
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsights into colistin-mediated fluorescence labelling of bacterial LPS†
Saurodeep Mandal a,
Dipanwita Patrab,
Sukhendu Mandalb,
Gourab Kanti Dasa and
Prithidipa Sahoo*a
a,
Dipanwita Patrab,
Sukhendu Mandalb,
Gourab Kanti Dasa and
Prithidipa Sahoo*a
aDepartment of Chemistry, Siksha Bhavana, Visva-Bharati, Santiniketan, 731235, West Bengal, India. E-mail: prithidipa.sahoo@visva-bharati.ac.in; saurodeep@outlook.com; gourabkanti.das@visva-bharati.ac.in
bDepartment of Microbiology, University of Calcutta, Kolkata 700019, West Bengal, India. E-mail: dipanwitapatra35@gmail.com; smmicrobio@caluniv.ac.in
First published on 17th January 2024
Abstract
Gram-negative bacterial infections are becoming untreatable due to their ability to mutate, and the gradual development of their resistance to the available antimicrobials. In recent times colistin, a drug of last resort, started losing its efficacy towards multidrug-resistant bacterial infections. Colistin targets bacterial endotoxin lipopolysaccharides (LPS) and destabilises the cytoplasmic membrane by disrupting the outer LPS membrane. In this study, we have tried to label the bacterial LPS, the main constituent of the cytoplasmic membrane of bacterial cells, to try to understand the interaction mechanism of LPS with colistin. The chemosensor, naphthaldehyde appended furfural (NAF) that selectively recognises colistin can label LPS, by showing its fluorescence signals. The computationally derived three-dimensional structure of LPS has been introduced to speculate on the possible binding mode of colistin with LPS, and this was also thoroughly studied with the help of quantum mechanics and molecular dynamics energy minimisation. Fluorescence microscopy and FE-SEM microscopic studies were also used to observe the change in the structural morphology of colistin-sensitive and resistant Salmonella typhi in different experimental conditions.
Introduction
The Gram-negative bacterial pathogens are continuously gaining antimicrobial resistance, and escape antibiosis by most of the available antibiotic regimens. Other than antibiotic therapy, there are very few treatment options which exist to combat the pathogenesis by Gram-negative organisms. Furthermore, there remain a limited number of drugs in the reserve list that may be effective against these strains of bacteria.1,2 In the recent past, carbapenems were considered to be the reliable and safe options for the treatment of severe Gram-negative multidrug-resistant bacterial infections, but pathogens such as Pseudomonas aeruginosa, Acinetobacter baumannii, and Klebsiella pneumoniae gradually developed carbapenem resistance and the scope of its treatment has depreciated systematically.3 Furthermore, colistin is a polycationic antibiotic4 that belongs to the polymyxin class and is effective in combating most multidrug-resistant Gram-negative bacterial infections5 by interacting with the membrane lipopolysaccharide (LPS).6 The LPSs have been the subject of intense study for over half a century.7–9 The LPS is the prototypical lipoglycan with an overall net negative charge and is the primary component of the outer membrane of nearly all the Gram-negative bacteria, which acts as a potential immune-stimulator inducer, and confers stability and integrity to the outer LPS membrane.10–14From the literature, the structure, function, and biogenesis15–17 reveal that LPS is composed of three subunits, namely the hydrophilic polysaccharide, the O antigen part, and the hydrophobic lipid A region. The lipid A domain is responsible for the endotoxic activity of the Gram-negative bacteria.18 The first proposition explains the plausible mode of action of colistin that crosses over the outer membrane via ‘self-promoted uptake’ mechanism.19–22 It proceeds through a two-step mechanistic pathway where the initial binding takes place and permeabilisation of the outer membrane induces the concomitant displacement of Ca2+ and Mg2+ ions from the anionic groups23–25 of LPS in a competitive manner.26 Destabilisation can manifest as transient ‘openings’ in the outer membrane that allow the passage of colistin, or it can be connected to an augmentation of the fluidity of the outer membrane. Consequently, by destabilising the cytoplasmic membrane and disrupting the outer LPS membrane, the inner cellular contents are released, and as a result, the bacteria are killed. Recent biophysical studies of outer membrane models27–30 have revealed many interesting phenomena about the colistin–LPS membrane interactions, but at the atomistic level resolution is not easily achieved.31–34
Although the LPS is the initial target, the exact mode of action of colistin remains unclear. A group of researchers believe that the LPS sugar residues are responsible for the interaction, whereas many of them think that the lipid A headgroups are responsible for the colistin–LPS interaction.35–46 One of the potent bactericidal mechanisms for colistin is thought to be inhibiting the antiendotoxin activity of the lipid A domain of LPS by interacting and neutralising the LPS molecules.47 Many previous researchers have used molecular simulation studies to reveal that there was colistin insertion into a hydrophobic membrane, and translocation of colistin through a hydrophobic membrane core.36,41,43,44,46 Although the simulation studies gave the broader picture over a relative time scale, it is very difficult to perceive an idea about the nature of the interaction from the context of a membrane system. Hence, in this work we tried to investigate the binding mechanism of colistin with LPS, using quantum mechanics by choosing isolated molecules, and using density functional theory (DFT) as a tool to generate a three-dimensional structure of LPS. In this context, some spectrophotometric and electron microscopic analyses were performed, which lead us to examine the quantum mechanics in a unidirectional way. As a continuation of our previous work where we were able to synthesise a fluorescent probe of naphthaldehyde appended furfural (NAF)48 that possesses the ability to selectively detect colistin, this probe has also been utilised for indirect labelling of LPS.
