
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnusual reactivity of 2,2-diphenyl-1-picrylhydrazyl (DPPH) with Fe3+ controlled by competing reactions†
Arlette Danelle Djitieu Deutchouaab,
Yannick Ngueumaleua,
Rossel Wendji Liendjia,
Sarrah Sonita Poungoue Hangaa,
Bruno Boniface Ngueloa,
Gustave Kenne Dedzo *a and
Emmanuel Ngamenia
*a and
Emmanuel Ngamenia
aLaboratory of Analytical Chemistry, Faculty of Sciences, University of Yaoundé I, B. P. 812, Yaoundé, Cameroon. E-mail: kennegusto@yahoo.fr
bDepartment of Processing and Quality Control of Aquatic Products, Institute of Fisheries and Aquatic Sciences at Yabassi, University of Douala, B. P. 7236, Douala, Cameroon
First published on 3rd January 2024
Abstract
2,2-Diphenyl-1-picrylhydrazyl (DPPH) is a stable organic free radical widely used in various fields as a model free radical. There is scarce information about the stability of this compound in the chemical environments in which it is used. Side reactions between DPPH and other species can alter the precision of experiments that use DPPH, such as the evaluation of antioxidant properties amongst others. Following recent investigations highlighting reactions between DPPH and metal cations or Lewis acids, a quantitative reaction between DPPH and Fe3+ in acetonitrile was studied in the present work. UV-Vis spectroscopy and electrochemistry were used to monitor the reaction. The results obtained indicate a fast and multistep reaction between Fe3+ and DPPH that can be simplified as a simple redox reaction with the formation of Fe2+ and DPPH+. The reaction mechanism proposed follows complex steps involving two competing phenomena: a disproportionation which accelerates the reaction and an oxidation process that slows it down through DPPH regeneration.
Introduction
2,2-Diphenyl-1-picrylhydrazyl (DPPH) is an organic compound that has an unpaired electron which is stabilized by the particular chemical structure of the molecule.1 The exceptional stability of this radical compound explains its use in many fields that require stable free radicals (standards for use in electron paramagnetic resonance spectroscopy, inhibition of the proliferation of free radicals in certain environments, etc.).1–5 From the point of view of a fundamental understanding, the study of this compound allows better knowledge of the reactivity of free radicals, recognized for their crucial role in the biochemistry of living organisms and the occurrence and understanding of certain diseases such as cancer.6–8 Meanwhile, the use of DPPH as a model free radical for the evaluation of the antioxidant properties of compounds is undoubtedly the most common application. Among the variety of existing methods, the DPPH spectrophotometric test is indeed the most widely used because of its simplicity and ease of implementation.1,9,10 This test consists of monitoring the decrease of the absorption intensity of the DPPH free radical in the presence of the antioxidant being tested. Despite the appreciable accuracy of this test, studies recently performed on DPPH stability reveal that this radical is capable of reacting under certain conditions, even in the absence of antioxidants. This is the case of its reactivity in the presence of oxygen or sunlight.11 However, these side reactions remain slow enough to be neglected during the spectrometric DPPH assay, characterized as it is by short analysis times (less than one hour of reaction between the