowing down the rate of photo-induced charge transfer. The overall results show that changes in experimental parameters, such as catalyst type, pollutant concentration, catalyst dose, light source, irradiation time, and chemical composition of the dyes, affect the photocatalytic degradation efficiency of the various studies. Additionally, the catalysts' chemical makeup, multiple textures, and morphological characteristics have a significant impact on how effective they are. It's crucial to design core–shell materials that have a suitable balance of hydrophilicity and hydrophobicity.
Finally, it can be concluded that the g-C3N4, S-g-C3N4, ZnO–S-g-C3N4 nanomaterials exhibit significant catalytic activity for the removal of SO dye in light as compared to dark, as it can be seen from their percent removal values provided in Table S3.† However, it is concluded that under alike experimental conditions, SO could be readily degraded by ZnO–S-g-C3N4 compared to g-C3N4 and S-g-C3N4.
The photocatalytic discoloration of a dye is supposed to be caused by the following mechanism. When a catalyst is exposed to UV light, electrons are moved from the valence band to the conduction band. Consequently, an electron–hole pair is created;61
| Catalyst + hv → ecb− + hvb+ | (5) |
| H2O + hvb+ → ˙OH + H+ | (6) |
| O2 + ecb− → O2−˙ | (7) |
The combination of the electron and the hole created in the first step is prevented by this reaction.
The dye discoloration is caused by the ˙OH and O2˙ that are created in the manner described above, which can then interact with the dye to form additional species.63
| O2−˙ + H2O → H2O2 | (8) |
| H2O2 → 2 ˙OH | (9) |
| ˙OH + dye → dyeox | (10) |
| Dye + e − cb → dyered | (11) |
| HO˙ + dye → intermediates → CO2 + H2O | (12) |
The powerful oxidizing species known as ˙OH degrades the organic pigment non-selectively into H2O, CO2 and inorganic ions.64,65
In addition, the oxidation reaction occurs on the VB of ZnO in the ZnO–S-g-C3N4 photocatalytic system, while the reduction reaction happens on the CB of S-g-C3N4,66 indicating that the ZnO–S-g-C3N4 heterojunction retains its strong redox properties. We also conclude that the heterojunction generated by S-g-C3N4 and ZnO is a Z-scheme heterojunction (Scheme 2), also the composite is a photocatalyst with high separation efficiency of photogenerated electron–hole pairs and high redox ability.46,48,67 A few years back, Kang's group proposed a direct Z-scheme heterojunction photocatalyst based on g-C3N4/ZnO in binary form. Mesoporous ZnO nano-triangles@g-C3N4 nanofoils (ZnO-nt@g-C3N4) were created using varying g-C3N4 concentrations. Under solar light irradiation, the photocatalyst ZnO-nt@g-C3N4 (20%) demonstrated 100% degradation of Rh B dye in 60 minutes.68 Wang et al. subsequently presented a Z-scheme mechanism mediated by oxygen defects in exfoliated g-C3N4 nanosheets connected to oxygen-defective ZnO (OD-ZnO) nanorods, i.e., CN/OD-ZnO heterojunction with variable wt% of g-C3N4.69 In the case of the GCN-ZnO0.4 sample, the increased surface area can result in decreased recombination of photogenerated charge carriers as well as more active sites during the photocatalytic reaction, which can produce more photogenerated electrons.70 Scheme 2 illustrates a schematic representation of the mechanisms of oxidative species formation in a photocatalytic investigation. Therefore, in the case of the ZnO–S-g-C3N4, both increased surface area and improved light absorption ability can result in superior degradation efficiency toward the SO dye.