Results and discussion
Absorption and fluorescence experiments
Extensive UV-vis and fluorescence studies were performed in order to obtain a clear perspective on the possible interactions between NAF, colistin, and LPS. Fig. 1A shows a decrease in the 384 nm absorption band of NAF upon a gradual interaction with colistin. Upon sequential addition of LPS with that solution, an increase in absorbance was observed. Moreover, an increase in the intensity of absorption was witnessed at about 262 nm, which was possibly due to the intrinsic absorption band of LPS (Fig. S1, ESI†). A similar phenomenon was noted in the fluorescence titration experiment, where an initial augmentation in fluorescence intensity arose from the robust interaction between NAF and colistin. Nevertheless, with the gradual introduction of LPS into the solution, it began to engage in competitive interactions with the NAF–colistin complex. This manifested as an initial enhancement in intensity (up to the addition of 2 μl of LPS (0.066 mg ml−1)), followed by an abrupt quenching in the fluorescence intensity of NAF at 510 nm (Fig. 1D). Interestingly, no significant change was observed at the 384 nm band while the premixed colistin and LPS (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) was titrated against NAF (Fig. 1B). Although at higher concentrations of the mixture, a reasonable decrease in absorbance was noticed and a simultaneous enhancement of intensity at 262 nm was perceived. Fig. 1E shows the significant increase in fluorescence of the premixed colistin and LPS against NAF during the fluorescence titration. The enhancement of the fluorescent intensity obtained from Fig. 1E became reduced when compared to that of Fig. 1D. This phenomenon showed that the free colistin can interact with NAF more easily, whereas the pre-conjugated colistin–LPS found it difficult to interact with the NAF due to the steric hindrance. The absorbance and fluorescence titration of NAF and LPS was achieved, and is shown in Fig. 1C and E, respectively. No significant change at the 384 nm absorption band was observed. Interestingly, at the higher concentration of LPS, a decrease in absorbance of 0.13 a.u. was witnessed, whereas the substantial increase of the absorbance at about 262 nm was impressive.
:1 v/v) was titrated against NAF (Fig. 1B). Although at higher concentrations of the mixture, a reasonable decrease in absorbance was noticed and a simultaneous enhancement of intensity at 262 nm was perceived. Fig. 1E shows the significant increase in fluorescence of the premixed colistin and LPS against NAF during the fluorescence titration. The enhancement of the fluorescent intensity obtained from Fig. 1E became reduced when compared to that of Fig. 1D. This phenomenon showed that the free colistin can interact with NAF more easily, whereas the pre-conjugated colistin–LPS found it difficult to interact with the NAF due to the steric hindrance. The absorbance and fluorescence titration of NAF and LPS was achieved, and is shown in Fig. 1C and E, respectively. No significant change at the 384 nm absorption band was observed. Interestingly, at the higher concentration of LPS, a decrease in absorbance of 0.13 a.u. was witnessed, whereas the substantial increase of the absorbance at about 262 nm was impressive.
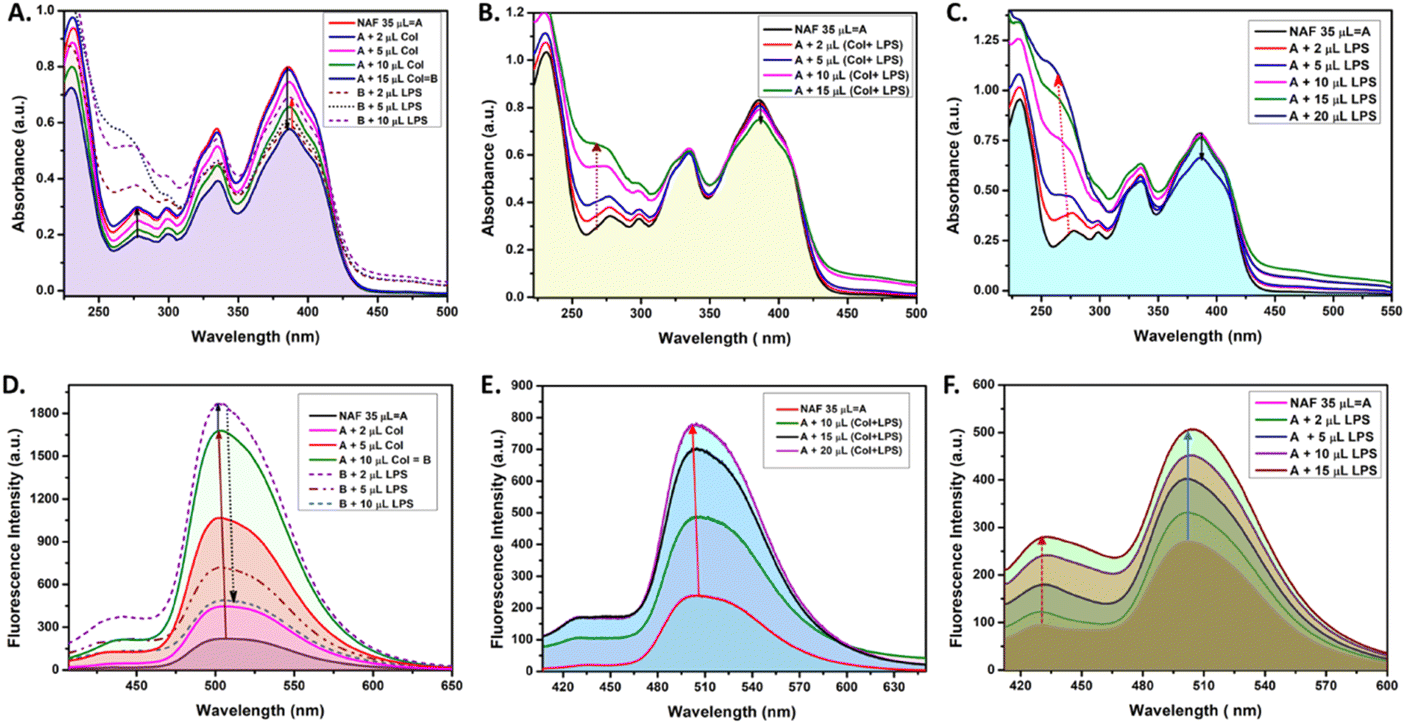 | ||
| Fig. 1 The UV-visible and fluorescence spectral studies: (A) and (D) NAF (0.017 mg mL−1 stock) with sequential addition of colistin (0.21 mg mL−1 stock) and LPS (1.718 mg mL−1), (B) and (E) NAF with simultaneous addition of colistin and LPS (1:1 v/v), and (C) and (F) NAF with LPS. | ||
After careful analysis of the data obtained from the UV-vis and fluorescent titration experiments, some important observations surfaced. The NAF can easily interact with colistin in its native state when compared to the pre-conjugated colistin–LPS. This phenomenon was observed due to the ease of interaction and as a result an increase in luminescence intensity was comparably high when colistin and LPS were added sequentially to NAF (Fig. 1A). When the pre-conjugated colistin–LPS was titrated against NAF, it encountered steric hindrance while interacting with colistin, thus resulting in an increase in fluorescence which was low when compared to that added sequentially. To some extent, NAF can directly interact with LPS because the increase in fluorescence was observed but it was difficult to conclude because of the fact that the LPS possesses an intrinsic absorption band at 262 nm which showed a remarkable change during titration with NAF, and the fluorescence intensity which increases at 450 nm plays a significant role. Another preliminary conclusion can be drawn that the binding affinity of colistin towards LPS is much higher when compared to NAF, and the ratio of possible interaction of colistin and LPS was 1:1 (Fig. 1B, E, S2 and S3, ESI†). A quantitative assessment of fluorescence signals was undertaken to elucidate the reliability and robustness of the findings (Fig. S4, ESI†).