antioxidant and the DPPH is usually necessary). Other works have focused on the reactivity of DPPH in the presence of several non-antioxidant chemical species. In acetonitrile for example, the Cu2+ ion was reactive enough to oxidize DPPH.12 Moreover, in the same solvent, Lewis acids such as protons or Sc3+ promotes DPPH disproportionation.12,13 Considering the reactivity of DPPH in the presence of Cu2+ and acids, it has been clearly demonstrated that in acetonitrile, these compounds strongly interfere with the evaluation of antioxidant properties.12 These investigations were extended in the present work, by studying DPPH reactivity in acetonitrile, in the presence of a compound combining both oxidizing and Lewis acid properties. The Fe3+ cation represents a good candidate for this purpose because of its acidity and oxidizing properties.14,15 In this work, the reactivity of DPPH in acetonitrile in the presence of a controlled amount of Fe3+ was studied. The reaction was monitored by UV-Vis spectroscopy and by electrochemistry at stationary and rotating glassy carbon electrodes. The present work is expected to shed more light on free radical reactivity in the presence of metal cations and also to evaluate the interference of these ubiquitous chemical species in natural living organisms when determining antioxidant properties.Materials and methods
Chemicals
2,2-Diphenyl-1-picrylhydrazyl (DPPH) (99%), tetrabutylammoniumhexafluoro phosphate (TBAHFP) (98%), copper perchlorate (Cu(ClO4)2·6H2O) (98%) and iron perchlorate (Fe(ClO4)3·xH2O) (98%) were purchased from Sigma-Aldrich. Acetonitrile (99.9%) was obtained from Prolabo.Electrochemical experiments
The electrochemical monitoring of DPPH reactivity in the presence of Fe3+ was performed in a standard three electrode electrochemical cell, with 0.03 M TBAHFP in acetonitrile as the electrolytic solution. A glassy carbon electrode (3 mm of diameter) was used as the working electrode. Prior to any set of experiments, this electrode was rinsed successively with acetone and deionized water before being polished with a soft pad using an aqueous suspension of alumina (particle diameter 0.05 μm). The electrode was then rinsed thoroughly with distilled water, dried in the open air, and directly used for electrochemical experiments performed at room temperature (25 °C) under atmospheric pressure. A platinum plate (1 cm2) was used as the counter electrode and a homemade silver wire covered by a porous film of silver chloride as the pseudo-reference electrode. The potentials reported in this work using this electrode were referenced to the ferrocene signal (anodic peak at 371 mV and cathodic peak at 291 mV) recorded in similar experimental conditions. These electrodes were connected to a μ-Autolab potentiostat (Metrohm) controlled by the GPES (General Purpose Electrochemical System) software.For a typical electrochemical experiment, the initial concentration of DPPH in the electrolytic solution was set at 0.1 mM and controlled amounts of Fe3+ were added to the solution using stock solutions prepared in acetonitrile. The signals were recorded at the stationary electrode by cyclic voltammetry at 50 mV s−1 or by linear sweep voltammetry at 50 mV s−1 on a rotating electrode (900 rpm).
UV-Visible spectroscopy monitoring of DPPH stability