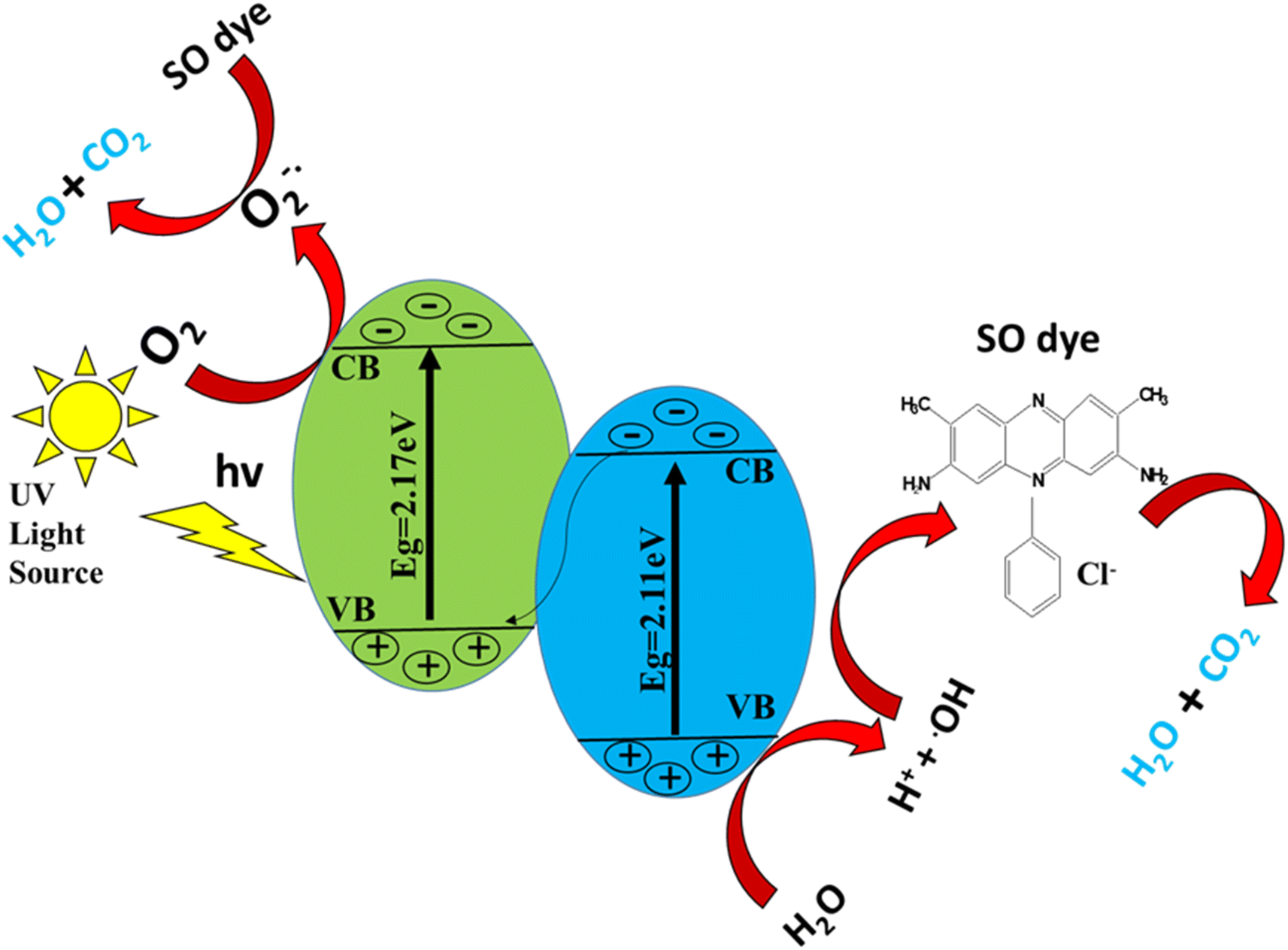 | ||
| Scheme 2 Schematic diagram for UV-light photocatalytic mechanism showing the generation of oxidative species and degradation of contaminants with the help of a catalyst. | ||
4. Conclusion
The g-C3N4, was prepared by the calcination of urea, while S-g-C3N4 was synthesized by calcination of the mixture of urea and thiourea in a 3:1 ratio. Finally the ZnO–S-g-C3N4 nanocomposite was synthesized using the liquid exfoliation technique, and distilled water was used as exfoliating solvent. Numerous characterization techniques such as UV-Visible spectroscopy, FTIR, XRD, EDX, SEM and TEM were used for the as prepared nanomaterials; in order to determine crystallinity, morphology, optical properties, and phase purity. Additionally, the direct-bandgap energy of the nanocomposites was determined, and it agrees strongly with the previously published work. The nanocomposites were employed for catalytic and photocatalytic applications toward Safranin-O dye degradation. According to a comparison of recent findings with those from earlier research, various experimental factors, including the type of catalyst and pollutant, their quantity, the reaction medium's pH, temperature, the quality of the light, and the presence of additional precursors in solution, have a significant impact on the photodegradation of dyes. Overall, it is confirmed that the ZnO–S-g-C3N4 and S-g-C3N4 nanocomposites have an active catalytic and photocatalytic performance as compared to pure g-C3N4, as indicated by their k-values. According to the kinetic evaluation, this removal procedure follows a pseudo first-order kinetics. In fact, the suggested approach creates new avenues for the development of effective electrocatalysts for water splitting, which yields hydrogen as a clean energy source. Lastly, we suggest that these materials can be used to degrade the pollutants and pathogens from waste water with high efficiency and stability, also it can be used to enhance the electroflocculation process and contribute to the development of a sustainable approach for advanced wastewater treatment.