The chemosensor NAF is capable of labelling the LPS of Gram-negative bacteria
We attempted to collect data from Gram-negative bacterial species in the test samples after archiving the positive results found in the fluorescence studies. Thus, we conducted fluorescent microscopic experiments using Salmonella typhi as our test organism. Before proceeding, we ensured that our test organism had become colistin resistant and then conducted field emission scanning electron microscopy (FE-SEM) studies. The mcr-1 and eptB genes are known to be responsible for the modifications by phosphoethanolamine and 4-amino-4-deoxy-L-arabinose cationic groups of the LPS, and that they are key factors for the development of intrinsic resistance (Fig. S5, ESI†). In addition, the mcr-1 gene protects the cytoplasmic membrane from the colistin mediated disruption.49Colistin alters cellular morphology of S. typhi
Alteration in cellular morphology of the test organism S. typhi has also been observed from the SEM micrographs when the cells are treated with colistin. The cellular morphology of the colistin treated colistin-resistant S. typhi and the untreated wild type S. typhi remained intact (Fig. 2A and C) whereas the colistin treated cells of the wild-type S. typhi exhibited an altered morphology (Fig. 2B), particularly in the cellular membrane, and it was anticipated that it was damaged severely due to the induction of crystallinity in the outer membranes.50 The unaltered morphology of the colistin treated bacteria shown in the FE-SEM micrograph ensured that the variety of S. typhi had developed colistin resistance.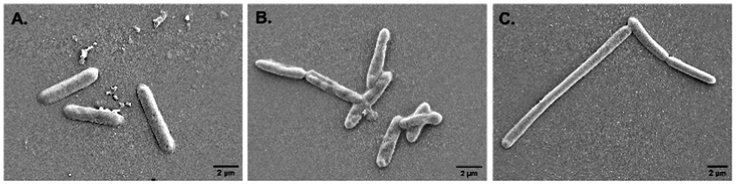 | ||
| Fig. 2 The FE-SEM micrographs of (A) wild type S. typhi, (B) colistin treated wild type S. typhi, and (C) colistin resistant S. typhi. | ||
Thus, after ensuring the species had developed colistin resistance, we conducted the fluorescence labelling experiments using a fluorescence microscope. It was evident from the cellular imaging experiments that the chemosensor NAF was readily permeable in bacterial cells. The NAF selectively recognizes colistin. The colistin-resistant S. typhi and Escherichia coli were treated with 8 μg per ml colistin followed by treatment with NAF.
It was found that the cells showed a bright fluorescence signal when treated with colistin followed by NAF (Fig. 3 and S6, ESI†). Also, the cells showed a weak fluorescence signal when the premixed colistin and NAF was used to treat the cells. However, there were no fluorescence signals found in the fluorescence microscopy when only the NAF was used to treat the cells. It was quite evident from the results of previous studies48 that NAF selectively binds with colistin. Whereas colistin has been reported to bind with the LPS of the Gram-negative bacteria.51 Thus, from the results it was inferred that in the labelled cells the colistin interacted with the impregnated LPS and the bound LPS was recognized by the NAF.
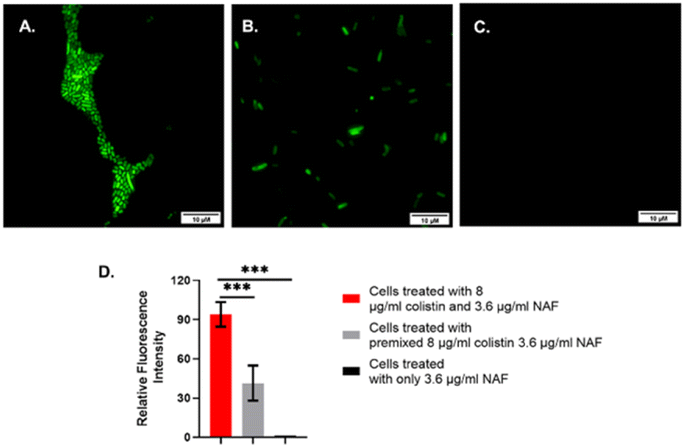 | ||
| Fig. 3 Fluorescence microscopy of colistin resistant S. typhi. The images are representative of (A) cells treated with 8 μg mL−1 colistin and 3.6 μg mL−1 NAF, (B) cells treated with premixed 8 μg mL−1 colistin and 3.6 μg mL−1 NAF, and (C) cells treated with only 3.6 μg mL−1 NAF. The fluorescent images were captured using a fluorescein isothiocyanate (FITC) filter and 100× magnification, (D) the relative fluorescence intensity of the previously mentioned images were quantified using ImageJ v1.54d software and a graph was obtained. | ||
While using colistin sensitive bacteria, in the optical density (OD) assay, both colistin and colistin + NAF exhibited the same minimal inhibitory concentration (MIC) value against S. typhi, with an MIC of 4 μg ml−1 for each. Consequently, the experimental results indicated that the NAF did not affect the antimicrobial activity of colistin against S. typhi (Fig. S7, S8 and Table S1, ESI†).