For the UV-Vis experiments, 2 mL of 0.1 mM DPPH prepared in acetonitrile was introduced into a 1 cm × 1 cm quartz cuvette. For rigorous comparison with the results obtained in the electrochemistry experiments, this solution also contained 0.03 M TBAHFP. The UV-Vis spectrum of this solution was recorded in the spectral range 280 nm to 900 nm using a GENESYS 10S UV-Vis (ThermoScientific) spectrophotometer. Controlled amounts of Fe3+ were added to the solution, homogenized with a glass rod for about 2 min and the UV-Vis spectrum recorded in the same wavelength range (280 nm to 900 nm).Determination of the theoretical residual DPPH percentages in solution as a function of the concentration of Fe3+ added
The theoretical percentage of DPPH in solution was determined using eqn (1).
 | (1) |
Results and discussion
The DPPH UV-Vis spectra recorded in the presence of increasing amounts of Fe3+ (Fig. 1) displayed a progressive decrease in the intensities of the bands characteristic of DPPH (at 519 nm and 328 nm). Meanwhile, a new absorption band at 384 nm assigned to DPPH+ formed. At an equal concentration of DPPH and Fe(ClO4)3, only the band at 384 nm remained present.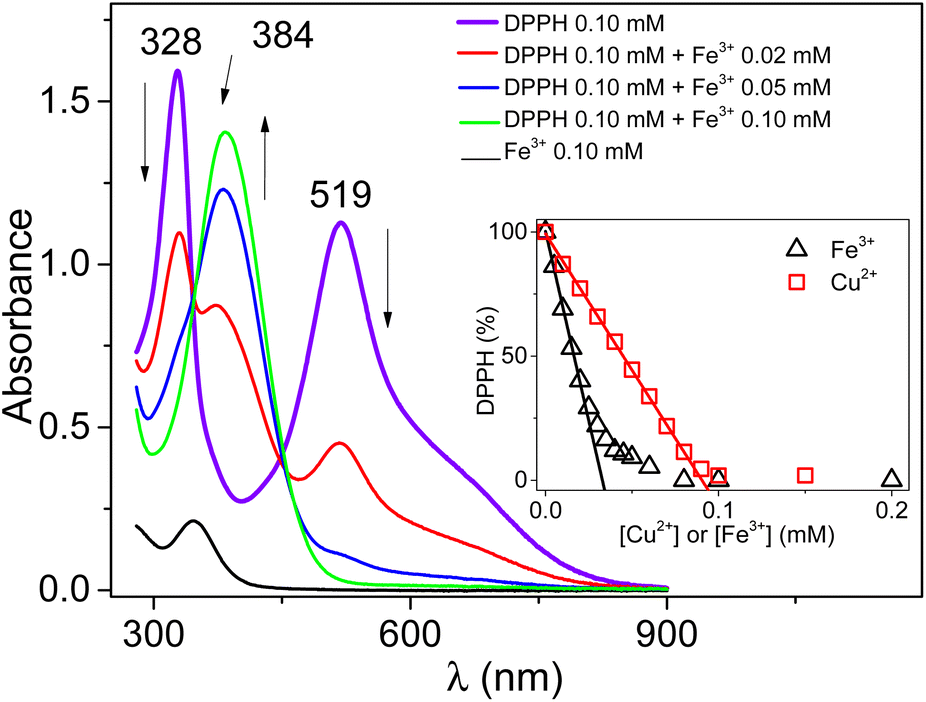 | ||
| Fig. 1 UV-Vis spectra of 0.1 mM DPPH in 0.03 M TBAHFP acetonitrile solution, in the presence of controlled amounts of Fe(ClO4)3 (0 mM, 0.02 mM, 0.05 mM, and 0.10 mM). | ||
This result indicates a quantitative chemical reaction between Fe3+ and DPPH in acetonitrile. A similar result was recently obtained while reacting DPPH with Cu2+.12 Fig. S1† presents a series of UV-Vis spectra of DPPH recorded in the presence of increasing amounts of Cu2+ and demonstrates that Cu2+ can oxidize DPPH to yield DPPH+ with subsequent formation of Cu+. It thus seems that Fe3+ reacts similarly with DPPH in acetonitrile. However, the plot of the residual percentage of DPPH (determined from the intensity of the band at 519 nm) as a function of the amount of Fe3+ added followed a different trend from the linear one obtained with Cu2+ (see the inset of Fig. 1).
For Fe3+, a fast and linear decrease in the concentration range 0 mM to 0.02 mM was followed by a gradual less important decrease of the DPPH percentage at higher concentrations of Fe3+. The slope of the linear section (−3051 ± 77% mM−1) was approximately 3 times greater than that obtained in the presence of Cu2+ (−1097 ± 9% mM−1). This indicates that the reactivity of DPPH in acetonitrile in the presence of Cu2+ or Fe3+ does not follow the same mechanism.
The electrochemical behavior of DPPH in the presence of Fe3+ was studied in order to better understand the mechanism involved during the reaction between these two chemical species. The electrochemical signal of DPPH in acetonitrile, obtained using cyclic voltammetry at a stationary electrode, is characterized by two fast and reversible monoelectronic systems (Fig. 2). These systems were assigned to the reversible reduction of DPPH to DPPH− (EPC = 0.017 V and EPA = 0.095 V) and the reversible oxidation of DPPH to DPPH+ (EPA = 0.640 V and EPC = 0.560 V).16–18
 | ||
| Fig. 2 Electrochemical signals of 0.1 mM DPPH in 0.03 M TBAHFP acetonitrile solution recorded using a glassy carbon electrode at 50 mV s−1. Cyclic voltammogram at stationary electrode (black curve) and linear sweep voltammogram at rotating (900 rpm) electrode (red curve). | ||
Fig. 2 also depicts the DPPH signal at a rotating electrode, showing two well-defined waves (oxidation at E1/2 = 0.60 V and reduction at E1/2 = 0.05 V) confirming the oxidizing and reducing properties of the DPPH radical.