Author contributions
All authors have contributed to this study at different stages. Azmat Ali Khan and Abbas Khan: study design, method design, analytical protocol design, writing, reviewing, and editing. Abbas Khan, Sumayya Khan, Nasrullah Shah and Asaad Khalid: experimental assistant, discussion during writing, and reviewing. Ajmal Khan, Ahmed Al-Harrasi, and Afnan Jan: characterization of the materials, experimental assistant, and data analysis of some results. Abbas Khan, Ajmal Khan, and Faheem Nawaz: reviewing, and editing. All authors read and approved the final version of this manuscript.Conflicts of interest
No potential conflict of interest was reported by the author(s).Acknowledgements
This study was funded by the Deputy deanship for Research and Innovation, Ministry of Education in Saudi Arabia (project number ISP23-81). The author would like to thanks to Abdul Wali Khan University Mardan Pakistan and University of Nizwa for general support of this work. The authors extend their appreciation to the Deputyship for Research and Innovation, Ministry of Education in Saudi Arabia for funding this research work through the project with number: ISP23-81.References
- S. Kumar, V. R. Battula and K. Kailasam, Carbon, 2021, 183, 332–354 CrossRef CAS.
- M. A. Qamar, S. Shahid, M. Javed, M. Shariq, M. M. Fadhali, O. Madkhali, S. K. Ali, I. S. Syed, M. Y. Awaji and M. Shakir Khan, Catalysts, 2022, 12, 1388 CrossRef CAS.
- X. Xiao, J. Wei, Y. Yang, R. Xiong, C. Pan and J. Shi, ACS Sustainable Chem. Eng., 2016, 4, 3017–3023 CrossRef CAS.
- M. Humayun, W. Pi, Y. Yuan, L. Shu, J. Cao, A. Khan, Z. Zheng, Q. Fu, Y. Tian and W. Luo, J. Colloid Interface Sci., 2021, 599, 484–496 CrossRef CAS PubMed.
- M. Verma and A. Haritash, J. Environ. Chem. Eng., 2019, 7, 102886 CrossRef CAS.
- M. A. Qamar, M. Javed, S. Shahid, M. Shariq, M. M. Fadhali, S. K. Ali and M. S. Khan, Heliyon, 2023, 9, e12685 CrossRef CAS PubMed.
- D. R. Paul, S. Gautam, P. Panchal, S. P. Nehra, P. Choudhary and A. Sharma, ACS Omega, 2020, 5, 3828–3838 CrossRef CAS PubMed.
- N. R. Vempuluru, C. K. Krishnan, R. Parnapalli, J. Velusamy, S. Marappan, S. Pitchaimuthu, M. Murikinati and S. M. Venkatakrishnan, Ceram. Int., 2021, 47, 10206–10215 CrossRef CAS.
- A. BaQais, M. Shariq, E. Almutib, N. Al-Qasmi, R. Azooz, S. K. Ali, K. Hassan and M. Iqbal, Eur. Phys. J. Plus, 2023, 138, 1–9 CrossRef.