Computational studies
To identify the possible interaction mechanism, we proceeded with computational studies where the energy minimised structure of LPS was generated to understand the possible binding modes of LPS with colistin and the colistin–NAF conjugate. It was observed that the lipid chains in LPS were protruding outwards in order to avoid steric crowding and hence minimise the energy required to obtain the stable conformation of LPS. Moreover, the core oligosaccharide domain of LPS was packed more closely and several intramolecular hydrogen bonds provide substantial stability to the structure (Fig. S9, ESI†). The isosurface of the computationally predicted structure of LPS shows the grooves and possible accessible binding pockets of LPS (Fig. S10, ESI†).To avoid the complexity of the system counter stabilising cations (Mg2+ and Ca2+) have been neglected in the calculations. The truncated structure of the LPS shows that it binds colistin with anionic phosphate groups present in the lipid A domain (Fig. 4 and S11, ESI†). The potential energy diagram obtained from energy minimisation using the GROMACS MD simulation ensured that all the fragmented structures had been properly optimised (Fig. S12, ESI†).
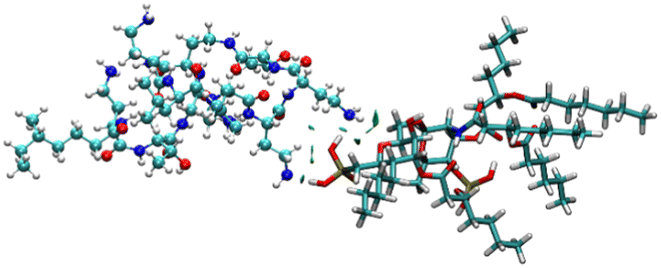 | ||
| Fig. 4 The NCI isosurface for the colistin–LPS complex obtained from the non-covalent interactions, where the RDG cut-off is 0.8, coloured over −0.1sign(λ2)ρ + 0.1 a.u. The sphere colour codes are: cyan = carbon, white = hydrogen, yellow = phosphorus, blue = nitrogen, red = oxygen. | ||
The anionic phosphates play a crucial role in the previous interaction. As the free amines of colistin are cationic in nature (protonated to increase the charge), they are easily attached to the counter anion sulfate molecule giving stability to the colistin in its native state. During the interaction with bacterial LPS, the –NH3+ groups of colistin bind with the anionic phosphates of the glucosamine disaccharide of the lipid A domain to attain a stable counter ionic conjugate system of colistin and LPS. It was observed that both NAF and LPS can interact with colistin simultaneously, because the binding site of colistin with NAF differs from that of the interaction region with LPS (Fig. 5).
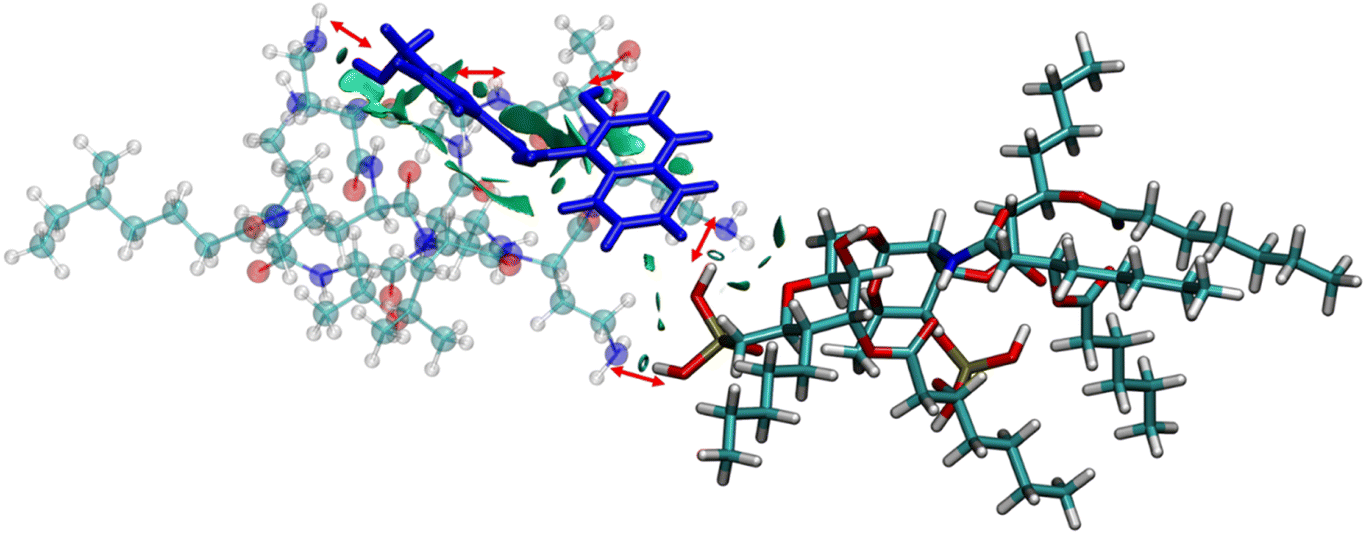 | ||
| Fig. 5 The NCI isosurface for the NAF–colistin–LPS complex obtained from the non-covalent interactions, where the RDG cut-off is 0.8, coloured over −0.1sign(λ2)ρ + 0.1 a.u. The sphere colour codes are: cyan = carbon, white = hydrogen, yellow = phosphorus, blue = nitrogen, red = oxygen. | ||
Calculating the inhomogeneous electron distribution at the interaction regions by considering the reduced density gradient (RDG) ‘s’ and the electron density ‘ρ’, it has been found that the reduced gradient was zero at the bond critical points:52
| s = [1/3√{2(3π)2}] × [|∇ρ|/ρ4/3] | (1) |
The characteristic signal peaks appear (for any weak interaction) on a ‘s’ versus ‘ρ’ plot at a low-density low-gradient region and after analysing the Laplacian or the second derivative of the density, the eqn (2) was obtained:
| ∇2ρ = λ1 + λ2 + λ3, λ1 ≤ λ2 ≤ λ3 | (2) |
After specifically identifying the major site-specific interactions, we tried to identify the nature of the attractive interactions. The NAF, colistin, and LPS were split into different fragments such as colistin–LPS, NAF–colistin, and NAF–colistin–LPS (Fig. 6).