The signals recorded on a rotating glassy carbon electrode in the presence of controlled amounts of Fe3+ in 0.1 mM DPPH solution (Fig. 3) reveal several findings.
 | ||
| Fig. 3 Electrochemical signals at equilibrium recorded by linear sweep voltammetry at rotating glassy carbon electrode (900 rpm) at 50 mV s−1, of DPPH 0.1 mM in TBAHFP 0.03 M acetonitrile solution, in presence of controlled amounts of Fe(ClO4)3 (0 mM, 0.02 mM, 0.05 mM, and 0.10 mM). | ||
Firstly, the presence of Fe3+ was followed by a significant decrease of the reduction current (at EC1/2 = 0.05 V) and the DPPH oxidation current (at EA1/2 = 0.60 V). The formation of a DPPH+ reduction wave (at EC1/2 = 0.56 V) that increases in intensity with the concentration of Fe3+ was also observed.
Secondly, the presence of an oxidation wave at EA1/2 = 0.95 V that increases in intensity with the concentration of the metal cation was seen. This signal was similar to the oxidation wave of reduced DPPH (DPPH-H).12,16
Thirdly, a new reduction wave at 0.2 V with an intensity that increased with the concentration of Fe3+ added to the solution was observed. A similar signal was recently observed in acetonitrile during the electrochemical reduction of DPPH in the presence of Zn2+. This signal was assigned to the electrochemical formation of the compound Zn(DPPH)2.12
The cyclic voltammograms recorded under identical experimental conditions, but at a stationary electrode (see ESI, Fig. S2†), confirmed these observations.
It thus appears that the presence of Fe3+ in a DPPH solution results in the formation of DPPH+ and a new reducing compound analogous to DPPH-H. A similar result was reported by Nakanishi et al. (2014) during the reaction between DPPH and Sc3+ (disproportionation of DPPH to yield DPPH+ and Sc(DPPH)2+).13 More recently, the disproportionation of DPPH in acetonitrile in the presence of protons (to yield DPPH+ and DPPH-H) was also reported.12 Thus, the acidity of Fe3+ promotes the disproportionation of DPPH with the formation of DPPH+ and DPPH−. DPPH−, being a Lewis base, would react with Fe3+ to form a compound with structure [Fe(DPPH)n]3−n, analogous to Sc(DPPH)2+, Zn(DPPH)2 or DPPH-H according to eqn (2).
| Fe3+ + nDPPH− → [Fe(DPPH)n]3−n | (2) |
Thus, DPPH disproportionation is expected to occur in the presence of Fe3+ following eqn (3).
| Fe3+ + 2nDPPH → nDPPH+ + [Fe(DPPH)n]3−n | (3) |
However, the disproportionation reaction proposed in eqn (3) does not take into account the experimental data reported (variation of the absorbance at 519 nm as a function of the concentration of Fe3+ added to the solution in the inset of Fig. 1). According to eqn (3), a linear decrease was expected with a slope depending on the stoichiometry of the reaction (i.e. the experimental value of n). Moreover, considering that the value of n should be greater than or equal to 1, the DPPH percentage should be zero for n = 1, when [Fe3+] = 1/2 [DPPH], as was recently observed in acetonitrile, in the presence of HClO4.12 The experimental data show (inset of Fig. 1) that the DPPH percentage decay was not linear and the consumption of the radical compound was complete when [Fe3+] = [DPPH], suggesting an equimolar reaction. Consequently, the reaction between DPPH and Fe3+ certainly proceeds via a complex mechanism.
This could be explained by considering two main hypotheses: (i) the disproportionation of DPPH in the presence of Fe3+ is more favorable compared to a potential classical oxidation reaction as observed in the presence of Cu2+.12 This hypothesis is supported by the high acidity of Fe3+;14,15 (ii) the oxidizing properties of Fe3+ are sufficient to promote the oxidation of the reduced DPPH (DPPH− is produced during the disproportionation) to DPPH (even if bound to Fe3+), according to eqn (4).
| [Fe(DPPH)n]3−n → DPPH + [Fe(DPPH)n−1]3−n | (4) |
Therefore, the reaction between Fe3+ and DPPH can be simplified by the balance of eqn (3) and (4) (eqn (5)).
| Fe3+ + (2n−1) DPPH → nDPPH+ + [Fe(DPPH)n−1]3−n | (5) |
In order to identify the reaction that better reflects these experimental observations, the theoretical variations of DPPH percentages in solution were calculated based on the stoichiometry of eqn (5), as a function of the concentrations of Fe3+ added during the measurements. The values of n explored were limited to 1, 2 and 3. Fig. 4 presents the expected variations of the theoretical DPPH percentages in solution as a function of the concentration of Fe3+ added.