- X. Dong and F. Cheng, J. Mater. Chem. A, 2015, 3, 23642–23652 RSC.
- L. Jiang, X. Yuan, Y. Pan, J. Liang, G. Zeng, Z. Wu and H. Wang, Appl. Catal., B, 2017, 217, 388–406 CrossRef CAS.
- J. Fu, B. Zhu, C. Jiang, B. Cheng, W. You and J. Yu, Small, 2017, 13, 1603938 CrossRef PubMed.
- M. Humayun, H. Ullah, J. Cao, W. Pi, Y. Yuan, S. Ali, A. A. Tahir, P. Yue, A. Khan and Z. Zheng, Nano-Micro Lett., 2020, 12, 1–18 CrossRef PubMed.
- A. Hussain, N. Ali, S. Ali, J. Hou, I. Aslam, H. Naeem, M. Boota, M. Ul-Hussan, J. Yin and X. Wang, Res. Chem. Intermed., 2022, 48, 2857–2870 CrossRef CAS.
- F. Raziq, M. Humayun, A. Ali, T. Wang, A. Khan, Q. Fu, W. Luo, H. Zeng, Z. Zheng and B. Khan, Appl. Catal., B, 2018, 237, 1082–1090 CrossRef CAS.
- M. Javed, M. Abid, S. Hussain, D. Shahwar, S. Arshad, N. Ahmad, M. Arif, H. Khan, S. Nadeem and H. Raza, Dig. J. Nanomater. Biostruct., 2020, 15, 1097–1105 CrossRef.
- Y. Zheng, Y. Liu, X. Guo, Z. Chen, W. Zhang, Y. Wang, X. Tang, Y. Zhang and Y. Zhao, J. Mater. Sci. Technol., 2020, 41, 117–126 CrossRef CAS.
- W. Pi, M. Humayun, Y. Li, Y. Yuan, J. Cao, S. Ali, M. Wang, H. Li, A. Khan and Z. Zheng, Int. J. Hydrogen Energy, 2021, 46, 21912–21923 CrossRef CAS.
- J. Liu, H. Wang and M. Antonietti, Chem. Soc. Rev., 2016, 45, 2308–2326 RSC.
- L. Tan, J. Xu, S. Li, D. Li, Y. Dai, B. Kou and Y. Chen, Materials, 2017, 10, 484 CrossRef PubMed.
- G. M. Veith, L. Baggetto, L. A. Adamczyk, B. Guo, S. S. Brown, X.-G. Sun, A. A. Albert, J. R. Humble, C. E. Barnes and M. J. Bojdys, Chem. Mater., 2013, 25, 503–508 CrossRef CAS.
- J. Wang, H. Zhao, B. Zhu, S. Larter, S. Cao, J. Yu, M. G. Kibria and J. Hu, ACS Catal., 2021, 11, 12170–12178 CrossRef CAS.
- R. Liu, W. Yang, G. He, W. Zheng, M. Li, W. Tao and M. Tian, ACS Omega, 2020, 5, 19615–19624 CrossRef CAS PubMed.
- V. Ragupathi, P. Panigrahi and N. G. Subramaniam, Optik, 2020, 202, 163601 CrossRef CAS.
- R. Liu, Z. Chen, Y. Yao, Y. Li, W. A. Cheema, D. Wang and S. Zhu, RSC Adv., 2020, 10, 29408–29418 RSC.
- A. Mittal, B. Mari, S. Sharma, V. Kumari, S. Maken, K. Kumari and N. Kumar, J. Mater. Sci.: Mater. Electron., 2019, 30, 3186–3207 CrossRef CAS.
- C. Retamoso, N. Escalona, M. González, L. Barrientos, P. Allende-González, S. Stancovich, R. Serpell, J. Fierro and M. Lopez, J. Photochem. Photobiol., A, 2019, 378, 136–141 CrossRef CAS.
- A. Hassani, P. Eghbali and Ö. Metin, Environ. Sci. Pollut. Res., 2018, 25, 32140–32155 CrossRef CAS PubMed.
- A. Hassani, M. Faraji and P. Eghbali, J. Photochem. Photobiol., A, 2020, 400, 112665 CrossRef CAS.