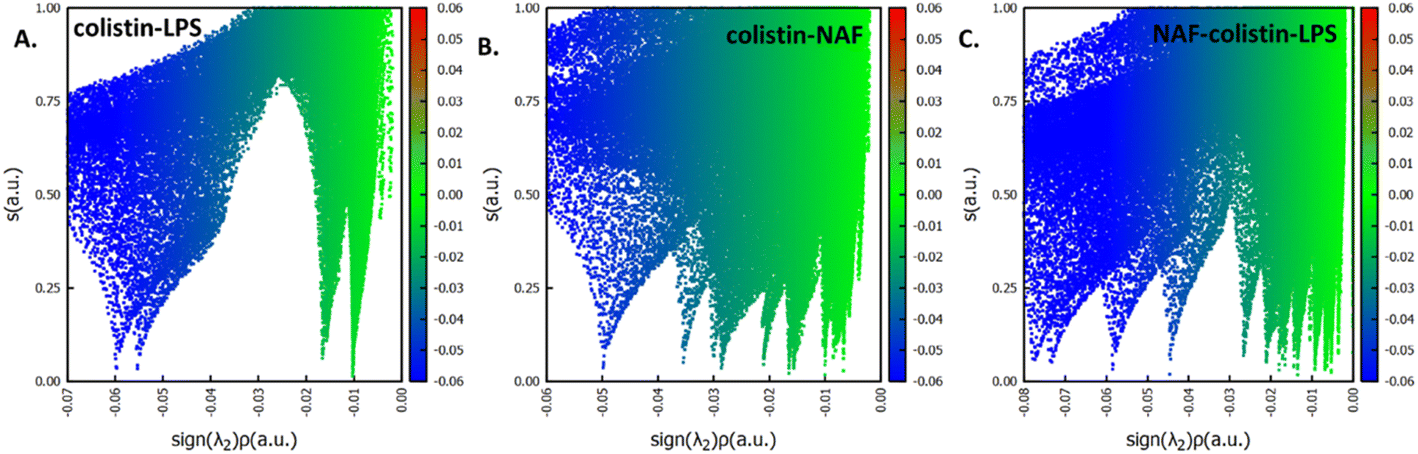 | ||
| Fig. 6 Plots of reduced density gradient (‘s’) on the ‘Y-axis’ and electron density multiplied by the sign of the second Hessian eigenvalue (sign(λ2)ρ) on the ‘X-axis’ for (A) colistin–LPS, (B) colistin–NAF, and (C) colistin–LPS–NAF. | ||
To avoid complications in the calculation, intramolecular interactions have been neglected and we focused only on attractive interaction regimens (i.e., the negative region sign(λ2)ρ axis or the low density low gradient region). The colistin–LPS conjugate (Fig. 6A) shows two signals that appeared at about −0.06 a.u., whereas the NAF–colistin fragment significantly possessed multiple interactions at −0.03, −0.04, and −0.05 a.u. (Fig. 6B). From the NAF–colistin–LPS complex, it was been seen that the colistin–LPS interactions became stronger in nature as the signals were shifted to the −0.07 and −0.08 a.u. regions. At the same time, the NAF–colistin peak shifted from −0.03 to −0.04 a.u. due to its increased attractive interaction (Fig. 6C). Shifting of the peaks that had appeared towards the more negative region of the sign(λ2)ρ axis clearly indicated that the NCIs present in the system had become stronger upon complexation. The natural bond orbital analysis (NBO) and electrostatic potential charge analysis (ESP) ensured that the oxygen atoms O1 (−0.890 MK, −1.292 NBO) and O2 (−0.908 MK, −1.299 NBO) of the phosphate were more negatively charged than the N1 (−0.709 MK, −0.787 NBO) and N2 (−0.629 MK, −0.776 NBO) (Fig. S13 and S14, ESI†). It should be noted that the –NH3+ groups of colistin give two of their protons to anionic LPS for the neutralisation of the LPS fragment of the complex. The most fascinating fact is that the colistin binds with LPS through an electrostatic interaction, but the binding gets stronger and the whole system becomes stabilised through the NCIs. The colistin and NAF interaction is purely non-covalent hydrogen bonding in nature.
Experimental section
We have classified the Experimental section into three major parts: the UV and fluorescent experiments, the computational calculations, and the microbiological experiments.Materials and methods
All the reagents were purchased from commercial suppliers and were used without further purification. Colistin sulfate was purchased from SRL Chemicals, and LPS from Salmonella typhosa (Sigma L6386) was obtained from Sigma-Aldrich. The NAF probe (Fig. 7) was synthesised using our previously reported protocol.48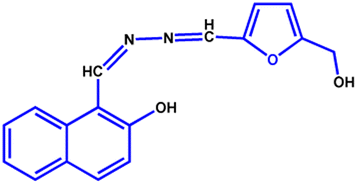 | ||
| Fig. 7 Structure of the NAF probe. | ||
UV-vis and fluorescence experiments
The fluorescence spectra were recorded on a Hitachi F-7100 spectrophotometer. The absorption spectra were recorded on a Shimadzu UV-3101PC spectrophotometer. A stock solution of NAF (0.017 mg ml−1) was prepared in acetonitrile–water. Colistin stock solutions (0.21 mg ml−1) and LPS stock solutions (1.718 mg ml−1) of different concentrations were prepared in Millipore water. The experiments were carried out in acetonitrile:water (2:5, v/v), buffered with 1 mM phosphate buffer (pH 7.0). During the titration, each time 35 μl of a 0.017 mg per ml solution of NAF was placed in a Starna quartz optical cell (sub-micro) of 3 mm optical path length, z = 15 mm, and then colistin and LPS solutions were added incrementally into the quartz optical cell using a micropipette. For a simultaneous discharge, premixed LPS–colistin solution was gradually added and titrated against NAF. The spectral data were recorded immediately after the addition of the colistin–LPS solution.