 | ||
| Fig. 4 Variations of theoretical DPPH percentages as a function of the concentration of Fe3+ added. Experimental data (green empty stars) were obtained by following 0.1 mM DPPH absorbance at 519 nm at increasing Fe3+ concentrations in the range 0 mM to 0.1 mM. | ||
As expected, the decrease of the DPPH percentages becomes increasingly fast at higher values of n (slopes of −1000% mM−1, −3000% mM−1 and −5000% mM−1 when the values of n were 1, 2 and 3, respectively). For Fe3+ concentrations in the range 0 mM to 0.02 mM, the experimental data overlapped almost perfectly with the case of n = 2 (slope of −3051 ± 77% mM−1 instead of −3000% mM−1). This is evidence that the reaction between DPPH and Fe3+ in this domain proceeds mainly via disproportionation (4 moles of DPPH for 1 mole of Fe3+) (eqn (6)), followed by the oxidation of reduced DPPH (DPPH−) by Fe3+ (eqn (7)). The combination of these two equations yields the overall reaction eqn (8).
| 4DPPH + Fe3+ → 2DPPH+ + Fe(DPPH)2+ | (6) |
| Fe(DPPH)2+ → Fe(DPPH)+ + DPPH | (7) |
| 3DPPH + Fe3+ → 2DPPH+ + Fe(DPPH)+ | (8) |
For Fe3+ concentrations higher than 0.02 mM, there was a gradual decrease of the slope of the curve, reflecting a substantial modification of the reaction mechanism. This modification tends to increase the amount of Fe3+ needed to react with DPPH. This phenomenon can be rationalized by considering the oxidation of the compound Fe(DPPH)+ by Fe3+ according to eqn (9).
| Fe(DPPH)+ + Fe3+ → DPPH + 2Fe2+ | (9) |
Three hypotheses support this explanation:
(i) The reaction of eqn (9) regenerates DPPH, which would perfectly explain the decrease of the slope of the experimental curve;
(ii) This reaction would be slower than the disproportionation reaction because the DPPH− bound to Fe2+ necessarily becomes less reducing (the electron being involved in the dative bond with the metal). This also explains the over voltage associated with the oxidation of compound Fe(DPPH)+ during the electrochemical measurements (Fig. 2);
(iii) The reaction of eqn (9) occurred quantitatively when Fe(DPPH)+ was found in appreciable amounts in solution, as indicated by the shape of the curve of Fig. 3. Disproportionation thus accelerates DPPH consumption, while the redox reaction between Fe3+ and reduced DPPH slows down the entire process. By combining eqn (8) and (9) that summarize the antagonistic processes, the chemical reaction between these two compounds is finally a simple oxidation of DPPH to DPPH+ by Fe3+ (eqn (10)).
| DPPH + Fe3+ → DPPH+ + Fe2+ | (10) |
This equation is perfectly in agreement with the overall stoichiometry of the reaction corresponding to 1 mole of DPPH for 1 mole of Fe3+, as depicted in the inset of Fig. 1.
To confirm this mechanism, the presence of Fe2+ in the medium after reaction between Fe3+ and DPPH was checked using the 2,2′-bipyridine (BiPy) colorimetric test. Bipy can react with Fe2+ to form a dark red complex.19,20 An excess of BiPy was thus added to a mixture containing equimolar amounts of DPPH and Fe3+ (Fig. 5). The violet solution of 0.1 mM DPPH (Fig. 5(i)) became yellow after addition of 0.1 mM Fe3+, indicating the presence of DPPH+ (Fig. 5(ii)). The solution then turns from yellow to orange-red after adding an excess of BiPy (Fig. 5(iii)). This confirmed the formation of the complex between Fe2+ and BiPy.