- I.-H. Tseng, Y.-M. Sung, P.-Y. Chang and C.-Y. Chen, Polymers, 2019, 11, 146 CrossRef PubMed.
- W. Wang, M. Zhang, B. Zhao, L. Liu, R. Han and N. Wang, Pigm. Resin Technol., 2022, 51, 91–100 CrossRef CAS.
- D. R. Paul, R. Sharma, P. Panchal, R. Malik, A. Sharma, V. K. Tomer and S. Nehra, J. Nanosci. Nanotechnol., 2019, 19, 5241–5248 CrossRef CAS PubMed.
- X. Sun, X. Wang, Y. Liu, Y. Lian, L. Meng and Z. Su, J. Water Process Eng., 2022, 47, 102747 CrossRef.
- M. Benedet, G. A. Rizzi, A. Gasparotto, O. I. Lebedev, L. Girardi, C. Maccato and D. Barreca, Chem. Eng. J., 2022, 448, 137645 CrossRef CAS.
- A. Kumar, P. Kumar, C. Joshi, M. Manchanda, R. Boukherroub and S. L. Jain, Nanomaterials, 2016, 6, 59 CrossRef PubMed.
- R. Augustine, E. A. Dominic, I. Reju, B. Kaimal, N. Kalarikkal and S. Thomas, RSC Adv., 2014, 4, 24777–24785 RSC.
- H. Karimi-Maleh, K. Ahanjan, M. Taghavi and M. Ghaemy, Anal. Methods, 2016, 8, 1780–1788 RSC.
- M. M. Obeid, H. R. Jappor, K. Al-Marzoki, I. A. Al-Hydary, S. J. Edrees and M. M. Shukur, RSC Adv., 2019, 9, 33207–33221 RSC.
- N. Aslan, Türk Doğa ve Fen Dergisi, 2022, 11, 95–101 CrossRef.
- H. Starukh and P. Praus, Catalysts, 2020, 10, 1119 CrossRef CAS.
- S. Muhammad, A. Ali, S. Zahoor, X. Xinghua, J. Shah, M. Hamza, M. Kashif, S. Khan, B. K. A. Khel and A. Iqbal, Acta Sci. Appl. Phys., 2023, 3, 33–48 Search PubMed.
- S. Shoran, A. Sharma and S. Chaudhary, Environ. Sci. Pollut. Res., 2023, 1–15 Search PubMed.
- H. Qin, Y. Zuo, J. Jin, W. Wang, Y. Xu, L. Cui and H. Dang, RSC Adv., 2019, 9, 24483–24488 RSC.
- X. Yang, B. Tang, T. Wu and X. Cao, J. Mater. Civ. Eng., 2019, 31, 04019141 CrossRef CAS.
- C. Zhang, Z. Ouyang, Y. Yang, X. Long, L. Qin, W. Wang, Y. Zhou, D. Qin, F. Qin and C. Lai, Chem. Eng. J., 2022, 448, 137370 CrossRef CAS.
- L. Yan, H. Gao and Y. Chen, ACS Appl. Nano Mater., 2021, 4, 7746–7757 CrossRef CAS.
- S. Benedoue, M. Benedet, A. Gasparotto, N. Gauquelin, A. Orekhov, J. Verbeeck, R. Seraglia, G. Pagot, G. A. Rizzi and V. Balzano, Nanomaterials, 2023, 13, 1035 CrossRef CAS PubMed.
- X. Guo, J. Duan, C. Li, Z. Zhang and W. Wang, J. Mater. Sci., 2020, 55, 2018–2031 CrossRef CAS.
- M. Yaseen, A. Khan, M. Humayun, S. Farooq, N. Shah, S. Bibi, Z. A. Khattak, A. U. Rehman, S. Ahmad and S. M. Ahmad, Macromol. Mater. Eng., 2023, 2200695 CrossRef CAS.
- D. Tamilselvi, N. Velmani and K. Rathidevi, Egypt. J. Chem., 2019, 62, 785–795 Search PubMed.
- Z.-Y. Zhao, M.-H. Wang and H.-P. Zhang, J. Mater. Sci.: Mater. Electron., 2016, 27, 1777–1782 CrossRef CAS.