Computational calculations
The initial three-dimensional structure was generated using Molecular Mechanics 2 (Allinger's MM2 Force Field).54 After obtaining the energy-minimised geometry up to a significantly considerable point, further quantum mechanical calculations were performed. For the quantum mechanics the Gaussian 16 package55 was utilised. The LPS structure was energetically minimised using the Becke-3-parameter-Lee–Yang–Parr (B3LYP), exchange–correlation functional. Incorporating a hybrid DFT model where Hartree–Fock exchange, local exchange, gradient exchange correction, local correlation, and gradient correlation corrections were included in the calculations, the chosen basis set was 6-31G for energy optimisation of the LPS structure. After obtaining the three-dimensional conformational structure in order to perform calculations based on quantum mechanics parameters, a truncated sub-structure of the lipid A region was generated. The most probable colistin binding possibilities were also estimated using the B3LYP/6-31G(d,p) level of theory incorporating the conductor like polarisable continuum model (CPCM) solvation model and water as a solvent. The possibility of interactions among NAF, colistin, and LPS were also studied. Structural modifications of LPS due to the development of colistin resistance were also studied to some extent. Using NCIPLOT53,56 and NBO, the pure hydrogen bonding interactions and electrostatic interactions were differentiated. After the static calculations, the complex structures have been involved in molecular dynamics (MD) simulations using GROMACS57 software package. All the molecules were parametrised using Automated Topology Builder (ATB) 3.0 (ref. 58) web-server incorporating the GROMOS 54A7 force field. All the necessary steps were performed and a SPC water explicit solvent model was incorporated for solvation. The energy minimisation was performed and the results have been incorporated in this paper.Bacterial cell culture
The bacterial strains (S. typhi, colistin resistant S. typhi, E. coli MTCC 1687) were grown in Luria–Bertani (LB) medium. The culture was grown overnight at 37 °C inside a culture incubator.Development of colistin resistant S. typhi
In order to develop a resistant variety of S. typhi, the wild type of bacterial strain was grown in Luria–Bertani (LB) broth supplemented with 2 μg per ml colistin and incubated overnight at 37 °C with shaking at 180 rpm. This culture was used as inoculum for further growth of the bacterial cells on an LB plate supplemented with 10 μg per ml colistin. The colonies that appear on the colistin containing plate were treated as colistin resistant S. typhi.Sample preparation for FE-SEM micrographs
To investigate the alteration of the cellular morphology of the test organisms: S. typhi, colistin resistant S. typhi, and E. coli MTCC 1687 after treatment with 4 μg per ml (MIC) colistin followed by incubation at 37 °C for 24 h with shaking (180 rpm). Both the colistin treated and untreated samples were subjected to centrifugation and fixed by 2.5% glutaraldehyde solution in phosphate buffer for 30 min. The fixed cells were again centrifuged and washed three times with 0.1 M phosphate buffer (pH 7.2). The samples were placed under the microscope and the images were captured. The FE-SEM microscopy [Zeiss-Gemini 450 (Gemini 2)] was performed according to the standard protocol.59Fluorescent microscopic studies
Colistin resistant S. typhi cells were treated with 8 μg per ml colistin for 20 min followed by washing with 1× phosphate buffered saline (1× PBS). The colistin treated cells were further treated with 3.6 μg per ml NAF for 20 min and washed twice with 1× PBS and then air dried for observation under an Olympus IX73 fluorescence microscope using the FITC filter with excitation at 384 nm and emission at 510 nm, and with 100× magnification. A parallel control set was also prepared where the cells were treated with only 3.6 μg per ml NAF for 20 min followed by washing with 1× PBS and the same further microscopic analyses were carried out.Determination of the minimal inhibitory concentration (MIC)
The MIC was determined in a 96-well microplate using the microdilution method in a Mueller–Hinton broth (HiMedia) medium. The S. typhi (5 × 105 CFU ml−1) was added to the broth containing different concentrations of colistin and colistin + NAF, and then incubated overnight at 37 °C. Next, the OD was measured using a microplate reader at 600 nm. The MIC was determined as the lowest concentration of antibiotic capable of inhibiting the growth of bacteria.Statistical analysis
Conclusions
This study focuses on unravelling the interactions of colistin with the bacterial endotoxin, LPS, in the context of the luminescence property of the chemosensor NAF. Results from previous studies concluded that the binding affinity of colistin towards LPS was greatly compromised when using fluorescent-tagged colistin. As far as colistin is concerned, its antimicrobial efficacy is directly correlated with the binding affinity towards LPS. The major advantage of utilising NAF as a non-covalently bound chemosensor-based marker over a covalently integrated colistin-tagged fluorescent molecule is that it does not influence the binding affinity of colistin towards LPS, because the interaction regions are completely different. The limitation of non-covalently tagged NAF–colistin conjugate, when applied in living bacterial colonies, is that there is a chance for certain NAF–colistin conjugates to break apart before reaching the final target LPS. This happens because the colistin possesses the freedom to overcome the binding affinity of NAF while binding with LPS. However, from the aspect of computational calculations, modelling of the whole system gives a broader idea of the relative time scale, but it is very difficult to perceive an idea about the nature of the interaction from the context of a membrane system as a whole or an ensemble of multimolecular systems. While performing the quantum mechanical investigation, the momentary interaction and stabilisation could be well understood by considering an isolated molecule of LPS and colistin in better resolution. As the primary target locus of colistin is the lipid A region, we have focused on studying interactions in a very specific manner. The truncated structure of the LPS shows that it binds colistin with anionic phosphate groups present in the lipid A domain. The anionic phosphates play a crucial role in the previous interaction. The NBO analysis and NCI-plot analysis give insights into the electrostatic neutralisation and stabilisation by the non-covalent attachment between colistin and LPS. Previous MD studies have shown that there is a chance of colistin aggregation, and the possible interaction ratio differs in each experiment. Results from the experimental investigations, as well as computational calculations from our study, helped us to conclude that the colistin and LPS interaction is 1:1 in isolated states. The studies also indicate that NAF may act as a suitable probe to quantify the colistin–LPS affinity. A better understanding of the specific interactions provides useful insight into the re-purposing of existing antibiotics and their precise mechanism of action in order to develop new generation, anti-microbial agents.