 | ||
| Fig. 5 Images of (i) 0.1 mM DPPH, (ii) mixture 0.1 mM DPPH + 0.1 mM Fe3+ after 5 minutes, (iii) tube (ii) + 100 μL of 1 M 2,2′-bipyridine, (iv) 0.1 mM Fe3+ (v) tube (iv) + 100 μL of 1 M 2,2′-bipyridine, (vi) 0.1 mM Fe2+ and (vii) tube (vi) + 100 μL of 1 M 2,2′-bipyridine. | ||
Therefore, it is reasonable that the transformation presented in eqn (9) is the one ideally describing the reactivity of DPPH in the presence of Fe3+ in acetonitrile. However, it is the intermediate steps (disproportionation of DPPH and oxidation of DPPH− by Fe3+) that govern the rate of the reaction. In other words, during the reaction between DPPH and Fe3+, disproportionation ensures a fast transformation of DPPH, while DPPH− oxidation slows down the entire process by regenerating DPPH, as summarized in Scheme 1.
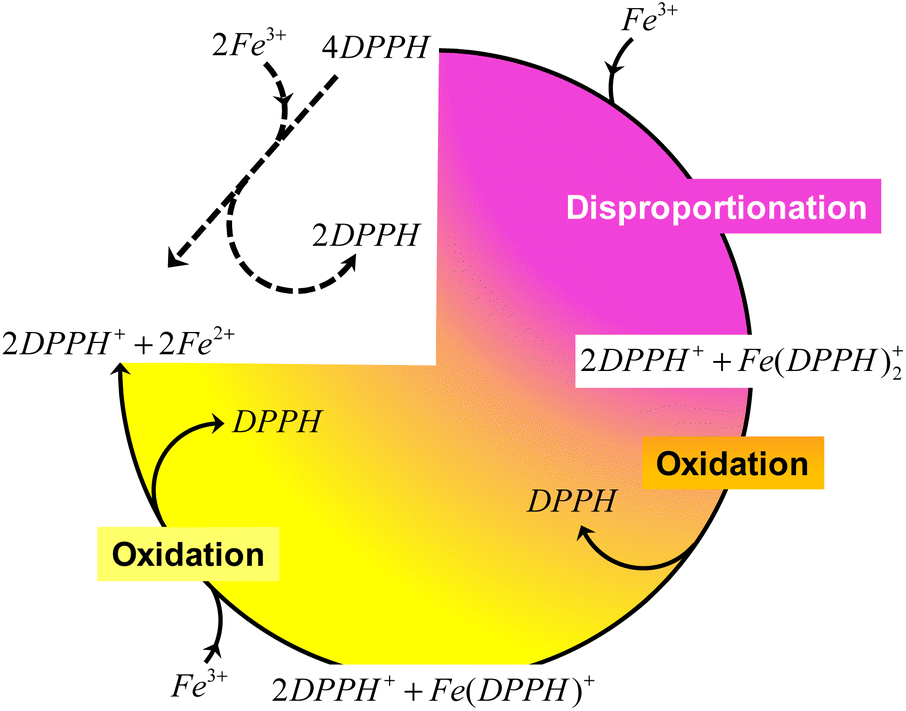 | ||
| Scheme 1 Suggested reaction mechanism between DPPH and Fe3+ in acetonitrile. | ||
It appears that under certain conditions, free radicals can be oxidized by metal cations such as Fe3+. In the case of the reactions involved during the antioxidant properties evaluation, Fe3+ could be mistakenly perceived as an antioxidant. Thus, antioxidant extracts containing Fe3+ would erroneously present greater antioxidant properties when determined in acetonitrile.
Conclusion
In the present work, the quantitative and fast reaction between the radical DPPH and Fe3+ ions in acetonitrile was reported. Regarding the antioxidant DPPH spectrophotometric test, Fe3+ behaves like a powerful antioxidant in acetonitrile, as was recently reported for Cu2+ and acids.12 A mechanism elucidating the reaction between DPPH and Fe3+ was proposed. This reaction follows a complex mechanism involving two competing phenomena: a disproportionation reaction accelerates the reaction but is then slowed down by an oxidation process which regenerates DPPH. Additional investigations with other metal cations in order to verify if such a reaction is specific to Fe3+ should be considered. The effect of the analytical solvent also represents another interesting investigation to be conducted. It is indeed known that the solvation effects of the solvent strongly affect the reactivity of chemical species, especially metal cations which can be easily solvated.Author contributions
Arlette Danelle Djitieu Deutchoua, conceptualization, methodology, data curation, investigation, validation, writing – original draft, writing – review & editing; Yannick Ngueumaleu, data curation, formal analysis, investigation; Sarrah Sonita Poungoue Hanga, formal analysis, investigation; Liendji Wendji Rossel, formal analysis, investigation; Bruno Boniface Nguelo, formal analysis; Gustave Kenne Dedzo, conceptualization, methodology, supervision, data curation, investigation, validation, writing – original draft, writing – review & editing; Emmanuel Ngameni, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the International Science Program (ISP) – Sweden through funding provided to the African Network of Electroanalytical Chemists (ANEC).Notes and references
- M. C. Foti, J. Agric. Food Chem., 2015, 63, 8765 CrossRef CAS.