- M. Benedet, A. Gallo, C. Maccato, G. A. Rizzi, D. Barreca, O. I. Lebedev, E. Modin, R. McGlynn, D. Mariotti and A. Gasparotto, ACS Appl. Mater. Interfaces, 2023, 15, 47368–47380 CrossRef CAS PubMed.
- V. Mariyappan, N. Karuppusamy, T.-W. Chen, S.-M. Chen, J. Jesuraj, M. Akilarasan, B.-S. Lou and J. Yu, Process Saf. Environ. Prot., 2023, 169, 776–787 CrossRef CAS.
- M. Javed, M. A. Qamar, S. Shahid, H. O. Alsaab and S. Asif, RSC Adv., 2021, 11, 37254–37267 RSC.
- H. Li, Y. Wu, Z. Xu and Y. Wang, Chem. Eng. J., 2023, 147508 Search PubMed.
- Y. M. Shanbhag, M. M. Shanbhag, S. J. Malode, S. Dhanalakshmi, K. Mondal and N. P. Shetti, Biosensors, 2022, 12, 552 CrossRef CAS PubMed.
- L. Kong, Y. Liu, L. Dong, L. Zhang, L. Qiao, W. Wang and H. You, Dalton Trans., 2020, 49, 1947–1954 RSC.
- M. Yaseen, M. Humayun, A. Khan, M. Idrees, N. Shah and S. Bibi, Molecules, 2022, 27, 5343 CrossRef CAS PubMed.
- M. Yaseen, S. Farooq, A. Khan, N. Shah, L. A. Shah, S. Bibi, I. U. Khan and S. Ahmad, J. Chin. Chem. Soc., 2022, 69, 1637–1653 CrossRef CAS.
- P. Kumar, E. Vahidzadeh, U. K. Thakur, P. Kar, K. M. Alam, A. Goswami, N. Mahdi, K. Cui, G. M. Bernard and V. K. Michaelis, J. Am. Chem. Soc., 2019, 141, 5415–5436 CrossRef CAS PubMed.
- M. Humayun, M. He, W. Feng, C. Jin, Z. Yao, Y. Wang, W. Pi, S. Ali, A. Khan and M. Wang, Sol. Energy, 2021, 215, 121–130 CrossRef CAS.
- S. Ali, M. Humayun, W. Pi, Y. Yuan, M. Wang, A. Khan, P. Yue, L. Shu, Z. Zheng and Q. Fu, J. Hazard. Mater., 2020, 397, 122708 CrossRef CAS PubMed.
- H.-B. Fang, Y. Luo, Y.-Z. Zheng, W. Ma and X. Tao, Ind. Eng. Chem. Res., 2016, 55, 4506–4514 CrossRef CAS.
- M. Kashif, S. Muhammad, A. Ali, K. Ali, S. Khan, S. Zahoor and M. Hamza, J. Xi’an Shiyou Univ., 2023, 19, 521–544 Search PubMed.
- Z. Chen, T. Ma, Z. Li, W. Zhu and L. Li, J. Mater. Sci. Technol., 2024, 179, 198–207 CrossRef.
- G. Panthi and M. Park, Int. J. Mol. Sci., 2023, 24, 15021 CrossRef CAS PubMed.
- M. Benedet, G. A. Rizzi, A. Gasparotto, N. Gauquelin, A. Orekhov, J. Verbeeck, C. Maccato and D. Barreca, Appl. Surf. Sci., 2023, 618, 156652 CrossRef CAS.
- K. Vignesh, S. Kang, B. S. Kwak and M. Kang, Sep. Purif. Technol., 2015, 147, 257–265 CrossRef CAS.
- J. Wang, Y. Xia, H. Zhao, G. Wang, L. Xiang, J. Xu and S. Komarneni, Appl. Catal., B, 2017, 206, 406–416 CrossRef CAS.
- M. Wu, Y. Gong, T. Nie, J. Zhang, R. Wang, H. Wang and B. He, J. Mater. Chem. A, 2019, 7, 5324–5332 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra07247a |
| This journal is © The Royal Society of Chemistry 2024 |