Author contributions
SM performed the fluorescence and absorption experiments including computational calculations, and analysed the data. The manuscript has been written by SM with contributions from all authors. DP performed all the biological experiments. SuM interpreted the data of all the biological experiments and edited the manuscript. GKD helped to interpret the computational data. PDS conceptualised the research project, wrote and edited the manuscript. All the authors have given their approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
PS acknowledges the Department of Chemistry, Visva Bharati University for use of the infrastructural facilities. SM acknowledges UGC, India for their Fellowship, and DST-PURSE (Grant No. SR/PURSE Phase2/42 (G & C)) for the FE-SEM facility. GKD acknowledges use of the PARAM Shakti server under the National Supercomputing Mission, IITKGP, GOI. All the authors thank the Department of Chemistry, Visva Bharati, and the Department of Microbiology, University of Calcutta for extending the use of their infrastructural facilities.References
- W. H. Organization, Antimicrobial Resistance—Global Report on Surveillance, 2014, vol. 2014 Search PubMed.
- Review on Antimicrobial Resistance, Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations, 2014 Search PubMed.
- N. Gupta, B. M. Limbago, J. B. Patel and A. J. Kallen, Clin. Infect. Dis., 2011, 53, 60–67 CrossRef PubMed
.
- S. Biswas, J. M. Brunel, J. C. Dubus, M. Reynaud-Gaubert and J. M. Rolain, Expert Rev. Anti Infect. Ther., 2012, 10, 917–934 CrossRef CAS PubMed
- M. E. Falagas and S. K. Kasiakou, Clin. Infect. Dis., 2005, 40, 1333–1341 CrossRef CAS PubMed
- J. Li, R. L. Nation, J. D. Turnidge, R. W. Milne, K. Coulthard, C. R. Rayner and D. L. Paterson, Lancet Infect. Dis., 2006, 6, 589–601 CrossRef CAS PubMed
- I. L. Bennete and L. E. Cluff, Pharmacol. Rev., 1957, 9, 427–475 Search PubMed
- The History of Pyrogen Research, ed. O. Westphal, U. Westphal and T. Sommer, American Society for Microbiology, Washington, DC, 1977 Search PubMed
- B. Beutler and E. T. Rietschel, Nat. Rev. Immunol., 2003, 3, 169–176 CrossRef CAS PubMed
- B. Bertani and N. Ruiz, EcoSal Plus, 2018, 8 DOI:10.1128/ecosalplus.ESP-0001-2018
- A. L. Olins and R. C. Warner, J. Biol. Chem., 1967, 242, 4994–5001 CrossRef CAS PubMed
- A. B. Schromm, K. Brandenburg, H. Loppnow, U. Zähringer, E. T. Rietschel, S. F. Carroll, M. H. J. Koch, S. Kusumoto and U. Seydel, J. Immunol., 1998, 161, 5464–5471 CrossRef CAS
- R. E. Bishop, Contrib. Microbiol., 2005, 12, 1–27 CAS
- C. Alexander and E. T. Rietschel, J. Endotoxin Res., 2001, 7, 167–202 CAS
- F. Di Lorenzo, K. A. Duda, R. Lanzetta, A. Silipo, C. De Castro and A. Molinaro, Chem. Rev., 2022, 122, 15767–15821 CrossRef CAS PubMed
- M. Caroff and A. Novikov, OCL, 2020, 27, 31–40 CrossRef CAS
- Bacterial Lipopolysaccharides, Structure, Chemical Synthesis, Biogenesis and Interaction with Host Cells, ed. Y. A. Knirel and M. A. Valvano, Springer, Vienna, 1st edn, 2011 Search PubMed
- F. F. Andrade, D. Silva, A. Rodrigues and C. Pina-Vaz, Microorganisms, 2020, 8(11), 1716–1727 CrossRef CAS PubMed
- R. E. W. Hancock, Annu. Rev. Microbiol., 1984, 38, 237–264 CrossRef CAS PubMed
- R. E. W. Hancock and A. Bell, Eur. J. Clin. Microbiol. Infect. Dis., 1988, 7, 713–720 CrossRef CAS PubMed
- R. E. W. Hancock, Lancet, 1997, 349, 418–422 CrossRef CAS PubMed
- L. Zhang, P. Dhillon, H. Yan, S. Farmer and R. E. W. Hancock, Antimicrob. Agents Chemother., 2000, 44, 3317–3321 CrossRef CAS PubMed
- R. A. Dixon and I. Chopra, Antimicrob. Agents Chemother., 1986, 29, 781–788 CrossRef CAS PubMed
- M. Teuber and J. Bader, Arch. Microbiol., 1976, 109, 51–58 CrossRef CAS PubMed
- A. Wiese, M. Münstermann, T. Gutsmann, B. Lindner, K. Kawahara, U. Zähringer and U. Seydel, J. Membr. Biol., 1998, 162, 127–138 CrossRef CAS PubMed
- A. Z. Bialvaei and H. S. Kafil, Curr. Med. Res. Opin., 2015, 31, 707–721 CrossRef CAS PubMed
- F. G. Dupuy, I. Pagano, K. Andenoro, M. F. Peralta, Y. Elhady, F. Heinrich and S. Tristram-Nagle, Biophys. J., 2018, 114, 919–928 CrossRef CAS PubMed
- M.-L. Han, T. Velkov, Y. Zhu, K. D. Roberts, A. P. Le Brun, S. H. Chow, A. D. Gutu, S. M. Moskowitz, H.-H. Shen and J. Li, ACS Chem. Biol., 2018, 13, 121–130 CrossRef CAS PubMed
- N. Paracini, L. A. Clifton, M. W. A. Skoda and J. H. Lakey, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E7587–E7594 CrossRef CAS PubMed