- N. D. Yordanov and A. Christova, Appl. Magn. Reson., 1994, 6, 341 CrossRef CAS.
- K. Suzuki, T. Watanabe and S. Murahashi, Angew. Chem., 2008, 47, 2079 CrossRef CAS.
- B. Wen-Long, Z. Zhen, C. Xin, W. Xiao, Z. Qiang, X. Zhi-Xin, L. Yu-Si, C. Bao-bao, W. Kai-Xue and C. Jie-Sheng, Energy Storage Mater., 2020, 31, 373 CrossRef.
- I. Nakanishi, S. Yamashita, T. Shimokawa, M. Kamibayashi, E. Sekine-Suzuki, M. Ueno, Y. Ogawa, T. Ozawa and K. Matsumoto, Org. Biomol. Chem., 2018, 16, 1272 RSC.
- V. Sindhi, V. Gupta, K. Sharma, S. Bhatnagar, R. Kumari and N. Dhaka, J. Pharm. Res., 2013, 7, 828 CAS.
- A. M. Pisoschi and A. Pop, Eur. J. Med. Chem., 2015, 97, 55 CrossRef CAS.
- R. Amorati, S. Menichetti, C. Viglianisi and M. C. Foti, Chem. Commun., 2012, 48, 11904 RSC.
- M. S. Blois, Nature, 1958, 181, 1199 CrossRef CAS.
- A. L. Dawidowicz, D. Wianowska and M. Olszowy, Food Chem., 2012, 131, 1037 CrossRef CAS.
- B. Ozcelik, J. H. Lee and D. B. Min, J. Food Sci., 2003, 68, 487 CrossRef.
- Y. Ngueumaleu, A. D. D. Deutchoua, S. S. P. Hanga, R. W. Liendji, G. K. Dedzo and E. Ngameni, Phys. Chem. Chem. Phys., 2023, 25, 5282 RSC.
- I. Nakanishi, T. Kawashima, K. Ohkubo, T. Waki, Y. Uto, T. Kamada, T. Ozawa, K. Matsumoto and S. Fukuzumi, Chem. Commun., 2014, 50, 814 RSC.
- F. C. Hawthorne, Phys. Chem. Miner., 2012, 39, 841 CrossRef CAS.
- I. D. Brown, Acta Crystallogr., 1988, 44, 545–553 CrossRef.
- A. D. D. Deutchoua, R. Siegnin, G. K. Kounaitze, G. K. Dedzo and E. Ngameni, ChemistrySelect, 2019, 4, 13746 CrossRef CAS.
- E. Solon and A. J. Bard, J. Am. Chem. Soc., 1964, 86, 1926 CrossRef CAS.
- A. W. Taylor, S. Puttick and P. Licence, J. Am. Chem. Soc., 2012, 134, 15636 CrossRef CAS PubMed.
- G. Xu, N. Li, Y. Sun, C. Gao, L. Ma, P. Song and L. Xia, Chem. Eng. J., 2021, 414, 128741 CrossRef CAS.
- M. K. Bera, T. Mori, T. Yoshida, K. Ariga and M. Higuchi, ACS Appl. Mater. Interfaces, 2019, 11, 11893 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section; Fig. S1, UV-Vis spectra of DPPH solution in presence of controlled amounts of copper perchlorate and Fig. S2, cyclic voltammograms of DPPH in presence of controlled amounts of iron(III) perchlorate. See DOI: https://doi.org/10.1039/d3ra07106e |
| This journal is © The Royal Society of Chemistry 2024 |