- Y. J. Oh, B. Plochberger, M. Rechberger and P. Hinterdorfer, J. Mol. Recognit., 2017, 30, e2605 CrossRef PubMed
- Z. Z. Deris, J. D. Swarbrick, K. D. Roberts, M. A. K. Azad, J. Akter, A. S. Horne, R. L. Nation, K. L. Rogers, P. E. Thompson, T. Velkov and J. Li, Bioconjugate Chem., 2014, 25, 750–760 CrossRef CAS PubMed
- J. Salazar, M. Alarcón, J. Huerta, B. Navarro and D. Aguayo, Arch. Biochem. Biophys., 2017, 620, 28–34 CrossRef CAS PubMed
- M. P. Ryder, X. Wu, G. R. McKelvey, J. McGuire and K. F. Schilke, Colloids Surf., B, 2014, 120, 81–87 CrossRef CAS PubMed
- S. Halder, K. K. Yadav, R. Sarkar, S. Mukherjee, P. Saha, S. Haldar, S. Karmakar and T. Sen, SpringerPlus, 2015, 4, 672 CrossRef PubMed
- N. A. Berglund, T. J. Piggot, D. Jefferies, R. B. Sessions, P. J. Bond and S. Khalid, PLoS Comput. Biol., 2015, 11, e1004180 CrossRef PubMed
- D. Jefferies, P.-C. Hsu and S. Khalid, Biochemistry, 2017, 56, 1672–1679 CrossRef CAS PubMed
- L. Fu, M. Wan, S. Zhang, L. Gao and W. Fang, Biophys. J., 2020, 118, 138–150 CrossRef CAS PubMed
- G. M. Ongwae, K. R. Morrison, R. A. Allen, S. Kim, W. Im, W. M. Wuest and M. M. Pires, ACS Infect. Dis., 2020, 6, 1427–1435 CrossRef CAS PubMed
- M. S. Feigman, S. Kim, S. E. Pidgeon, Y. Yu, G. M. Ongwae, D. S. Patel, S. Regen, W. Im and M. M. Pires, Cell Chem. Biol., 2018, 25, 1185–1194 CrossRef CAS PubMed
- D. E. S. Santos, L. Pol-Fachin, R. D. Lins and T. A. Soares, J. Chem. Inf. Model., 2017, 57, 2181–2193 CrossRef CAS PubMed
- X. Jiang, N. A. Patil, M. A. K. Azad, H. Wickremasinghe, H. Yu, J. Zhao, X. Zhang, M. Li, B. Gong, L. Wan, W. Ma, P. E. Thompson, K. Yang, B. Yuan, F. Schreiber, L. Wang, T. Velkov, K. D. Roberts and J. Li, Chem. Sci., 2021, 12, 12211–12220 RSC
- J. Li, R. Beuerman and C. S. Verma, Biochim. Biophys. Acta, Biomembr., 2020, 1862, 183297 CrossRef CAS PubMed
- X. Jiang, Y. Sun, K. Yang, B. Yuan, T. Velkov, L. Wang and J. Li, Comput. Struct. Biotechnol. J., 2021, 19, 3885–3891 CrossRef CAS PubMed
- X. Jiang, K. Yang, B. Yuan, M. Han, Y. Zhu, K. D. Roberts, N. A. Patil, J. Li, B. Gong, R. E. W. Hancock, T. Velkov, F. Schreiber, L. Wang and J. Li, J. Antimicrob. Chemother., 2020, 75, 3534–3543 CrossRef CAS PubMed
- X. Jiang, K. Yang, M.-L. Han, B. Yuan, J. Li, B. Gong, T. Velkov, F. Schreiber, L. Wang and J. Li, ACS Infect. Dis., 2020, 6, 2698–2708 CrossRef CAS PubMed
- X. Jiang, K. Yang, B. Yuan, B. Gong, L. Wan, N. A. Patil, J. D. Swarbrick, K. D. Roberts, F. Schreiber, L. Wang, T. Velkov and J. Li, J. Biol. Chem., 2020, 295, 15902–15912 CrossRef CAS PubMed
- N. S. Ly, J. Yang, J. B. Bulitta and B. T. Tsuji, Antimicrob. Agents Chemother., 2012, 56, 3453–3456 CrossRef CAS PubMed
- S. Mandal, A. Dey Bhowmik, A. Mukhuty, S. Kundu, K. N. Truong, K. Rissanen, A. Chattopadhyay and P. Sahoo, Sci. Rep., 2022, 12, 9307 CrossRef CAS PubMed
- A. B. Janssen and W. van Schaik, PLoS Genet., 2021, 17, e1009262 CrossRef CAS PubMed
- S. Manioglu, S. M. Modaresi, N. Ritzmann, J. Thoma, S. A. Overall, A. Harms, G. Upert, A. Luther, A. B. Barnes, D. Obrecht, D. J. Müller and S. Hiller, Nat. Commun., 2022, 13, 6195 CrossRef CAS PubMed
- A. Sabnis, K. L. H. Hagart, A. Klöckner, M. Becce, L. E. Evans, R. C. D. Furniss, D. A. I. Mavridou, R. Murphy, M. M. Stevens, J. C. Davies, G. J. Larrouy-Maumus, T. B. Clarke and A. M. Edwards, eLife, 2021, 10, e65836 CrossRef CAS PubMed
- P. Bandyopadhyay, S. Ray and M. M. Seikh, Phys. Chem. Chem. Phys., 2019, 21, 26580–26590 RSC
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed
- N. L. Allinger, J. Am. Chem. Soc., 1977, 99, 8127–8134 CrossRef CAS
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed
- M. Abraham, A. Alekseenko, C. Bergh, C. Blau, E. Briand, M. Doijade, S. Fleischmann, V. Gapsys, G. Garg, S. Gorelov, G. Gouaillardet, A. Gray, M. E. Irrgang, F. Jalalypour, J. Jordan, C. Junghans, P. Kanduri, S. Keller, C. Kutzner, J. A. Lemkul, M. Lundborg, P. Merz, V. Miletić, D. Morozov, S. Páll, R. Schulz, M. Shirts, A. Shvetsov, B. Soproni, D. v. d. Spoel, P. Turner, C. Uphoff, A. Villa, S. Wingbermühle, A. Zhmurov, P. Bauer, B. Hess and E. Lindahl, GROMACS - 2023.3., DOI:10.5281/zenodo.7852189
- A. K. Malde, L. Zuo, M. Breeze, M. Stroet, D. Poger, P. C. Nair, C. Oostenbrink and A. E. Mark, J. Chem. Theory Comput., 2011, 7, 4026–4037 CrossRef CAS PubMed
- P. Das, S. Kundu, P. K. Maiti, S. Mandal, P. Sahoo and S. Mandal, Sci. Rep., 2022, 12, 10176 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra07107c |
| This journal is © The Royal Society of Chemistry 2024 |